
Abstract
Nitrite, previously considered physiologically irrelevant and a simple end product of endogenous nitric oxide (NO) metabolism, is now envisaged as a reservoir of NO to be activated in response to oxygen (O2) depletion. In the first part of this review, we summarize and compare the mechanisms of nitrite-dependent production of NO in selected bacteria and in eukaryotes. Bacterial nitrite reductases, which are copper or heme-containing enzymes, play an important role in the adaptation of pathogens to O2 limitation and enable microrganisms to survive in the human body. In mammals, reduction of nitrite to NO under hypoxic conditions is carried out in tissues and blood by an array of metalloproteins, including heme-containing proteins and molybdenum enzymes. In humans, tissues play a more important role in nitrite reduction, not only because most tissues produce more NO than blood, but also because deoxyhemoglobin efficiently scavenges NO in blood. In the second part of the review, we outline the significance of nitrite in human health and disease and describe the recent advances and pitfalls of nitrite-based therapy, with special attention to its application in cardiovascular disorders, inflammation, and anti-bacterial defence. It can be concluded that nitrite (as well as nitrate-rich diet for long-term applications) may hold promise as therapeutic agent in vascular dysfunction and ischemic injury, as well as an effective compound able to promote angiogenesis. Antioxid. Redox Signal. 17, 684–716.
I. Introduction
A. Nitrite is the Cinderella molecule in biological signaling
In bacteria, nitrite is a well-known source of nitric oxide (the NO• radical, hereinafter NO) under anaerobic conditions (380). In eukaryotes, on the other hand, for years nitrite has been considered physiologically irrelevant and a simple end product of endogenous NO metabolism (113, 185); only in the last decade, the relevance of nitrite as a source of NO has emerged.
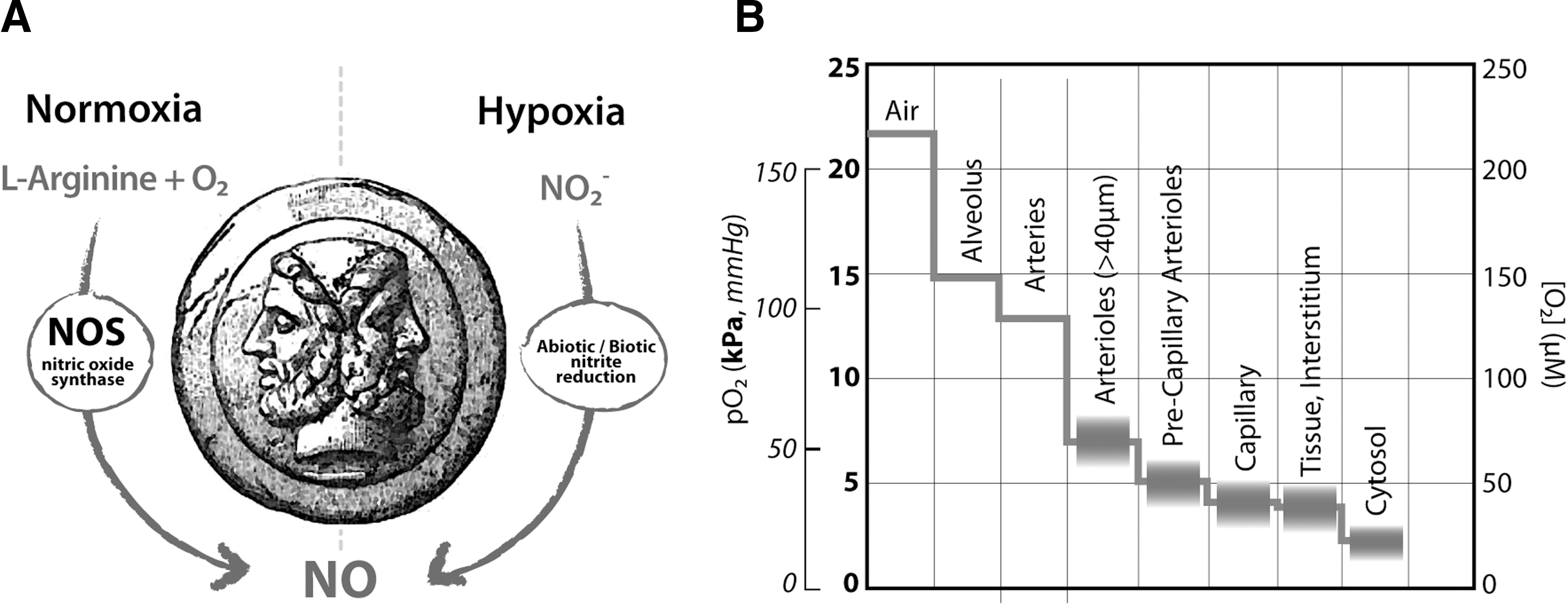
In mammals, NO is mainly synthetized by the enzyme nitric oxide synthase (NOS) via oxidation of the aminoacid L-arginine; this reaction requires oxygen (O2) as an essential substrate (Fig. 1A), with a Km in the 5–20 μM range (in vitro) (5, 199, 287). The exact O2 threshold level at which NOS-dependent NO generation is compromised and fails to signal is unknown mainly due to uncertainties on the in vivo value of the Km for O2 of NOS enzymes (220). Organs and tissues are characterized by their own unique normoxic status (57, 352), given that the local oxygen pressure (pO2) is a key component of the physiological state of an organ and results from the balance between O2 consumption and delivery (Fig. 1B). However, in the human body, NO is also produced under hypoxic conditions (352); under these conditions, NO formation is not blocked by NOS inhibitors (74, 131, 382) and nitrite reduction is found to be enhanced (44, 131). These lines of evidence suggest that, in eukaryotes, an NOS-independent system exists, able to ensure sufficient NO formation when O2 supply is limited (Fig. 1A), and identify nitrite as a main source of NO under hypoxic conditions. As a consequence, nitrite has recently obtained novel relevance and continuously expanding popularity.
This review summarizes and compares the molecular mechanisms of nitrite-dependent NO production in selected bacteria and in eukaryotes. Moreover, the review analyses the involvement of nitrite in human physiology and the possible therapeutic applications of this molecule. As will be detailed in the review, the nitrite reductase (NiR) activity is carried out by enzymes and proteins with intrinsically different cellular roles and biochemical features, spread across the two kingdoms; the wide distribution of the NiR activity highlights the importance of nitrite in cellular homeostasis.
II. Bacterial NiR and Their Significance
In this section we describe the reaction mechanism of two different classes of bacterial enzymes involved in the reduction of nitrite and the significance of this activity for human health, with particular attention to host–pathogen interactions. Three examples are reported: AniA from Neisseria (section A), NirBD from Mycobacterium tuberculosis (section B), and cytochrome cd1 from Pseudomonas aeruginosa (section C).
The human body is heavily colonized by bacteria. These bacteria are frequently exposed to anoxia and to NO (and, thus, also to nitrite) generated by the host. Dealing with these two environmental factors often forces the bacteria to metabolic changes to (i) maintain growth and survive in the absence of O2, and (ii) detoxify the free radical NO and/or the toxic compound nitrite. While microbial colonization is part of the normal physiology of the human body, the body can also be exposed to pathogenic bacteria such as P. aeruginosa, M. tuberculosis, and the pathogenic Neisseria species N. meningitidis and N. gonorrhoeae. The ability to reduce nitrite can therefore confer to these species a selective advantage in the host–pathogen arms race to survive in an O2-limited and nitrite-rich environment.
Two distinct classes of NiR are responsible for the reduction of nitrite in bacteria. The first group comprises the enzyme NiR (EC 1.7.2.1), which reduces nitrite to NO during denitrification, the anaerobic respiratory process widely found in both autotrophic and heterotrophic microrganisms, in which oxidized nitrogen compounds such as nitrate and nitrite are used as electron acceptors for energy production (380). Denitrification has been implicated in the virulence of several bacterial species, including Brucella (12), Pseudomonas (342), and Neisseria (17).
Depending on the bacterial species, the NiR enzyme might be a copper-containing protein or a hemoprotein (cytochrome cd1 nitrite reductase [cd1
NiR]), encoded by the nirK and nirS genes, respectively. The reaction catalyzed by these enzymes is
The second group of NiR includes two quite distinct enzymes catalyzing the reduction of nitrite to ammonia. The first type is the (NAD(P)H)-dependent enzyme NirBD (EC 1.7.1.4), which reduces nitrite to ammonium coupled to the oxidation of either NADH or NADPH. It is a flavin-dependent enzyme that also contains a specialized heme cofactor named siroheme (119, 326); this enzyme participates in assimilatory nitrite reduction in bacteria but also in algae, fungi, and higher plants. The reaction catalyzed by NirBD NiR is
The other class of enzymes able to convert nitrite to ammonia is the multiheme NiR (cytochrome-ammonia forming) NrfA (EC 1.7.2.2). The enzyme also reduces NO and hydroxylamine to ammonia, and sulfite to sulfide. The reaction catalyzed is:
In the following paragraphs three relevant examples of bacterial NiR strategic for the survival of pathogens within the infected host are reported.
A. Neisseria and the copper NiR AniA
The three closely related bacterial species, N. meningitidis, N. gonorrhoeae, and Neisseria lactamica, colonize mucosal surfaces in humans. N. gonorrhoeae is the causative organism of the sexually transmitted disease, gonorrhoea, one of the most frequently reported communicable diseases; N. meningitidis does occasionally cause severe, life-threatening illness known as meningitis, whereas N. lactamica is a common, harmless commensal of children.
N. gonorrhoeae is an obligate human pathogen that colonizes O2-limited environments of the genitourinary tract. As in N. meningitidis, it conserves energy during electron transfer from physiological substrates via a membrane-bound respiratory chain to a single terminal cytochrome oxidase, cytochrome cbb 3 (65, 266, 319). When the O2 supply is growth-limiting, the bacterium produces a truncated denitrification pathway in which nitrite is reduced to NO by the anaerobically induced outer membrane protein AniA, a copper-containing NiR of the NirK family (34, 188, 231). NO is then reduced to N2O by a single-subunit nitric oxide reductase (Nor) B subunit (NorB) that receives electrons directly from ubiquinol (153). Therefore, AniA and NorB cooperate together to facilitate the anaerobic growth of gonococci: the nirK and norB genes are differentially controlled by a group of transcriptional regulators that respond to changes in the levels of O2, nitrite, and NO (146, 163, 164).
The crystal structure of the soluble domain of AniA shows that the protein adopts a fold typical of copper-containing bacterial NiR (34): a tightly packed trimer of identical subunits, containing a type I and a type II copper atoms (Fig. 2).
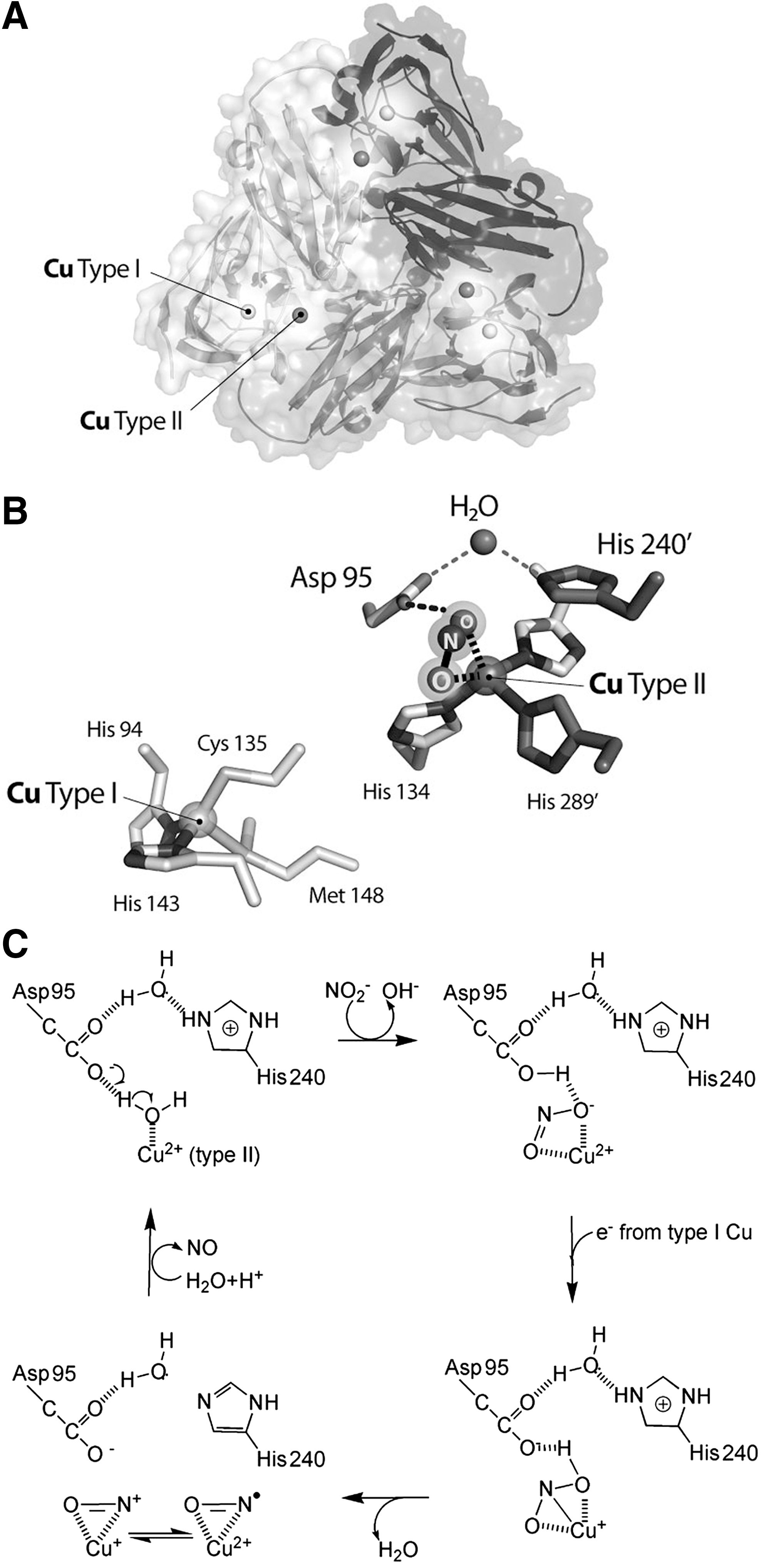
The catalytic mechanism of CuNiRs requires one electron and two protons to convert nitrite into NO and water (2, 7, 249) (Fig. 2C). The protons are donated by a conserved active-site aspartate, which hydrogen bonds directly to the nitrite molecule, and by a histidine residue; these aminoacids are linked through a solvent-bridged hydrogen bond. The aspartate and histidine residues are conserved in all known CuNiR sequences and correspond to Asp97 and His240 in AniA (Fig. 2B). The most likely electron donors to AniA are the CcoP subunit of cytochrome oxidase cbb3 and, by sequence similarity to the CcoP subunit, also the cytochrome c5 (10, 152).
