
Abstract
Introduction
Innovation
This is the first description that acute hyperoxia induces mitochondrial matrix ROS prior to cytosolic ROS. Our data suggest that redox signals within one subcellular compartment can affect signaling within another subcellular compartment, and provide evidence that even a few minutes of hyperoxia exposure can have a significant impact on the cell signaling milieu. These results are clinically relevant because positive-pressure ventilation with oxygen remains widely used for delivery room resuscitation of infants worldwide, and oxygen is one of the most widely used therapies in all of medicine.
In utero, pulmonary arteries (PA) are constricted, allowing only 10% of the cardiac output to circulate through the fetal pulmonary vascular bed. Multiple pathways contribute to the regulation of pulmonary vascular tone in the perinatal period. In particular, increases in endothelial nitric oxide synthase (eNOS) expression, endogenous nitric oxide (NO) production, and soluble guanylate cyclase (sGC) expression at birth contribute to increased cyclic GMP (cGMP) and relaxation of pulmonary vascular tone (2, 3, 7, 8). Phosphodiesterase 5 (PDE5) down-regulates endogenous NO signaling by hydrolyzing cGMP. PDE5 expression normally decreases after birth, further contributing to an increase in cGMP (6, 12). Previous studies demonstrate that ventilation of newborn sheep with persistent pulmonary hypertension of the newborn (PPHN) with 100% O2 for 24 h increases contractile responses to norepinephrine, but these changes can be prevented by pretreatment with a single dose of an antioxidant, recombinant human superoxide