
Abstract
Introduction
Gas sensor proteins that contain metal centers are suitable for answering these questions because the metal centers can be used as spectroscopic probes in various techniques such as ultraviolet/visible (UV/Vis) spectroscopy, electron paramagnetic resonance (EPR), resonance Raman spectroscopy, circular dichroism spectroscopy, Mössbauer spectroscopy, and X-ray absorption spectroscopy. These spectroscopic techniques can provide detailed information on the reactivity of the metal center with a gas molecule and on the structure around the metal center. X-ray crystallography is another important technique for studying gas sensor proteins. This article focuses on recent findings obtained using these spectroscopic methods regarding the structure–function relationships of bacterial gas sensor proteins.
Cyclic AMP Receptor Protein/Regulatory Factor for Fumarate and Nitrate Reduction Family of Transcriptional Regulators Acting as Gas Sensor Proteins
The cyclic AMP receptor protein (CRP)/regulatory factor for the fumarate and nitrate reduction (FNR) family of transcriptional regulators activate or repress target genes in response to a number of intracellular and exogenous signals. Some of them use a prosthetic group such as a heme or an iron–sulfur (Fe–S) cluster to sense oxygen (O2), carbon monoxide (CO), or nitric oxide (NO). A transcriptional regulator for the CO oxidation operon (CooA) is a CO sensor protein in this family; it is activated when CO binds to the heme (2). Recent findings regarding CooA have been reported in previous reviews (2,3). The present article focuses on FNR from Escherichia coli, which is an O2 sensor protein in the CRP/FNR family of transcriptional regulators. The three-dimensional structure of FNR has not yet been determined, although X-ray crystal structures of several members of the CRP/FNR family (including CRP and CooA) have been reported (Fig. 1). FNR is thought to have a similar structure to CRP and CooA because there is sufficient sequence similarity.
FNR is a global transcriptional regulator that controls the expression of more than 100 genes in aerobic/anaerobic metabolism in response to O2 availability (16). FNR uses Fe–S clusters as active sites for sensing O2 and it plays a key role in regulating O2-dependent FNR activity. Under anaerobic conditions, FNR exists as a homodimer with each monomer containing a [4Fe–4S]2+ cluster. The [4Fe–4S]2+ cluster in FNR is O2 labile and it is converted into a [2Fe–S]2+ cluster on exposure to O2 both in vitro and in vivo (16). Prolonged exposure to O2 results in the loss of the cluster through the formation of the apo form of FNR (16). This results in FNR dissociating into monomers, thereby losing its sequence-specific DNA binding activity. FNR activity is regulated via a monomer–dimer equilibrium in response to O2 (16).
While UV/Vis spectroscopy, EPR, and Mössbauer spectroscopy have indicated that the [4Fe–4S]2+ cluster is converted into a [2Fe–2S]2+ cluster on exposure to O2 both in vitro and in vivo (16), the detailed reaction mechanism is not obvious. Crack et al. (6,7) recently proposed a mechanism for FNR cluster conversion (Fig. 2). Cluster conversion is a multistep process in which the first reaction is oxidation of the [4Fe–4S]2+ cluster to the [4Fe–4S]3+ form, which is rapidly converted into a [3Fe–4S]+ cluster with the release of a Fe2+ ion (Equation 1 in Fig. 2) (6,7). The [3Fe–4S]+ cluster is a relatively stable intermediate that can be observed by EPR (7). The [3Fe–4S]+ cluster converts into the [2Fe–2S]2+ cluster, during which one Fe3+ and two S2− ions are released (Equation 2 in Fig. 2). The overall reaction is expressed by Equation 3 in Fig. 2.
