
Abstract
Mitochondria are highly dynamic, multifunctional organelles. Aside from their major role in energy metabolism, they are also crucial for many cellular processes including neurotransmission, synaptic maintenance, calcium homeostasis, cell death, and neuronal survival.
Mitochondria and Parkinson's Disease
The discovery of disease-associated genetic mutations has provided an opportunity to model the pathological and clinical features of familial disease in animals and other model organisms. PD-related genes harboring pathogenic mutations have been introduced into a variety of model organisms (34, 115, 182). The most widely used species for modeling familial PD are mice, since they are amenable to genetic manipulation and they have a complex nervous system mediating highly coordinated movement. Moreover, mice have a sufficiently long lifespan (up to 3 years) suitable for evaluating disease or phenotype progression and for modeling the chronic nature of disease in humans. In addition to rodents, insight has also been derived from the development and characterization of invertebrate models in the fruit fly, Drosophila melanogaster, and the nematode worm, Caenorhabditis elegans (16, 66). Although invertebrate models lack the complexity to rigorously evaluate clinical aspects of the disease, they have proved useful for exploring the function of PD-related proteins in evolutionary conserved pathways. Ideally, a disease model should replicate the major features of the human disease; that is, the late-onset and chronically progressive phenotype, impairment of the nigrostriatal dopaminergic system with accompanying motor dysfunction resulting from neuronal degeneration, and the presence of intraneuronal LB-like proteinaceous inclusions (8). Even though none of the models described to date meet all of the above criteria they have proven valuable in dissecting cellular processes such as protein aggregation, autophagy, apoptosis, and mitochondrial function in which the pathogenic proteins are reported to be implicated.
An intriguing observation that emerged from the characterization of these PD-related proteins is that they are associated directly or indirectly with mitochondria, participating in important functions within these organelles (Table 1). PTEN-induced kinase 1 (PINK1), parkin, and DJ-1 either localize to mitochondria (PINK1, partially DJ-1) or are translocated to damaged mitochondria (parkin) and can regulate mitochondrial activity and morphology. α-Synuclein, a predominantly cytoplasmic protein, can be found in mitochondria under certain conditions. Similarly, leucine-rich repeat kinase 2 (LRRK2) is associated in part with the outer mitochondrial membrane although little is known about the physiological function of this putative kinase. These observations suggest that mitochondria could potentially be important in the pathophysiology of PD.
IMM, inner mitochondrial membrane; IMS, intermembrane space; LRRK2, leucine-rich repeat kinase 2; OMM, outer mitochondrial membrane; PINK1, PTEN-induced kinase 1.
The first evidence that mitochondria might play a role in PD emerged from the original discovery that the mitochondrial Complex-I inhibitor, 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine (MPTP), can cause acute and irreversible parkinsonian symptoms in humans (92) and was later demonstrated in mice and non-human primates. The active metabolite of MPTP, MPP+, which concentrates in dopaminergic neurons due to its high affinity for the dopamine transporter (DAT), is suggested to inhibit Complex-I of the mitochondrial electron transport chain. Complex-I inhibition and the obstruction of normal electron transfer can lead to increased generation of reactive oxygen species (ROS) and to reduced ATP production. Additional mitochondrial toxins (i.e., rotenone, paraquat/maneb) were subsequently described to induce selective neuronal loss and protein aggregation similar to MPTP in rodents (176, 205). Toxin-based animal models are widely used for inducing acute and chronic dopaminergic neuronal loss to study the mitochondrial-related pathways potentially leading to PD (12, 32, 44, 176). Although the contribution of mitochondrial toxins to PD risk is unclear, animal models based upon these toxins provide compelling evidence that nigral dopaminergic neurons are selectively vulnerable to impaired mitochondrial activity.
An important role for mitochondria in PD pathology is further supported by studies on postmortem human tissues. Mitochondrial Complex-I activity is impaired in the substantia nigra (157), frontal cortex (138), and platelets derived from PD patients (65). Interestingly, mitochondrial DNA (mtDNA) deletions accumulate in nigral dopaminergic neurons of aged individuals and sporadic PD subjects, which are associated with deficits in normal mitochondrial activity (9, 86). Nigral dopaminergic neurons are considered to exhibit increased sensitivity to respiratory chain impairments due to the increased levels of oxidative stress resulting from dopamine metabolism (102). A heightened oxidative burden may only explain a proportion of the selective neuronal vulnerability observed in PD brains, as not all dopaminergic neuronal populations are equally sensitive or affected in PD (i.e., ventral tegmental area neurons are relatively spared in PD), and nondopaminergic neurons with a lower oxidative burden also degenerate in PD. What can be surmised at this point is that nigral dopaminergic neurons in PD brains exhibit some alterations in mitochondrial activity, and that dopaminergic neurons are particularly susceptible to mitochondrial insults. However, what is not clear at this juncture is whether impairments of mitochondrial activity are a primary cause or consequence of dopaminergic neuronal death in PD.
Aside from their important role as the energy factory of cells, mitochondria are also critically involved in many cellular processes, including calcium homeostasis, the response to stress and the induction of cell death. Mitochondria are highly dynamic organelles with a range of morphologies depending on the type, health and activity of particular cells. Mitochondrial morphology is regulated by the balance of highly coordinated fission and fusion events. These dynamic changes in morphology are necessary for maintaining healthy mitochondria and are tightly regulated since a misbalance in fission and fusion can result in inappropriate fragmentation or elongation of mitochondria (36). Dynamic changes in morphology are also required for the appropriate subcellular localization of mitochondria to meet the changing energetic demands of cellular subcompartments. In neurons, mitochondrial biogenesis takes place in the soma, whereas the precise localization of mitochondria is regulated by anterograde and retrograde transport along microtubules and actin filaments within axons and dendrites (46, 105).
It is evident that mitochondria are vital for the proper function of neuronal cells, which are particularly sensitive to mitochondrial dysfunction due to their high energy requirements for synaptic maintenance and transmission (98). Accumulating evidence from human cases and animal models suggests that mitochondrial dysfunction is implicated in the pathophysiology of PD but these observations raise a number of important questions. (i) Is mitochondrial impairment required for the development of PD or do mitochondrial deficits arise as a consequence of the ensuing pathological process within degenerating neurons? (ii) Is it possible to use genetic animal models to order the pathogenic events resulting from a genetic mutation, and therefore place mitochondrial dysfunction in context? (iii) Is there sufficient evidence in genetic animal models to support a primary role for mitochondrial dysfunction in propagating the pathology of PD? (iv) What specific role do these PD-linked proteins normally play in mitochondrial maintenance and function? The answers to all of these questions are closely related and best answered through careful studies in genetic animal models where one can monitor the progression of phenotypes and disease resulting from a defined genetic perturbation associated with familial PD. In this next section, we critically evaluate the in vivo evidence obtained from several genetic animal models potentially implicating mitochondrial dysfunction in the pathogenic process leading to PD.
Models of Autosomal Dominant PD: α-Synuclein (PARK1/4)
α-Synuclein is a small cytosolic protein of 140 amino acids that is expressed throughout the central nervous system, predominantly in neurons where it is enriched at presynaptic nerve terminals in close proximity to synaptic vesicles (108). The physiological function of α-synuclein remains elusive although its localization to nerve terminals along with the demonstration that it can associate with vesicular and membranous structures suggests a potential role in regulating the release and recycling of synaptic vesicles (102). The protein has attracted much scientific interest after the discovery that missense mutations (A53T, A30P, and E46K) in the α-synuclein (SNCA) gene as well as genomic multiplications of a region containing the SNCA locus cause PD with autosomal dominant inheritance (49). More recently, genome-wide association studies have demonstrated that common variation in the SNCA gene is associated with an increased risk of PD (156, 164). α-Synuclein was the first gene identified to cause familial PD and the development of antibodies that specifically detect the protein revealed that fibrillar α-synuclein is a major constituent of LBs and Lewy neurites within surviving neurons of sporadic and certain familial forms of PD (169). These observations provide strong evidence that α-synuclein has a primary pathogenic role in idiopathic PD.
α-Synuclein possesses great conformational plasticity depending on the cellular milieu. In its native state, in aqueous solutions, it is soluble, monomeric and unfolded; however, upon binding to phospholipids its structure shifts to an α-helical conformation. More importantly, α-synuclein has the propensity to self-aggregate into higher-ordered structures of oligomers, protofibrils, and fibrillar β-pleated sheet structures encountered in LBs in PD and other α-synucleinopathies. Accumulating evidence suggests that the misfolded forms of the protein mediate neuronal dysfunction and degeneration since the overexpression of α-synuclein in transgenic animal and cell models results in the accumulation of intracellular aggregates concomitant with neurodegeneration (Table 2) (43, 63). In vitro studies demonstrate that the pathogenic mutations of α-synuclein accelerate the oligomerization/fibrillization process, strengthening the hypothesis that the pathogenicity of α-synuclein is mediated by the formation of neurotoxic aggregates, even though it is still controversial whether the oligomeric or fibrillar forms of α-synuclein are most important for toxicity (24, 60). Moreover, conditions of elevated oxidative and nitrosative stress, which increase as a result of aging as the antioxidant machinery of cells becomes less effective or due to mitochondrial dysfunction, have the potential to enhance the propensity of α-synuclein to aggregate (53, 168). Additionally, increased oxidative stress and mitochondrial dysfunction can aggravate the accumulation of misfolded proteins since these conditions lead to impairment of protein degradation pathways in cells (67). Conversely, as will be discussed below, the abnormal accumulation of α-synuclein is suggested to trigger mitochondrial dysfunction; therefore, it appears that there is a vicious cycle that ultimately leads to the demise of neuronal cells.
