
Abstract
Introduction
B cells are designed to produce antibodies (Ab). Fulfilling this task entails many formidable difficulties, including the sequential recombination of immunoglobulin (Ig)-genes, allelic exclusion, isotype switch, somatic mutation, assembly and expression of functional B cell receptors, and finally the secretion of large quantities of antigen-specific antibodies of the most suited class (22).
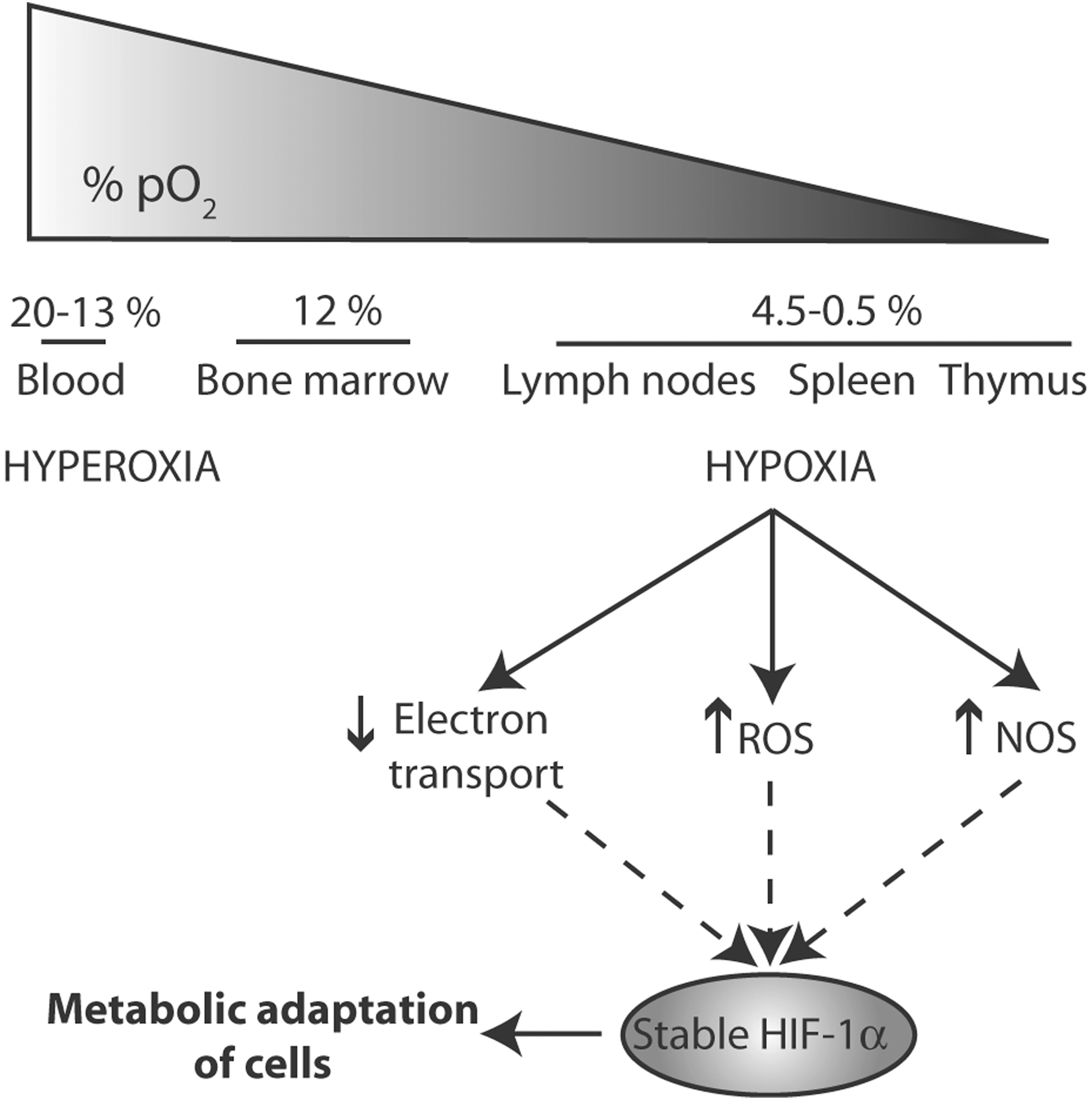
In addition to cytokines and chemokines, a key yet still poorly recognized role is played in these processes by the physicochemical features of the B cell microenvironment, including metabolism (17), oxygen tension, and redox state. These parameters vary considerably in the different organs crossed by B lymphocytes during their journeys from the bone marrow to the periphery. As shown in Figure 1, the O2 tension is highest in the blood, intermediate in the bone marrow where the first differentiation steps occur, and very low in lymph nodes, where mature resting B cells are stimulated by antigens to become Ab producing cells. Interestingly, the molecular oxygen (O2) levels and redox status are tightly entangled (42). Thus, mitochondrial reactive oxygen species generation at Complex III is induced by hypoxia in spite of the lower O2 levels and mitochondrial respiration. In turn, ROS trigger hypoxia-induced transcriptional programs through activation and stabilization of hypoxia-induced Factor 1α (HIF-1α) (12). Interestingly, HIF-1α not only induces the expression of several genes involved in the adaptation to hypoxia but also upregulates antioxidant genes.