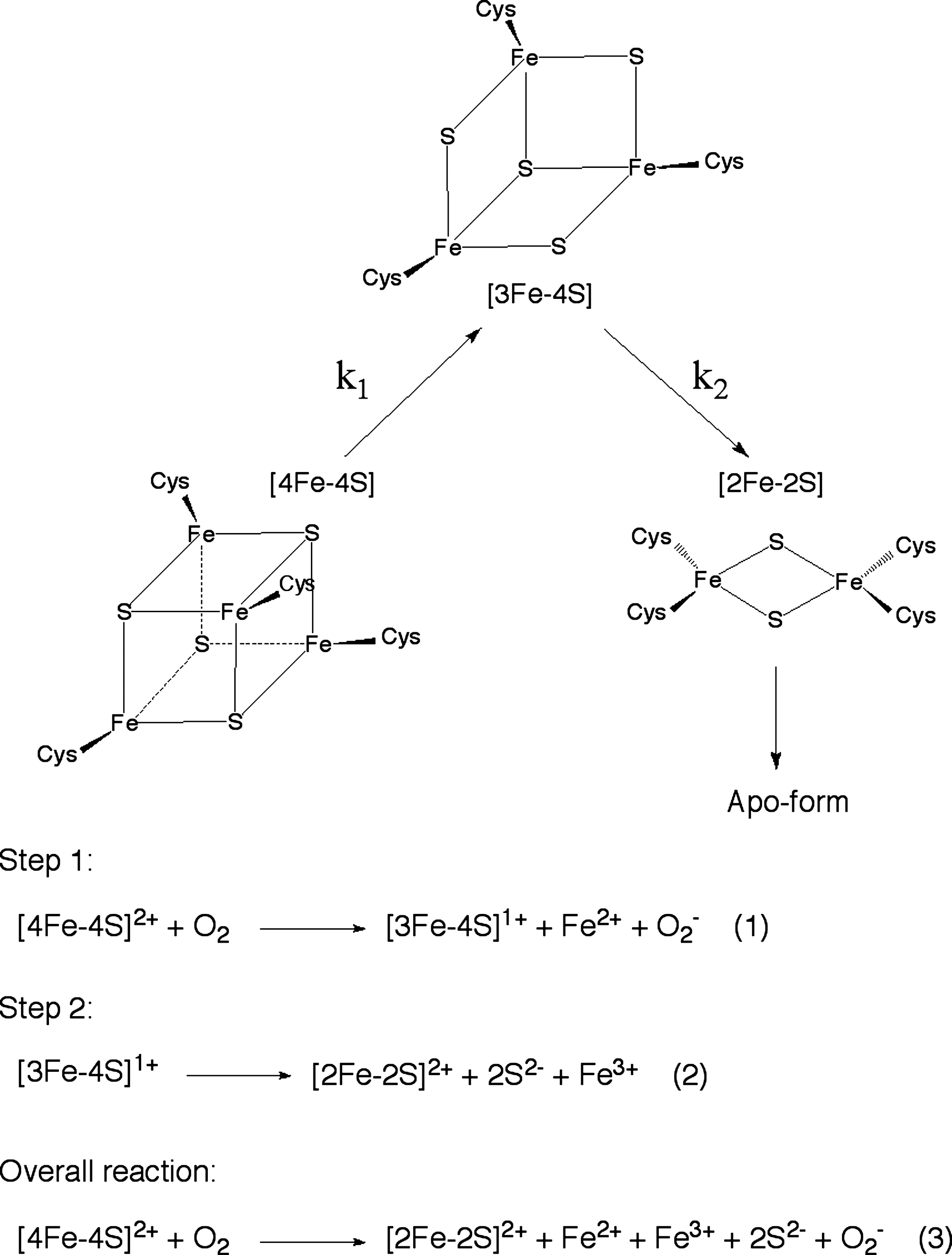
The reaction rate constants k 1 and k 2 (Fig. 2) are estimated to be 278 M−1 s−1 and 0.0087 s−1 at 21°C, respectively (7). The superoxide ion generated during the first step is thought to be readily dissipated into hydrogen peroxide and O2 by the significant dismutase activity of FNR (7). As E. coli contains superoxide dismutase and catalase, the superoxide/hydrogen peroxide may be rapidly reconverted to the primary signal molecule, O2, which would provide a feedback mechanism that would amplify the sensitivity of the [4Fe–4S]2+ cluster in FNR to O2 (7).
The [2Fe–2S]2+ cluster is relatively stable in vitro in both the presence and absence of air, having a half-life of several hours (41). However, apo-FNR is the predominant form of FNR in cells grown under aerobic conditions (41). It has been suggested that FNR is cycled between active and inactive states in vivo, where the Isc system, an Fe–S cluster assembly machinery, plays a major role in Fe–S cluster biogenesis in FNR (8,24).
Rrf2 Family of Transcriptional Regulators Acting as Gas Sensor Proteins
The NO-responsive transcriptional regulator (NsrR) is a member of the Rrf2 family of transcriptional regulators that is conjectured to be a master regulator in NO metabolism, acting as an NO sensor in both gram-positive and gram-negative bacteria (35,40,44). NsrR regulates the expression of genes that encode denitrification and/or NO detoxification systems. NsrR has a characteristic cysteine motif, Cys–X4–7–Cys–X5–Cys, in the C-terminal region (44). The cysteines in this motif are postulated to be ligands of the Fe–S cluster in NsrR. The fourth ligand of the Fe–S cluster in NsrR has not yet been identified experimentally, but a possible candidate is a conserved His located in the predicted helix-turn-helix motif in the N-terminal region (44).
UV/Vis, resonance Raman, and EPR analyses have revealed that anaerobically purified NsrR from Bacillus subtilis (Bs-NsrR) contains a [4Fe–4S]2+ cluster (46). Only the [4Fe–4S] cluster–containing Bs-NsrR exhibits high-affinity binding to the target DNA, which indicates that the [4Fe–4S] cluster is essential for Bs-NsrR to be functional (19). Bs-NsrR contains three Cys residues at positions 92, 100, and 106; they are assumed to be the ligands of the [4Fe–4S] cluster.
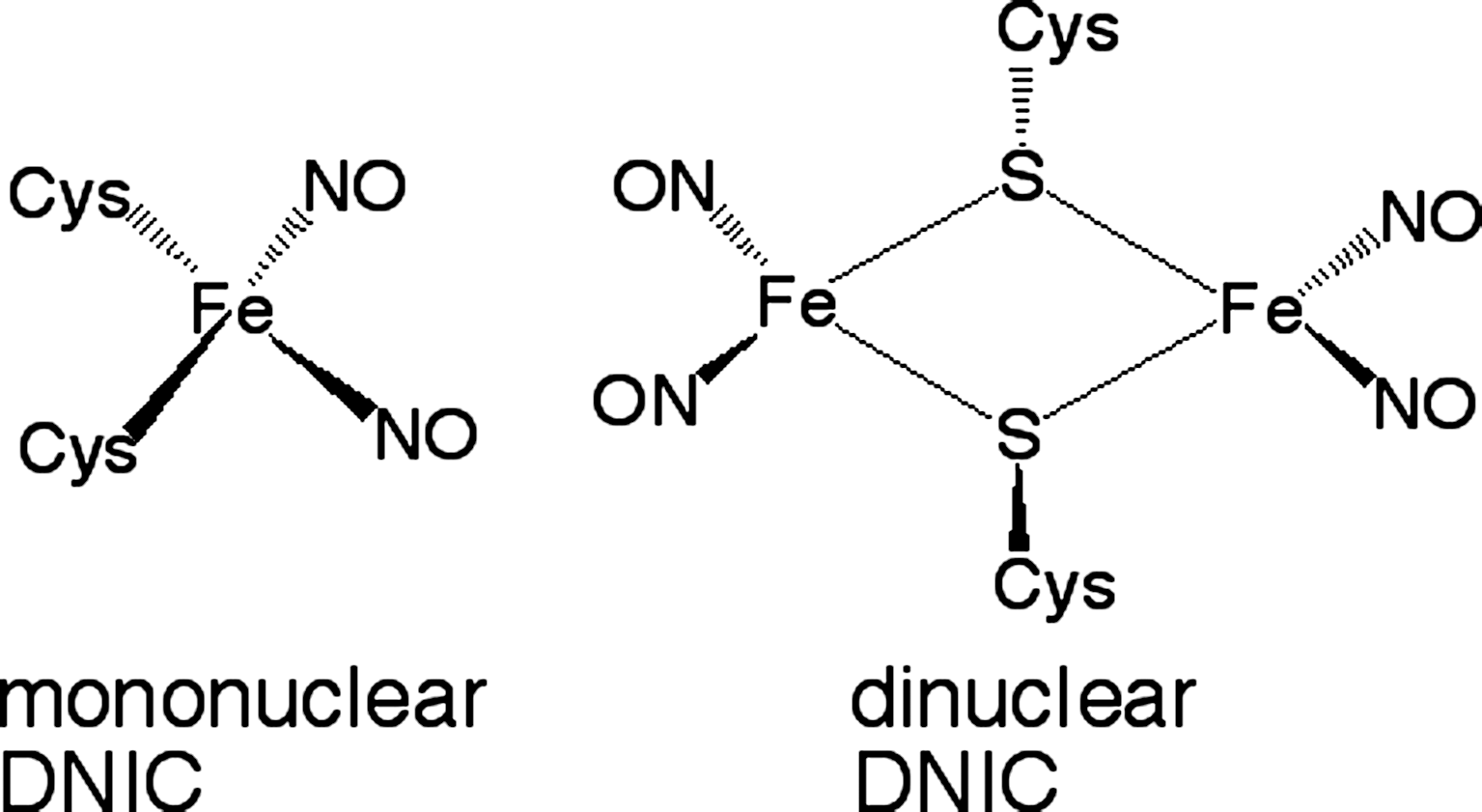
Nakano et al. (28) reported that Bs-NsrR is responsible for NO-dependent up-regulation of the ResDE regulon. The DNA-binding activity of Bs-NsrR is inhibited on exposure to NO, causing derepression (28). Multiple dinitrosyl iron complexes (DNICs) are formed when Bs-NsrR is exposed to NO, as characterized by UV/Vis spectroscopy and EPR (46) (Fig. 3). On the formation of DNICs, the ligands coordinated to the [4Fe–4S] cluster dissociate from the cluster and the structure of the cluster core changes, which results in the conformational changes of Bs-NsrR. The formation of DNICs is expected to be the first step in Bs-NsrR sensing NO. However, it is not clear whether Bs-NsrR–containing DNICs are in the physiological inactive state because DNIC formation is observed for purified Bs-NsrR only in vitro and not in vivo. As the apo form of Bs-NsrR without the Fe–S cluster exhibits weak DNA-binding activity (19), the apo form may be the physiological inactive form. Whole-cell EPR analysis using E. coli or B. subtilis cells expressing Bs-NsrR would be helpful for determining whether DNICs formed on exposure to NO are sufficiently stable to be the physiological inactive form.