α-syn, α-synuclein; DAergic, dopaminergic; ETC, electron transport chain; N.D., not determined; PrP, prion protein; TH, tyrosine hydroxylase; tTA, transactivator; WT, wild-type.
α-Synuclein is detected diffusely in the cytoplasm and can be associated with membranous structures; subcellular localization studies in rodent brain suggest that a fraction of the protein is associated with the mitochondrial membrane or can be transported into mitochondria (74, 95, 210) under conditions of increased cellular stress and cytosolic acidification (23), whereas another study demonstrated mitochondrial localization of α-synuclein in dopaminergic neurons of postmortem PD brains proposing that there is a putative mitochondrial targeting sequence in the amino-terminus of the protein (37). Overexpression of α-synuclein in cellular models causes mitochondrial depolarization and compromises mitochondrial activity (69, 175). The result of mitochondrial dysfunction caused by expression of either wild-type (WT) or A53T α-synuclein is induction of oxidative stress indicated by oxidative modifications of mitochondrial proteins, increased levels of calcium and nitric oxide, and mitochondrial-dependent apoptosis (134, 175). Findings from cellular models indicate that one mechanism to explain α-synuclein-induced toxicity is by interfering with normal mitochondrial function.
Over the last decade numerous transgenic and viral-based models of α-synuclein have been generated mostly in rodents in an effort to model dominantly inherited parkinsonian neurodegeneration (Table 2). Most models have focused on the human disease-associated A53T mutation, and to a lesser extent the A30P mutation or WT protein, whereas a broad spectrum of heterologous promoters have been employed to drive overexpression of transgenes in the nervous system. In some of these models, α-synuclein filaments and inclusions resembling LBs have been detected in various neuronal regions accompanied by progressive motor deficits (34, 115, 182). The formation of α-synuclein-positive inclusions is dependent on the promoter used to drive expression of the transgene, with the prion protein (PrP) promoter being more successful in recapitulating this key feature of disease in mice. More specifically, expression of human A53T α-synuclein in mice produces a progressive motor impairment that is accompanied by the accumulation of fibrillar α-synuclein, muscular atrophy, neuronal loss, and axonal degeneration (54, 94). In contrast to transgenic models, the viral-mediated overexpression of human α-synuclein targeted to the substantia nigra is sufficient to induce progressive dopaminergic degeneration in rat models (77). Correspondingly, overexpression of human α-synuclein in Drosophila causes motor dysfunction, cytoplasmic inclusions of α-synuclein, and selective loss of dopaminergic neurons (42). In fact, the biggest skepticism concerning transgenic mouse models expressing full-length human α-synuclein variants is that they fail to elicit the progressive and selective degeneration of the nigrostriatal dopaminergic pathway. This failure may relate to promoter choice, transgene copy number, developmental compensatory processes, and/or intrinsic differences between mice and rats. However, mouse models of α-synuclein can recapitulate some cardinal features of PD and neuronal loss, and are therefore fundamental for elucidating the molecular mechanism(s) through which mutant α-synuclein induces neurodegeneration.
Mitochondrial dysfunction and abnormalities in morphology have been reported in several transgenic models overexpressing human α-synuclein (Table 2). In a Drosophila model overexpressing A30P or A53T α-synuclein, proteomic profiling revealed dysregulation of actin cytoskeletal, membrane, and mitochondrial proteins (200, 201), whereas similar alterations in the expression of proteins involved in mitochondrial function and actin dynamics were reported in mice overexpressing A30P α-synuclein (56). The first detailed description of mitochondrial dysfunction in a transgenic model of α-synuclein was reported on a mouse line expressing A53T α-synuclein under the mouse PrP promoter (109). The transgenic mice developed mitochondrial degeneration and cell death in the spinal cord and brainstem. In addition to morphological changes in mitochondria, electron-dense α-synuclein inclusions were observed in dendrites, axons, or even within mitochondria, together with a reduction of Complex-IV activity and mtDNA damage (109). It would be of interest to determine the timing of mitochondrial abnormalities in this mouse model and whether they are first evident before disease-onset and neuronal degeneration. Similarly, a detailed ultrastructural analysis focused on mitochondria from transgenic mice expressing both A53T and A30P mutations of α-synuclein under either the chicken β-actin promoter or the mouse tyrosine hydroxylase promoter. This study reported changes in mitochondrial morphology, whereas mitochondrial damage appeared to be more extensive on a parkin null background, indicating that the two proteins have additive effects on mitochondria. Moreover, the α-synuclein transgenic mice show decreased Complex-I activity restricted to the substantia nigra, but in the absence of nigral degeneration or an overt neurological phenotype (170). The observation of compromised mitochondria in the absence of pathological or behavioral abnormalities could be due to variation in the pattern or level of transgene expression since the chicken β-actin promoter drives transgene expression predominantly in glial cells in this model and consequently glial cells exhibit more pronounced mitochondrial pathology than neurons (158). Furthermore, this study suggests that mitochondrial impairments can exist before overt neuronal degeneration, implying that they may not necessarily develop as a consequence of the degenerative process. Oxidatively modified mitochondrial proteins with decreased enzymatic activity have been detected in mice expressing A30P α-synuclein driven by the neuronal Thy-1 promoter, thus reinforcing the concept that a potential mechanism underlying α-synuclein-induced mitochondrial dysfunction is through elevation of oxidative stress levels (145). An alternative pathway for the pathogenic effects of α-synuclein was suggested by a study on mice lacking α-synuclein. Mice with ablation of the SNCA gene fail to develop an overt neurological phenotype but do exhibit alterations in the composition of phospholipids occupying the inner mitochondrial membrane and display compromised function of the electron transport chain (41). Correspondingly, incubation of recombinant α-synuclein with isolated rat brain mitochondria in vitro results in alterations to the mitochondrial membrane (6). Intriguingly, in cultured cells and C. elegans, α-synuclein can interact with the outer mitochondrial membrane and inhibit mitochondrial membrane fusion, resulting in mitochondrial fragmentation, and ultimately in mitochondrial activity impairment. This inhibition is suggested to occur through direct binding of α-synuclein to the mitochondrial membrane and is not dependent on interactions with fusion and fission proteins (74, 125). Recently, it was suggested that negative regulation of mitochondrial Complex-I activity by α-synuclein is a physiological function rather than a pathological event. Brains isolated from transgenic mice overexpressing A53T α-synuclein under the PrP promoter display decreased Complex-I activity irrespective of age or the accumulation of oligomeric or insoluble α-synuclein (101).
Several lines of evidence indicate that α-synuclein is implicated in autophagy pathways. Oxidatively modified α-synuclein as well as the A53T and A30P variants were shown in cellular studies to impair chaperone-mediated autophagy (CMA) by blocking lysosomal receptors, thereby leading to the impaired lysosomal degradation of α-synuclein as well as additional CMA substrates (27, 110). Similarly, the overexpression of α-synuclein was suggested to impair macroautophagy via inhibition of autophagosome synthesis, which could impair the clearance of damaged or dysfunctional mitochondria by autophagy degradation (termed mitophagy) (196). However, evidence from cellular models and mice expressing A53T α-synuclein driven by the rat TH promoter suggest that overexpression of α-synuclein may inappropriately induce mitophagy, thus leading to mitochondrial dysfunction (20, 21).
