
Abstract
Introduction
Innovation
Current research reveals that the mitochondrial DRP1 protein is necessary for ROS signaling in hypothalamic glucose sensing mechanism. Our results suggest that the mitochondrial DRP1 protein may be a subtle upstream actor in glucose-induced mROS signaling in the hypothalamus, thereby promoting the effects of glucose in vivo. These results highlight a new mechanism involved in glucose detection which is primordial for both the control of food intake and energy metabolism. Further investigations will be necessary to determine the role of DRP1 in metabolic disorders associated with defective hypothalamic glucose detection.
Mitochondria are dynamic organelles that continually change form. Different sets of proteins control mitochondria dynamics through fission and fusion mechanisms (10). Fusion mechanisms are under control of optic atrophy 1 (OPA1) and mitofusins (MFNs). The fission machinery involves fission protein 1 (FIS1) and dynamin-related protein 1 (DRP1). DRP1, which is mainly cytoplasmic under basal conditions, translocates to the mitochondrial membrane where it forms a contractile “ring” involved in the division of the mitochondria. Mitochondrial fusion and fission are on a tightly regulated equilibrium to maintain mitochondrial morphology in the cell and are critical for cell function (7).
Mitochondrial morphology is associated with mROS production and energy status. Nutrients and hormones can alter mitochondria morphology (39, 41). For instance, a recent study showed the involvement of mitochondrial dynamics in high glucose-induced ROS production (40). Acute exposure of hepatic and muscular cell lines to high glucose concentrations induced a rapid and transient mitochondrial fission. This mitochondrial fission is associated with an increase in ROS levels. In contrast, inhibiting mitochondrial fission was shown to abolish high glucose-induced ROS levels. Thus, morphological changes of mitochondria appeared to be an upstream causal factor for ROS production under acute hyperglycemic stimulation. This suggests that mitochondrial dynamics act as a regulating factor of mitochondrial activity.
