
Abstract
Introduction
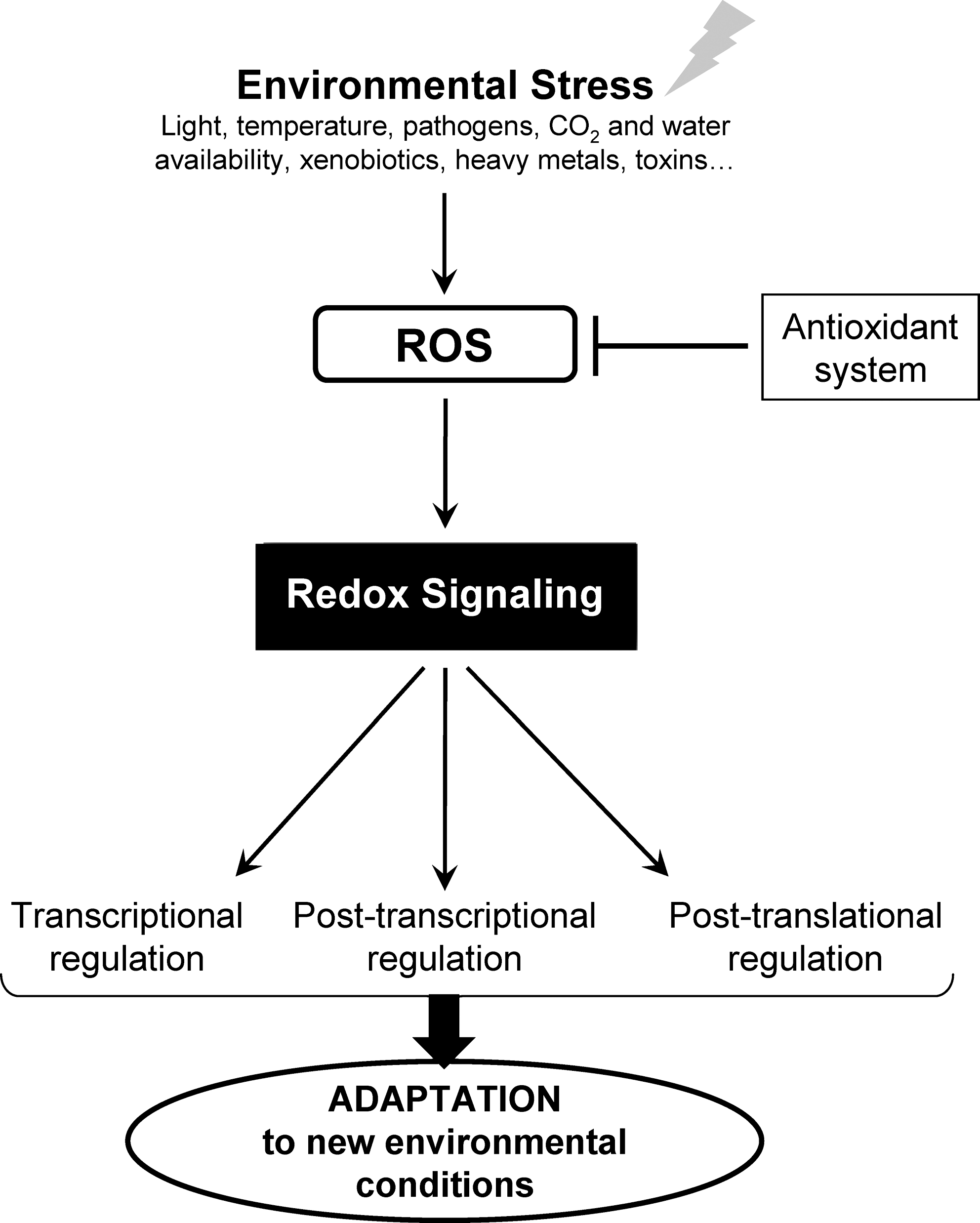
In the ROS- and RNS-dependent signaling pathways, protein thiols play a central role, as they couple the changes in the intracellular redox state to biochemical responses. In response to ROS or RNS, redox-sensitive cysteines undergo a diverse spectrum of thiol modifications, thereby modulating protein function. Protein thiol reactivity is largely determined by the cysteine's structural environment and its pK a value. Most protein thiols have pK a values >8.0, which render the thiol group predominantly protonated and largely nonreactive at intracellular pH (64). By contrast, thiol groups of redox-sensitive cysteines present much lower pK a values ranging from 3 to 6 (12). The low pK a values of these redox-sensitive cysteines may depend on different local factors (63). Acidic thiols exist as highly reactive thiolate anions (-S-) under physiological pH conditions and, in contrast to their protonated counterparts, are strongly susceptible to oxidation by ROS and RNS (Fig. 2). Reactive cysteines can be oxidized by ROS into sulfenic (–SOH), sulfinic (–SO2H), or sulfonic (–SO3H) acids. Cysteine oxidations into sulfenic acids are reversible, whereas oxidations into sulfinic and sulfonic acids are generally considered irreversible. However, recent studies have shown that in the particular case of 2-cys peroxiredoxins (PRXs), the sulfinic form of the catalytic cysteine could be reduced to the sulfenic acid form by sulfiredoxin or sestrin (11, 89, 205). Oxidative modifications of protein cysteines can also involve formation of disulfides with other protein thiol groups (intra- or intermolecular protein disulfide bonds) or with low-molecular-weight thiols such as glutathione (S-glutathionylation), cysteine (S-cysteinylation), or cysteamine (S-cysteaminylation). In addition, in the presence of RNS, cysteines can be modified by either S-nitrosylation or S-glutathionylation. Importantly, most of these redox post-translational modifications (PTMs) are fully reversible and mainly controlled by two major protein families: thioredoxins (TRXs) and glutaredoxins (GRXs), both of which play a major role in redox signaling.
In plants, many metabolic pathways are regulated by TRX-dependent dithiol/disulfide exchange reactions in redox-regulated proteins (13, 105, 127, 181). TRX dependent regulation of chloroplast enzymes in response to light/dark conditions has been known in plants for several decades (127, 181, 182, 204). Although some TRX-regulated chloroplast enzymes are activated by thiol oxidation (140), most are activated by disulfide reduction, the latter involving increased reduction state of the TRX pool mediated by light-driven reduction of ferredoxin and ferredoxin-thioredoxin reductase (FTR) (105). In chloroplasts, five major types of TRXs are present (f, m, x, y, and z) (24, 105, 106). TRX x, TRX y, and TRX z are preferential electron donors to several antioxidant enzymes, such as PRXs, methionine sulfoxide reductases (MSRs), or glutathione peroxidases (GPXs) (25, 36, 191), while f- and m-type TRXs are more specifically involved in the light-dependent regulation of chloroplast metabolism, including the Calvin cycle (105). Other plant cell compartments such as the cytosol and mitochondria also contain TRX isoforms (h and o) whose redox state is dependent on the flavoenzyme NADPH-thioredoxin reductase (NTR), which is not directly linked to light conditions. Over the last decade, numerous proteomic-based approaches led to identification of ca. 400 putative TRX targets implicated in nearly all plant cell processes [reviewed in ref. (111, 127, 133)].
Besides TRX-dependent regulation of the redox state of proteins, glutathionylation is emerging as a redox post-translational modification that may play a role in cell regulation and signaling. This modification involves glutathione and consists in the formation of a mixed disulfide bridge between an accessible free thiol on a protein and the cysteine thiol of glutathione, the most abundant (1–10 mM) low-molecular-weight thiol in most living organisms (48, 174). Due to the abundance of glutathione, glutathionylation is, by far, the major form of S-thiolation. Glutathionylation can affect the functions of numerous proteins by modifying their activity, their subcellular localization, their stability, or their interactions with partner proteins (33, 62, 127, 174, 183). Although protein glutathionylation occurs under basal conditions (34), it is particularly promoted in response to oxidative or nitrosative stress and can not only play a protective role against irreversible and deleterious oxidation of sensitive cysteine residues but also result in protein-specific functional changes. The exact mechanisms leading to protein glutathionylation in vivo remain unclear, whereas the reverse reaction, named deglutathionylation, is likely catalyzed by GRXs.
