
Abstract
Breast Cancer–General Knowledge
Breast cancer is one of the major causes of cancer mortality in women. The incidence of breast cancer is still increasing worldwide. It is estimated that in the year 2011, there will be about 200,000 new cases of invasive breast cancer in the United States alone. These data translate to 1 in 8 (∼12%) women developing some form of breast cancer, over the course of their lifetime. About 5–10% of breast cancer cases are due to familial/inherited mutations within the BRCA1 or BRCA2 genes. Further, about 20–30% of women diagnosed with breast cancer have a family history of cancer. However, the most important risk factor for breast cancer is increased age (over 45 years old). Other risk factors can be prevented or modified, such as hormone use, low physical activity, alcohol consumption, diet, obesity, and smoking (2).
Breast cancer, like most cancers, is highly variable with great clinical heterogeneity. Gene profiling and high-throughput expression technologies have prompted the classification of breast cancer into molecular subtypes, based on conventional markers, such as estrogen receptor (ER+), progesterone receptor (PR+), and human epidermal growth factor receptor (EGFR) 2 (Her2/Neu+) (28). In addition, classification can also be based on clinical outcome, response to therapy, and varied biological properties. Classification of breast cancer into subtypes is important to determine overall therapy, the clinical course of action, and patient prognosis.
The Tumor Microenvironment
Tumors are complex structures composed of multiple cell types, with the most prominent among these being the actual cancer cells, which exhibit a high or unlimited proliferative capacity. Cancer cells are surrounded by other cell types, which help to create and maintain the tumor microenvironment. There is an emerging interest in the role of the tumor microenvironment, since early studies have shown that the normal mammary microenvironment is capable of reversing the malignant nature of breast cancer cells, by inducing differentiation (23, 24). In the last decade the tumor microenvironment has received renewed attention for its role in supporting and even promoting the invasive cancer phenotype (Fig. 1).
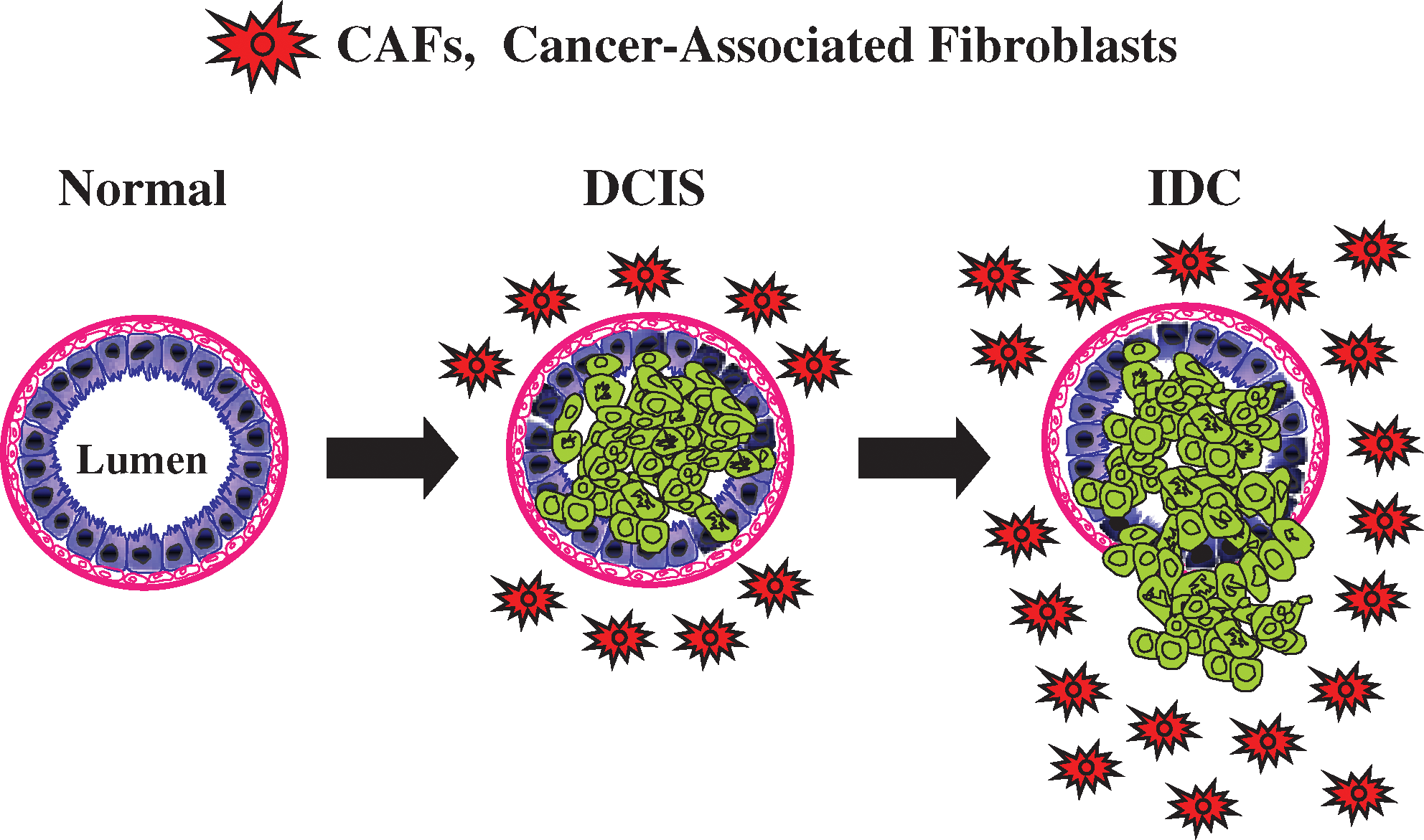
The tumor microenvironment consists of immune cells (lymphocytes, natural killer cells, and antigen presenting cells), stromal cells (including myofibroblasts), and the vasculature (53). Together, these key components are known as the tumor stroma and account for nearly 50% of a tumor's mass (1). Myofibroblasts and/or cancer-associated fibroblasts (CAFs) are cells whose origin is yet not well defined, but it has been suggested that they arise from progenitors, such as mesenchymal stem cells (MSCs) derived from the bone marrow (BM) or from other differentiated cells, such smooth muscle cells, preexisting fibroblasts, and preadipocytes (83). MSCs are precursor cells that go on to become osteoblasts, chondrocytes, adipocytes, and fibroblasts, where they function in bone, cartilage, fat, and muscle morphogenesis, respectively (9). During injury, MSCs are dispatched to the site of injury, where they function in renewal and promote regeneration (9).
It is now well-recognized that cancer can be understood as “an injury that does not heal” (29, 104). Rudolf Virchow, in 1863, hypothesized that tumorigenesis requires a chronic irritation or a previous nonhealing wound (114), suggesting that cancer itself also promotes the attraction of mesenchymal cells to the tumor site (9). In support of this notion, CAFs have been shown to contribute to tumor growth by affecting cancer survival, (i) by generating antiapoptotic factors (91), (ii) by contributing to the vasculature by secreting vascular endothelial growth factor and angiopoetin, (iii) by promoting cell motility and metastasis through secretion of chemokines, such as CCL5 (54) and matrix metalloproteinases (MMPs), and (iv) by blocking the immune response through secretion of IL-6, IL-10 and TGF-beta (115).
Paracrine communication between CAFs and neighboring cancer epithelial cells is a crucial factor for the continued growth of the tumor mass (108). The tumor microenvironment also plays a role in resistance to radio/chemotherapy (3). In order to become more mobile, it is now accepted that cancer cells need to acquire mesenchymal-associated features, in a process known as an epithelial–mesenchymal transition (EMT) (127). CAFs seem to be playing a role in regulating the EMT, by either facilitating or inducing the EMT in cancer cells (42, 112, 113). This makes CAFs key player(s) in tumor progression, invasion, and metastasis.
The Warburg Effect
In the early 1920s, Otto Warburg, a Nobel Laureate (1931), formulated a hypothesis to explain the “fundamental basis” of cancer, based on his observations of tumor behavior where cancer cells displayed a metabolic shift toward glycolysis (116). Warburg stated that:
“Cancer, above all other diseases, has countless secondary causes. But, even for cancer, there is only one prime cause. Summarized in a few words, the prime cause of cancer is the replacement of the respiration of oxygen in the normal body cells by a fermentation of sugar.”
In this hypothesis, referred to as the “Warburg Effect,” cancer cells produce energy via the conversion of glucose into lactate, despite the presence of oxygen, a process known as aerobic glycolysis. Anaerobic glycolysis, in contrast, is initiated under hypoxic conditions in normal cells or when cancer cells adapt to hypoxia (22, 58, 110). Normal cells under aerobic conditions utilize glucose to produce pyruvate, which then gets oxidized in the mitochondria via the tricarboxylic acid (TCA) cycle into carbon dioxide, a process called oxidative phosphorylation (37).
Warburg's observations contrasted with the observations of Louis Pasteur. Sixty years before Warburg's time (1861), Pasteur described how in normal cells, the presence of oxygen normally inhibits glycolysis (47). The shift toward glycolysis, as a consequence of diminished oxygen, is also known as the “Pasteur effect” (85). By convention, aerobic glycolysis is less efficient for energy production (two ATP produced per molecule of glucose) compared to mitochondrial oxidative phosphorylation (36 ATP produced per molecule of glucose). Thus, aerobic glycolysis became a hallmark of the cancer phenotype.
Despite nearly 85 years since it was first described, the Warburg effect remains a paradox within the scientific community and much effort has been invested to explain the basis for these observations. Warburg attributed this metabolic shift to mitochondrial dysfunction in cancer cells (116). However, several studies from the early 1970s found no evidence for a respiratory defect in cancer cells (30, 35, 75). For example, electron micrographs of tumors revealed the presence of mitochondria in epithelial cancer cells, which contrasted with the neighboring fibroblasts (84). With the advent of cell culture techniques, investigations in tumor biology and respiratory chain activity have been carried out, using immortalized cancer cell lines (101, 105, 111). These studies demonstrated that the increased growth rate of cancer cells is a consequence of an increased glycolytic rate (glycolytic shift), prompting a decrease in oxidative phosphorylation and mitochondrial density. Simonnet et al., in particular, concluded that tumor cells with increased mitochondrial dysfunction exhibited a more aggressive phenotype (105).