A limitation in the studies on the role of ROS and redox signaling in B cell activation/differentiation has long been the difficulty to reliably measure hypoxia, ROS formation and tissue redox remodeling in vivo. Recently developed staining techniques allow monitoring redox changes in lymphoid organs during lymphocyte activation/differentiation (10, 54). Comparing these in vivo studies with the results obtained from experiments in vitro has provided new insights on the role of redox signaling in B lymphocyte differentiation.
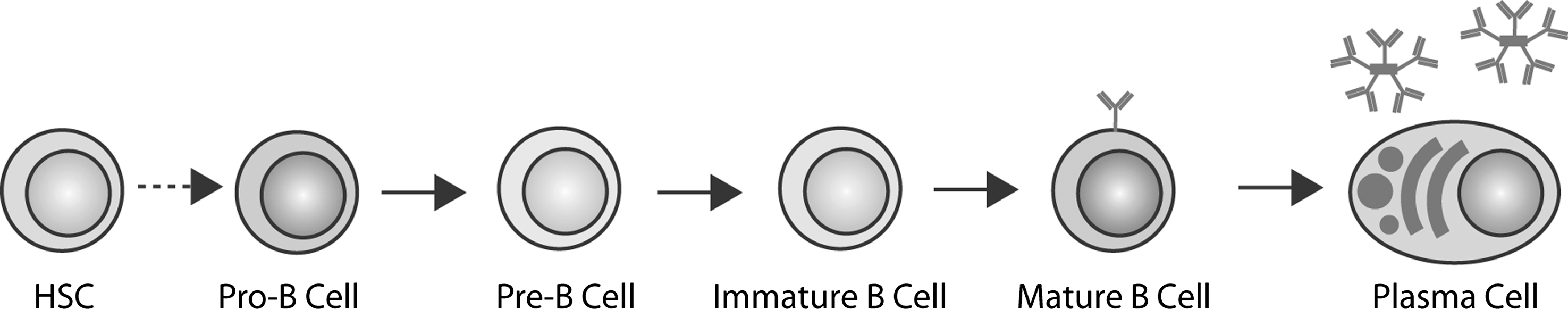
The main steps of the development of a hematopoietic stem cell in the bone marrow to a short-lived plasma cell in the periphery are summarized in Figure 2. Analyzing all the immunological events that occur throughout this long path goes beyond the aims of this review. Here we will discuss a few paradigmatic examples in which redox regulation plays a role, focusing our attention on how the successful outcome of the complex B lymphocyte differentiation process depends on a finely tuned interaction between intra- and extracellular redox reactions.
Redox and B Lymphocyte Development
The processes of somatic recombination and mutation necessary to generate high affinity antibodies are intrinsically error prone. As a consequence, numerous checkpoints are present along B cell development imposing survival/progression/death decisions based on the successful accomplishment of each crucial step. Moreover, at the end of the differentiation line, most plasma cells are short lived to limit antibody responses.
The hormetic nature of oxygen and its reactive species, essential for survival but toxic when in excess, seems well suited to regulate these crucial life/death decisions. Indeed, evidence is accumulating for a role of ROS and redox remodeling at many levels of B cell existence and progression.
Following the pioneering work of the Tonks and Rhee labs showing that intracellular ROS control tyrosine kinase signaling (36, 49), it became clear that ROS generation is essential for Toll-like receptors (TLR) and—at the mature B cell stage—B cell receptors (BCR) to transmit their signals upon binding of cognate ligands. The intense utilization of the antibody factory, forced to produce thousands of Ig molecules per second at the plasma cell level, entails massive redox reshaping and ROS production that likely contribute in limiting their lifespan (11).
Intracellular Sources of ROS During B Lymphocyte Differentiation
Although the occurrence of redox changes during B to plasma cell differentiation is well supported, the underlying mechanisms remain largely unknown.
A first unsolved problem concerns where, how, and why ROS are produced. As the physiological generation of ROS occurs as a byproduct of respiration, mitochondria are traditionally considered the major intracellular source of ROS. However, other intracellular organelles can generate ROS, under both physiological and stress conditions.