The aim of this study was to determine whether mitochondrial dynamics protein DRP1, involved in fission mechanisms, could constitute an upstream actor in VMH mROS production, and consequently play a role in VMH glucose sensing. We hypothesized that a DRP1 mitochondrial translocation occurs in VMH in response to a transient cerebral hyperglycemia. This process will be necessary for the mROS-induced physiological responses.
Results
Transient cerebral glucose increase triggers the recruitment of VMH DRP1 to mitochondrial membranes
Under basal conditions, most of the cellular DRP1 is located in the cytosol. Fusion is induced when DRP1 is relocalized to the mitochondrial membrane (34). We evaluated VMH DRP1 cellular localization in response to an intracarotid glucose bolus injection (9 mg/kg). Intracarotid glucose injection toward the brain significantly increased DRP1 level (263% of controls) in VMH mitochondria extract 1 minute post-injection. No significant modification of increased mitochondrial DRP1 was detected in the cortex and thalamus (data not shown). In addition, the nonmetabolizable glucose analog L-glucose did not increase mitochondrial DRP1 (Fig. 1A). These data suggest that DRP1 is rapidly relocalized to the mitochondria in response to increased glucose. Total protein levels of fusion proteins (OPA1 and MFN2), fission proteins (DRP1 and FIS1), and mitochondrial inner membrane protein CypD, a mediator of mitochondrial permeability transition pore opening, were unchanged in NaCl- or glucose-injected rats (Fig. 1B). The analysis of mitochondrial morphology did not reveal significant modification in VMH areas of glucose-injected rats compared to NaCl-injected rats (Supplementary Fig. S1B; Supplementary data are available online at
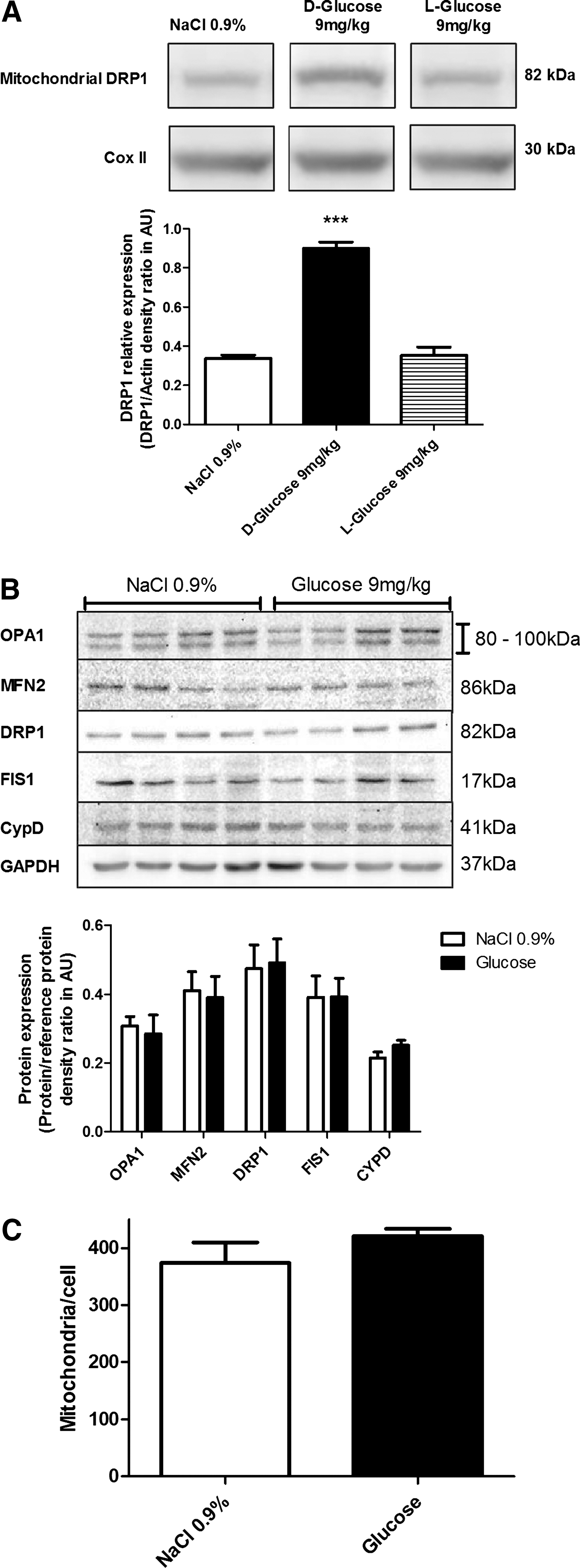
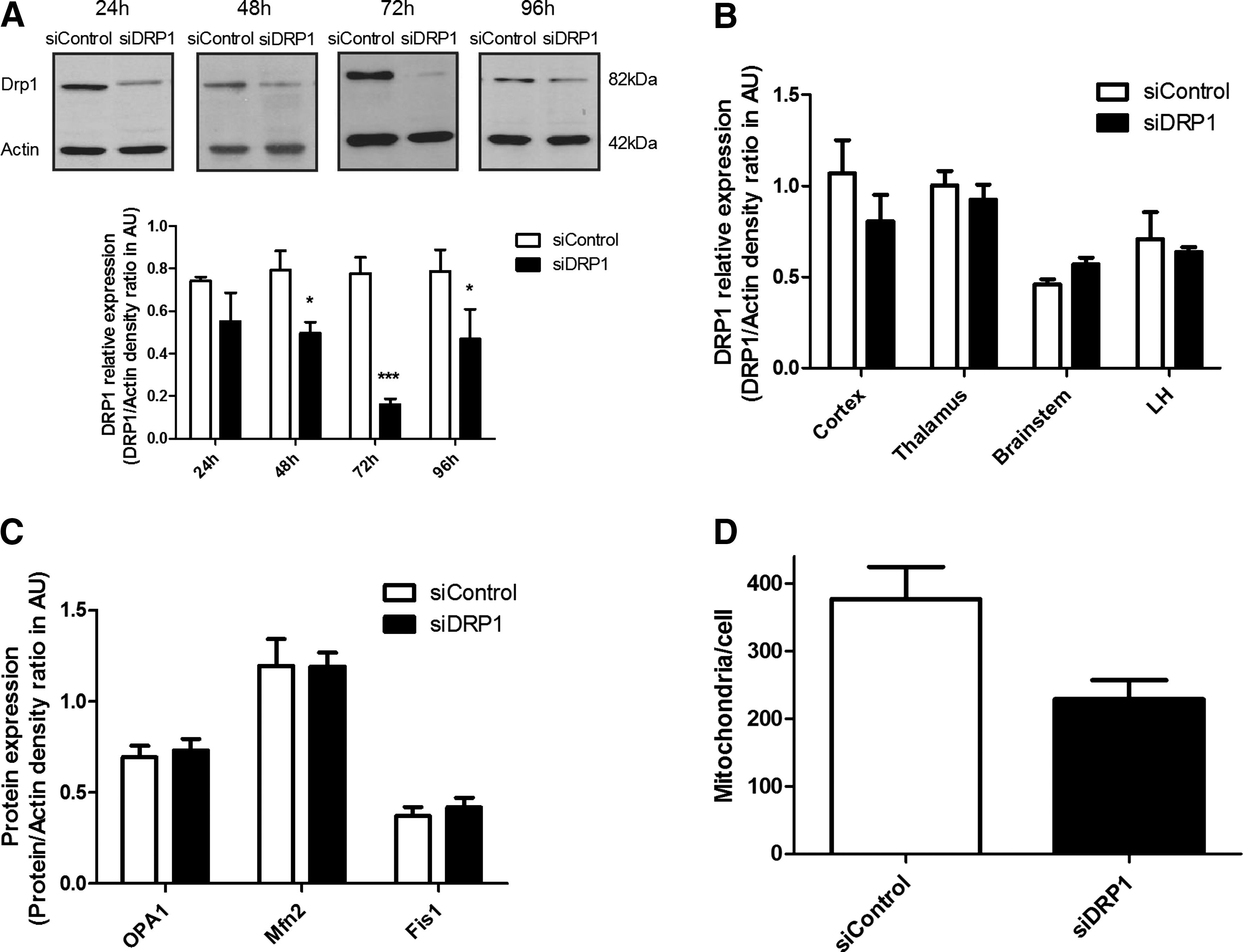
DRP1 downregulation inhibits mitochondrial fission in vivo
Downregulation of DRP1 by RNA interference was used to investigate its role in hypothalamic glucose sensing. Therefore, siRNAs directed against DRP1 or siRNA controls were injected into the VMH, and DRP1 levels estimated by Western blot in the ventromedial part of the hypothalamus that includes ARC. DRP1 extinction reached 28.9±5.8%, 35.1±9.9%, 76.6±2.5%, and 40.7±23.3% at 24, 48, 72, and 96 h post-injection, respectively (Fig. 3A). No modification in DRP1 expression was observed in the cortex, thalamus, brainstem, or lateral hypothalamus (LH) 72 h post-injection (Fig. 3B). No modification in other proteins expression involved in