To date, this reversible post-translational modification has been mainly studied in human cells where it plays a major role in numerous fundamental processes and is implicated in a broad spectrum of human diseases, including cancer, diabetes, and several neurodegenerative, cardiovascular, muscular, or pulmonary diseases (10, 120, 129). Recently, S-glutathionylation started to be recognized as an emerging redox regulation mechanism in photosynthetic organisms, but the physiological implications of the modification have not yet been clearly established (174), and much investigation is needed to clarify the actual importance of protein S-glutathionylation in these organisms.
In this article, we provide a global overview of glutathionylation in photosynthetic organisms with particular emphasis on the most recent studies. We first rapidly present the importance of glutathione in plants and describe the mechanisms of protein glutathionylation and deglutathionylation with a particular focus on the functions and catalytic activities of GRXs. Then, we describe the methods employed for detection and identification of glutathionylated proteins in photosynthetic organisms and review the targets and the possible physiological functions of protein glutathionylation as a mechanism of protection, regulation, regeneration, and signaling. Finally, we discuss the emerging role of this modification in protein redox regulation and redox signaling in photosynthetic organisms.
Glutathione
Glutathione is a major low-molecular-weight thiol in most organisms. Glutathione is a tripeptide (γ-
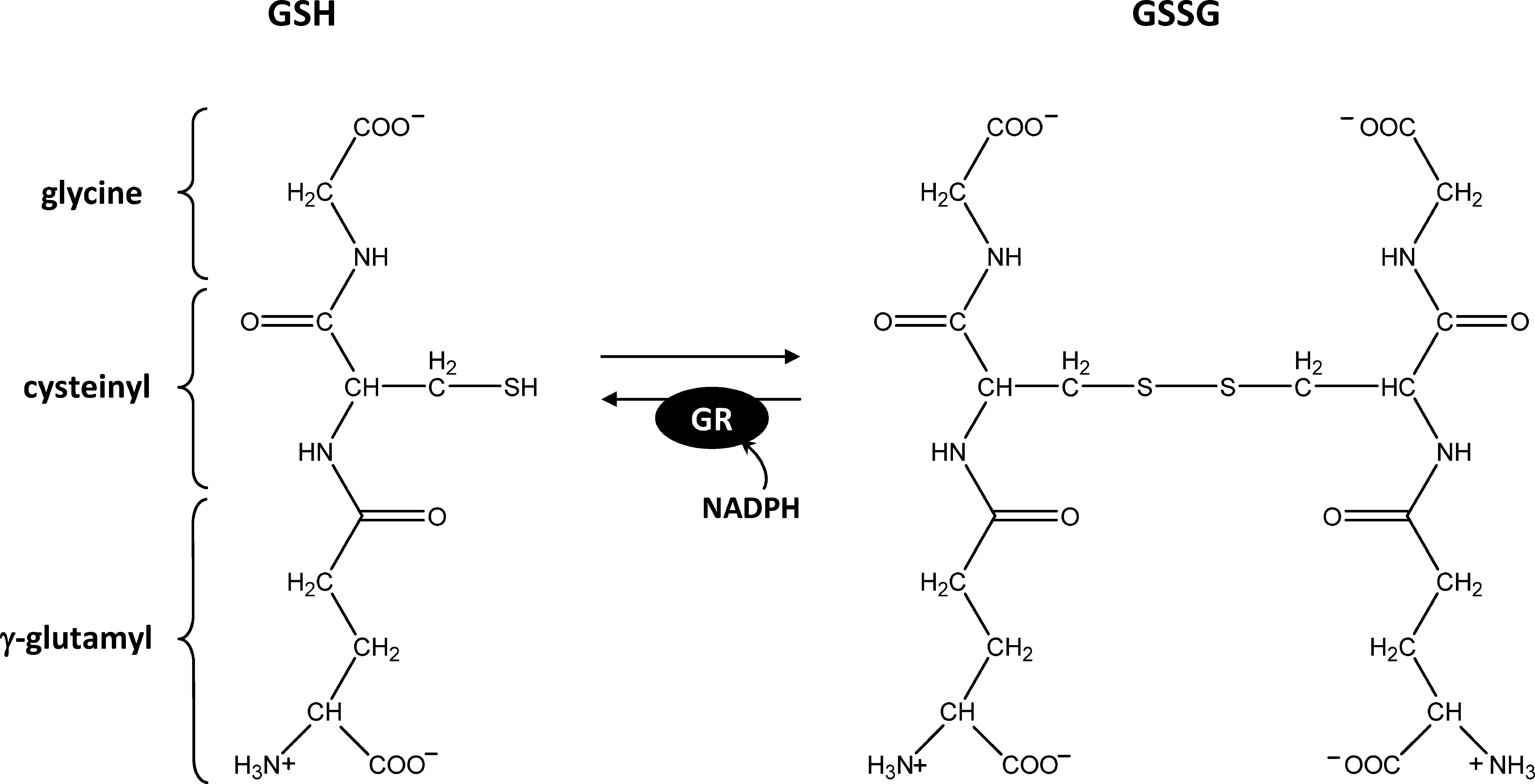
Glutathione can exist in a reduced (GSH) or oxidized (GSSG) form. GSSG is continuously regenerated into GSH by glutathione reductase (GR) using NADPH as a reductant (Fig. 3). The E m value of the couple GSH/GSSG is −240 mV at pH 7.0 (123). Since GSSG reduction leads to the formation of two GSH molecules, the actual redox potential of glutathione depends on both the total glutathione concentration and the GSH/GSSG ratio (123, 135). As a consequence, at physiological millimolar concentrations of GSH, the E m corresponds to GSH/GSSG ratios between 102 and 103. The pK a value of the cysteine residue of glutathione is between 8.6 and 9, a value higher than the pK a of 8.3 for free cysteine (92). The glutathione pool is maintained highly reduced by GR, and plant extracts usually contain ca. 5% GSSG, although the subcellular distribution of this GSSG is uncertain (49). Recently, the use of a redox sensitive green fluorescent protein (GFP) supposed to report the in vivo glutathione redox potential in the cytosol of Arabidopsis cells yielded values close to −320 mV (85, 122), suggesting a much higher GSH/GSSG ratio than reported in plant extracts, apparently around 105. Such conditions could allow glutathione to play a signaling role, as a minor increase in GSSG concentration would result in a huge variation of the glutathione redox potential [see ref. (49) for a detailed discussion].
The multiple functions of glutathione in photosynthetic organisms have been extensively reviewed recently (48, 123, 142, 148, 153, 174). As the main free soluble thiol of low molecular weight, glutathione is classically considered as constituting a redox buffer that maintains the intracellular environment in a reduced state. Glutathione plays a major role in oxidant detoxification. It can react directly with ROS and RNS or provide electrons to diverse antioxidant enzymes. GSH can not only react with nitric oxide (NO) to form nitrosoglutathione (GSNO), a molecule acting as an NO reservoir and NO donor, but can also participate in glutathionylation (see below). Glutathione also exerts a major antioxidant function through reduction of ascorbate via the ascorbate-glutathione cycle (50). Glutathione can also participate in ROS detoxification by reduction of different types of peroxidases. In mammals, GPXs containing a catalytic selenocysteine are reduced by GSH. Plants lack selenoproteins and contain cysteine-based GPXs that are rather reduced by TRXs than by GSH (83, 138). However, this is not true for all photosynthetic eukaryotes, as green algae contain several selenoproteins, including GSH-dependent GPXs (36, 146). Nevertheless, glutathione can also serve as an electron donor for some glutathione-S-transferases (GSTs) or some GRXs exhibiting GPX activities (31, 40, 102). In addition, GSH has been reported to reduce either directly or through GRXs several PRXs (47, 58, 172) and to participate in the regeneration of MSR-B1 (191). The regeneration mechanism of these enzymes most likely involves glutathionylated intermediates as detailed next. Glutathione also participates in the detoxification of xenobiotics via glutathione-S-transferases (32, 42) and the detoxification of heavy metals, as a substrate for phytochelatin biosynthesis (27).
In plants, glutathione participates in PCD and in resistance against pathogens (38, 48, 134). These roles are most likely linked to interaction of glutathione with salicylic acid (SA) and jasmonic acid (JA) signaling, including redox regulation of the transcription factors NPR1 and TGA1 mediated by GSNO-dependent nitrosylation (113, 189). Glutathione has also been implicated in other cell processes such as the cell cycle (159, 198, 200), cell differentiation (74, 75), flowering (149, 150), or accumulation of anthocyanins (207). However, for almost all these functions, the exact role of glutathione is still unclear, and the molecular mechanisms remain to be delineated. In many cases, glutathionylation could play an important role.