Studies in toxin-induced animal models further demonstrate that α-synuclein has a significant impact on mitochondrial function with several studies reporting that overexpression of α-synuclein increases the susceptibility of neurons to mitochondrial toxins. Specifically, several studies of mutant α-synuclein transgenic mice reported enhanced MPTP-induced dopaminergic neuronal degeneration accompanied by the accumulation of α-synuclein aggregates and mitochondrial abnormalities (131, 167, 207). Interestingly, MPTP administration to non-human primates produced eosinophilic inclusion bodies reminiscent of LBs found in the brains of PD subjects (45, 85). However, it should be noted that the effect of α-synuclein overexpression on MPTP toxicity in transgenic mouse models has produced inconsistent results that might be attributed to differences in the transgene expression levels or patterns between different genetic models (150, 198). In contrast, α-synuclein knockout (KO) mice are resistant to the neurotoxic effects of MPTP as well as malonate and 3-nitropropionic acid (31, 81, 178). This would imply that α-synuclein potentially lies downstream of mitochondrial dysfunction induced by MPTP exposure and may be required for mediating MPTP-induced neuronal cell death. The overexpression of human WT or A53T α-synuclein can complement the MPTP-resistant phenotype of α-synuclein KO mice, suggesting that human and mouse α-synuclein serve similar functions in MPTP-induced dopaminergic toxicity (178). α-Synuclein KO mice exhibit normal sensitivity to neurotoxicity induced by 2′NH2-MPTP, an analog of MPTP selective for serotoninergic and noradrenergic neurons, indicating that the effects of α-synuclein on MPTP-like compounds are specific to nigral dopaminergic neurons (178). In rats, chronic infusion with rotenone, a potent Complex-I inhibitor, elicits selective dopaminergic cell loss along with the accumulation of inclusions containing ubiquitin and α-synuclein (12). This potentially suggests that α-synuclein aggregation lies downstream of mitochondrial impairment. Rotenone, unlike MPP+, is highly lipophilic and can enter any cell type, thereby suggesting that nigrostriatal dopaminergic neurons are selectively vulnerable to mitochondrial impairment. It has not been possible to explore the effects of rotenone in genetic mouse models of PD since mice unlike rats are not consistently sensitive to rotenone-induced neurotoxicity.
Collectively, combined evidence from studies in vitro and in transgenic animal models suggests that α-synuclein aggregation and mitochondrial dysfunction may be two interrelated events with one producing the other in a vicious cycle that may precipitate neuronal degeneration (Fig. 1). At this stage, however, it is not known whether mitochondrial dysfunction mediates α-synuclein-induced neurodegeneration, whereas it is clear that the overexpression of human α-synuclein can lead to mitochondrial damage in vivo.
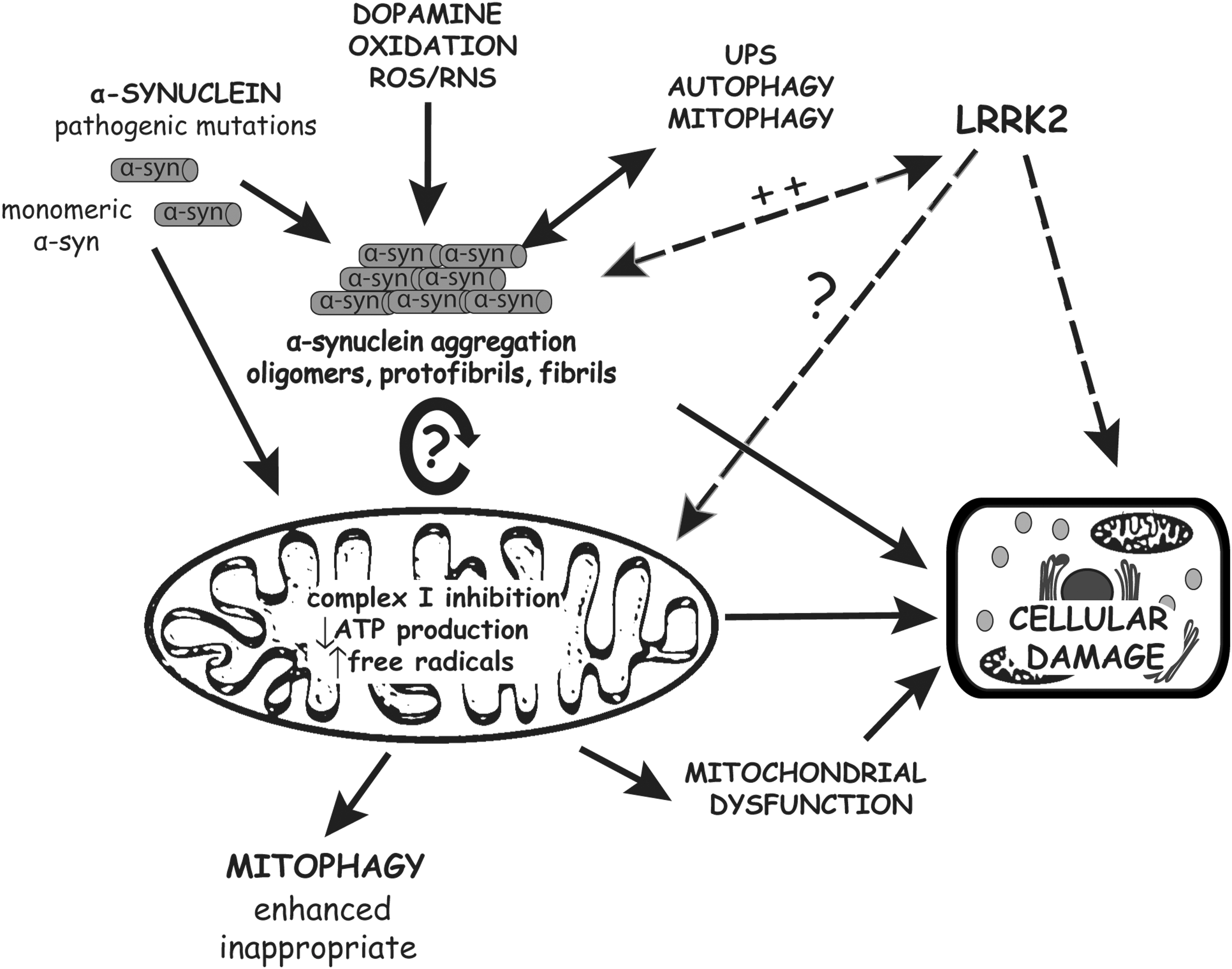
Models of Autosomal Dominant PD: LRRK2 (PARK8)
Missense mutations in the LRRK2 gene (PARK8, OMIM 607060) are the most common cause of familial PD and are also found at variable frequencies in idiopathic PD cases depending on ethnicity, indicating that LRRK2 plays an important role in the pathophysiology of PD (25). The pathological mechanisms through which autosomal dominant LRRK2 mutations lead to neurodegeneration remain to be elucidated. LRRK2 encodes a large protein of ∼280 kDa consisting of several highly conserved functional domains including a serine/threonine kinase domain similar to the mixed-lineage and RIP kinase families, Ras of Complex (Roc) GTPase domain, C-terminal of Roc (COR) domain, and LRRK2-specific, ankyrin, leucine-rich, and WD40 repeat domains suggesting that this protein plays a major role in cell signaling potentially acting as a protein scaffold (11, 117, 213).
PD-linked mutations tend to cluster within the Roc GTPase, COR, and kinase domains of the protein, whereas no deletions or truncations have been reported. This observation together with the dominant inheritance of LRRK2 mutations is suggestive of a toxic gain-of-function mechanism. The most common mutations encountered in familial PD alter the kinase and GTPase activity of LRRK2. The G2019S mutation in the kinase domain, which is the most prevalent variant described to date, enhances the kinase activity of the protein (192), whereas neurotoxicity induced by LRRK2 variants in culture models and in vivo critically depends on kinase activity (62, 93, 165, 193). However, to date putative substrates or potential regulators of LRRK2 kinase activity in vivo are not well characterized.
Despite the defined domain architecture of LRRK2 its biological function remains obscure. Unraveling the normal physiological role of LRRK2 is critical for understanding the molecular pathways that are perturbed due to familial mutations. Current evidence suggests a role for LRRK2 in the regulation of cytoskeletal dynamics and neurite outgrowth and complexity. Silencing of LRRK2 expression in primary neurons enhances the length and branching of neuronal processes (28, 107), whereas overexpression of human LRRK2 variants oppositely reduces neuritic complexity (107, 135, 148). The effects of LRRK2 on neuronal process morphology further support a toxic gain-of-function mechanism, rather than a dominant-negative effect or haploinsufficiency, for the pathogenic actions of familial mutations. LRRK2 has been proposed to regulate neurite outgrowth and morphology via phosphorylation of moesin/ezrin/radixin (ERM) protein family members, which are known to mediate the interaction of plasma membrane proteins with the actin cytoskeleton to determine cell morphology (72, 135). Primary cultures derived from transgenic mice overexpressing G2019S LRRK2 reveal the enhanced phosphorylation of ERM proteins localized to neuronal filapodia during early neurite extension, which resulted in increased filamentous actin and inhibition of neurite outgrowth, whereas deleting LRRK2 produced opposite effects (135). Also, it has been shown that LRRK2 associates with tubulins through its GTPase domain and can influence microtubule assembly (47) potentially through phosphorylation of β-tubulin (55). The overexpression of human LRRK2 in transgenic mice enhances tubulin polymerization and impairs microtubule assembly (99). Disturbances in the microtubule network would affect intracellular transport and potentially microtubule-based transport of mitochondria. Since neuronal cells are highly polarized, they rely heavily on the microtubule network for transporting proteins, vesicles, and organelles along axons and dendrites. Alterations in microtubule or actin filament transport due to LRRK2 mutations, and potentially abnormal mitochondrial transport, may contribute to pathogenic pathways leading to PD.