mitochondrial dynamics was found, showing that only DRP1 expression was affected (Fig. 3C). Because the greatest decrease in DRP1 levels occurred 72 h post-injection, we chose this time to evaluate the impact of VMH DRP1 siRNA on glucose sensing. The impact of DRP1 siRNA injection on mitochondrial morphology was evaluated by fluorescent microscopy imaging 72 h after siRNA injections. No mitochondrial morphological differences was measured between siDRP1 and siControl animals (Supplementary Fig. S2), only a decreased number of mitochondria per cell was observed (376.8±45.6 mitochondria/cell for siControl vs. 229.1±27.7 for siDRP1, p<0.05) (Fig. 3D). 72 h after the siRNA injection, both siDRP1 and siControl rats exhibited a similar food intake and weight gain as before injection (data not shown).
Downregulation of DRP1 in VMH decreases glucose sensitivity due to the loss of mROS signaling and impacts both insulin secretion and the satiating effect of glucose
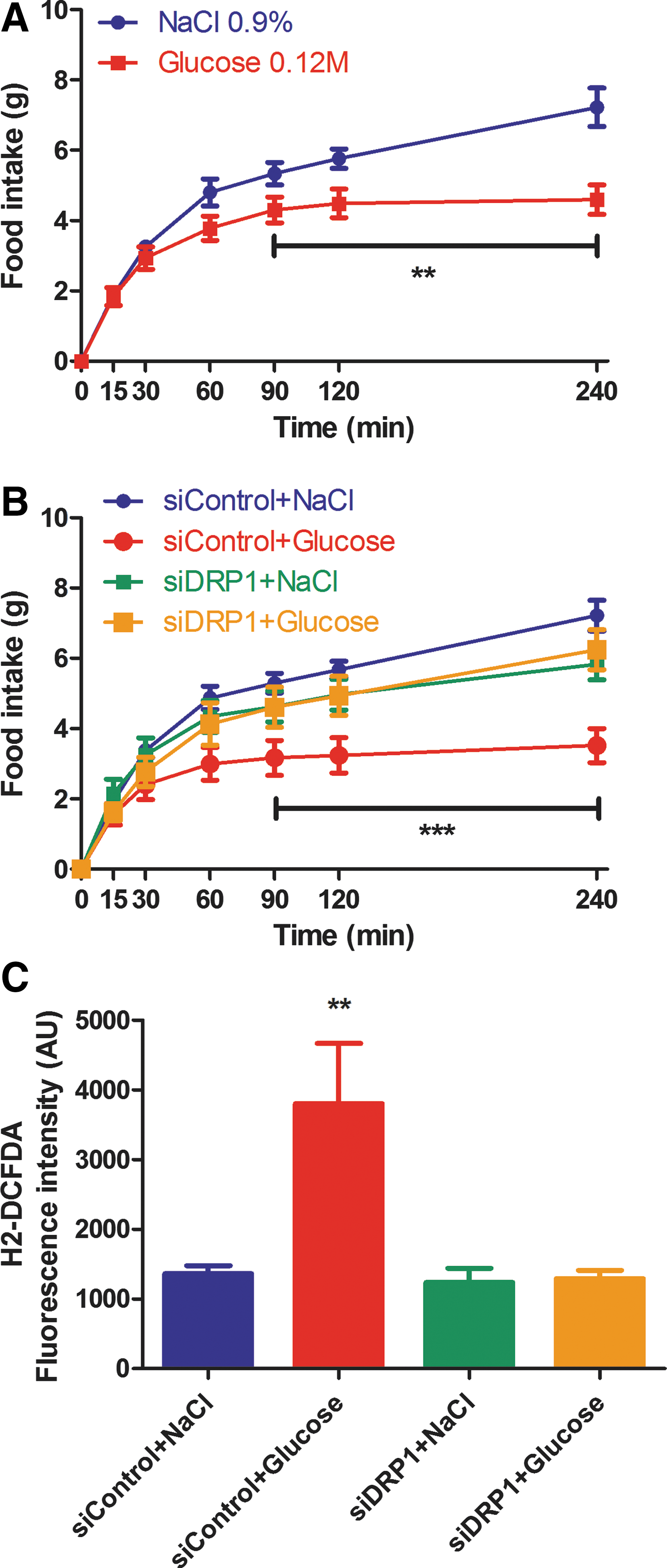
To decipher the role of DRP1 in VMH glucose sensing, we evaluated the consequences of its downregulation in the detection of an increased glucose concentration through two different tests. The first test involved the hypothalamic pancreatic axis and the parasympathetic control of insulin secretion. The second test allowed the measurement of the satiating effect of glucose on fasted rats with a refeeding test. The role of DRP1 inhibition was first determined in response to intracarotid glucose injection-induced insulin secretion (22). Intracarotid glucose injections had no effect on peripheral blood glucose level in both siControl and siDRP1 groups (Fig. 4A). However, intracarotid glucose injection-induced insulin secretion was significantly inhibited in siDRP1-treated rats (for siControls, delta insulin level 103.2±20.4 mU/L; siDRP1=37.8±7.2 mU/L) (Fig. 4B). In addition, impaired insulin secretion was accompanied with a complete inhibition of mROS production induced by a glucose load (siControl+NaCl: 17033±3540 AU; siControl+Glucose: 37797±6211 AU; siDRP1±NaCl: 13139±1469 AU; siDRP1+Glucose: 18156±1974 AU) (Fig. 4C). No difference was found in other brain regions such as the cortex (Fig. 4D) or thalamus (Fig. 4E). To assume that decreased insulin secretion was not the consequence of impaired pancreatic β-cell function, ex