
Abstract
Iron is a crucial factor for life. However, it also has the potential to cause the formation of noxious free radicals. These double-edged sword characteristics demand a tight regulation of cellular iron metabolism. In this review, we discuss the various pathways of cellular iron uptake, cellular iron storage, and transport. Recent advances in understanding the reduction and uptake of non-transferrin-bound iron are discussed. We also discuss the recent progress in the understanding of transcriptional and translational regulation by iron. Furthermore, we discuss recent advances in the understanding of the regulation of cellular and systemic iron homeostasis and several key diseases resulting from iron deficiency and overload. We also discuss the knockout mice available for studying iron metabolism and the related human conditions. Antioxid. Redox Signal. 18, 2473–2507.
I. Introduction
Another major group of iron-containing proteins, the iron–sulfur cluster (ISC) proteins, is essential for mitochondrial respiration, the citric acid cycle, nucleotide metabolism, translation, and a wide range of other cellular functions. They are found both in the mitochondria and the cytosol with ISCs existing as three different types (i.e., [2Fe–2S], [3Fe–4S], and [4Fe–4S] types), depending on the numbers of iron and sulfur atoms in the cluster (338).
Nonheme iron is a cofactor for many enzymes involved in the oxidoreductase/electron transfer processes, gene expression and repair, homeostasis and signal transduction, and in a range of other enzymatic reactions. Iron is necessary for neurotransmitter synthesis [as a cofactor of tyrosine hydroxylase, the first enzyme in catecholamine synthesis (120), and as cofactor of tryptophan hydroxylase, the first enzyme in serotonin synthesis (242)], and nucleotide synthesis [as a cofactor for eukaryotic (class Ia) ribonucleotide reductase (194)], the regulation of inflammation via the action of lipoxygenases (167), tyrosine metabolism [as a cofactor of phenylalanine hydroxylase, the first enzyme in phenylalanine breakdown (99)], as a cofactor of prolyl hydroxylase in collagen metabolism and many other 2-oxoglutarate-dependent oxygenases that regulate the cellular response to hypoxia (see below), nucleic acid repair and modification, fatty acid metabolism, and chromatin modification (230).
However, iron can also play the role of a foe, as it can act as a pro-oxidant by catalyzing the formation of reactive oxygen species, as described below. The most famous pro-oxidant reaction of iron, the Fenton reaction (104), results in the formation of highly damaging hydroxyl radicals from hydrogen peroxide, according to the equation,
Additionally, the one-electron reduction of dioxygen by Fe2+ can generate superoxide anions (O2·−) according to the following reaction:
These superoxide anions can then be dismutated, either enzymatically by superoxide dismutases (SODs) or nonenzymatically, to yield H2O2, according to the following reaction:
Additionally, or if SOD activity is rate limiting, superoxide anions can reduce the trace amounts of labile aqueous Fe3+ (i.e., low-M
r iron) to form dioxygen and regenerate Fe2+:
The sum of Reaction 1 (Fenton reaction) and Reaction 4 is known as the Haber–Weiss reaction (145):
The Haber–Weiss reaction illustrates that in the presence of catalytic amounts of redox-active aqueous low-M r iron, which can become increasingly prevalent under conditions of iron overload (see Section VII.B), hydrogen peroxide may provide a ready source of damaging hydroxyl radicals in the presence of a reductant such as superoxide that is required to regenerate the reduced form of the metal. Other abundant cellular reductants (e.g., ascorbate and GSH) can reduce ferric ions in an analogous manner to superoxide in Reaction 4 (40, 362). Thus, cellular reductants such as ascorbate and GSH, which typically function in an antioxidative capacity, can have a pro-oxidant activity in the presence of catalytic concentrations of labile iron. In a cellular, tissue, or organ context, the iron-catalyzed formation of highly reactive and damaging reaction oxygen species such as the hydroxyl radical, which is driven by cellular reductants, is primarily responsible for the ability of labile iron to cause oxidative stress. As indicated in Section VII.B, this iron-induced oxidative stress underpins much of the initial and ongoing pathology of diseases of iron overload (e.g., hemochromatosis) (271).
In conclusion, since too much or too little iron can compromise cell viability, cellular iron homeostasis must be tightly controlled (150).
II. Uptake of Iron by Mammalian Cells
In its physiological form, extracellular iron is typically complexed by chelators or iron-binding proteins. The most important iron complexants are Tf and citrate. Although the basic mechanisms involved in the uptake of iron from Tf are reasonably well understood, the mechanisms of non-Tf-bound iron (NTBI) uptake by mammalian cells remain ill defined.
A. Tf-dependent uptake of iron
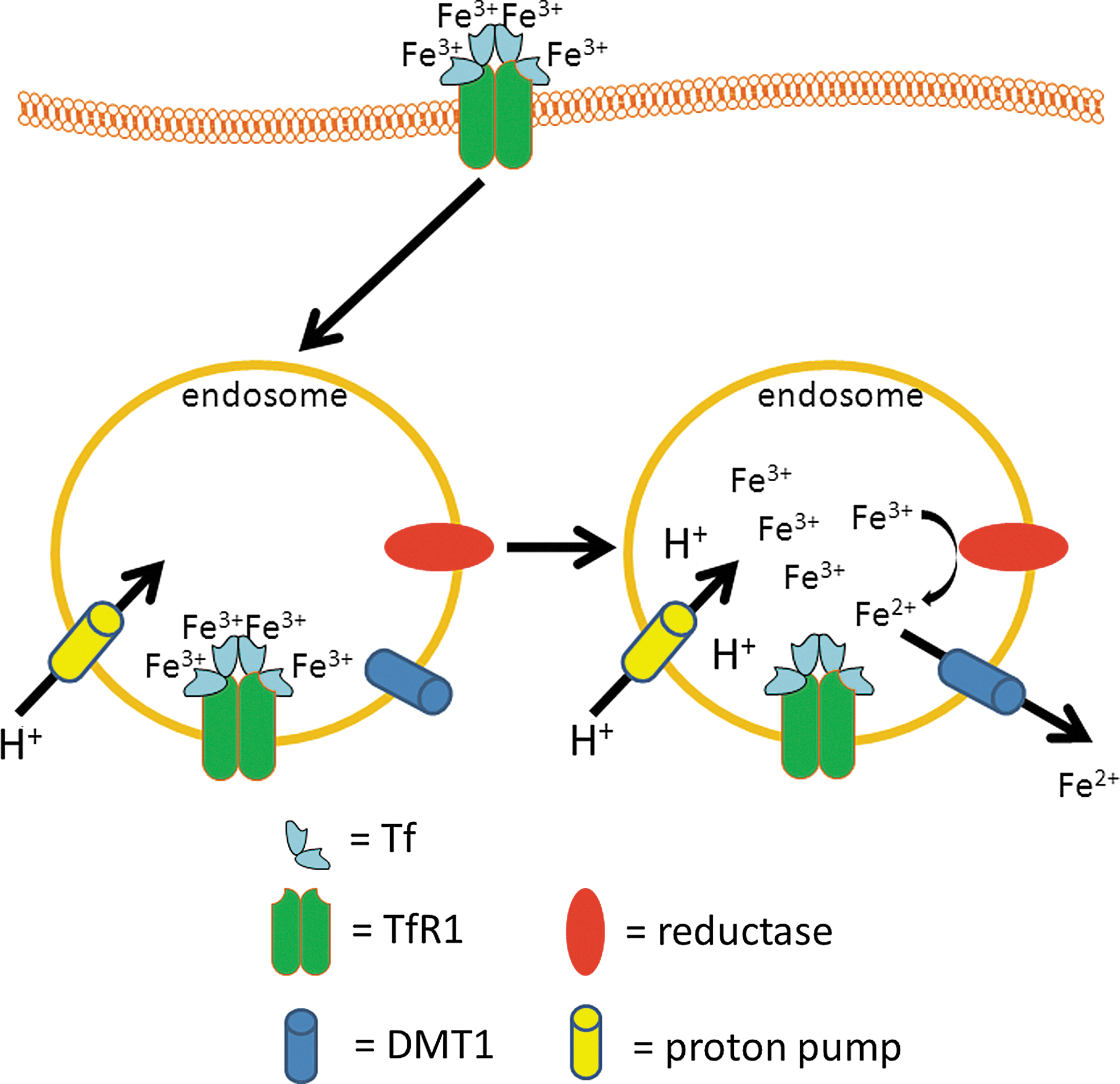
In a healthy person, virtually all plasma iron is tightly bound to Tf. Iron that is taken up by enterocytes (see Section III.A) is released into the blood stream and oxidized to the ferric state [most likely by the multi-copper ferroxidase hephaestin, which is a transmembrane multi-copper ferroxidase of the basolateral membrane of enterocytes, or by the homologous soluble multi-copper ferroxidase, ceruloplasmin, which is abundant in plasma (398)]. The ferric iron that is formed by the action of these ferroxidases is then specifically bound to serum Tf, a 80-kDa glycoprotein that is mainly synthesized by the liver (263). Each Tf molecule can bind one or two Fe3+ ions (15) to form one molecule of mono- or diferric Tf (holo-Tf), respectively. However, under normal conditions where serum Tf saturation is ∼20%–30% (83), the majority of Tf-bound iron is probably in one or other of the two distinct monoferric forms [i.e., in which the Fe3+ ion is bound preferentially to either the N- or C- lobe of Tf, depending on the chemical form in which the Fe is presented to apo-Tf (2)] (263). Tf binds Fe3+ with a high affinity, which depends on the pH, with maximal binding occurring at pH 7.4 (affinity constant at atmospheric pCO2 for each binding site ∼1020 M −1) (2). Holo-Tf binds to the integral membrane protein Tf receptor 1 (TfR1). TfR1 is a glycoprotein of about 95 kDa (237), which forms a homodimer that is linked by disulfide bonds (174), and is capable of binding two molecules of holo-Tf (214). TfR1 is expressed by most cells, with the exception of mature erythrocytes (98) and possibly oligodendrocytes, microglia, and astrocytes in vivo (259). The affinity of TfR1 for diferric Tf at pH 7.4 is about 2000-fold higher than for apo-Tf (385) and about 20-fold higher than for either of the monoferric Tf forms. The entire complex is then endocytosed via receptor-mediated endocytosis in clathrin-coated pits (131). The pH in the endosome drops after internalization, due to the activity of an ATP-dependent proton pump: the vacuolar-type H+-ATPase (V-ATPase), in the endosomal membrane (299). Due to the resulting acidic environment [i.e., a lumen pH of 5.3–5.6 (240)], the ferric ions dissociate from Tf, whereas apo-Tf remains tightly bound to TfR1 (181, 282). Before the transport of iron from the endosome to the cytoplasm by the Fe(II)-selective proton-coupled divalent metal transporter 1 (DMT1; also known as divalent cation transporter 1 [DCT1] or natural resistance-associated macrophage protein 2 [Nramp2]) (113, 139, 367), ferric iron that is released from Tf must first be reduced to ferrous iron (279, 409). The ferrireductases involved in this process include the six-transmembrane epithelial antigen of the prostate-3 (Steap3) (283), at least in erythroid precursors, and/or potentially the members of the cytochrome b 561 family (204). It should also be noted that other Fe(II) transporters have been proposed to contribute to iron mobilization from Tf-cycle endosomes, including ZRT/IRT-like protein 14 (ZIP14) (432), although their relative contributions in different cell types remain to be established. Finally, the complex of apo-Tf and TfR1 is then recycled back to the plasma membrane, and the apo-Tf dissociates from TfR1 at the slightly alkaline pH of the extracellular space (26). A summary of this pathway is given in Figure 1.