The Fe–S cluster in Bs-NsrR is also reactive toward O2. Exposure of anaerobically purified Bs-NsrR to O2 gradual degrades the [4Fe–4S] cluster via [3Fe–4S] and [2Fe–2S] intermediates (46). Kommineni et al. (19) recently reported that apo- and holo- ([4Fe–4S] cluster–containing) Bs-NsrR interacts with two distinct classes of DNA targets that have different recognition sequences. Their results suggest that O2 functions as another physiological effector of Bs-NsrR by producing the apo form. Given that DNICs and apo forms are selectively produced by the reactions of Bs-NsrR with NO and O2, respectively, these two forms may have different conformations, which Bs-NsrR could use to discriminate between NO and O2. Further studies are required to test these hypotheses.
Heme-Nitric Oxide/Oxygen Binding Protein/Domain as a Heme-based Gas Sensor Protein
Soluble guanylate cyclase (sGC) is an NO receptor protein responsible for NO-dependent signal transduction in mammalian cells. It is a heterodimeric protein composed of two homologous subunits, α and β (20). The N-terminal region of the β subunit binds a heme with a histidine as the proximal ligand. Heme-binding domains similar to the N-terminal heme-binding domain of sGC have recently been identified using sequence profile searches of bacteria; they have been termed Heme NO Binding (HNOB) domains (14). HNOB domains have also been referred to as Heme-Nitric oxide/Oxygen binding (H-NOX) domains because it is apparent that some of these domains can bind not only NO but also O2 (15,32). Bacterial H-NOX proteins display two principal architectures, namely stand-alone proteins and multidomain proteins consisting of H-NOX fused to other functional domains such as the methyl-accepting chemotaxis receptor domain (14). Stand-alone H-NOX domains are most frequently found in a predicted operon with a histidine kinase, a receiver domain/protein of a two-component system, a diguanylate cyclase containing a GGDEF domain, a phosphatase, or a cyclic diguanylate phosphodiesterase (14). H-NOX domains/proteins regulate these enzymatic activities in response to O2 or NO.
H-NOX from Shewanella oneidensis (So H-NOX) is a member of the stand-alone H-NOXs. The gene encoding So H-NOX (SO2144) is followed by a soluble sensor histidine kinase gene (SO2145) (14,34). Purified So H-NOX has a five-coordinate Fe2+-heme that exhibits a Soret peak at 427 nm (34). His103 serves as the proximal ligand of the heme in So H-NOX (10). The reduced form with the Fe2+-heme [So H–NOX(Fe2+)] is highly stable and resistant to oxidation, which suggests that the resting state of So H–NOX is the Fe2+ form (reduced form) in vivo (34). When So H–NOX(Fe2+) reacts with NO, NO-bound So H-NOX [So H-NOX(Fe2+–NO)] is formed with a five-coordinate nitrosyl heme in which His103 is dissociated from the heme iron (Fig. 4). While CO also binds to the heme in So H-NOX(Fe2+) to form a CO-bound So H-NOX with a six-coordinate CO-bound heme (So H-NOX(Fe2+–CO)), O2 does not bind to the heme in So H-NOX.