vivo glucose-induced insulin secretion was evaluated in islets of siControl and siDRP1-injected animals. Islets of siDRP1-treated rats had a similar basal insulin secretion (in 5.5 mM glucose) to siControl islets, and they did not show any impairment in pancreatic insulin secretion in response to 16.7 mM glucose (siControl: 6.67±0.17 ng/ml/10 islets, siDRP1: 7.15±0.22 ng/ml/10 islets) (Fig. 4F). Because intracarotid glucose injection-induced insulin secretion involves the activation of parasympathetic nerves (2, 13, 16), we evaluated the islets' sensitivity to a cholinergic agonist. Islets were stimulated with a muscarinic cholinergic receptor agonist (acetyl-methylcholine bromide) in 5.5 mM glucose. SiDRP1 animals showed increased insulin secretion in response to the acetyl-methylcholine bromide (siControls: 3.84±0.10 ng/ml/10 islets; siDRP1: 4.54±0.21 ng/ml/10 islets) (Fig. 4G).

We next evaluated the effect of siDRP1 treatment on VMH glucose infusion-induced decrease in food intake after refeeding. First of all, this effect was evaluated in animals without siRNA treatment. In control animals, VMH glucose infusion attenuated increased food intake after refeeding following an overnight fast (NaCl group: 7.22±0.54 g and Glucose group: 4.59±0.42 g at the end of the test) (Fig. 5A). VMH glucose infused-SiDRP1 animals showed a significant reduction in food intake inhibition during 4 h of refeeding compared to SiControl rats, while food ingestion in response to NaCl VMH infusion was similar in both siControl and siDRP1-treated animals (siControl+Glucose: 3.52±0.48 g; siDRP1+Glucose: 6.25±0.56 g; siControl+NaCl: 7.22±0.43 g; siDRP1+NaCl: 5.83±0.44 g) (Fig. 5B). As for intracarotid glucose-induced insulin secretion, we examined if decreased food intake induced by VMH glucose infusion involved impaired VMH ROS production in siDRP1 rats. In siControl animals, VMH glucose infusion increased mROS production threefold the level of NaCl in infused-SiControl group (siControl+NaCl: 1362±117 AU; siControl+Glucose: 3800±864 AU) (Fig. 5C). However, mROS production was not increased in glucose-infused siDRP1-treated rats (siDRP1+NaCl: 1234±204 AU; siDRP1+Glucose: 1291±119 AU) (Fig. 5C). These results suggest that the satiating effect of VMH glucose infusion is dependent on mROS production through mitochondrial DRP1-dependent fission.