There is evidence that some Tf can be taken up by cells via TfR1-independent endocytosis. Holo-Tf binds to low-affinity-binding sites, perhaps including those provided by Tf receptor 2 (TfR2, see below) (130, 184, 185), on the surface of hepatocytes from where it is endocytosed and the iron is released into the cytosol (382). There is also evidence for fluid-phase endocytosis (i.e., pinocytosis) of Tf as a contributor to cellular holo-Tf uptake (382). The pathways of TfR1-independent holo-Tf uptake are summarized in Figure 2.
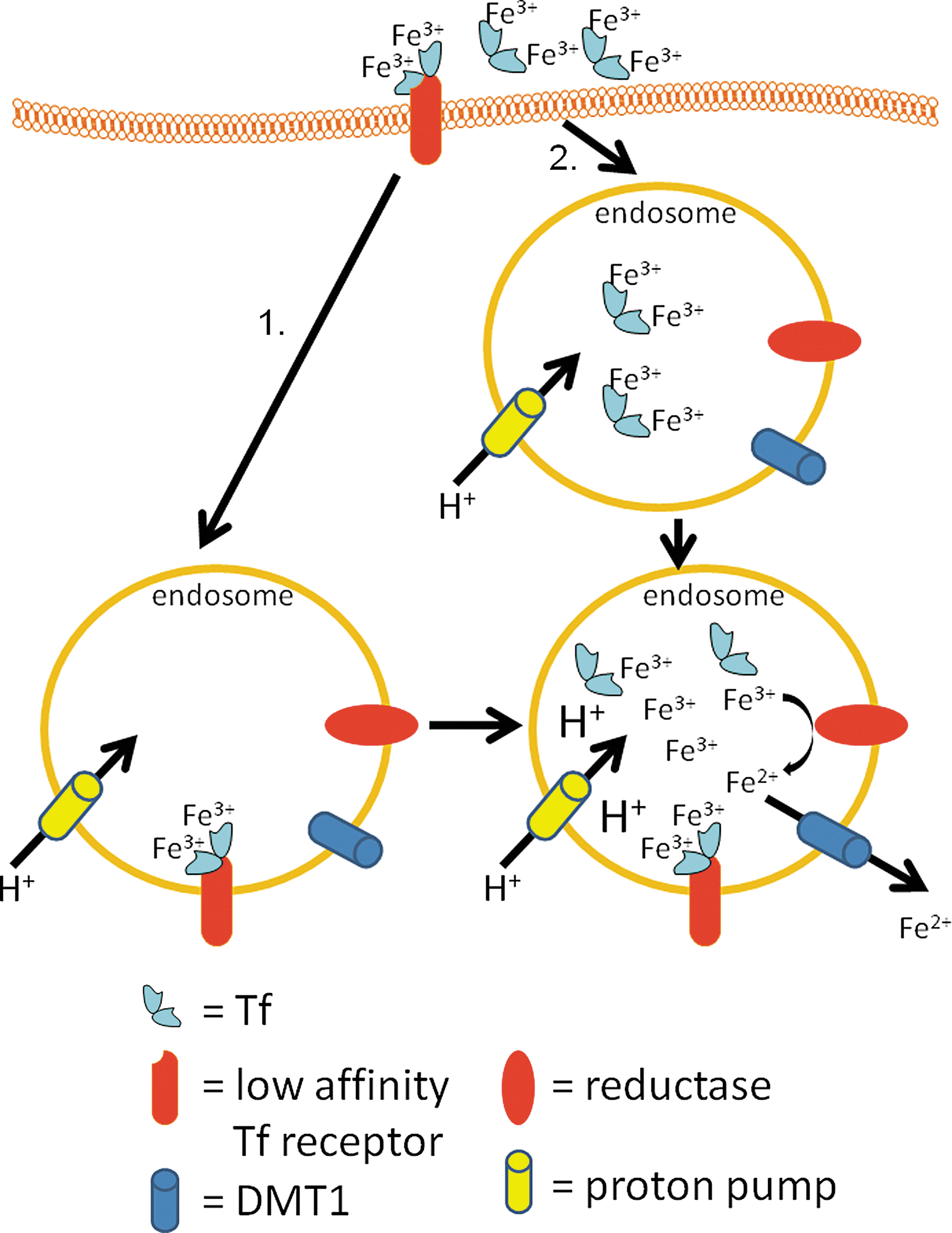
As mentioned above, a second transferrin receptor, TfR2, has been described (185). Its gene yields two transcripts, α and β (185). While TfR2-α results in a membrane-bound protein (185), the TfR2-β transcript lacks the transmembrane domain of TfR2-α, probably resulting in a secreted and soluble receptor (185). TfR2 binds Tf with a much lower affinity (more than one order of magnitude less) than TfR1 (184). As discussed in depth below (see Section VI.C), it appears that the major role played by TfR2 is the sensing of systemic iron levels and the modulation of hepatic hepcidin production rather than iron uptake per se (129). This is supported by data showing that while TfR1 expression is typically upregulated in situations of cellular iron deficiency and downregulated in situations of iron overload (149), TfR2 expression is not regulated by the cellular iron status. However, TfR2 expression is regulated by the cell cycle, with the highest expression occurring in the late G1 phase (184). TfR2 is also necessary for the protective role of Tf against Fas-induced hepatocyte apoptosis (222). That TfR2 probably does not play a major role in iron uptake is also highlighted by the fact that it cannot compensate for loss of TfR1, as TfR1 knockout mice are not viable (224). On the other hand, the overexpression of TfR2 in variant Chinese hamster ovary (CHO) cells that are devoid of endogenous hamster TfRs (i.e., CHO-TRVb cells) results in increased uptake of Tf and iron from 55Fe-Tf, suggestive of some role for TfR2 in iron uptake (130, 185).
B. Non-Tf-dependent uptake of iron
As discussed above, virtually all iron in plasma is bound to Tf under physiological conditions. However, in diseases resulting in iron overload (see Section VII.B), Tf can become saturated with iron, such that excess plasma iron will be present in the circulation as NTBI [originally referred to non-Tf iron)] (152, 153). The precise biochemical nature of NTBI is ill-defined (36). Although literally referring to all iron that is not specifically bound to Tf, the term NTBI is usually reserved for uses referring to a putative low-M r pool of iron that is bound to small organic chelators, such as citrate and organophosphates (e.g., ATP) (36). This pool of NTBI is present at variable concentrations in extracellular biological fluids such as plasma and interstitial fluid (36, 153). Notably, there are a multitude of other possible sources of plasma iron that will not be bound to Tf or exclusively to small chelators. These sources will include proteins such as serum ferritin, which may be released into extracellular fluids constitutively or as an acute-phase reactant, or albumin, which has a relatively weak affinity for ferric iron, but may be a significant iron carrier in plasma because of its high concentration (i.e., ∼0.5 mM) (36). It is in the original sense of the term (i.e., low-M r plasma iron that may or may not be weakly adsorbed to abundant non-Tf plasma proteins such as albumin) that we will discuss NTBI throughout the remainder of this review.
Although NTBI uptake may be particularly relevant in the face of iron-overload diseases such as hereditary hemochromatosis (HH), hypotransferrinemia, and the hemolytic anemias (e.g., β-thalassemia; see Section VII.B) (4, 87), in which plasma iron levels increase and may exceed the Tf-binding capacity, low levels (<1 μM) of NTBI have been documented in healthy individuals, but may rise up to 10–20 μM under conditions of severe iron overload (36). Computer simulations suggest that most of the plasma NTBI is complexed by citrate (195), and experimental data support this notion (135). Most, if not all, cells have the capacity to take up NTBI, and such uptake is well documented in vivo (65, 81) and for a range of cell types, including hepatocytes (16), erythroid cells (264), intestinal epithelial-like cells (Caco-2 cells) (210), primary astrocytes (206), neurons and microglia (25), skin fibroblasts and cervical carcinoma (HeLa) cells (177, 270), monocytic (U937) cells (245), and erythroleukemia (K562) cells (164, 203).