Interestingly, aniA is the most highly induced gene during anaerobic growth; expression of AniA during infection has been detected immunologically, supporting the induction of nitrite reduction in vivo (54, 67, 297). The ability to reduce nitrite during O2-limited growth appears therefore to confer a selective advantage for the survival of pathogenic Neisseria in the human host (66). The analysis of aniA expression has been recently extended to the biofilm mode of growth of the bacterium; biofilms are organized bacterial communities in which the cells are embedded in a self-produced extracellular polymeric substance, attached to a surface. Biofilms formed by pathogens play an important role in the infection of living tissues and are responsible for the resistance to antibiotics and to the host immune system (45). N. gonorrhoeae readily forms biofilms over abiotic surfaces, over primary and transformed cervical epithelial cells and over cervical tissues in vivo. Expression of AniA in biofilms is induced over time; this evidence shows that a combination of anaerobic/aerobic respiration is used by Neisseria to support growth in the biofilm and that nitrite appears to be the preferred substrate for anaerobic respiration (110, 111).
Interestingly, Aspholm et al. (10) reported the identification of a single-nucleotide polymorphism (SNP) unique in the species N. meningitidis that leads to truncation of the c-type heme protein CcoP, an essential component of cytochrome oxidase. This SNP was found in all strains of N. meningitidis but not in strains of N. gonorrhoeae and N. lactamica. Although this mutation results in the truncation of an essential component of the cytochrome cbb3 oxidase, it also conditionally affects nitrite consumption, providing evidence that an alteration in the circuitry of respiratory electron-transfer networks is associated with N. meningitidis speciation.
B. M. tuberculosis and NirBD NiR
M. tuberculosis, the etiologic agent of tuberculosis, is a facultative intracellular pathogen that can persist within the host; the bacterium may lie dormant in the human body for decades, only progressing to active disease in 5%–10% of individuals. One of the primary host defense mechanisms against mycobacterial diseases involves the formation of a granuloma-like structure. Immune containment by granuloma formation creates a microenvironment in which nutrient limitation, low pH, reactive nitrogen and O2 species, and reduced O2 tension are believed to be factors that coincide with the establishment of chronic infection. Under these conditions, M. tuberculosis changes its metabolism and, to survive, it can utilize various nutrients, including nitrate, as a source of nitrogen. Assimilation of nitrate requires the reduction of nitrate via nitrite to ammonium, which is then incorporated into metabolic pathways (89, 143); assimilation of nitrite is therefore essential for the survival of M. tuberculosis in vitro and in vivo.
The second step in nitrate assimilation is the reduction of nitrite to ammonium, catalyzed by the siroheme-dependent NADH-NiR, encoded by the nirBD operon; this enzyme is known to catalyze nitrate assimilation in various bacteria and fungi (208). The NirB protein is produced by Mycobacterium throughout infection, as recently shown in a proteomic study, together with the proteins required for nitrate/nitrite transport (such as NarX) and nitrate reduction (narGHJI and narK2X operons) (192).
In M. tuberculosis, NirBD has been proposed to be involved in the assimilatory reduction of nitrite (228). A recent study suggests, however, that the NirBD complex is not required for nitrate-dependent protection from acid-induced death under hypoxia (325); under these conditions the nitrite produced upon nitrate reduction through the NarGH complex is not further reduced into ammonium by the NirBD complex. Thus, in Mycobacterium, protection from nitrite toxicity at acidic pH, is most likely achieved by exporting nitrite outside the cell, likely through predicted nitrite extrusion proteins, including NarK3 and NarU (69). An alternative role for the nirBD-encoded NiR enzyme has been proposed for Escherichia coli and other enterobacteria; in these microrganisms, NirBD is induced under anaerobic conditions and it is involved in detoxifying nitrite that accumulates from nitrate respiration (126).
C. P. aeruginosa and cd1NiR
The ubiquitous gram-negative bacterium P. aeruginosa is an opportunistic pathogen responsible for both acute and chronic infections. P. aeruginosa is an etiologic agent common in several infections, including those affecting ears (94), burn wounds (244), and eyes (169). In addition, P. aeruginosa chronic lung infection is the major cause of death in cystic fibrosis (CF) patients, a genetic disease affecting 1/2500 newborn in Europe (99). P. aeruginosa is frequently resistant to conventional antibiotic therapy and to the host antimicrobial effector mechanisms. A major problem in the control of P. aeruginosa infection is given by the sessile, biofilm-mode of growth adopted by this bacterium in many infection sites, and typically in CF lung chronic infections (137, 139). P. aeruginosa survives in the low O2 environment of the airway mucus of CF patients by using anaerobic metabolism and forming robust biofilms (246). The stagnant mucus overlaying the CF lung epithelium constitutes a nitrate-rich microaerobic/anaerobic environment (Fig. 3); nitrate in CF mucus is generated in part by the host inflammatory response to infection via NO. In this environment, P. aeruginosa produces energy from nitrate also using the metabolic pathway of denitrification (6, 139) (Fig. 3). Four reductases are involved in this process (380), namely, nitrate reductase (Nar), NiR, Nor, and nitrous oxide reductase (N2OR), whose expression is tightly regulated, being the intermediate NO a cytotoxic compound. Genetic mutants lacking nar and nir genes show swarming defects and reduced virulence (342).

The molecular mechanisms controlling enhanced biofilm formation during anaerobic growth are not clearly defined. Low concentrations of NO have been shown to promote biofilm dispersion (15); on the other hand, Yoon and coworkers (369) have shown that P. aeruginosa PAO1 grown anaerobically is more elongated than that grown aerobically and is defective in cell division. Elongated cells easily form highly cohesive clumps, thus yielding a robust biofilm. Cell elongation is dependent on the presence of NiR and is repressed in P. aeruginosa PAO1 in the presence of an NO antagonist (2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide [carboxy-PTIO]); these evidence suggests a link between cell elongation, NO, and anaerobic respiration (369). Importantly, the nonelongated NiR-deficient mutant failed to form biofilm, while the wild-type PAO1 is highly elongated and formed robust biofilm.
In addition to its role in anaerobic growth of P. aeruginosa, the NiR activity controls other important aspects of pathogenesis even under conditions where O2 is apparently not limiting, including motility, initiation of biofilm formation, and virulence. As an example, a recent study has demonstrated that the NO produced by P. aeruginosa NiR regulates the activity of type III secretion system (343), an apparatus whereby cytotoxic effector proteins are directly secreted into the host cell cytoplasm after contact of the bacterium with a target cell.
Therefore, in P. aeruginosa, pathogenesis, biofilm formation, and denitrification, expecially nitrite reduction, are closely related. The enzyme responsible for nitrite reduction to NO is P. aeruginosa cytochrome cd1 nitrite reductase (Pa-cd1 NiR), a homodimer containing one c-heme and one d1 -heme group in each subunit (Fig. 4A). Electrons are transferred from the soluble electron donor cytochrome c551 to the c-heme moiety of the enzyme (347) and thereby internally to the d1 -heme (Fig. 4B); in the active site (Fig. 4C), the substrate nitrite binds to the heme iron and is reduced to NO (380). The d1 -heme (3,8-dioxo-17-acrylate-porphyrindione) (Fig. 4B) is a partially saturated macrocycle with a set of oxo, methyl, and acrylate substituents, unique to the cd1 NiRs (4, 380) and synthesized by a specialized pathway present only in denitrifiers (strongly induced in P. aeruginosa upon nitrite treatment). Other hemes in which the porphyrin ring is partially saturated are the d heme in E. coli and siroheme of bacterial and plant sulfite and NiR (119, 326).
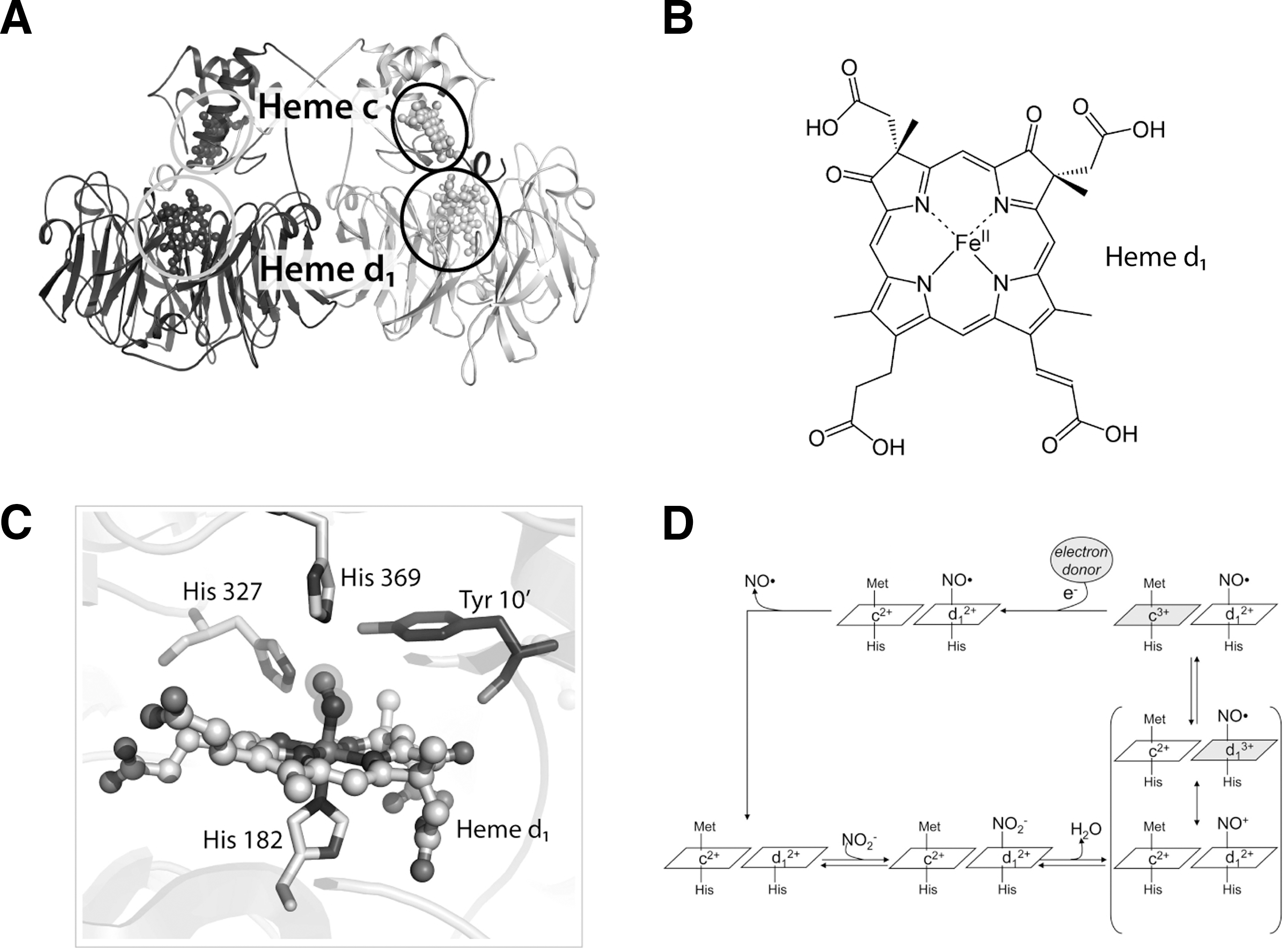
Nitrite reduction to NO is the physiologically relevant activity of cd1 NiR (289, 365); the expression of cd1 NiR is induced by low O2 tension and presence of nitrogen oxides (380). NO is produced efficiently by Pa-cd1 NiR (turnover number=6 s−1 at pH 7.0) (292) and the activity is pH dependent with an optimum between pH 5.8 and 6.5 (292, 365). The current knowledge on the individual steps in the catalytic cycle is summarized below and described in Figure 4D.
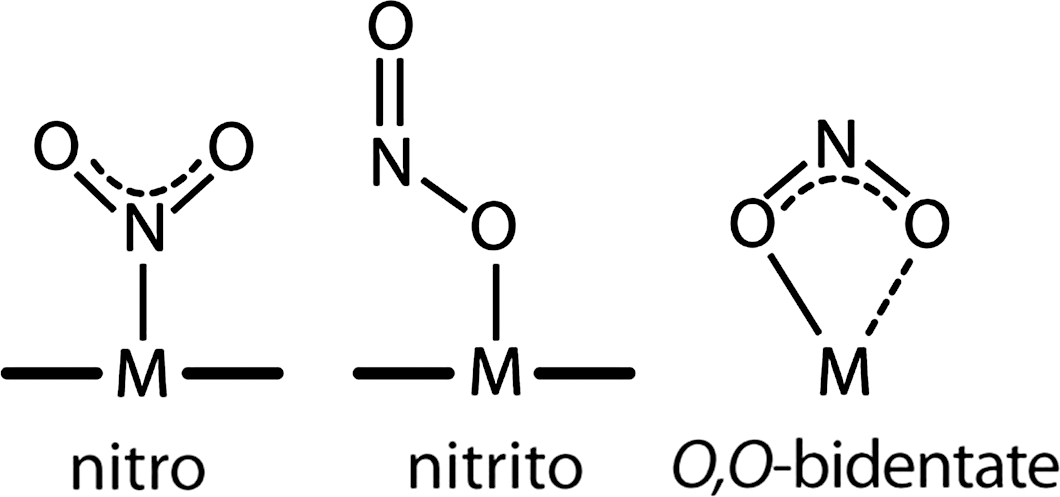
In cd1 NiR, nitrite binds to the ferrous (Fe2+) d1-heme with high affinity (Km=6 μM) (81); binding of nitrite to the ferrous iron is expected in the reaction mechanism since this state of the iron has to supply the electron needed for the reduction process (Fig. 4D). In principle three different coordination modes of nitrite to monometallic centers are possible: the N-nitro, the O-nitrito, and the O,O-bidentate mode (Fig. 5) (364). The latter one is only observed in copper-containing bacterial NiR (16) and synthetic copper complexes (203). The N-nitro binding mode is observed in synthetic iron porphyrin nitrite complexes (64, 257, 363). Nitrite is thought to bind via the N-atom in Pa-cd1 NiR forming the so-called nitro-complex; this binding mode is inferred from the crystal structure of the nitrite-bound derivative of the homologous enzyme from Paracoccus pantotrophus (358). This observation agrees well with data on other heme-containing NiR (76, 105) and on synthetic iron porphyrin nitrite complexes in which, regardless of the iron oxidation state, the N-binding mode is observed (see Fig. 5) (64, 257, 363). The nitrite N-binding mode also agrees with the current mechanism of reduction of nitrite by cd1 NiRs, thought to occur via a double protonation of a terminal O atom of the nitrite molecule. Theoretical calculations have suggested that the O-binding mode is also possible for cd1 NiR (315) and for other hemoproteins such as hemoglobin (Hb) (272). Although nitrite can bind through the O-binding mode (the so-called nitrito mode) to the heme of myoglobin (Mb) and Hb (73, 367, 368), in the case of the d1 -heme there is no experimental evidence that such O-binding mode may occur.