The histidine kinase SO2145 exhibits constitutive self-kinase activity in vitro, but its activity is altered by So H-NOX(Fe2+–NO) in a concentration-dependent manner (34). When 1 μM of SO2145 was used in an activity assay, So H-NOX(Fe2+–NO) inhibited the self-kinase activity of SO2145 with an IC50 value of 9 μM, whereas no inhibition was observed for So H-NOX(Fe2+) (10). The inhibitory effect of CO was 10-fold less potent than that of NO. The IC50 value for So H-NOX(Fe2+–CO) was 84 μM (10). Pull-down assays have revealed that there is a protein–protein interaction between So H-NOX and the histidine kinase SO2145. However, this interaction is independent of the ligation state of the So H-NOX because both the So H-NOX(Fe2+–NO) and the So H-NOX(Fe2+) are pulled down (34). These results suggest that NO binding to So H-NOX in the So H-NOX/SO2145 complex changes the conformation of the complex, which inhibits the self-kinase activity of SO2145.
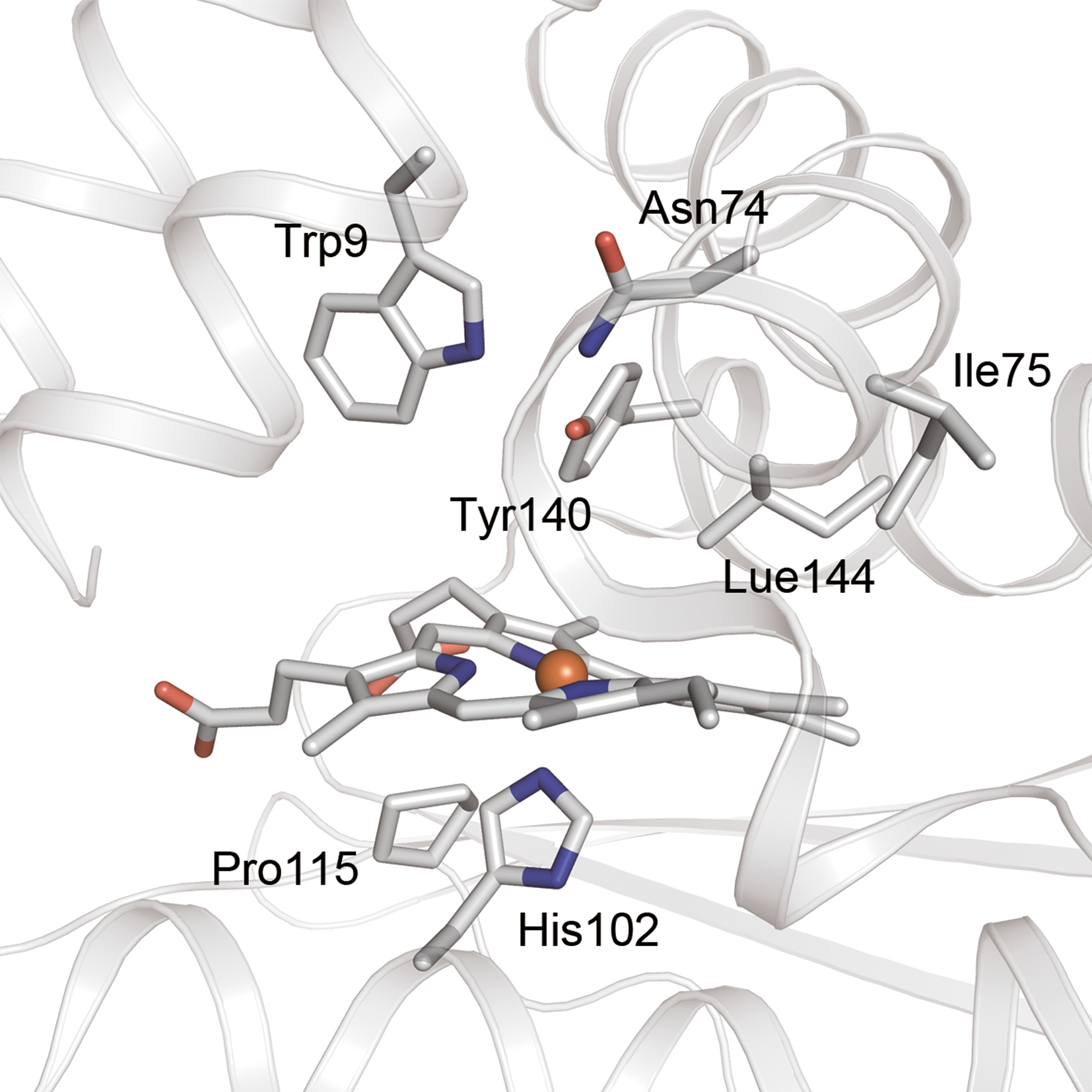
The crystal structures of H-NOX domains have a highly distorted heme in which the major distortion modes are saddling and ruffling (23,29,30). The out-of-plane heme distortions found in Tt H-NOX show a large deviation from planarity (30) (Fig. 5). This distortion is caused by a steric interaction between the pyrrole D ring of the heme and Pro115 that is within van der Waals contact; this has been confirmed by the crystal structure of the P115A mutant (30). Resonance Raman spectroscopy has also revealed that the P115A mutation relaxes the heme distortion (43). Pro115 is conserved among all H-NOX proteins, suggesting that the heme distortion is conserved across the entire family (30). When NO binds to the heme in H-NOX, the planarity of the heme is altered, triggering intramolecular signal transduction.