As a consequence of the Warburg effect, cancer cells would produce large amounts of lactate. The excess lactate is then released to the extracellular environment which results in a decrease in extracellular pH. This acidic microenvironment is thought to favor the metastatic potential of some cancer cells (46, 102). Another recent explanation for this paradox proposes that such a metabolic shift is beneficial for proliferating cells that require, in addition to energy in the form of ATP, macromolecules such as nucleotides, amino acids, and lipids. In this explanation, glycolytic intermediates promote increased growth rate through the incorporation of carbon sources, such as glucose, into the biomass and thus contribute to a balance between catabolic and anabolic needs of actively proliferating cells (110). Despite extensive research, there is still no consensus on why cancer cells would use an inherently inefficient process to meet their ever-increasing energy demands.
Autophagy
In 1963, Christian de Duve, a Nobel Laureate, first coined the phrase auto-phagy, derived from the Greek words “auto” and “phagy,” meaning “self” and “eating.” Autophagy is an evolutionarily conserved survival pathway involving degradation of cytoplasmic constituents, the recycling of ATP, and the maintenance of cellular biosynthesis, during nutrient deprivation or metabolic stress (17). Autophagy plays an important homeostatic role, mediating removal, digestion, and recycling of damaged organelles (73, 74). Autophagy is activated in response to a range of stimuli, including nutrient depletion, hypoxia, and activated oncogenes. Autophagy has also been shown to play a dual role: (i) it results in consumption of cellular components and subsequent cell death (5); (ii) it is able to maintain cancer cell survival under conditions of metabolic stress, and it may confer resistance to radiation and chemotherapy (4, 25).
Autophagy can provide a survival advantage to tumors by overcoming metabolic stress during tumorigenesis. Tumor cells are frequently exposed to metabolic stress owing to hypoxia and nutrient deprivation and autophagy can help maintain essential cellular functions through its recycling capacity. In addition, there is an increase in hypoxia-inducible transcription factor 1-alpha (HIF1-alpha), which promotes autophagy (through BNIP3) and angiogenesis (7, 128). HIF1-dependent expression of BNIP3 is essential for the selective autophagy of mitochondria, also called mitophagy. Hypoxia can also activate autophagy through HIF1-independent pathways (26). Understanding the mechanisms of autophagy in different cells of the tumor microenvironment is an essential prerequisite for developing an autophagy-activating or autophagy-inhibiting treatment strategy.
Caveolin-1 and CAFs
Caveolins are a family of scaffolding proteins that are involved in several cellular processes, such as cholesterol homeostasis, vesicular transport, and the regulation of signal transduction (18, 120). The Caveolin-1 (Cav-1) gene is located in close proximity to the D7S522 locus, which is frequently deleted in various human cancers, including tumors of the breast, colon, ovary, and head and neck (120). Cav-1 mRNA and protein are downregulated or absent in primary human cancers and numerous cancer cell lines (56). Cav-1 is a negative regulator of cytokine receptor signaling and various studies indicate contrasting tumor suppressor and tumor promoter functions, depending on the tumor cell type studied (43, 120). Tumor microarray (TMA) analysis of invasive breast carcinomas identifies Cav-1 overexpression to be associated with a basal-like phenotype, predicting a worse prognosis in breast cancer patients (95, 103) and hepatocellular carcinomas (129). Cav-1 is also reduced in human ovarian cancer suggesting a role as tumor suppressor (118).
We have shown that Cav-1 expression is significantly reduced in human breast CAFs, compared with matched normal mammary fibroblasts obtained from the same patients (70). In this study, CAFs were isolated from 11 breast cancer patients, 8 of which showed a significant downregulation of Cav-1 at the protein level, but not at the transcriptional level (70). To validate a correlation between decreased Cav-1 expression in CAFs and mammary stromal fibroblasts lacking Cav-1, we performed in vitro experiments to demonstrate a direct cause–effect relationship (108). Human CAFs and mammary stromal fibroblasts obtained from Cav-1−/− null mice share many characteristics such as hyperproliferation, increased collagen production, and activated TGF-beta signaling, the upregulation of muscle-related genes, and the ability to contract/retract (108). Further, genome-wide expression profiling showed common transcriptional characteristics between the human breast CAFs and the Cav-1−/− mammary stromal fibroblasts (86 –89, 108). These shared phenotypic characteristics are hallmarks of a constitutive myofibroblastic phenotype.
CAFs are suggested to play an important role in mediating paracrine signaling to mammary epithelial-derived tumor cells (96, 97). In support of this, we recently showed that conditioned media prepared from Cav-1−/− mammary stromal fibroblasts promotes an EMT, blocks the typical 3D acinar structure formation of epithelial cells, and induces instead a mesenchymal appearance, with the induction of smooth muscle actin expression (108). The EMT is a major characteristic of cancer progression toward a metastatic phenotype (50, 108). In agreement with the above findings, in in vivo transplant studies, there is a two-fold increased growth of mammary tumor cells, when these are implanted in the mammary fat pad of Cav-1−/−-deficient mice (122). Taken together, these studies directly demonstrate the association between mammary stromal fibroblasts lacking Cav-1, and the cancer-associated fibroblastic phenotype (70, 108). Thus, Cav-1−/−-deficient mammary fibroblasts represent a valid experimental model for the study of CAF biology in cancer progression (108).
Cav-1 in Human Tumor Samples: New Stromal Biomarker Discovery
Although Cav-1 appears to have both tumor-suppressive and promoter activity in different cancers, studies have suggested that Cav-1 inhibits breast cancer cell migration and metastasis (14, 34, 39, 40, 56, 107, 120). Cav-1-null mice carrying the MMTV-PyMT transgene develop an increased number of dysplastic lesions throughout the mammary gland and lung metastasis is dramatically enhanced (119 –121). Further, Cav-1 expression levels are significantly lower in human breast cancer cells, than in their normal mammary epithelial cell counterparts (60).
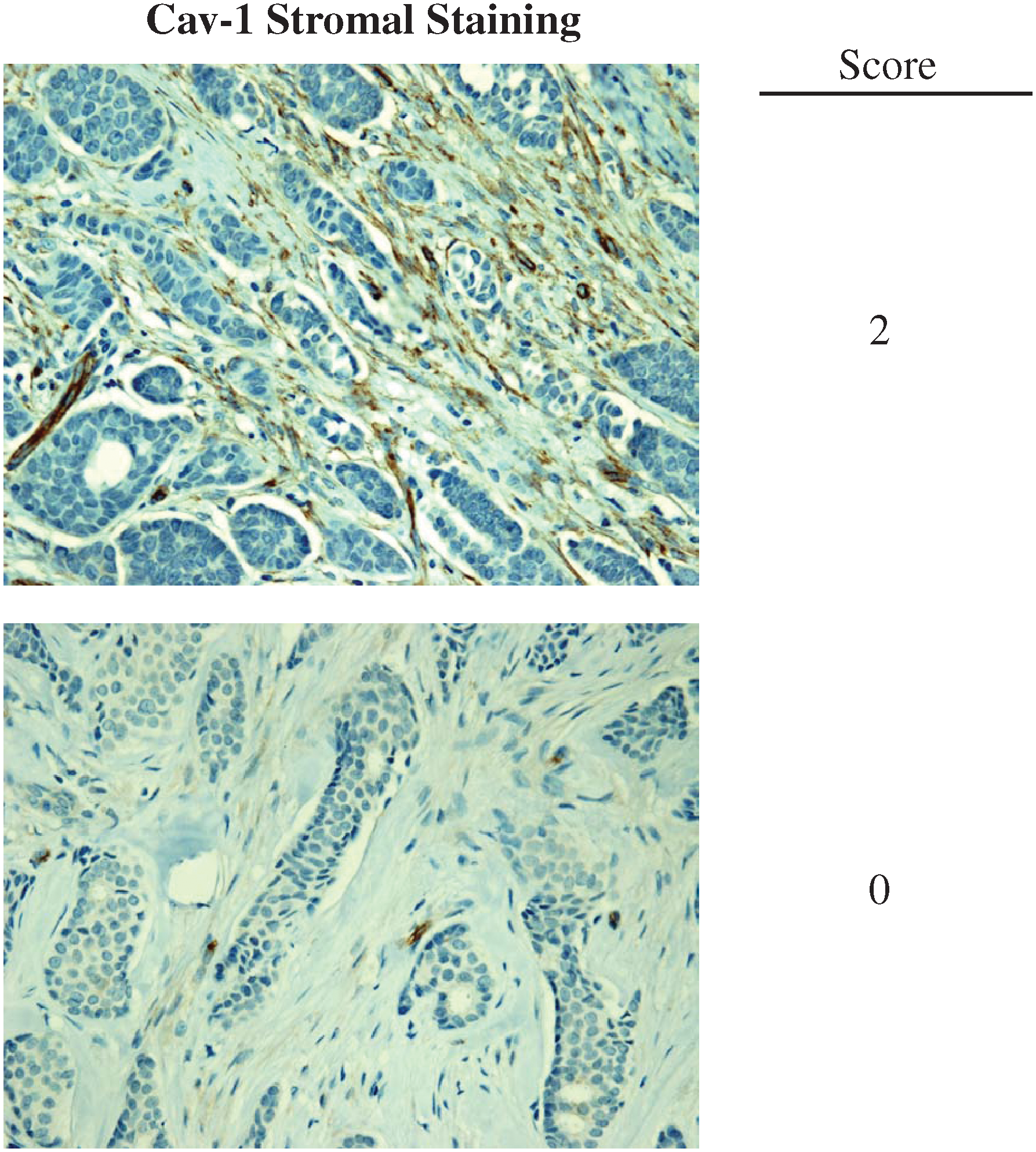
All of this experimental and clinical evidence supports the idea that Cav-1 is a tumor suppressor in patients with breast cancer. To test whether loss of stromal Cav-1 could serve as a biomarker for prognostic and diagnostic analysis, several independent studies were carried out using various human breast cancer cohorts. In one study, a cohort of 78 ductal carcinoma in situ (DCIS) patients was evaluated for the presence of Cav-1 in the breast cancer tumor stroma. These patients were treated with wide excision, but without any chemo- or radiotherapy, thus ruling out other variables due to treatment. These findings directly demonstrated that a loss of stromal Cav-1 is a powerful predictor of DCIS recurrence and progression to invasive breast cancer. Moreover, analysis of the DCIS lesions revealed that a loss of stromal Cav-1 showed increased inflammatory cells at the lesion site. On the other hand, ER+ patients with high levels of Cav-1 expression in their tumor stroma showed no recurrence and no progression (123). Thus, in this study, the prognostic value of Cav-1 as a biomarker in early breast lesions was established, indicating that stromal Cav-1 could be used nearly 15–20 years in advance, to predict the onset of DCIS recurrence and progression to invasive breast cancer (123).