VMH mitochondrial respiration of DRP1-inhibited rats exhibits a decreased activity in response to substrates
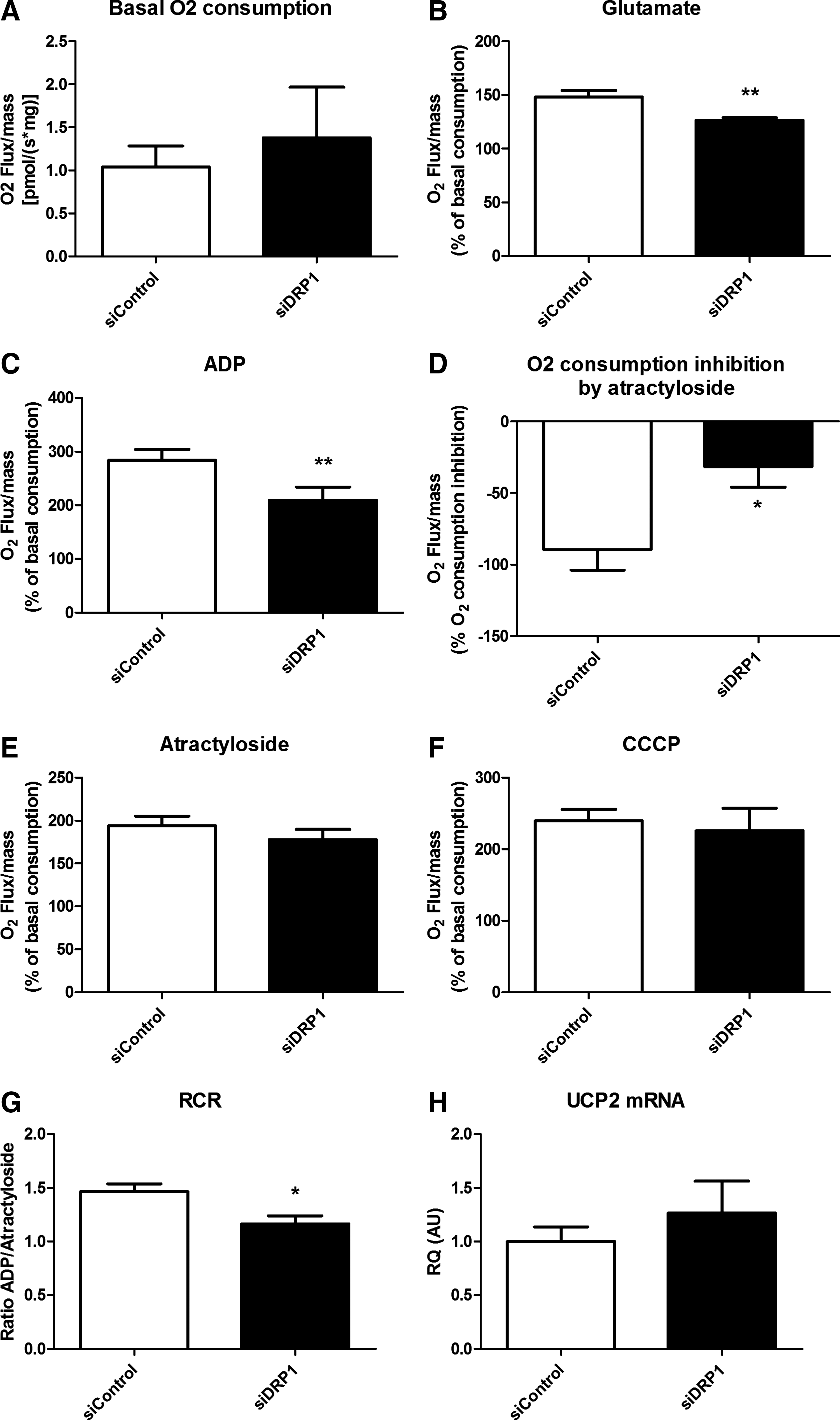
To explore the VMH mitochondrial function, oxygen consumption was analyzed on permeabilized hypothalamic tissue. Basal respiratory activity was similar between siControl and siDRP1 mitochondria (Fig. 6A). Substrate-driven respiration determined by glutamate stimulation was significantly decreased in siDRP1 rats (siControls: 148.0±5.9% of basal consumption; siDRP1: 126.3±2.6%) (Fig. 6B). State 3 (substrate/ADP-driven) respiration was assessed with saturating ADP concentration (Fig. 6C). In these conditions, the O2 flux was decreased in siDRP1 rats (siControls: 283.83±20.49% of basal O2 flux; siDRP1: 209.45±24.22%). Carboxyatractyloside (CAtr), an ATP–ADP exchange inhibitor, was then added to obtain the ADP-independent resting state 4. Respiratory inhibition was significantly attenuated in siDRP1 rats (siControl: −89.72±14.15%; siDRP1: −31.62±14.26%) (Fig. 6D), highlighting a lower activity of Complex V, which resulted in an identical O2 consumption in both groups after CAtr stimulation (siControls: 194.11±11.14% of basal O2 consumption; siDRP1: 177.83±11.9%) (Fig. 6E). Finally, the calculated Respiratory Control Ratio (RCR, which represents the ratio of state 3/state 4) was significantly decreased in siDRP1 rats (siControls: 1.46±0.07; siDRP1: 1.16±0.07) (Fig. 6F), indicating a loss of coupling in mitochondria. The maximal respiratory capacity induced by carbonyl cyanide m-chlorophenylhydrazone (CCCP) was not different between siControl and siDRP1 rats (siControl: 239.79±15.75% of basal O2 consumption; siDRP1: 225.71±31.49%) (Fig. 6G). To assess the role of mitochondrial uncoupling proteins (UCPs) on the respiratory modification, we quantified the mRNA level of uncoupling protein 2 (UCP2), which is a major rodent hypothalamic UCP (33) and did not find any difference between groups (Fig. 6H). These results reveal that siDRP1 rats present a decreased capacity of substrate-driven respiration linked to decreased coupling respiration.