Mechanisms of Protein Glutathionylation
Protein glutathionylation is a dynamic process. Theoretically, several mechanisms can lead to protein glutathionylation, but the precise mechanisms occurring in vivo are not completely clarified. Although S-glutathionylation can either occur spontaneously or be catalyzed by specific enzymes, the relevance of the enzyme-dependent glutathionylation reactions remains to be established under physiological conditions (33, 55).
Thiol-disulfide exchange
One of the most widely studied mechanisms of glutathionylation involves a spontaneous thiol/disulfide exchange between a protein cysteine thiol and GSSG (Fig. 4). This mechanism implies that the glutathionylation rate is primarily influenced by the redox state of the glutathione pool. In most cells and subcellular compartments, the glutathione pool is maintained highly reduced by GR with GSH/GSSG ratios reaching 105 in Arabidopsis cytosol as measured with redox-sensitive GFP (69, 85, 90, 122). Although stress conditions have generally the effect of decreasing this ratio to some extent (50), these conditions would hardly lead to any substantial glutathionylation of proteins by thiol/disulfide exchange, because the K ox of most protein cysteines (i.e., the GSH/GSSG ratio at which 50% of the protein is in the glutathionylated form) is around 1, a very unlikely change in the intracellular GSH/GSSG ratio even under artificial oxidative stress (55, 185, 201). Therefore, although glutathionylation by thiol/disulfide exchange can lead to glutathionylation in vitro and is a useful method to study the biochemical and structural consequences of protein glutathionylation, this mechanism is, for most proteins, not likely to contribute significantly to glutathionylation in vivo. For example, during the oxidative burst in human neutrophils, a massive increase in protein glutathionylation is observed without any increase in GSSG concentration (17). Some exceptions to these general considerations may, however, exist. Indeed, some human proteins, such as the c-Jun transcription factor (96) or caspase 3 (82), appear to undergo GSSG-mediated glutathionylation at physiological GSH/GSSG ratios and GSSG concentrations. Interestingly, a particular type of chloroplastic GRX of poplar named GRXS12 has a very stable mixed disulfide (30) and a K ox of 309 at pH 7.0, suggesting that it might be glutathionylated in vivo by thiol/disulfide exchange at GSH/GSSG ratios that could occur under physiological stress conditions (212).
Activated thiols
The mechanisms of glutathionylation that are more likely to occur in vivo involve activated thiol derivatives such as sulfenic acids (–SOH), S-nitrosylated thiols (–SNO), thiyl radicals (–S•), or thiosulfinates (–S(O)SR).
Sulfenic acids
Protein glutathionylation can be mediated by a two-electron oxidation of the protein thiol to sulfenic acid, followed by a reaction with GSH (Fig. 4). Protein sulfenates are the major products formed by thiols on contact with nonradical oxygen-derived species such as H2O2 and ONOO−. In principle, GSH could also be converted to glutathione sulfenate (GSOH) and then react with protein thiols, but the low acidity of GSH thiol (pK a 8.6–9) is likely to limit this reaction in vivo. With the exception of several ROS scavenging enzymes such as PRXs, GPXs, NADH, peroxidases, or MSRs, which are able to stabilize sulfenic acids within their active site (157), sulfenic acids are unstable molecules that are either rapidly converted by further oxidations to sulfinic (–SO2H) or sulfonic (–SO3H) acids or react with vicinal thiols to form disulfides. In most cases, sulfinic and sulfonic acid forms are irreversible and lead to the degradation of the overoxidized proteins (161).
Glutathionylation being reversible, it could constitute a mechanism of protection of protein cysteine residues from irreversible oxidation, as demonstrated in vitro for photosynthetic glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (213). Many proteins have been suggested to be regulated by sulfenic acid formation (157, 158), but in most cases, the experiments were performed in the absence of GSH. Albeit in some cases, both the sulfenic and glutathionylated forms may be present in vivo and may have distinct consequences on protein activities, as shown for the OxyR transcription factor (94); in the cell, most sulfenic acids would be converted to glutathionylated thiols (57). This was recently shown for protein tyrosine phosphatase 1B (PTP-1B), initially suggested to be regulated by hydrogen peroxide through sulfenic acid formation (37) and later shown to undergo glutathionylation after reaction of GSH with the sulfenic form (4, 5). Similarly, the Bacillus subtilis organic peroxide sensor OhrR was initially suggested to be inactivated by sulfenic acid formation (54), but it was recently demonstrated that the sulfenic forms are converted to S-thiolated forms after a reaction with low-molecular-weight thiols (101). Therefore, it has been suggested that although protein sulfenates may be the initial form of oxidative modification for many proteins, in most cases, protein glutathionylation is expected to serve as the more stable intermediate in redox signaling (55).
Nitrosylated thiols
During nitrosative stress, S-glutathionylation can occur after a reaction of a protein nitrosothiol with reduced glutathione or, alternatively, by a reaction of GSNO with a protein thiol (Fig. 4). GSNO is a relatively stable molecule considered as an NO reservoir that can trigger both nitrosylation and glutathionylation (78). The ability of GSNO to trigger protein glutathionylation was reported, either in vivo or in vitro, for many proteins from nonphotosynthetic organisms such as c-Jun (97), GAPDH (18, 65, 131), or protein disulfide isomerase (PDI) (196). Recently, several plant proteins were found to behave similarly, including glycine decarboxylase (152) or plastidial GRXS12 (212). However, the protein properties or conditions favoring nitrosylation versus glutathionylation on reaction of protein thiols with GSNO remain largely unclear (65), and there is still little information about the alternative glutathionylation mechanism mediated by the reaction between a nitrosylated protein thiol and GSH.
Thiyl radicals and thiosulfinates
One electron oxidation of a protein thiol or GSH, for example, by the hydroxyl radical (•OH), leads to the formation of a protein thiyl radical (PS•) or a glutathione thiyl radical (GS•) that can form a radical mixed disulfide by a reaction with GSH or a protein thiol, respectively. This radical mixed disulfide will then transfer an electron to oxygen to form the superoxide anion (O2 •-), leaving a glutathionylated protein (Fig. 4). Thiyl radicals are likely to be formed under oxidative or nitrosative stress conditions (93, 100, 117) and are among the shortest lived activated thiols (203). Several proteins have been shown to undergo thiyl-mediated glutathionylation in vitro, and the reaction was suggested to be catalyzed by GRXs (56, 186).
The thiosulfinate derivative of GSH, GS(O)SG is a product of GSNO decomposition that is highly reactive with protein thiols and can lead to protein glutathionylation (protein-SSG, Fig. 4). Thiosulfinates were proposed to mediate the glutathionylation of rat brain neurogranin (81, 107), tyrosine hydroxylase (178), and matrix metalloproteinases (151).
Finally, even if thiyl radicals and thiosulfinates can trigger glutathionylation in vitro, it remains unclear whether these short-lived and highly reactive species significantly contribute to protein glutathionylation in vivo.
Other mechanisms and catalysis of protein glutathionylation
Disulfide bonds with compatible redox potentials can be reduced by GSH, leading to glutathionylation of one of the two thiols. When the disulfide bond is intramolecular, the glutathionylated form constitutes a transient form that will either lead to the fully reduced form after a reaction with a second GSH molecule or return to the disulfide form after a nucleophilic attack of the second cysteine on the glutathionylated thiol (see below for further details). However, when the disulfide bond is intermolecular and contributes significantly to the stability of the multimer, glutathionylation of one subunit by a reaction of GSH with the disulfide might lead to a dissociation of the subunits and lead to the formation of a stable glutathionylated form (not shown).