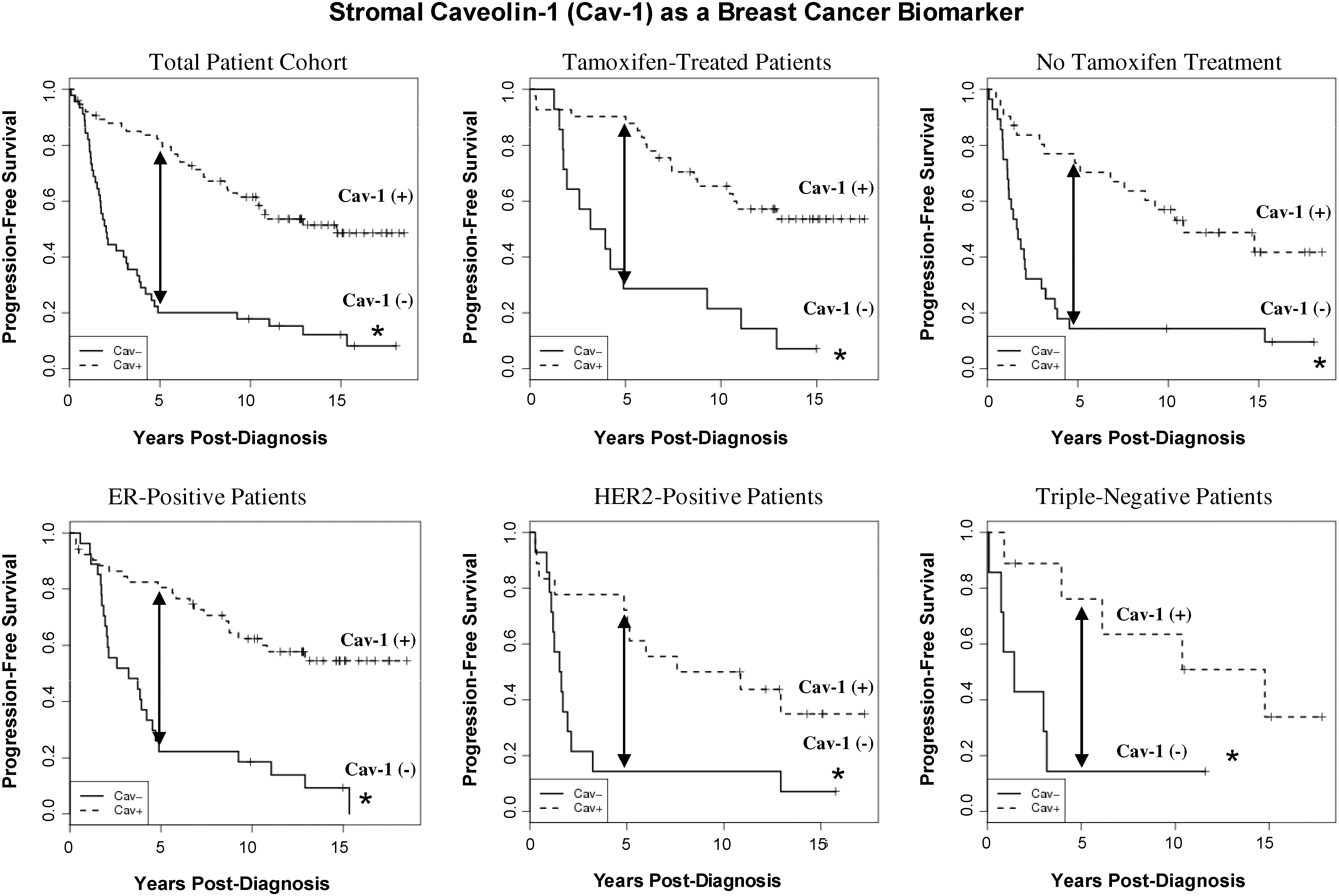
Importantly, the predictive value of a loss of stromal Cav-1 is independent of epithelial marker status, indicating that it is a useful biomarker in all the most common epithelial subtypes of human breast cancer, including ER+, PR+, HER2+, and triple-negative breast cancers (Figs. 2 and 3). For example, a TMA of 154 patients with invasive carcinoma was analyzed to determine the correlation of loss of stromal Cav-1, with patient outcome, and other clinical and pathological variables. Importantly, an absence of stromal Cav-1 correlated with early tumor recurrence, lymph node metastasis, and advanced tumor stage. In addition, an absence of stromal Cav-1 expression was particularly predictive in lymph node positive (LN+) patients. In this group of LN+ patients, high stromal levels of Cav-1 were associated with a 5-year survival rate of >80%. In contrast, an absence of stromal Cav-1 was predictive of a 5-year survival rate of ∼7%, an 11.5-fold stratification (125). In addition, a loss of stromal Cav-1 was positively correlated with treatment failure in ER+ patients who received tamoxifen, indicative of resistance to endocrine-related therapies (125).
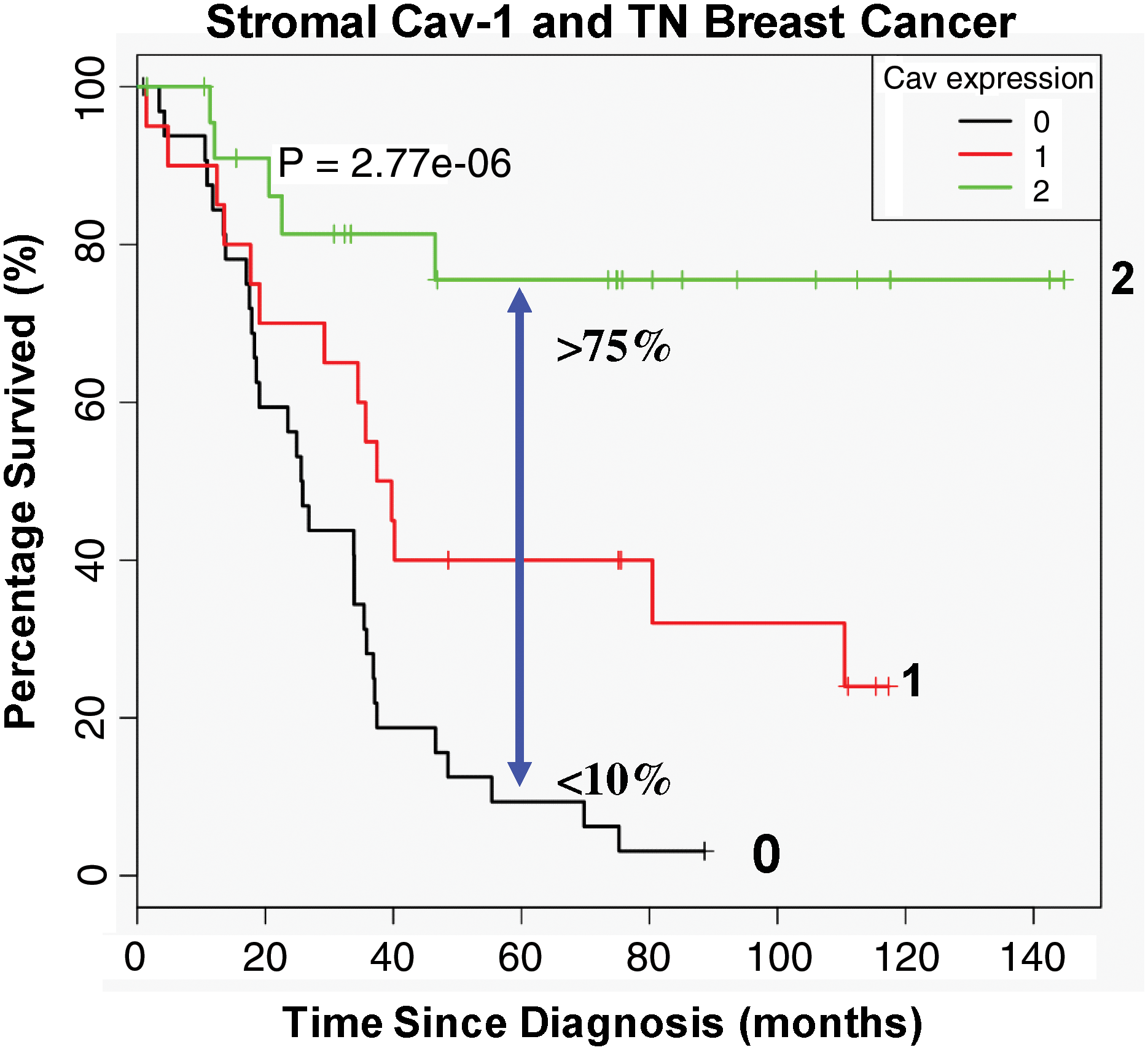
To further scrutinize the importance of Cav-1 as a predictive tool in late-stage carcinoma, a TN (triple negative; ER−; PR−; HER2−) cohort was specifically tested for Cav-1 expression and correlated this with the overall survival of patients based on 12 years of accumulated outcome data. In this context, TN patients with high stromal Cav-1 had a 5-year survival rate >75%, while <10% survived when Cav-1 was absent in the tumor stroma (124) (Fig. 4). Further, after comparison with other established TN and basal breast cancer markers such as cytokeratin 5/6 (CK5/6) (+) and EGFR (+), a loss of stromal Cav-1 was substantiated as a robust and valuable prognostic factor.
