
Abstract
Introduction
Free radicals and reactive oxygen species (ROS) play a major role in normal biological functions. They include the bactericidal action of neutrophils and macrophages, cellular signal transduction, and activation of transcriptional factors, and they are formed as intermediates in many enzyme-catalyzed reactions. However, they can also react rapidly with biomacromolecules such as nucleic acids, lipids, and proteins and lead to the loss/change of their biological functions. Therefore, they have been associated with the etiology and/or progression of a number of diseases and with aging (for reviews, see [6, 101, 123, 127]). Free radicals and ROS are formed as unavoidable byproducts of normal electron transport processes and as products of reactions between metabolites, such as hydrogen peroxide or alkyl peroxides and trace amounts of iron or copper. Thus, it is necessary for a functionally healthy cell to maintain an elegant regulatory mechanism to prevent the accumulation of harmful concentrations of ROS yet allow the accumulation of ROS to mediate their diverse physiological functions. To this end, cells rely on several enzymes including peroxiredoxins (Prxs), a family of ubiquitously expressed heme- and selenium-free thiol-specific peroxidases. These enzymes possess multiple functions in stress protection as antioxidants, molecular chaperones, and regulators of signal transduction. Prxs appear to play a role in maintaining a transient and cellular location specific environment with an elevated level of ROS for signaling activation of cascades, as well as in eliminating ROS from inducing cytotoxicity.
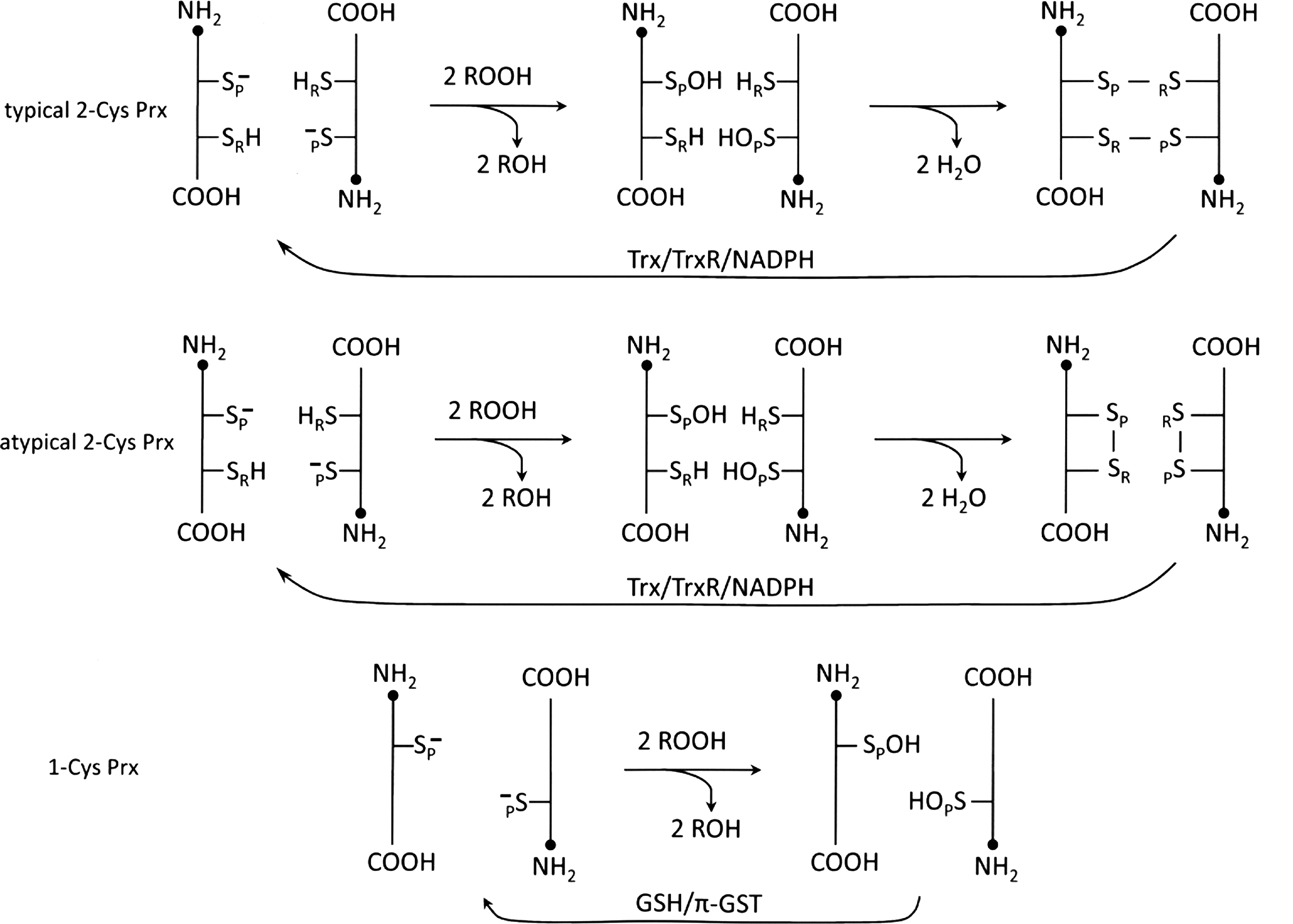
Prxs, found in all kingdoms with multiple isoforms, are among the most abundant proteins in most cells. This family of antioxidants was initially identified, purified, and characterized as “protector” proteins in Saccharomyces cerevisiae (62, 63). Without knowing its function, one member of this family, “torin,” was isolated from red blood cells early on and later identified as Prx II (46). The correct annotation for the first time was “thioredoxin peroxidase”–reducing peroxides with the use of thioredoxin (Trx) (9), and later “peroxiredoxin”—reflecting an expansion of family members using other than Trx as an electron donor such as GSH, tryparedoxin, and the 57-kDa component of alkylhydroperoxide reductase (AhpF) (12, 53, 85, 103). Prxs are not confined to cytosol. They are also found in mitochondria, chloroplasts, nuclei, peroxisomes, and extracellular regions (28, 40, 41, 69, 103, 105, 121, 126, 136). This family of peroxidases exerts their antioxidant function by reducing H2O2, organic hydroperoxides and peroxynitrite using intracellular reducing equivalents containing thiol (48, 104). Based on sequence and structural homology, Prxs have been proposed to comprise six subfamilies, namely, Prx A, B, C, D, E, and F (29). However, another classification, based on the structure, number, and location of conserved Cys residues and their contribution to peroxidase catalysis, is more commonly accepted and is preferentially applied to mammalian Prxs. In this classification, Prxs are grouped into three subfamilies (typical 2-Cys, atypical 2-Cys, and 1-Cys) (Fig. 1) (103, 105, 136). Mammalian cells express six Prx isoforms (Prx I–VI), but all isoforms are not necessarily expressed in one cell at the same time. The typical 2-Cys Prx subfamily consists of Prx I–IV. These four 2-Cys Prxs share >70% amino acid sequence identity (103), and they possess both conserved N- and C-terminal Cys residues. The conserved N-terminal low pKa Cys is referred to as the peroxidatic Cys (SP) while the conserved cysteine residue at the C-terminal region is the resolving cysteine (SR). Most Prx proteins with the SP in reduced state form a head-to-tail homodimer, which in turn serves as a building block for a doughnut-shaped decamer, composed of five homodimer pairs (Fig. 2) (115, 134). During peroxidase catalysis, the peroxidatic Cys is oxidized by either H2O2, organic peroxide (ROOH), or peroxinitrite (ONOO



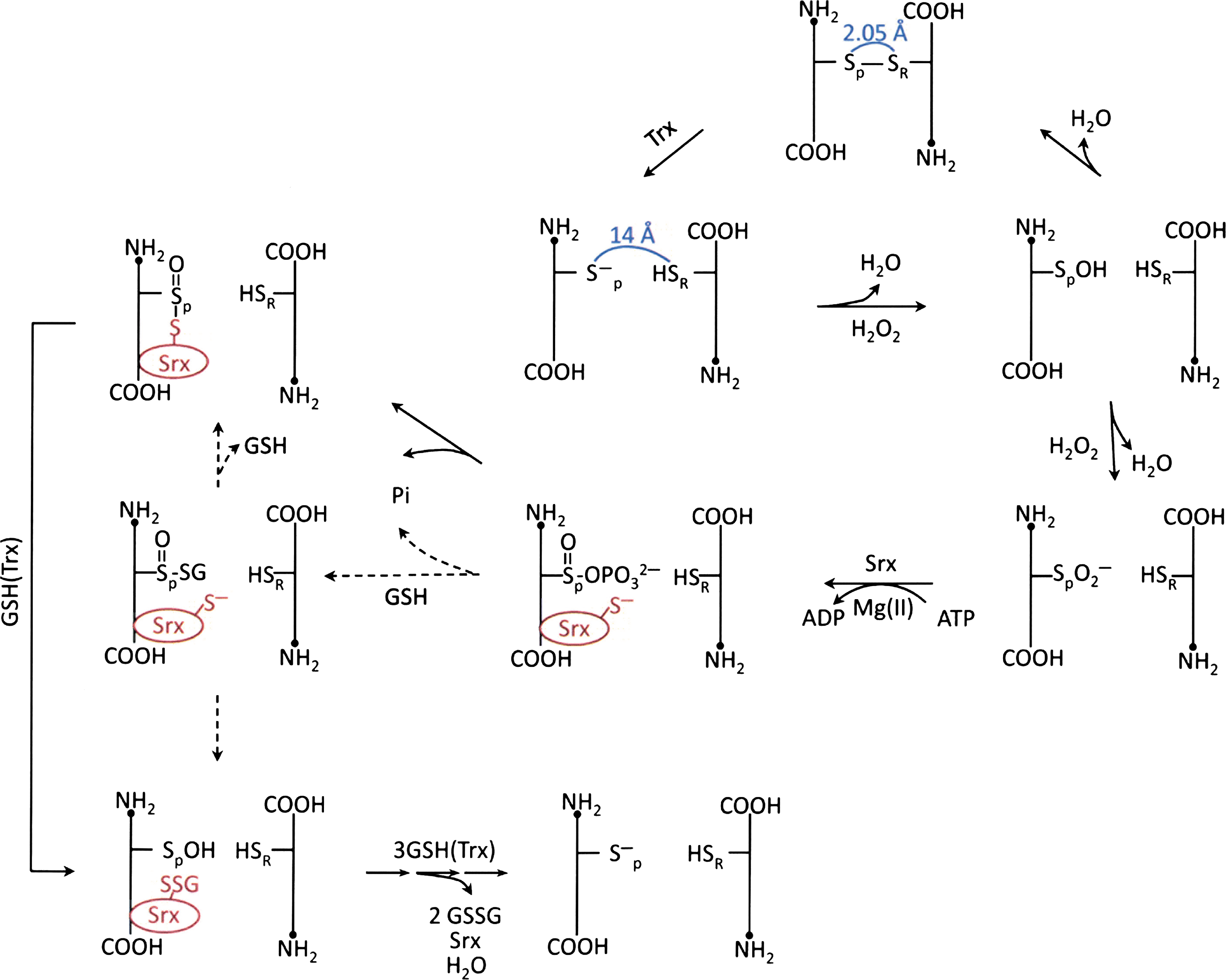