dismutase (rhSOD) or catalase (18, 37). Additionally, studies in healthy newborn lambs demonstrate that 30 min of resuscitation with 100% O2 attenuates the vasorelaxation response to exogenous NO, suggesting that the NO-sGC-cGMP-PDE5 signaling pathway is impaired (17). We reported that exposure of primary FPASMC to 95% O2 for 24 h decreases cGMP responsiveness to exogenous NO and increases PDE5 activity and expression (6). Similarly, ventilation of newborn sheep with 100% O2 for 24 h increases PDE5 activity and expression and decreases cGMP in resistance PA (6, 7). However, we do not understand the mechanism by which hyperoxia regulates PDE5 in the PASMC, and there is no information about the critical threshold or duration of O2 exposure necessary to activate PDE5 and inhibit cGMP signaling. The present study sought to determine the mechanism by which hyperoxia increases PDE5 activity and decreases the cGMP response to exogenous NO in FPASMC.
Results
Brief hyperoxia exposure increases FPASMC PDE5 activity
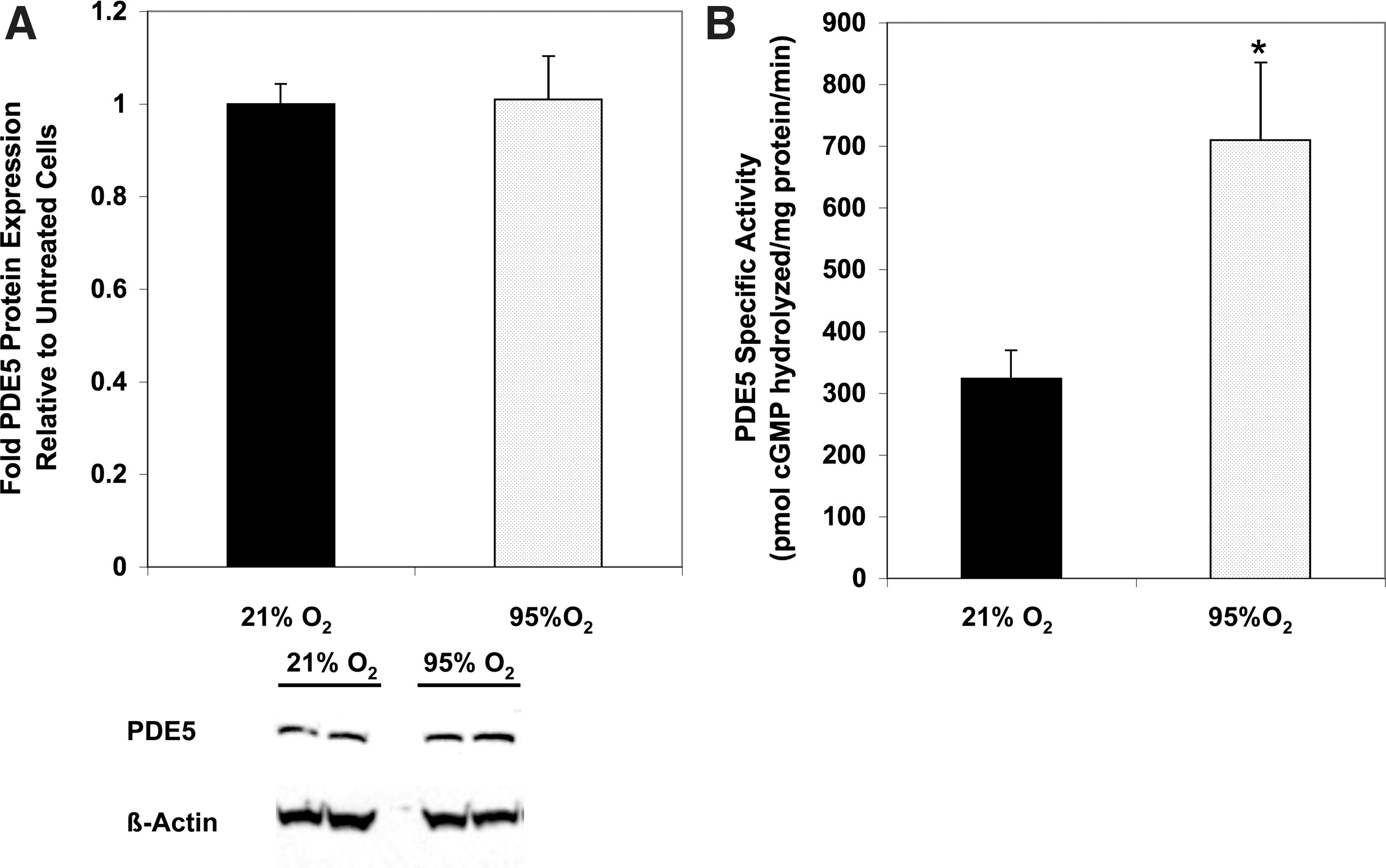
We previously demonstrated that 24 h of 95%–100% O2 increases PDE5 protein expression and activity in isolated FPASMC and resistance PA from mechanically ventilated neonatal sheep (6). However, the effects of shorter O2 durations on PDE5 were not evaluated. Figure 1 illustrates that 95% O2/5% CO2 for 30 min does not affect PDE5 protein expression, but does increase PDE5 activity relative to normoxic (21% O2/5% CO2) FPASMC within 20 min after exposure (Fig. 1 and Supplementary Fig. S1A; Supplementary Data are available online at
Brief hyperoxia decreases cGMP response to exogenous NO