In most instances, iron uptake from NTBI can be blocked by extracellular iron (II) chelators [e.g., (164, 177, 203)], suggesting that the iron (III) present in the extracellular milieu has to be reduced to iron (II) before uptake. Soon after the description of trans-plasma membrane electron transport (tPMET) [for a recent review see Ref. (89)] by Crane (80), it was hypothesized that a plasma membrane ferricyanide reductase is responsible for NTBI reduction before uptake by mammalian cells (17, 205, 233) (Fig. 3). Identification of the enzymes responsible for iron uptake thus far has had limited success. The discovery of duodenal cytochrome b 561 (Dcytb) as a putative ferrireductase in duodenal enterocytes resulted in the suggestion that this enzyme is responsible for NTBI reduction before uptake (247). Dcytb is a member of the cytochrome b 561 family, which exists in all eukaryotic kingdoms (366). Cytochrome b 561 (also known as chromaffin granule cytochrome b 561 [CGcytb]) is best known for catalyzing trans-membranous electron transfer from cytosolic ascorbate to intravesicular ascorbate free radicals (AFR) in neuroendocrine secretory granules (115). Other members of this mammalian family of redox enzymes include lysosomal cytochrome b 561 (Lcytb) (366), stromal cell-derived receptor 2 (SDR2) (388), and gene product 101F6 (257). Dcytb was originally proposed to play a crucial role in direct NTBI reduction at the brush-border membrane of duodenal enterocytes before their uptake of the resulting ferrous iron by a ferrous iron-selective transporter such as DMT1 (247). However, the hypothesis that Dcytb plays a crucial role in iron absorption has been challenged by the observation that Dcytb knockout mice do not present with iron deficiency (140). Importantly, as the latter study only examined liver iron stores, rather than directly measuring enteric iron absorption in control vs. knockout animals, it remains possible that changes in enteric iron absorption may still occur in the absence of Dcytb activity, although clearly not sufficient to lower liver iron stores over the duration of the experiments conducted (140). The results of Gunshin et al. (140) do suggest that the role of Dcytb in iron absorption is dispensable, and may be supplemented by the action of another ferrireductase and/or nonenzymatic reduction by chemical reductants such as ascorbate, which is synthesized by mice, but not by humans. However, as Dcytb expression is iron-regulated (171, 211, 329, 332), and as expression of Dcytb clearly stimulates iron uptake in vitro (210), Dcytb is likely to play some role in cellular iron uptake from NTBI.
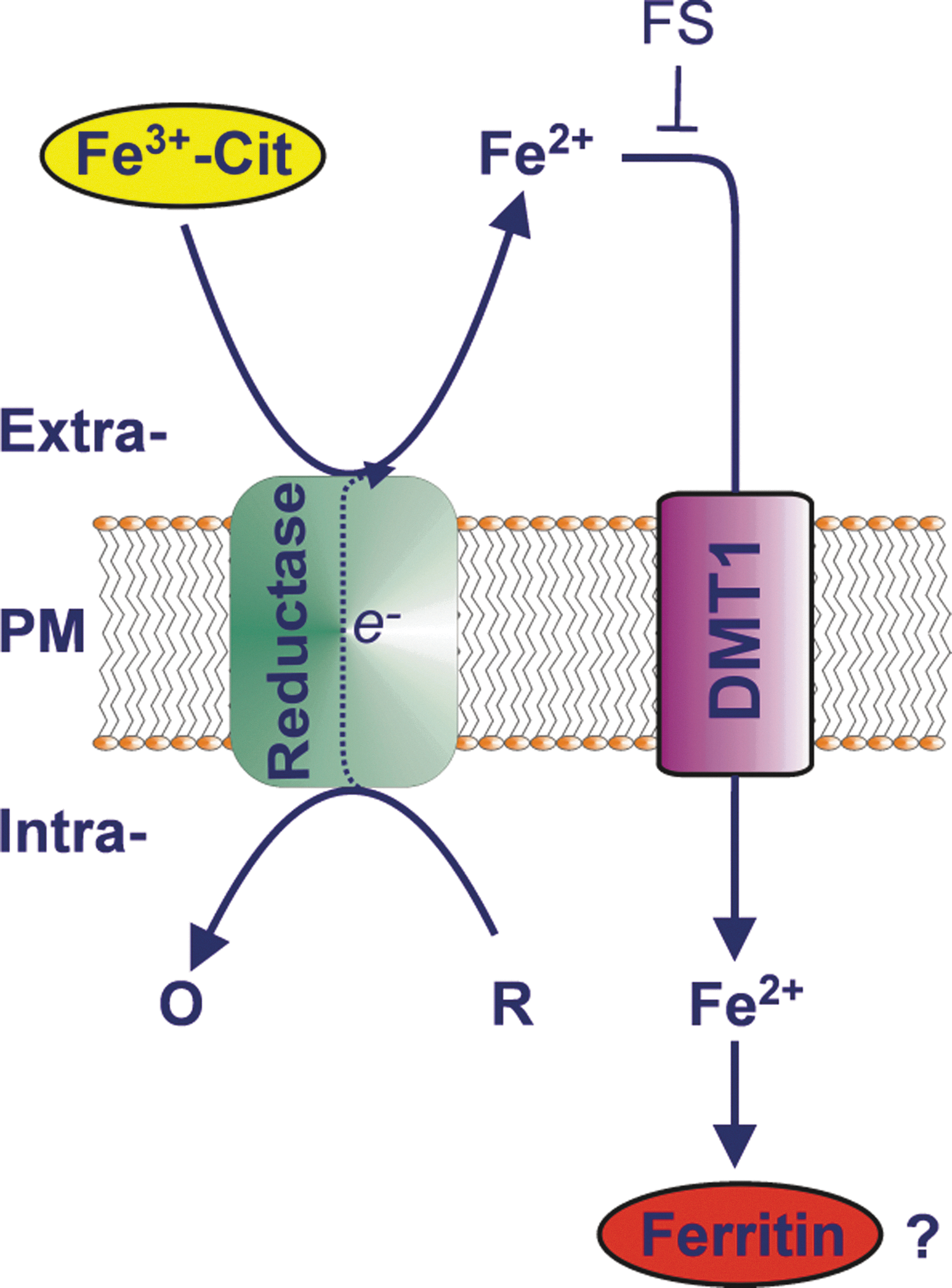
We have recently challenged the notion that NTBI is reduced by an ascorbate-stimulated plasma membrane ferrireductase activity before cellular uptake of the resulting ferrous iron (203, 204, 206). We demonstrated that extracellular ascorbate oxidase (AO), an enzyme that selectively and rapidly degrades extracellular but not intracellular ascorbate, abolished the ascorbate-stimulated reduction of ferric citrate, and greatly inhibited the ascorbate-stimulated rate of iron uptake, by various cell types that had been preloaded with ascorbate (203, 206). This sensitivity clearly indicates a requirement for extracellular ascorbate in the reduction of NTBI before uptake. Importantly, our data further suggest that it is ascorbate that is exported from the cells that directly reduces low-M r ferric citrate complexes to iron (II) for cellular uptake, with the latter occurring via a ferrous iron-selective transporter such as DMT1 (109, 114, 206). Our current model of NTBI uptake by cells is presented in Figure 4. An alternative, but analogous, mechanism of shuttle-based tPMET (204, 205) by which iron (II) might be generated at the cell surface from iron (III) is via reduction of iron (III) by superoxide radicals that exit cells via anion transporters (124, 234).
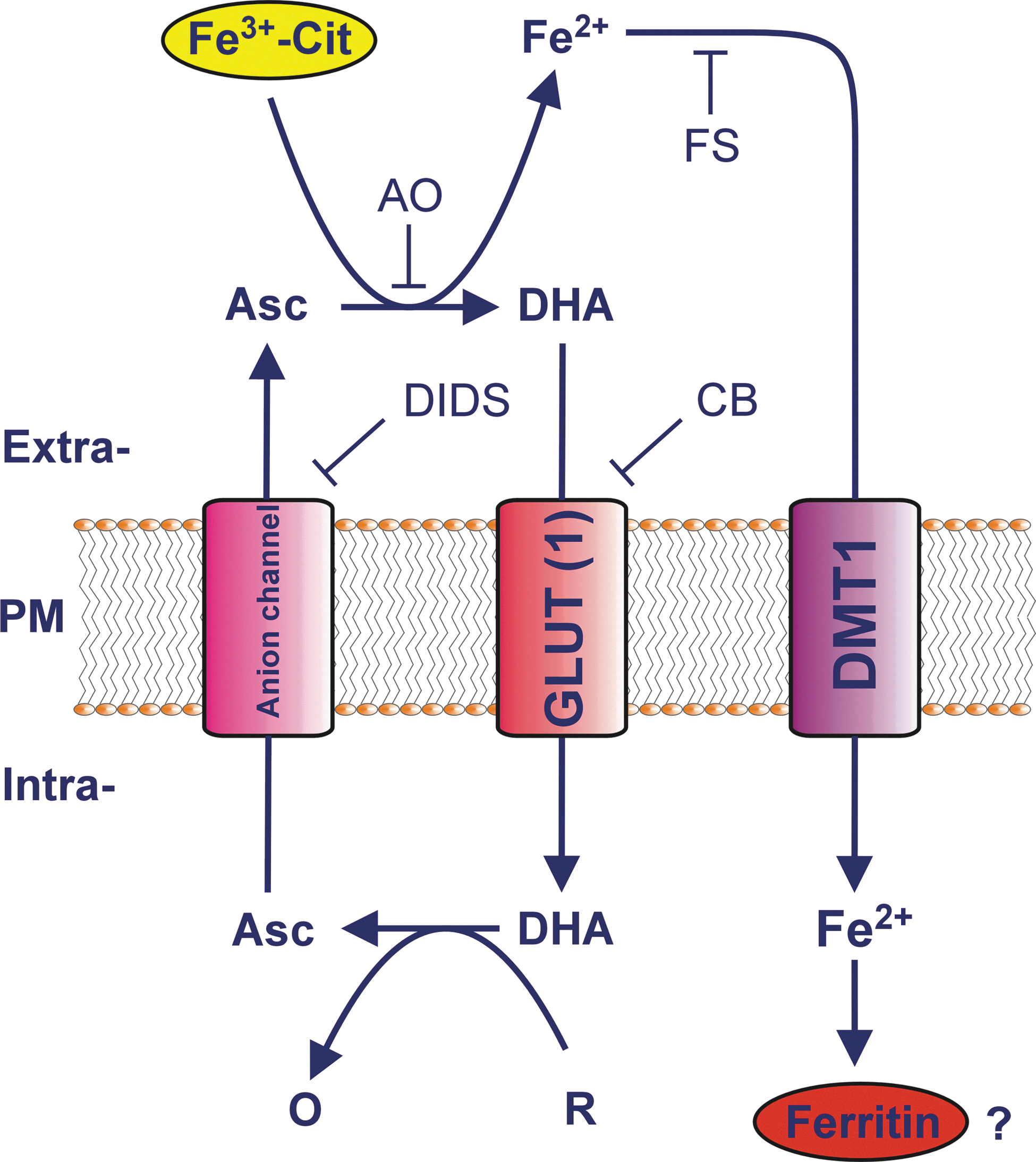
If an ascorbate-stimulated reductase (e.g., Dcytb) is not involved in direct NTBI reduction before uptake of Fe(II), the role of such an activity in NTBI uptake remains to be established. We have suggested that the most likely role of such a reductase is to reduce extracellular AFR at the expense of intracellular ascorbate and thereby bolster extracellular ascorbate in the face of extracellular metal-dependent ascorbate oxidation. However, this hypothesis has yet to be thoroughly tested.