To identify and characterize the signaling pathways that are activated in a Cav-1–negative tumor microenvironment, the fibroblastic stroma was laser-capture microdissected from four cases showing high stromal Cav-1 expression and seven cases with a loss of stromal Cav-1. Transcriptional analysis identified 238 gene transcripts that were upregulated and 232 gene transcripts that were downregulated in the stroma of tumors showing a loss of Cav-1 expression (p≤0.01 and fold-change ≥1.5). Gene set enrichment analysis revealed that these transcripts were associated with “stemness,” inflammation, DNA damage, aging, oxidative stress, hypoxia, autophagy, and mitochondrial dysfunction in the stroma of Cav-1–negative patients (126) (Fig. 5). These results are consistent with previous biomarker studies showing that increased expression of autophagy markers, such as ATG16L, in the tumor stroma is specifically associated with metastatic tumor progression and/or poor clinical outcome (82).

The importance of a loss of stromal Cav-1 in regard to breast cancer invasiveness (metastasis) and survival was recently independently validated by two other research groups, using different antibody probes and distinct breast cancer patient populations, in Australia, Argentina, and Korea (57, 106). Similarly, in Japan, a third research group showed that a loss of Cav-1 in the stroma (coupled with positive expression of Cav-1 in the tumor cells) is significantly correlated with an unfavorable prognostic outcome in breast cancer (98).
Moreover, a loss of stromal Cav-1 could serve as a universal or widely applicable biomarker in other epithelial cancers, since similar observations were also now validated in advanced and metastatic prostate cancer (27). In this cohort, a loss of stromal Cav-1 strictly correlated with a high Gleason score, the current gold standard for predicting clinical outcome in prostate cancer patients (27).
The Reverse Warburg Effect Drives Tumor-Stroma Metabolic Coupling
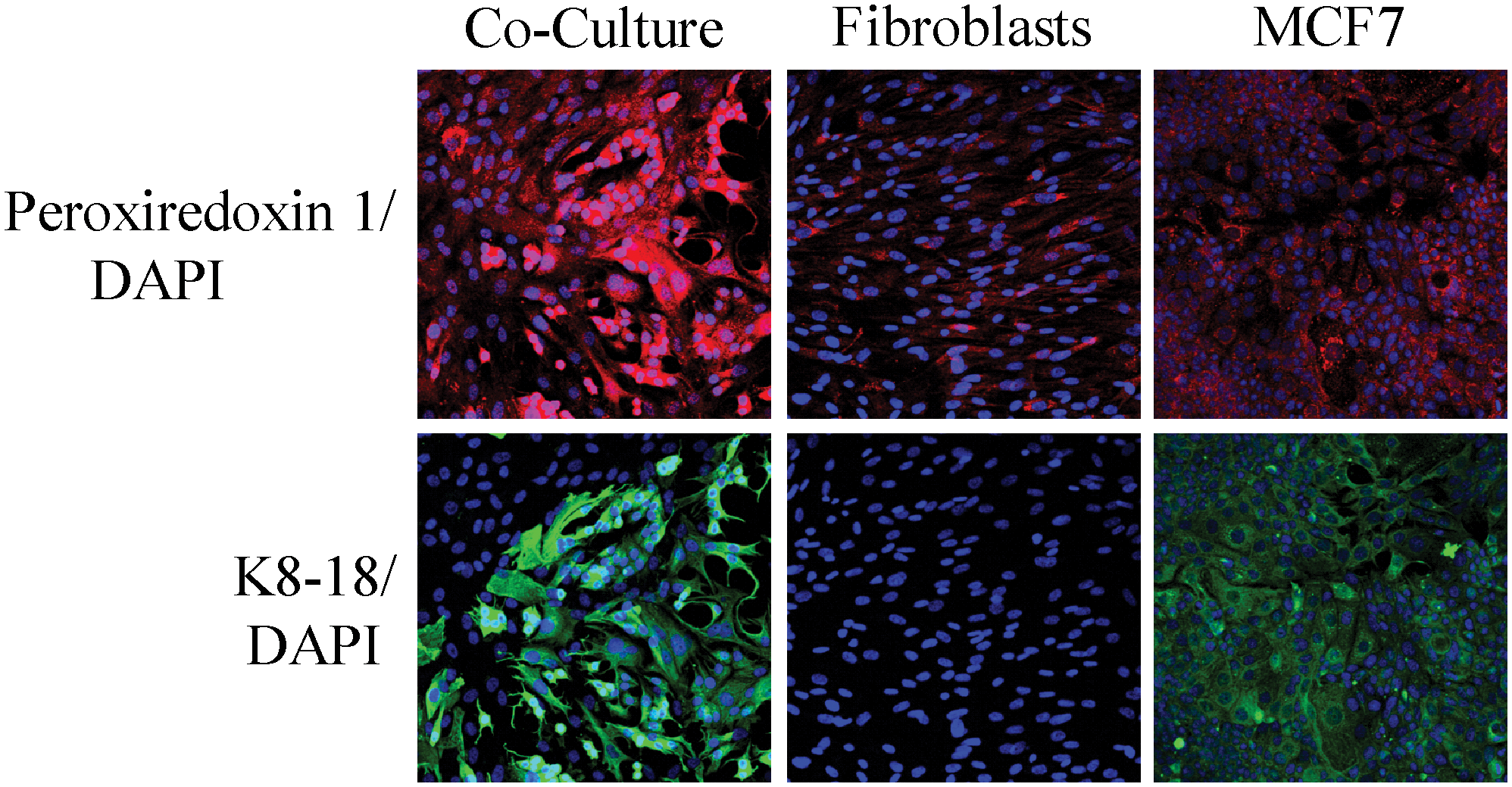
Given the powerful predictive value of a loss of stromal Cav-1 in human breast and prostate cancer patients, we decided to pursue the discovery of new biomarkers that were associated with a lack of Cav-1 expression in the tumor stroma cells. As an initial approach to identify upregulated proteins in the Cav-1−/− stromal cells, we carried out an unbiased proteomics analysis using BM mesenchymal stromal cells derived from Cav-1−/− null and wild-type mice (89). We chose this approach as it has been shown that CAFs at tumor sites originate from precursor stromal cells of the BM (6, 44, 72). We examined both cell lysates to identify proteins within cells, as well as conditioned media, to identify secreted proteins. In addition, gene expression profiling using the Affymetrix exon array was carried out to provide independent validation of the proteomics data. Analysis of this new model of CAFs (Cav-1−/−-deficient stromal cells) specifically showed the upregulation of glycolytic enzymes (PKM2, LDH-A), myofibroblastic markers (vimentin, calponin), extracellular matrix (ECM) proteins (collagen, SPARC), signaling molecules (annexins A1, A2, and RhoGDI), oncogenes (EF1-delta), liver-specific proteins (albumin, fetoprotein), and antioxidant enzymes (catalase, peroxiredoxin-1) (89). The biological relevance of these observations was further validated by directly immunostaining breast cancer tissue sections derived from patients with a loss of stromal Cav-1 (89).