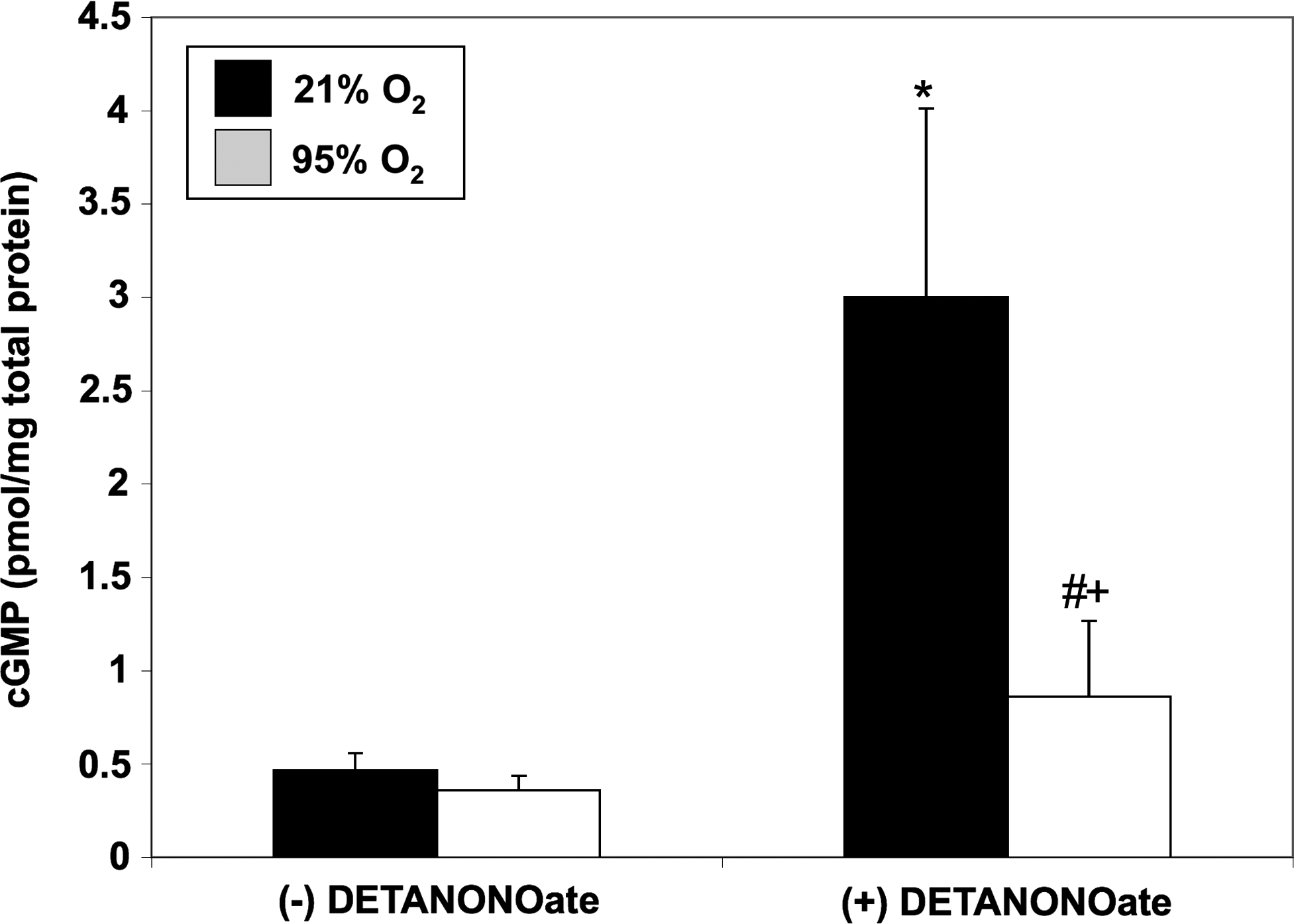
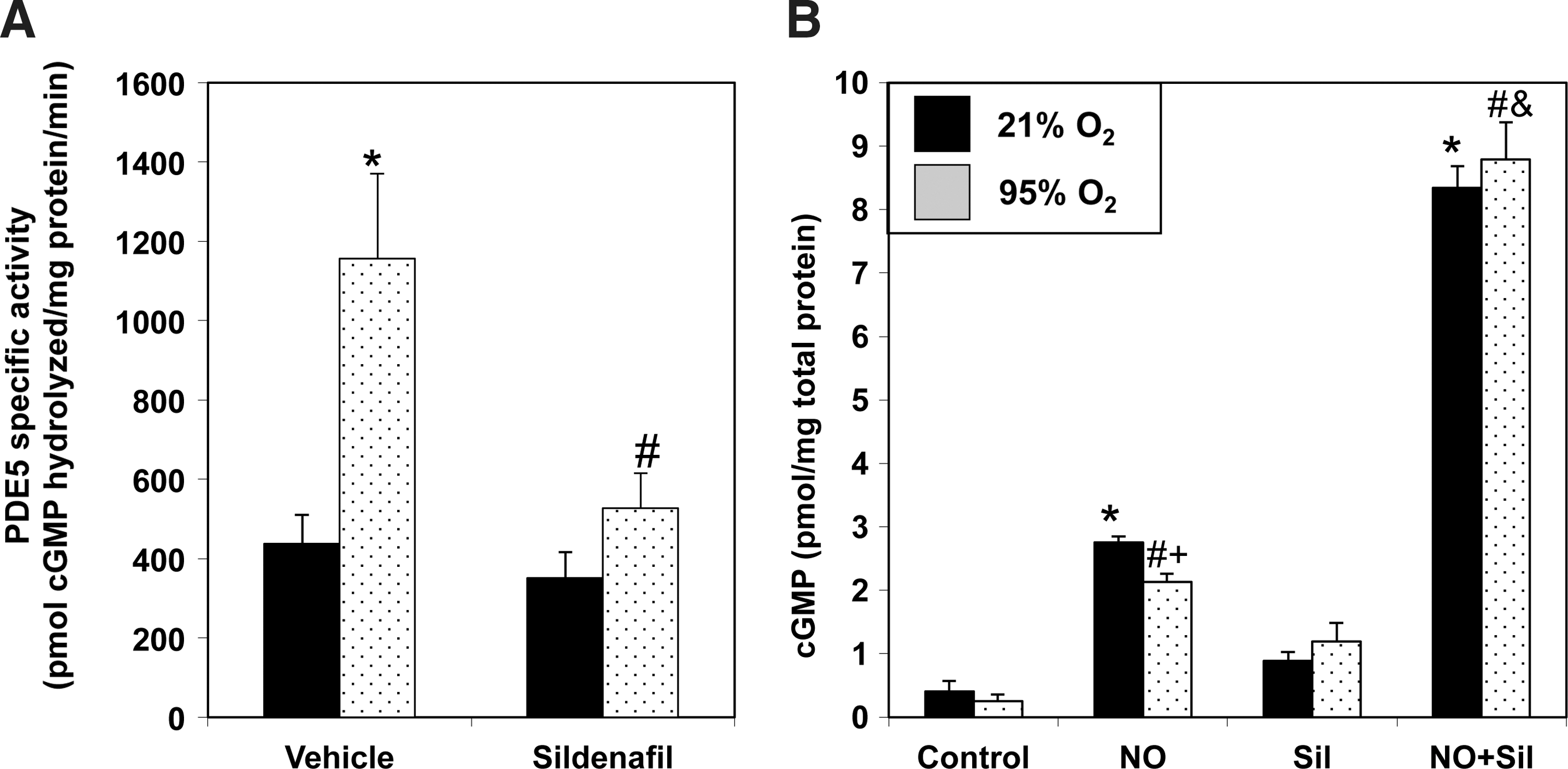
PDE5 hydrolyzes cGMP and down-regulates NO signaling. We sought to determine if hyperoxia-mediated PDE5 induction would attenuate the cGMP response to an exogenous NO stimulus in FPASMC. After 30 min of exposure to 95% O2/5% CO2, the cGMP response to the NO donor, DETANONOate, was significantly blunted as compared to normoxic FPASMC (Fig. 2). Exposure to 95% O2/5% CO2 for 4 h similarly blunted the cGMP response to DETANONOate (Supplementary Fig. S3). The cGMP response to NO depends on sGC and PDE5 activities. Thus, we sought to determine if hyperoxia-induced PDE5 activity was solely responsible for this blunted cGMP response to NO. Pretreatment with sildenafil (100 nM) significantly decreased PDE5 activity in hyperoxia-exposed FPASMC relative to untreated FPASMC (Fig. 3A) and restored normal cGMP response to exogenous NO (Fig. 3B). These findings indicate that a significant component of the attenuated NO response after brief hyperoxia is due to increased PDE5 activity. Of note, sildenafil augments the cGMP response to exogenous NO in normoxia as well, likely due to the fact that PDE5 largely regulates the NO-stimulated cGMP pool, not basal cGMP (6).
Brief hyperoxia increases mitochondrial, but not cytosolic oxidation
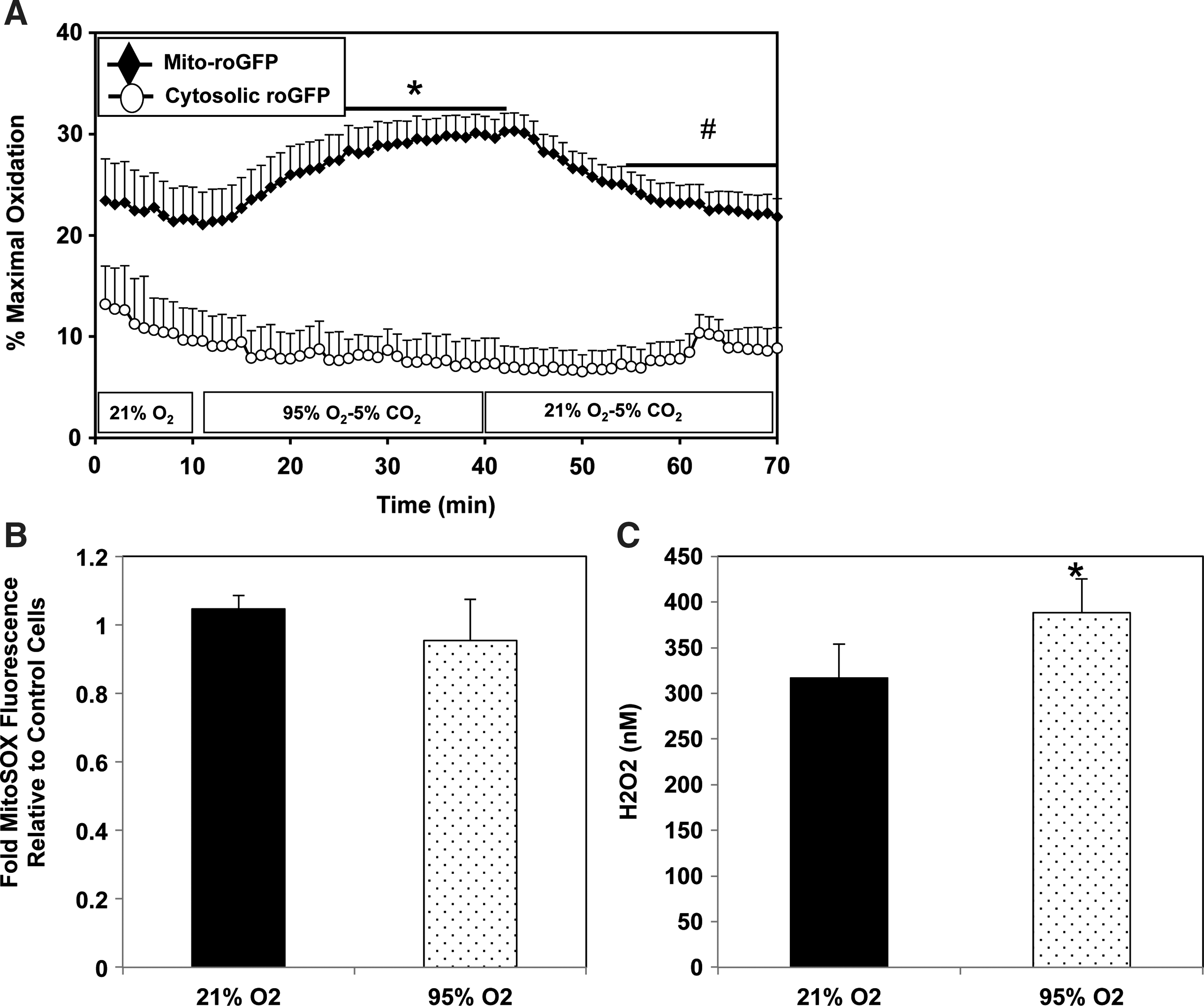
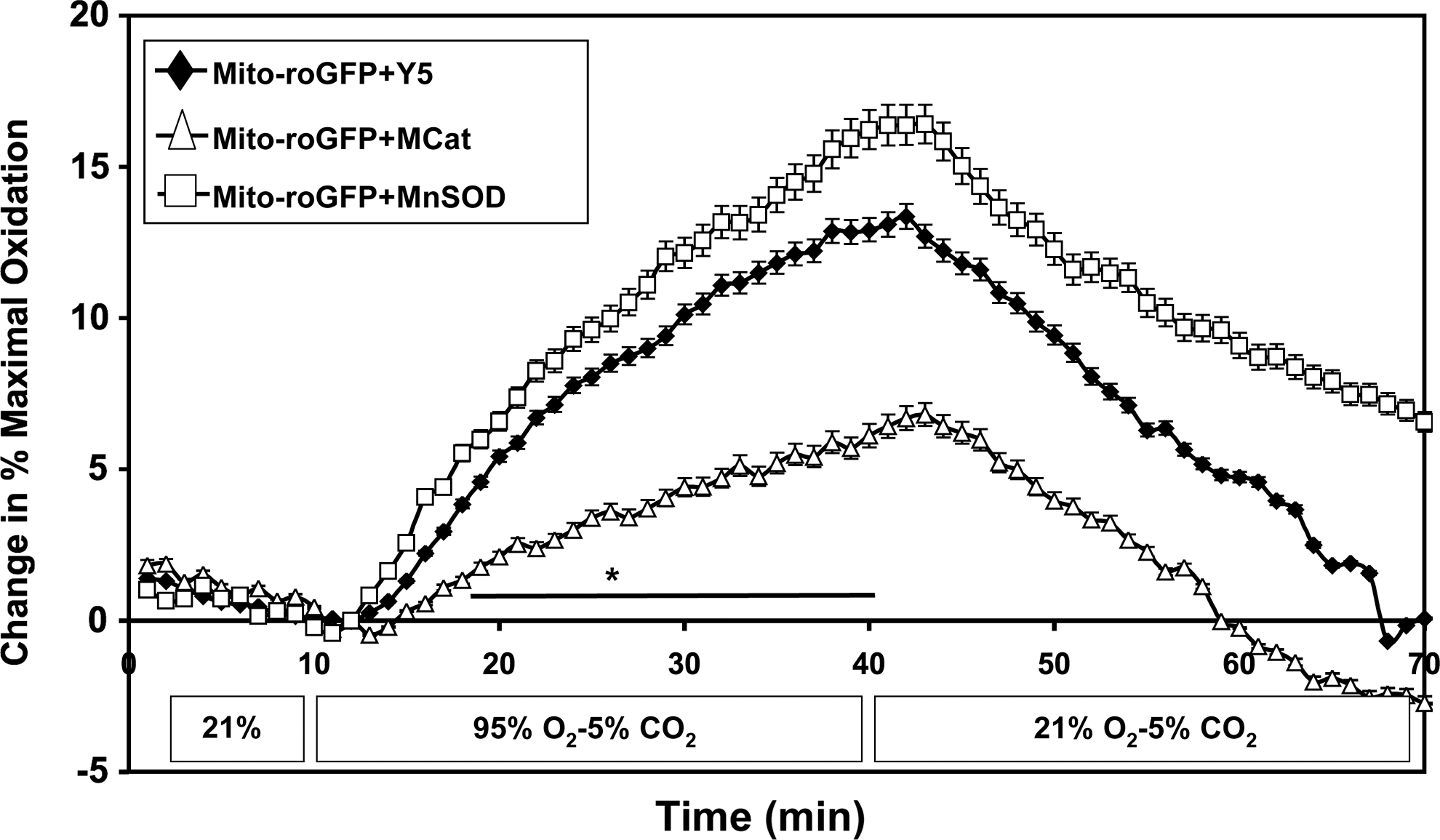
We previously demonstrated that FPASMC exposure to 95% O2/5% CO2 for 24 h increases reactive oxygen species (ROS) in the cytosol and the mitochondrial matrix (6, 9). We sought to determine the temporal relationship between the increase in ROS in one