The high affinity for nitrite (and other anions such as cyanide) (166, 323) of the ferrous (Fe2+) d1
-heme is a peculiar and physiologically relevant feature of all cd1
NiRs. This behavior is remarkably different from that observed in the b-type heme-containing proteins in which the negatively charged molecules (nitrite and cyanide) usually bind much better to the ferric (Fe3+) iron. The much higher affinity for nitrite of the d
1-heme can be partially explained with the presence of two electron-withdrawing carbonyl groups on the d
1 heme ring (Fig. 4B). Two conserved histidines (His327 and His369) in the active-site pocket (261) (Fig. 4C) also contribute to the stabilization of the nitrite anion (
In Pa-cd1 NiR, nitrite can displace the NO bound to the ferrous enzyme (293), allowing the enzyme to enter a new catalytic cycle (Fig. 4D); therefore, the high affinity of the active-site ferrous d1 -heme for nitrite (see above) actively contributes to NO dissociation during the catalytic cycle. In agreement with this observation, if the affinity of Pa-cd1 NiR for nitrite is decreased (by mutation of a conserved active-site residue) the fully reduced-NO bound derivative accumulates (81, 293). The observation that NO and nitrite can compete for binding may suggest that the formation of dinitrogen trioxide (N2O3) could in principle occur, for example, in a reaction similar to that proposed for Hb (23). This event is, however, highly unlikely, mainly because during catalysis the d1 -heme is maintained in the reduced state by internal electron transfer from the c-heme. Moreover, ferric d1 -heme has low affinity for nitrite (329), a feature that likely limits further reaction with free NO to produce N2O3.
In the catalytic cycle of Pa-cd1 NiR, the formation of a complex between NO and the reduced d1 -heme might slow down product release (11). Trapping of ferrous hemes by NO is highly likely, given the high affinity of this ligand for Fe2+ (182, 245). However, we have clearly shown that the rate constant of NO dissociation from the reduced d1 -heme is fast (up to 70 s−1) not only for Pa-cd1 NiR (292) but also for the cd1 NiR from P. pantotrophus (294). Consequently, the affinity of reduced Pa-cd1 NiR for NO is relatively low (∼10−7M) and the ferrous enzyme is not firmly inhibited by NO (292, 293). Noteworthy, nitrite reduction can still be monitored after preincubation of reduced Pa-cd1 NiR with NO (292).
The rapid dissociation of NO is largely controlled by the d1 -heme cofactor itself, as recently shown using a complex of the d1 -heme and apomyoglobin (294). This evidence underscores that the d1 -heme has evolved to have low affinity for NO, as compared with other ferrous hemes. The results on the d1 -heme suggest that the reactivity of porphyrins with nitrite and NO can be modulated very extensively by the functional groups present on the heme macrocycle—information of general significance in light of the emerging important biological functions of nitrite as a source of NO under hypoxic conditions.
III. Nitrite Reduction in Mammals
In this section we first describe the sources and the distribution of nitrite in mammalian tissues (section A) and than we discuss the chemical (abiotic) and biochemical (biotic) mechanisms of nitrite reduction in mammals (section B).
A. Sources, levels, and distribution of nitrite
There are several sources of nitrite in mammals, both endogenous and exogenous. The endogenous source of nitrite is mainly the oxidation of the NO produced by NOS (Fig. 6) (241). The oxidation of NOS-derived NO is slow in vitro, but is enhanced in plasma mainly by the copper protein ceruloplasmin (314) and in membranes by the accumulation of NO in the lipid bilayer by preferential partition (210).

The main exogenous source of nitrite is dietary nitrate, which is converted to nitrite by commensal bacteria in the mouth or intestines (Fig. 6). Dietary nitrate levels are particularly high in specific fruits and vegetables; as an example, one serving of spinach, lettuce, or beetroot contains more nitrate than that generated endogenously over a day by the oxidative metabolism of NO carried out by all isoforms of NOS (220). A considerable variability in the nitrate concentrations of plants is observed depending on different species, environmental conditions, nutrient availability, insect damage, and application of nitrogen-based fertilizers. On the other hand, only a small amount of nitrite is contained in dietary food (345); nitrite is used for meat preservation, to cure flavor and color and to enhance meat appearance (59, 219). In general, nitrite intake varies from 0 to 20 mg/day (270).
Nitrates and/or nitrites are also contained in drinking water (<10 mg/L in the absence of bacterial contamination) (42). Other environmental sources of nitrate and nitrite include cigarette smoke (258) and car exhausts (219). These and other environmental pollutants contain volatile nitrogen oxides, some of which are converted to nitrate or nitrite in the body (219). The relative contribution from these different sources of nitrite during normal conditions is variable (218). Nitrite levels show considerable variation between individuals and are significantly affected by dietary habits and lifestyle (216): for example, plasma nitrite and nitrate may be lowered by about 50% by dietary restriction (288).
Dietary nitrate is rapidly absorbed from the gastrointestinal tract into the blood stream, and distributed throughout the body, mixing itself with the endogenous nitrate (219) (Fig. 7). Some nitrate is excreted and concentrated 10-fold in salivary glands (354): the amount of nitrate secreted in saliva is up to 25% of plasma nitrate level (320). The dorsal face of tongue harbors a specialized flora of symbiotic facultative anaerobic bacteria expressing enzymes that can reduce nitrate to nitrite (306) (Fig. 6). These bacteria use nitrate as an alternative terminal electron acceptor during respiration to produce ATP (356). The biologic effect of ingested nitrate, as well as the concomitant increase in plasma nitrite, is abolished after avoiding swallowing of saliva (216, 354) or by the use of an antibacterial mouthwash (135, 274). Therefore, if mammals were germ free, the endogenous or ingested nitrate would not be metabolized because of the lack of the enzymatic machinery for its reduction (42, 219).
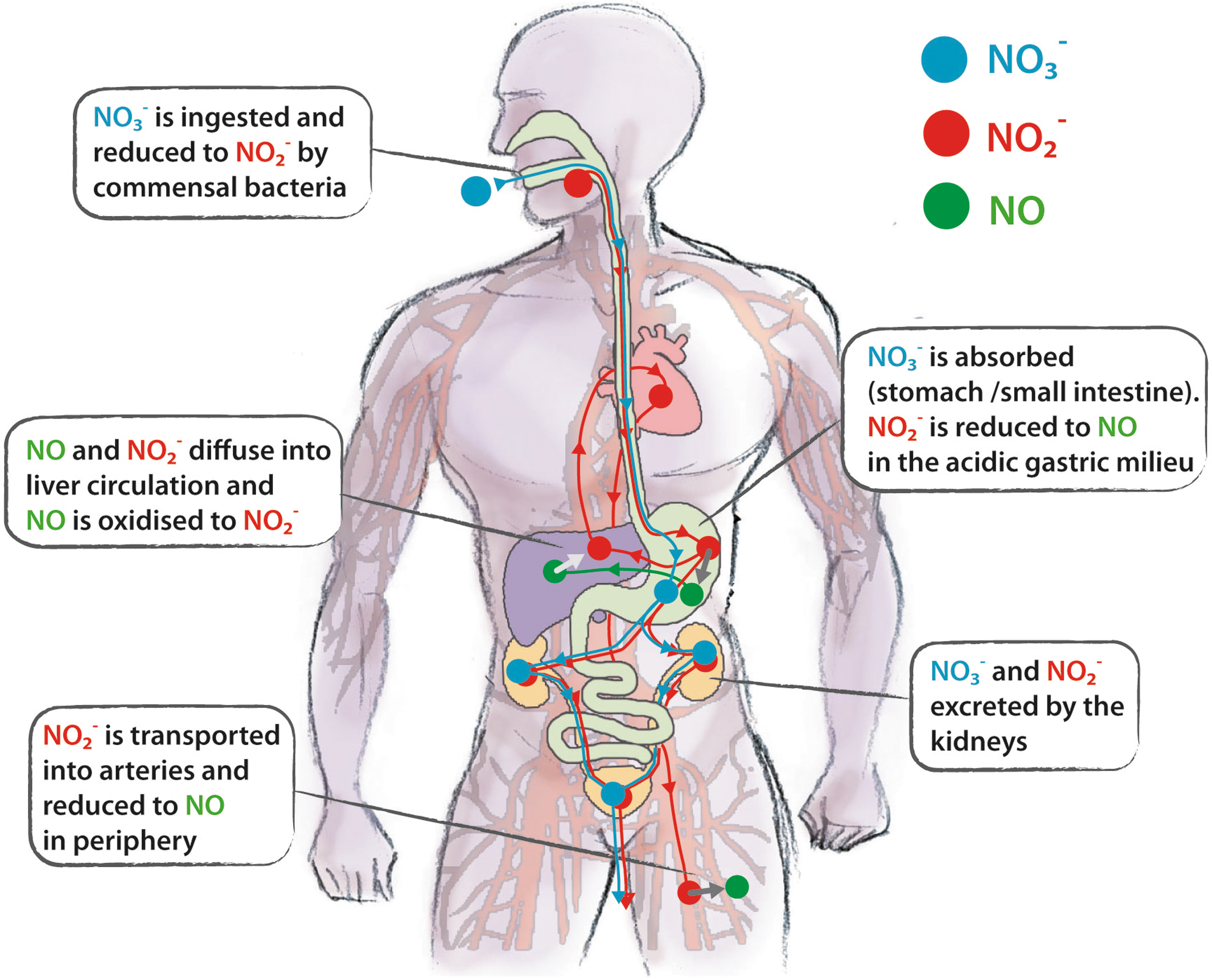
The metabolic fate of dietary nitrite in humans is depicted in Figure 7. After ingestion, nitrite reaches the stomach, where it is reduced to NO. In the acidic gastric milieu, nitrite is protonated to form nitrous acid (HNO2), which then decomposes to NO and other nitrogen oxides (27, 221). Low pH and reducing compounds, such as ascorbic acid and polyphenols, enhance this reaction (28, 122). The level of NO gas in the stomach can be considerable (more than 100 ppm) (356). Most of the salivary nitrite escapes from this conversion and diffuse in systemic circulation (216); nitrite is transported in the arterial circulation to resistance vessels, where the low O2 tension favors the reduction of nitrite to NO, causing vasodilatation and lowering the blood pressure (354).
Plasma levels of nitrite are usually in the range 0.1–0.6 μM with a mean of 0.345±0.017 μM (128, 186, 216) (Table 1). The plasma concentration of nitrate is much higher (20–50 μM); as previously described, in the whole blood, nitrite is rapidly oxidized to nitrate (97, 241) (Fig. 6). Nitrate and nitrite increase greatly in saliva, plasma, and urina after a nitrate load (216).
Data are taken from (186). Concentration of free nitrite (μM) in plasma in a variety of species has been measured using three different analytical approaches: flow injection analysis technique, reductive gas phase chemiluminescence, and sensitive high-pressure liquid chromatography techniques. Data are given as mean±standard error of the mean (SEM).
Each tissue has a different concentration of nitrite (Table 2). As an example, inside the erythrocyte the levels are higher than in plasma (44, 86); given that the hematocrit represents between 40% (children) and 50% (adult males) of total blood volume, the erythrocytes contribute the largest nitrite pool in whole blood.
The levels of nitrite in the body show significant variability due to differences in dietary habits, lifestyle (e.g., tobacco consumption), and physical exercise (37, 255, 265, 375). Circulating nitrite may be significantly enhanced in individuals suffering from an infection (332, 333) and markedly lowered during pregnancy. Plasma levels of nitrite also depend on NOS activity (186): in the presence of different NOS inhibitors, a change in vascular resistance occurs, paralleled by a reduction in plasma nitrite by 30%±8% (186).
It is important to mention that the stationary levels of free nitrite or NO may also be influenced by the buffering effect of reduced glutathione (GSH), one of the main antioxidants present in cells and tissues. Under acidic conditions, nitrite reacts with GSH to form S-Nitrosoglutathione (GSNO), an endogenous S-nitrosothiol (SNO) (187, 337); on the other hand, GSNO may be formed by reaction of GSH with N2O3, with production of nitrite and protons, or, to a lower extent, with peroxynitrite (ONOO–) (25, 183, 344). GSNO, in turn, is one of the most relevant biological molecules to carry out nitrosation reactions under physiological conditions (9, 373).
B. Mechanisms of nitrite reduction
1. Abiotic nitrite reduction: solution chemistry of nitrite
Nitrite is the conjugate base of HNO2 [pk∼3.1–3.2 (83)]. Therefore, at physiological pH, HNO2 is essentially in the deprotonated form, the NO2
−, according to the following equation:
At lower pH values, HNO2 decomposes to various nitrogen oxides, including N2O3, which can dissociate to NO and NO2 (K=2×10−5 M) (335).
Moreover, the reduction of nitrite in aqueous solution strictly depends upon pH, according to the following reaction:
Therefore, the so-called disproportionation of nitrite to NO, promoted by acidic pH, includes these reactions; this abiotic pathway is responsible for the production of NO in the gastric milieu of humans (27, 221, 230). The disproportionation of nitrite to NO is enhanced in the presence of reducing agents such as ascorbate (vitamin C) (213, 239) and other compounds such as polyphenols (122, 271, 295). Interestingly, disproportionation of nitrite to NO in biological tissues is enhanced under ischemic conditions when the pH drops from the normal value (7.4) to values as low as 5.5 (304, 382).
2. Biotic nitrite reduction
Several examples of NiR activities have been found in blood, tissues, and mitochondria. All of them can be ascribed to proteins, which, under aerobic conditions, play an O2-dependent biological role but turn into NiR under hypoxic conditions.
a. Hemoglobin
Hb is the heme-containing metalloprotein involved in O2 transport in humans. It is located in the erythrocyte, where it binds and releases O2 in response to the partial pressure of this gas. The heterotetrameric adult HbA (alpha2-beta2) exists in two quaternary conformations, R-state and T-state, which display different affinity for the heme ligands, including O2 and nitrite. Hb has been assigned a central role in the physiological reduction of nitrite: Hb binds and reacts with nitrite in both deoxygenated (deoxygenated hemoglobin [deoxyHb]) and oxygen-bound forms (oxygenated hemoglobin [oxyHb]), yielding different intermediate species and products, as detailed below.
(1) Reaction of deoxyHb with nitrite
The ability of deoxyHb to act as an NiR and produce NO was known since the pioneering work of Brooks (36). In blood, the reaction of nitrite with deoxyHb has been proposed to represent a source of NO bioactivity, according to the following reaction (129):
The Hb-dependent NiR activity is allosterically controlled by the quaternary structure of the protein (Fig. 8) (53, 129). The bimolecular rate constant of the reaction varies as the allosteric conformation of Hb changes: the R-state Hb has a bimolecular rate constant of 6 M –1 s–1, whereas the constant for T-state Hb is 0.12 M –1 s–1, giving the reaction an average bimolecular rate constant of 0.35 M –1 s–1 (pH 7.4, 25°C) (156). The R-state Hb is a better reductant due to a more negative redox potential and/or to a more accessible heme pocket relative to T-state Hb (30, 77, 302).