Insight into the physiological role of LRRK2 is provided also by localization studies demonstrating the association of LRRK2 with membranous and vesicular intracellular structures, including the endoplasmic reticulum, Golgi complex, axonal microtubules, endosomes, lysosomes, and the mitochondrial outer membrane and matrix (14, 192). Therefore, LRRK2 could potentially be implicated in mitochondrial function or transport. Accordingly, cultured human fibroblasts derived from PD subjects with G2019S mutations exhibit deficits in mitochondrial activity and increased mitochondrial elongation and interconnectivity (121). Whether LRRK2 can regulate mitochondrial fission and fusion events is not clear but is plausible given the preferential association of active LRRK2 dimers with membranous compartments (10). Neuronal toxicity induced by mutant LRRK2 is mediated through mitochondria-dependent apoptosis involving cytochrome c release, apoptosome formation and caspase-3 activation (70). In a yeast model, LRRK2-induced cytotoxicity can be suppressed by deletion of CCE1, a mitochondrial endonuclease (199). Therefore, there is some evidence supporting a role for mitochondrial dysfunction in mediating the pathogenic actions of familial LRRK2 mutations, at least in cell culture models.
A pathogenic role for LRRK2 in regulating mitochondrial function is also supported by studies in invertebrate model organisms. Expression of human WT LRRK2 in C. elegans protects dopaminergic neurons after exposure to the mitochondrial toxin, rotenone, whereas G2019S LRRK2 was less protective (152). In this context, the G2019S mutation may act through a loss-of-function mechanism. Drosophila expressing human G2019S LRRK2 developed retinal degeneration, selective loss of dopaminergic neurons, and motoric deficits (100). However, in contrast to worms, flies expressing familial LRRK2 mutants have increased susceptibility to toxicity induced by chronic rotenone exposure (130, 186). LRRK2 KO mice exhibit normal sensitivity to MPTP-induced neurotoxicity (2), suggesting that LRRK2 may normally function independent of Complex-I activity or MPTP-related toxicity, or could lie in a pathway located upstream of mitochondrial dysfunction. The sensitivity of LRRK2 transgenic mice to MPTP toxicity has not yet been reported. Therefore, the effects of LRRK2 expression upon neuronal toxicity induced by inhibitors of the mitochondrial transport chain in different model organisms are contrasting and require further clarification.
Driven by their success in inducing nigral dopaminergic cell loss, a cardinal feature of PD pathology, viral-mediated gene transfer models for LRRK2 have been developed in rodents (Table 2). Adeno-associated viral (AAV2)-mediated gene transfer of the human LRRK2 kinase domain alone in the substantia nigra of adult rats produced phospho-tau-positive spheroidal inclusions in neuronal processes, increased apoptotic markers in dopaminergic neurons, and altered neurite and mitochondrial morphology (107). This should be interpreted with caution since the isolated kinase domain alone is not known to be functional and therefore pathogenic effects are likely to be independent of kinase activity (72). Expression of human G2019S LRRK2 via striatal delivery of human adenoviral vectors in adult rats results in the progressive loss of nigral dopaminergic neurons accompanied by transient and mutation-independent neuritic pathology composed of hyperphosphorylated tau (39). Similarly, herpes simplex virus amplicon-mediated delivery of human G2019S LRRK2 to the striatum of mice led to nigral dopaminergic neurodegeneration (93). Viral-mediated gene transfer models are an attractive choice for evaluating the effects of familial LRRK2 mutants on the nigrostriatal dopaminergic system, the potential interaction with other PD-associated proteins, and the molecular pathogenic pathways that may involve abnormal mitochondrial function. Presently, the contribution of mitochondria to LRRK2-related disease in viral-based models is not known.
Numerous transgenic mice have been developed that overexpress human or mouse LRRK2 with the majority based upon bacterial artificial chromosome (BAC)-driven expression (Table 2). In one BAC transgenic model, the overexpression of human R1441G LRRK2 causes progressive and L-Dopa-responsive motor deficits, reduced striatal dopamine release, reduced density of nigral dopaminergic neurites, and discrete axonal pathology accompanied by hyperphosphorylated tau (97). BAC models expressing G2019S LRRK2 or R1441C knockin mice collectively exhibit abnormalities in nigrostriatal dopaminergic transmission, motor performance and processing of tau protein (96, 111, 181). Transgenic mice expressing human G2019S LRRK2 from a CMV-enhanced PDGF-β promoter display modest age-dependent nigral dopaminergic degeneration concomitant with the abnormal accumulation of autophagic vacuoles and condensed mitochondrial aggregates (148). Several studies have attempted to investigate whether LRRK2 and other PD-related genes converge on common pathways regulating mitochondrial integrity. It was first reported that LRRK2 interacts with parkin in cultured cells (166). In Drosophila, the overexpression of parkin protects dopaminergic neurons from G2019S LRRK2-induced degeneration after aging or exposure to rotenone (130), suggesting that parkin is located downstream of LRRK2 in a pathway potentially regulating mitochondrial homeostasis. LRRK2 also regulates A53T α-synuclein-induced pathology in transgenic mice. Neurodegeneration, α-synuclein accumulation, and cytopathological abnormalities induced by the overexpression of human A53T α-synuclein in inducible transgenic mice are enhanced by the coexpression of human WT or G2019S LRRK2, whereas the deletion of LRRK2 reduces these phenotypes (99). Of the cytopathological abnormalities, LRRK2 expression regulated Golgi fragmentation, abnormal mitochondrial morphology, and tubulin assembly induced by α-synuclein expression, suggesting that LRRK2 expression can further impair microtubule-based trafficking pathways and may potentially exacerbate the α-synuclein-induced disruption of ER-Golgi trafficking (26). This study further suggests that α-synuclein and LRRK2 potentially act in a synergistic manner to produce abnormal mitochondrial morphology in bigenic mice (99). Considering the differential sensitivity of α-synuclein and LRRK2 KO mouse models to MPTP-induced neurotoxicity, it is reasonable to hypothesize that if both proteins participate in a common pathogenic pathway, α-synuclein would be located downstream of LRRK2 and mitochondrial function, with LRRK2 upstream of mitochondrial function. There may also exist parallel pathways and feedback regulatory mechanisms for both proteins.
Overall, the involvement of LRRK2 in mitochondrial function, and mitochondria in LRRK2-dependent neurodegeneration, awaits further clarification (Fig. 1). LRRK2 is localized in part to mitochondria in neurons but is dispensable for MPTP-induced toxicity, whereas the expression of familial LRRK2 mutations produces subtle alterations in mitochondrial morphology and integrity in vivo. Given that LRRK2 localizes to the outer mitochondrial membrane and contains a small GTPase domain, and similar GTPases such as dynamin-related protein 1 (Drp1), mitofusin 1/2 (Mfn1/2), and optic atrophy 1 (OPA1) critically regulate mitochondrial fission and fusion events (82), a potential role for LRRK2 in regulating mitochondrial dynamics is plausible and warrants attention. Mitochondrial fission requires GTP hydrolysis and such GTPases are recruited to the fission machinery and regulated by scaffold proteins containing WD40 repeat domains (194). LRRK2 contains numerous protein–protein interaction domains, including WD40 repeats, and could have a similar scaffold function upon mitochondrial membranes. Insight into the mitochondrial involvement of LRRK2 could be elaborated upon by exploring the conditions regulating its association with mitochondria, including the effects of enzymatic activity, familial pathogenic mutations, interacting proteins, and cellular stress. Moreover, it would be of interest to investigate the potential involvement of LRRK2 in autophagy pathways, and by association mitophagy. Overexpression of G2019S or R1441C LRRK2 leads to increased autophagy and accumulation of autophagic vacuoles in mammalian cells (1, 141), whereas expression of LRRK2 GTPase variants in yeast promotes the accumulation of autophagic vacuoles and impairs endocytic vesicular trafficking (199). In G2019S and R1441C LRRK2 transgenic mice there is an accumulation of autophagic vacuoles in brain regions with highest transgene expression (148). It is anticipated that abnormalities in autophagy pathways could have deleterious effects on the normal removal of damaged or dysfunctional mitochondria by mitophagy (17). Accordingly, in G2019S LRRK2 transgenic mice there is evidence for the accumulation of condensed mitochondrial aggregates reminiscent of preapoptotic mitochondria undergoing autophagic degradation (148). Further characterization of existing and new LRRK2 animal models is necessary pertaining to mitochondrial function, dynamics and integrity to firmly clarify a role for mitochondrial abnormalities in mediating LRRK2-related neuronal degeneration. At present, however, there is modest in vivo evidence from genetic models supporting a pathogenic effect of LRRK2 mutations on mitochondrial shape and integrity.