During the reduction of H2O2 or ROOH, the oxidation of the peroxidatic cysteine in Prxs may not stop at its sulfenic acid state. When the sulfenic intermediate is further oxidized to form a sulfinic acid (Cys-SO2H) derivative, or oxidized to an irreversible sulfonic acid (Cys-SO3H) derivative, Prx loses its peroxidase activity (98, 138). However, in the case of the typical 2-Cys Prxs, their sulfinic derivatives can be reduced to sulfenic acid, catalyzed by the sulfiredoxin (Srx), conserved only among eukaryotes, in the presence of MgATP (7, 131, 132) (Fig. 5). This unique Srx-mediated reactivation of the inactive sulfinic derivative of Prx in eukaryotes would constitute a mechanism for cells to accumulate sufficient quantities of H2O2 to induce signal transduction at a specific cellular location in a time-dependent manner. This notion is sometimes referred to as a floodgate hypothesis (102, 135). In addition, when typical 2-Cys Prxs are hyperoxidized, they form higher-order oligomeric structures. These high molecular weight complexes have been shown to gain a molecular chaperone activity (55, 72). The chaperone activity is capable of protecting protein substrates from thermally induced aggregation, resembling the heat shock proteins that also form well-ordered oligomers. Thus, the cellular protective effect of Prx is not limited to the removal of H2O2 or ROOH; it also directly protects proteins from irreversible denaturation.
Protein Glutathionylation of 2-Cys Prxs
Using proteomic analysis, Prx I in human T lymphocytes and atypical 2-Cys Prx, Prx V, in rat hepatocytes and human hepatoma cells were found to undergo glutathionylation when the cells were treated with a millimolar concentration range of H2O2 or with 1 mM of diamide, which generated a significantly higher level of glutathionylated proteins (37, 38). However, since diamide, a strong oxidant that promotes mixed disulfide formation with thiols, such as GSH, that would not occur under natural conditions (66), the Prx glutathionylation induced by diamide may not be physiologically relevant. Recently, it was reported that Prx I was glutathionylated in A549 (human lung carcinoma) and HeLa cells after treatment with micromolar concentration of H2O2 (88). To identify the potential glutathionylation sites, purified recombinant human Prx I or its C83S/C173S, C52S/C173S, and C173S/C83S mutants were treated with GSSG in Tris buffer at pH 7.4 and incubated for 18 h at 4°C. The glutathionylated Prx I products were analyzed using reverse phase high-pressure liquid chromatography coupled with an electrospray mass spectrometric method. These in vitro studies revealed that among the four cysteine residues found in Prx I, three of them, including the peroxidatic (Cys52) and the resolving (Cys173) cysteine, and Cys83 were found glutathionylated. Structure studies of 2-Cys Prxs have revealed that the peroxidatic Cys is located ∼14 Å apart from the resolving Cys in the homodimer of Prx and that Cys83 is located at the dimer–dimer interface (47, 115). The potential physiological function of Cys83 glutathionylation is discussed below. Glutathionylated Prxs have also been observed as reaction intermediates in the catalytic mechanism of peroxidase activity of human 1-Cys Prx VI (75), 2-Cys Prxs from Schistosoma mansoni (113), poplar Prx, a homolog of mammalian Prx V (30, 92, 109), as well as rice Grx (OsGrx) (68).
Deglutathionylation of protein-GSH mixed disulfides (protein-SSG) is known to be efficiently catalyzed by the thio-disulfide oxidoreductase, Grx. The catalytic action of Grx accounts for most of the deglutathionylating activity observed in mammalian cells (23, 59). Among the Grx isozymes, the mammalian cytosolic Grx I is the most thoroughly characterized deglutathionylating enzyme. It catalyzes the deglutathionylation of multiple protein substrates. However, the efficiency of Grx I–mediated deglutathionylation varies ∼100-fold among protein-SSG substrates studied so far (80). This suggests that substrate recognition plays an important role in cellular redox regulation. In addition to Grx I, other enzymes may also participate in catalyzing the deglutathionylation of some specific glutathionylated proteins under physiological conditions. In fact, cultured cells overexpressing human Srx have been shown to have diminished protein glutathionylation levels induced by a diazeniumdiolate, PABA/NO (33). In addition, purified recombinant Srx protein, known to catalyze the reduction of cysteine sulfinic derivative (see next section for detail), has been shown to reduce the levels of the PABA/NO/GSH-induced glutathionylated actin and protein tyrosine phosphatase 1B (PTP1B) (33). In view of the fact that protein glutathionylation is a sensitive response to the increase in H2O2 levels, it would not be surprising that reversible glutathionylation of Prx could be involved in Prx-mediated signal transduction and physiological redox signaling because these events are associated with a specific local generation of ROS.