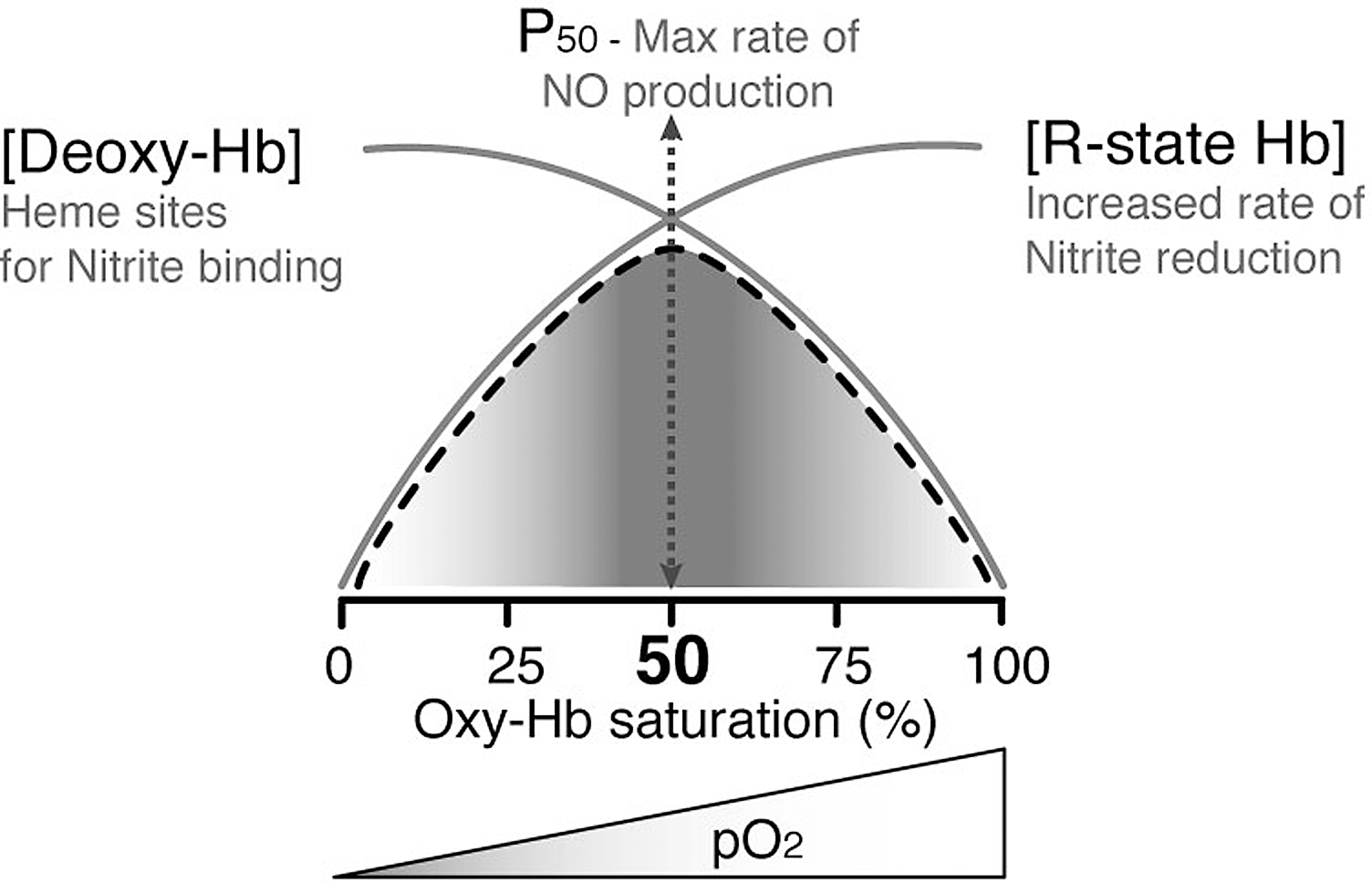
The maximal rate of nitrite reduction is reached at the pO2 value of about 35 μM, which corresponds to the P50 (i.e., the pO2 at which Hb is half-saturated) (Fig. 8). The observed kinetics of the reaction is the combination of two processes, where the rate of nitrite reduction increases with increasing O2 fractional saturation in parallel with the increased R-state character of Hb; at high fractional saturation, the concentration of deoxyHb (one of the substrates) decreases, thereby slowing down the rate. This peculiar chemistry has been described as an allosteric autocatalytic reaction (130, 156), where autocatalysis is controlled by the allosteric transition of the Hb tetramer from the T- to the R-state. Stabilization of Hb in either the T- or R-state (by chemical cross-linking) has recently confirmed this interpretation (53).
Accordingly, fetal Hb, in which the γ-subunits replace the β-subunits and the R-state is favored, shows an increased efficiency of nitrite reduction (30). Conversion of nitrite to NO in the fetus is thus favored (i) by the molecular properties of fetal Hb and (ii) by the fetal arteriovenous oxyHb concentrations (∼75% to 45%) that fall in the range where nitrite reduction is maximal. The reaction of Hb with nitrite is potentially of great importance for the fetus because it provides an O2-sensitive mechanism for NO production in the vasculature and may contribute to mantain the low resistance to blood flow, characteristic of the fetal circulation. However, taking into account that tissues are generally more effective than blood in reducing nitrite to NO (113, 204) (see also below), it remains to be demonstrated whether reduction in blood or in tissues is more important for the low vascular resistance in fetal circulation.
The exact molecular mechanism of nitrite reduction by deoxyHb is still a question of debate. A possible reaction pathway has been suggested (251, 252, 290, 291, 301) and is summarized in the following scheme:
In this scheme, the initial intermediate is the nitrite-associated Hb, while the second one has an electron delocalized between the heme iron and the ligand (Hb2+NO+ ⇆ Hb3+NO) and is stable even in the presence of a large excess of oxyHb and deoxyHb, particularly at low nitrite/Hb ratios found in vivo. The release of NO in this reaction is facilitated by excess nitrite and is conformationally regulated: the R-state quaternary conformation favors the formation of the second intermediate, whereas the T-state quaternary conformation favors the release of NO from the intermediate. This observation has been explained in terms of a conformational effect on the distal heme pocket, involving hydrogen bonding of nitrite to the distal histidine (291).
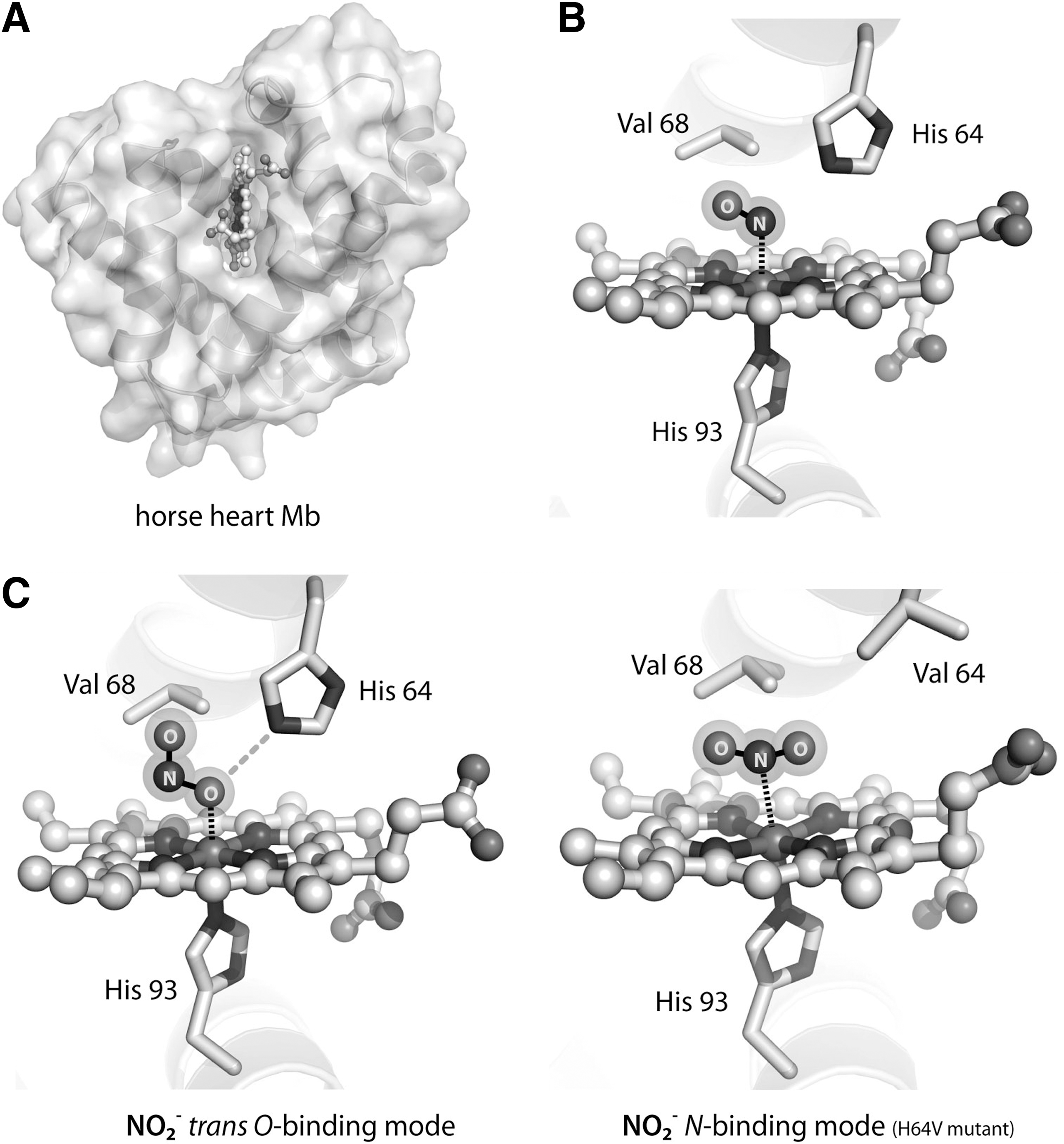
Despite the relevance of the interaction between nitrite and the heme iron, no crystal structure of the ferrous (Fe2+) iron–nitrite complex of heme globins (including Hb) is available. The only adducts for which crystal structures are available are the nitrite-bound ferric (Fe3+) complex of Hb at 1.8 Å resolution (368) and of Mb at 1.2 Å resolution (73) (Fig. 9). Interestingly, in both cases, the nitrite molecule is bound in the O-nitrito mode (Fig. 5); the ligand is stabilized by a hydrogen bond to the distal histidine residue. The hydrogen bonding capability of the distal heme pocket is crucial to direct the binding mode of nitrite: substitution of His64 with Val in Mb leads to the reorientation of the nitrite molecule to the N-nitro binding mode (Fig. 9C) and decreases the rate of nitrite reduction (366). Altogether, the existence of the N- and O-binding to hemoproteins (see also section on bacterial NiR) may suggest that the efficiency of nitrite reduction could be a function of the binding mode of the nitrite molecule. Thus, in principle, the mechanism of the reaction with deoxyHb should involve the formation of a nitrite complex of ferrous heme (either N- or O-bound), followed by proton transfer by a nearby histidine. In the case of the N-nitro complex, dehydration of nitrite then yields the Hb (Fe3+)-NO derivative; on the other hand, starting from the O-nitrito complex the final derivative is the Hb (Fe3+)-OH complex, and thus NO is released. Regardless of the mechanism employed, one of the products of the reaction is methemoglobin (MetHb or Hb3+), whose accumulation is associated with the pathological status named methemoglobinemia (see section on nitrite therapy for detailed discussion). However, metHb can be recycled to Hb, thus yielding an enzymatic conversion of nitrite into NO (179), by the NADH-cytochrome b5 reductase system. There are two forms of the NADH-cytochrome b5 reductase in humans: a soluble, erythrocyte-restricted form, which is active in metHb reduction, and a ubiquitous membrane-associated form involved in lipid metabolism. Genetic alterations of these genes are associated with congenital methemoglobinemia due to an enzyme defect in the reductase activity (179).
In summary, deoxyHb reacts with nitrite to produce NO. The main conundrum of the reaction of deoxy Hb with nitrite as a source of bioactive NO lies in the expectation that the NO produced is likely to be either immediately oxidized to nitrate by reaction with oxyHb or trapped by the excess of deoxyHb, yielding a stable ferrous–nitrosyl complex (koffNO=10−3/10−5 s−1) (182, 245). Possible chemical tricks to overcome this problem include the oxidative denitrosilation carried out by nitrite itself and the nitrite anhydrase activity of Hb forming N2O3, which may diffuse out of the erythrocyte, later forming NO or acting by nitrosylating a thiol. Both possibilities are analyzed in more detail below.
(2) Reaction of nitrite with oxyHb and oxidative denitrosylation
Nitrite can react with oxyHb in a complex reaction to produce metHb and nitrate (184, 190). Thus, by reacting with oxyHb, the majority of NO and nitrite end up as nitrate, which may serve as another storage form of these N-oxides.
The reaction of nitrite with oxyHb does not produce NO directly; however, as it will become clearer below, it is relevant to analyze the mechanism of this reaction and the possible crosstalk with the reduction of nitrite with deoxyHb, given that, under oxygenated conditions, the two reactions (i.e., with deoxyHb and oxyHb) compete with one another (184).
In the reaction of nitrite with oxyHb, the rate of metHb production is about 0.5–1 M−1s−1 (345). This reaction has an autocatalytic kinetics, as it is initially slow (in the lag or induction phase) but then enters a rapid autocatalytic phase involving radical-mediated chain reactions and branching steps (96, 98, 180, 209, 351). The reaction can be divided into at least two steps, that is, initiation and propagation: the initiation step yields metHb and hydrogen peroxide (H2O2), which then react to produce the FerrylHb (FeIV=O) radical. In the second step, oxidation of Ferryl-Hb by nitrite produces the nitrogen dioxide (NO2•) radical, which reacts again with oxyHb to produce nitrate and ferrylHb, resulting in an autocatalitic loop (propagation step) (180).
The intermediate species of the nitrite/oxyHb reaction, most probably (NO2•), can also oxidize (Fe2+)Hb-NO, thus releasing NO in the so-called oxidative denitrosylation (22, 136). However, given that the nitrite levels in the erythrocytes and plasma are in the sub-micromolar range, it is unlikely that the reaction can proceed to the autocatalytic step in vivo, and thus low levels of NO2• are produced.
(3) The nitrite anhydrase reaction of Hb
An attractive hypothesis to explain NO bioactivity from nitrite is the formation of a carrier molecule, less reactive and more easily diffusible, which can reach the target tissue and be converted again to NO. A likely candidate is N2O3, which can produce NO (see below) and is also able to form nitrosothiols (357) adding an extra possibility of chemical signaling. Formation of N2O3 might be readily explained by the nitrite anhydrase reaction of MetHb:
Different possibilities for metHb-catalyzed formation of N2O3 have been proposed, involving initial formation of an Fe3+–nitrite (23) or Fe3+–NO (116) complex. A recent theoretical study of the various mechanistic alternatives (151) shows that both pathways of Fe3+-mediated N2O3 formation are energetically feasible. N2O3 back-conversion to NO has also been proposed to be mediated by Hb (23).
The formation of N2O3 is supported experimentally by the formation of S-nitrosothiols in vivo and in vitro (23, 212, 252, 311). It is thus likely that Hb may function in vivo as a nitrite anhydrase and that N2O3 may be an important player in nitrite-mediated NO and S-nitrosothiol signaling (see also section IV).
b. The other globins: Mb and neuroglobin
Mb is one of the most extensively studied hemoproteins; it is a monomeric globin bearing a single b-heme group. Mb concentration in human skeletal and cardiac muscle is as high as 200–500 μmol/kg wet tissue (362).