Based on the results of NMR structural analyses for the CO-bound forms of wild-type and H103G mutant So H-NOX, Erbil et al. (10) have proposed a mechanism for intramolecular signal transduction triggered by ligand binding in So H-NOX. The H103G mutant lacks the original proximal His residue on the F helix, which is a mimic of the NO-bound heme in H-NOX in which the Fe–His bond is cleaved (Fig. 4). In the case of So H-NOX, the NO-bound and CO-bound forms are the active and inactive forms, respectively, as described above. Particularly striking differences between the CO-bound forms of wild-type and H103G mutant are observed in the ruffling and doming modes of the heme (10). The H103G mutant exhibits less heme doming and ruffling than the wild-type, suggesting that the heme becomes more planar in the active (kinase-inhibitory) conformation (10). The loss of the proximal Fe–His bond also initiates a rotation of the N-terminal subdomain relative to the proximal one, indicating that the heme and the N-terminal subdomain move in concert away from the proximal F helix (10). X-ray crystal structure analyses corroborate these conformational changes, including the relaxation of the heme distortion (29).
Heme-Based Gas Sensor Proteins using a PAS Domain
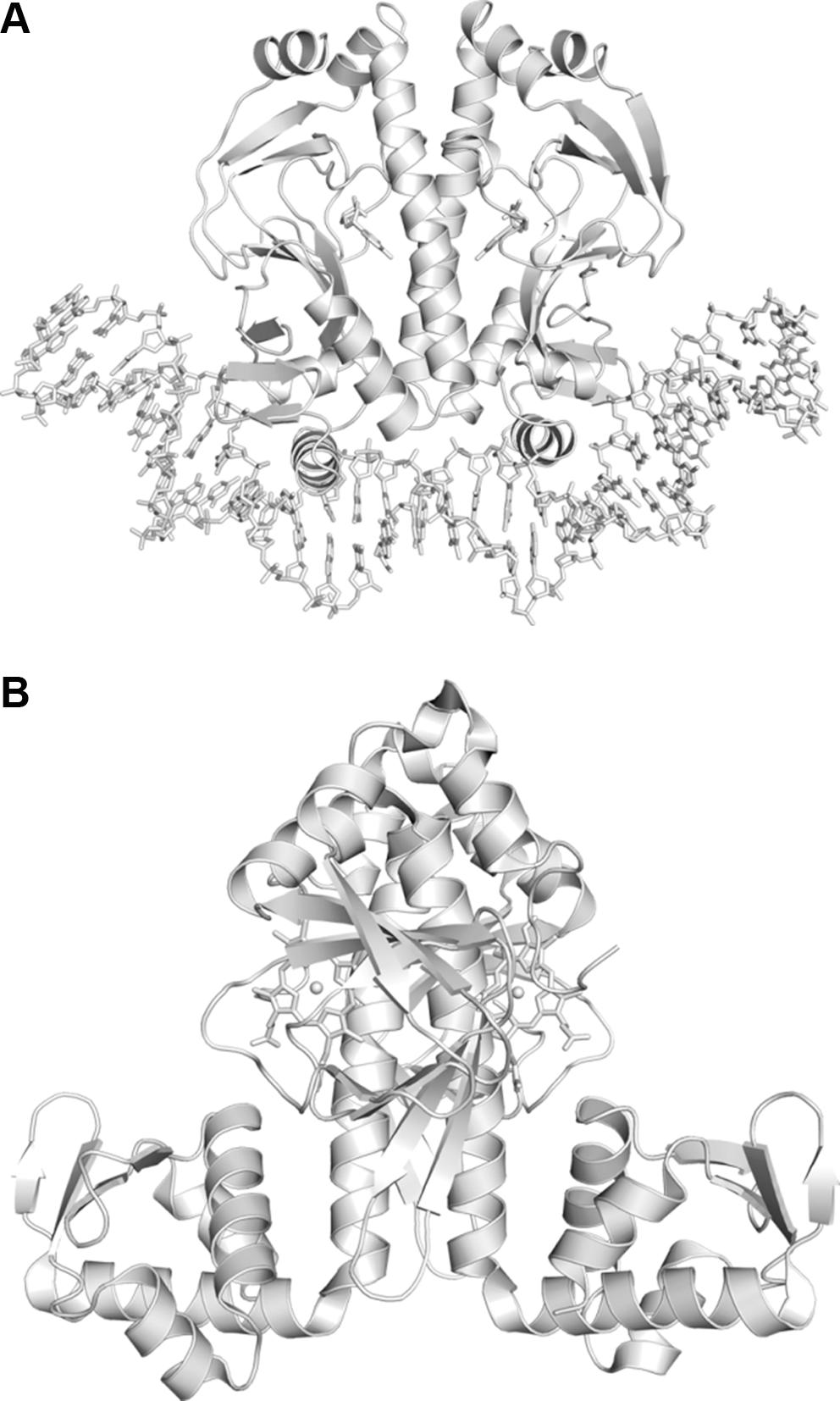
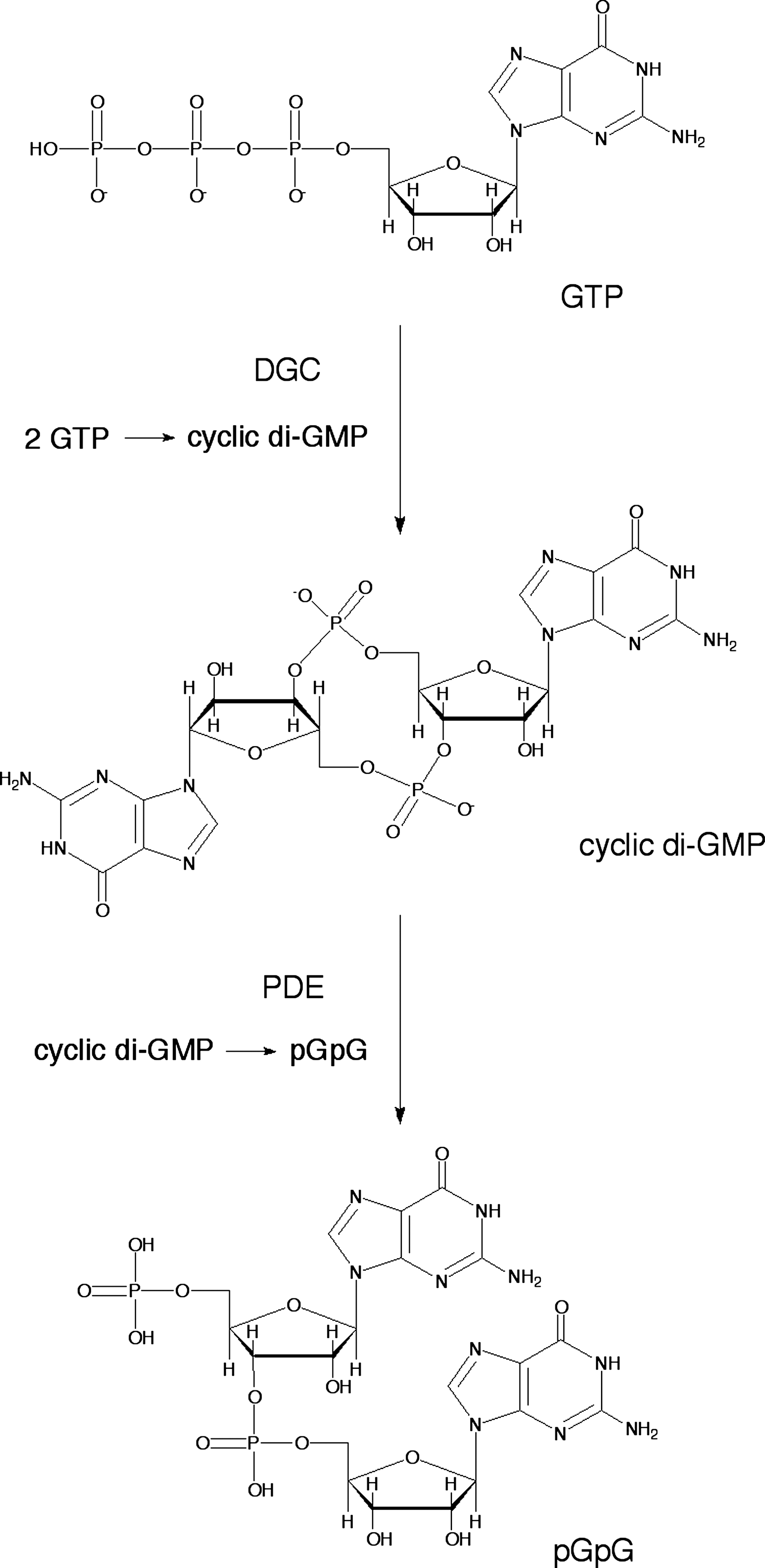
PAS domains are widespread components of signal transduction proteins in which they function as versatile sensors and interaction modules (25). Several PAS domains bind cofactors either covalently or noncovalently to sense chemical or physical signals. PAS domains that function as gas sensors containing a heme (11) or Fe–S cluster (26) are known. Most PAS domains make up a part of larger proteins, and they are covalently linked to the effector and other domains. The direct oxygen sensor from E. coli (EcDos) is one of the most extensively studied bacterial gas sensors of those that adopt a heme-containing PAS domain. EcDos contains two PAS domains in tandem and a phosphodiesterases (EAL) domain in the N- and C-terminal regions, respectively. Only the first PAS domain binds a heme that functions as a sensor domain, whereas the EAL domain exhibits phosphodiesterase (PDE) activity with a cyclic di-GMP substrate (45). This review primarily focuses on recent developments regarding EcDos since the biochemical and biophysical properties of EcDos studied before 2006 have been well summarized in a previous review (38).