Role of Glutathionylation in Srx-Catalyzed Reduction of Sulfinic Acid Derivative of 2-Cys Prx
Sulfiredoxin, Srx, was discovered as an enzyme that catalyzes the reduction of the cysteine sulfinic acid derivative of typical 2-Cys Prx (7, 131). However, Srx has also been shown to exhibit glutathionyl mixed disulfide oxidoreductase activity (33, 88). Using glutathionylated Prx I as substrate, steady state kinetic analysis showed that the deglutathionylation of Cys173 and Cys83 were preferentially catalyzed by Srx, whereas Grx I was more favorable for deglutathionylating Cys52. Furthermore, site-directed mutagenesis at the C-terminal tail of Prx I, a potential interacting site between Prx I and Srx, coupled with binding and deglutathionylation activity studies revealed that Srx is a specific deglutathionylating enzyme for Prx I due to its favorable affinity for the typical 2-Cys Prxs (58, 88). Mechanistic studies revealed that Srx, glutathionylated at Cys99, is an intermediate for the deglutathionylation catalyzed by Srx (88). This more recent finding is inconsistent but more plausible than a previous report (33), which indicated that Cys99 was not glutathionylated even though it was required for the deglutathionylation reaction. Experimental evidence in support of glutathionylated Srx as a reactive intermediate for the Srx-catalyzed deglutathionylation of Prx I includes products analysis of an on-going Srx-catalyzed deglutathionylation of the glutathionylated Prx I (C83S/C173S) mutant. The results showed the formation of the glutathionylated Srx at Cys99 and the deglutathionylated Prx I (C83S/C173S) as well as the unreacted Srx and glutahionylated Prx I (C83S/C173S) (88). These findings are consistent with the observation that Cys99 of Srx, exhibiting a pKa of 7.3 (14), is easily glutathionylated and rapidly deglutathionylated by GSH (88). The physiological relevance of Srx catalyzing the deglutathionylation of typical 2-Cys Prxs was demonstrated by the effect of Srx in regulating the level of glutathionylated Prx I induced by 10 μM of H2O2 in A549 cells. The results from these experiments showed that the level of H2O2-induced glutathionylated Prx I was substantially elevated in small interfering RNA-mediated Srx-knocked down cells, whereas the reverse was observed in Srx-overexpressed cells. However, glutathionylation of Prx V, not known to bind to Srx, was not affected by the change in Srx expression levels (88).
The findings that Cys52 of Prx I and Cys99 of Srx can easily undergo glutathionylation/deglutathionylation suggest that protein glutathionylation could be involved in the mechanism by which Srx-catalyzes the reduction of sulfinic acid derivative of typical 2-Cys Prxs (see Fig. 5). Crystallographic analysis reveals that the hyperoxidized form of 2-Cys Prx exists as a stable decameric structure with each active site buried, and its peroxidatic and resolving cysteine are located ∼14 Å apart. With this structure, the sulfinic moiety of the peroxidatic cysteine is not accessible to the active site of Srx because of the restriction imposed by the YF and GGLG motifs in Prx (115). Therefore the catalytic mechanism must involve a Srx-induced structural change in 2-Cys Prx. The X-ray crystallographic structural analysis of the Srx-Prx complex revealed that binding of ATP-Srx complex to the sulfinic derivative of Prx II indeed induces the unfolding of the Prx II active site such that its Cys51–SO2
Glutathionylation Regulates the Oligomerization and Catalytic Activities of Prx I
Structural studies reveal that 2-Cys Prxs exist in equilibrium between the head-to-tail homodimers and decamer (115, 136). Hyperoxidation of the peroxidatic cysteine would shift the equilibrium toward the formation of decamers or higher molecular weight complexes (55, 82). The structural change from lower molecular weight oligomers, including dimers, to higher molecular weight complexes is accompanied with a change of enzymic function, from peroxidase to molecular chaperone (55). On one hand, shifting the equilibrium to higher molecular weight forms need not be accompanied by the loss of their peroxidase activity, unless hyperoxidation is involved. On the other hand, the rapid and dynamic structural equilibrium that also occurs during the peroxidase catalytic assay provides a reason for a careful reevaluation of the relationship between oligomeric structure change and alteration in enzymic activity.
The effect of protein glutathionylation on the oligomeric status of Prx I has been investigated using sedimentation velocity methods and recombinant Prx I as well as its C52S/C173S and C83S mutants (89). In view of the fact that the cellular concentration of Prx I is ∼15–60 μM (11, 89), the concentration used for the sedimentation velocity study was set mainly at 50 μM. Under these conditions, 97% of the wild-type (WT) Prx I is present as a decamer. Concentration-dependent study revealed that the dissociation constant for the WT Prx I decamer is in the submicromolar range. However glutathionylated WT Prx I showed a different sedimentation velocity profile with a distinctly different sedimentation coefficient reflecting that glutathionylation disrupts the oligomerization of Prx I in such a way that the decameric population was totally eliminated at 50 μM protein concentration. Instead, the glutathionylated Prx I was converted mainly to small oligomeric species, with dimers as the predominant species present in the state of dynamic equilibrium (89) (see Fig. 6). Study with yeast Prx revealed that peroxidase activity is not required for Prx to exhibit molecular chaperone activity (55). This observation is in agreement with a report showing that the reduced form of human Prx I, and Prx I (C52S/C173S), a peroxidase inactive double mutant, exist as decamers and possess molecular chaperone activity (89). However, glutathionylation of these two Prxs led to the loss of chaperone activity and concomitantly converted the decameric structure to mainly dimers (89). It should be pointed out that the result obtained with the Prx I (C52S/C172S) mutant revealed that monoglutathionylation of Prx I at Cys83 is sufficient to convert the decameric enzyme, which possesses chaperone activity, to its dimeric state, which loses the molecular chaperone activity. It has been reported that Cys83 is located at the putative dimer–dimer interface and it plays a role in stabilizing the hydrophobic interaction required for the decamer formation (78, 115, 136). This notion is consistent with the observation that the dissociation constant for the high molecular weight oligomer of Prx I (C83S) mutant is significantly higher than that found for WT and the (C52S/C173S) double mutant (89).