Despite the fact that glutathionylation can occur spontaneously, especially in the presence of ROS/RNS, several enzymes could also catalyze protein glutathionylation. In animal systems, GST was shown to promote the glutathionylation of the sulfenic form of 1-cys-PRX (116, 160) and possibly other protein sulfenates (193 –195). Although GRXs are the main enzymes contributing to deglutathionylation reactions in cells (detailed next), human GRX1 and GRX2 were also shown to enhance the rate of glutathionylation of several proteins in the presence of thiyl radicals (55, 56, 186). GRXs were proposed to stabilize GS• as an enzyme disulfide anion radical intermediate (GRX1-SSG•−), thereby facilitating GS-radical recombination with a protein thiyl radical (55, 186). Finally, the quiescin sulfhydryl oxidase (QSOX) was also proposed as a potential catalyst of protein glutathionylation (55). The possible role of similar enzymatic mechanisms of glutathionylation in plants is still completely undefined.
Conclusion
Overall, all the mechanisms of glutathionylation just described are possible, and most of them have been demonstrated in vitro but the one prevalent in vivo remains unknown. With regard to specificity, it is unclear what features contribute to the sensitivity of a given cysteinyl residue to protein glutathionylation though the accessibility, the reactivity, and the microenvironment of the cysteine are likely to play a major role (34).
Deglutathionylation Reactions and GRXs
Glutaredoxins
GRXs are small disulfide oxidoreductases, ubiquitous in aerobic organisms, belonging to the TRX family and bearing in their active site a characteristic four-amino acids motif with one or two reactive cysteines (CXXC/S) (104, 171). They are considered to represent the major deglutathionylating enzymes in cells. Historically, Mannervick's group first discovered a thioltransferase enzyme in mammalian cells (2). Two years later, a structurally and functionally similar enzyme of Escherichia coli was characterized by Arne Holmgren and named GRX on the basis of its GSH-dependent reduction activity of oxidized ribonucleotide reductase (79). Two decades after its discovery, GRX was described as a specific glutathionyl-mixed disulfide oxidoreductase (67). Indeed, GRXs exhibit a very high selectivity for protein-SSG substrates compared with other protein mixed disulfides such as protein-SSCys and catalyze deglutathionylation much more efficiently than other disulfide oxidoreductases such as TRX or PDI (26, 91, 147, 155, 214). Since protein glutathionylation appears to constitute an important mechanism of redox regulation, the role of GRXs as deglutathionylating enzymes may be crucial for redox signaling pathways.
GRXs were initially classified, based on their active site sequence, into two groups, dithiol (CPYC) and monothiol (CGFS) (164). A much greater diversity of GRX sequences is found in plants compared with nonphotosynthetic organisms. Approximately 30 different GRX isoforms have been identified in land plants, whereas lower photosynthetic organisms contain much less GRX genes (six in the eukaryotic green alga Chlamydomonas reinhardtii and three in the cyanobacterium Synechocystis sp. PCC6803). In land plants, three distinct classes of GRXs (CPYC, CGFS, and CC) have been defined based on phylogenetic analyses and on their active site structure (104, 168, 171) (Fig. 5). Additional classes of GRXs were also defined more recently (29, 167) but will not be further considered here, as they are restricted to specific organisms or distantly related to GRXs.
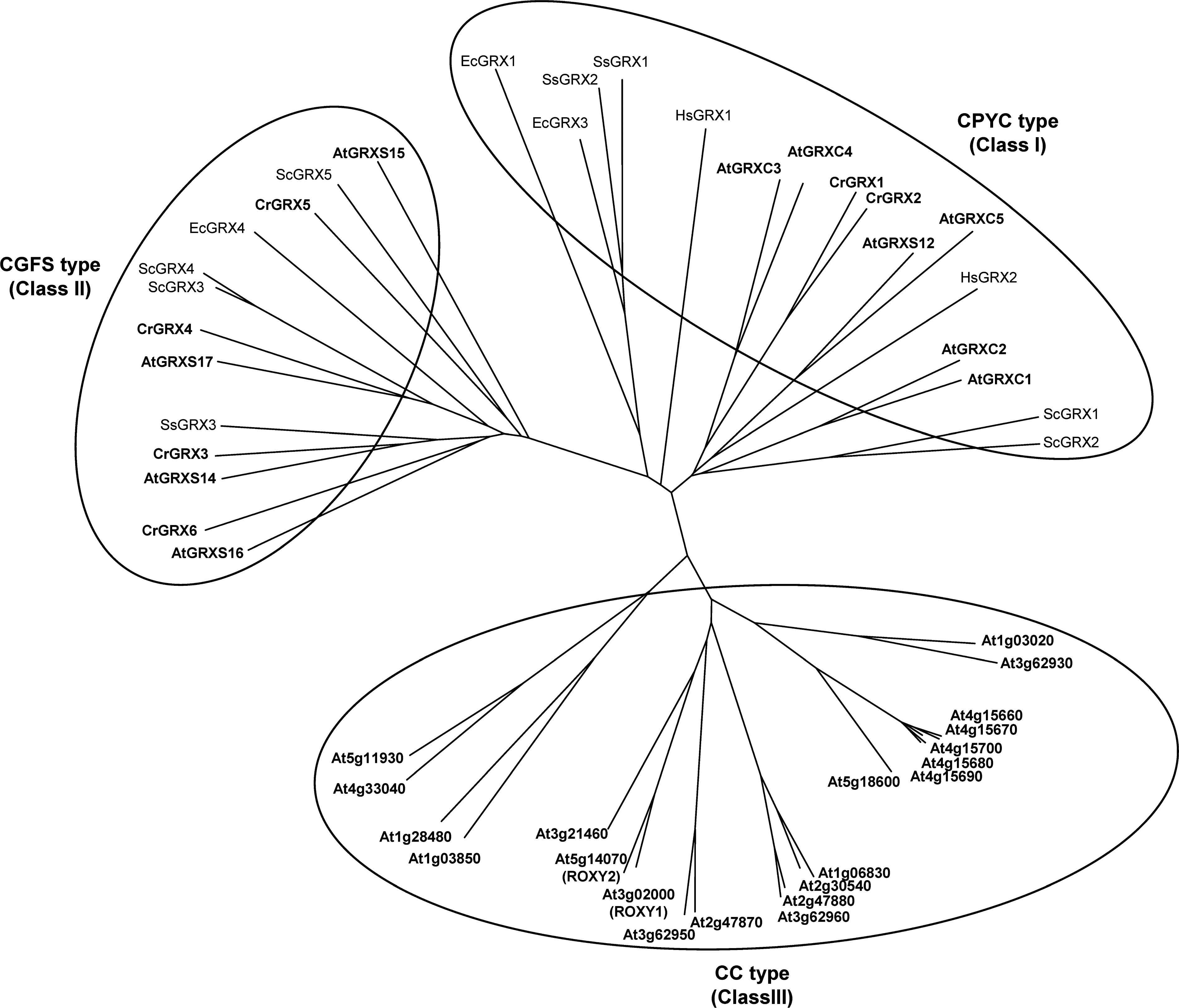
Class I (CPYC type), which includes proteins with CXXC/S active sites other than CGFS and CCXX, is the most extensively studied class and contains classical dithiol GRXs such as E. coli GRX1 and GRX3, yeast GRX1 and GRX2, and human GRX1 and GRX2. Arabidopsis contains six genes belonging to this class (Table 1). All Class I GRXs are typically reduced by glutathione and apparently catalyze both protein disulfide reduction and deglutathionylation (29, 46, 57, 60, 77, 174, 214). Class II (CGFS type), identified more recently, corresponds to proteins with a CGFS active-site. Both Arabidopsis and Chlamydomonas contain four CGFS GRXs (Table 1). Despite the presence of only one active-site cysteine, some CGFS-type GRXs have been shown to contain an intramolecular disulfide involving a partially conserved C-terminal cysteine (87, 190, 214). These proteins do not apparently catalyze disulfide reduction but were reported to catalyze protein deglutathionylation. CGFS-type GRXs are apparently not reduced by GSH but rather by NTR or FTR.
The acronyms include reference to the organism (At: Arabidopsis thaliana; Cr: Chlamydomonas reinhardtii).
Active site sequences: GRXC2: CPFC; GRXC3: CPYC; GRXC5: CSYC.
Orthologs from Arabidopsis and Chlamydomonas are grouped and supposed to have similar physiological reductant, activity, and mechanism.
With correction of exon 2 based on cDNA sequencing (insertion of KVHVFM between residues D63 and K64).
ND, not determined.