The physiological function of EcDos, which is a phosphodiesterase for cyclic di-GMP (Fig. 6), is regulated by exogenous ligand binding to the heme. Thus, the PDE activity is enhanced when O2 binds to the Fe2+ heme (42,45). Binding of CO and NO to the heme cause a similar enhancement in the PDE activity (42,45), indicating that EcDos cannot discriminate between these gas molecules. Positive cooperativity in O2 binding is observed for EcDos (21,45). Lechauve et al. (21) have reported that the second O2 molecule binds to EcDos with a sixfold higher intrinsic affinity than the first O2 molecule. They proposed the following mechanism. When Met95 is replaced by O2, it is flipped out of the heme pocket in one subunit, which modifies the dimer interface and presumably reduces the Met95 affinity for heme of the other subunit, thereby increasing the second O2 binding affinity. The PDE activity also varies nonlinearly on the fraction of the O2-saturated heme, which enables activation of EcDos in a narrow range of O2 concentrations (18,45).
X-ray crystal structural analyses reveal that O2 binding to the heme also results in rearrangement of the hydrogen-bonding networks around the heme. The Arg97 residue is rotated by about 180° on O2 binding, and it forms a hydrogen bond with the heme-bound O2 (31) (Fig. 7). This hydrogen bond is essential for stable O2 binding to the heme. In Arg97 mutants, the stable O2-bound EcDos is not formed due to rapid autoxidation and/or low O2 affinity (42). The propionate groups of the heme also rotate on O2 binding, which may trigger the formation of a hydrogen bond between Asn84 and Tyr126 through the heme propionate-6 hydrogen-bonding network (9). These hydrogen-bonding networks have been suggested to be responsible for the intramolecular signal transduction from the sensor to enzymatic domains (9).