Models of Autosomal Recessive PD: PINK1 (PARK6)
Mutations in the PINK1 gene (PARK6, OMIM 605909) represent the second most common genetic cause of autosomal recessive early-onset PD (184). PINK1 encodes a 581 amino acid protein with a C-terminal serine/threonine kinase catalytic domain and an amino-terminal mitochondrial targeting sequence and short transmembrane domain. The kinase domain is oriented toward the cytosol suggesting that PINK1 substrates are cytosolic or mitochondrial-associated (163, 211). Primary fibroblasts isolated from PD patients carrying PINK1 mutations exhibit respiratory activity impairment, increased lipid peroxidation and enhanced sensitivity to oxidative stress (68), which is suggestive of a protective antioxidant role for PINK1. Most PD-linked mutations are located within the kinase domain and result in impaired kinase activity and decreased neuroprotective function of PINK1.
To investigate the physiological function of PINK1 and its potential neuroprotective role, several PINK1-deficient Drosophila lines were generated (Table 3) (22, 137, 203). Flies lacking PINK1 exhibit shorter lifespan, male sterility, apoptotic muscle degeneration and dopaminergic neuronal degeneration. At the level of mitochondria, all fly lines reveal prominent defects including enlarged and swollen mitochondria, fragmented cristae, decreased ATP production, and increased vulnerability to oxidative stress and respiratory complex inhibitors (rotenone and paraquat) (22, 137, 203). Overexpression of WT, but not kinase-inactive PINK1 rescued the aforementioned mitochondrial phenotypes, suggesting that kinase activity is crucial for its function in regulating mitochondrial integrity (22). Similarly, in C. elegans, loss of PINK1 resulted in reduced mitochondrial cristae length, increased oxidative stress, and paraquat sensitivity, and deficits in neuronal outgrowth. It is noteworthy that this phenotype can be suppressed in the absence of lrk-1, the C. elegans ortholog of human LRRK1/LRRK2, suggesting an antagonistic function of the two kinases in stress response and neuronal activities (154). Additional evidence for the role of PINK1 in maintaining mitochondrial morphology was provided by a second worm model in which α-synuclein overexpression inhibited mitochondrial fusion and resulted in mitochondrial fragmentation. This phenotype was suppressed by coexpression of PINK1, parkin, or DJ-1, but not their pathological PD-linked mutations (74). In zebrafish models, PINK1 knockdown produced a severe developmental phenotype and neuronal loss, including dopaminergic neurons, accompanied by increased ROS levels and caspase-3 activation suggestive of mitochondrial dysfunction (4, 153).
ΔΨ, mitochondrial membrane potential; CAT, catecholamine; DA, dopamine; KO, knockout; LTD, long-term depression; LTP, long-term potentiation; ox., oxidative; resp., respiratory; RNAi, RNA interference; ROS, reactive oxygen species.
Deletion of PINK1 in mice did not result in obvious pathological phenotypes such as motor deficits or dopaminergic neuronal loss, but subtle changes in long-term potentiation (LTP) and long-term depression (LTD) were observed (Table 3) (79, 212). Consistent with Drosophila models, PINK1 null mice exhibited significant functional impairment of mitochondria and alterations in mitochondrial morphology (50), elevated production of ROS, and increased sensitivity to oxidative stress (79). Analysis of PINK1 null mice revealed a significant increase in the number of enlarged mitochondria even though there are no gross alterations in mitochondrial number. A decrease in mitochondrial respiratory chain and aconitase activity was detected selectively in the striatum of young mice which extended to the cerebral cortex in aged mice, potentially indicating that aging exacerbates mitochondrial dysfunction in this model (50).
Complementing the in vivo studies, analysis of PINK1 knockdown in cultured human and mouse dopaminergic neurons showed morphological changes and functional impairment of mitochondria, including increased sensitivity to oxidative stress (staurosporine) and lysosomal pathology. Moreover, PINK1-deficient neurons reveal a decrease in long-term viability and increased caspase-3 activation and apoptosis (197). One potential mechanism connecting PINK1 deficiency with increased sensitivity to mitochondrial-dependent apoptosis is through impaired calcium homeostasis (48). PINK1 knockdown led to reduced mitochondrial calcium efflux, accumulation of mitochondrial calcium, increased ROS production, and respiratory impairment. Mitochondrial calcium overload lowers the threshold for opening of the mitochondrial permeability transition pore with the consequence of further impairment of mitochondrial activity and apoptotic cell death (48).
Collectively, data from animal models clearly suggest that PINK1 is important for maintaining normal mitochondrial function although its precise role in mitochondria remains obscure (Table 3). To fully understand the mechanism of PINK1 action within mitochondria, it will be necessary to identify substrates of phosphorylation under normal and pathological conditions. Putative substrates of PINK1 kinase activity include the protease HtrA2/Omi (142), Hsp90 (190), and TNF receptor-associated protein 1 (TRAP1) (146). HtrA2/Omi is a mitochondrial serine protease that translocates to the cytosol during apoptosis and interacts with inhibitor of apoptosis proteins. It was shown that HtrA2/Omi is phosphorylated indirectly in a PINK1-dependent manner upon activation of the p38/MAPK stress pathway (142). HtrA2/Omi-deficient mice reveal mitochondrial dysfunction with several PD-like features, suggesting that it functions to mediate neuroprotection rather than promoting apoptosis. Moreover, HtrA2/Omi is hypophosphorylated in brain tissue from PD subjects harboring PINK1 mutations (142), further supporting a functional link between PINK1 and HtrA2/Omi. Of note, a G399S mutation in Omi/HtrA2 has been identified in German subjects with sporadic PD (171) although common variation in the Omi/HtrA2 gene does not alter the risk for developing PD (87). TRAP1 is a mitochondrial chaperone that is phosphorylated by PINK1 in cellular models. PINK1-mediated phosphorylation of TRAP1 suppress mitochondrial cytochrome c release upon oxidative stress (146), suggesting that phosphorylation may enhance the activity of TRAP1. Current evidence suggests that PINK1 mediates mitochondrial protection in cellular and animal models (Table 3). The identification of putative substrates of its kinase activity, such as HtrA2/Omi or TRAP1, have broadened our understanding of the pathogenic processes resulting from loss-of-function mutations but the precise mechanisms remain to be clarified.
Models of Autosomal Recessive Parkinsonism: Parkin (PARK2)
Parkin was the first gene identified as a genetic cause of autosomal recessive juvenile-onset parkinsonism (AR-JP, PARK2, OMIM 600116) (78). Since this first observation more than 100 homozygous or compound heterozygous parkin mutations have been identified in PD subjects (191) which represent up to 50% of recessive familial PD cases and about 20% of early-onset PD (103). Parkin encodes a 465 amino acid protein which is predominantly localized in the cytoplasm. Parkin contains an N-terminal ubiquitin-like domain and a catalytic C-terminal RING box domain. Parkin functions as an E3 ubiquitin protein ligase mediating the covalent attachment of ubiquitin monomers or linked chains to protein substrates, which may serve to target proteins for proteasomal degradation or modulate their function in a number of nondegradative pathways (116). An early hypothesis proposed that loss-of-function parkin mutations cause impaired ubiquitination and proteasomal degradation of substrates leading to abnormal substrate accumulation and neurotoxicity. However, there is limited evidence supporting such a mechanism with only a few substrates, such as CDCrel-1, Pael-R, AIMP2/JTV-1, and FBP1, modestly accumulating in brains from AR-JP patients (124) or parkin KO mice (83, 84, 139). Viral-mediated expression of AIMP2 and CDCrel-1 in the substantia nigra of rodents induces dopaminergic neuronal loss, providing some support for their neurotoxic actions in vivo (38, 84). An intriguing recent study has identified PARIS as a new substrate of parkin which undergoes ubiquitination and proteasomal degradation (162). PARIS functions as a transcriptional repressor and its accumulation in the absence of parkin leads to the repression of PGC-1α expression, the master regulator of mitochondrial biogenesis. The conditional deletion of parkin in adult mice causes nigral dopaminergic loss in a PARIS-dependent manner. Further, PARIS overexpression in mice leads to the selective loss of substantia nigra dopaminergic neurons, which could be rescued by overexpression of parkin or PGC-1α. This important study lends support to the toxic substrate hypothesis and further implicates PGC-1α and mitochondria as important downstream targets in neurodegeneration due to parkin inactivation in PD (162).