There is also some evidence for the direct uptake of iron (III) by astrocytes by an as-yet-to-be established mechanism (206, 386). Two possible mechanisms for iron (III)-selective uptake have been described in the literature, but both are still elusive. They are the β3-integrin/mobilferrin pathway (69, 70) and the trivalent-cation-selective pathway (11). Another possibility that remains to be thoroughly demonstrated is the possibility that TfR2 itself may facilitate uptake of NTBI (130), although whether the reported increase in iron uptake is a direct or indirect effect of TfR2 overexpression remains unclear. It is also possible that iron (III) is internalized by fluid-phase endocytosis before its intraluminal reduction and mobilization to the cytosol, which would be consistent with the observed insensitivity of iron uptake to membrane-impermeant iron (II) chelators (206), although there are currently no data to support this hypothesis.
Finally, NTBI can be taken up as heme (i.e., iron–protoporphyrin IX). Heme forms a complex with hemopexin (378), which is an ∼63-kDa glycoprotein (370) with a high affinity for heme and one heme-binding site per hemopexin molecule (157). The heme/hemopexin complex is then recognized by hemopexin receptors, believed to be the low-density lipoprotein receptor, LRP/CD91, at the cell surface (163), and possibly toll-like receptors 2/4 (227). The hemopexin receptor/hemopexin/heme complex is then internalized by receptor-mediated endocytosis, and afterward, the hemopexin normally recycles intact (355). The in vivo hepatic uptake of intravenous heme–hemopexin occurs rapidly (i.e., within 5 min) (356, 357). Heme delivered by hemopexin acts in transcriptional regulation or is degraded by heme oxygenases (HOs; i.e., HO1 and/or HO2) within endocytic vesicles, and the nascently mobilized iron (i) enters the transit or labile iron pool (LIP), where it contributes to regulatory processes; (ii) is stored in ferritin; (iii) is used in regulation; (iv) is used for metabolism; or (v) is exported (see below for further discussion of these processes). Reviews on the hemopexin system have been recently published in this journal (278) and, more comprehensively, elsewhere (243, 354). Hemopexin helps to scavenge heme that is released or lost during the turnover of heme-containing proteins such as hemoglobin (e.g., during erythrophagocytosis or hemolysis; see Section III.D), and additionally protects cells from heme toxicity (142, 392). In this capacity, the hemopexin system can be seen to work alongside the CD163- and haptoglobin-mediated uptake of hemoglobin by splenic macrophages. The endocytosis of hemopexin activates the JNK/SAPK, NF-κB, and PKC pathways, while free heme does not (353, 354). Neuroprotection by hemopexin has been shown in mouse models of transient ischemia (225) and in intracerebral hemorrhage (57). Additionally, hemopexin prevents death from severe sepsis (209). Cells that do not express hemopexin receptors would require an alternative pathway for heme uptake, and heme carrier protein 1 (HCP1; also known as the proton-coupled folate transporter [PCFT]; SLC46A1) has been proposed as an alternative pathway for heme uptake (335). In fact, this protein has been suggested to be the intestinal heme transporter responsible for dietary iron uptake from heme in carnivores and omnivores (335) (see Section III.A). Importantly, a later article has questioned this hypothesis and has provided evidence that PCFT/HCP1 is much more efficient in transporting folate than heme (301).
III. Intracellular Transport of Iron
As iron has the potential to damage cells, iron transport, as well as iron uptake and storage, must be tightly regulated. In this section, we discuss the transport of iron across the enterocyte, the transport across the blood–brain barrier, and finally, intracellular iron transport into mitochondria.
A. Iron transport across the enterocyte
Dietary iron is present in two forms: heme and nonheme iron (146). Dietary heme iron is absorbed more efficiently than nonheme iron (see below), is derived from ingested heme-containing proteins such as hemoglobin and myoglobin, and is released by low-pH proteolytic activity in the stomach. Heme is thought to be absorbed across the duodenal brush border, but the mechanisms involved are poorly understood. While multiple heme transporters may be expressed at the enterocyte apical membrane, one candidate is PCFT/HCP1 (335). This protein is highly expressed at the enterocyte apical membrane, is expressed apically under conditions of iron deficiency, and is located intracellularly in endosomes during iron repletion (335). These observations suggest that HCP1 apical expression, and presumably heme transport activity, is negatively regulated by iron status. Heme binding to HCP1 induces receptor-mediated endocytosis, with the imported heme appearing in intracellular vesicles (417). The heme is then oxidized by HO1 and/or HO2 (302), and the contained iron is thought to be released in a similar manner to the hemopexin system (see above, section II.B). The resulting mobilized iron probably then enters the same cytoplasmic pool of labile iron as for nonheme iron (146) (see below). As indicated above, the finding that PCFT/HCP1 transports folate with a far greater affinity than heme (301) suggests that heme transport is not the major function of PCFT/HCP1, although the protein may nonetheless function as an intestinal heme transporter.
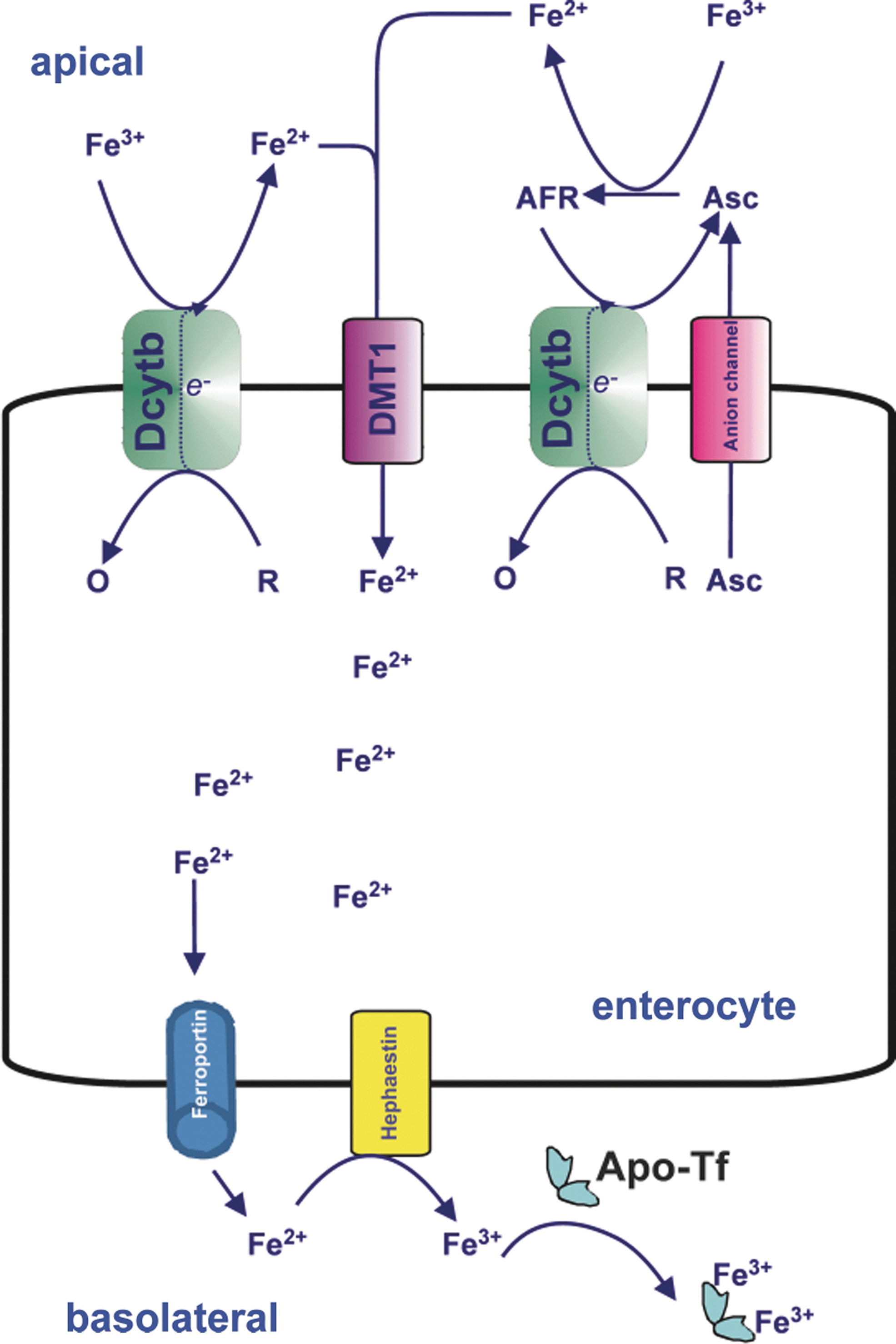
Dietary nonheme iron, which includes all other forms of dietary iron, is liberated as free or low-M r iron from carrier molecules (e.g., ferritin) within the acidic environment of the stomach. This iron remains soluble as long as the environment remains acidic and reducing, the latter of which will promote formation of ferrous iron (146). Importantly, ferrous iron is vastly more soluble than ferric iron [10−1 M vs. 10−18 M at physiological pH, respectively (29)], and so a redox equilibrium in favor of ferrous iron formation will promote absorption. Once liberated, this low-M r iron is mainly taken up by the brush border of duodenal enterocytes (97). Most nonheme iron in the duodenal lumen is probably present as low-M r chelates of iron (III). As discussed in section II.B., before iron can be transported by DMT1 at the enterocyte apical membrane, it must be reduced to iron (II). At present, the common view is that this reduction is mediated by apical membrane Dcytb (247) and/or possibly other reductases. However, there is evidence that the reduction of nonheme iron in the extracellular milieu may be achieved by nonenzymatic ferrireduction with the help of endogenous reductants such as ascorbate (10, 203, 245) and/or superoxide (124). However, due to its higher reducing capacity per molecule (2 e −/molecule of ascorbate compared to 1 e −/molecule of superoxide), ascorbate may be more significant on a quantitative basis. Amino acids such as cysteine may also be involved in reducing ferric to ferrous iron in the gut (146). The iron (II) formed is then thought to be transported into the enterocyte via DMT1 (139) or ZIP14 (294). There is evidence that at least half the iron transported across the enterocyte uses a vesicular pathway of transcytosis (235). Iron that reaches the basolateral membrane by poorly understood mechanisms can then be transported into the circulation by the only-known iron export protein, ferroportin (FPN1; also known as SLC40A1, metal transporter protein 1 [MTP1], or iron-regulated transporter 1 [IREG1]) (1, 91, 246). The trans-membrane ferroxidase, hephaestin, is associated with ferroportin and is believed to oxidize the exported iron (II) back to iron (III), probably immediately subsequent to its export by ferroportin, after/during which it is complexed to Tf for transport through the circulation (398). A summary of iron transport across the enterocyte is given in Figure 5.