Mb has been implicated not only in the storage and facilitation of O2 diffusion (236, 360, 361), but also in the scavenging of NO to protect mitochondrial respiration (39, 118). The major mechanism of attenuating intracellular NO bioactivity in cardiac muscle is the reaction of MbO2 with NO to give metmyoglobin (metMb) and nitrate. Mb-deficient (myo−/−) mice are more sensitive to endogenously formed and exogenously applied NO; regeneration of metMb by metMb reductase to Mb and subsequent association with O2 leads to reformation of MbO2 available for another NO degradation cycle.
Therefore, the reactivity of Mb with different ligands depends upon O2 concentration: under normal O2 levels, Mb mainly acts as an O2/NO binding protein. Accordingly, Mb displays a functional relevance in O2 supply and NO scavenging on the whole animal level: loss of Mb leads to impaired myocardial contractile function and exercise endurance (233).
When the O2 concentration decreases to a value around the P50 of Mb (3–4 μM), the protein becomes significantly deoxygenated; under these conditions, Mb is able to reduce nitrite to bioavailable NO in the red muscle and in the heart (312). Therefore, Mb comes into play as an NiR mainly under hypoxic or ischemic conditions (144). Mb has distinct properties from Hb as an NiR: first of all, it has a very low redox potential, and therefore it reduces nitrite ∼50 times faster than T-state Hb (154). Moreover, since Mb is a monomer without allosteric behavior, the reaction of nitrite with deoxyMb is a second-order reaction with a bimolecular rate constant of 6–12 M −1s−1 (between 25°C and 37°C, pH 7.4); the products of the reaction are equimolar amounts of metMb (Fe3+) and iron-nitrosyl-Mb (312). Using an Mb-knock out mouse model, Hendgen-Cotta and coworkers (145, 285) provided unequivocal evidence that deoxyMb reduces nitrite to form NO that regulates mitochondrial respiration and cardiac contractility during hypoxia and ischemia/reperfusion.
Interestingly, both neuroglobin (Ngb) (41) and cytoglobin (273) have low heme redox potential and high O2 affinity, suggesting similar properties as Mb in terms of nitrite reduction. This hypothesis has been recently investigated for Ngb, a highly conserved hemoprotein that evolved from a common ancestor to Hb and Mb. Ngb possesses a six-coordinate heme with proximal and distal histidines ligands; coordination of the sixth ligand is reversible. Gladwin and coworkers have recently shown that deoxygenated human Ngb reacts with nitrite to form NO (330). This reaction is regulated by two redox-sensitive surface thiols (cysteine 55 and 46) controlling the fraction of five-coordinate heme together with nitrite binding and NO formation. Replacement of the distal histidine by leucine or glutamine leads to a stable five-coordinate geometry; these Ngb mutants reduce nitrite to NO ∼2000 times faster than the wild type, whereas mutation of either Cys-55 or Cys-46 to alanine stabilizes the six-coordinate structure and slows down the reaction. The NiR activity of Ngb was found to inhibit cellular respiration via NO binding to cytochrome c oxidase (Cox), thus suggesting that Ngb may function as a physiological oxidative stress sensor and a post-translationally redox-regulated NiR. Therefore, NO generation by Ngb is controlled by the transition from six-to-five-coordinate heme state (138, 330). The authors also speculate that the six-coordinate heme globin superfamily may serve a function as primordial hypoxic and redox-regulated NO-signaling proteins. This hypothesis is in agreement with the observation that also mitochondrial cytochrome c can act as an NiR only in the five-coordinate state.
c. Xanthine oxidoreductase and aldehyde oxidase
In tissues, nitrite-derived NO production is due to the activity of both xanthine oxidoreductase (XOR) and aldehyde oxidase (AO) (aldehyde: O2 oxidoreductase), belonging to the family of molybdenum-containing hydroxylases (232), classified under a single EC number (EC 1.2.3.1). These enzymes require, for their catalytic activity, flavin adenine dinucleotide (FAD) and a particular form of organic molybdenum, known as the molybdenum cofactor (MoCo). MoCo is a molybdopterin in eukaryotes, while it is a molybdopterin nucleotide in prokaryotes.
XOR enzymes have been isolated from a wide range of organisms, from bacteria to human, and catalyze the hydroxylation of a wide variety of purine, pyrimidine, pterin, and aldehyde substrates. The mammalian enzymes, which catalyze the hydroxylation of hypoxanthine and xanthine, the last two steps in the formation of urate, are synthesized as the dehydrogenase form (xanthine dehydrogenase [XDH]) and exist mostly as such in the cell but can be readily converted to the oxidase form (xanthine oxidase [XO]) by reversible oxidation of critical cysteine residues (535 and 992) or limited proteolysis (108, 149). Conversion of XDH to XO is enhanced by hypoxic conditions and ischemia (299). XDH shows a preference for NAD+ reduction at the FAD reaction site, whereas XO fails to react with NAD+ and exclusively uses dioxygen as its substrate, leading to the formation of superoxide anion and H2O2.
Mammalian XO is a complex homodimer (Fig. 10); in addition to MoCo, two different [2Fe–2S] centers and one FAD are present in the enzyme (35, 148). The ability of XO to catalyze, under normoxic conditions, the reduction of nitrate is well recognized (93, 120, 235); evidence for Nar activity in an endothelial NO synthase–deficient and germ-free mice highlights the contribution of XOR to the overall nitrite levels and nitrite homeostasis (Fig. 6) (167). Expression of XOR in the liver is increased in germ-free mice compared to conventional animals, which may explain the apparently greater tissue Nar activity observed in the germ-free animals (155), representing a compensatory response to uphold nitrite homeostasis in the absence of commensal bacteria.

The XO-catalyzed reduction of nitrite to NO has also been reported over the last decade (132, 235, 376). This activity has been proposed to be a major source of NO in tissues (204) and to exert a protective role during myocardial infarction and ischemia-reperfusion (I/R) damage (13, 353). In rat and mouse models of pulmonary hypertension, sodium nitrite is converted to biologically active NO via reduction in large part by XOR; in these model systems, NO production was attenuated by allopurinol (200 μM), an inhibitor of XOR (379).
The reaction catalyzed by XO is
During the reduction of nitrite, one O2 atom is abstracted from the nitrite molecule, resulting in the production of NO: under anaerobic conditions, NADH and xanthine are used as reducing agents. Involvement of the molybdenum site of XO in nitrite reduction was shown by the fact that alloxanthine inhibits xanthine oxidation competitively with nitrite (132). Moura and coworkers have recently shown that the molybdenum metal center is the direct electron donor to nitrite (225). The Km for nitrite is in the mM range and the reduction is dependent on the concentration of O2, which acts as a competitive inhibitor (207). Despite these lines of evidence, under hypoxia and anoxia, the acidosis and the increased concentrations of xanthine and NADH will probably be sufficient to support the XO-mediated nitrite reduction physiologically, as supported by in vivo studies (13, 102, 353).
Interestingly, also the mammalian AO was shown to catalyze the reduction of nitrite to NO. AO is a cytosolic enzyme that plays an important role in the biotransformation of drugs and xenobiotics (371). AO is present in highest levels in the liver but is also broadly distributed in other tissues, such as lung, blood vessels, heart, and kidney (26, 247). Similarly to XOR, also AO contains two iron–sulfur clusters, a flavin cofactor, and a molybdopterin cofactor. The Km for nitrite of AO is 3 mM (205) but the affinity for NADH (Km=24 μM) is much higher than that of XOR (Km=0.9 mM). Therefore, this pathway would be predicted to better retain its nitrite reduction ability in the presence of O2: AO-mediated NO generation could exceed the NO generation from XOR in the lung and approach that from XOR in the heart and liver under anaerobic conditions.
d. Other mammalian proteins acting as NiR
In the last decade, multiple proteins, besides those discussed above, have been implicated in nitrite reduction to NO. As an example, nitrite reduction by mitochondria under low O2 concentrations has been reported and ascribed by different authors to Cox (mitochondrial respiratory complex IV), to mitochondrial complex III (191) and to the soluble electron carrier cytochrome c.
Castello et al. (60) have reported that yeast and rat liver mitochondria produce NO at O2 concentrations below 20 μM. This NO production is nitrite dependent, is carried out by Cox in a pH-dependent fashion, and is accompanied by an increase in protein tyrosine nitration. The ability of Cox to reduce nitrite in yeast can be modulated by O2 by altering the subunit composition of the complex: the presence of the isoform COX5b of subunit V, preferentially expressed at low O2 tensions, enhances NO production (61). The authors suggest a positive feedback mechanism in which mitochondrially produced NO induces expression of COX5b, whose protein product then functions to enhance the ability of Cox to produce NO in hypoxic/anoxic cells. The NO generated in the mitochondria by Cox might be released from cells, thereby reaching external targets (280). Interestingly, it has been reported that the nitrite-derived NO synthesis catalyzed by Cox is enhanced by low intensity light, offering a new explanation for the increase in NO bioavailability experienced by tissue exposed to light (14, 279).
In agreement to what has been observed for the six-coordinate hemoprotein Ngb previously discussed, also cytochrome c is able to reduce nitrite to NO when in the five-coordinate state (22, 62). These data ascribe a possible role for cytochrome c as an NiR, possibly relevant in the hypoxic, redox, and apoptotic signaling pathways within the cell.
Other possible NiR at the tissue level include the ubiquitous enzyme carbonic anhydrase (CA), a crucial player in CO2 transport (1), and rat liver cytochrome P450s (206), a family of proteins involved in the metabolism of xenobiotics (including organic nitrates). In the latter case, mammalian cytochrome P450 reductase (CPR) and cytochrome P450 cooperate to function in a sequential manner to produce nitrite and then NO and nitrosothiols, serving as the link between organic nitrates and NO-mediated signaling.
Altogether, these lines of evidence reinforce the idea that multiple proteins may function as NiR under low O2 tension; however, the precise regulatory pathways controlling these activities is still far from being understood.
IV. Significance of Nitrite in Human Health and Disease
In this section we first discuss the involvement of nitrite in vasodilation, focusing on its role as a source of NO (section A); then, we analyze the cytoprotective effects of NO in the I/R injury and the role of nitrite as endocrine reservoir of NO (section B); finally, we briefly describe the other NO-independent physiological activities of nitrite (section C).
A. Nitrite in vasodilation
The primary function of vasodilation is to increase blood flow in the body to tissues that need it most; if the supply of O2 is not sufficient, for example, in a working muscle or under hypoxia, an increase of O2 delivery must occur, to selectively distribute the blood according to variable needs (90, 303, 339).
Recent evidence suggests that nitrite is a putative physiological signaling molecule with a potential role in hypoxic vasodilation, signaling, and cytoprotection after I/R (130). Several studies have suggested and confirmed the involvement of nitrite in vasodilation in humans, also in the presence of NOS inhibitors (74); the effect of nitrite has been confirmed in mouse, rat, sheep, and primates (29, 88, 338). Interestingly, intra-arterial infusion of nitrite during normoxia and hypoxia has shown that arteries are modestly affected under normoxic conditions, but are potently dilated under hypoxic conditions (224).
Nitrite-dependent effects on vasodilation are mostly mediated by NO, which stimulates soluble guanylate cyclase (sGC), thereby increasing cGMP levels, activating cGMP-dependent protein kinase and producing smooth muscle relaxation (130, 241). A physiologic NO-dependent post-translational regulation of vascular sGC in mammals involving S-nitrosylation as a key mechanism has recently been suggested (264). NO can also relax smooth muscle by cyclic GMP-independent mechanisms, including the direct modifications of sarco/endoplasmic reticulum calcium ATPase (SERCA), the enzyme essential for the control of intracellular free Ca2+ levels. NO modification causes, in smooth muscle cells, reduction of intracellular Ca2+ levels and, consequently, vascular relaxation (331). It has been demonstrated that NO, via ONOO– and N2O3 formation, can adduct GSH to SERCA cysteine thiol; this modification predominantly activates SERCA and refills Ca2+ stores in sarcoplasmic reticulum. This NO-dependent mechanism of SERCA modification is crucial for proper vascular relaxation; in altered redox-state background such as in atherosclerotic arteries, NO does not stimulate SERCA activity because of the irreversible oxidation of the target cysteine thiol by the high levels of oxidants accompanying the disease (331).
A key role in the control of hypoxic vasodilation by nitrite has been assigned to the erythrocyte (100, 107, 130), mainly due to the NiR activity of deoxyHb, previously discussed (74, 91, 170). An open question is how NO escapes the erythrocytes to exert its vasodilatatory effect. Different potential mechanisms could be operative (130): one hypothesis suggests that the NO escape is inefficient, but sufficient to regulate vascular tone due to lipophilicity and potency of the molecule. Another hypothesis is that the erythrocyte membrane provides a potential NiR metabolon, a system that would concentrate chemical reactants, nitrite, protons, and deoxyheme with highly hydrophobic channels. A third solution is that nitrite reduction produces intermediate(s) that could facilitate the transport of NO, such as N2O3. Finally, the formation of S-nitrosothiols can also occur (130, 170). The formation of Hb derivatives, such as the S-nitrosated Hb, as a possible reservoir of NO bioactivity has been proposed (172, 321), but was recently critically analyzed and disputed by in vivo experiments in rats (168).
Nitrite-dependent vasodilation at low pH may also occur in a protein-independent pathway. For more than a century, it was known that acidic conditions allow vascular smooth muscle relaxation; indeed, to date, it has been suggested that abiotic reduction of inorganic nitrite to NO, a phenomenon known as acidic metabolic vasodilation, regulates local blood flow under hypoxia or ischemia (213, 238).
B. Nitrite-based cytoprotection in I/R injury
I/R injury is the major cause of death and illness in the Western World (281). This injury consists of multifaceted cellular events that take place on the recovery of O2 delivery after a period of hypoxia. The heart, the kidneys, and the brain are among the organs that are the most quickly damaged by loss of blood flow. Insufficient blood supply to the myocardium can lead to myocardial ischemia infarction; timely restoration of the blood flow to the acute ischemic myocardium is essential to reduce morbidity and mortality of the patients. However, the process of reperfusion after an ischemic episode can paradoxically lead to a unique form of damage, termed myocardial reperfusion injury. As initially observed by Hearse et al. (141), the reoxygenation required during reperfusion of ischemic myocardium generates reactive oxygen species (ROS), which trigger cellular injury (340, 341).
One of the most effective strategies, when applicable, to preserve tissues from I/R damage is the ischemia preconditioning, consisting of short periods of ischemia followed by reperfusion before a long ischemic period (250, 281); this setting restricts its potential clinical utility to planned acute myocardial I/R injury such as coronary artery bypass graft surgery, cardiac transplantation, and percutaneous coronary intervention (355). The ischemia preconditioning guarantees the activation of the protective signaling pathway (ROS-induced, see below) at the time of the reperfusion, which is able to reduce tissue infarction significantly (32, 281); the same protective effect (reduction of infarct size and attenuation in inflammatory response) can be obtained by ischemic postconditioning (378).