compartment and the other. Oxidation within subcellular compartments was assessed using the ratiometric thiol sensor, roGFP. Under normoxic conditions, the mitochondrial matrix exhibited a more oxidized state compared with the cytosol (Fig. 4A). During exposure to 95% O2/5% CO2, no change in the cytosolic oxidative state was detected within 30 min. In contrast, oxidation within the mitochondrial matrix increased significantly within 15 min, reaching maximal oxidation within 30 min. Upon return to 21% O2/5% CO2, matrix oxidation decreased progressively, reaching baseline levels within 16 min. Similarly, no change in cytosolic oxidative state was observed after 4 h under 95% O2/5% CO2, whereas the mitochondrial matrix oxidant status increased compared with normoxia (Supplementary Fig. S4). We sought to determine whether increased mitochondrial oxidation was due to superoxide or H2O2. We demonstrated no change in fluorescence of MitoSOX, a mitochondrially targeted superoxide sensor (Fig. 4B). We did detect increased intracellular H2O2 by Amplex Red fluorescence (Fig. 4C) but could not localize the source to any specific subcellular compartment.
To determine if overexpression of antioxidant enzymes would abrogate this hyperoxia-induced mitochondrial oxidative response, we overexpressed MnSOD and mito-catalase using recombinant adenoviruses (Supplementary Figs. S5 and S6) (35). Interestingly, MnSOD overexpression did not abrogate, but instead enhanced the hyperoxia-induced oxidation of mito-roGFP (Fig. 5). However, mito-catalase expression significantly blunted the response, as compared to cells treated with empty adenovirus (Fig. 5).