B. Iron transport across the blood–brain barrier
In a similar manner to iron transport into the cell, iron transport across the blood–brain barrier is tightly regulated (260, 261). It is believed that the major route of iron import into the brain is that of Tf-bound iron across the luminal membrane of the capillary endothelium (31, 239, 260). This uptake follows the pathway discussed under Section II.A. Other pathways suggested to play a role in iron transport across the blood–brain barrier are the lactoferrin receptor/lactoferrin pathway (101, 106, 371) and the glycosylphosphatidylinositol (GPI)-anchored melanotransferrin/soluble melanotransferrin pathway (265). However, significant involvement of the latter pathway has been disputed, at least in the rat, as most of the administered iron bound to soluble melanotransferrin was retained in the liver and kidney and did not reach the brain (309). Moreover, as melanotransferrin knockout mice are viable and fertile, show no physical abnormalities, and develop normally, melanotransferrin clearly does not play a crucial role in iron metabolism (331). As discussed for other cell types under Section II.B, there is also some evidence for NTBI transport across the blood–brain barrier (39, 88). It has been suggested that the release of NTBI into the interstitial fluid (262) follows the same mechanism as the release of iron from the enterocyte into the circulation (319). However, as brain iron uptake appears to be regulated by TfR1 expression in brain capillary endothelial cells, a likely route for iron uptake across the blood–brain barrier is the initial uptake of Tf-bound iron by these cells followed by the ferroportin-dependent efflux of NTBI into the interstitial space on the abluminal side of the endothelium (318). Astrocytes have been proposed to take up iron (II) that is released by endothelial cells through their end-foot processes that are in close contact with the capillary endothelia (93, 239). Likely, Fe2+ transporters in brain cells such as astrocytes and neurons include DMT1 (151, 419) and ZIP14 (24, 127), although the contribution of DMT1 to neuronal iron uptake under physiological conditions has recently been questioned (289). Interestingly, while DMT1 is typically considered to be a proton symporter that strictly requires cotransport of protons with Fe2+ ions (e.g., in its capacity as an Fe2+ transporter in the endosomal Tf cycle), there is evidence that questions this assumption. Indeed, it has been demonstrated that while DMT1 Fe2+ conductance is optimal at acidic pH values (425), at higher extracellular pH values (e.g., pH 7.4), DMT1 has an Fe2+ transport activity that is uncoupled from proton transport (236). This observation is important as it solves the apparent paradox of a proton-coupled transporter (DMT1) having transport activity at pH values> pH 7 where proton concentrations are less than hydroxide ion concentrations. This may be particularly relevant for DMT1's proposed role in iron uptake by brain cells such as astrocytes (94, 206, 386), which are located in an extracellular fluid of a pH of ∼7.2 under physiological conditions. The redistribution of iron within the brain parenchyma remains unclear, but probably involves subsequent complexing of Fe(III) to Tf that is produced and secreted by oligodendrocytes, followed by the Tf-TfR1-dependent iron uptake by brain cells that express TfR1 (e.g., neurons) (318).
C. Iron transport to mitochondria
Mitochondria play an important role in iron metabolism, as they are the site of heme synthesis (215) and the major site for the biogenesis of ISCs (420). As many of the proteins depending on these iron-containing groups play vital roles in cellular metabolism, the deregulation of mitochondrial iron metabolism often leads to severe disease outcomes. In fact, many neurodegenerative diseases are caused by such disruptions in mitochondrial iron processing (156). The role of mitochondria in cellular iron homeostasis has been recently reviewed in this journal (159); thus, we give here only a brief summary. Iron is taken up by the mitochondria by one or more of the following mechanisms: (i) direct uptake of iron (II) from the cytosol, driven by the mitochondrial membrane potential (ΔΨm) (207); (ii) uptake of a chelator-inaccessible low-M r iron pool from the cytosol (343); and/or (iii) by a kiss-and-run mechanism, at least in hemoglobin-synthesizing cells such as reticulocytes, in which Tf-laden endosomes make brief contact with the outer mitochondrial membrane (311, 339).
Iron is transported across the inner mitochondrial membrane by a mitochondrial-specific iron transport protein, mitoferrin (Mfrn1) (334). Although the precise biochemistry of Mfrn1-mediated iron import by mitochondria is unknown, Mfrn1 interacts with the inner mitochondrial membrane ATP-binding cassette transporter, Abcb10, which is highly expressed in the erythroid mitochondria and increases Mfrn1 stability and mitochondrial iron import. This interaction probably promotes efficient heme synthesis by “funneling” iron directly to ferrochelatase, the enzyme that inserts iron into protoporphyrin IX to form heme. Indeed, a complex forming between Mfrn1, Abcb10, and ferrochelatase has recently been discovered (52). As a more in-depth discussion of mitochondrial iron transport and handling, as well as the disease states resulting from their derangement, is beyond the scope of this review, we refer readers to several recent reviews (159, 311, 336).
D. Macrophage-mediated recycling of red cell iron via erythrophagocytosis
As discussed above, ∼80% of body iron is found within the hemoglobin of the red blood cell population (197a). While only 1–2 mg of dietary iron is typically required per day to balance losses, ∼25 mg of iron is required for the erythropoiesis that leads to the daily production of 200 billion new red blood cells (197a). The majority of this iron demand of erythropoiesis derives from the highly efficient recycling of hemoglobin-derived iron from senescent and damaged red blood cells, which occurs at a rate of ∼2×106 red cells/s (337). Reticuloendothelial cells (also known as the mononuclear phagocyte system), which include splenic macrophages and Kupffer cells of the liver, play this crucial recycling role (43, 197a). After engulfment of a senescent erythrocyte, the red blood cell is dismantled, and the hemoglobin is released intravesicularly (33). Proteolytic activity releases the heme, and HO1 is involved in the liberation of the contained iron (197a). The iron is then thought to be released from the phagocytic vesicle lumen by a process that is analogous to iron recycling by the autophagic degradation of ferritin and, as such, requires the action of a ferrous-selective trans-membrane iron transporter (201). Regarding the identity of this transporter, recent data strongly suggest the involvement of Nramp1, a homolog of DMT1 that is expressed exclusively in phagocytic cells such as macrophages and neutrophils (359). Instructively, Nramp1 knockout mice accumulated iron within the liver and spleen during erythrophagocytosis, whereas wild-type mice efficiently recycled erythrocyte-derived iron to the bone marrow and nascent red cells (359). In fact, even more recent data suggest that DMT1 may provide some functional overlap with Nramp1 in macrophage-mediated erythrophagocytosis (360). While RAW264.7 macrophages lacking either functional DMT1 or Nramp1 experienced moderate reductions in the iron-recycling efficiency, those with a deficiency in both proteins experienced markedly more severe reductions in their iron-recycling efficiency (360).
The iron released from the phagocytic lumen probably then enters a common transit pool of iron, which is typically referred to as the LIP (see Section IV.A below for further discussion). This iron is released to the plasma by ferroportin to be complexed to Tf for delivery to the erythropoietic bone marrow [for a recent review of macrophage-mediated iron release, see Ref. (43)], and macrophage ferroportin expression is induced by erythrophagocytosis (191). There is also evidence that macrophage-derived ferritin iron may also serve as a source of iron for erythroid development, particularly in the absence of holo-Tf (219).
IV. Intracellular Iron Storage
A. Labile iron pool
In the cytosol, most iron is stored in ferritin. However, the uptake of iron (cf. Section II) does not appear to involve this protein. As a result, iron uptake (at least in nonerythroid cells) results in the transient existence of some cytosolic iron that is loosely bound (i.e., labile) and readily accessible to iron chelators (37). This iron was termed the labile iron pool by Greenberg and Wintrobe (133) in 1946 and the transient iron pool by Jacobs (170) in 1977. The LIP is defined as the redox-active, chelator-accessible component of intracellular iron (180) and can be determined by measuring the quenching of fluorescence of intracellularly located metallosensors such as calcein. It can consist of iron (II) and iron (III) (206). The LIP is likely to be in a dynamic equilibrium with low-M r chelators such as citrate (180) and organophosphates, or bound with moderate (i.e., micromolar) affinities to intracellular proteins that may function as iron chaperones, such as the poly(rC)-binding protein 1 (PCBP1) that delivers cytosolic iron to ferritin (340). However, no convincing data exist on the exact biochemical nature of the LIP and its complexants. From a clinical perspective, the LIP is redox-active and can generate oxidative stress through the Fenton- and Haber–Weiss-type reactions (see Section I), and it has been implicated in the pathogenesis of many diseases, including coronary heart disease (199), cancer (310), muscle fatigue (306), and osteoporosis (384). In addition to a cytosolic LIP, the existence of a mitochondrial and nuclear LIP has been suggested (37).