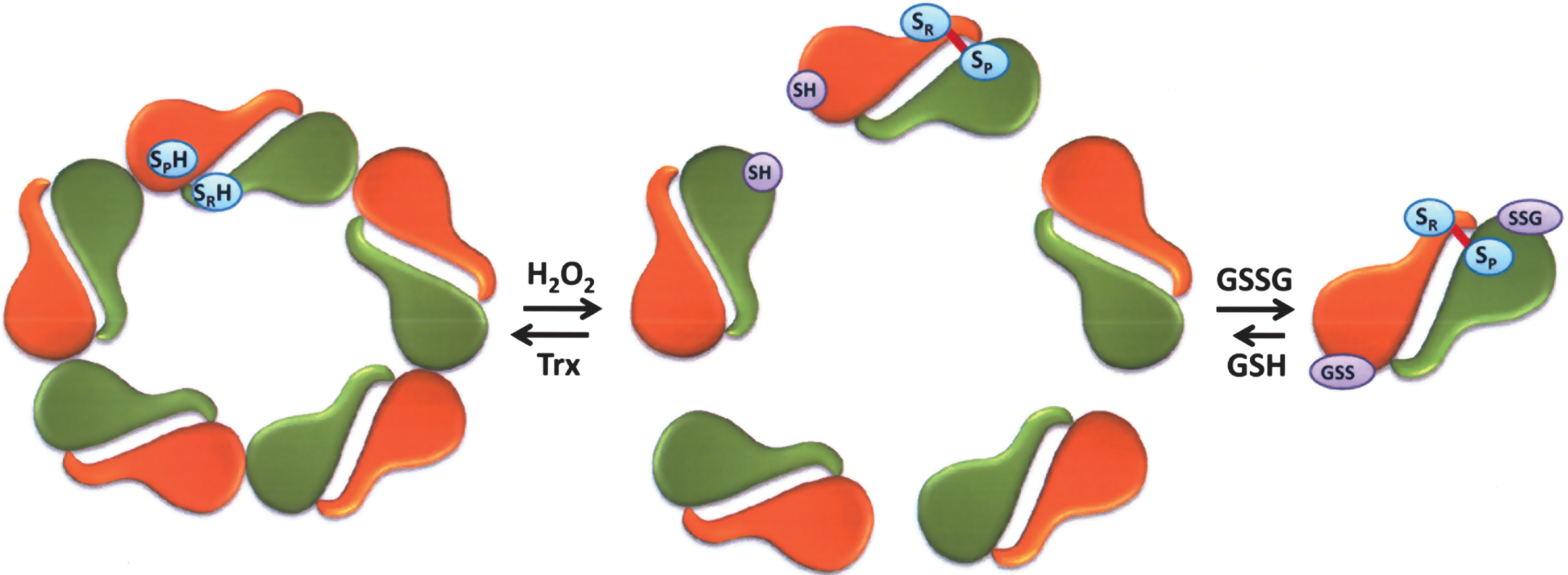
In view of the fact that glutathionylation of Cys83 of Prx I is sufficient to convert the decameric Prx I to its dimeric structure (Fig. 6), the biological relevance of Cys83 glutathionylation was assessed in a comparative study on glutathionylation of Prx I and Prx II in HeLa cells. Prx I and Prx II show 78% identity in their amino acid sequences and share similar peroxidase catalytic mechanism, except Prx II does not contain Cys83 (103). The glutathionylation reaction was monitored using biotinylated GSH ethyl ester (BioGEE), a cell membrane permeable form of GSH, followed by cell treatment with 10–50 μM H2O2. The results of this experiment revealed that glutathionylation occurred mainly on Prx I but not Prx II. This implies that Cys83 in Prx I is probably easily accessible for glutathionylation with BioGEE, relative to the peroxidatic and the resolving cysteine residues in either Prx I or Prx II (89). In other words, glutathionylation of Cys83 in Prx I may play a regulatory role in shifting Prx I from its decameric to dimeric form, and consequently causing a loss of its molecular chaperone activity.
When human cytosolic Prx I (cPrx I) was treated with H2O2, the concentration of its higher molecular weight complexes increased and its peroxidase activity correlated positively with the diminution of its low molecular weight oligomers. The dimer exhibited the highest peroxidase activity (55). In the case of molecular chaperone activity, the opposite correlation was observed. The dimer exhibited no chaperone activity while the decameric cPrx I or its higher molecular weight complexes exhibited high chaperone activity (55). However, an opposite conclusion indicating that the decamer possesses a higher peroxidase activity than that of the dimer has been reported based on kinetic study. In this study, kcat values of 55.1, 75.8, 25.0, and 31.5 s−1 were obtained for the reduction of H2O2 catalyzed by the wild-type alkyl hydroperoxide reductase (AhpC), and its T77V (favoring decamer), T77I and T77D (both favors dimer formation) mutants, respectively (91). However, the concentration of the WT AhpC used, 40–100 nM, was too low to maintain a meaningful population of decamers because the sedimentation velocity studies revealed that at 10 μM of WT AhpC, more than 25% of the protein was present as dimer. In addition the wide variation in the value of Km for S128W/NTD indicates that the mutation causes structural changes in AhpC and alters the kinetic properties of the mutants. Thus, the mutation does not simply change the distribution between the decamer and dimer population (91). In essence, the peroxidase activity of the reduced decamer remains to be established.
Mammalian Prx I and Prx II are mainly localized in the cytoplasm. Relative to Prx I, Prx II is a much less efficient peroxidase; however, Prx I is more susceptible to hyperoxidation and hence more readily gains chaperone activity compared with Prx II (18, 117). On the other hand, Prx I appears to have precise and versatile built-in machinery to regulate its peroxidase activity in response to local need for H2O2. Prx I and Prx II have been shown to associate with lipid rafts where signaling molecules such as NADPH oxidase, receptor tyrosine kinases, Src family kinases, and tyrosine phosphatases are found. Here, Prx I and Prx II would prevent the uncontrolled accumulation of H2O2, which in turn could activate signaling cascades. However, when cells were stimulated with growth factors, Prx I, but not Prx II, was transiently inactivated via a Src kinase–mediated tyrosine phosphorylation at Tyr194 (133). This finding provides an alternative mechanism for the “floodgate hypothesis,” involving phosphorylation rather than hyperoxidation as the mean of inactivating the peroxidase activity. Although both Prx I and Prx II are associated with lipid rafts, inactivation of Prx I, which is more efficient as a peroxidase relative to Prx II, might be sufficient to allow local accumulation of H2O2 for signal propagation. The role of raft-associated Prx II remains to be investigated. Prx I has also been shown to be inactivated when phosphorylated at Thr90 by cyclin B-dependent kinase 1 (Cdk1) (13). The Cdk1-catalyzed Prx I phosphorylation was observed during mitosis, but it was virtually undetectable during interphase. Consistent with this unique regulatory function of Prx I observed during growth factor stimulation and cell cycle progression, mice lacking Prx I had an increased susceptibility to cancer, while those deficient in Prx II did not (70, 84). Prx I has also been shown to play a role in tumor suppression (8). The local accumulation of H2O2 is likely to cause glutathionylation at Cys83 of Prx I molecules that are localized at a site where the ROS level is elevated; for example, a lipid raft during growth factor signaling. The differential susceptibility of Prx I and Prx II to phosphorylation, glutathionylation, and hyperoxidation suggests that these enzymes and modes of post-translational modification may play distinct roles in cell signaling.
What could be the functional role of Cys83 glutathionylated Prx I? Kinetic studies of the reaction of peroxidatic Cys of Prx and H2O2 suggest that its rate constant principally depends on collision efficiency, which results from steric accessibility and reactivity of the peroxy group (36). It is reasonable to expect five active dimers remove the H2O2 remnant more effectively than a decamer or a higher-order oligomer. This increase in peroxidase activity would occur after a lag phase due to the glutathionylation reaction and could provide a transient accumulation of H2O2 to complete its signaling function prior to being efficiently removed to prevent unwanted signal transduction. Recently Morinaka et al. (83) reported that the oligomeric Prx I structure, induced by relatively high levels of H2O2, is essential for Prx I binding and activating mammalian Ste20-like kinase-1 (MST1) kinase. Activation of MST1-kinase induces cellular apoptosis via a p53-mediated pathway. Thus, H2O2 appears to induce both activation and inhibition of apoptotic pathways through oligomeric structural change by hyperoxidation and via a dimeric structural change induced by glutathionylation, respectively. Considering the fact that Cys83 glutathionylation is induced by a lower range of H2O2 relative to that capable of causing hyperoxidation, it is reasonable to speculate that Cys83 glutathionylation functions as a built-in safety switch to prevent hypersusceptible initiation of the apoptotic pathway. In addition Cao et al. (8) revealed that Prx I inhibits tumorigenesis by binding and consequently protecting phosphatase and tensin homolog (PTEN) from oxidation-induced inactivation. Thus, Cys83 glutathionylation may also function as a sensor for low levels of H2O2 and respond by binding to PTEN to prevent unwarranted cell growth. The structural analysis by these authors indicates that PTEN likely binds to the Prx I dimer, which could well be the glutathionylated Prx I. Together, these two reports indicate that dimer/oligomer structure specific Prx I binding proteins such as MST1 kinase and PTEN regulate apoptosis and cell cycle, respectively, suggesting a possible link of glutathionylation with those signaling pathways.