MitoTEMPO and mito-catalase block hyperoxia-induced increases in PDE5 activity
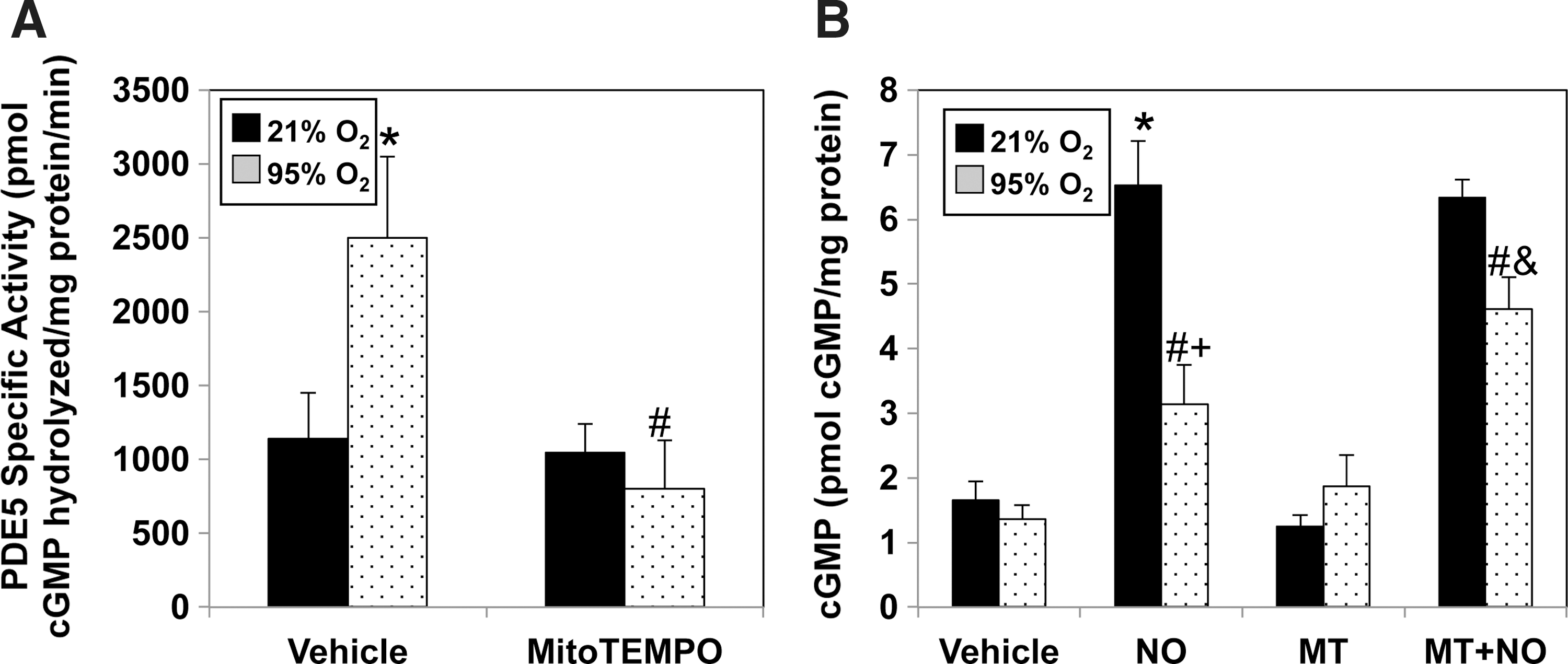
To determine if mitochondrial oxidation directly increased PDE5 activity, we utilized the mitochondrially targeted antioxidant, MitoTEMPO, which scavenges mitochondrial superoxide and decreases mitochondrial H2O2 (5). Pretreatment with MitoTEMPO significantly blunted the hyperoxia-mediated increase in PDE5 activity (Fig. 6A) and significantly improved cGMP accumulation to exogenous NO in comparison to vehicle-treated FPASMC (Fig. 6B).
We next determined if overexpression of antioxidant enzymes would block hyperoxia-induced increases in PDE5 activity and restore cGMP responsiveness to exogenous NO. MnSOD overexpression did not abolish hyperoxia-induced increases in PDE5 activity (Fig. 7A), nor did it improve cGMP response to NO in hyperoxia-exposed FPASMC (Fig. 7B). Surprisingly, MnSOD overexpression in normoxic FPASMC led to a small but significant decrease in the cGMP response to exogenous NO (Fig. 7B). However, mito-catalase overexpression in FPASMC in 95% O2/5% CO2 blocked the increase in PDE5 activity compared to FPASMC infected with empty virus (Fig. 7C), and improved cGMP response to exogenous NO (Fig. 7D).

Hyperoxia augments PDE5 activity in a cGMP-dependent protein kinase I alpha–dependent manner
Cyclic GMP-dependent protein kinase (PKG)-mediated phosphorylation has been reported to increase PDE5 activity, and we have shown that 24 h of hyperoxia increases PDE5 phosphorylation (6, 25). In the present study, we demonstrate increased PDE5 phosphorylation at the previously characterized PKG phosphorylation site after 30 min of hyperoxia (Fig. 8A). DT-3, a peptide inhibitor of cGMP-dependent protein kinase I alpha (PKGIα), significantly blunted the hyperoxia-mediated increase in PDE5 activity (Fig. 8B). Likewise, DT-3 significantly improved the cGMP response to exogenous NO in hyperoxia-exposed FPASMC in comparison to vehicle-treated FPASMC (Fig. 8C).

Hyperoxia increases PDE5 activity in small PA in adult mice
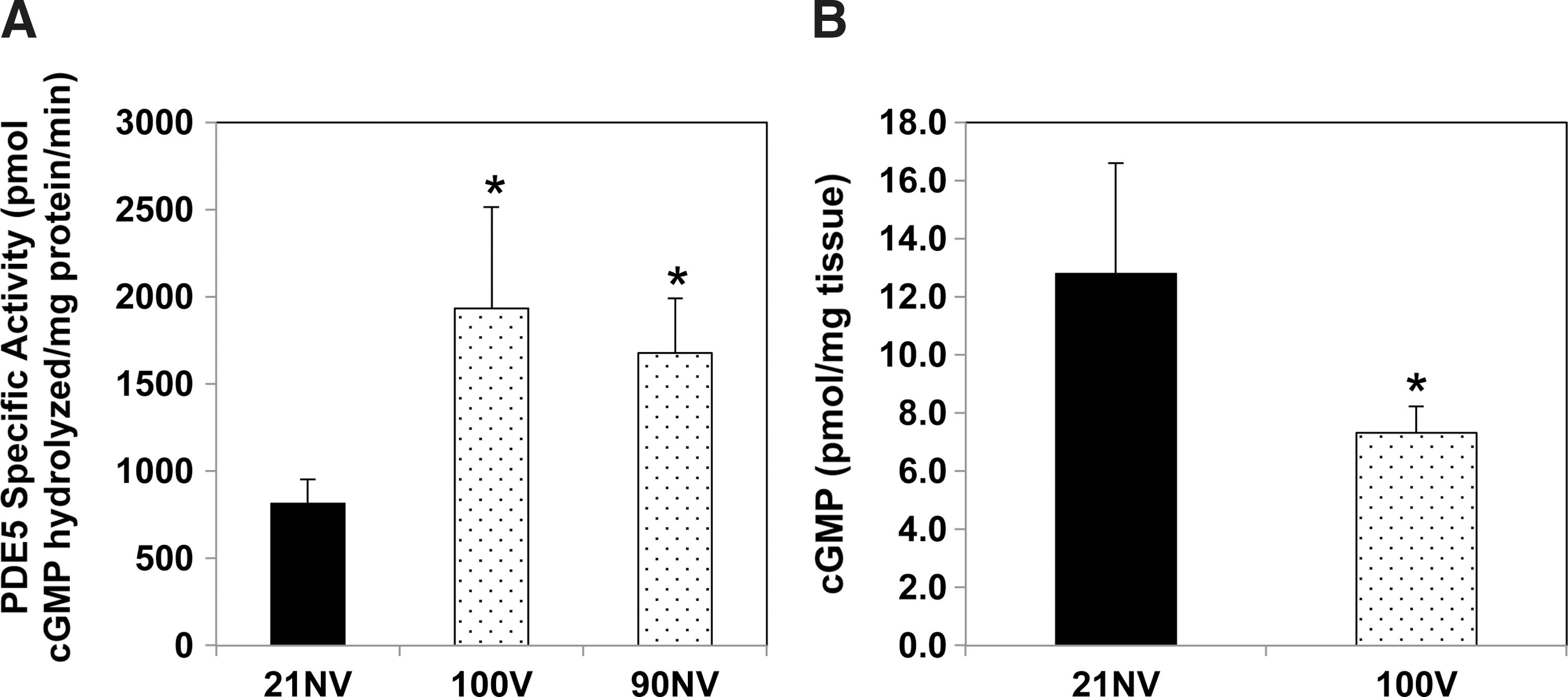
To determine the relevance of our findings in a whole animal model, we exposed C57Bl6 young adult male mice to hyperoxia both with and without mechanical ventilation. Both mechanical ventilation with 100% O2 and exposure to 90% O2 without mechanical ventilation for 45 min led to a similar increase in small PA PDE5 activity (Fig. 9A), suggesting that hyperoxia, not mechanical ventilation, is directly responsible for increased PA PDE5 activity. While isolated PA consist of multiple cell types, we also confirmed that hyperoxia exposure for 30 min increased PDE5 activity in isolated adult mouse PASMC (Supplementary Fig. S7). Similarly, mice that were mechanically ventilated with 100% O2 had decreased total lung cGMP relative to room air nonventilated controls (Fig. 9B).