B. Cytoplasmic iron storage
Most iron storage occurs in hepatocytes and, in the shorter term, in reticuloendothelial macrophages. After transiently entering the LIP in nonerythroid cells, the majority of the iron (70%–80%) is incorporated into ferritin (422). Ferritin is a water-soluble molecule composed of 24 subunits that form a hollow sphere accommodating up to 4500 atoms of iron as a mineralized core consisting of ferric, phosphate, and hydroxide ions (9, 147). Mammals express two major ferritin subunits: H-ferritin (heavy; also known as FTH1) and L-ferritin (light; also known as FTL), which by differing combinations form a wide range of different isoferritins with tissue-specific distribution (9). When iron (II) binds to ferritin, it is readily oxidized by the intrinsic ferroxidase activity of the H-ferritin in an oxygen-dependent reaction to iron (III) (which prevents any cellular Fenton reactions from occurring), after which L-ferritins, which are devoid of ferroxidase activity, facilitate nucleation, and mineralization of the iron center (28, 147). The major mechanism of iron release from ferritin appears to be by proteolysis of the protein (196). However, reductive mobilization reactions that can operate independently of protein degradation have also been proposed (92). Incomplete ferritin degradation is believed to result in the formation of hemosiderin, a protein mainly found in the lysosomes of reticuloendothelial macrophages (110) (see Section III.D). Hemosiderin is capable of binding iron, but less capable of releasing it than ferritin (281). A small fraction of the cellular ferritin is transported into the nucleus [which appears to occur by a mechanism not involving a nuclear localization signal (41)], where it may play a protective role against DNA damage (42).
C. Mitochondrial iron storage
Some mammalian tissues express a mitochondrial-specific ferritin that has a high level of sequence identity with H-ferritin. Mitochondrial ferritin, which is nuclear encoded, has a long N-terminal extension of about 60 amino acids that contains a mitochondrial targeting sequence (223), and forms a homopolymer in the mitochondrial matrix (75). The highest expression of mitochondrial ferritin is observed in the testis, whereas it appears to be completely absent from the iron-storage organs, the liver and spleen. Moreover, its expression level appears to be better correlated with mitochondrial number rather than to cellular iron content (95). Similar to cytosolic H-ferritin, mitochondrial ferritin has ferroxidase activity and stores the iron as iron (III) (75). The mitochondrial ferritin gene is intron-less and, in contrast to those encoding cytosolic ferritins, does not contain an iron-responsive element (IRE; see next section). Consequently, its expression is not post-transcriptionally regulated by intracellular iron concentration (95). As mitochondria may contain significant levels of redox-active iron and are a major source of cellular free radicals (159, 311), mitochondrial ferritin is likely to play a protective role; a notion supported by the finding that in yeast cells lacking the gene for the yeast frataxin homolog (Yfh1), respiratory function can be rescued by expressing human mitochondrial ferritin (48). Similarly, mitochondrial ferritin expression rescues mammalian cells in which frataxin expression has been downregulated by small-interfering RNA (siRNA) (424).
V. Cellular Roles of Iron
Many of the proteins involved in the import, export, and sequestration of iron are themselves regulated by cellular iron concentration. In the next section, we discuss the various levels at which cellular regulation in response to iron occurs. Although the post-transcriptional regulation of genes involved in cellular iron metabolism is the primary homeostatic response to changes in intracellular iron status (see Sections V.B and VI.A), many genes, including many iron metabolism genes, are also regulated at the transcriptional level by iron and other factors. This transcriptional regulation typically occurs in response to combinations of cellular iron status, cellular oxygen status, and/or cytokine signaling.
A. Regulation of transcription by iron
Several genes involved in iron metabolism have been reported to be regulated in an iron-dependent manner at the transcriptional level. These include the transcription of the Dcytb gene (also known as CYBRD1), which lacks an IRE sequence in its mRNA and is upregulated in mice deprived of iron (247). This finding is a strong argument in favor of Dcytb involvement in iron uptake. However, as discussed in Section II.B, the exact mechanism by which Dcytb is transcriptionally regulated by iron is still not known.
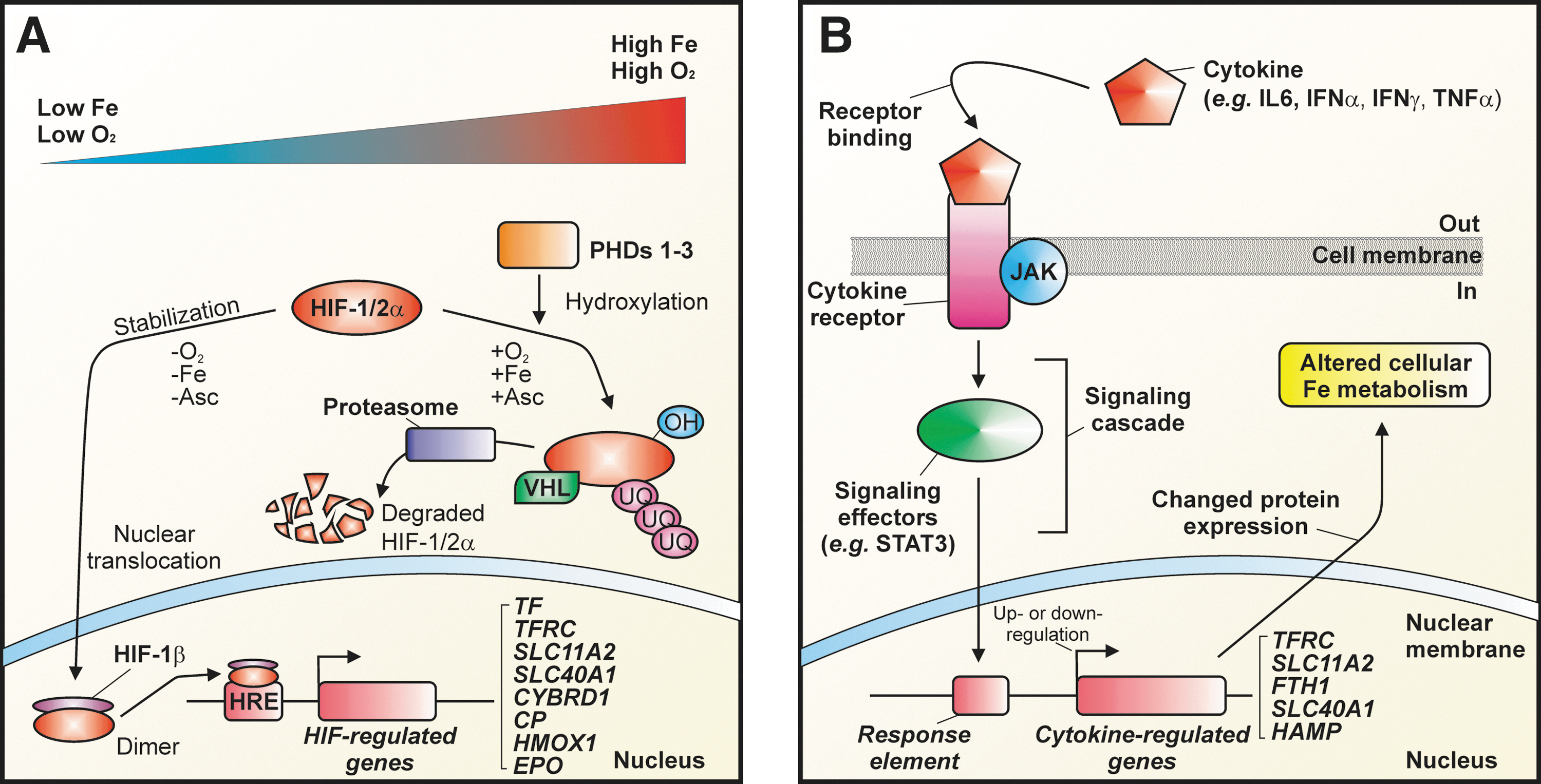
Low oxygen pressure, as well as low intracellular iron concentration, activates hypoxia-inducible factor (HIF)-1- and/or HIF-2-regulated transcription by the increased formation of heterodimers of HIF-1α or HIF-2α and the constitutively expressed HIF-1β subunit (also known as the aryl hydrocarbon receptor nuclear translocator) (50). HIF-1α is ubiquitously expressed, whereas HIF-2α has a more restricted tissue distribution (414). The HIFα/β heterodimers form transcription factors that regulate a wide range of genes encoding proteins that are important for cellular oxygen homeostasis and the response to hypoxia (Fig. 6A). Both HIF-1α and HIF-2α are post-translationally regulated at the level of protein degradation in an oxygen-dependent and iron-dependent manner by a specific class of 2-oxoglutarate-dependent dioxygenases: the prolyl-4-hydroxylase domain-containing iron-dependent prolyl hydroxylases (PHDs 1–3) and the asparaginyl hydroxylase, a factor inhibiting HIF [for a recent review see Ref. (258)]. The PHD-type hydroxylases, which are believed to be similar to those involved the targeting of iron-regulatory protein (IRP)2 for degradation, are typically fully active under conditions of normoxia and iron repletion and hydroxylate HIFα proteins at specific proline residues (112, 287). Importantly, it is the strict dependence of these hydroxylases on iron that is presumed to be largely responsible for the ability of cellular iron levels to regulate HIF-regulated gene expression (60, 144, 258). The hydroxylated α-subunits of HIF are then targeted for ubiquitination by the E3 ubquitin ligase and von Hippel-Lindau tumor suppressor protein (VHL), which earmarks the proteins for degradation by the proteasome (50, 68, 166, 169, 244). Under hypoxic conditions, the hydroxylated α-subunits are stabilized and bind as HIFα/β heterodimers to hypoxia-response elements (i.e., the consensus sequence 5′-XCGTG-3′, where X=A, T, or G in either the 3′ or 5′ regions flanking the gene) in a variety of genes [e.g., the gene encoding erythropoietin (EPO) (50, 217)]. This leads to increased erythropoiesis and therefore an increased demand for iron, as well as the regulation of other key proteins involved in iron metabolism, including Tf (315), TfR1 (23, 231, 368), DMT1 (226, 241, 300, 332), ferroportin (246), ceruloplasmin (267), Dcytb (212, 247, 332), and possibly hepcidin (291) (Fig. 6A). Notably, recent data question the hypothesis that HIFs directly downregulate hepatic hepcidin transcription by binding to the HAMP promoter (394).