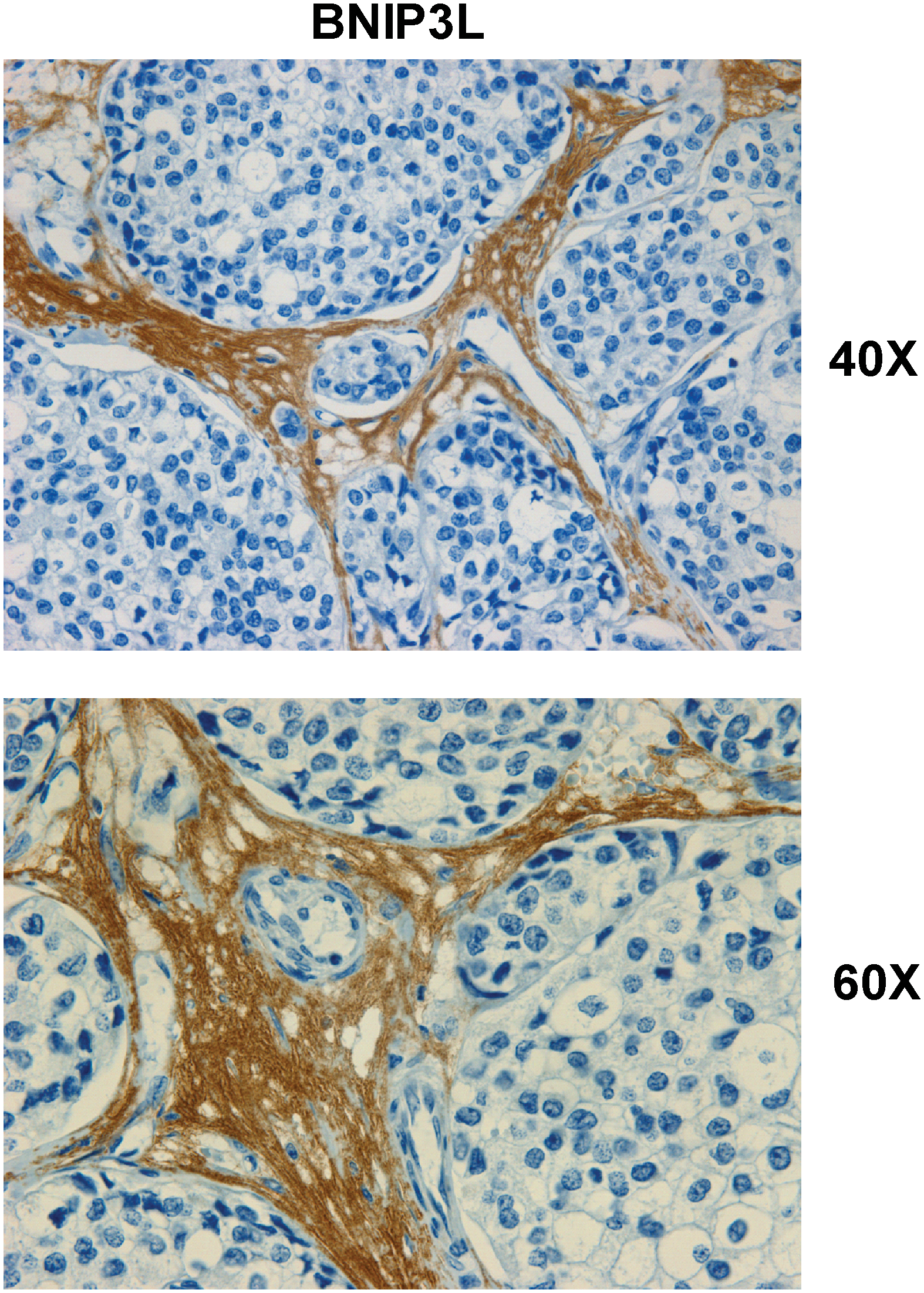
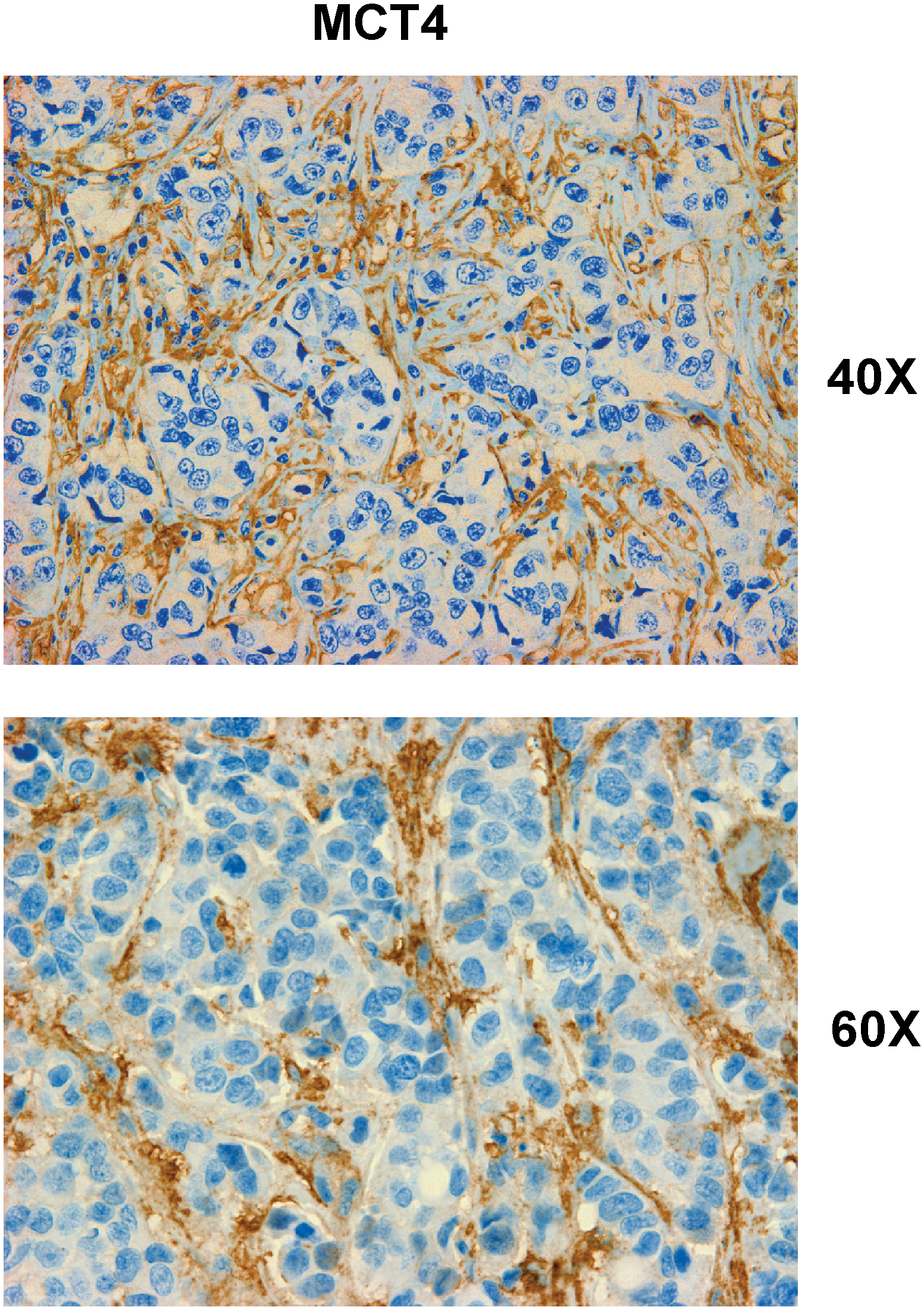
The most important observation from this study is that a panel of glycolytic markers (such as PKM2, LDH, enolase, and fructose-bisphosphate aldolase A) were all seen to be upregulated in Cav-1–deficient stromal cells, both at the protein and mRNA transcript level, which led us to consider that the Warburg effect might actually originate in the tumor stroma, and not in epithelial cancer cells (89). This has important physiological consequences, as it means that glycolytic cancer associated fibroblasts would function as factories to convert glucose to L-lactate, a high-energy metabolite. In turn, the L-lactate could then be transferred to oxidative cancer cells, which then would generate large amounts of ATP, via the TCA cycle, and oxidative mitochondrial metabolism. In this scenario, epithelial cancer cells are parasites that hijack neighboring normal stromal cells as a fuel supply. To distinguish this new idea from the conventional Warburg effect, we have coined the term, the “reverse Warburg effect,” where cancer cells steal energy via aerobic glycolysis in the tumor stroma (Figs. 6 and 7) (89). Consistent with the idea that Cav-1–deficient tumor stroma is highly glycolytic, it stains positively for BNIP3L (a marker of mitochondrial dysfunction; Fig. 8) and monocarboxylate transporter 4 (MCT4) (a marker of L-lactate production and secretion; Fig. 9) in human breast cancer samples (68, 117).
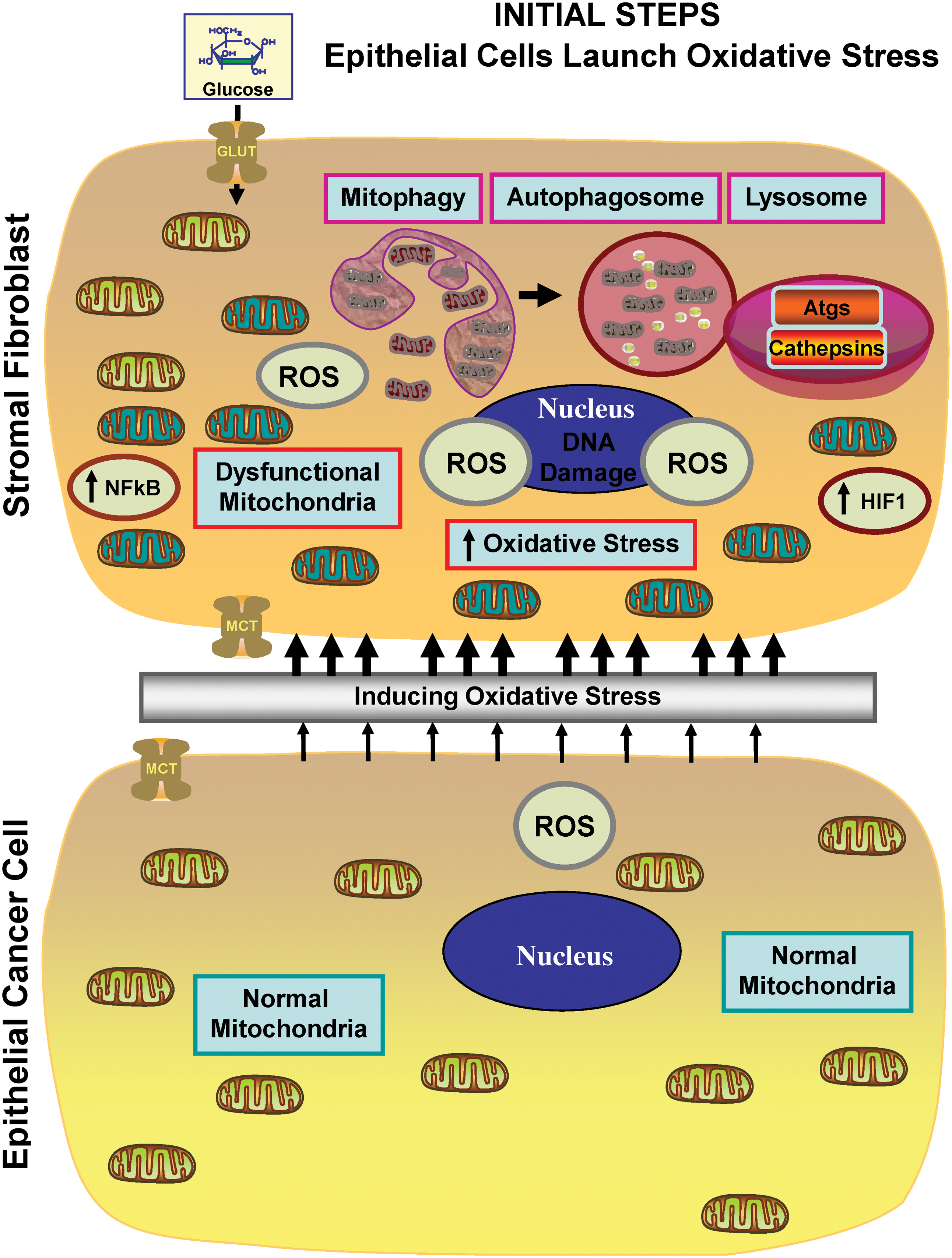

A more in-depth analysis of the Cav-1–deficient transcriptional data obtained from the above study (89) was performed using a bioinformatics approach to reinterrogate and intersect our transcriptional data, with previously published data sets, to identify specific transcriptional patterns (87). In this context, several interesting and relevant transcriptional signatures and biological processes emerged, which included gene sets associated with aging, inflammation, DNA damage, oxidative stress, nitric oxide (NO) overproduction, glycolysis, and mitochondrial dysfunction. Similarly, we also observed the upregulation of key reactive oxygen species (ROS)–producing enzymes, and the critical targets of transcription factors normally associated with oxidative stress, such as NFkB (innate immune response) and HIF1-alpha (hypoxia, glycolysis, and mitochondrial dysfunction) (87).