Heme-Based Gas Sensor Proteins using a GAF Domain
GAF domains represent one of the largest and most widespread domain families. They exhibit a variety of functions, including binding of small molecules and protein–protein interactions (12). They are distantly related to the PAS domains (discussed in the previous section), which is another superfamily of the same basic fold. Both families are involved in many signal transduction pathways and protein regulatory and sensory systems (1). Several gas sensor proteins have a GAF domain that binds a heme, Fe–S cluster (27), or nonheme iron (4) as the active site for gas sensing. DosS and DosT from Mycobacterium tuberculosis are the most extensively studied among them; we focus on them in this article.
The sensor kinases DosS (also known as DevS) and DosT activate the transcriptional regulator DosR (also known as DevR) in response to both hypoxia and nonlethal levels of NO. The DosS/DosR and DosT/DosR two-component signal transduction systems regulate the expression of the dosR regulon that is responsible for early adaptation to these stimuli, as well as for initiating the entrance of M. tuberculosis into a nonreplicating persistent state. DosS and DosT each contain two tandem GAF domains and a histidine kinase domain in their N-terminal and C-terminal regions, respectively. In both DosS and DosT, the first GAF domain (GAF-A) contains a heme, whereas the second GAF domain (GAF-B) does not (37,39). The second GAF domain (GAF-B) has been suggested to assist the formation of a better-defined distal heme pocket of the GAF-A domain through interdomain interactions within DosS (22). The activation of the response regulator DosR occurs through autophosphorylation of either DosS or DosT followed by transfer of the phosphate to DosR (13,36).
The kinase activities of DosS and DosT depend on the coordination state of the heme. For both DosS and DosT, kinase activity is observed when the heme is in a five-coordinate ferrous (Fe2+) form, or CO- or NO-bound forms (Fe2+–CO or Fe2+–NO), but is strongly inhibited in an oxy (Fe2+–O2) form (39,47). The activities of DosS and DosT are inhibited by about 84% and 98%, respectively, on O2 binding to the ferrous heme (39). These results indicate that both of DosS and DosT are direct O2 sensor proteins.