Mitochondria
Mitochondrial respiration produces ROS through Complexes I and III. A considerable fraction of the oxygen consumed may end up into superoxide anion, depending on the cell type and activity (20, 44). The large basal production of ROS by Complexes I and III makes it difficult to determine whether mitochondrial ROS increase, and have a role, in B cell activation/differentiation. An inhibitor of the electron-transport chain, rotenone, decreases but does not abolish ROS production both in resting B cells and upon lipopolysaccharide (LPS) stimulation (54), indicating that ROS are simultaneously produced by different sources. At the mitochondrial level, the redox enzyme p66Shc, that catalyzes the reduction of O2 to hydrogen peroxide (H2O2) (18), may conceivably be implicated in ROS production by B lymphocytes. Indeed, a number of evidence indicate that p66Shc is involved in the maintenance of lymphocyte homeostasis. Firstly, its deficiency results in age-related splenomegaly and spontaneous T- and B-cell activation and proliferation (15). Secondly, p66Shc promotes B-cell apoptosis by uncoupling the BCR from v-akt murine thymoma viral oncogene homolog (Akt)/extracellular-signal-regulated kinase (Erk)-dependent survival pathways. Finally, p66Shc expression is impaired in chronic lymphocytic leukemia (CLL) B cells (7).
Oxidative folding
Although there is evidence for redox regulation of endoplasmic reticulum (ER)-related functions such as protein folding and secretion (11), which enzymatic systems actually generate ROS in the early secretory compartment during lymphocyte differentiation, is still unclear. Among ER enzymes, ER-oxidoreductin 1 (Ero1) oxidases produce stoichiometric amounts of H2O2 for every disulfide bond formed in vitro (19, 56), which, presumably, also occurs in the cellular context (14). Therefore, huge amounts of ROS should be generated during Ig biosynthesis in Ab secreting cells. Ero1-derived ROS have been proposed to affect cell physiology and induce stress, a situation that manifests itself most clearly in cell types with high secretory activity, such as plasma or pancreatic β-cells (11, 40).
The role of Ero1 flavoproteins as sources of ROS in differentiating B cells is however questioned by the finding that protein synthesis inhibitors only modestly affect ROS levels in LPS stimulated B lymphocytes (4, 54), suggesting that neither Ig biosynthesis nor Ig oxidative folding are responsible for the strong ROS increase. That ROS are generated at high levels even when Ig production is blocked is only apparently odd. Rather, it is in agreement with previous findings that, on activation, B cells expand their metabolic capacity and secretory machinery well before IgM secretion starts (51). The increased energetic metabolism in antibody producing cells and the massive expansion of the secretory apparatus may account for ROS production before Ig production starts. In the absence of Ig synthesis, the involvement of Ero1 is unlikely. However, O2 − • can be generated by other ER-resident enzymes, such as nicotinamide adenine dinucleotide phosphate (NADPH) cytochrome p450, and b5 family proteins ((58) and references therein) and the less studied microsomal NADH oxidoreductase (28).
NADPH oxidases
NADPH oxidases (NOX) are complex enzymes that transfer electrons across biological membranes to O2 generating superoxide. First discovered in phagocytes, isoforms of NOX and dual oxidase (DUOX) are present in other cell types, with important consequences upon signaling (3). NOXes are localized mainly in the plasma membrane and secretory granules. However, increasing evidence indicate the presence of active NOX enzymes in various subcellular compartments, including specialized plasma membrane domains and nucleus. The compartmentalization of NOX is likely to influence the function of both the signaling mechanisms that activate the specific oxidases, and the regulatory pathways they control. Among NOX enzymes, B lymphocytes express NOX2 and DUOX1 (3, 34, 41). DPI, a NOX inhibitor, decreases ROS generation upon B cell stimulation (54) and impairs B-cell activation/differentiation as well as the efficiency of IgM secretion, indicating a role for NOX-dependent ROS at various steps along the transition of B lymphocyte to plasma cell (4, 54). This result is in line with the previous suggestion that H2O2 may act as a secondary messenger in the initiation and amplification of signalling after BCR triggering (34) (see below). Mutations in gp91phox, the prototypic NOX expressed by phagocytes, or associated proteins are the cause of chronic granulomatous disease (55). Although patients affected by this disease have a clear defect in macrophage and neutrophil function, they do not display the general block in B lymphocyte development that one would expect from molecules playing a crucial role in lymphocyte activation. The same has been found in gp91phox–deficient mice (37). Other enzymes must hence be at work in cells of the B lineage, including DUOX1 (41). Nonetheless, murine gp91phox−/− B cells generate fewer ROS after BCR ligation (37) and are hyper-responsive to BCR stimulation. Since gp91phox−/− B cells stimulated with LPS proliferate at normal levels, ROS could help sustain negative cell cycle regulation, acting basically on Ag-specific functions of B cells (37).