Parkin is suggested to have neuroprotective effects through the regulation of a number of cellular processes or pathways (116), including mtDNA transcription/replication, or mitochondrial activity, morphology, biogenesis, and protection against mitochondria-dependent cell death. In proliferating cells, transcription and replication of mtDNA was enhanced by parkin overexpression and reduced by parkin knockdown, whereas parkin was associated with mitochondrial transcription factor A (TFAM) and enhanced its transcriptional activity (89). One of the initial indications supporting a role for parkin in regulating mitochondrial activity was the observation that parkin is protective against mitochondrial swelling and cytochrome c release induced by ceramide treatment in PC12 cells (30). A role for parkin in mitochondrial function was also provided by studies with parkin-mutant leukocytes (123) and fibroblasts (122) derived from PD subjects which display alterations in mitochondrial morphology and decreased mitochondrial respiratory activity linked with a significant decrease in ATP production and the collapse of mitochondrial membrane potential. Parkin normally resides in the cytoplasm but can translocate to depolarized or damaged mitochondria to mediate their removal by mitophagy in cooperation with PINK1 and potentially other factors (128). Parkin-deficient Drosophila models develop age-dependent muscle degeneration, male sterility, reduced lifespan, and dopaminergic neuronal degeneration, reminiscent of PINK1 deficiency in flies (Table 4) (61, 140). This phenotype was accompanied by mitochondrial dysfunction involving swollen mitochondria with fragmented cristae, decreased activity of respiratory Complexes I and IV, increased reactive oxygen/nitrogen species, as well as increased vulnerability to oxidative stress and mitochondrial toxins (61, 140, 195). In mice, parkin deficiency was reported to cause reduction in abundance of proteins involved in mitochondrial function and antioxidant capacity of cells (several subunits of complexes I and IV and peroxide reductases) (133). In accordance with these data, functional assays revealed impairment in the respiratory capacity of mitochondria isolated from the striatum of parkin null mice and increased protein and lipid peroxidation (133, 179). However, toxin-based models reported contradictory effects on mitochondrial sensitivity in parkin null mice. Primary cultures of skeletal muscle derived from parkin KO mice result in greater sensitivity to the Complex-I inhibitor, rotenone (151), whereas parkin null mice are normally sensitive to MPTP-induced neuronal toxicity (179). These studies may highlight differential sensitivity between muscle and brain cells, or different mechanisms of rotenone and MPTP toxicity. While parkin KO mice fail to recapitulate key aspects of PD pathology (58, 71, 188), they do reproduce some mitochondrial phenotypes that are also evident in Drosophila parkin models (Table 4). Whether mitochondrial dynamics are perturbed in brains from parkin null mice remains to be determined. Further, unlike fly models, the germline inactivation of parkin in mice fails to induce neuronal loss despite modest mitochondrial impairment and damage, supporting a causal role for mitochondrial dysfunction in manifesting neurodegeneration in PD. A recent study has demonstrated that the conditional inactivation of parkin in the substantia nigra of adult mice appears sufficient to induce the progressive degeneration of dopaminergic neurons (162). It will be of interest to assess mitochondrial activity and morphology in this model both before and after the onset of neuronal loss.
DA neurons, dopaminergic neurons; het., heterozygous; hom., homozygous; PD, Parkinson's disease.
Current evidence indicates a broad neuroprotective function for parkin, and a requirement of parkin for the maintenance of mitochondrial function and dynamics. The molecular mechanism leading from parkin inactivation to neurodegeneration is not clear. The observation that PGC-1α expression is regulated by PARIS in a parkin-dependent manner provides a tantalizing new pathway (162). However, parkin itself can directly regulate mitochondrial proteins, suggesting that multiple pathways may lead to mitochondrial impairment after parkin inactivation (Fig. 2) (19). Assessing the relative contribution and sequential order of these pathogenic mitochondrial pathways to parkin-linked disease is necessary to untangle the molecular events that precipitate neurodegeneration in the absence of parkin.
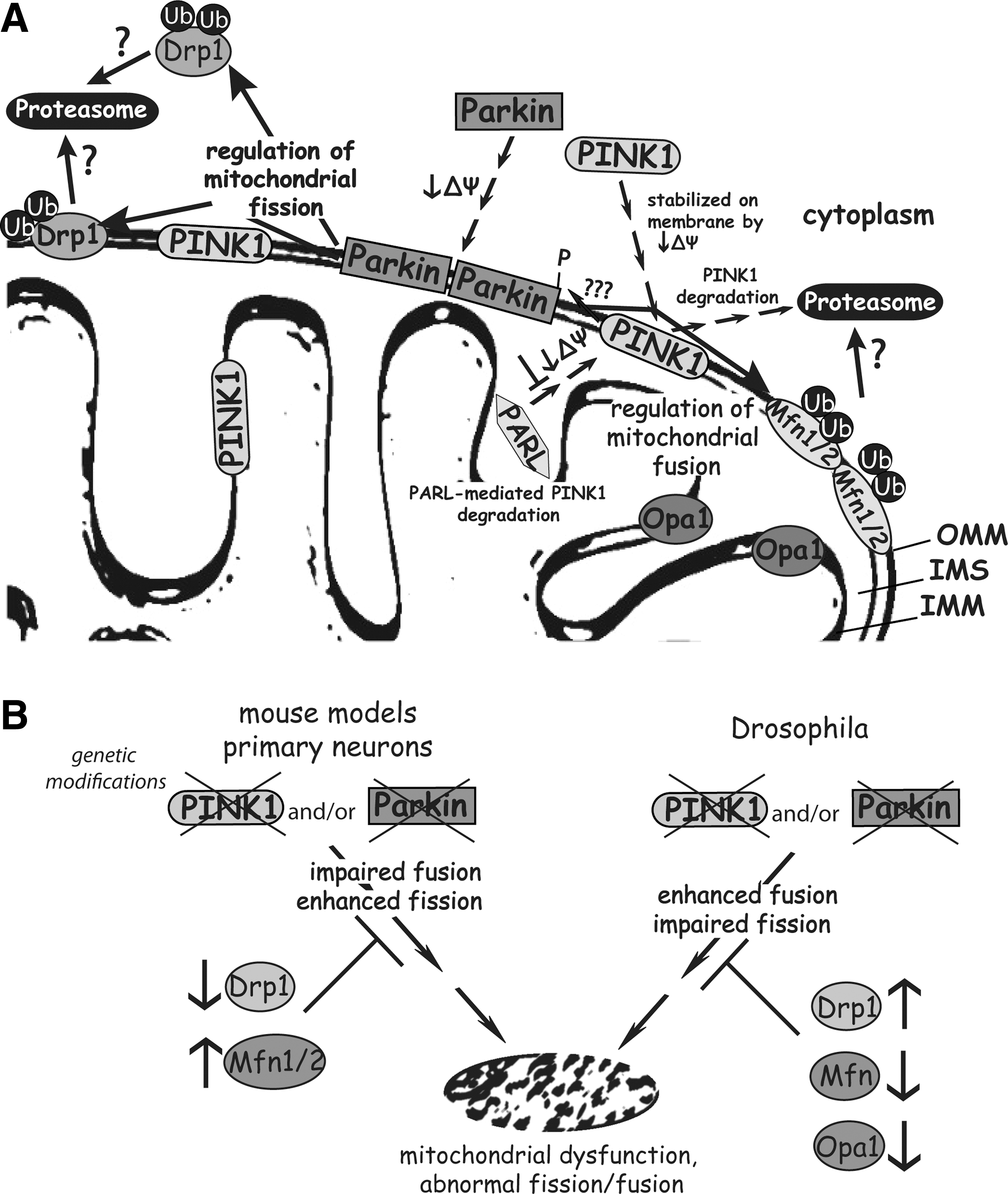
PINK1/Parkin Pathway and Mitochondria
The observation that PINK1 and parkin null flies present with similar phenotypes suggested that these two proteins might act in a common molecular pathway (Fig. 2). This was supported by evidence showing that PINK1/parkin double mutant flies do not exhibit additive phenotypes compared to single mutants (22). Moreover, overexpression of parkin in PINK1 mutant flies rescues the mitochondrial morphology defects and male sterility, whereas the overexpression of PINK1 in parkin null flies failed to rescue the mitochondrial phenotype. These observations support a common pathway for PINK1 and parkin with PINK1 acting upstream of parkin (22, 137, 203).
Parkin is normally a cytoplasmic protein but has been shown to translocate to depolarized or damaged mitochondria in numerous studies in Drosophila or mammalian cellular models (127, 132, 187). Under physiological conditions, PINK1 is unstable and present at low levels on the outer mitochondrial membrane due to its rapid, voltage-dependent proteolytic cleavage by presenilin-associated rhomboid-like (PARL) protease, and subsequent proteasomal degradation. In damaged mitochondria with a loss of membrane potential, PARL activity is inhibited and PINK1 becomes stabilized and accumulates on the outer mitochondrial membrane (73, 129). Interestingly, elevated levels of PINK1 on the mitochondrial membrane are suggested to trigger the translocation of parkin to mitochondria in a kinase activity-dependent manner potentially through direct phosphorylation of parkin at Thr-175 (76, 159). Additional studies, however, have failed to demonstrate the phosphorylation of parkin by PINK1 (187). The mechanism underlying the PINK1-dependent translocation of parkin to the mitochondrial outer membrane is unclear. The kinase domain of PINK1 is localized to the outer membrane of mitochondria where it may be exposed to the cytosol (211), suggesting that PINK1 may phosphorylate cytosolic as well as mitochondrial substrates. Accordingly, PINK1 and parkin have been shown to directly interact in cells potentially in a kinase-dependent manner (76, 116, 161, 208). In vivo evidence for a direct PINK1/parkin interaction or the functional consequences of this interaction are still lacking.