CPYC and CGFS GRXs are present in almost all genomes of aerobic prokaryotes and eukaryotes (1, 106, 109, 168, 171, 179). By contrast, the third class of GRXs (CC type), corresponding to proteins with a CCXC or CCXS active-site, is specific to land plants, as no counterparts are present in nonphotosynthetic organisms or lower photosynthetic eukaryotes (1, 106, 202, 215). With ∼20 members in most plant genomes (21 in Arabidopsis), the CC-type gene family represents the most extended subgroup. Two CC-type GRXs from Arabidopsis, designated ROXY1 and ROXY2, were recently shown to play a role in floral development (108, 136, 202, 208, 209). ROXY1 interacts with TGA transcription factors, an interaction also shown for other CC-type GRXs that were suggested to participate in JA/SA responses (139). Nevertheless, the biochemical properties of CC-type GRXs have not yet been investigated and, therefore, the molecular basis of their functions remains unknown, as well as their ability to catalyze deglutathionylation. The classification of GRXs has been described in detail in several recent reviews and is summarized in Table 1 (9, 29, 57, 76, 104, 109, 124, 127, 167, 168, 171, 174).
One of the best documented physiological functions of GRXs in plants is their role in oxidative stress responses. For example, they are directly involved in the regeneration of thiol peroxidases, PRXs and MSRs (47, 58, 102, 170, 172, 191, 199). An Arabidopsis knock-out mutant lacking CGFS GRXS14 was shown to be hypersensitive to oxidative damage (22). Overexpression of a poplar GRXC4 (class I) in E. coli was also reported to confer resistance to oxidative stress (176). In tomato, modulation of the expression of a CGFS GRX gene named SlGRX1 affected the sensitivity to oxidative and salt stresses and the tolerance to drought stress (68). More recently, Arabidopsis lines with altered expression of CGFS GRXS17 suggested an essential role for this GRX in redox homeostasis and hormone perception (23).
In addition, many studies indicate that GRXs from plants and diverse organisms including humans, yeast, and Synechocystis are able to assemble a [2Fe-2S] cluster (3, 28, 86, 88, 156, 175). In yeast, mitochondrial CGFS GRX5 appears to be required for the assembly/biogenesis of iron sulfur clusters (76, 165). Several plant GRXs are able to complement the yeast GRX5 mutant and restore iron sulfur cluster assembly/biogenesis (3, 21, 22). A poplar GRX was shown to transfer rapidly and quantitatively its bound [2Fe-2S] to apoferredoxin (3). Alternatively, it was proposed for human mitochondrial GRX2 that the binding of the cluster may be a means for temporarily inactivating the protein by sequestering active site residues, reducing conditions being able to restore the GRX activity by releasing the cluster (86). Clearly, further work is required to determine the function of iron-sulfur bound GRX.
Overall, all these studies suggested an important role for GRXs in photosynthetic organisms. Further studies, mainly based on manipulation of in vivo GRX levels, will be required in order to establish the functions of GRXs and especially the importance of their deglutathionylation activity for redox signaling networks.
Mechanisms of deglutathionylation by GRXs
Theoretically, GRXs containing two active site cysteines could catalyze the reduction of disulfide bonds on target proteins, following a TRX-like dithiol mechanism similar to that employed by TRXs. Indeed, the ability of plant GRXs to catalyze reduction of protein disulfide bonds has been proved by the reduction of the pro-substrate insulin [insulin assay; (60, 214)]. However, GRXs appear poorly efficient in these assays, especially compared with TRXs. Moreover, to date, no physiological target containing a GRX reducible disulfide has been identified in plants, either in vitro or in vivo. Therefore, the function of GRXs as protein disulfide reductases is questionable. By contrast, GRXs are known to specifically interact with the GSH moiety of proteins containing glutathione-mixed disulfides and to catalyze protein deglutathionylation very efficiently.
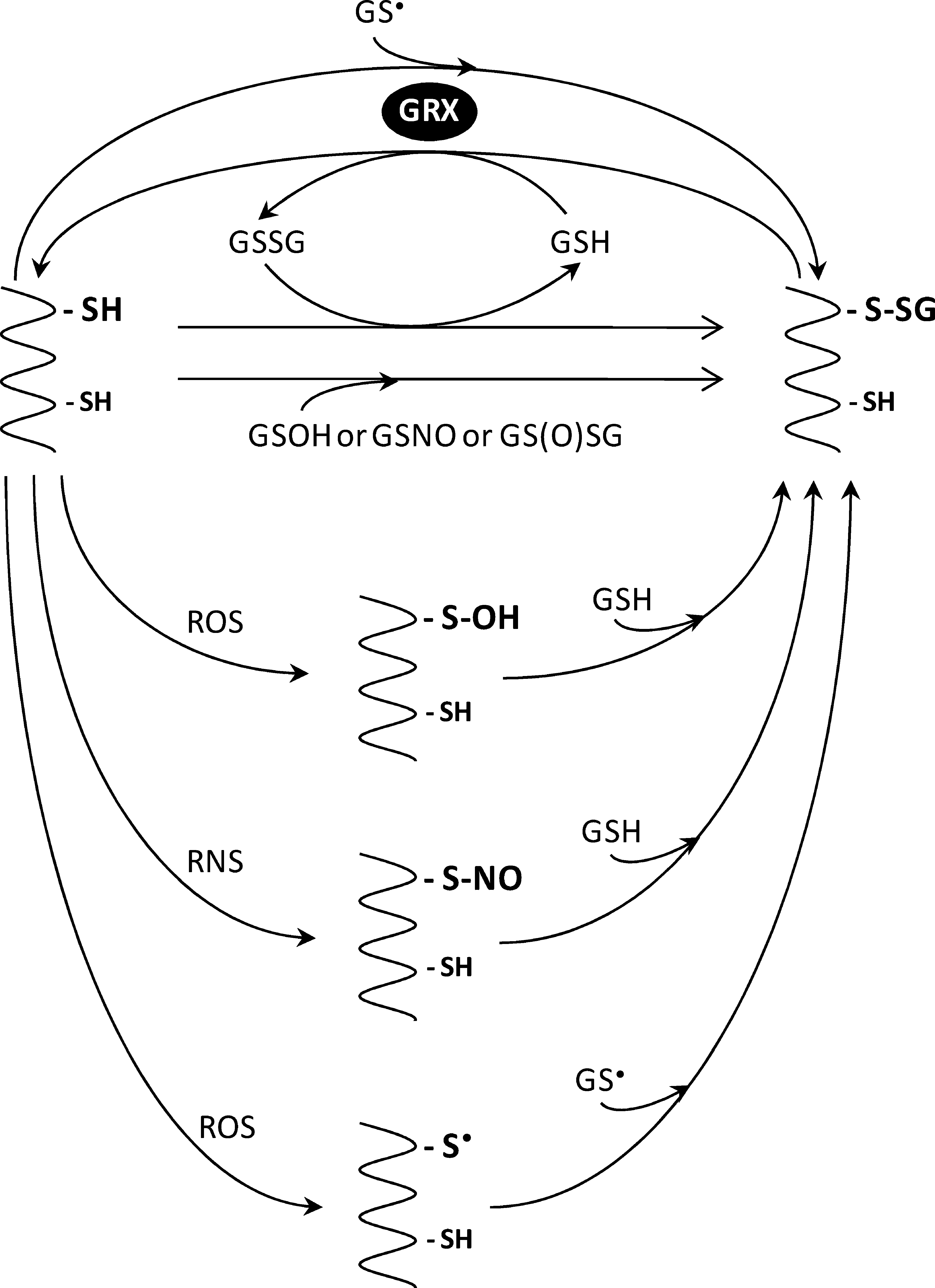
Reduced GRXs can catalyze deglutathionylation through either a monothiol or a dithiol mechanism (Fig. 6). The first step is common to both mechanisms and consists of a nucleophilic attack of the glutathione-mixed disulfide by the most reactive cysteine of GRX (the N-terminal active site cysteine) and results in the release of a deglutathionylated substrate (protein-SH) and the formation of a glutathionylated GRX intermediate (GRX-SSG). In the monothiol mechanism, the second step consists of the reduction of GRX-SSG by a second molecule of GSH to form GSSG and reduced GRX (Fig. 6A). This latter reaction usually constitutes the rate-determining step of the overall process. In the dithiol mechanism, the intermediate GRX-SSG instead of being reduced by GSH is attacked by a nearby cysteine of GRX, which, acting as a resolving cysteine, leads to the formation of oxidized GRX (containing an intramolecular disulfide) and GSH (Fig. 6B).