Reversible Phosphorylation, Nitrosylation and Acetylation Regulating The Catalytic Activities of Prx I and Prx II
In addition to protein glutathionylation, the catalytic activity of Prxs can be regulated by other forms of covalent modifications. For example, the peroxidase activity of Prx I and Prx II can be regulated by phosphorylation of a specific threonine or tyrosine residue (see Fig. 7). Both Prx I and Prx II are phosphorylated by cyclin-dependent kinases (Cdks) at the Thr90 and Thr89 of Prx I and Prx II, respectively, in a Cdk-specific phosphorylation site consensus sequence, Thr-Pro-Lys-Lys. Thr90 of Prx I is phosphorylated by cyclin B-dependent kinase Cdk1 and leads to the inhibition of its peroxidase activity (13). Notably the inactive Thr90 phosphorylated Prx I, as well as the Prx I (T90D) mutant, which mimics the effect of Thr90 phosphorylation in Prx I, exhibit significant structural changes as indicated by changes in protein surface hydrophobicity monitored by binding of bis-ANS, and by changes in tryptophan fluorescence and circular dichroism spectra. These structural changes shift the Prx I structure to higher molecular weight oligomers, which possess molecular chaperone activity (54). Although Cdk1 also phosphorylates Prx II at Thr89, albeit at lower catalytic efficiency, Prx II has been shown to bind Cdk5/p35, which in turn phosphorylates its Thr89 when cells were treated with 1-methyl-4-phenylpyridinium ion (MPP+) (97). Like Prx I, this Thr phosphorylation also leads to inhibition of Prx II peroxidase activity. Considering the crystal structure of human Prx I (47), introducing a negatively charged phosphate to Thr90 in Prx I or Thr89 in Prx II, which is closely located to their respective peroxidatic Cys, would affect the charge–charge or charge–dipole interaction rendering the reactive cysteine insensitive to H2O2. Recently, Woo et al. (133) reported that Prx I is phosphorylated at Tyr194 and diminishes its peroxidase activity when cells were stimulated by platelet-derived growth factor or epidermal growth factor. Contrary to the Thr and Tyr phosphorylation leading to the inhibition of peroxidase activity, Ser32 phosphorylation has been shown to enhance the peroxidase activity of Prx I (142). T-cell–originated protein kinase catalyzes the phosphorylation of Ser32, which in turn prevents UVB-induced apoptosis in RPMI7951 melanoma cells through decreasing the intracellular H2O2 level by an increase in peroxidase activity of Prx I (142). As mentioned earlier, Thr90/89 phosphorylation of Prx I/Prx II induces a structural change and leads to a switch in the enzymic activity, from peroxidase to molecular chaperone. However oligomeric structural change and activity switching were not observed in Prx I phosphorylated at Tyr194 or Ser32 (133, 142). These are important observations with respect to the mechanism of cell signaling. Together, these studies reveal that the regulation and switching of the dual functions of 2-Cys Prxs appear to be mediated by post-translational modifications such as sulfinylation and phosphorylation. Both of these modifications are induced by the elevation of intracellular H2O2 leading to the activation or inactivation of the peroxidase activity of Prx I and II to provide a mechanism for transient depletion or accumulation of H2O2 which leads to downstream signaling events (43, 105, 133, 135).
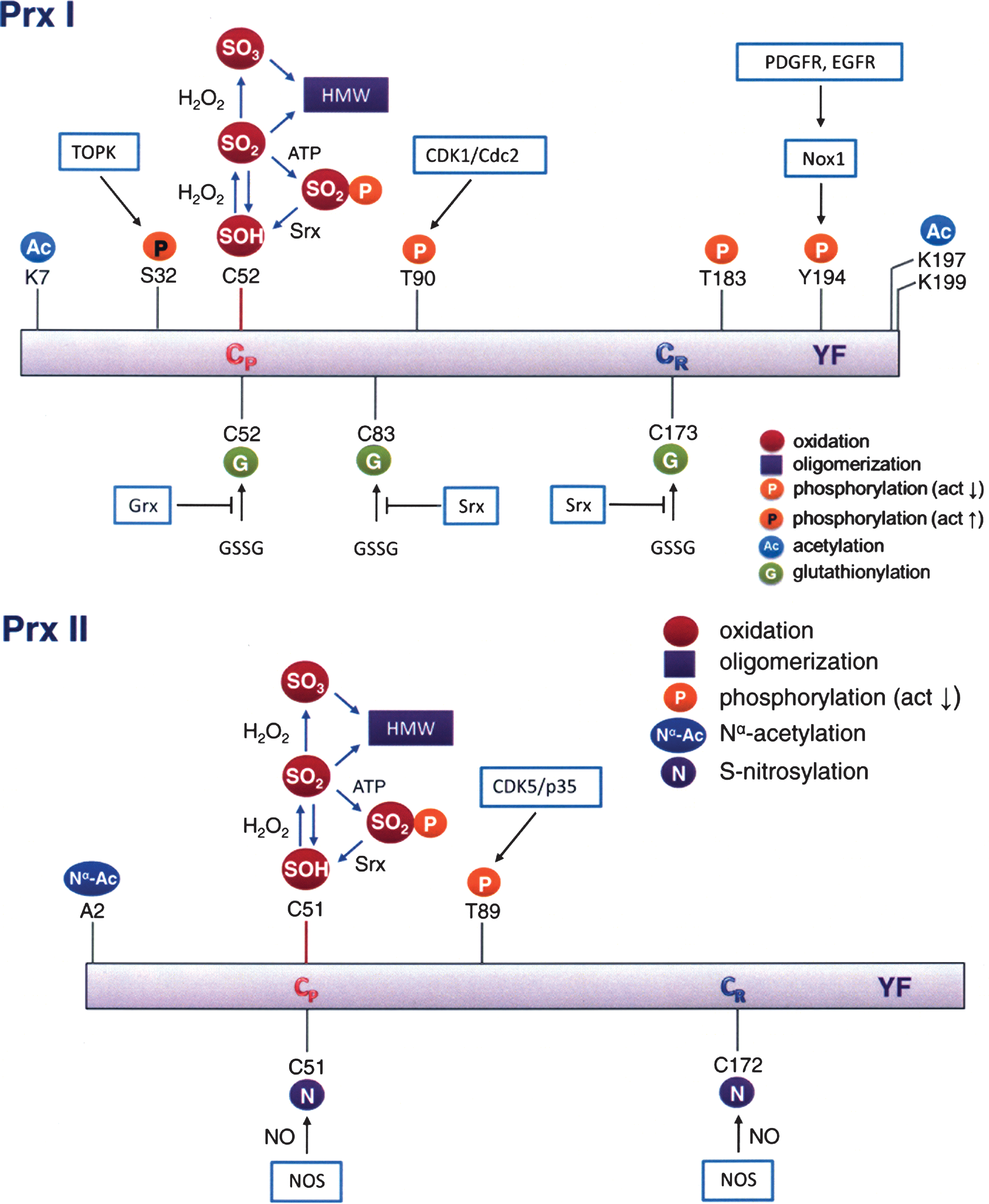
Prxs have also been shown to undergo protein nitrosylation and acetylation. Protein S-nitrosylation involves covalent attachment of a NO group to a cysteine thiol and it is known to regulate the function of many target enzymes, transcription factors, and ion channels, such as Parkin (24, 139), N-methyl-D-aspartate receptors (22), protein disulfide isomerase (106), nuclear factor-κB (77), and a number of other proteins (128, 141). Prx II has been shown to undergo S-nitrosylation by reaction with nitric oxide at its peroxidatic and resolving cysteine residues, inhibiting its peroxidase activity (32). Interestingly, these authors also found that the level of nitrosylated Prx II in the brain of patients with Parkinson's disease was elevated. Direct linkage between S-nitrosylation of Prx II and the neuronal cell death observed in Parkinson's disease remains to be established. Furthermore, Prx I has also been shown to undergo reversible N-acetylation (90, 117), with histone deacetylase 6 catalyzing the deacetylation reaction (90). The acetylation of Prx I led to an increase in their peroxidase activity and enhanced their resistance to hyperoxidation. Investigation by Seo et al. (117) revealed that when hPrx I and hPrx II cDNAs were overexpressed in Escherichia coli, Prx I was found N-acetylated while Nα-terminal acetylation (Nα-Ac) occurred exclusively on Prx II after demethionylation at its N-terminus. While N-acetylation of Prx I has been implicated in neurodegenerative disorders (90), Nα-terminal acetylation of Prx II leads to a significant structural change and blocks Prx II from irreversible hyperoxidation without altering its peroxidase activity. It should be pointed out that Nα-terminal acetylation is regulated by the availability of acetyl CoA (140) and is generally considered an irreversible and co-translational process. The facts that (i) the stability of Prx II is protected by Nα-terminal acetylation (117), (ii) Prx II has been shown to be involved in mediating cellular apoptosis (50, 73), and (iii) metabolic regulation of protein Nα-acetylation has been reported (140) together suggest a possible linkage between Nα-terminal acetylation of Prx II and the metabolism. However, the correlations between Prx acetylation and specific cellular events remain to be elucidated.