How does a loss of stromal Cav-1 have such powerful effects on gene expression profiling? One idea is that this is mostly secondary to the overproduction of NO, which acts as a mitochondrial “poison.” Cav-1 normally functions as known negative regulator of NO production, via the tonic inhibition of nitric oxide synthase (NOS). Thus, a loss of stromal Cav-1 would drive NO overproduction. In turn, NO overproduction can directly drive mitochondrial dysfunction, as it interferes with proper oxidative phosphorylation, leading to increased mitochondrial ROS production, and driving oxidative stress which mimics hypoxia (also known as pseudohypoxia). This would then account for the observed increases in gene profiles associated with aging, DNA damage, inflammation, glycolysis, as well as NFkB and HIF1-alpha activation (87).
Previously, we and others have shown that Cav-1 is a negative regulator of NOS (41, 52, 100). In our working model, Cav-1 downregulation leads to increased NOS activity leading to excess NO production. NO at normal levels is an important signaling molecule for modulating inflammatory response (16); however, in excess amounts, accumulation of NO can be deleterious to cells by promoting ROS production (8).
It has been shown that excess NO causes mitochondrial dysfunction through increased nitrosative stress and the production of peroxynitrite (10, 45, 55, 99), and we demonstrated that Cav-1−/− stromal cells have increased protein tyrosine-nitration, which is an functional indicator of excess of NO production and oxidative stress. We showed this at the whole cell level via immunofluorescence to detect nitro-tyrosinated proteins (87). In particular, we also showed via immunoprecipitation studies of BM-derived stromal cells and siRNA-treated fibroblasts that mitochondrial components (such as complex-I) undergo increased tyrosine nitration in the absence of Cav-1 (87). Increased tyrosine nitration of mitochondrial components, such as complex-I, is indicative of mitochondrial dysfunction and oxidative stress (10, 15, 45, 99).
In accordance with this view, previous studies have shown that Cav-1−/− null mice have functional mitochondrial defects and reduced mitochondrial reserve capacity (18 –20). For example, Cav-1−/− white adipose tissue was shown to have mislocalized mitochondria and abnormal triglyceride hydrolysis, which is an indication of an inability to utilize fatty acids in the mitochondria (19). In a separate study, looking at brown adipose tissue (BAT), it was shown that mice lacking Cav-1 have defects in thermoregulation, since metabolic restriction (starvation) coupled with cold treatment caused Cav-1 null mice to be unable to maintain their body temperature (20). This is an indication of a defect in mitochondrial function where fatty acid liberation and fatty acid beta-oxidation (a mitochondrial process) does not occur in these mice (20). Further, electron microscopy revealed defects in mitochondrial morphology with enlarged and dilated mitochondria in the BAT of Cav-1−/− mice (20). Similarly, another study demonstrated that Cav-1−/− null mice that have undergone partial hepatectomy are unable to regenerate their liver, resulting in lethality (36). This fatal outcome is prevented when these mice are provided with increased glucose, but not fatty acids. As such, it appears that Cav-1–deficient mice are strictly dependent on glucose and aerobic glycolysis for energy production, rather than mitochondrial function and beta-oxidation (36). Mitochondrial dysfunction in Cav-1–deficient mice may also be caused due to increased cholesterol accumulation in mitochondrial membranes, which leads to a loss of efficiency of mitochondrial respiratory chain and increased ROS production (13). Therefore, these examples demonstrate that a Cav-1 deficiency leads to mitochondrial dysfunction in various cell types, under stressful conditions.
As Cav-1−/−-deficient mice represent a “whole body” animal model of mitochondrial dysfunction in the tumor microenvironment, this could lead to the development of new strategies for anticancer therapy (87). For example, we could target the tumor stroma with inhibitors of glycolysis and mitochondrial oxidative phosphorylation. To validate this working hypothesis, we carried out in vivo studies using conventional inhibitors of glycolysis (2-deoxy-glucose [2DG]) and mitochondrial metabolism (metformin; a mitochondrial complex I inhibitor), to block the two arms of energy production and test the mitochondrial reserve capacity of Cav-1−/− null mice (87). For this purpose, mice were treated daily with both inhibitors for up to 2 weeks. Importantly, Cav-1−/− null mice were extremely sensitive to the treatment; 80% of the mice died within 24 h, and 100% died within 48 h (87). However, all wild-type mice survived the entire treatment period (87). Thus, a Cav-1 deficiency is synthetically lethal with energy restriction, possibly explaining the tumor suppressor effects of caloric restriction. It is important to note that treatment with 2DG alone or metformin alone was not lethal in Cav-1 null mice, indicating that the combination was indeed required for effective chemical induction of energy restriction.
In further support of these findings, a separate study from our laboratory used a novel human tumor xenograft model to mechanistically address the reverse Warburg effect in vivo (12). Aggressive human breast cancer cells, MDA-MB-231, were coinjected in nude mice with WT or Cav-1–deficient stromal fibroblasts. Fibroblasts lacking Cav-1 specifically promoted tumor growth, with a ∼2.5-fold increase in both tumor weight and volume (12). Interestingly, treatment of this Cav-1–deficient xenograft model with a combination of two glycolysis inhibitors, namely 2DG and dichloro-acetate (DCA), reduced tumor growth by ∼4.5-fold (12). Thus, this demonstrates a promising new therapeutic strategy for breast cancer patients who lack stromal Cav-1 expression (12).
Further, several follow-up studies from our laboratory have now validated and contributed to a better mechanistic understanding of this hypothesis. Well-established fibroblast cell lines were used to show that tumorigenesis is enhanced in the presence of glycolytic fibroblasts versus fibroblasts that use oxidative respiration (71). These glycolytic fibroblasts showed an increase in vimentin expression and a loss of Cav-1, consistent with both myofibroblastic differentiation and the reverse Warburg effect, respectively (71). Interestingly, these glycolytic fibroblasts also dramatically promoted tumor growth, with a ∼4-fold increase in tumor mass and ∼8-fold increase in tumor volume, as assessed using the MDA-MB-231 xenograft model (71).
Using an informatics approach, we next compared our gene profiling data from Cav-1–deficient MSCs (89) with existing transcriptional data from human breast cancer tumor stroma (38). This strategy allowed us to show that these two data sets have considerable overlap, indicating that Cav-1–deficient stromal cells have a similar transcriptional profile to bona fide tumor stroma from human breast cancers (88). In addition, these two gene profiles (from Cav-1–deficient stromal cells and the tumor stroma) showed significant overlap with the Alzheimer's disease brain gene set, consistent with the idea that all three gene sets are associated with inflammation and oxidative stress (88). These data also suggest that loss of Cav-1 expression in CAFs could be used as a surrogate marker for oxidative stress, aerobic glycolysis, and inflammation in the tumor microenvironment (88).
In this context, it is interesting to note that the brain also uses a form of metabolic coupling, analogous to the one we have proposed for the reverse Warburg effect. This idea was first proposed ∼10–15 years ago, and is known as Neuron-Glia metabolic coupling (62 –64, 90, 109). In this model, astroctyes display a glycolytic shift and secrete lactate and pyruvate, high-energy metabolites, which directly feed neurons. Thus, the shuttling of high-energy lactate from one cellular compartment to another is a normal physiological mechanism, which tumors have copied, resulting in epithelial-stromal metabolic coupling.
Another prediction of this hypothesis is that CAFs should express MCT4, a monocarboxylate transporter that has been implicated in lactate efflux. MCF7 breast cancer cells were cocultured with normal fibroblasts and it was shown that breast cancer cells specifically induced the expression of MCT4 in CAFs; whereas in monoculture, neither cell type expressed MCT4 (117). Further experiments showed that the induction MCT4 in CAFs is due to oxidative stress or pseudohypoxia and treatment with antioxidants such as N-acetylcysteine (NAC) was sufficient to block this upregulation (117). This points toward the idea that accumulation of MCT4 in CAFs is a marker of oxidative stress. Interestingly, MCT1 (another lactate transporter) was found to be specifically upregulated in MCF7 breast cancer cells, when cocultured with fibroblasts. Similar results were obtained with primary human breast cancer samples. In human breast cancers, MCT4 is specifically expressed in CAFs (Fig. 9), but MCT1 was selectively expressed in the epithelial cancer cells, within the same tumors, suggesting that expression of both transporters in separate cellular compartments could confer a survival advantage for human tumors (117). These findings provide the first evidence for the existence of a stromal-epithelial lactate shuttle in human tumors (117).
MCT1 has also been found to be overexpressed in lung cancer cell lines (51), colorectal carcinomas (94), and during progression to invasive cervical carcinoma (93), suggesting that cancer cells may be utilizing lactate in more than conventional ways. These findings all seem to point to the occurrence of the reverse Warburg effect. MCT1, but not MCT4, has also been reported to be upregulated in a set of patients with basal-like breast carcinoma (92). This suggests that breast epithelial cancer cells are not glycolytic, but instead they import lactate possibly to be used as fuel for the TCA cycle and oxidative phosphorylation as described by the reverse Warburg effect (Table 1) (12).
Note that mRNA transcripts associated with oxidative mitochondrial metabolism are upregulated in human breast cancer cells, relative to adjacent stromal cells. Note also that breast cancer cells are not undergoing nutrient starvation, hypoxia, or accelerated aging (oxidative stress), as similarities with the down (DN) gene sets are observed. Modified and reproduced with permission from Bonuccelli et al. (12).
OXPHOS, oxidative phosphorylation; PR, progesterone receptor; TCA, tricarboxylic acid.