Under normoxic conditions, the mitochondrial respiratory chain produces a small amount of ROS and reactive nitrogen species, which are scavenged by different antioxidant enzymatic systems and compounds (24). On the other hand, after ischemia, reperfusion of ischemic tissue leads to an exceptional production of ROS: this oxidative burst depletes or damages the pool of antioxidants available, causing tissue injury (106, 281, 377). More in detail, during ischemia, the electron transport chain functions as a reservoir of electrons since O2 availability is limited. This high reductive potential, particularly for complex I and III, promotes incomplete O2 reduction, and thus radical formation, when a massive entrance of O2 occurs during reperfusion (47). Incomplete O2 reduction during reperfusion leads to the production of superoxide anion, H2O2, and hydroxyl radical to levels that are 1 to 2 orders of magnitude higher than those detected in nonischemic background (381).
It should be mentioned that other enzymes partecipate in ROS generation during I/R injury, such as NADPH oxidase, NOS (during substrates depletion), and XO; in the latter case, during ischemia, ATP is predominalty metabolized as hypoxanthine and xanthine, which reacts with XO (whose formation from XDH is favored in postischemic heart) upon reoxygenation, resulting in superoxide generation (381).
In the last years, many studies demonstrated that NO production is associated with cytoprotection against I/R injury in many organs, such as the heart, liver, lungs, and kidneys (174, 275). One of the first evidence associating NO with cardioprotective effect was obtained 20 years ago by using acidified nitrite, which was shown to exert a significant protective action during ischemia and reperfusion injury (173). A relative decrease in NO bioavailability appears to be central in the pathogenesis of this injury, and therapeutic strategies aimed at replacing NO by inhalation, nitrite/nitrate anion supplementation, or via donor drugs represent a novel means for ameliorating I/R injury (275, 318).
The mechanism activated during ischemic postconditioning to minimize ROS-dependent tissue damages during I/R injury has been extensively studied in last two decades (91); even though the molecular details controlling this phenomenon remain to be deciphered, Bolli and coworkers demonstrated that NO plays a prominent role in mediating the cardioprotective response (33). The authors demonstrated that a brief ischemic stress causes a burst of NO production as well as·O2 –production, which in turn could then react to form ONOO–; this reaction triggers a complex signal cascade involving the protein kinase C (PKCɛ) and the transcription factor NF-kB (33). ONOO– formation prevents further ROS production occurring via the Fenton reaction and, despite the cytotoxic effects observed in the presence of excess of ONOO–, its decomposition produces intermediates, which are scavenged by NO itself (359). This reaction ultimately results in production of the nitrosating species N2O3, which in turn contributes to the overall NO-dependent cellular response (359). As demonstrated both in vivo and in vitro by using chemical models, this nitrosative stress provide an optimal antioxidant environment rather than toxic species, indicating that NO acts to counterbalance oxidative stress, beside its role in controlling signaling events via ONOO– formation (85, 328).
NO modulates a plethora of processes during I/R injury, including inflammation, ROS formation, and apoptosis (Fig. 11). NO can also exert its cytoprotective role by inhibiting caspase through S-nitrosation and via cGMP to avoid the release of cytochrome c from mitochondria, thus preventing apoptosis (91). NO inhibits calcium overloading, responsible for the opening of a nonselective mitochondrial permeability transition pore, which uncouples oxidative phosphorylation and worsens ions/energetic homeostasis, leading to cell death by necrosis or apoptosis (277). At the same time, it is known that NO reversibly inhibits complex IV (40, 72) and, more importantly, complex I (38). Inactivation of complex I involves mainly S-nitrosation of critical thiols (68); since this modification is reversible, the inhibition of complex I is slowly reduced over time, thus allowing the respiratory chain to gradually recover the normal electron transfer between complexes, without the deleterious instigation of oxidative damage (248) (see below). Inhibition of complex I activity seems to be crucial for cytoprotection during I/R injury being the inhibited form of this enzyme unable to generate ROS (47) and involved in limiting calcium overload (277). Moreover, as mentioned above, the respiratory chain is directly involved in the induction of some hypoxic nuclear genes (hypoxic signaling); as proposed recently (60), mitochondrially produced NO functions in a signaling pathway to the nucleus via ONOO−, which may directly or indirectly modify specific proteins and activate hypoxic signaling.
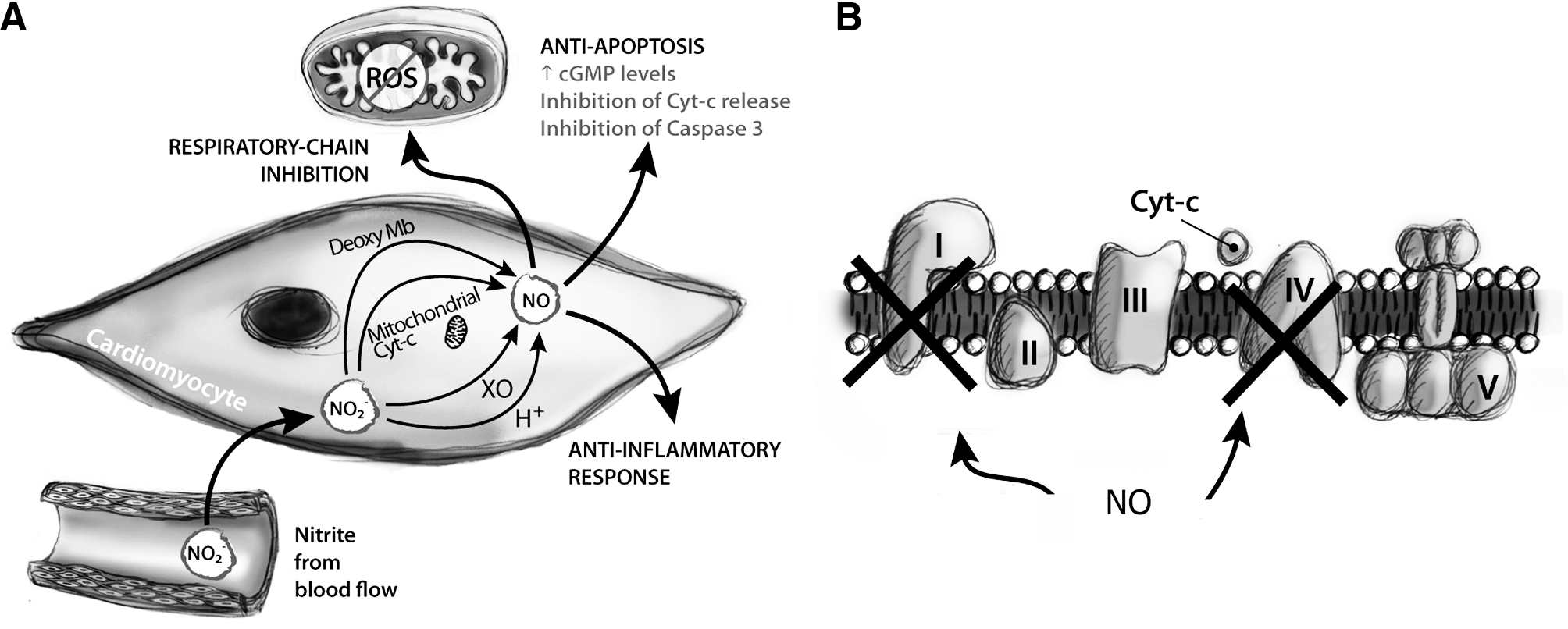
Thus, the concept of reversible ROS inhibition in mitochondria during the early phases of reperfusion may represent an intriguing therapeutic chance to minimize tissue damages caused by I/R injury. In hypoxia, it is plausible that nitrite functions as an alternative NO source under those conditions. As previously reported, Zweier and coworkers in 1995 demonstrated for the first time the pivotal role of nitrite as source of NO (382); the finding that nitrite may act as endocrine reservoir of NO has prompted many studies demonstrating the capability of nitrite to mediate potent cytoprotection (via NO) in a number of organs and animal models of I/R injury [see (102)], also by modulating mitochondrial electron transfer (313).
The pathway by which nitrite forms NO in hypoxic tissue remains to be determined; several mechanisms have been proposed/identified over the years (281).
Two groups suggested the involvement of XO on the basis of reduced efficacy after treatment with allopurinol, an XO inhibitor (336, 353). Other groups have shown the involvement of Mb in nitrite conversion to NO and thus in cytoprotection. Addition of nitrite to rat heart homogenates containing both Mb and mitochondria resulted in NO generation and inhibition of respiration; these effects were blocked by Mb oxidation with ferricyanide but not by the addition of XO inhibitor allopurinol (312). Moreover, intracoronary application of nitrite in wild-type and myo(−/−) mice (285) clearly showed that the NO generated by reaction of deoxyMb with nitrite is functionally relevant and leads to a downregulation of cardiac energy status, not observed in mice lacking Mb. The involvement of other hemoproteins and mitochondrial heme-containing complexes (such as cytochrome P450 and complex IV) in nitrite reduction has also been demonstrated (281).
However, even though the strategy of NO production is not unequivocally identified, enough NO seems to be produced from nitrite either directly within the mitochondria (22, 60, 61) or in the cytoplasm, as it occurs in myocytes where deoxyMb acts as an NiR (75, 145).
The discovery that nitrite may act as an endocrine reservoir of NO has suggested the idea that in the future this anion may represent an alternative strategy for an effective NO-based therapy by acting as an NO prodrug (181, 324). An overview of the main results foused on nitrite-mediated cytoprotection and more in general on the state-of-the-art of nitrite therapy is reported in chapter V.
C. Other activities of nitrite
To date, the majority of studies have described the nitrite effect as NO-dependent, but a novel NO-independent role of nitrite has been recently suggested, which may involve nitrite-mediated nitrosation without passing through a free NO intermediate (113). Nitrite-mediated nitrosation may protect tissues against inflammation (349).
Finally, a global role of nitrite in hypoxic signaling has been described: it affects cyclic-GMP production, cytochrome P450 activity, heat shock protein 70, and heme oxygenase-1 expression in a variety of tissues (43). These cellular activities of nitrite, in addition to its stability and abundance in vivo, suggest that this anion has a distinct and important role in mammalian biology, perhaps by serving as an endocrine messenger and synchronizing agent.
All these unexpected roles of nitrite in human physiology have opened the research to the possibility of therapeutic application associated with hypoxia and vasoconstriction.
V. Nitrite Therapy
In this section we discuss the advantages and the applications of nitrite-based therapy. We first analyze its feasibility and the pharmacokinetics of nitrite (section A); than, we discuss the tolerance and the toxicity of the compound (section B); finally, we described the most recent results concerning the use of nitrite in therapy (section C).
In 2006, David Lefer, a leader in I/R injury research, stated in an Editorial Focus that “the field of nitrite chemistry and biology is a truly exciting area of research that is certain to expand in the near future and lead to a dramatically improved understanding of the physiology of NO synthases and NO in terms of cytoprotection” (202).
It is now clear that reduction of nitrite to form NO under both physiological and pathological conditions is a challenging topic in medicinal chemistry (50). The potential of nitrite-based therapy is indeed clearly demonstrated by the rapidly growing number of studies on animal models of injury, the review articles (58, 91, 177, 248, 259, 316), and special issues (such as volume 89, issue 3, of Cardiovascular Research) on this topic, by the clinical trials aimed at investigating the action of nitrite in protection [ClinicalTrials.gov and (181)] and by the number of patents (e.g., uspto.gov).
The state of the art on the activity of nitrite under physiological and pathological states and on the translation of nitrite and nitrate research for clinical applications has been presented, from 2005 and every 2 years, during a dedicated international meeting held in Atlanta (for info on past meetings, see
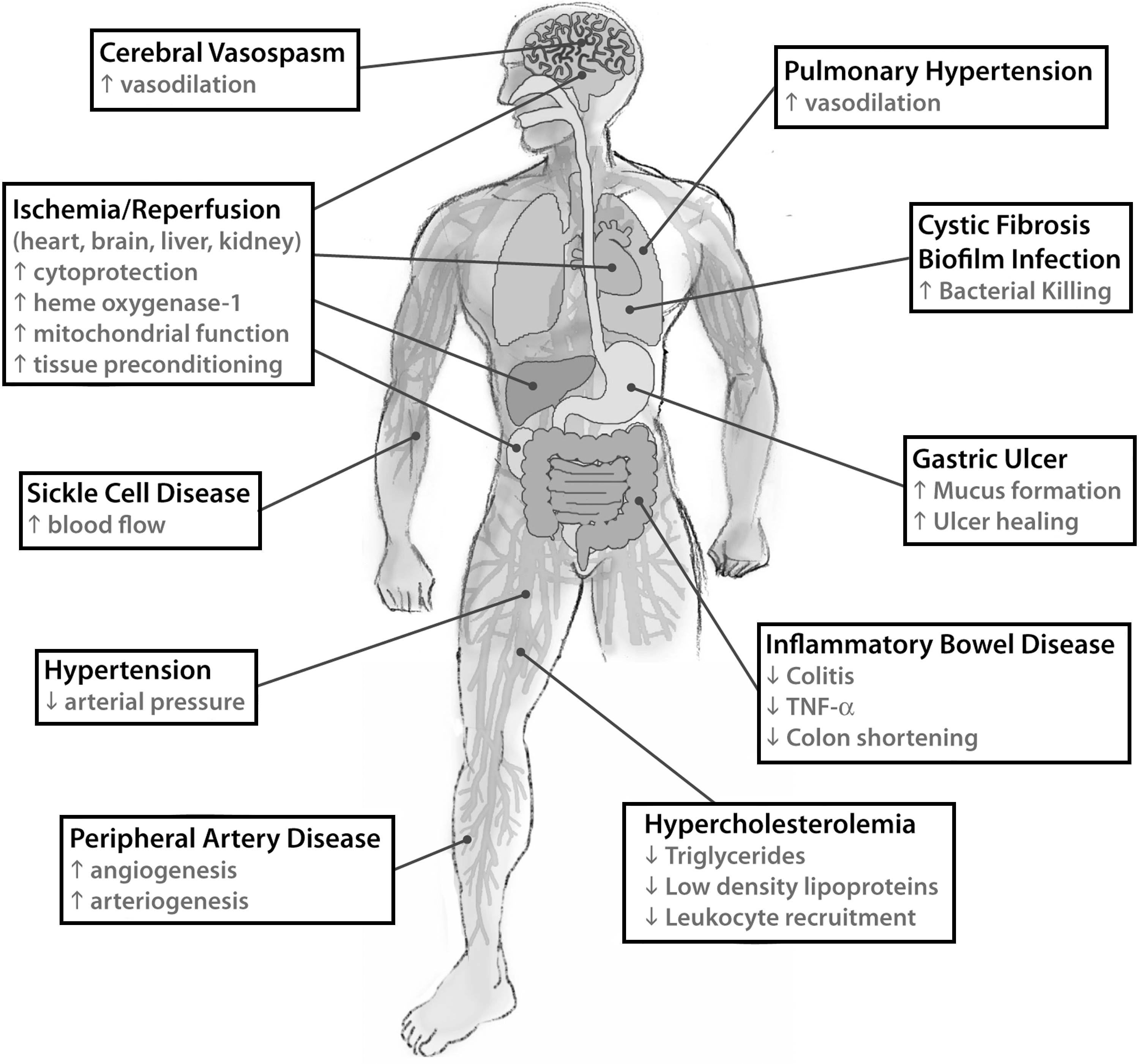
The advantage to use nitrite therapy instead of NO therapy is closely related to the high stability of nitrite, which can be transported in the circulation and stored in tissues. Moreover, since nitrite reduction occurs under conditions close to those of injured tissues (ischemia, hypoxia, and low pH), nitrite-dependent NO release targets injured tissues preferentially, thereby reducing the risk of systemic side effects (51). A summary of the possible effects of nitrite-based therapy is given in Figure 12.