Discussion
Using a ratiometric sensor to assess thiol protein oxidation status in subcellular compartments of FPASMC, we find that hyperoxia induces a selective increase in mitochondrial matrix oxidation within 15 min; this response is attenuated by catalase overexpression targeted to the same compartment. Additional studies with Amplex Red demonstrate an increase in intracellular H2O2 with hyperoxia exposure, but these studies cannot localize the H2O2 within a specific subcellular compartment. Taken together, these findings indicate that hyperoxia acutely increases the generation of H2O2 in the mitochondrial matrix compartment. Over a similar time interval, no change in the cytosolic oxidation state was observed, suggesting that the matrix is the compartment where oxidants are first generated in response to hyperoxia. This finding contrasts recent reports utilizing similar roGFP tools showing that acute hypoxia increases cytosolic but not mitochondrial oxidant status in rat PASMC (36). Collectively, these findings indicate that hyperoxia and hypoxia generate ROS by distinct mechanisms in subcellular compartments, which explains why they evoke different signaling responses in the pulmonary circulation.
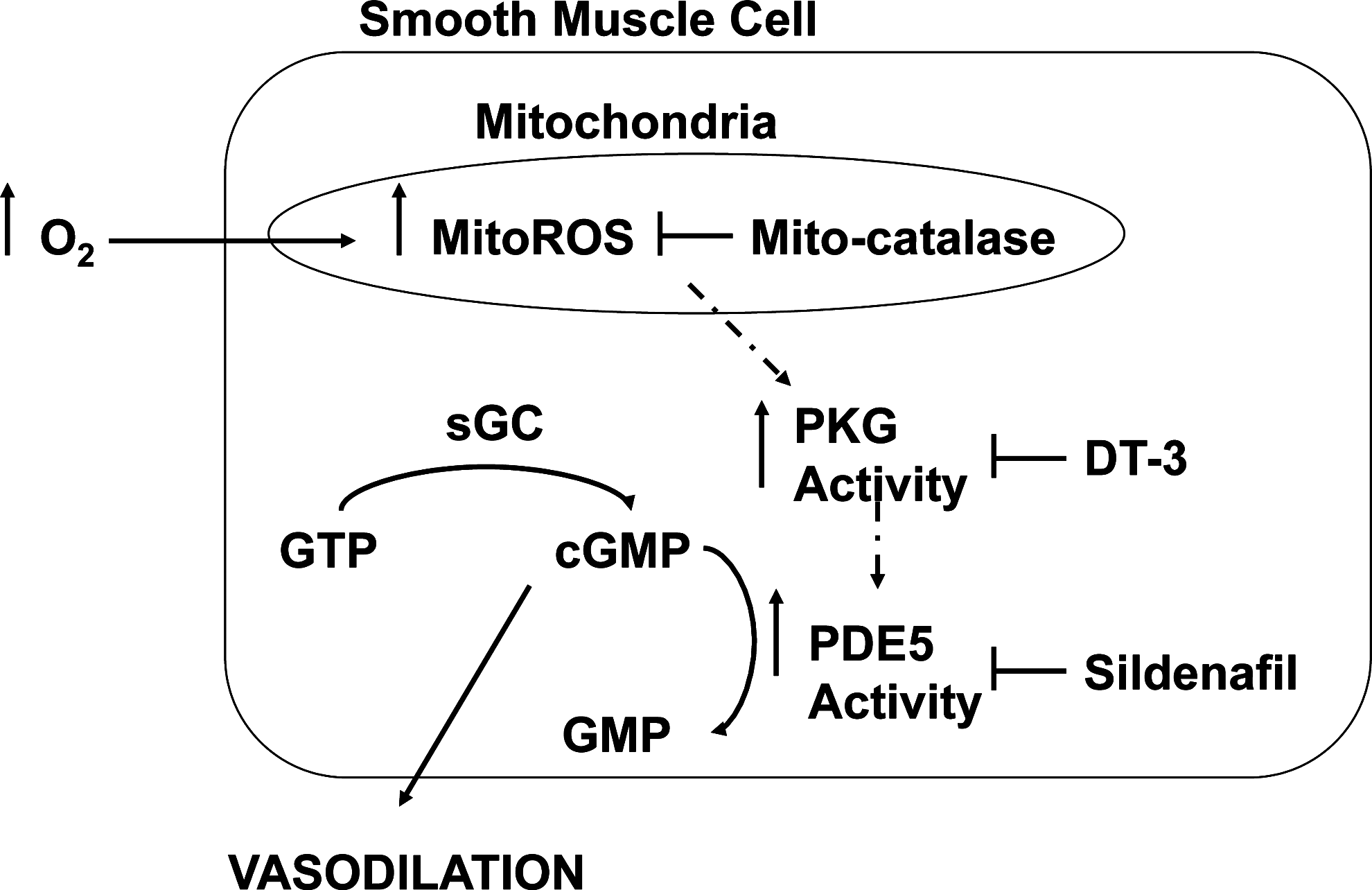
The present study also shows that supraphysiologic oxygen increases PDE5 activity in as little as 20 min. Hyperoxia also decreases the cGMP response to exogenous NO, which can be prevented by sildenafil, a PDE5-specific inhibitor. Thus, the hyperoxia-mediated decrease in cGMP response to exogenous NO is due to increased PDE5 activity. Additionally, sildenafil may further impact cGMP response to exogenous NO by inhibiting PDE1, which is important in adult pulmonary hypertension, or by decreasing ROS through other mechanisms (10, 13, 29, 31). A mitochondrially targeted antioxidant, MitoTEMPO, attenuated the hyperoxia-induced increase in PDE5 activity and restored cGMP responsiveness to NO, but MitoTEMPO can decrease both superoxide and H2O2 in mitochondria, and thus does not identify a specific oxidant (5). Mitochondrial expression of catalase, but not MnSOD, attenuated hyperoxia-induced PDE5 activity and restored cGMP responsiveness to NO, implicating H2O2 as the critical oxidant for impacting PDE5. Finally, DT-3, a PKGIα inhibitor, prevented the hyperoxia-induced increase in PDE5 activity and restored cGMP responsiveness to NO. Thus, mitochondrial matrix ROS, specifically H2O2, is responsible for the induction of PDE5 activity through a PKGIα -dependent mechanism (Fig. 10).