B. Post-transcriptional regulation by iron
Many iron metabolism proteins are iron-regulated at the post-transcriptional level. Proteins whose corresponding mRNAs contain an IRE in the 5′-untranslated region (5′-UTR) are positively regulated in response to an increase in cellular iron concentration (viz. the LIP). The typical IRE contains a hairpin structure containing a conserved consensus motif (Fig. 7) to which one of two IRP isoforms (IRP1 and IRP2) can bind (the two IRP isoforms are discussed in detail below). IREs are heterogeneous sequences of 28–30 nucleotides in the noncoding 5′ and 3′ UTRs of specific mRNAs that typically encode proteins involved iron and/or oxidative metabolism (see below). These sequences form stem–loop secondary structures that can be contacted by IRPs at multiple sites. The main IRE consensus motif for IRP binding is the conserved sequence, 5′-CAGUGX-3′ (where X=U, C or A), which occurs within a six-residue hairpin loop atop a stem containing an unpaired cytosine (Fig. 7) (319, 376, 395). IRPs bind to IREs with high affinity in iron-depleted cells, either suppressing the translation of the mRNA (i.e., mRNAs in which the IRE is located in the 5′-UTR; e.g., H- or L-ferritin), or by enhancing the mRNA stability against nuclease attack (i.e., mRNAs in which the IRE is located in the 3′-UTR; e.g., TfR1 and DMT1). In either case, ultimately it is the de novo synthesis of the encoded protein that is regulated (Fig. 8) (401).

A single functional IRE exists in the 5′-UTRs of the mRNAs for H- and L-ferritin (324), ferroportin (246), erythroid δ-aminolevulinic acid synthase (eALAS) (79), mitochondrial aconitase (mAcon) (433), and the iron–protein subunit of Drosophila melanogaster succinate dehydrogenase (192). An IRE-like structure was also described for the 5′-UTR of α-synuclein (119). If this turns out to be a functional IRE, it would offer a mechanism for increased α-synuclein expression in response to an increased iron load in the substantia nigra of aged brains.
TfR1 mRNAs contain multiple IREs that are located in the 3′-UTR (49, 268). IRP binding to 3′-UTRs results in stabilization of the mRNA under conditions of iron depletion, while loss of IRP binding under conditions of iron repletion leads to rapid mRNA degradation by nucleases (49) (Fig. 8). DMT1 can be encoded by one of four variant mRNA transcripts depending on whether translation starts from exon 1A or 1B (i.e., 5′-end processing variants) and whether or not the 3′-UTR contains an IRE (i.e., 3′-end processing variants). Two of the four DMT1 mRNA splice variants contain a single IRE in their 3′-UTR, which preferentially binds IRP1, leading to mRNA stabilization as described for TfR1 mRNA (138). While the exon 1A form of DMT1 is ubiquitous, the exon 1A form is present mainly in the duodenum and kidney, with the+IRE forms contributing to the post-transcriptionally iron-regulated expression of DMT1 (160). In addition, potential IREs have been documented in the 3′-UTRs of the mRNAs for glycolate oxidase (193), myotonic dystrophy kinase-related Cdc42-binding kinase α (67), and a splice variant of cell division cycle 14A mRNAs (325).
Importantly, the different IREs are bound by the IRPs with a high, but varying, affinity [i.e., K D ∼20–100 pM (266)]. For example, IRP2 binds the H-ferritin and L-ferritin IREs more tightly than it binds the mAcon IRE (186), and consequently, the post-transcriptional regulation of ferritin is more sensitive than mAcon to changes in intracellular iron (58, 327). This has the important consequence that in response to changes in intracellular iron, ferritin translation will be regulated more rapidly than translation of mAcon (327, 376). This behavior has been termed combinatorial control (376), and it provides cells with a means to fine-tune cellular responses to changes in the intracellular iron milieu (376).
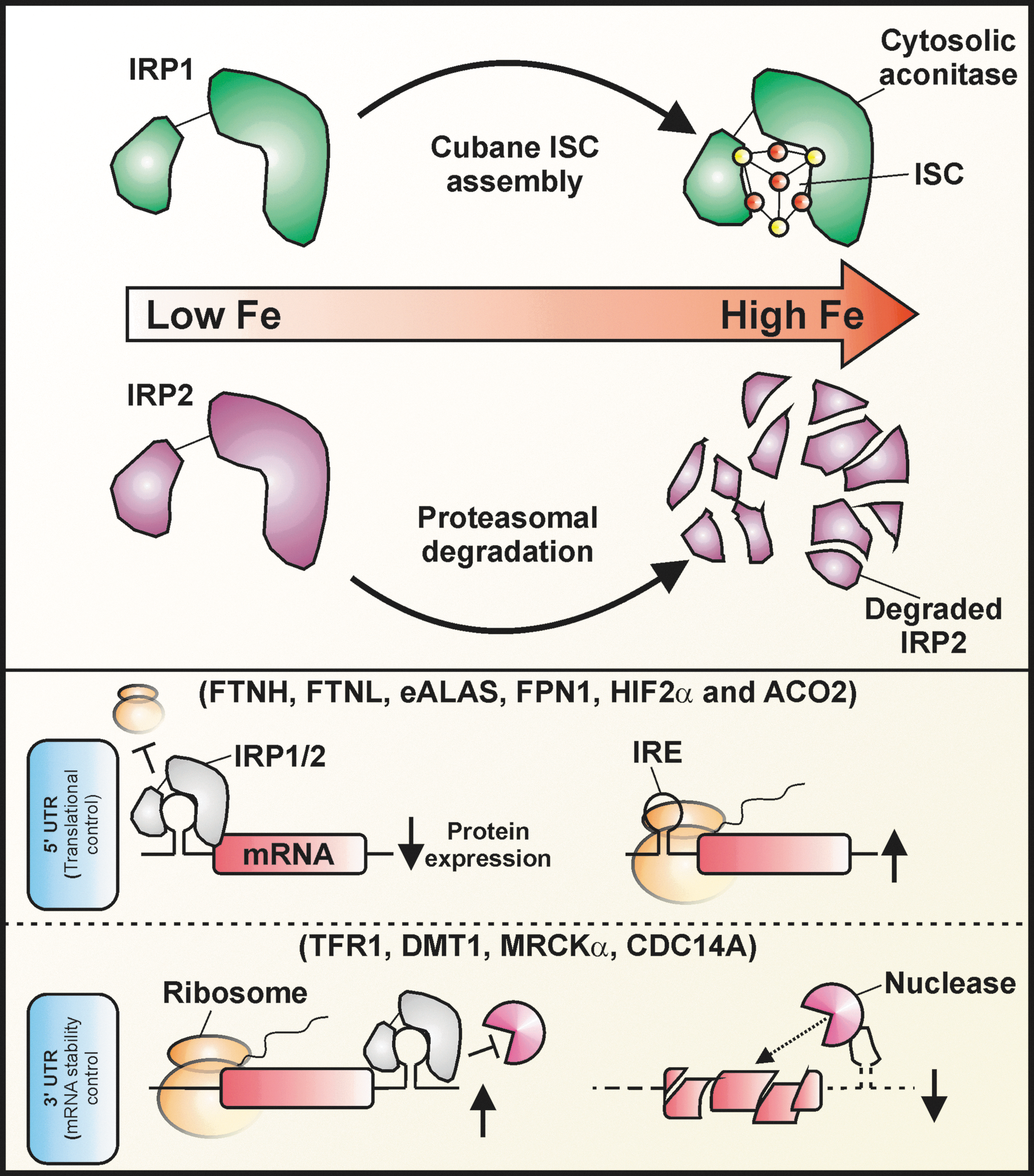
As indicated above, two homologous IRE-binding proteins have been identified (viz. IRPs 1 and 2) that possess a high degree of amino acid sequence identity [i.e., 64% in humans (269)] [for a recent review, see Ref. (307)]. IRPs 1 and 2 are members of the aconitase gene family, and are probably derived from gene duplication events. Despite their homology, the molecular responses of IRPs 1 and 2 to iron levels are intrinsically different. IRP1 is an intriguing bifunctional protein that responds to intracellular iron primarily through an ISC-switch mechanism (395). Under conditions of increased cellular iron, which can be potentiated by reductants such as ascorbate (380), IRP1 loses its IRE-binding activity by acquiring a cubane ISC ([4Fe-4S] cluster) (319). The acquisition of this [4Fe-4S] cluster converts IRP1 into a cytosolic aconitase: an enzyme capable of catalyzing the stereospecific isomerization of citrate to isocitrate via cis-aconitate (296). Conversely, when intracellular iron levels are low, and/or in response to various ISC-destabilizing oxidants such as nitric oxide (30) and hydrogen peroxide (44), the [4Fe-4S] cluster within IRP1 disassembles and reveals the protein's latent IRE-binding activity. These dual activities of IRP1 are mutually exclusive. In addition, IRP1 can also be modulated by selective protein degradation under certain circumstances (66, 406). For instance, Clarke et al. (66) observed that an IRP1 mutant, in which all cluster-ligating cysteines were mutated to serine (thereby preventing assembly of the [4Fe–4S] cluster), was subject to iron-dependent protein degradation in cell culture models. These authors also observed that iron-dependent IRP1 degradation occurred in two different mouse models (i.e., the SOD1 −/− mouse and the ABCBlv/Y mouse) with perturbed ISC metabolism (66). Similarly, Wang et al. (406) observed that an IRP1C437S mutant, which fails to form a [4Fe-4S] cluster, is subject to iron-dependent protein degradation in transfected H1299 cells. Moreover, the inhibition of normal ISC biogenesis in HeLa cells by the siRNA-mediated knockdown of the mitochondrial cysteine desulfurase, NFS1, sensitized endogenous IRP1 to iron-dependent degradation (406). The inhibition of this degradation by the proteasome inhibitors MG132 and lactacystin strongly suggests the involvement of the ubiquitin–proteasome pathway (406). Evidence also suggests that, in addition to regulating the conversion of IRP1 to a cytosolic aconitase (296), phosphorylation of IRP1 potentiates the iron-dependent regulation of IRP1 abundance (105). Although the regulation of ISC formation in IRP1 may be the normal physiologic regulator of IRP1's IRE-binding activity, in the face of defective ISC biogenesis, IRP1 half-life can be regulated (66, 404).