Currently, NO therapy has been exploited via inhaled NO (iNO) gas from pressurized tanks; this approach is, to date, the preferred and the only approved NO treatment for acute pulmonary hypertension (78, 92, 372) even though it is inconvenient and onerous (92, 160). Alternatively, other strategies imply compounds containing either NO or an NO precursor in a stable form (178, 195), which may represent a clinical promise once it will be possible to control carefully the sustained delivery (49).
However, contrary to NO, nitrite allows multiple adiministration strategies, including oral, topical, intravenous, intraperitoneal, and aerosolized.
A. Pharmacokinetics and feasibility
It is known that nitrite intake occurring via nitrate as dietary source leads to its rapid absorption in the duodenum and jejunum and distribution in the whole body (18, 181, 296) (Fig. 7). Studies focused on nitrite administration/infusion have been obtained recently on both nonhuman primates (87) and human volunteers (87, 158). These experiments demonstrated that infused nitrite is a rapid vasodilator at physiological concentration and that nitrite-induced effects in terms of forearm blood flow are immediate (ranging from 15 to 60 sec, depending on the dose) (87). Upon infusion, nitrite levels increased to micromolar levels and then decayed with an apparent biological half-life of 42 min, while nitrite-induced hypotension lasted for 3 h (87).
A detailed study focused on the pharmacokinetics of ingested nitrite by human volunteers has shown that nitrite is able to reach the systemic circulation (158).
Very recently, the outcomes of a completed clinical trial, developed to determine the safety and feasibility of prolonged sodium nitrite infusion, have been published (278); this study provided pharmacokinetic and toxicity parameters needed for using nitrite as a therapeutic agent, such as maximal tolerated dose and dose-limited toxicity for long-term intravenous infusion of sodium nitrite (266.9 and 445.7 mg/kg/h, respectively) and the mean half-life of nitrite in plasma and whole blood (45.3 and 51.4 min, respectively). More interestingly, the small increase of metHb seen in this study was asymptomatic and the decrease of blood pressure was transient (278). This study suggests that prolonged intravenous infusion of sodium nitrite (at doses within the maximal tolerated dose) is safe and should provide the proper amount of nitrite required to exert its therapeutic role (278).
B. Tolerance and toxicity
It is worth to notice that, contrary to organic nitrate class of drugs such as nitroglycerin (327), continous sodium nitrite administration in nonhuman primates does not induce tolerance (87), that is, decrease of sensitivity of vascular smooth muscle to further vasorelaxation. In 2002 Ignarro underlined how nitroglycerine tolerance limits the chronic use of such compounds to treat angina pectoris and how “the discovery either of ways to avoid tolerance or of new NO-generating drugs that do not cause tolerance” is of great interest (161). Nitroglycerin tolerance arises likely from the inhibition of the enzymes involved in nitroglycerin bioactivation, such as the mitochondrial aldehyde dehydrogenase, cytochrome P450, and glutathione-S-transferase, which function also as nitroglycerin reductases (63, 121, 237). The capability of nitrite to bypass the enzymatic tolerance may represent a convenient alternative to organic nitrate therapy, as wished by Ignarro about 10 years ago.
Since more than 95% of orally ingested nitrite is absorbed, its toxicity during extended exposure to nitrite was evaluated. The intake of nitrite has been historically associated with poisoning due to methemoglobinemia (52, 229); this condition can interfere with the ability of blood cells to carry O2 when metHb concentrations reach 20%–30% of total Hb concentrations. In healthy individuals without anemia, acquired methemoglobinemia causes few symptoms at 15% metHb, while levels of 20%–30% metHb cause headache, fatigue, and syncope; at levels of ∼50% dys-rhythmias, coma and death occur (165). Infantile methemoglobinaemia has been associated with feeding reconstituted with well waters rich in nitrate (70, 95).
However, after intravenous administration of nitrite, the maximum percent metHb in blood was between 3% and 12% and ∼4% after oral administration (87, 158); in both cases the maximum metHb level was reached about 0.8–1.2 h after intravenous or oral administration (87, 158). These results indicate that nitrite levels able to induce vasodilation do not cause clinically significant methemoglobinemia, probably thanks to the coupled methemoglobin reductase activity of the NADH-cytochrome b5 reductase, previously discussed.
Oral administration of nitrite has been causally associated with gastrointestinal cancer (229). It has been postulated that, under strong acidic conditions, swallowed salivary nitrite may nitrosate via HNO2 secondary amines ingested in food to form nitrosamines (309); some of these nitrosamine have been found to be carcinogenic in animal and in epidemiological studies (20, 189, 211). However, the direct involvement of swallowed salivary nitrite [via nitrate reduction, (101)] in promoting gastrointestinal cancer has not been demonstrated (189); all available data indicate that there is not a direct causative role of nitrate in gastric cancer (229), possibly because food ingestion promotes an increase in pH to a value that hampers secondary amines nitrosation, a reaction preferentially occurring at pH 2.2–3.5 (19).
In conclusion, all data available to date on pharmacokinetics and toxicity of nitrite strongly support the use of nitrite therapy as a strategy to bypass the limitations of the NO therapy (51).
C. Effects of nitrate/nitrite administration and the therapeutic potential
Nitrite-based therapy has been used to control vasodilation and cytoprotection after hypoxic damage, but also inflammatory disease and host defense processes (181, 229). Accordingly, experimental data can be clustered into three main groups: • Nitrite as a cardiovascular drug • Nitrite use for inflammatory diseases • Acidified nitrite as a tool against bacterial pathogens
A summary on these topics is reported below, with special attention to the use of nitrite therapy in cardiovascular diseases.
1. Nitrite as a cardiovascular drug
A growing number of studies reported the effects of nitrite administration on protection against I/R injury (176, 201, 317, 353), platelet function inhibition (222, 354), vasodilation (74, 131), reduction of blood pressure (127, 197), and increase of cGMP levels (43). All these results are in agreement with the evidence that most of the cardiovascular risk factors are associated with reduced NO availability in the vasculature (150, 256). Most of the studies indeed demonstrated that the protective effect of nitrite or dietary nitrate is significantly lowered by the presence of NO scavenger such as carboxy-PTIO and Hb (176, 194, 222).
Webb and coworkers (353) assembled 7 years ago a specially adapted NO-collecting chamber placed around isolated perfused rat heart to measure NO concentration, using the ozone chemiluminescence detection method previously developed by other groups (115, 186); this chamber was also employed to measure NO production in homogenized myocardium (rat and human). In both experimental settings, a significant NO production was observed upon addition of sodium nitrite (10–100 μM) at pH 5–6, a condition typical of myocardial ischemia (353). Nitrite significantly reduced infarct size (∼50%); the cytoprotective effect is abolished in the presence of NO scavengers (353).
a. Ischemia/reperfusion injury
The efficacy of nitrite administration in limiting I/R damages has been investigated extensively in myocardial and brain ischemia, as much as in liver and kidney; the main outcome of these studies is that the amount and the way of administration of nitrite are crucial to exert its protective role as therapeutics upon ischemic damage.
Duranski and collegues performed studies in mice to evaluate the cardioprotective effects of acute nitrite therapy in the setting of coronary artery occlusion and reperfusion, exploring different nitrite dosages (2.4–1920 nmol) (102). The administration of nitrite into the left ventricular cavity 5 min before reperfusion significantly limited myocardial infarct size, in a dose-dependent manner with a maximal protective effect at 48 nmol of nitrite (102). Low doses of intravenous nitrite improved microvascular perfusion, reduced apoptosis, and improved contractile function (133). Nitrite can also exert potent cytoprotective effects after I/R injury on liver in a dose-dependent fashion by limiting serum increase of the liver transaminases, hepatocellular injury, and apoptosis (102).
The effect of dose and duration of nitrite/NO exposure is critical, resulting in a precise setting and timing for NO-mediated cytoprotection in I/R pathophysiology. Different experimental protocols may give contradictory results, as shown in studies focused on the treatment of ischemic brain in rats by two different research groups. The group of Jung and coworkers (176) observed a significant reduction in infarct size, while Schatlo and collegues did not observe the dose-protective effect of nitrite in ischemic brain (307). These contradictory results may arise from differences in the experimental setting employed by the two research groups, in terms of both duration of nitrite administration and age of the animals.
The route of administration may also explain discrepancies in studies on the efficacy of nitrite therapy. A clear example is represented by the results obtained on a rat model of I/R-induced renal injury by two different groups, both using the same amount and timing of nitrite application. Basireddy and collegues observed that the intravenous or intraperitoneal administration of nitrite did not provide protection upon I/R induced renal injury (21), in contrast to its beneficial effects in cardiac and hepatic I/R injury (102, 133, 353). Tripatara et al. confirmed this result but were also able to attenuate the degree of renal dysfunction, reperfusion injury, glomerular dysfunction, and tubular injury upon topical administration of nitrite (336).
Oral nitrite administration in the treatment of I/R injury after transplantation was shown to improve cardiac allograft outcomes in rats by lowering allograft inflammation, apoptosis, and alloimmune responses (374). Nitrite administration resulted in prolonged allograft survival, while reduced oral intake of NOx promoted rejection (374). Accordingly, administration of iNO (80 ppm) to patients undergoing orthotopic liver transplantation inhibits hepatic I/R injury (196). The concentration of NO metabolites (including nitrate and nitrite) was significantly increased in these patients, in agreement with other studies on iNO administration (253); Lang and coworkers (196) suggested that, in their study, the extrapulmonary effects of iNO were mainly due to nitrite.
b. Cardiovascular disorders
The therapeutic potential of nitrite in the field of cardiovascular diseases has been also evaluated for disorders associated with reduced NO availability, such as chronic tissue ischemia (31), sickle cell disease (SCD) (227), and pulmonary arterial hypertension (PAH) (79). Nitrite therapy has been studied also as a strategy to control vasodilation and hypertension (127, 181, 354).
Chronic tissue ischemia is mainly caused by peripheral arterial disease (PAD) (181, 267), responsible for hypertension, atherosclerosis, and diabetes (181). Currently, the treatment of such pathological status is based on the employment of anti-platelet agents and statins (267). The capability of nitrite to promote angiogenesis as a therapeutic strategy to treat PAD and more in general chronic tissue ischemia is a challenging issue; since NO plays an important role in stimulating angiogenesis (71), the effect of nitrite therapy in controlling ischemia-induced angiogenesis has been analyzed (194). Nitrite was found to improve angiogenesis in terms of increase in the quantity of endothelial cells and in the vascular density (194). A dose-dependent effect of nitrite in the increase of ischemic tissue blood flow in mice was observed (310). The capability of nitrite to restore ischemic blood flow was significantly impared by the presence of NO scavenger such as carboxy-PTIO (194). Interestingly, the capability of nitrite to augment angiogenesis was also demonstrated in the neural regeneration stimulated upon prolonged nitrite therapy in the ischemic brains (177).
The possibility to employ nitrite as source of NO in managing PAD has been supported also indirectly; pharmacological treatment of permanent tissue ischemia with the antiplatelet drug dipyridamole augments ischemic tissue reperfusion, promotes endothelial nitric oxide synthase (eNOS) activity, and, as a consequence, increases nitrite levels in ischemic muscle tissue (269). Nitrite reduction may enhance the vascular growth observed upon dipyridamole treatment (346), confirming the presence of a nitrite/NO endocrine system able to produce benefits not only locally but also in distal tissues (267).
As mentioned above, the reduced NO bioavailability also represents one of the pathological trait of SCD, a genetic disease due to a point mutation of the beta-chain of Hb (227); these patients accumulate huge amounts of free Hb, which reduces NO bioavailability (286, 348). The possibility to develop therapeutic strategies aimed at optimizing NO delivery has been considered at least 10 years ago (242); a recent phase I/II study demonstrated that sodium nitrite infusion into the brachial artery augmented in a dose-dependent manner the mean venous plasma nitrite concentration and stimulated forearm blood flow (223). Since this treatment was well tolerated (lack of hypotension and significative methemoglobinemia), nitrite could represent a plausible NO donor for future clinical trials in SCD (223).
The very efficient NO scavenging property of oxyHb (175) represents a limitation in using blood substitutes, where a secondary hypertension is associated with scavenging of NO by Hb-based oxygen carriers (HBOCs). A novel therapeutic strategy to attenuate this side effect has been proposed based on the use of nitrite as an adjunct therapy to attenuate HBOC-dependent hypertension (298).
Impaired NO bioavailabilty plays an important role also in the development of PAH, a chronic and progressive disease characterized by a persistent elevation of pulmonary artery pressure that progressively leads to right heart failure and ultimately death (79). Currently, iNO therapy is only approved for use in infants who have respiratory distress syndrome (140) even though the short-term effects and the elevated cost of such therapy made researchers to explore novel strategies (79). The administration of nebulized nitrite to treat hypoxic pulmonary hypertension in newborn lambs decreases the pulmonary artery pressure without measurable effect on mean arterial blood (159). More recently, the beneficial effects of nebulized nitrite under hypoxia or monocrotaline-induced PAH were confirmed in mice and rats (379). This therapeutic approach completely prevented or reversed PAH and pathological right ventricular hypertrophy and failure (379).
The evidence that nitrite may really target pulmonary circulation preferentially under hypoxic conditions has been verified on healthy human volunteers (162). The study was done in a hypoxic chamber (inspired O2, 12%; room air as control) where each volunteer received an infusion of sodium nitrite (1 μmol/min). During hypoxia, nitrite increased forearm blood flow (+36%) and reduced pulmonary arterial pressure (12%–17%). Interestingly, pulmonary, but not systemic, arterial effects persisted 1 h after stopping the infusion, in conjunction with plasma nitrite levels lowered to the baseline values (162).
It should be pointed out that, even though rapid nitrate administration is largely ineffective in the pathological settings discussed above (as most of these studies used nitrate as negative control), recent data report that sustained delivery of nitrate (over days/weeks) may represent an alternative strategy to increase plasma nitrite and achieve protection. Dietary nitrate can enhance exercise performance in PAD patients (179), representing a chance for these patients, where plasma nitrite levels could be indicative of vascular health and function (3).
The effects of dietary nitrate have been also analyzed in different pathological backgrounds. Dietary supplementation of nitrate was used in eNOS-deficient mice, which presents several features of the metabolic syndrome (55), a cardiovascular disorder associated with reduced NO bioavailability from eNOS, with increased cardiovascular risk and type 2 diabetes (117, 243). Dietary nitrate treatment (7–10 weeks) reduced both fat accumulation and circulating triglycerides; moreover, it reversed the prediabetic phenotype by improving glucose tolerance and reducing fasting blood glucose (55).