Discussion
We recently highlighted that the hypothalamic glucose-sensing mechanism requires mROS production to trigger both electrical ARC activation and insulin secretion (22). Moreover, this signaling is altered in the Zücker obese, insulin-resistant rat model (11). Here, we show for the first time the importance of mitochondrial dynamic protein DRP1 in glucose-induced hypothalamic mROS production and energy metabolism regulation. We found the following: (a) DRP1-dependent mitochondrial relocalization is induced by increased central glucose; (b) VMH DRP1 downregulation blocks glucose-induced mROS signaling, as well as central glucose-induced insulin secretion and central glucose-induced food intake decrease; and (c) decreased glucose-induced mROS signaling is consistently associated with altered mitochondrial respiratory activity.
The fission process is mainly dependent on DRP1, a cytoplasmic protein relocalized to the mitochondrial membrane under various stimuli (36). Here, we show that increased glucose level rapidly (1 min) promotes DRP1 translocation from the cytosol to the mitochondria in the VMH in vivo. The ability of glucose to induce in vivo DRP1 translocation to the mitochondria is consistent with previous studies showing that mitochondrial fragmentation in high glucose conditions requires mitochondrial fission machinery in cell culture (27, 36, 40). In our conditions, no significant alteration of mitochondrial morphology was shown. This is consistent with the fact that only some VMH neurons are glucose sensitive (8). Moreover, up to date only in vitro mitochondria have been morphologically measured, suggesting that quantification on tissue remains uncertain. Altogether, our results suggest that the mitochondrial fission mechanism may be a subtle upstream actor in glucose-induced mROS signaling in the hypothalamus, thereby promoting the effects of glucose in vivo (11, 22).
To explore this mechanism, a downregulation of DRP1 in the VMH was performed by RNA interference. Using this strategy, we determined a maximal extinction (80%) of DRP1 expression 3 days after injection. At this time, there was no compensation through the other main mitochondrial dynamics actors (OPA1, MFN2, and FIS1). VMH mitochondrial morphology was not significantly changed. This might be due to a decreased activity of fusion mechanisms to maintain a normal morphology or to the difficulties in measuring mitochondria in vivo (22, 29 –31). However, mitochondria per cell were decreased, suggesting the fusion process predominates. The down expression of DRP1 protein led to a defect in glucose-induced ROS production and physiological responses. This might be explained only if a lack of the DRP1 relocalization to the mitochondrial membrane occurred.
We then explored the impact of VMH DRP1 inhibition on the hypothalamic glucose-sensing mechanism. We previously showed that an intracarotid injection of a glucose load that does not alter peripheral blood glucose level induces a transient insulin secretion. In addition, we also showed that this intracarotid glucose load-induced insulin secretion is hypothalamic mROS production-dependent (22). In this study, there was no increased mROS production in siDRP1-treated animals 1 min post-glucose injection. This result suggests that the mitochondrial DRP1-dependent fission is an early and rapid event preceding mROS production. This suggestion is consistent with a study by Yu et al. (40) and previous studies on mitochondrial fission showing the ability of mitochondria to split rapidly and re-fuse (28).
As previously described, we found that intracarotid glucose injection-induced insulin secretion was altered in VMH siDRP1-treated rats. This result could be the consequence of altered β-cell function. To gain further insight into the control of insulin secretion in transient DRP1-deficient rats, their freshly isolated islets were compared to control ones for both their intrinsic response to glucose and to a classical muscarinic receptor agonist. This approach is used as a marker of parasympathetic control, as previously described (12, 25). Regarding islet response to high glucose, there was a normal response of the VMH DRP1-deficient rats, showing that direct glucose stimulation was not altered. However, the muscarinic islet activation through a cholinergic agonist triggered a hyper response. This response reflects an increased sensitivity of the muscarinic receptor and is classically observed when parasympathetic tone decreases, enhancing either the activity and/or the number of receptors on the islets (12, 25). We can conclude that parasympathetic activity was decreased. Indeed, the in vivo response of the islets to a central glucose load showed a drastic drop of insulin release, which is explained by this downregulated parasympathetic tone.
The satiating role of glucose in DRP1-deficient animals was completely abolished. This effect was concomitant with an absence of VMH mROS production. Together with the defective response to central glucose-induced insulin secretion, these results reinforce the role of the fission protein DRP1 in VMH glucose-induced mROS signaling as well as a role in hypothalamic glucose sensing. Finally, the exploration of mitochondrial respiration highlighted changes induced by DRP1 down-expression in the VMH. The results demonstrate that the loss of mROS production in response to glucose load is due to decreased capacity of the mitochondrial electron transfer chain (ETC) to oxidize the substrates, associated with decreased coupled respiration. Indeed, the RCR (ratio between O2 flux of ADP-stimulated respiration to ATP synthase-inhibited respiration, which represents the coupling activity of the ETC) was decreased. These observations are consistent with others showing modifications of coupling activity when mitochondrial dynamics are altered (7, 40). ROS are produced by electron leakage during mitochondrial metabolism. The rate of ROS formation is enhanced as mitochondrial metabolism increases when mitochondria are well coupled (9, 37). Classical observations indicate that active mitochondria are more condensed and have an electron dense matrix, which favors mROS production (14). This suggests that physiological mitochondrial fragmentation upon glucose increase triggers contracted and condensed mitochondria. Dynamic changes of the internal structure (mitochondrial cristae and the complexes' arrangement) are tightly associated with functional respiration (26, 39). These explanations are consistent with previous observations describing decreased glucose oxidation associated with mitochondrial dynamics disruption in peripheral tissues of obese patients (3, 4). Regarding hypothalamic glucose sensing, altered mROS signaling has been shown in vivo in obese, insulin-resistant rat model (11). These animals exhibit hypersensitivity to glucose that leads to insulin secretion in response to a lower glucose bolus injection compared to lean rats. Since DRP1 appears as a crucial element in mROS signaling in response to glucose, it suggests that deregulated mitochondrial dynamics through DRP1 might be present in this model. Further investigations will determine whether DRP1 plays a role in hypothalamic glucose sensing dysfunction of obese rats. Studies on pancreatic beta-cells show that cellular alteration seen during chronic hyperglycemia is fission dependent. Molina and colleagues found that beta-cell mitochondria become fragmented and lose their ability to undergo fusion (29). These data suggest that deregulation of hypothalamic DRP1 in response to excessive nutrients exposure might be involved in impaired nutrient sensitivity in models of obesity or Type 2 diabetes.