To date, cross-talk and integrated control, as well as the hierarchy among various post-translational modifications of Prx proteins have not been identified. This could be due in part to the high intracellular concentration of Prx proteins relative to other signaling or antioxidant proteins. As a consequence, a small fraction of modified Prxs are sufficient to accomplish specific cellular needs. Therefore, single or combinatorial modifications of Prx could occur at a given time at various locations in cells. This variety of modifications may explain the wide range of intracellular functional roles of Prx proteins.
Protein Glutathionylation Provides Reducing Equivalents to Peroxidase Catalysis
The catalytic cycle for the peroxidase activity of all Prxs begins with the same oxidation of conserved peroxidatic cysteine, SP, to its sulfenic derivative, SP-OH, by the reduction of peroxide to its corresponding alcohol. The SP-OH derivative will in turn form an intermolecular disulfide bond with the resolving cysteine, SR, in the case of mammalian typical 2-Cys Prxs (105), an intramolecular disulfide bond in the case of mammalian atypical 2-Cys Prxs (31, 65), or a disulfide bond with GSH, Grx, or Trx in certain Prxs from plants, bacteria, or worms (29, 94, 108). Reduction of these intermediates can be accomplished by the Trx/TrxR/NADPH system, the Grx/GSH/glutathione reductase (GR)/NADPH system (49), or (Trx or GSH)/TrxGR (TGR)/NADPH system (2). In this context, Schistosoma mansoni, a causative agent of schistosomiasis that lacks both TrxR and GR, possesses TGR to mediate electron donors via both the Trx and the GSH systems. The three Prxs (Prx1, Prx2, and Prx3) found in S. mansoni are 2-Cys Prxs that use the reducing equivalents from both Trx and GSH. Among them, Prx1 shows high specificity for Trx, while Prx2 and Prx3 exhibit significantly higher catalytic efficiency with GSH as reducing agent. The GSH binding affinity is conferred by the C-terminal 22 amino acid residues (113). However, a majority of Prxs exhibiting GSH-dependent peroxidase activity are 1-Cys Prxs isolated from plants, bacteria, and pathogens. A Prx from Haemophilus influenza Rd (PGdx), identified as a chimeric protein composed of a Prx and a Grx in its N-terminal and C-terminal region, respectively, has been purified and characterized (92). The peroxidase activity of PGdx requires the GSH redox cycle, but not the Trx/TrxR/NADPH system. Assays with H2O2 as substrate yielded a kcat/Km value of 5×106 M−1 s−1, comparable with that of the major peroxidase system of E. coli, AhpC (116). In addition, mass spectrometric analysis revealed that Cys49 of PGdx is the peroxidatic cysteine, and it was found glutathionylated in the enzyme purified from PGdx over-expressing E. coli. (92). Sequence analysis shows that the C-terminal region of PGdx shares strong homology with Grx 3 of E. coli, which has a higher activity as reductant of glutathionylated proteins than E. coli Grx 1 and 2 (3). This Grx-homologous domain contains the CXXC motif of Grx at cysteine residues 180 and 183. Interestingly, Grx has also been shown to function, like Trx, as an electron donor to reduce the sulfenic derivative of an oxidized poplar phloem Prx (109). Based on protein analysis showing Cys49 can be glutathionylated and the kinetic study displaying a sigmoidal kinetics as a function of increasing the concentration of GSH, a catalytic mechanism for the peroxidase activity of PGdx was proposed with glutathionylated Cys49 and Cys180 as reaction intermediates (92) (see Fig. 8). In this mechanism, the sulfenic acid formed at Cys49 reacts with GSH to form a glutathionylated PGdx at its N-terminal Prx region. Reaction 3 shows the deglutathionylation of the Prx region by Cys180 of the C-terminal Grx region. This Cys180 glutathionylated intermediate is in equilibrium with the formation of a disulfide bridge between Cys180 and Cys183 at the active site of the Grx region as shown in reaction 6. This is an unproductive side reaction. Altogether this mechanism indicates that at a low GSH concentration, only a relatively small fraction of the total enzyme population remains reduced and active. However, when the concentration of GSH is increased until reaction 6 becomes insignificant, the Grx region would be fully active. This proposed mechanism could explain the observed sigmoidal kinetics and is consistent with data showing the sigmoidicity was not detected with a Cys183 mutant of PGdx (92).
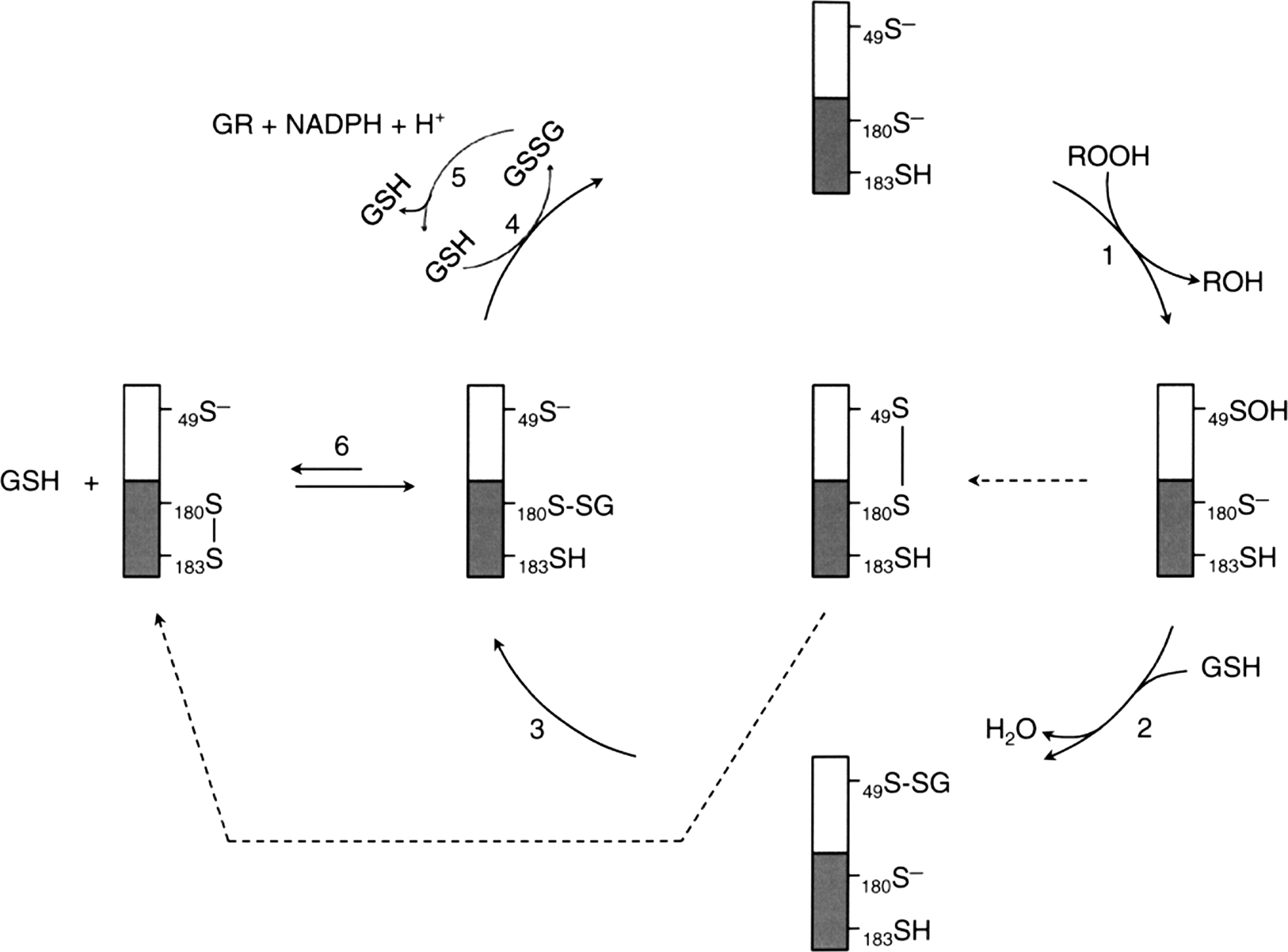
Protein Glutathionylation of 1-Cys Prx and Its Peroxidase Catalytic Cycle
Peroxiredoxin VI was first isolated as a nonselenium GSH peroxidase purified from the bovine ciliary body. It catalyzes the reduction of both H2O2 and organic hydroperoxides using GSH and yet does not have any GSH S-transferase (GST) activity (120). A 28-kDa protein was purified from rat olfactory epithelium whose N-terminal sequencing showed that it has a high homology with the nonselenium GSH peroxidase previously purified from bovine ciliary body. Furthermore, their substrate specificity and enzymic activity are comparable (95). cDNA identification and the deduced protein sequence confirmed that this protein belongs to the Prx family (Prx/Prdx). The Prx VI isolated from rat and bovine contains only the single conserved Cys47. However, unlike the rat and bovine Prx VI that possess only one cysteine residue, an additional nonconserved Cys is found in human (C91) and mouse (C201) proteins. In human Prx VI, Cys91 can be substituted by serine without affecting the peroxidase activity (61). Like the other Prxs, Prx VI contains a typical Trx fold with ∼80 amino acids arranged in four antiparallel β sheets sandwiched between two α helices (21). This region is essential for the peroxidative function of the enzyme. Furthermore, the sequence surrounding the peroxidatic cysteine in Prx VI and typical 2-Cys Prx I to IV are very similar, PVCTTE and FVCPTE, respectively. However, despite Prx VI sharing the structural and functional properties with other members of the Prx family, it has important characteristics that