Nitrate/nitrite-derived NO may also control blood pressure (197). Dietary supplementation with sodium nitrate (0.1 mmol/Kg per day for 3 days) in healthy volunteers decreases the mean arterial pressure (by 3.2 mmHg) and increases plasma levels of both nitrate and nitrite (197). The effect of dietary nitrate has been recently analyzed in a model of renal and cardiovascular disease (56); chronic dietary nitrate supplementation attenuates hypertension and prevents the development of cardiac and renal damage (56).
Dietary nitrite therapy may play an important role in preventing cardiovascular disease associated with aging, such as vascular endothelial dysfunction and large elastic artery stiffening (316). Nitrite supplementation in old and young mice showed that the treatment ameliorated carotid artery endothelial dysfunction and reduced large elastic artery stiffness in old mice to a level similar to that of young mice, confirming the translational potential of nitrite administration to prevent cardiovascular disorders associated with aging (316).
2. Nitrite in inflammatory diseases
There is growing experimental evidence that inorganic nitrite therapy confers substantial benefit to numerous pathophysiological conditions includes experimental colitis, hypercholesterolemia, and gastric ulcer.
Colitis is a chronic inflammatory disorder of the intestinal tract characterized by mucosal inflammation of the large bowel (217) with excessive production of cytokines, adhesion molecules, and ROS (334). In a colitis model in mice (263), colonic nitrite levels decrease with concomitant increase in colonic TNF-α expression followed by increased iNOS and heme oxygenase-1 expression (262). Oral sodium nitrite administration (6.4–7.2 mg/mouse/day) attenuates acute inflammatory flares of colitis by preserving tissue NO bioavailability, as suggested by the increase in nitrite colonic levels and decrease in TNF-α expression (262).
Nitrite has shown a novel role as a therapeutic agent also to contrast microvascular inflammation and endothelial dysfunction associated with hypercholesterolemia (322). The hypercholesterolemia proinflammatory phenotype is characterized by a decline in NO bioavailability and increase of ROS production. Nitrite administration is able to restore NO biochemical homeostasis and significantly attenuates hypercholesterolemia-mediated leukocyte recruitment, which plays a key role in controlling vascular inflammation (262).
A recent whole genome array analysis (268), combined with previous data of nitrite-dependent induction of angiogenesis (194), suggests that nitrite therapy may also facilitate vascular remodeling during chronic tissue ischemia by modulating inflammatory gene expression. Nitrite-treated animals (330 μg/kg/day) present a significant decrease in inflammatory gene expression concomitant with an increase in genes that function to protect and preserve skeletal muscle tissue (268). Moreover, nitrite administration upregulates several genes involved in innate immune responses, such as the macrophage migration inhibitor factor and the heat shock protein 90 class B member (Hsp90ab1) (269). Since Hsp90ab1 promotes eNOS activation, the final outcome is NO production in ischemic tissue (123). On the other hand, nitrite also significantly downregulates other genes associated with innate immunity, for example, IL-10 (269). Several studies (84, 124, 254, 300) suggest that IL-10 expression acts in an antiangiogenic manner in several pathological events, including chronic tissue ischemia; therefore, the nitrite therapy likely augments reperfusion in chronic tissue ischemia caused by this regulation of innate inflammation response.
3. Acidified nitrite as a tool against bacterial pathogens
As mentioned before, under acidic conditions, sodium nitrite forms HNO2 that, in the presence of O2, generates reactive nitrogen intermediates, including ONOO–, N2O3, NO2•, and NO (276). These small molecules readily cross cell membranes, where they can react with reduced thiols to form nitrosothiols, which are thought to be important in microbial killing (112).
A common perception of gastrointestinal host defence to pathogens ingested with food and water is that it depends entirely on immune factors secreted in the saliva and gastric acidity itself. However, for many gastrointestinal pathogens such as Campylobacter, Shigella, and Salmonella, even prolonged immersion in strong acid at pH 2, to emulate the normal fasting stomach, exerts only a bacteriostatic effect (104). Failure to kill these organisms means that viable pathogens would pass to the small intestine if the defence depended on acid alone. Salivary and gastric nitrites, converted to reactive nitrogen compounds under acidic conditions, may enhance the killing of pathogens in the stomach (103, 104, 283). An in vitro study (283) demonstrated that normal stomach acidity in combination with physiological concentrations of nitrite are required to kill Clostridium difficile spores that were not killed in acidic buffers or gastric content; moreover, nitrite enhances the killing of several nosocominal pathogens, including vancomycin-resistant Enterococcus spp., methicillin-resistant Staphylococcus aureus, and Candida glabrata (283). These data suggest that interventions to limit the overuse of acid-suppressive medications could potentially have an impact on multiple pathogens. For example, supplementing the diets of hospitalized patients with nitrates could bolster gastric defenses by increasing levels of acidified nitrite (103, 104).
Recent data (226) suggest that the nitrite could be used in the treatment of chronic bacterial infections caused by opportunistic pathogens such as P. aeruginosa, S. aureus, and Burkholderia capacia in the airways of CF patients. The most dangerous form of P. aeruginosa is the mucoid P. aeruginosa characterized by a mutation within the mucA gene and consequently by the overproduction of alginate, an exopolysaccharide that inhibits the diffusion of O2 and antibiotics. As previously mentioned, mucoid P. aeruginosa biofilms are inherently resistant to antibiotics (134) and phagocytic neutrophils (48). Acidified nitrite (∼15 mM) kills the mucoid form of P. aeruginosa (370), inhibits nonmucoid planktonic P. aeruginosa PAO1, and kills S. aureus and, unexpectedly, planktonic and biofilm communities of Burkolderia cepacia (226). Although the concentration of acidified nitrite necessary for the antimicrobial effect is quite high, it is worth mentioning that 10 to 40 mg/kg of acidified nitrite are typically used in the cure of meat products (308), suggesting that humans have a high tolerance for nitrite, as discussed above. Therefore, acidified nitrite could be used for the clinical treatment of CF patients with lung infections.
The acidified nitrite antimicrobial activity is also effective on nosocomial mycoses (8) that represent an increasing problem among critical care patients (193). The susceptibility of Candida albicans, Candida glabrata, Candida tropicalis, Cryptococcus neoformans, Aspergillus fumigatus, and a Rhodotorula species; the lack of induction of resistance; and the possibility of a direct aerosol administration of acidified nitrite (8) provide a novel potential method for treating fungal infections.
Effective antimicrobial strategies are also important in industrial settings, including food processing environment, water treatment, and contact lens industry. One of the most used antimicrobial agents in disinfection processes is the oxidizing agent H2O2, which causes microbial death by protein denaturation (125). However, the antimicrobial efficacy of H2O2 can be significantly enhanced by addition of acidified nitrite (142). This strategy could find an application in disinfection of contact lenses that are a main predisposing factor of blinding keratitis caused by the highly resistant Acanthamoeba spp. (147, 282). H2O2 (3%) used as a contact lens disinfectant is effective against Acanthamoeba cysts, but an exposure time of at least 4–6 h (157) and/or extensive neutralization are required before use of the contact lenses. On the other hand, if sodium nitrite is added to a 3% (v/v) H2O2 solution in one-step systems, significant killing of Acanthamoeba cysts is rapidly obtained (157).
VI. Conclusions and Open Questions
It is widely accepted nowadays that nitrite is a biologically relevant molecule: in bacteria, it may function as a terminal acceptor in electron transport chains and it may act as a key signal in many host–pathogen interactions, by producing NO. The role and biological fate of nitrite in humans is also closely interwoven with the homeostasis of NO (Fig. 13). Nitrite is not only an endocrine reservoir of NO under hypoxic conditions, but also an O2-dependent treasure house of NO [see also (80)].
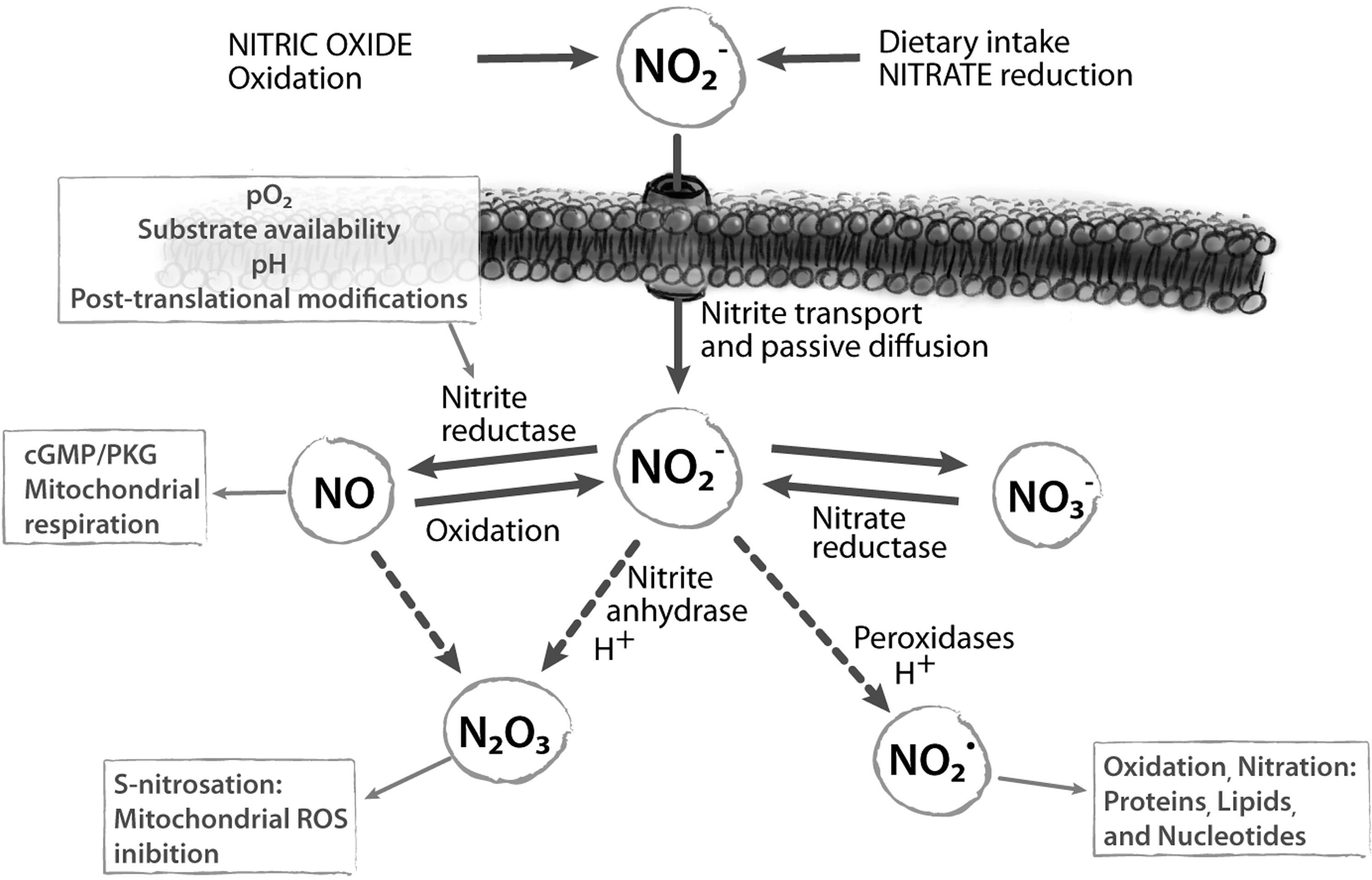
In mammals, a wide variety of proteins are able to act as NiR both in vitro and in vivo. It is possible that more proteins will be identified in the near future, which may accomplish the moonlighting function of NiR under O2 shortage conditions. We propose that this redundancy is not fortuitous for several reasons. First of all, it may be considered an inheritance of a preaerobic past where N-oxides were available as terminal electron acceptors to produce energy (82, 114); second, it may represent an inherent property of the eukaryotic cell, which has several rescue pathways to escape harmful conditions such as O2 depletion and redox unbalance.
It is very intriguing to notice that, in the bacterial kingdom, the heme-containing NiR mostly use specialized porphyrins, such as heme d1, to avoid trapping of the enzyme in an irreversibly inhibited NO complex, thus escaping the kinetic trap faced by mammalian hemoproteins such as Hb and Mb. Indeed, a major conundrum on to the relevance of these hemoproteins, expecially Hb, in the nitrite/NO homeostasis is still the question of how the NO molecule can escape the trap of ferrous hemes. A recent mass spectrometry study has confirmed that, after addition of millimolar levels of nitrite to suspensions of deoxygenated red cells, no significant accumulation of extracellular NO can be monitored; these data support the hypothesis that the autocapture of NO by deoxyHb precludes efflux of NO from the erythrocyte (234). As recently suggested by other authors (349), it would be helpful to find an experimental setup able to uncouple the effect on the Hb/nitrite system from the effect on the intrinsic NO homeostasis within the erythrocyte.
The capability of Mb to reduce nitrite and its role in cytoprotection under hypoxic conditions is well established. Many experiments can still be planned on six-coordinate globins or on cytochromes: these hemoproteins may well serve the intriguing function of redox-controlled NiR, as recently suggested (22, 330), after switching their heme coordination from six- to five-coordinate.
Another important issue to be investigated in detail is the mechanism of nitrite transport across the cell membrane. Whereas this topic is well studied in bacteria [see (240)], the literature data on eukaryotic cells are mainly restricted to the erythrocyte, where the transport mechanism seems to involve both
In conclusion, evaluation of the contribution of blood-based and tissue-based nitrite reduction as a source of NO bioactivity seems to indicate that the tissue sources may play a more important role. A recent theoretical analysis of NO and O2 transport around an arteriole, including superoxide generation and NiR activity in blood and tissue, confirms that NiR activity in blood should not be a very effective mechanism for conserving NO due to the strong scavenging activity of deoxyHb (46). These authors suggest that the NiR activity in tissue is much more effective in increasing NO bioavailability in the vascular wall and contributes progressively more NO as tissue hypoxia becomes more severe.
Finally, nitrite-based therapy has received a growing attention in the last years: several clinical trials are ongoing or planned in the next future [see e.g., (278)]. Nitrite is a molecule that naturally occurs in the human body in blood and tissues (in nanomolar or micromolar concentrations, respectivey); this confers a great advantage to nitrite-based therapy, which is safer than other strategies employing organic nitrates or NO donors. However, the interaction of therapeutic nitrite with other drugs is still poorly understood and this aspect will require further studies to assess possible toxic effects.
The variability of nitrite administration protocols, nitrite uptake rates, and final biological effects suggests that an effective nitrite therapy will require personalized protocols on the basis of the pathology and individual patient, considering also gender and race. Interestingly, even though further studies are needed, it was recently suggested that dietary nitrate may be as effective as nitrite administration for long-term therapy (214) and that it is also able to influence basal cell functions [such as mitochondrial activity (198)].
Footnotes
Acknowledgments
Funds from the Ministero della Università e Ricerca of Italy [RBRN07BMCT, 20094BJ9R7], from the Sapienza University of Rome, and from Fondazione Italiana Fibrosi Cistica to F.C. are gratefully acknowledged.