Collectively, this work demonstrates for the first time in vivo that DRP1-dependent mitochondrial fission is essential for glucose-induced mROS signaling in hypothalamic glucose sensing. Further investigations to determine which metabolic or hormonal factors trigger changes in mitochondrial dynamics and mROS production will help us to better understand the mechanisms involved in the central control of energy homeostasis.
Methods
Animals: Diet and experimental procedures
Male Wistar rats (7-weeks-old; Charles River, Lyon, France) were individually housed in a controlled environment (12 h light/dark cycle, light on at 7:00
Cannula implantation and siRNA injection
In anesthetized animals, intracerebral double guide stainless-steel cannulae (26-gauge (Plastics One, Roanoke, VA) were inserted bilaterally into the VMH according to stereotaxic coordinates (anterior-posterior: −3.14 mm vs. bregma; medial-lateral: 0.25 mm; dorsal-ventral: 9.4 mm). Cannula location was confirmed by bromophenol blue injection after experimentation. The animals were allowed to recover for 6 days and were handled daily. On the day of the experiment, a double-stranded 21-nucleotide small interfering RNA (siRNA) obtained from GeneCust (Lux) was transduced using the JetSI system (OZYME, Saint-Quentin-en-Yvelines, France) (15) and injected (100 pM) through the cannula guide to knockdown the expression of DRP1. The siRNA sequence efficiently targeting rat DRP1 (Accession N° BC085843) corresponds to the coding region 258–278 relative to the first nucleotide of the start codon (siRNA, sense strand: 5’-AACUCAGAGCAGUGGAAAGAG-UdTdT-3’). Knockdown of DRP1 in the VMH was measured 24 h, 48 h, 72 h and 96 h post-injection.
Intracarotid injection of glucose towards the brain
Experiments were performed as previously described (10). Plasma insulin level was determined using the ultrasensitive ELISA kit (Eurobio, Courtaboeuf, France).
Mitochondrial isolation
After euthanasia by cervical dislocation, brains were quickly removed and immediately immersed in ice-cold MB+ buffer (10 mM HEPES, 210 mM mannitol, 1 mM DTT, 240 mM sucrose, protease inhibitor cocktail tablet (Roche, Meylan, France). VMH, cortex, and thalami were dissected, immersed for 15 min in MB+ buffer and homogenized with a Dounce homogenizer (7.5 μl/10 mg tissue). Homogenates were resuspended in 125 μl/10 mg tissue of MB+ buffer and centrifuged (1000 g, 10 min, 4°C). Supernatants were centrifuged (12,000 g, 10 min, 4°C). The remaining mitochondrial pellet was resuspended for Western blot analysis.
ROS level measurement
One minute after the carotid or VMH glucose injection, rats were decapitated, VMH, thalami, and cortex were harvested and frozen and stored at −80°C. ROS were assessed as previously described (11).
Western blot analysis
Proteins were separated on 10% SDS-PAGE. Antibodies against OXPHOS, Actin, DRP1, OPA1, FIS1, or MFN2 were used. After transfer and blocking, membranes were probed in 1% nonfat milk prepared in TBS-T with 1/1,000 mouse anti-OXPHOS (Mitosciences, Euromedex, Mundolsheim, France), 1/100,000 mouse anti-Actin (Millipore, Molsheim, France), 1/10,000 mouse anti-DRP1 (BD Biosciences, Le Pont-De-Claix, France), 1/500 mouse anti-OPA1 (BD Biosciences), 1/500 rabbit anti-FIS1 (Clinisciences, Montrouge, France) or 1/500 mouse anti-MFN2 (Abnova, Montluçon, France) overnight at 4°C. Specific bands of the proteins were detected using a goat anti-mouse (1/10,000 in TBST-1X) or anti-rabbit (1/10,000 in TBST-1X) peroxidase-conjugated secondary antibody (Amersham, Saclay, France) incubated for 1 h at room temperature. Bands were revealed with a chemiluminescence kit (Amersham) and were exposed to a ChemiDoc XRS+ system (BioRad, Marnes-la-coquette, France) for densitometry analysis.