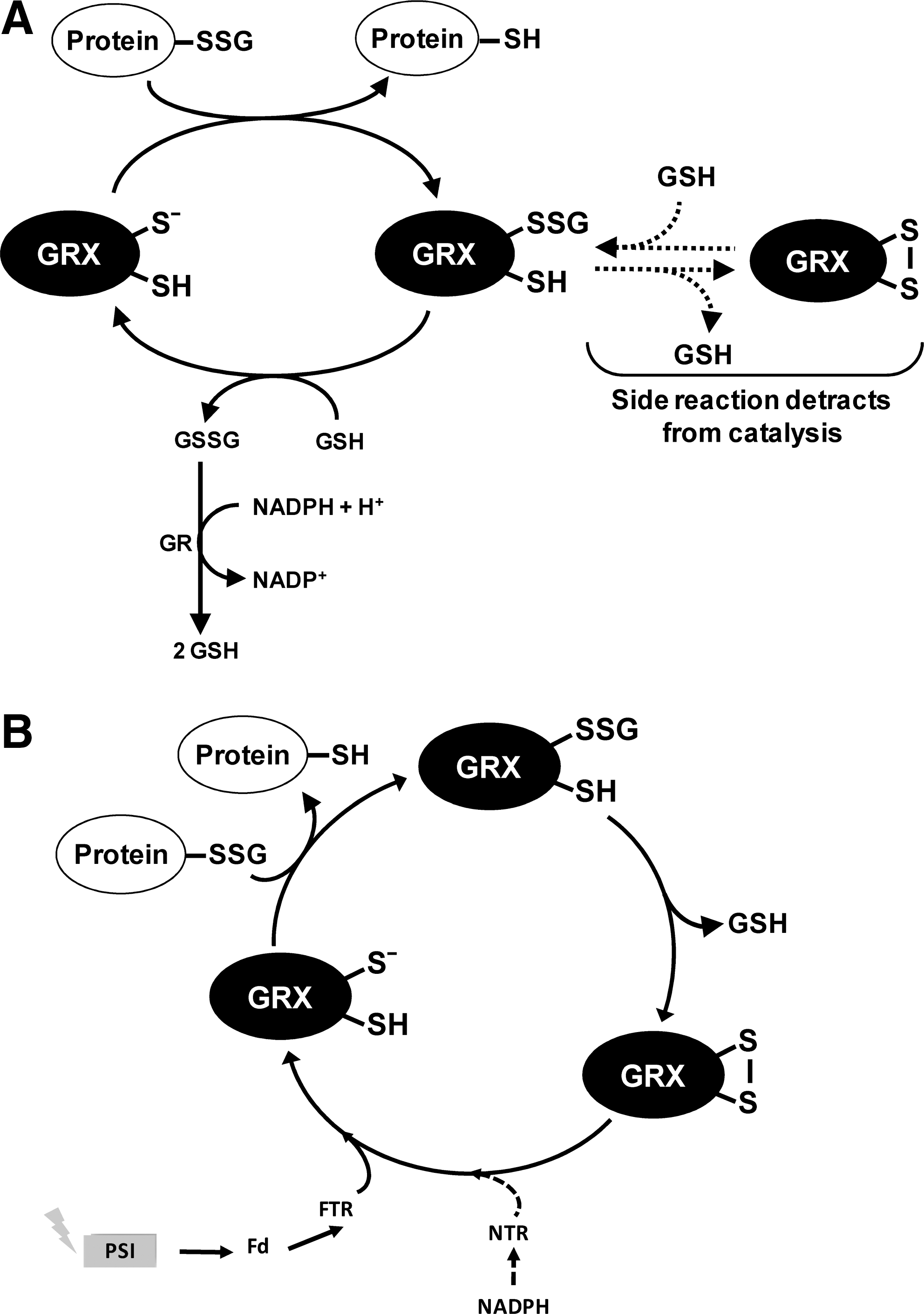
CPYC-type GRXs from human, yeast, and E. coli were found to catalyze deglutathionylation through a monothiol mechanism requiring only the most N-terminal cysteine of the GRX active site. Indeed, GRX monocysteinic mutants, where the second active site cysteine has been replaced by serine (CPYS active site), retain their ability to catalyze deglutathionylation (14, 56, 87, 155, 210). A natural monocysteinic variant such as chloroplastic GRXS12 is also an efficient catalyst of protein deglutathionylation (30).
However, due to the presence of the second active-site cysteine in most CPYC-type GRXs, a side reaction can lead to the formation of the intramolecular disulfide form of GRX from the GRX-SSG intermediate (Fig. 6A). This intramolecular disulfide can be reduced by GSH to return to GRX-SSG. Consequently, the side reaction can compete with the turnover of the GRX-SSG intermediate, thus decreasing the catalytic rate of the GRX-catalyzed deglutathionylation reaction. Quite expectedly, substitution of the C-terminal cysteine to serine increased the activity of several CPYC GRXs [e.g., human GRX1, yeast GRX1, and recently in Chlamydomonas GRX1 (39, 56, 87, 210, 211); Gao and Lemaire, unpublished results]. Examples are known, however, in which the same mutation had detrimental effects on the activity (e.g., E. coli GRX1 and GRX3, yeast GRX2, and poplar GRXC4), suggesting that the C-terminal cysteine may have a positive role in the monothiol reaction performed by the N-terminal cysteine, in addition to its essential role in the side reaction (14, 39, 145, 155, 169). E. coli GRX1 provides a possible explanation for this effect, as the second active-site cysteine of this GRX was suggested to determine the specificity for glutathione in the second step of the monothiol reaction (177). Whether similar effects might explain the inhibition observed in other CPYS mutants is still to be resolved.
Several studies revealed that plant class I GRXs can catalyze the deglutathionylation of artificial substrates such as glutathionylated β-mercaptoethanol (β-ME-SSG, hydroxyethyl disulfide assay), showing kinetic parameters similar to those determined for other GRXs from bacteria to humans (30, 60, 214). It was also observed that plant GRXs are able to catalyze the reduction of dehydroascorbate (DHA) even if the in vivo contribution of GRXs in the regeneration of ascorbate from DHA is debatable, as the turnover numbers appear much lower than the values observed for dehydroascorbate reductases (DHARs) (30, 60, 214). Plant GRXs were also found to catalyze deglutathionylation of glutathionylated plant protein substrates. Both plastidial A4-GAPDH from Arabidopsis and isocitrate lyase (ICL) from Chlamydomonas could be deglutathionylated and thereby reactivated by two classical CPYC GRXs of Chlamydomonas (GRX1, GRX2), (60, 214). These studies showed that deglutathionylation rate depended on both the nature of the glutathionylated substrate (GAPDH vs. ICL) and the GRX involved (GRX1 vs. GRX2). The latter effect was correlated with the reactivity of the catalytic cysteine of GRXs. In fact, the pK a of GRX1 (3.9) was lower than that of GRX2 (4.8), and the activity of GRX1 was consistently higher. These properties are strikingly similar to those observed in human GRX1 and GRX2 that also have about one pH unit difference in the pK a of their catalytic cysteines and show different catalytic activities (56). Further studies are required to delineate the structural and molecular determinants of the reactivation of glutathionylated proteins by GRXs and to investigate whether these differences may have a functional importance in vivo.
Among CGFS-type GRXs, Chlamydomonas GRX3 has been reported to catalyze deglutathionylation (214), most likely through a dithiol mechanism (Fig. 6B), like yeast GRX5, which belongs to the same class (190). These GRXs contain an intramolecular disulfide bond formed between the active site cysteine and a partially conserved C-terminal cysteine that is not reduced by GSH but rather by TRX reductases. For CrGRX3, the reduction of the disulfide bond occurs through a light-dependent reaction involving ferredoxin and FTR (214). However, other CGFS-type GRXs lack the ability to catalyze deglutathionylation or do not seem to form an internal disulfide (45, 88). Recently, a biochemical characterization of two CGFS-type from Chlamydomonas, CrGRX5 and CrGRX6, revealed that they are not active as deglutathionylating enzymes (60). It remains to be established what could be the physiological role of these CGFS GRXs.