Recently, the mitochondrial fusion factors, Mfn1 and Mfn2 (51, 57, 144, 147, 174, 214), the mitochondrial fission factor Drp1 (189), voltage-dependent anion-selective channel 1 (VDAC1) (52), and multiple proteins of the outer mitochondrial membrane (19) were proposed to be substrates of the E3 ligase activity of parkin (Fig. 2). In mammalian cells, VDAC1, a protein localized to the outer mitochondrial membrane, was shown to be ubiquitinated via Lys-63- and Lys-27-linked chains in a parkin-dependent manner after mitochondrial depolarization (52). Moreover, VDAC1 and p62/SQSTM1 are required for PINK1/parkin-mediated mitophagy (52). The adaptor protein p62/SQSTM1 links ubiquitination with autophagy through simultaneously binding to ubiquitinated proteins and LC3-II, a component of the autophagosomal membrane (52). Therefore, mitochondrial depolarization induces the parkin-mediated ubiquitination of VDAC1, which leads to p62 recruitment and the autophagic clearance of damaged mitochondria. A recent study demonstrates that p62 is required for mitochondrial clustering but does not promote parkin-dependent or mitochondrial depolarization-induced mitophagy. Further, parkin was shown to recruit p62 independent of VDAC1 with proteins other than p62 likely required for mitophagy (126). Thus, there is some discrepancy concerning the precise order of events and requirement of VDAC1 and p62 for PINK1/parkin-mediated mitophagy.
A role for the PINK1/parkin pathway in mediating the autophagic clearance of damaged mitochondria is supported by the accumulation of abnormal large, swollen mitochondria in PINK1, and parkin null fly models (Fig. 2). Therefore, the PINK1/parkin pathway may serve to regulate mitochondrial dynamics through promoting fission or inhibiting fusion events. Fission and fusion are crucial for the maintenance of mitochondrial function and are controlled by evolutionary conserved fusion-promoting (Mfn1, Mfn2 and OPA1) and fission-promoting (Drp1, Fis1) factors. In contrast to Drosophila models, studies in mammalian neuronal cells reveal excessive mitochondrial fragmentation induced by deletion or knockdown of PINK1 and parkin, which can be rescued by enhancing mitochondrial fusion through the overexpression of Mfns and OPA1, or a dominant-negative Drp1 mutant (29, 104, 155). Further, parkin and PINK1 are able to suppress mitochondrial fragmentation induced by Drp1. In mammalian cells, the PINK1/parkin pathway may oppositely promote mitochondrial fusion or inhibit fission (Fig. 2). The contrasting roles of the PINK1/parkin pathway in fly and mammalian models upon mitochondrial morphology are difficult to reconcile at present. Further complicating this issue is the consistent observation that parkin mediates the ubiquitination and degradation of Mfn1 and Mfn2 in both fly and mammalian cells (35, 51, 57, 144, 147, 174, 204), implicating a role for parkin in negatively regulating mitochondrial fusion. An elegant recent study has proposed that the parkin-mediated degradation of Mfns induced by mitochondrial damage may serve to limit the refusion of damaged mitochondria with healthy mitochondria, thereby promoting the selective isolation and autophagic removal of damaged mitochondria (173). Recently, parkin was also shown to promote the ubiquitination and proteasomal degradation of Drp1 supporting a role for parkin in regulating mitochondrial fission (189). In Drosophila models, heterozygous deletion of Drp1 dramatically reduced the viability of PINK1 or parkin null flies, whereas mitochondrial phenotypes were rescued by overexpression of Drp1 or by loss-of-function mutations to Mfn and OPA1 (35, 143, 204). Therefore, the PINK1/parkin pathway may serve to regulate mitochondrial dynamics and mitophagy in different model systems and it is likely that the precise effects are context-dependent, that is, cell or tissue type, mitochondrial damaging agents, etc. What is becoming clear is that the PINK1/parkin pathway is required for the selective elimination of damaged or dysfunctional mitochondria via autophagic degradation with a potential role for mitochondrial dynamics in regulating this process (Fig. 2). A revealing study has shown that parkin may function somewhat indiscriminately to promote the proteasomal degradation of mitochondrial outer membrane proteins, with 26S proteosomal activity being critical for parkin-mediated mitophagy (19). Further clarification of the molecular mechanism underlying PINK1/parkin-mediated mitophagy is required together with clarification of the discrepancies between fly and mammalian models.
Models of Autosomal Recessive PD: DJ-1 (PARK7)
Missense mutations, deletions and truncations in the DJ-1 gene (PARK7, OMIM 606324) represent rare causes of autosomal recessive, early-onset PD (15). DJ-1 encodes a protein of 189 amino acids, which belongs to the ThiJ/PfpI superfamily. DJ-1 exists as an obligate homo-dimeric protein and missense mutations impair dimerization and/or protein stability leading to enhanced degradation by the proteasome (106, 114, 120). The precise physiological function of DJ-1 remains elusive, with present data supporting a role as a redox-sensitive molecular chaperone, an RNA-binding protein or an antioxidant peroxiredoxin-like peroxidase (Fig. 3) (3, 172, 185, 206). Under normal conditions DJ-1 is localized throughout the cytoplasm of neurons with a small proportion localized to the mitochondrial matrix and intermembrane space (209). Oxidative stress promotes the redistribution of DJ-1 from the cytoplasm to the mitochondrial outer membrane (18), involving the oxidation of Cys-106. A mutation in this residue prevents the oxidation-induced mitochondrial translocation of DJ-1 and the protective effect of DJ-1 against mitochondrial-dependent cell death (18). Analysis of brain samples from sporadic PD patients reveal a shift of DJ-1 into more acidic forms, suggesting elevated levels of cysteine oxidation and oxidative stress in PD brains (5).

To understand the physiological role of DJ-1 and its pathological relevance to PD, several DJ-1-deficient Drosophila models have been developed (Table 5) (112, 113, 118, 136, 202). In contrast to mammals, Drosophila possess two DJ-1 orthologs, DJ-1α and DJ-1β. Drosophila DJ-1α is expressed predominantly in the testis, whereas DJ-1β is expressed ubiquitously. Loss-of-function DJ-1β mutants demonstrate defects in locomotion, assessed by climbing ability, however, without loss of dopaminergic neurons. At the level of mitochondria, these flies showed an increased number of enlarged mitochondria and increased sensitivity to hydrogen peroxide and paraquat (136). In contrast, a second model with deletion of DJ-1β did not result in paraquat sensitivity possibly due to a compensatory neuroprotective up-regulation of DJ-1α expression in the brain (112). DJ-1α knockdown in flies by transgenic RNA interference (RNAi) resulted in the cellular accumulation of ROS, hypersensitivity to oxidative stress, and degeneration of dopaminergic and photoreceptor neurons (202). These pathological phenotypes are surprising given the restricted expression pattern of DJ-1α compared to DJ-1β. DJ-1α/β double mutant flies do not exhibit any major abnormalities or decreased lifespan, are viable and fertile, but they are more sensitive to paraquat and rotenone exposure (113). These studies overall suggest that DJ-1 activity is selectively involved in protection from oxidative insult in vivo.
MPTP, 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine.
DJ-1 null mice, similar to PINK1 and parkin null mice, fail to develop PD-related pathology such as dopaminergic neuronal degeneration, striatal dopamine depletion or protein inclusion formation (Table 5). DJ-1 KO mice exhibit subtle nigrostriatal pathway abnormalities including mild deficits in motoric function, increased striatal dopamine reuptake, and impaired corticostriatal LTD (59, 75, 78). DJ-1 null mice are more sensitive to MPTP-induced neurotoxicity, and this hypersensitive phenotype is suppressed by viral-mediated overexpression of DJ-1 (75). Moreover, inactivation of DJ-1 in mice led to amplification of oxidative stress resulting in oxidation of mitochondrial matrix proteins, specifically in dopaminergic neurons (64). These observations suggest a neuroprotective function for DJ-1 during mitochondrial insult. Mitochondria isolated from young DJ-1 KO mice reveal increased ROS levels and markedly decreased aconitase activity without additional deficits in other mitochondrial activities (3). Accordingly, DJ-1 has been proposed to function as an atypical peroxiredoxin-like peroxidase to scavenge mitochondrial hydrogen peroxide (3). An age-dependent up-regulation of mitochondrial manganese superoxide dismutase and glutathione peroxidase levels and mitochondrial glutathione peroxidase activity is observed in mitochondria isolated from brains of old DJ-1 KO mice, suggesting a compensatory mechanism that is invoked in the absence of DJ-1 that may explain the lack of an overt neurological phenotype in aged DJ-1 KO mice (3). Such compensatory mechanisms could arise from the ability of DJ-1 to associate with RNA molecules, including mRNAs producing proteins participating in glutathione metabolism (185).