Phagocyte NOX function is supported by voltage-gated proton currents, which prevent membrane depolarization, caused by the transport of electrons from the intracellular NADPH to extracellular or phagosomal superoxide. Without the charge compensation provided by proton channels, NOX activity would eventually inhibit itself (35). The hydrogen voltage-gated proton channel 1 (HVCN1) modulates also BCR signal strength (Fig. 3). In fact, HVCN1-deficient B cells show pronounced alterations in their ROS-generating activity, which result in attenuated BCR signaling and downstream metabolism (6). The discrepant phenotypes of gp91phox−/− and hvcn1−/− could be due to the fact that ROS measurements in gp91phox−/− B cells miss a minor but physiologically relevant local ROS production close to the BCR. Actually what makes these studies difficult is that ROS can be produced by various systems in multiple locations and are counterbalanced by antioxidant defense mechanisms. These could represent a way of fine-tuning redox homeostasis in B cells when they need to develop their most important physiologic feature, antibody production. The correlation between ROS generation and B cell activation/proliferation remains a key issue: the function of NOXes, DUOXes, and HVCN1 in selectively regulating BCR-dependent proliferation and differentiation clearly deserves further investigations.

How Do B Cells React to Oxidative Stress?
Elaborate antioxidant defense mechanisms—largely coordinated by transcription factor NF-E2-related factor 2 (Nrf2)—allow cells to neutralize excess ROS and handle oxidative stress (Fig. 4). These include scavenging enzymes, oxidoreductases, and small nonprotein molecules such as reduced glutathione (GSH) (see (4) and references therein). Mice or plants lacking individual factors display no or mild phenotype, implying a high degree of redundancy (4,5) likely reflecting an evolutionary pressure to cope with oxidative stress. Indeed, the presence of ROS is an inevitable aspect of life under aerobic conditions. Furthermore, ROS have been exploited as signaling molecules in several fundamental processes (16). A fast and efficient control is therefore required not only to avoid oxidative stress but also to ensure the correct outcome of these processes. This is particularly true in cells of the innate and adaptive immune systems (26, 45, 46).

In B cells, an important antioxidant defense is played by peroxiredoxin 4 (Prx4) and other peroxidases residing in the secretory pathway. Prx4 increases dramatically during plasma cell differentiation, paralleling Ig production, and counteracts in part the strong increase of ROS in the ER (4). Consistent with the notion that Prx4 metabolizes Ero1α-derived H2O2 (60), increased levels of hyperoxidized Prx4 are detected under conditions of augmented Ero1α activity in cells (47). Since Prx4 couples the degradation of H2O2 to the generation of disulfide bonds (60), its dramatic upregulation in Ig secreting cells may reflect an adaptive response to the increased oxidative demand. However, Prx4−/− mice have only a marginal defect in Ig production (4), implying the existence of additional pathways.
In addition to the antioxidant defenses described above, a crucial role in B cell responses and other pathophysiologic processes, including inflammation and tumor development and progression, is played by the cysteine/cystine redox cycle (13, 25). The cycle is basically composed by system xc-; the redox couple cystine/cysteine; and intracellular oxido-reductases including thioredoxin (Fig. 5A). System xc- is a Nrf2-regulated, oxidative stress-inducible, cystine/glutamate antiporter that imports oxidized cystine releasing equimolar glutamate into the extracellular space. It consists of two protein components, the 4F2 heavy chain, necessary for membrane targeting of the heterodimer, and the xCT protein, responsible for transport activity (13).