make it unique among the Prx family members. First, unlike both typical (Prx I to IV) and atypical (Prx V) 2-Cys Prx family members, Prx VI has only one conserved cysteine located at the N-terminal region (Cys47) involved in the peroxidase activity (136). In addition, the oxidation of peroxidatic cysteine (Cys47) to Cys47–SOH in Prx VI does not form a disulfide bond despite the fact that the enzyme forms a homodimer via its C-terminal domain involving probably hydrophobic interaction (21). A crystal structure study revealed that in the oxidatively inactivated Prx VI homodimer, the cysteine in each monomer exists as cysteine-sulfenic acid, positioned far apart from others and located in the bottom of a relatively narrow pocket. Generally, sulfenic acids are unstable and highly reactive (25, 26). However, it was found that cysteine sulfenic acid was quite stable in native Prx VI even under aerobic conditions. Further oxidation of the cysteine sulfenic acid of bovine Prx VI to higher oxidation states was observed only under denaturing conditions (96). Transformation of sulfenic acids proceeds through thiosulfinate formation by the self- condensation reaction of two RSOHs or oxidation to sulfinic acid. Sulfenic acids can also react with R-SH and R-NH2 to give disulfides and sulfenamides, respectively (25, 26). However the distance between the cysteine residues in the oxidatively inactivated dimer form and their location do not allow Prx VI to dimerize through disulfide bond formation in the native form. Instead, Prx VI, which exists as an obligatory dimer, forms a homodimer through hydrophobic interaction. A distinctive property of Prx VI from other Prxs is that Trx is not involved in the catalytic cycle (35, 61). Nevertheless, it is worth noting that this is not a general rule that can be applied to all 1-Cys Prx. For example, Prx1p, a 1-CysPrx of budding yeast Saccharomyces cerevisiae is located in the mitochondria, and it is induced when the cellular respiratory pathway is in operation, as well as when yeast is responding to oxidative stress. Contrary to human Prx VI, yeast Prx1p showed peroxidase activity in vitro using the S. cerevisiae mitochondrial Trx system as electron donor (93).
The role of GSH in the catalytic cycle of Prx VI has been controversial for some time. The peroxidase protein purified from bovine ciliary body was shown to be dependent on GSH (120). Later on, translated protein in vitro with a bovine cDNA was also found to utilize GSH as a reductant for the peroxidase activity (122). In contrast to the previous study using the protein from in vitro translation system, an in vitro assay with the recombinant human or bovine protein indicated that GSH was not an effective reductant (61, 95). The controversy surrounding the role of GSH in the Prx VI catalytic cycle was recently put to rest, and the further detailed catalytic cycle is summarized in following text and depicted in Fig. 9. It has been shown that the partial purification of Prx VI from bovine lung revealed the presence of the π isozyme of GST (πGST) in active preparations. Additional experiments showed that the heterodimerization of Prx VI and πGST led to the glutathionylation of the cysteine sulfenic acid followed by the spontaneous reduction of the mixed disulfide leading to the restoration of the peroxidative activity of the enzyme (75). These results were later elegantly confirmed by a study showing that purified recombinant human Prx VI is able to form a complex with GSH-loaded πGST leading to its full activation (99). Interestingly, Prx VI can form a complex with πGST even in the presence of S-methylglutathione showing that the heterodimer formation does not require the reducing power of GSH. Under these conditions, the protein has no peroxidase activity. However, the yield of the heterodimer is very low in the absence of GSH suggesting that GSH binding to πGST induced a conformational change that favors the heterodimer formation between πGST and Prx VI. In addition, this Prx VI homodimer does not have any peroxidase activity even in the presence of GSH, showing that GSH alone cannot activate the inactive form of Prx VI. This result suggests that the heterodimerization with πGST, a GSH carrier, is a key step in overcoming the inaccessibility of oxidized cysteine residue in inactive human Prx VI (see Fig. 9). Immediately after the heterodimer formation, Prx VI is glutathionylated at the peroxidatic cysteine residue, but the enzyme is still inactive. This step is followed by the disulfide bond formation between glutathionylated Cys47 of Prx VI and Cys47 of πGST. In fact, among all πGST mutants tested, only Cys47Ser πGST mutant failed to form a heterodimer with WT PrxVI as revealed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis in the presence or absence of DTT, suggesting that Cys47 is involved in forming the intersubunit disulfide bond between the two enzymes. The Prx VI in the heterodimer is inactive. Its activation requires reduced GSH to regenerate the active Prx VI capable of reducing hydroperoxide and phospholipid hydroperoxides, another important characteristic that sets Prx VI apart from other Prx proteins. However, it is not known whether the regenerated active form of Prx VI is a monomer or a homodimer. Unlike human Prx VI, 1-Cys D-Prx, the plant and pathogenic bacteria counterpart of human Prx V, is activated by the Grx/GSH system and exists as an active homodimer. Upon reduction of hydroperoxide, the cysteine residues in the dimer active site are oxidized to sulfenic acids. Unlike human Prx VI, this step is followed by the glutathionylation of the sulfenic acid by GSH leading to a conformational change that destabilizes and dissociates the noncovalent homodimer to monomers (see Fig. 10). The glutathionylated monomers of 1-Cys-D Prx are then reduced back by either DTT or the Grx/GSH system and lead to the regeneration of their homodimer (86). This redox-dependent dimer–monomer switch is similar to decamer–dimer switch observed in 2-Cys Prx I that is responsible for the toggling of its peroxidase-chaperone activity (89).