Oxidative Stress Promotes Tumor-Stroma Coevolution
To understand the mechanism(s) underlying a loss of Cav-1 expression in the tumor stroma, we created a novel coculture system using fibroblasts and cancer cells. In this model system, MCF7 human breast cancer cells are cocultured with immortalized fibroblasts under low-mitogen conditions for a period of up to 5 days. Most importantly, under these conditions of coculture, MCF7 cells downregulated the expression of Cav-1 in adjacent CAFs (Fig. 10). Mechanistically, a loss of Cav-1 in stromal fibroblasts was associated with certain phenotypic changes, such as the increased expression of myofibroblast markers, the increased secretion of ECM proteins, and activated TGF-beta signaling, consistent with a CAF phenotype (67). We speculated that a loss of Cav-1 was due to increased protein degradation or turnover. In accordance with this hypothesis, pretreatment with chloroquine (a lysosomal inhibitor) prevented the loss of stromal fibroblast Cav-1 (67). These results indicate that Cav-1 is downregulated via lysosomal degradation during autophagy, a well-known stress response.
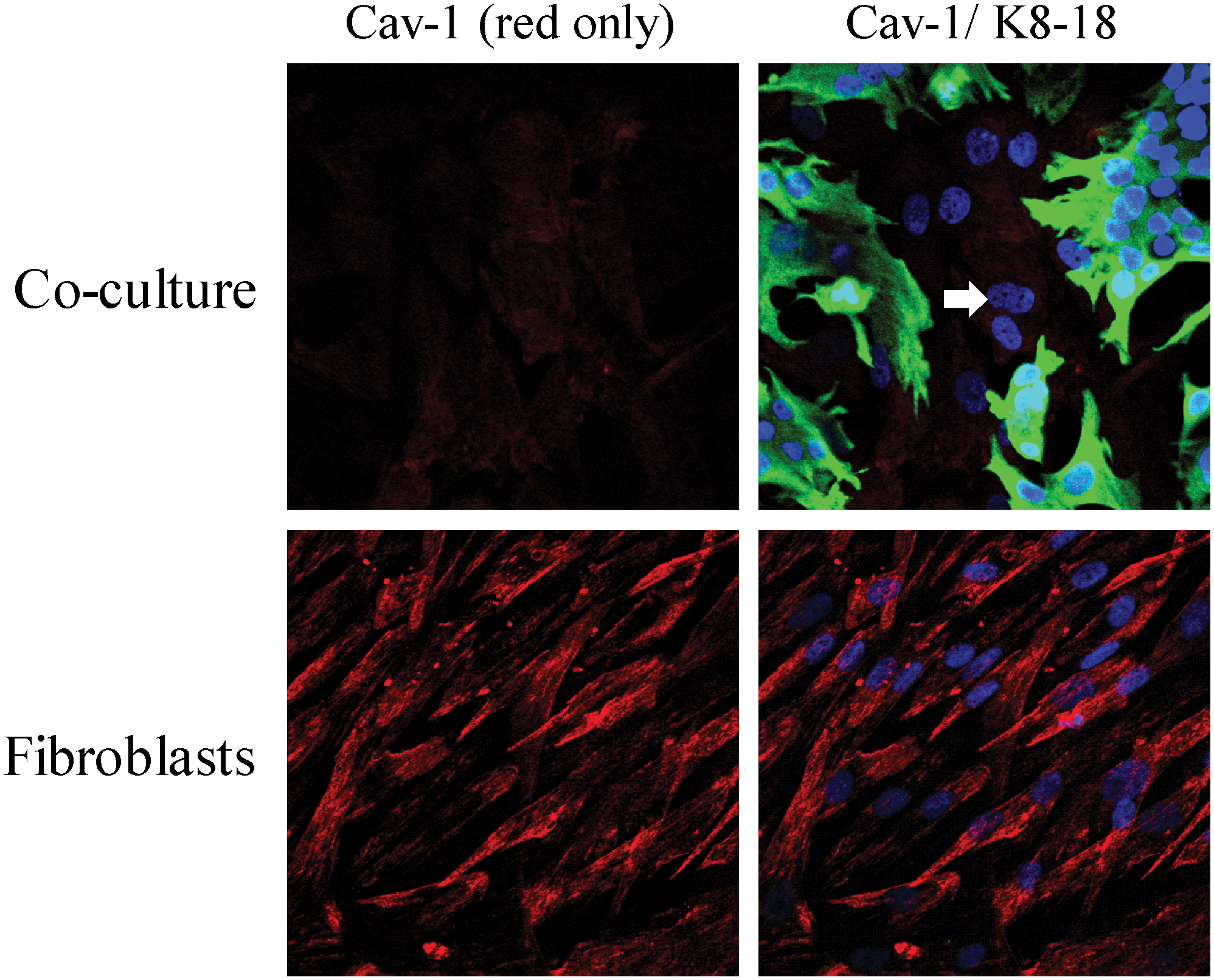
What is the nature of the stressor? How do cancer cells activate autophagy in adjacent stromal fibroblasts? One possibility is that cancer cells induce oxidative stress in fibroblasts via a paracrine mechanism. To test this hypothesis directly, we treated MCF7–fibroblast cocultures with antioxidants and monitored the expression of Cav-1 in stromal fibroblasts. In this context, pretreatment with antioxidants (such as NAC, metformin, and quercetin) was indeed sufficient to prevent the loss of Cav-1 in fibroblasts, as predicted (Figs. 11 –13). Thus, MCF7 cancer cells induce the autophagic degradation of Cav-1 via paracrine oxidative stress (66).
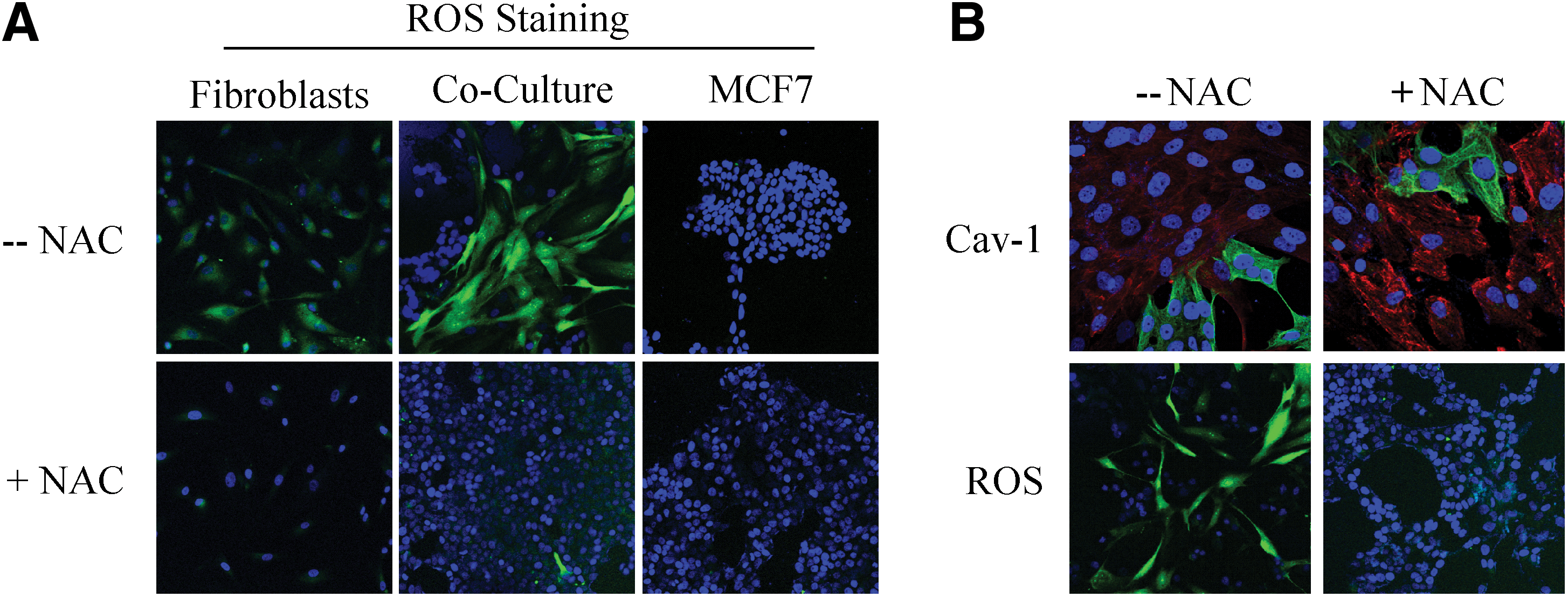

To better understand the mechanism(s) by which cancer epithelial cells promote the CAF phenotype, we further examined the role of oxidative stress as an initial trigger. Using the coculture system, we showed that Cav-1 downregulation is dependent on ROS in a feed-forward mechanism, where increased ROS leads to decreased Cav-1 and decreased Cav-1 leads to increased ROS (66). To validate the effect of ROS alone, homotypic cultures of fibroblasts were treated with a glutathione synthase inhibitor (namely buthionine sulfoxide [BSO]) to synthetically increase ROS (66). BSO treatment caused Cav-1 downregulation in a dose-dependent manner (66). Furthermore, we showed that under coculture conditions, the increase in ROS production in fibroblasts leads to mitochondrial dysfunction and a switch toward glycolytic metabolism, both specifically in fibroblasts (66). ROS production in fibroblasts also affected the cancer epithelial cells, via a bystander effect, causing genomic instability and aneuploidy; however, mitochondrial function and mass were both increased in the cancer cells (66). To validate that the observed mitochondrial biogenesis in cancer cells came as a consequence of the coculture with glycolytic fibroblasts, MCF7 cancer cells alone were treated with L-lactate to mimic the coculture conditions (66). Remarkably, L-lactate treatment was indeed sufficient to induce mitochondrial biogenesis in MCF7 cancer cells (66, 68).