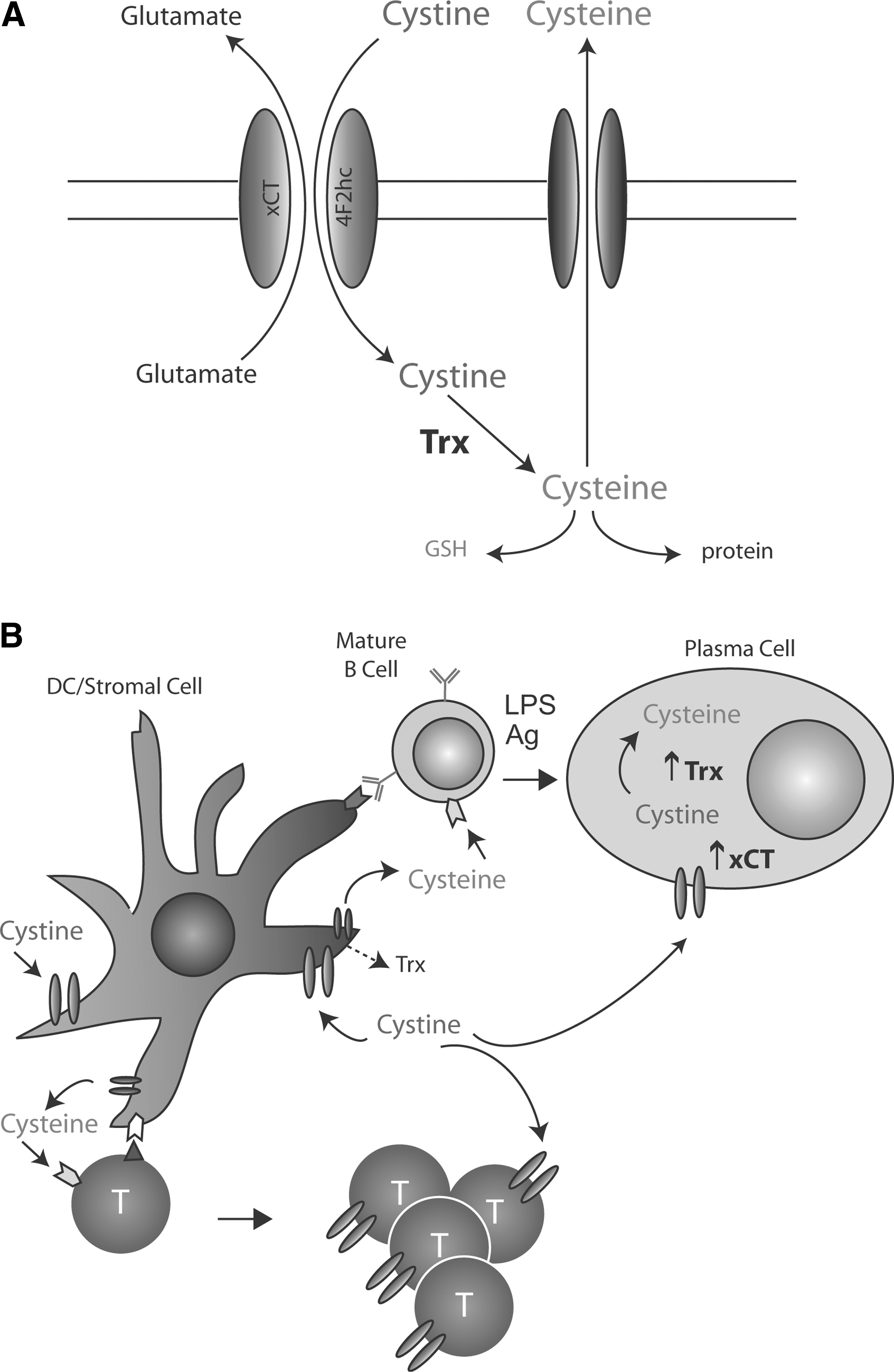
While cysteine is more abundant in the reducing milieu of the cytosol, cystine prevails extracellularly. Once internalized, cystine is reduced to cysteine by thioredoxin. Internalized cysteine was long thought to be merely used for protein and GSH synthesis. However, increasing evidence indicates that some cysteine is actively released extracellularly via the neutral amino acid transport systems. There, cysteine is rapidly oxidized to cystine, and consequently taken up by system xc-. If this cystine/cysteine cycle persists, a steady state is reached, and cysteine remains reduced due to a switch of the extracellular redox state from a oxidizing to a reduced state.
In vitro evaluations of cysteine release by immune and tumor cells following an oxidative hit revealed that the levels of extracellular cysteine may reach surprising amplitudes. For instance, the 24-hour spent medium of 106 primary human monocytes contains up to 15 μM/ml. These cells secrete more than three-fold more cysteine if exposed to LPS (Fig. 5B), which induces ROS generation, and a consequent antioxidant response entailing further upregulation of the cystine–cysteine cycle (45, 46). In monocyte-derived dendritic cells (DC), the cycle is even more active, reaching up to 100 μM/ml of cysteine/106 DC/24h (1). Treatment of human melanoma cells with arsenic, a pro-oxidant drug that upregulates xCT, results in the release of similar cysteine levels and a consequent switch of the oxidizing physiologic extracellular redox to a reduced state (53). Therefore, the cystine/cysteine redox cycle affects also the redox state of the microenvironment, with important pathophysiological consequences (38).