The increase in NO and cGMP and decrease in PDE5 activity are important for the normal pulmonary vascular transition after birth (2, 6, 12). In healthy near-term sheep as well as sheep with PPHN, mechanical ventilation with 100% O2 for 24 h increased ROS, increased PDE5 protein expression and activity, and decreased steady-state cGMP levels in the resistance PA (6 –8). In PPHN lambs, treatment with an antioxidant, either rhSOD or catalase, significantly decreased PDE5 activity and increased cGMP levels in the resistance PA suggesting that these changes were oxidant-mediated (7, 9). In isolated FPASMC, exposure to 95% O2 for 24 h decreased cGMP responsiveness to exogenous NO and increased PDE5 expression and activity. These changes were rescued by pretreatment with the chemical antioxidant N-acetyl-cysteine, suggesting that oxygen-mediated regulation of PDE5 is ROS dependent (6). While our previous data suggested that both cytosolic and mitochondrial ROS were increased after 24 h of hyperoxia, we recently demonstrated that pretreatment of FPASMC with mitochondrially targeted catalase (mito-catalase) blocked PDE5 activity induced by 24 h of 95% O2, suggesting that hyperoxia-induced changes in PDE5 activity were due to mitochondrial ROS (9). The present study suggests that the increase in mitochondrial H2O2 precedes changes in cytosolic ROS and that mitochondrial H2O2 is the key trigger of elevated PDE5 activity.
When animals or cells are exposed to prolonged hyperoxia, responses may involve post-translational modifications to proteins and changes in gene expression. However, many infants are only exposed to hyperoxic gas mixtures briefly, during delivery room resuscitation or due to mild respiratory distress after birth. While O2 is a well-known pulmonary vasodilator, recent animal data indicate that even brief exposure to 100% O2 may adversely affect pulmonary vascular tone, while activating markers of oxidative stress (16, 17, 32). In human studies, delivery room resuscitation with 100% O2 results in lower Apgar scores and a longer time to first cry compared with infants resuscitated with 21% O2 (27, 28). Additionally, 28 days later, infants resuscitated with 100% O2 show increased oxidative stress markers, including low reduced-to-oxidized glutathione ratios (32). Further, in healthy and PPHN lambs, 30 min of resuscitation with 100% O2 suppressed pulmonary vascular relaxations to NO, suggesting impairment of NO-sGC-cGMP-PDE5 signaling (17, 19).
In the present study, we find that exposure to as little as 20 min of 95% O2 is sufficient to induce PDE5 activity in FPASMC. These hyperoxic effects are dose dependent, such that exposure to greater than 50% O2 was required to induce PDE5 activity (Supplementary Fig. S1). The induction of PDE5 activity mirrors the increase in mitochondrial ROS, and selective targeting of mitochondrial ROS abolishes the PDE5 response. These results suggest that hyperoxia-induced mitochondrial H2O2 triggers the increase in PDE5 activity, and it is likely that the intermediate signaling event is rapid and induces post-translational modification of PDE5 by activating PKG. It was previously shown that PDE5 expression and activity can be regulated in an ROS-dependent manner, but after exposures of 16 h to 2 weeks (6, 20, 21, 23). Studies of PDE5 activity in gastric and aortic smooth muscle indicate that PKG phosphorylates and activates PDE5 (22, 25). Indeed, both hyperoxia for 24 h and 30 min increases PDE5 phosphorylation at the previously characterized PKG phosphorylation site (Fig. 8A) (6). There is increasing evidence that PKG is regulated by cellular redox status, making it a likely candidate as the intermediary between ROS and PDE5 (4, 26, 30). While oxidant stress can potentially activate PKG by oxidizing cysteine residues in zinc-finger domains of the protein, the mechanism of PKG activation in the present study is perplexing because the oxidant stress occurred in the matrix but was not observed in the cytosol where the PKG acts on PDE5. One possible explanation is that PKG resides on the endoplasmic reticulum membrane near mitochondria, which could allow ROS leaking from the matrix to activate PKG locally in that domain, before the entire cytosolic compartment becomes oxidized (33, 39). An alternative possibility is that an intermediary signaling molecule translocates from the mitochondria to the cytosol to activate PKG.
Interestingly, MnSOD overexpression failed to abrogate the hyperoxia-induced mitochondrial roGFP response. Because roGFP is oxidized by H2O2 and MnSOD decreases the lifetime of superoxide but may indirectly increase the level of H2O2 present in the matrix, the effect on the roGFP response is not surprising. However, MnSOD also failed to abolish the activation of PDE5 in hyperoxia, indicating that mitochondrial matrix superoxide is not a critical intermediary. Interestingly, MnSOD overexpression decreased the cGMP response to NO, suggesting that it can contribute to the downstream effects of increased mitochondrial H2O2. Overexpression of mito-catalase blocked roGFP oxidation and PDE5 activation, indicating that H2O2 is required for both responses. Finally, the nonspecific mitochondrially targeted antioxidant MitoTEMPO also blocked hyperoxia-induced PDE5 activity. Thus, our data are consistent with previous studies showing that exogenous H2O2 is capable of activating PDE5 in FPASMC (6). Regardless of the source, H2O2 can inhibit eNOS (15), down-regulate sGC (38), and stimulate PDE5 (6) in the pulmonary vasculature.
We also demonstrate that brief exposure to hyperoxia increases PDE5 in small PA from adult mice. These in vivo data prove that our findings are relevant across species and developmental stages. Thus, while our research has been previously focused on the neonate, these new data may suggest that hyperoxic exposures may have detrimental effects on adults as well.