Methods for Identification and Detection of Glutathionylated Proteins
A number of methods have been developed to identify and analyze proteins undergoing glutathionylation. These methods, which range from the analysis of single purified proteins in vitro to large-scale proteomic studies, allowed identification of more than 200 targets in mammalian cells (35, 53, 62, 66, 95, 174). More recently, these techniques also allowed identification of more than 200 targets in photosynthetic organisms. These approaches have been extensively described in several recent reviews (33, 53, 59). Here, we will rapidly present these techniques with a special emphasis on proteomic approaches and their application to the identification of glutathionylated proteins in photosynthetic organisms.
The most popular technique for large-scale in vivo proteomic analysis of glutathionylated proteins is based on radiolabeling of the glutathione pool in cell cultures in the presence of 35S-cysteine and protein synthesis inhibitors. Radiolabeled proteins are visualized by fluorography after separation on 2D gel. The spots disappearing in the presence of reductants such as dithiothreitol, which correspond to glutathionylated proteins, are then identified by mass spectrometry. Originally developed for human T lymphocytes (51), this method was recently employed to identify S-thiolated proteins in the unicellular green alga C. reinhardtii (128). This study allowed in vivo identification of 25 S-thiolated proteins, mainly chloroplastic and involved in diverse metabolic pathways. On the other hand, attempts to employ this method with Arabidopsis cell cultures proved unsuccessful due to low levels of radiolabeling that precluded identification of S-thiolated proteins (43). This method has numerous drawbacks, the major one being the requirement of a pretreatment with protein synthesis inhibitors that will unavoidably perturb cell physiology. Moreover, this method cannot distinguish protein-SSG from other possible types of S-thiolation (such as S-cysteinylation), it is limited by the necessity to perform 2D gels and can only be used with cell cultures, thereby preventing studies on whole plants under physiological conditions. Finally, this approach can only detect proteins undergoing glutathionylation during treatment while some proteins might be already glutathionylated under basal conditions.
More recently, methods based on biotinylated glutathione (BioGSH/BioGSSG) or the membrane permeant biotinylated reduced glutathione ethyl ester (BioGEE) have also been developed. The presence of the biotin tag allows detection of glutathionylated proteins by multiple methods including immunoblotting with antibiotin antibodies or avidin-based affinity chromatography. The latter can be coupled to mass spectrometry for identification of glutathionylated proteins. The major drawback of the methods based on biotinylated glutathione is the fact that proteins are not glutathionylated by the cellular GSH itself but by an exogenous molecule that is chemically different and is also characterized by a greater steric hindrance. The presence of the biotin tag on the glutathione molecule might perturb the function of proteins interacting with glutathione molecule and especially GRXs (Zaffagnini and Lemaire, unpublished data). Another drawback, shared by the 35S labeling method, is that proteins glutathionylated under basal conditions are not detected. Originally used in mammalian systems (187), these methods have also been applied to the analysis of protein glutathionylation in photosynthetic organisms. BioGEE allowed visualization of 22 glutathionylated proteins after H2O2 treatment of Arabidopsis cell cultures, two of which, cytosolic triose phosphate isomerase (TPI) and chloroplastic fructose-1,6-bisphosphate aldolase (FBA), were identified (84). In the presence of BioGSSG, 9 other proteins that underwent glutathionylation in vivo in Arabidopsis dark-grown cell cultures after treatment with tert-butyl hydroperoxide were identified (43). In addition, in the same study, in vitro treatment of total cell extracts from Arabidopsis cell cultures with BioGSSG followed by streptavidin agarose affinity purification and 2D gels led to the identification of 72 proteins. A similar strategy allowed identification of more than 200 proteins in C. reinhardtii and the mapping of 56 glutathionylation sites (212). Indeed, a major advantage of biotinylated glutathione is to allow mapping of targeted cysteines using peptide affinity chromatography.
Several alternative methods have been employed. Commercial antiglutathione antibodies have proved useful for analysis of individual proteins, but a lack of specificity and sensitivity precludes using such antibodies for high-throughput proteomic analyses. Methods based on GRX reduction (70, 110), GST overlay (20), or immobilized glutathione (141) have also been employed occasionally but were not used in photosynthetic organisms.
Glutathionylated Proteins in Photosynthetic Organisms
Most studies on protein glutathionylation have been performed in mammals, but this post-translational modification has also recently emerged as a mechanism of redox signaling in photosynthetic organisms. To our knowledge, the first publications reporting the glutathionylation of plant proteins appeared in 2002. The first one identified birch PR-10c protein, which belongs to the family of intracellular pathogenesis-related (PR) proteins, as a target of glutathionylation (98). The functional significance of this modification remains unclear, as glutathionylation affected a nonconserved cysteine and had no effect on the ribonuclease activity of the protein. A second early study reported that four GST isoforms from Arabidopsis thaliana could undergo glutathionylation in vitro upon GSSG treatments (40). Glutathionylation of DHAR1 and DHAR2, two cytosolic GSTs possessing DHAR and glutathione-dependent thioltransferase activities, was proposed to constitute a key intermediate in the catalytic mechanism of DHA reduction. However, two isoforms belonging to the Lambda GST family (GSTL; cytosolic GSTL1 and chloroplastic GSTL2) were also glutathionylated under similar conditions although they possess only thioltransferase activity. Proteomic analyses have also suggested that further isoforms of GSTs could be glutathionylated in vitro (43), and the glutathionylation of the catalytic cysteine of a zeta-class GST (GSTZ1) was confirmed in vitro.
As just described, PTP-1B is a well-characterized target of glutathionylation in human cells (5, 163). A soybean PTP was also found to be glutathionylated in vitro in the presence of GSSG (41). However, compared with its mammalian counterpart that readily reacts with H2O2 to form a sulfenic acid on the catalytic cysteine, the plant PTP was insensitive to H2O2 but hypersensitive to GSSG. This inactivation of soybean PTP by GSSG was proposed to constitute a mechanism of protection of the enzyme under highly oxidizing conditions.
PRXs are thiol-dependent peroxidases that can be regenerated by different electron donors, including TRXs and GRXs (36, 173). Many PRXs have been identified among glutathionylated proteins in mammals (51 –53, 110, 116, 187) but also in photosynthetic organisms. For example, the in vitro glutathionylation of the peroxidatic cysteine of a poplar type II PRX was shown to trigger the dissociation of the noncovalent homodimers into monomers (144). In vitro treatments with BioGSSG also suggested that Arabidopsis 2-cys PRXs could undergo glutathionylation (43). In Chlamydomonas, 35S-cysteine labeling experiments revealed that both cytosolic and chloroplastic 2-cys PRXs are S-thiolated in vivo (128). In vitro, the glutathionylation of Chlamydomonas chloroplastic PRX led to a dimer/monomer switch. Glutathionylation could, thus, constitute a mechanism of regulation of the oligomerization/activity of PRXs (128, 144). Alternatively, glutathionylation might be an intermediate in the mechanism of reactivation of some PRXs, as previously observed for mammalian 1-cys PRX, which is converted to the glutathionylated form by interaction with GSTπ (116, 160). In the case of MSR-B1, the sulfenic acid formed on the catalytic cysteine after reduction of methionine sulfoxide reacts with GSH to give rise to a glutathionylated cysteine that can be reduced by GRX (191, 192).
Human TRX was shown to undergo glutathionylation in vivo, on an extra cysteine, distinct from the two active site cysteines (16, 51). The glutathionylation of human TRX appears to partially inhibit its oxidoreductase activity, probably by decreasing its reduction by NTR. These results on human TRX have prompted several groups to examine the ability of plant TRXs to undergo glutathionylation in vitro. An extra active site cysteine of poplar mitochondrial TRX h was shown to undergo glutathionylation in vitro with a concomitant destabilization of the active site disulfide that might perturb the function of this TRX (61). The analysis of all chloroplastic TRXs from Arabidopsis and Chlamydomonas revealed that only f-type TRXs could undergo glutathionylation in vitro (126). The glutathionylated residue is an extra cysteine, distinct from the two active-site cysteines, which is conserved in all f-type TRXs and is localized in the three-dimensional structure of the enzyme, close to the active site. The glutathionylation of TRX f strongly decreased its ability to activate A2B2-GAPDH and NADP-malate dehydrogenase likely by perturbing the interaction with FTR, as glutathionylated TRX f is less efficiently reduced in the light. This suggests that glutathionylation could affect all TRX f targets, which include many enzymes involved in carbon fixation by the Calvin cycle and other chloroplast metabolic pathways.