Since resting T, pre-B, and B lymphocytes do not express xc-, cysteine must be made available to sustain their activation and proliferation (1). The extracellular redox hence provides a rheostat for immune responses. In the case of T lymphocytes, the “wetnurse” has been shown to be the dendritic cell (1) (Fig 5B). Which cell(s) provide cysteine to B lymphocytes during their differentiation/activation remains to be firmly established. In B-CLL, lymph node stromal cells have been proposed to supply neoplastic B lymphocytes with thioredoxin, which protect them from apoptosis ((2) see below). Whether stromal cells make available reducing equivalents to normal B cells as well is still unknown (Fig. 5B). Interestingly, upon activation, both T and B lymphocytes undergo a rapid “weaning”, due to expression of system xc-. The transporter allows cystine uptake and therefore releases lymphocytes from the requirement of cysteine feeding. Moreover, activated B lymphocytes, but not T cells, start to release reduced cysteine themselves (54). It is conceivable that, as in myeloid cells, TLR stimulation upregulates the cystine/cysteine cycle via sequential ROS and antioxidant response waves (54), resulting in a shift of the redox balance toward the reductive side.
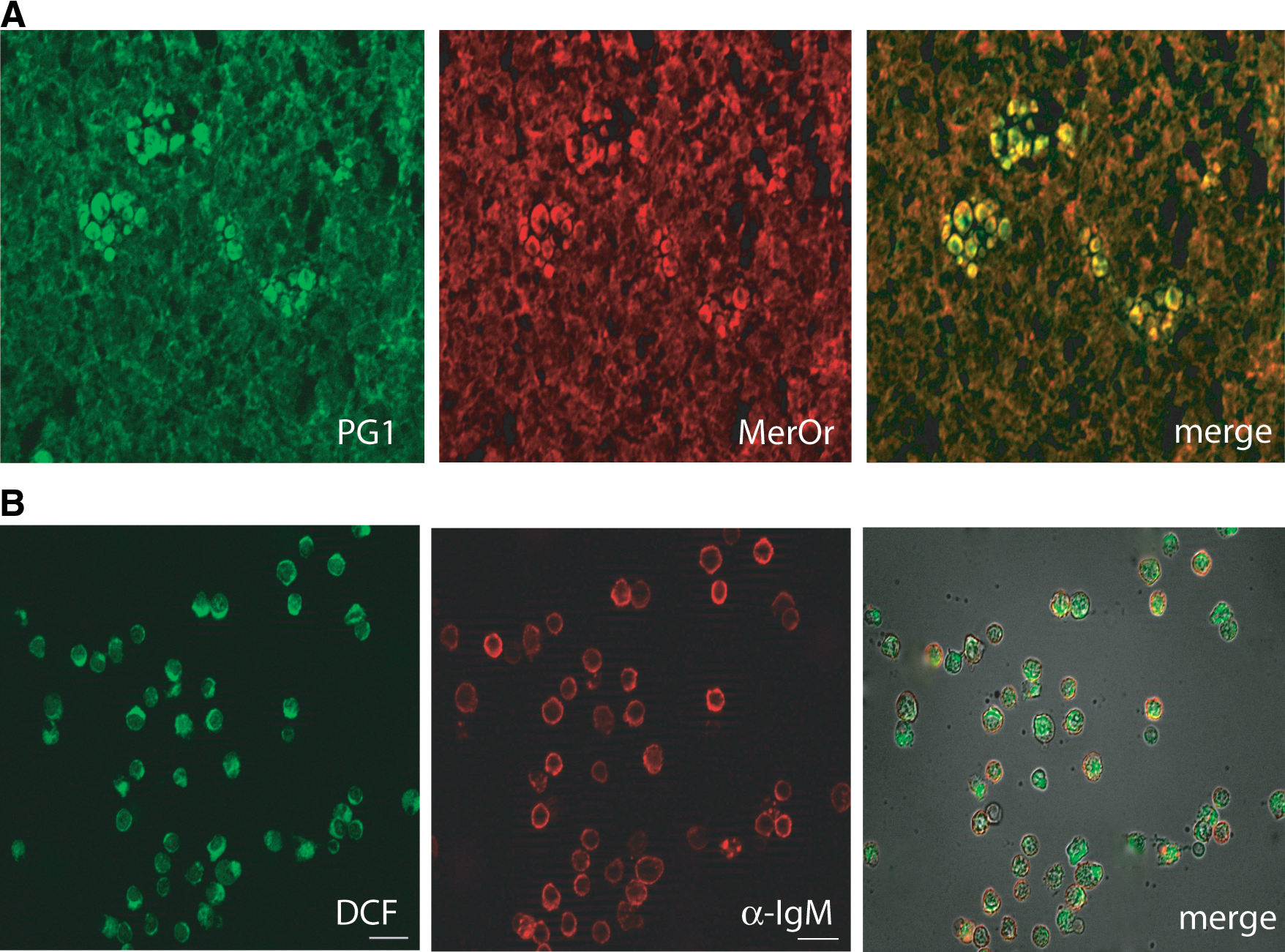
Remarkably, the redox remodeling characterized by an oxidative step followed by an antioxidant response is not restricted to selected immune cell populations stimulated in vitro but is evident also in vivo. By using biochemical quantification of nonprotein thiols (GSH and cysteine) and histochemical staining of frozen tissues, we demonstrated that lymphoid organs are generally more reduced than average tissues (10). Moreover, the thioredoxin and free thiol content of lymph-nodes increases dramatically during the immune response (10, 54) (Fig. 6). Interestingly, not only do DCs contribute to the generation of the reducing microenvironment in secondary lymphoid organs, confirming their feeding activity toward T cells (1), but also B lymphocytes dramatically increase thioredoxin and cysteine production upon activation, resulting in highly “reduced” germinal centers. This strong “reduction” of immune lymphoid tissues represents conceivably a response to an oxidative stress, since increased ROS levels are present in immunized mouse spleens (54).