Genetic interaction studies in Drosophila and in mammalian cells identified the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway as a specific modifier of DJ-1-associated cell death (75, 202). In mammalian cells, DJ-1 regulates the phosphorylation of Akt through the tumor suppressor PTEN (75). In Drosophila, down-regulation of DJ-1α results in impaired PI3K/Akt signaling which may regulate cellular ROS levels (202). Parkin deletion can also lead to impaired PI3K/Akt signaling (202), supporting a common pathway for parkin and DJ-1. Surprisingly, however, PINK1/parkin/DJ-1 triple KO mice develop normally without producing a PD-related phenotype (80). Inactivation of all three recessive PD-linked genes is not sufficient to cause nigral degeneration or PD-related pathology within the lifespan of mice, suggesting that these proteins may function in a neuroprotective capacity but are dispensable for neuronal development and maintenance (80). Whether the triple KO mice exhibit abnormalities in mitochondrial morphology or activity is not known but is warranted to provide further insight into the functional relationship between parkin, PINK1, and DJ-1 at the level of mitochondria. Evidence obtained from cellular and Drosophila models indicates that DJ-1 may work in parallel to the PINK1/parkin pathway to maintain mitochondrial function under conditions of oxidative stress. Phenotypes observed in PINK1 and parkin null flies could be compensated by the overexpression of parkin but not DJ-1 (203). In mammalian cells, the overexpression of parkin can rescue mitochondrial phenotypes due to DJ-1 loss-of-function such as mitochondrial depolarization, fragmentation, accumulation of the autophagosomal marker LC3, and increased sensitivity to rotenone, whereas DJ-1 is unable to rescue PINK1 null phenotypes (180). Thus, the PINK1/parkin and DJ-1 pathways may converge to preserve mitochondrial integrity.
The functional interaction of DJ-1 with α-synuclein has also been assessed using mouse models after the observation that DJ-1 can inhibit α-synuclein aggregation in vitro (7, 160). The deletion of DJ-1 in an A53T α-synuclein transgenic mouse model fails to influence disease-onset and neuropathology induced by A53T α-synuclein expression (54, 149). It appears therefore that DJ-1 expression is not required for mediating α-synuclein-induced neurotoxicity but could plausibly be located upstream of α-synuclein in a common pathway where it would be dispensable for α-synuclein-induced neurotoxicity. It is also possible that compensatory mechanisms could mask DJ-1-deficiency in these double mutant mice (149). The combined impact of DJ-1 and α-synuclein mutations on mitochondrial function in these mice has not been assessed.
The function of DJ-1 remains elusive although it has become clear from cell and animal models that DJ-1 can provide protection against oxidative insult, and may regulate the levels of mitochondrial ROS (Table 5 and Fig. 3). The mechanism for these actions is unclear and may involve chaperone, antioxidant and/or gene regulatory activities of DJ-1 operating within mitochondria but also more broadly in the cytoplasm to combat oxidative stress. DJ-1-linked models of PD show subtle signs of mitochondrial damage and enhanced sensitivity to mitochondrial stressors, but it is not yet clear whether these factors are required for the development of pathological phenotypes. In addition, DJ-1 null mice and fly models have largely failed to reproduce dopaminergic neuronal degeneration and other PD-related phenotypes suggesting that modest mitochondrial abnormalities in these models are not sufficient to elicit neuronal loss (Table 5). Further studies are required to determine the relative contribution of mitochondrial and cytoplasmic phenotypes to neuronal damage induced by DJ-1 mutations in PD.
Nonfamilial Mitochondrial Models
The accumulation of somatic mutations and deletions in mtDNA have been implicated in the pathogenesis of neurodegenerative diseases, including PD, and aging in general (9, 13, 86). The importance of maintaining mtDNA integrity was elegantly demonstrated in a mouse model lacking the TFAM, termed the MitoPark mouse (40). TFAM, a nuclear-encoded protein, is post-translationally transported to mitochondria where it acts as a DNA-binding protein essential for both transcription and maintenance of mtDNA. Conditional deletion of the TFAM gene selectively in dopaminergic neurons was achieved by crossing mice harboring a loxP-flanked TFAM allele with mice expressing Cre recombinase from the endogenous DAT promoter (40). Deletion of TFAM in DAT-positive neurons resulted in reduced expression of mtDNA and respiratory chain deficiency. Further, MitoPark mice successfully recapitulate some key aspects of PD-related pathology, including progressive, adult-onset dopaminergic neurodegeneration accompanied by levodopa-responsive motor deficits, and the formation of intraneuronal mitochondrial aggregates (40). MitoPark mice demonstrate the critical requirement of nigral dopaminergic neurons for intact mitochondrial function with age. The mice also offer a potential mechanism for how mtDNA deletions could lead to the development of PD. It is not clear whether other nondopaminergic neuronal populations are equivalently vulnerable in vivo to mtDNA deletions, since the MitoPark mice are based upon the conditional deletion of TFAM in dopaminergic neurons. It is also worth considering that familial PD is not known to be caused by mutations in TFAM suggesting that MitoPark mice, like MPTP toxin models, do not represent models of PD but instead potentially reveal the heightened sensitivity of nigrostriatal dopaminergic neurons to mitochondrial insults. Mutations in DNA polymerase γ (POLG), which is responsible for replication of mtDNA, are associated with parkinsonism (33), further suggesting that alterations in mtDNA could be important for the survival of dopaminergic neurons. Conditional knockin mice expressing a proofreading-deficient version of POLG accumulate mutations and deletions in mtDNA and develop phenotypes consistent with premature aging although neuronal degeneration and the viability of dopaminergic neurons in these mice have not been directly assessed (88, 183).
Concluding Remarks
Genetic animal models of familial PD based upon mutations known to cause disease have provided tremendous insight into the molecular pathogenic pathways that lead to PD. Such models provide an unparalleled opportunity to unravel molecular pathways that are critical for mediating neurodegeneration. A caveat of these mouse models is that they often lack PD-related pathology but they can provide instead essential information concerning the presymptomatic phase of disease before the onset of overt neurodegeneration where therapies are likely to be most effective. Significant evidence points to a role for mitochondrial dysfunction in the pathophysiology of PD. With genetic animal models it has been possible to ask whether mitochondrial dysfunction results from familial mutations and subsequently whether this impairment is necessary for mediating the neurotoxic actions of disease-associated proteins. It is now clear that mutations in α-synuclein, LRRK2, parkin, PINK1 and DJ-1 in animal models can produce varying levels of mitochondrial impairment, including alterations in activity (particularly ROS production), morphology and turnover. There is evidence that mitochondrial morphological changes may lead to alterations in mitochondrial activity and turnover regulated by the PINK1/parkin pathway and potentially other PD-related pathways. There is also evidence that familial mutations heighten the sensitivity of mitochondria to stress, and in some cases that these proteins are required for mediating the downstream detrimental actions of mitochondrial neurotoxins. What is not clear at present is whether mitochondrial impairment observed in these genetic animal models of PD is necessary for mediating pathological processes precipitating neuronal degeneration. The best evidence for this comes from Drosophila models where modulation of fission and fusion factors can rescue phenotypes induced by the deletion of PINK1 or parkin. Similar studies in mouse or rat models have not been forthcoming but should be imminently testable. The major problem here would be which phenotypes in mice to study and which are most relevant to PD. It may be necessary to focus less on traditional neurodegeneration, dopamine depletion and motoric deficits in existing animal models, with a greater emphasis on the role of mitochondria in more subtle phenotypes such as synaptic pathology or ultrastructural abnormalities. For example, there is evidence suggesting that α-synuclein may inhibit mitochondrial fusion and induce inappropriate mitophagy and it should be possible in rodent models to evaluate whether restoring mitochondrial fusion protects against neurodegeneration. It will take time to carefully determine whether mitochondrial impairment in genetic animal models is the most critical pathway determining the outcome of the pathological process, or whether other processes are more important. A role for mitochondrial dysfunction underlying PD certainly makes for a strong argument based on current evidence but further work is required to clarify which aspects of mitochondria become dysfunctional and when in the context of the entire pathological process. In future, it should be possible to begin to rationally test in animal models of PD whether restoring such mitochondrial deficits can provide therapeutic benefit.
Footnotes
Acknowledgments
This work was support by grants from the Swiss National Science Foundation (grant no. 310030_127478 to D.J.M.), Michael J. Fox Foundation for Parkinson's Research (D.J.M.) and Parkinson Schweiz (D.J.M.) and financial support from the EPFL (D.J.M.).