Several Calvin cycle enzymes were also reported to undergo glutathionylation in vivo: FBA was identified in Arabidopsis (84) but also phosphoglycerate kinase and ribose-5-phosphate isomerase in Chlamydomonas (128). However, the effect of glutathionylation on the activity of these enzymes is not yet known. The glutathionylation of the two higher plant GAPDH isoforms, the heterotetrameric A2B2-GAPDH and the homotetrameric A4-GAPDH, that participate in the Calvin cycle was also investigated (213). The NADPH-dependent activity of the major isoform, A2B2-GAPDH, is specifically light regulated by TRX f, while the A4 isoform is not (118). Arabidopsis A4-GAPDH is glutathionylated in vitro on its catalytic cysteine with a concomitant loss of enzyme activity. The enzyme is very sensitive to oxidants and is rapidly and irreversibly inactivated by H2O2. However, incubation of the enzyme with H2O2 in the presence of GSH leads to glutathionylation, most likely through a mechanism involving a sulfenic acid intermediate. Therefore, glutathionylation efficiently protects A4-GAPDH from irreversible oxidation, and glutathionylated A4-GAPDH was reported to be efficiently reactivated by GRXs (214). By contrast, the A2B2-GAPDH isoform and its higher oligomeric state A8B8-GAPDH were not found to undergo glutathionylation in vitro. Consequently, under conditions leading to protein glutathionylation in chloroplasts, the activity of both types of GAPDH is likely to be decreased; A4-GAPDH would be directly glutathionylated on its catalytic cysteine, whereas A2B2-GAPDH would be less efficiently activated by glutathionylated TRX f. More generally, the glutathionylation of TRX f and of several Calvin cycle enzymes suggest that this post-translational modification could constitute a new mechanism of regulation of photosynthetic metabolism. Such a mechanism could allow a fine tuning of the Calvin cycle allowing redistribution of the reducing power within chloroplasts under oxidative stress, thereby favoring ROS scavenging (105, 126).
In Arabidopsis, cytosolic GAPDH isoforms were also identified among glutathionylated proteins in vivo (43) and shown to be inactivated by glutathionylation of their catalytic cysteine in vitro (80). Similarly, cytosolic TPI was found to be glutathionylated in vivo, and this modification inactivates purified TPI in vitro (84). These results suggest that, as suggested in nonphotosynthetic organisms (184), glycolysis might be regulated by glutathionylation in plants.
Several protein chaperones were reported to be glutathionylated not only in vivo and in vitro in mammals but also in Arabidopsis and Chlamydomonas (43, 128). Among these, Chlamydomonas chloroplastic heat shock protein 70B (HSP70B) could be glutathionylated in vitro on a partially conserved cysteine residue located in the ATPase domain of the protein. This suggests that glutathionylation could affect the activity of this HSP70 isoform which has been implicated in the folding of chloroplast proteins and the protection of photosystem II under high light illumination (114, 180).
In photosynthetic organisms, the glutathionylation of a few additional enzymes involved in diverse pathways was also reported. Glutathionylation was shown to reversibly inactivate and to protect from irreversible oxidation Chlamydomonas ICL, a key enzyme of acetate assimilation (7, 128). An in vitro study also indicated that galactonolactone dehydrogenase, which catalyzes the terminal step of
Proteomic analyses allowed identification, either in vivo or in vitro, of many additional potential targets of glutathionylation in Arabidopsis and Chlamydomonas (43, 128, 212). These proteins are involved in diverse processes and metabolic pathways, including photosynthesis, oxidative stress responses, protein folding, amino-acid biosynthesis, lipid metabolism, translation, ATP metabolism, and cytoskeletal arrangements. Several of these proteins and processes were previously suggested to be regulated by glutathionylation in mammals. However, all these candidates await further characterization to determine the targeted residues, the effect of glutathionylation on their activity, and the functional importance of glutathionylation for the regulation of the corresponding cellular processes.
All these data strongly suggest that protein glutathionylation may play an important role in photosynthetic organisms under stress conditions. To summarize, glutathionylation appears to fulfill 3 main functions (Fig. 7): (1) Protection and/or regulation of proteins containing reactive cysteines such as GAPDH; (2) transduction of redox signals as observed for TRX f, and (3) regeneration of antioxidant enzymes by reduction of sulfenic acids in combination with GRXs, as in the case of MSR-B1, thereby reinforcing the importance of glutathionylation under oxidative stress conditions.

Multiple Interconnections
Emerging evidence suggest the existence of multiple interconnections between different types of post-translational redox modifications under the control of TRXs and GRXs. GRXs appear specifically involved in the control of glutathionylation, whereas TRXs, which are well established to regulate the oxido-reduction state of protein disulfide bonds, were recently suggested to catalyze denitrosylation reactions (8, 189). TRXs and GRXs were also reported to undergo both nitrosylation and glutathionylation on extra active-site cysteines that lead to reversible inactivation of their redox activities (16, 72, 73, 126). Moreover, the nitrosylated forms of several proteins, including human TRX1 and GAPDH, were found to function as trans-nitrosylating agents for different target proteins (99, 130, 137, 154, 206). These additional functions observed for human TRX and GAPDH might also occur for plant counterparts but remain to be established.
The reduction systems of TRXs and GRXs are also interconnected. A partial redundancy between the two redox systems has been observed in E. coli and S. cerevisiae (15, 197). A similar redundancy between TRX and glutathione was recently demonstrated in Arabidopsis (6, 121, 162), although some specificities could also be identified, such as for daylength-dependent redox signaling (125). Overall, these results revealed that NTR/TRX system and GSH redox system are strongly connected mainly via backup systems that allow a fine control of the redox state of the two major actors of these redox systems, that is, TRX and GSH.
In photosynthetic organisms, many pathways have been shown to be regulated by multiple redox PTMs, the Calvin cycle being a good example. Five enzymes of this pathway and two regulatory proteins of this pathway (Rubisco activase and CP12) are established targets of TRXs, but all Calvin cycle enzymes were identified among putative TRX targets eluted from monocysteinic TRX affinity columns (105, 111, 127). In addition, as just detailed, glutathionylation is also likely regulating the activity of Calvin cycle enzymes, either directly or indirectly through glutathionylation of f-type TRX. While the former is a light-dependent mechanism based on disulfide/dithiol exchange reactions controlled by TRX, the latter would constitute an alternative mechanism of regulation of this pathway occurring under light conditions and dependent on ROS production and glutathione. Therefore, it has been proposed that glutathionylation could constitute a new mechanism of regulation of photosynthetic metabolism allowing a fine tuning of the Calvin cycle in order to redistribute reducing power (NAPDH) and energy (ATP) within chloroplasts under oxidative stress, thereby favoring ROS scavenging (105, 126).
There is also a strong interplay between glutathionylation and nitrosylation (120), as recently illustrated by the demonstration that the activity of endothelial nitric oxide synthase is regulated by glutathionylation (19). Interestingly, several Calvin cycle enzymes were also reported to undergo nitrosylation in Arabidopsis (112, 166) suggesting the existence of an intricate and complex system for regulation of this pathway. GSNO is another element of this interplay, as this molecule is able to function as either a nitrosylating or a glutathionylating agent (as just described). GSNO treatment has been shown to induce both modifications but appears to specifically induce either nitrosylation or glutathionylation of a target protein thiol. In numerous organisms, the content of GSNO appears to be controlled by a specific enzyme named GSNO reductase, which uses NADH as an electron donor (115). Recently, Arabidopsis plants lacking GSNO reductase were found to contain a higher amount of protein nitrosothiols, indicating that this enzyme plays an important role in the control of nitrosothiol content (44). However, it is not known whether the absence of this enzyme also leads to an increase concentration of glutathionylated protein. Further studies are indeed required to determine the function and specificity of GSNO as thiol modifying agent.
Overall, the complex interplay between TRXs, GRXs, and the post-translational redox modifications under their control has only recently emerged. Efforts aimed at understanding these complex interconnections will have to be pursued to clarify the respective specific functions and redundancies of the different components of the redox signaling network. Unraveling the importance of this crosstalk will certainly constitute a major challenge for future studies.