A fascinating question is whether the antioxidant response to ROS production is only a defense mechanism aimed at restoring the redox homeostasis or it also exerts functional effects on B lymphocytes differentiation. Indeed, an active role for the antioxidant response has been already established in various processes of the innate and adaptive immunity, including IL-1β secretion (45, 46) and T cell activation (1). Similarly, in vitro exposure of B lymphocytes to exogenous antioxidants revealed that highly reducing conditions are detrimental in the early phases of B-cell activation whereas promote IgM secretion later on during differentiation (54). Therefore also the antioxidant response plays a role in B cell differentiation, as well as ROS, before considered only a waste product, which instead turned out to be crucial intracellular signaling molecules (34).
ROS as Second Messengers in Lymphocyte Activation
In the last decades, intracellular ROS, H2O2, and nitric oxide have been recognized as second messengers in the activation of numerous signaling pathways (16, 43). The molecular mechanisms for H2O2 signaling generally involve the reversible oxidation of low pKa protein cysteine thiols to sulfenic acid (-SOH) on specific target proteins. However, these protein cysteine thiols are also susceptible to irreversible oxidation at higher H2O2 concentrations, with the generation of sulfinic and sulfonic derivatives. Therefore, the level of ROS, as well as their intracellular diffusion, must be tightly controlled. How far does H2O2 diffuse within cells? Is it permeant to cell membranes? Spatial regulation and topological constraints are key issues (50) that need further mechanistic understanding. Also some of the antioxidants that damp down H2O2 signal propagation are spatially regulated, often via sophisticated molecular circuits that control their activity by site-specific post-translational modifications (52). For instance, Prx1 activity is inhibited by phosphorylation, so as to allow tyrosine kinase signaling while limiting it in space and time (57). Intriguingly, work in Zebrafish suggest that a H2O2 gradient promotes leukocyte migration to the wound sites (30). We can therefore envision ROS signaling as a dynamic process that occurs within cells between different organelles, as well as between cells over long distances (27).
In the BCR, membrane-bound Ig molecules and Ig-αβ heterodimers function as antigen-binding and signaling subunits, respectively (39). At least three PTKs (v-yes-1 Yamaguchi sarcoma viral related oncogene homolog (Lyn), spleen tyrosine kinase (Syk), and Bruton agammaglobulinemia tyrosine kinase (Btk)) and one protein tyrosine phosphatase (PTP), SHP-1, are involved in signal transduction from the BCR. The negative regulator SHP-1 is inhibited by ROS, which oxidize a cysteine residue in the catalytic site of the enzyme (see Fig. 3 legend for details). This explains why suitable doses of H2O2 can mimic antigen binding, triggering the same phosphorylation cascade as BCR cross-linking (34). Moreover, H2O2-treated B cells exhibit PTK-dependent inositol 1,4,5-trisphosphate (IP3) generation and Ca2+ release (33). The initial, low-intensity activation of BCR signaling is amplified by the Ca2+-dependent, self-sustaining feedback loop (41). Thus, oxidants and Ca2+ cooperatively regulate the strength and duration of BCR signaling, crucial parameters for determining the subtype of B cell generated from transitional, immature B cells (9).
Surprisingly, Lee et al. found that activation of effectors like Erk, c-Jun N-terminal kinase (JNK), p38, and Akt by the BCR was relatively insensitive to ROS depletion (23). Also chemokine receptor type 4 (CXCR4), which binds chemokines involved in B cell development and trafficking, activates MAP kinases via ROS-independent pathways, but its ability to activate Akt depends on ROS (23). In contrast, the surface receptor CD40, important to receive T cell costimulatory signals, activates JNK, p38 and Akt for the most part via ROS-dependent pathways that are sensitive to ROS scavengers such as N-acetyl-cysteine or ebselen. The role of HVCN1 in BCR signaling (see above, Fig. 3) confirms that oxidation is needed, the activation of Syk and Akt being impaired in HVCN1-deficient cells. Akt activation depends on production of phosphatidylinositol 3,4,5-triphosphate (PIP3) by PI3K (23). The discrepant results concerning Akt activation could be due to the fact that HVCN1 deletion probably affects directly PI3K in a ROS-independent fashion, while ROS scavengers modulate the general intracellular redox status and Akt only indirectly. However, not all signaling pathways downstream of Syk were equally impaired removing the proton flux that sustains electrogenic ROS production. Ca2+ mobilization and Erk activation were not impaired in the absence of HVCN1, possibly indicating different activation thresholds (6). These results imply that a complex interplay between distinct pathways regulates the outcome of a signal.