In conclusion, we propose a model in which hyperoxia induces mitochondrial matrix generation of H2O2, which diffuses from the mitochondria to rapidly activate PKGIα which resides in close proximity to the mitochondria. PKGIα phosphorylates and increases PDE5 activity, which in turn blunts the cGMP response to exogenous NO (Fig. 10). In view of the potential toxicity associated with hyperoxic therapy, the newest guidelines for neonatal resuscitation now suggest that oxygen be titrated to meet the patient's needs (14). However, it is not yet known whether these new guidelines will avoid the cellular dysfunction associated with hyperoxia, given that these events are triggered by such brief hyperoxic exposures.
Materials and Methods
Cell culture
Primary PASMC cultures were prepared from intrapulmonary arteries isolated from healthy 136-day gestation fetal lambs and maintained in culture as described in the online Supplementary Data (6). PASMC were exposed to hyperoxia in a sealed humidified chamber in 95% O2/5% CO2 (ProOx C-21, Biospherix, Lacona, NY) for 30 min followed by 30 min of recovery at 21% O2/5% CO2. Some cells were treated with DETANONOate (100 μM; Cayman Chemical, Ann Arbor, MI), sildenafil (100 nM; provided by Drs. Francis and Corbin), MitoTEMPO (25 nM; Enzo Life Sciences, Plymouth Meeting, PA), and/or DT-3 (15 μM, Calbiochem, Philadelphia, PA). Other cells were infected with an adenoviral construct expressing MnSOD (100 plaque-forming units [pfu]/cell; ViraQuest, North Liberty, IA) or mito-catalase (750 pfu/cell; University of Iowa, Iowa City, IA) or empty adenoviral vector, Y5 (100 or 750 pfu/cell; University of Iowa).
Hyperoxia exposure of mice and isolation of lungs and small PA
The study was approved by the Northwestern University Laboratory Animal Care Committee. Male C57Bl6 mice (Charles River, age 5–6 weeks) were placed into three experimental groups: 1) room air, nonventilated (21NV, n=5), 2) mechanically ventilation with 100% O2 and a TOPO Dual Mode Ventilator (Kent Scientific, Torrington, CT) for 45 min (100V, n=5), and 3) 90% O2 exposure for 45 min in an animal hyperoxia chamber (Biospherix; 90NV, n=5). After exposure, animals were euthanized, and lung tissue or PA protein was isolated as described in the online Supplementary Data (34).
Western blot analysis
FPASMC protein was harvested, and Western blot was performed as described in the online Supplementary Data (6). Membranes were blocked and incubated overnight with primary antibody at an appropriate dilution (1:333 for mouse anti-PDE5 [BD Transduction], 1:500 for rabbit anti-PhosphoPDE5 [Fabgennix, Frisco, TX], and 1:2000 for mouse β-actin [Sigma, St. Louis, MO]). The membranes were incubated with the appropriate secondary antibody conjugated to horseradish peroxidase (Pierce, Rockford, IL). Membranes were exposed via chemiluminescence (Pierce) using a Digital Science Image Station (Kodak, Rochester, NY). PDE5 expression was normalized to β-actin. Data are shown as fold relative to untreated control PASMC.
PDE5 activity assay
FPASMC and small PA were harvested for total protein and assayed the same day as described in the online Supplementary Data using a commercially available colorimetric cyclic nucleotide phosphodiesterase assay kit (Enzo Life Sciences) (6, 7). Each sample was read with or without sildenafil (100 nM). The difference between the picomoles of cGMP hydrolyzed per milligrams of total protein per minute with or without sildenafil represents the PDE5-specific cGMP-hydrolytic activity.
cGMP enzyme immunoassay
FPASMC and lung cGMP content was measured by enzyme immunoassay in duplicate using a commercially available kit (Cayman Chemical) as previously described (6, 7). Results are shown as picomoles of cGMP per milligram of protein for FPASMC samples and picomoles of cGMP per milligram of tissue for lung samples.
Detection of ROS
FPASMC were plated and exposed to 95% O2/5% CO2 for 30 min. Immediately after exposure, FPASMC were lysed, and intracellular H2O2 was measured via Amplex Red assay as described in the online Supplementary Data (Invitrogen, Grand Island, NY). Other FPASMC were loaded with MitoSOX (Invitrogen), a fluorescent mitochondrially targeted superoxide probe, prior to hyperoxia exposure. MitoSOX fluorescence was measured by fluorescence microscopy with Metamorph imaging software (Molecular Devices, Sunnyvale, CA) as described in the online Supplementary Data.
FPASMC were plated on collagen-coated coverslips and infected with 100 pfu/cell of cytosolic roGFP or mitochondrial matrix–targeted roGFP (mito-roGFP) adenoviral construct. RoGFP is a previously characterized ratiometric fluorescent probe with surface-exposed cysteine residues capable of forming disulfide bonds. Assessment of fluorescence ratios provides real-time measurements of cysteine thiol redox status in that subcellular compartment (6, 9, 11, 36). After infection (48 h), coverslips were mounted in a flow-through chamber and perfused with a balanced salt solution bubbled with 21% O2/5% CO2 for fluorescent imaging on an epifluorescence inverted microscope (Nikon, Japan) with a cooled CCD camera (Photometrics, Tucson, AZ) connected to a computer running Metafluor (Universal Imaging, Downington, PA) as previously described (36). The roGFP probe was excited using 400 and 485 nm, and emission at 525 nm was imaged. After establishing a stable baseline, the gas perfusion mixture was changed to 95% O2/5% CO2 for 30 min. After 30 min, the cells were returned to 21% O2/5% CO2 for 30 min. For calibration, the probe was fully reduced with dithiothreitol (1 mM, Sigma) and fully oxidized using t-butyl hydroperoxide (1 mM, Sigma). For each pair of images, a ratiometric image was obtained (485 nm divided by 400 nm image). For each condition, the cysteine thiol redox status was calculated as percent oxidized, by comparison to the values obtained for the fully reduced and fully oxidized conditions.
Statistical analysis
All data are expressed as mean±SEM. Results were analyzed by unpaired t-test or ANOVA with Bonferroni's post hoc analysis when appropriate using Prism software (GraphPad Software Inc., San Diego, CA). Statistical significance was set at p<0.05.
Footnotes
Acknowledgments
The authors thank Sharron Francis and Jackie Corbin, Vanderbilt University, for providing sildenafil, and Sylvia Gugino and Lyubov Czech for expert technical assistance. This work was supported by Northwestern University Alumnae Grant (K.N.F.), Neonatal Resuscitation Program Young Investigator Grant (K.N.F.), and National Institutes of Health grants: HL086715 and HL109478 (K.N.F.), HL054705 (R.H.S.), and HL035440 and HL079650 (P.T.S.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.