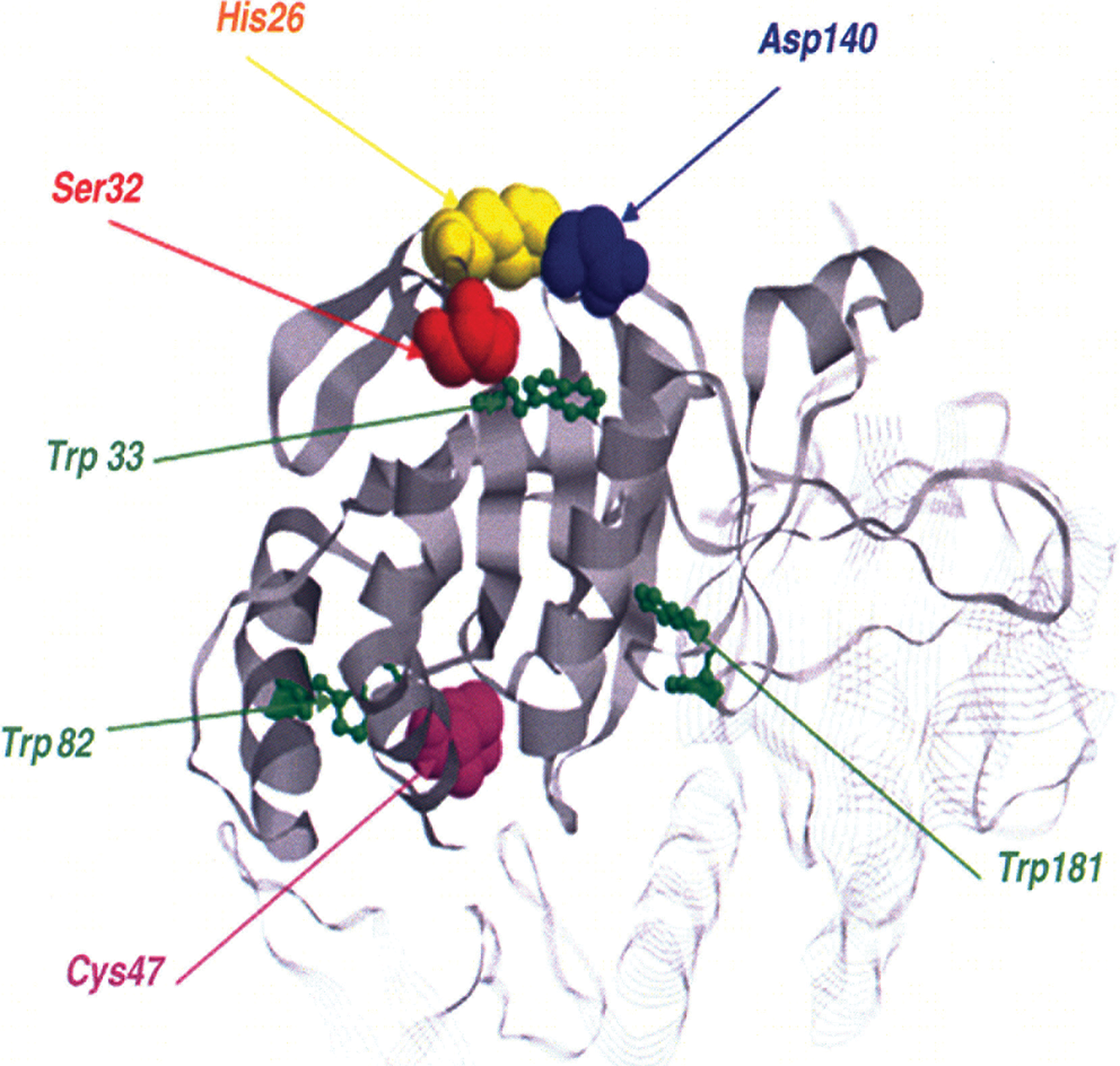
It should be pointed out that recombinant Prx VI protein expressed in bacteria and native protein purified from rat lung exhibited both peroxidase and phospholipase A2 activities (61, 64), making Prx VI a bifunctional enzyme with two separate active sites (Fig. 11). Mutagenic analysis revealed that the mutation of peroxidatic cysteine, Cys47, to serine in Prx VI abolishes the peroxidase activity but does not influence PLA2 activity (16). Likewise, the mutation of Ser32 to alanine affects the PLA2 activity without any effect on the peroxidase activity. Note that Ser32 together with His26 and Asp140 forms the catalytic triad of the PLA2 active site. In addition, it has been shown that PLA2 activity and lipid binding affinity at pH 7 were markedly enhanced by Thr177 phosphorylation without affecting its peroxidase activity (137). Recently, it has been reported that phospholipase A2 activity of Prx VI is required for NADPH oxidase 2 activation (15), a major source of ROS, paving the way for recognition of an important role of Prx VI, not only in antioxidant defense but also in cell signaling.
Concluding Remarks
The complexity of the diverse activities of Prxs and their regulatory mechanisms is attributed to their redox-sensitive structural changes and binding to their protein partners, as well as their various post-translational modifications. Functional diversities enable Prxs to function as signal modulators, tumor suppressors, and apoptotic regulators as well as broad-spectrum antioxidants. Prxs react rapidly with a wide range of intracellular peroxides to form a sulfenic acid derivative with their conserved peroxidatic cysteine, SP. However, reduction of the sulfenic intermediate differs among Prxs. They include formation of an intermolecular disulfide bond with a conserved resolving cysteine, SR, in a typical 2-Cys Prxs, an intramolecular disulfide bond in atypical 2-Cys Prx, and formation of a disulfide bond with GSH in human 1-Cys, or with GSH, Grx, or Trx in Prxs from plant, bacteria, or worms. Reduction of these disulfide intermediates is catalyzed respectively by Trx/TrxR/NADPH, Grx/GSH/GR/NADPH, (Trx or GSH)/TGR/NADPH, or the GSH/πGST systems. While Prxs have the capacity to remove peroxides efficiently, they also have an intrinsic vulnerability to be hyperoxidized to sulfinic or sulfonic derivatives and lose their peroxidase activity. The hyperoxidized Prxs favor the decamer/higher-order-oligomeric structures and result in the augmentation of molecular chaperone activity, which in turn protects the protein substrate from aggregation. Various reversible post-translational modifications such as hyperoxidations, phosphorylations, nitrosylations, glutathionylations, and acetylations are involved in the regulation of Prx activity. However, at this moment, their integrated control mechanism or their modification hierarchy is yet to be established.
Protein glutathionylation using GSH, the most abundant thiol in cells, as the donor molecule provides not only the reducing equivalents to peroxidase catalysis and protection of peroxidatic cysteine from irreversible hyperoxidation, it is also involved in Srx-mediated reactivation of hyperoxidized 2-Cys Prxs and in regulating the quarternary structure of Prx I. Considering that the cellular concentration of Prx I is in the range of 10–60 μM, and the decamer/dimer dissociation constant is in the submicromolar range, the reduced Prx I in the cells exists mainly as decamers. In response to low level peroxides, Cys83 of Prx I undergoes glutathionylation and leads to dimer formation. The peroxidase active dimeric Prx I in turn removes the excess peroxides more effectively; it could also bind to PTEN to protect it from oxidative inactivation and results in preventing tumorigenesis. However, when the peroxide levels are sufficiently high to induce hyperoxidation of Prx I, the hyperoxidized high molecular weight oligomers of Prx I have been shown to bind and activate MST1 kinase which in turn induces apoptosis via a p53-mediated pathway. Thus, this constitutes a mechanism for removing oxidatively damaged cells. Furthermore, the involvement of glutathionylation in the Srx-catalyzed reactivation of the inactive hyperoxidized 2-Cys Prx has been considered as a switching mechanism for maintaining a transient elevation of H2O2 levels for activating cell signaling cascades followed by the removal of ROS to prevent oxidatively induced cytotoxicity.
Footnotes
Acknowledgments
This work was supported, in the whole or in part, by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health. Prx research in H.Z.C. lab is supported by grant from the National Nuclear R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Sciences and Technology (2010-0018772) and by CNU grant (2009-2533). S.G.R. was supported by grants from the Korean Science and Engineering Foundation (National Honor Scientist Program grant 2006-05106 and Bio R&D program grant M10642040001-07N4204-00110).