Organelle movement and the cytoskeleton could play a central role in distributing ROS signals. Positive and negative signals mediated through Btk and SH2 (Src homology 2)-containing inositol phosphatase-1 (SHIP-1) regulate B cell membrane dynamics and spatio-temporal organization of surface BCRs via actin reorganization (24). The magnitude of aggregation and the mobility of aggregates regulate BCR signaling capability. This interplay between actin reorganization and signaling reveals a mechanistic basis for feedback regulation for BCR and other receptors. The development and utilization of more mutants deficient in ROS signaling and additional ROS imaging tools will allow the identification of additional molecular linkers and a better definition of the interplays between the actin cytoskeleton, ROS, and BCR signaling pathways.
The complexity and interdependence of redox networks and the inadequacy of robust readouts have hampered a detailed mechanistic dissection of ROS signaling. Innovative approaches with organelle-specific ROS reporters in vitro and in vivo may allow spatio-temporal monitoring of redox reactions.
(Dys)regulation of Redox Homeostasis in B Cell Malignancies
B cell malignancies recapitulate the different stages of B-cell maturation. B lymphoblastic leukemia/lymphoma are precursor B cell neoplasms, most lymphomas (follicular, Burkitt, Hodgkin, and diffuse large B cell lymphoma) derive from germinal center B cells, whereas CLL/small lymphocytic lymphoma and myeloma are the malignant counterparts of post-germinal centers cells that include both long-lived plasma cells and memory/marginal B-cells.
The redox state of the different B cell malignancies compared to the corresponding normal B cells is still poorly defined. Most studies are restricted to chronic B cell malignancies and focus on the redox response to therapy rather than on the baseline differences between normal and neoplastic B cells. In general, the effects of chemotherapy on redox homeostasis are strong, as in solid tumors (53). For instance,
Few are the available systematic analyses of the redox state of B cell malignancies from untreated patients. A transcriptional redox signature score that identifies diffuse large B-cell lymphoma patients with poor prognosis has been described (48). Malignant cells from these patients had decreased expression of catalase, glutathione peroxidase (GPx), Mn- superoxide dismutase (SOD), and thioredoxin-binding protein-2 (TBP-2, also known as VDUP-1 and TXNIP), a protein that inhibits thioredoxin (Trx) activity. The incongruity between fewer antioxidant enzymes and increased Trx is only apparent. The former likely results from the increased stress that characterizes neoplastic cells (8). The concomitant decrease of TBP-2 and consequent increase of Trx activity is possibly related to the poor prognosis of these B cell lymphomas, since Trx prevents apoptosis and promote cell growth (31). Interestingly, Trx has been implicated also in B-CLL cell survival (31, 32). However, B-CLL cells express very low levels of endogenous Trx. In this case, therefore, protection from apoptosis is likely provided by exogenous Trx, secreted by lymph node stromal cells (2).
It would be of great interest to assess the presence and the regulation of xc- in neoplastic B cells. A dysregulated expression of the transporter, allowing cystine internalization, may release resting B cells from the control exerted by the environment, thereby allowing unwanted proliferation. Also a better definition of the local, intercellular redox circuits may shed light on the biology of these malignancies and pave the way to novel therapeutic approaches.
Concluding Remarks
Over the last years, it has become clear that not only ROS production but also the antioxidant systems they elicit have profound impacts on the survival and differentiation of cells of the B lineage. An emerging point is that the antioxidant response, through enhanced activity of the cystine/cysteine redox cycle, goes beyond the cell border and increases the extracellular redox potential, generating a reduced microenvironment. Profound redox reshapings with an extracellullar impact are shared by other cell types, with implications on pathological processes (e.g., chronic inflammation, tumor progression, and drug resistance). A better understanding of these events may lead to new approaches exploiting redox modulation as a therapeutic tool.
Footnotes
Acknowledgments
We thank the members of our laboratories for helpful discussions and suggestions, Tina Scacciante and Raffaella Brambati for secretarial assistance, and the AIRC (IG and 5 x 1000 Program), Compagnia San Paolo, Regione Lombardia and Telethon for funding our research. We apologize to the many colleagues whose pioneering work could not be cited owing to the strict space limits.
Author Disclosure Statement
No competing financial interests exist.