O2 consumption measurement on permeabilized VMH
Oxygen consumption was measured using a respirometer (Oxygraph-2k, Oroboros Instruments, Innsbruck, Austria) as previously described (5, 11). Mitochondrial respiration was stimulated by the addition of 10 mM glutamate to achieve the state 2. Next, 1 mM ADP was added to achieve the state 3 respiration. Then, 0.5 μM carboxyatractylate (CAtr) was added to block ATP synthesis and achieve the state 4 respiration. Finally, 0.4 μM CCCP, a chemical uncoupler, was used to measure the maximal respiration.
Refeeding test in response to VMH glucose injection
After overnight fasting, glucose (0.12 M in 0.9% saline) was injected into the VMH (1 μl). Food consumption was measured 15, 30, 45, 60, 90, 120, and 240 min post-injection. For ROS measurements, brains were removed 1 min after glucose injection as described above.
RNA extraction, reverse transcription, and quantitative real-time PCR
cDNA were synthesized from total VMH RNA and amplified by PCR with UCP2 or 18S primers using the StepOnePlus Real-Time PCR System (Applied Biosystems, Villebon sur Yvette, France) [primer sequences: UCP2 forward 5’-3’:GGC-GGT-GGT-CGG-AGA-TA, reverse 5’-3’:GGC-AGA-AGT-GAA-GTG-GCA-AGG-G (19); 18S forward 5’-3’:CCA-TTC-GAA-CGT-CTG-CCC-TAT, reverse 5’-3’:GTC-ACC-CGT-CAC-CAT-G (19)].
Islet treatments and insulin measurement
Islets isolation and preincubation period were performed as previously described (14, 25). Over the preincubation period, islets were incubated in the different glucose concentrations (5.5 mM or 16.7 mM) for 30 min at 37°C. The acetylcholine receptor agonist acetyl-methylcholine bromide 0.1 mM (Sigma, Lyon, France) was assessed in 5.5 mM glucose. Incubation medium was harvested and stored at −20°C before insulin measurement. The release of insulin by the islets was measured using a radioimmunoassay kit (Cis-Bio International, Gif-sur-Yvette, France).
Immunohistochemistry
After either cerebral hyperglycemia or 72 h after VMH siRNA injection, brains were quickly frozen in isopentane, −40°C. 10 μm hypothalamic cryosections were fixed, then permeabilized for 10 min and blocked in PBS/0.5% Tween20/3% normal goat serum (Mitosciences) (38). After incubation with mouse anti-OXPHOS (1/2000 in PBS/0.5% Tween 20/3% NGS, Mitosciences) overnight at 4°C and washing, sections were incubated in goat anti-mouse alexa 488 (1/4000 in PBS/0.5% Tween20) for 1 h. Sections were mounted and examined under an Apotome fluorescence microscope (Zeiss). Specificity of labeling was ascertained with omission of primary antibody. Quantification of the number of mitochondria was performed after image acquisition. Alexa 488 fluorescent objects which correspond to mitochondria were detected using ImageJ software. The number of total objects per picture was divided by the number of total nuclei (labeled by DAPI).
Electron microscopy
Brain samples were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.2 at 4°C for 4 h, and post-fixed overnight with 1% osmic acid in 0.1 M phosphate buffer, pH 7.2. Samples were then dehydrated in acetone, stained with 2% uranyl acetate in acetone for 2 h at 4°C in a dark room, and embedded in epoxy resin (Araldite, Fluka). Ultrathin sections were contrasted with 2% lead citrate in water for 1 min, and were observed with a Hitachi 7650 transmission electron microscope.
Statistical analysis
Results are presented as mean±SEM. Statistical analysis was performed using Prism 4.0. Normality has been tested with Kolmogorov-Smirnov test. According to the result, unpaired t-test, unpaired t-test with Welch's correction (when equal variance was not assume) or Mann Whitney test (when normality was not assumed), one way ANOVA or two-way ANOVA analysis (followed by Bonferroni post test) were performed as detailed in figure legend. Differences are symbolized as *, **, or *** on graphic representations for p values of <0.05, 0.01, and 0.001, respectively.
Author Contributions
LC and CL designed and performed the experiments, interpreted data, and wrote the manuscript. CT-C and DB performed pancreatic islets experiments; CA, LC, and CB carried out histochemistry and pictures analysis; XF, CG, and GO helped on in vivo experiments. LC and EN performed RT- qPCR. BS and MR performed transmission electron microscopy. PB and LP contributed to discussion and reviewed the manuscript.
Footnotes
Acknowledgments
LC has a fellowship from the Ministere de la Recherche et de la Technologie. CL, LP, and PB have a grant from Agence Nationale de la Recherche (ANR-06-PHYSIO-Oox, “MetaDisMitoDyn”), and C.L has a grant from Alfediam (Prize Merck-Lipha 2009). We are indebted to A. Lefranc (CSGA, UMR 6265 CNRS, UMR 1324 INRA, Université de Bourgogne, 21000 Dijon, France) for assistance with the rats.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.