Taken together, these findings have allowed us construct a new two-compartment metabolic model for the paracrine interactions between CAFs and cancer epithelial cells (Figs. 6 and 7). In this model, cancer cells promote oxidative stress in neighboring CAFs via NO overproduction, leading to mitochondrial dysfunction and the loss of Cav-1. This triggering event then further promotes the production of ROS by the CAFs. In turn, such stromal ROS production has a bystander effect in the cancer cells, leading to increased DNA damage and aneuploidy. Mitochondrial dysfunction in CAFs causes autophagy and mitophagy, leading to the reverse Warburg effect, by increasing HIF1-alpha and NFkB activation. The reverse Warburg effect in the CAFs creates a nutrient-rich microenvironment to directly feed cancer cells, via metabolic coupling. In addition, cancer cells mount an antioxidant defense, by upregulating antiapoptotic proteins (TIGAR) and antioxidant enzymes (peroxiredoxin-1) (66, 68).
In order to maximize treatment benefits, new biomarkers (such as stromal Cav-1) will be required to predict which cancer patients will respond best to antioxidant-based therapies and/or autophagy inhibitors. Importantly, the recent study by Nechuta et al. shows that antioxidant therapy can significantly reduce breast cancer recurrence and mortality (76). Reductions in mitochondrial stress in both cancer and stroma cells may be beneficial for preventing tumor progression and metastasis. Ultimately, clinical adoption of this new strategy could change current thinking on how we treat cancer patients.
The Reverse Warburg Effect: A Consequence of Autophagy in the Tumor Stroma
To begin to understand metabolism within the tumor microenvironment, the mammary fat pads of WT and Cav-1–deficient mice were subjected to an unbiased metabolomic analysis. As a result of this comprehensive analysis, ∼200 metabolites were observed, and ∼100 of them were seen to be increased in Cav-1−/− null mice (86), consistent with a major catabolic phenotype. Of particular interest, there was a significant increase (∼3–4 fold) in the levels of 3-hydroxy-butyrate (a ketone body) and asymmetric dimethyl arginine (ADMA), which are metabolites that are indicative of mitochondrial dysfunction and oxidative stress in the mammary fat pads of Cav-1−/−-deficient mice (86).
These findings prompted us to reinterrogate the transcriptional profiling of Cav-1–deficient MSCs, with a focus on particular metabolic pathways, such as autophagy, mitophagy, and lysosomal enzyme expression (86). In addition, microRNAs (miRs) were also analyzed, since they have been implicated in both cancer initiation and progression (59). Interestingly, the top two miRs induced in Cav-1−/− mesenchymal stromal cells, miR31 and miR34c, are functionally associated with HIF1-alpha activation and oxidative stress, respectively (86). HIF1-alpha activation and oxidative stress are both known to be important for the induction of autophagy (7, 69). To further validate our findings, we next used transcriptional profiling data from human breast cancer tumors and showed that these processes are indeed present within human tumor stroma, as well (87). The most important observation in this study is that extensive catabolism in the tumor stroma could directly feed the anabolic growth of cancer cells, via this parasitic mechanism (86). We have termed this new paradigm “The Autophagic Tumor Stroma Model of Cancer” (Figs. 6 and 7) (86).
These findings suggest that inhibition of autophagy in the tumor stroma will stop energy transfer to epithelial cancer cells, thereby inhibiting tumor growth. In an analogous way, autophagy induction in epithelial cancer cells should also halt tumor growth, by preventing cancer cells from using recycled nutrients. This provides a rational explanation for the “Autophagy Paradox” (31) in cancer, since both blocking and promoting autophagy, with systemic therapies, has been experimentally shown to stop tumor growth.
In a follow-up study, we provided functional evidence to demonstrate that ketones and lactate fuel tumor growth and metastasis in vivo (11). A xenograft mouse model of human breast cancer (MDA-MB-231 cells) was used to study the impact of systemic administration of L-lactate and 3-hydroxy-butyrate (a ketone body) on tumor growth, progression, and metastasis (11). This study demonstrated that the two metabolites promote tumor growth and metastasis, independent of angiogenesis (11). In addition, it also demonstrated that epithelial cancer cells shift their metabolism toward mitochondrial oxidative phosphorylation, compared to the adjacent stroma cells which shift their metabolism toward glycolysis (11). Further, we suggest that oncology surgeons may wish to reconsider using Lactate Ringer's solution in cancer patients, as it may promote tumor recurrence and metastasis (11).
Further evidence for the autophagic tumor stroma model of cancer was observed in electron micrographs of cocultures of fibroblasts with MCF7 breast cancer epithelial cells. An abundance of lysosomes and autophagosomes actively degrading mitochondrial-like structures was observed within CAFs and, in parallel, the MCF7 cells displayed extensive and healthy mitochondria (68).
To further delineate the molecular mechanism(s) by which Cav-1 expression is controlled in this process, it was shown that hypoxic conditions were sufficient to induce the autophagic degradation of Cav-1. Also, siRNA-mediated downregulation of Cav-1 expression was sufficient to induce various autophagy/mitophagy and lysosomal markers (68). Similarly, we also showed that cancer cells activate HIF1-alpha– and NFkB-based luciferase reporters in adjacent CAFs, via a paracrine mechanism (68). Activation of proautophagic HIF1-alpha is sufficient to induce Cav-1 downregulation in fibroblasts under hypoxia (68). Interestingly, cancer cells under coculture conditions show reductions in apoptosis and the upregulation of the antiapoptotic protein TIGAR (68), indicating that they functionally benefit from the recycled nutrients provided by autophagic fibroblasts.
Cancer Cells Fertilize the Tumor Microenvironment with Hydrogen Peroxide
Until recently, it remained unknown how cancer cells induce oxidative stress in adjacent stromal fibroblasts. What is the triggering event? Is it growth factor mediated or due to the secretion of ROS directly from cancer cells?
To address this issue, we used the MCF7–fibroblast coculture system. We observed that cancer cells initially secrete hydrogen peroxide, which then triggers oxidative stress in neighboring fibroblasts (61). More specifically, at 2 days of coculture, most of the ROS production occurred in MCF7 cells (61). This ROS production was reduced to baseline levels by coincubation with extracellular catalase, identifying the predominant ROS as hydrogen peroxide (61). In contrast, by day 5, most of the ROS production occurred in the CAFs (61). Thus, oxidative stress is contagious and is propagated laterally from cancer cells to adjacent fibroblasts. Importantly, we observed using this MCF7–fibroblast coculture system that hydrogen peroxide secretion activates NFkB and HIF1 in CAFs, driving stromal inflammation and aerobic glycolysis, as well as autophagy. As such, hydrogen peroxide helps to produce glycolytic fibroblasts which feed hungry oxidative cancer cells, actively fertilizing the tumor microenvironment (61).
Most importantly, under coculture conditions, treatment with catalase (to neutralize hydrogen peroxide) dramatically induced apoptosis in cocultured MCF7 cancer cells (61). Mechanistically, this may explain why catalase therapy in preclinical models prevents both tumor recurrence and metastasis (48, 49, 77 –81), as it cuts off the fuel supply.
Consistent with these findings, MCT4 protein expression (a marker of aerobic glycolysis and lactate production) in CAFs is induced by oxidative stress and can be prevented with antioxidants, such as NAC (117) (Fig. 14).

Glutaminolysis and Autophagy in the Tumor Stroma: A Vicious Cycle
Previous studies have shown that cancer cells can become addicted to glutamine. After entering the cell, glutamine is converted to glutamate, enters the TCA cycle, which results in the production of ATP by oxidative phosphorylation (21). Ammonia is produced as a by-product of this reaction, which in turn can act as an inducer of autophagy (32, 33, 65). In our laboratory, we have shown that increased autophagy (as a result of a loss of stromal Cav-1) results in increased glutamine levels in the stromal compartment (86). The glutamine produced as a result of autophagy, in the tumor microenvironment, could then fuel the growth of adjacent cancer cells, while the ammonia by-product could help to maintain the autophagic production of glutamine. This vicious cycle fits well with the autophagic tumor stroma model of cancer, (86) where energy-rich recycled nutrients (L-lactate, ketones, and glutamine) fuel oxidative mitochondrial metabolism in cancer cells.
Conclusions
Here, we have highlighted two new models for understanding tumor metabolism, (i) the reverse Warburg effect and (ii) the autophagic tumor stroma model of cancer, which suggest that an intimate relationship exists between tumor cells and their associated fibroblasts. It is now clear that both cell types (epithelial cancer cells and fibroblasts) functionally create the cancer microenvironment via hydrogen peroxide and oxidative stress, even though prior studies did not recognize that these two compartments are metabolically coupled.
What these new models bring to “the table” is the establishment of new therapeutic avenues for fighting cancer. These new paradigms provide novel targets for the development of more potent anticancer therapies, either by blocking energy transfer between cancer cells and fibroblasts (using MCT inhibitors), or by preventing ROS production and oxidative stress (with powerful antioxidants), or by inhibiting autophagy in CAFs and promoting autophagy in cancer cells, thereby metabolically uncoupling tumor cells from their surrounding stroma.
In this context, existing drugs, such as metformin and chloroquine, antioxidant supplements, such as quercetin or NAC, can have a significant impact. Similarly, new drugs can be developed, as is the case for two new MCT1 inhibitors (from AstraZeneca). Importantly, these new metabolic models will foster the development of novel biomarkers, and corresponding therapies, to fulfill the goal of personalized cancer medicine.
Footnotes
Acknowledgments
Federica Sotgia and her laboratory were supported by grants from the W.W. Smith Charitable Trust, the Breast Cancer Alliance (BCA), and a Research Scholar Grant from the American Cancer Society (ACS). Michael P. Lisanti was supported by grants from the NIH/NCI (R01-CA-080250, R01-CA-098779, R01-CA-120876, and R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. Isabelle Mercier was the recipient of a Post-doctoral Fellowship from the Susan G. Komen Breast Cancer Foundation. Richard G. Pestell was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072, and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to Richard G. Pestell). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to Michael P. Lisanti). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to Michael P. Lisanti and Federica Sotgia). The Department specifically disclaims responsibility for any analyses, interpretations, or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the United Kingdom (to Anthony Howell) and an Advanced ERC Grant from the European Research Council.