Kim et al. (17) have suggested that the presence of both DosS and DosT paralogues in M. tuberculosis is not a functional redundancy, but that these paralogues play distinct roles in sensing changes in O2 tension. Their research indicates that DosT appears to first respond to a decline in O2 tension when M. tuberculosis is gradually transferred from aerobic to anaerobic conditions (17). DosS and DosT show equilibrium dissociation constants for O2 of 3 and 26 μM, respectively (39), which is consistent with the above results.
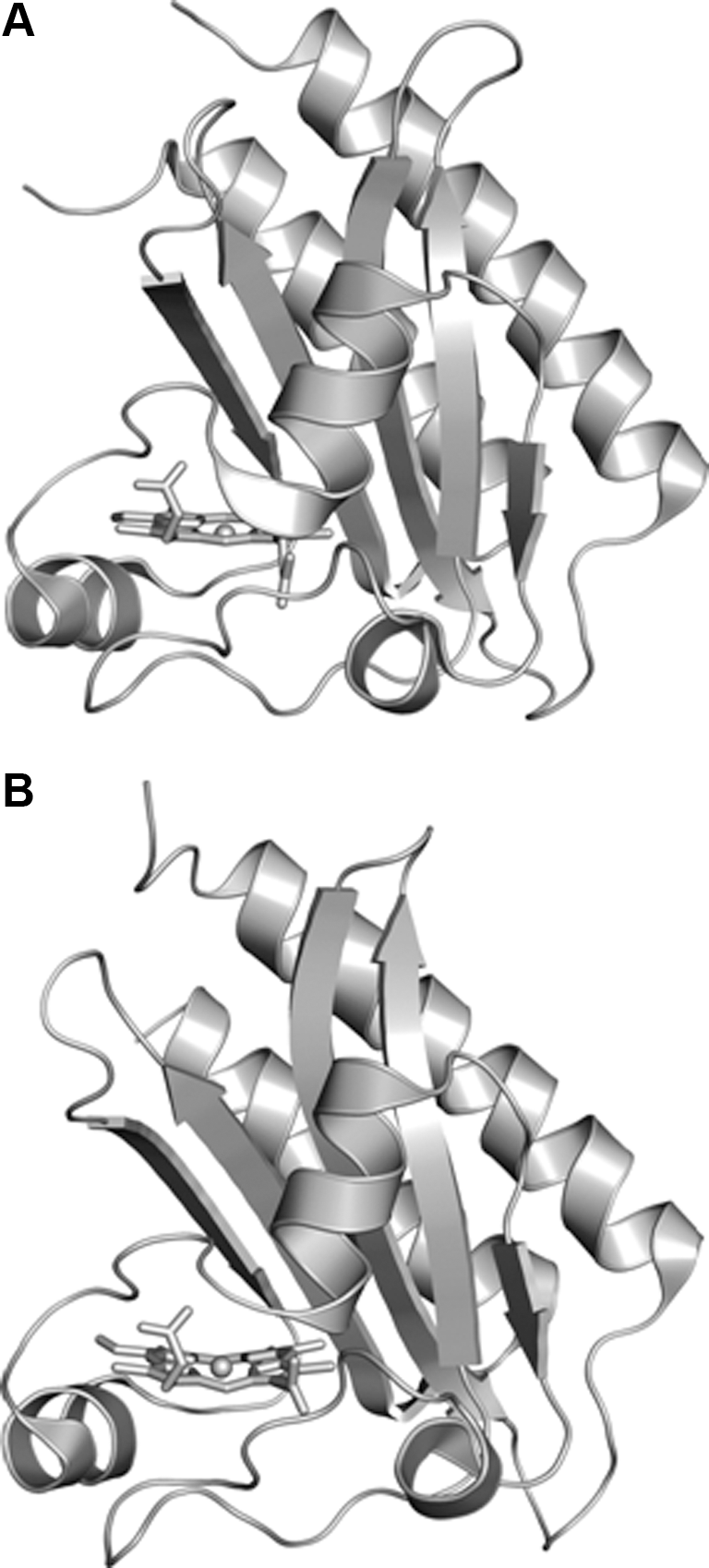
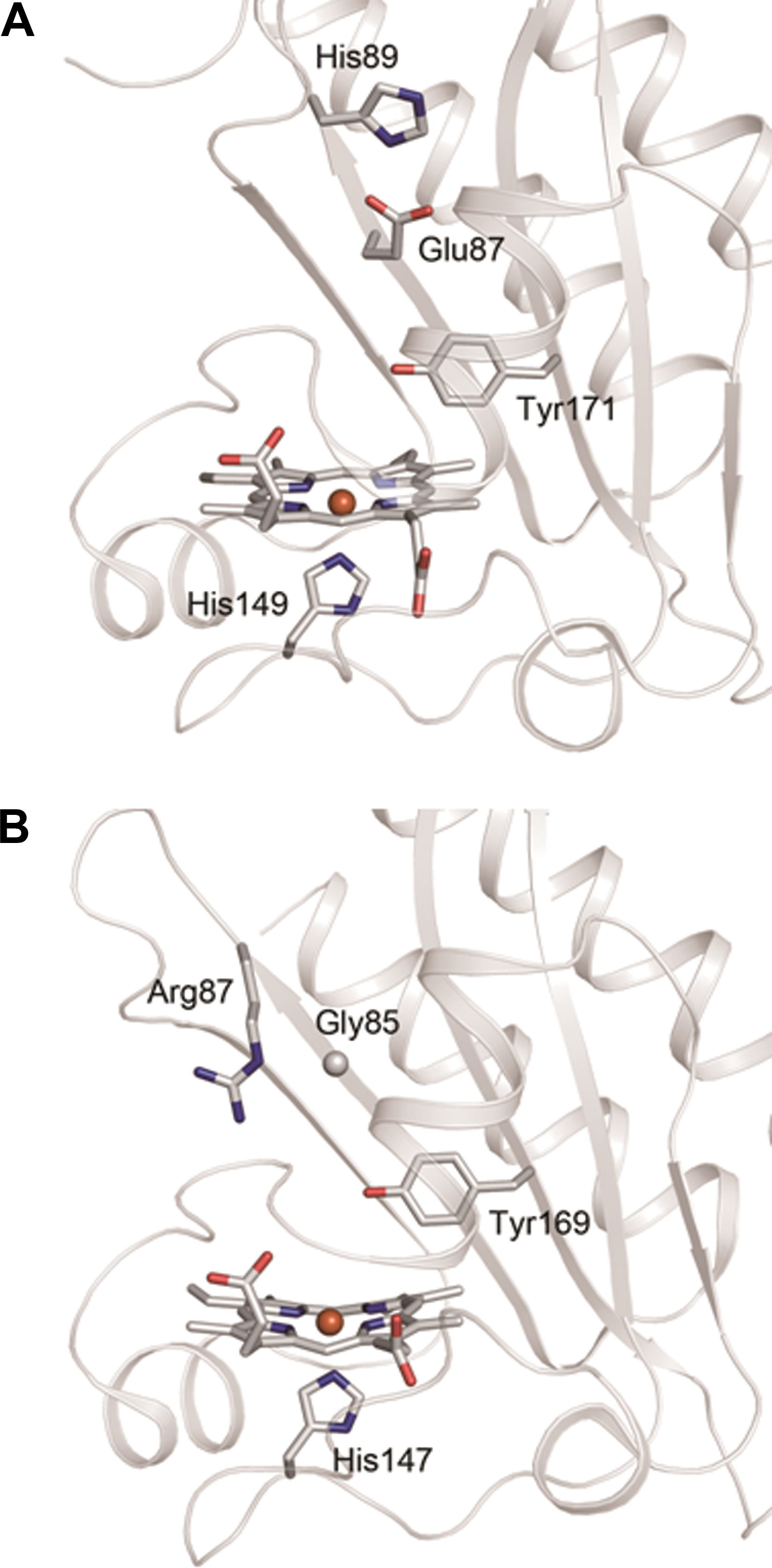
The crystal structures of the GAF-A domains of DosS (residues 63–210) and DosT (residues 61–208) have been reported (5,33) (Figs. 8 and 9). Yukl et al. (47) have proposed that interactions between Tyr171 and distal diatomic ligands turn the kinase activity on or off and that the mutation of Tyr171 to Phe disrupts the on–off switch, resulting in an inactive kinase in all states.
Summary and Conclusion
Heme and Fe–S clusters are widely used as the active sites for sensing gas molecules in bacterial gas sensor proteins. It has become apparent that these two prosthetic groups employ different mechanisms for sensing gas molecules. In Fe–S cluster–containing sensor proteins, structural changes to the cluster core trigger conformational changes in sensor proteins; this mechanism is unique to these sensor proteins. The structural change of the Fe–S cluster core responsible for regulating the physiological function is not found in Fe–S proteins that are not gas sensor proteins. The [4Fe–4S] cluster in FNR is converted to the [2Fe–2S] cluster via the [3Fe–4S] cluster on sensing O2. The detailed mechanism of this cluster conversion has been recently clarified, which is an important step toward fully understanding the structural–functional relationships of FNR.
In heme-based sensor proteins, gas molecules are sensed by reversible binding to the heme. Recent X-ray crystallographic and spectroscopic studies have revealed that conformational changes are induced by the reconstruction of hydrogen-bonding networks among the heme-bound gas molecule and the surrounding amino acid residues, which play an important role in gas discrimination and subsequent signal transduction. The conformational change induced by gas sensing is a crucial step in regulating the physiological functions of gas sensor proteins. These dynamic conformational changes induced by the binding of gas molecules to the heme are unique to heme-based gas sensor proteins; they are not observed in other heme proteins.
Footnotes
Acknowledgments
The author is supported by Japan Society for the Promotion of Sciences (Grant-in-Aid for Exploratory Research 23651086) and a grant from The NOVARTIS Foundation (Japan) for the Promotion of Sciences.