Unlike IRP1, the iron-dependent regulation of IRP2 abundance appears to be the primary mechanism by which this protein is regulated in response to iron (307, 319). Under conditions of iron repletion, IRP2 expression, and consequently the binding of IREs by IRP2, is diminished by an increase in iron-dependent degradation of the protein via the ubiquitin–proteasome pathway (141). IRP2 does not form ISCs and exhibits no aconitase activity, despite the possession of some of the cysteines required for this activity in IRP1. The mechanism by which IRP2 is targeted for degradation in an iron-dependent manner remains controversial. Relative to IRP1, IRP2 contains an additional cysteine-rich 73-amino-acid insertion at the N-terminus, which is encoded by an extra exon. The cysteine oxidation model postulates that site-specific, iron-dependent oxidation within this cysteine-rich region is required for the targeting of IRP2 for proteasomal degradation under conditions of iron repletion (168). However, several studies indicate that the 73-amino-acid domain is not necessary for this activity (29, 405). Another model proposes the involvement of heme-dependent oxidation of specific cysteines within a so-called heme regulatory motif in IRP2 (29, 165, 173, 418). A third model proposes that IRP2 is instead regulated by iron-dependent hydroxylases, in a similar manner to those discussed above (see Section V.A) in relation to HIFα hydroxylation activity (405). The strict iron dependence of these enzymes could explain, at least in part, the iron dependence of IRP2 degradation. The proposed dependence of IRP2 degradation on hydroxylases also helps explain the capacity of reductants such as ascorbate, α-tocopherol, and N-acetylcysteine to stimulate IRP2 degradation, rather than inhibit degradation as the protein oxidation models would predict (45). Reductants such as ascorbate stimulate the hydroxylating activity of iron-dependent 2-oxoglutarate-dependent dioxygenases (112, 363).
Both genetic ablation and silencing studies strongly suggest that IRP2 is the major IRP involved in the IRE-IRP system under normal circumstances. Mice with a targeted deletion of IRP2 (IRP2 −/−) develop microcytic anemia and progressive neurodegeneration associated with functional cellular iron depletion, which is caused by decreased expression of TfR1 and overexpression of ferritin in multiple tissues (213). Moreover, IRP2-deficient mice overexpress eALAS in erythroid cells, leading to >200-fold production of the mitochondrial heme precursor, protoporphyrin IX, compared to wild-type controls (72). Cells taken from Irp2 −/− mice show deregulated iron metabolism, but only when the oxygen tension is lowered from the standard 21%, found under normal cell culture conditions, to levels found in actual tissues (e.g., ∼3%–6%) (252). This is probably due to the fact that IRP1 is predominantly in the cytosolic aconitase form under conditions of low oxygen tension, but is converted to its IRE-binding form under conditions of raised oxygen tension (e.g., 21%). Importantly, while the combined ablation of both Irp1 and Irp2 in mice is embryonically lethal (358), the targeted deletion of Irp1 alone appears to deregulate iron metabolism only in brown fat and kidney (253). Importantly, Irp1 is highly expressed in these tissues and exceeds that of Irp2 (253). Moreover, mice that are homozygous for Irp2 ablation and heterozygous for Irp1 ablation (i.e., Irp2 −/−, Irp1+/ −) develop a more severe form of neurodegeneration than Irp2 −/− mice (358). Moreover, cells that are stably silenced for both IRP1 and IRP2 expression show a greater loss of IRE-binding activity, as well as perturbation of ferritin and TfR1 regulation in response to iron, than cells silenced for only IRP1 or IRP2 (408). Importantly, cells that are silenced only for IRP2 show perturbation of ferritin and TfR1 in response to iron, whereas cells silenced only for IRP1 do not (408). Taken together, these results indicate that there is some functional overlap between the two IRPs, and that IRP2 is clearly the dominant IRP in vivo. The ability of both IRPs to regulate ferritin expression similarly [although not equivalently (408)] in vitro is a probable result of the nonphysiologic oxygen tension employed under these conditions. That is, the supraphysiologic oxygen tension in room air experienced under typical cell culture conditions likely promotes the conversion of the cytosolic aconitase form of IRP1, which predominates in vivo, to its IRE-binding form.
Interestingly, the HIF2α transcript contains an IRE (326) and is post-transcriptionally regulated by IRP2 (434), which further illustrate the complex crosstalk between cellular and systemic regulators of iron metabolism (266).
C. Regulation of protein degradation by iron
In addition to the regulation of the expression of proteins involved in iron metabolism by both post-transcriptional/translational and transcriptional mechanisms, selective protein degradation is a crucial post-translational mechanism for regulating protein expression. As discussed above, the expression of IRP2 and IRP1 under certain circumstances as well as the HIFα proteins can be regulated at the level of protein degradation by the ubiquitin–proteasome system. The IRP2-targeting E3 ubiquitin ligase, FBXL5, is itself regulated by iron and oxygen. This protein contains a hemerythrin domain capable of binding iron and oxygen, and its rate of degradation is increased upon iron and/or oxygen binding (323). Essentially, this form of regulation modulates the expression of specific proteins by adjusting the protein half-life. As discussed further in Sections VI.B and C, the keystone role that is played by hepcidin in the regulation of systemic iron metabolism hinges on the stimulation of ferroportin degradation by the lysosome. Other iron metabolism proteins that are known to be regulated by changes in protein half-life include TfR2, whose half-life is extended in response to binding to diferric Tf (see below). Intriguingly, DMT1 expression has also recently been shown to be modulated by iron-dependent ubiquitination and degradation by the proteasomal (32, 117, 317) and/or lysosomal (117) degradative pathways. Interestingly, the proteasomal degradation of DMT1 can be triggered by hepcidin (32), providing yet another linkage between cellular and systemic iron homeostasis.
D. Regulation of enzyme activity by iron
Several enzymes contain iron as a prosthetic group. Especially, in the case of nonheme iron, the iron often can be readily dissociated from the protein under conditions of low cellular iron concentration. The loss of iron from ribonucleotide reductase, the rate-limiting enzyme in the de novo synthesis of all four deoxyribonucleotides, is believed to be one of the main reasons why iron depletion results in inhibition of cell growth and why iron chelators can have effective anticancer activity (423). Eukaryotic ribonucleotide reductases are composed of two different homodimers, R1 and R2. The catalytic site is located in R1, and the tyrosyl radical needed for the reaction is generated, stored, and delivered by R2, which also contains the iron atom. Although the enzyme can also bind manganese instead of iron, it appears to be dependent on iron for activity (155). Similar regulatory functions would be expected for most other nonheme iron enzymes listed in Section I.
VI. Regulation of Iron Homeostasis
Iron homeostasis is regulated at both the cellular and systemic levels, with substantive crosstalk across these levels (151, 261). Although mammals do not possess a regulated iron excretion pathway, iron is nonetheless continuously being lost through the sloughing of mucosal cells (e.g., gut epithelium and urothelium) and skin cells, or during bleeding and sweating. The typical daily amount of iron lost through these processes is ∼1–2 mg, which under normal circumstances is balanced by dietary intake (62). Crucially, systemic iron levels are ultimately regulated at the level of dietary absorption.
As discussed in Section III.A, the uptake of dietary iron across the duodenal epithelium involves numerous regulated steps. In both heme and nonheme iron uptake, the resulting intracellular iron (II) is then exported across the basolateral membrane and into the portal circulation of the blood stream via ferroportin. As detailed below, this efflux step is a critical regulatory checkpoint for iron absorption and consequently for systemic iron homeostasis. The ferroportin-mediated efflux of iron (II) is coupled to its extracellular reoxidation to iron (III) by hephaestin in the basolateral membrane. The homologous soluble ferroxidase, ceruloplasmin (148), which is an abundant plasma protein, but also occurs as a GPI-anchored form in the brain (see Section III.B), can also play a role in reoxidizing iron (II) upon or immediately after its efflux via ferroportin (148). This reoxidation of effluxed iron is considered to be crucial, as the major serum iron transporter, Tf, has two extremely high-affinity binding sites for iron (III) (see above) that have very low affinity for iron (II) (2, 3, 263).
Once bound to Tf, which occurs in normal adult plasma at concentrations of 20–30 μM (263, 421), Tf-bound iron is distributed via the circulation to the rest of the body. Importantly, the major determinant and indicator of systemic iron metabolism is the extent of saturation of plasma Tf, the latter of which can exist in apo-, mono-, and diferric forms (see above). As discussed below, the saturation level of Tf can signal for changes in the expression level of the hormone of iron metabolism, hepcidin, which then in turn affects the rate at which iron is transported via ferroportin into the plasma from several key cell types (i.e., duodenal enterocytes, reticuloendothelial macrophages, and hepatocytes). Additionally, liver iron levels (i.e., iron stores) can signal through the bone morphogenetic protein (BMP; see below) signaling pathway, while changes in serum Tf saturation (74, 305) appear to involve TfR2- and/or hereditary hemochromatosis protein (HFE)-dependent signaling. The various pathways of iron-dependent hepcidin regulation are discussed in detail in Section VI.C.
As discussed in detail in Section II.A, the uptake of Tf-bound iron is the major cellular iron uptake route under normal conditions. The uptake of iron by this pathway is tightly regulated by changes in the expression of the various proteins involved (e.g., TfR1 and DMT1). At the cellular level, iron levels can be homeostatically regulated by alterations in the expression and activity of proteins involved in the efflux (i.e., ferroportin) and intracellular storage of iron (i.e., via ferritins).
This section will examine both cellular and systemic iron homeostasis and provide an overview of recent advances made in these fields and remaining open questions.
A. Overview of cellular iron homeostasis
The regulation of cellular iron metabolism is coordinated by an intricate web of changes in the expression and/or activity of proteins involved in iron uptake, storage/utilization, and release. These changes that occur as a result of feedback loops that are activated in response to alterations in iron levels, oxygen tension, and/or oxidants are vital for (i) fine-tuning the rate at which iron is made available within the cell for metabolic usage; and (ii) minimizing the deleterious effects that result from an excess of redox-labile or free iron in sensitive subcellular compartments. The major mechanisms involved in this regulation occur at transcriptional, post-transcriptional, and post-translational levels. These tiers of regulation were discussed in detail in Sections V.A–C.
The IRE-IRP is the central regulator of cellular iron metabolism, and allows for rapid alterations in protein synthesis in response to fluctuations in intracellular labile iron concentrations. It is interesting to speculate that as the control of protein synthesis allowed by the IRE-IRP system occurs at the level of the translation in the cytosol, the response is probably more rapid than that permitted by pure transcriptional control (see below), due to the potentially rate-limiting requirements for the latter, including the initiation of transcription, mRNA processing in the nucleus, and export of mRNAs to the cytosol.
As indicated in Section V.A, the expression of proteins, including some of those involved in iron metabolism, can also be regulated transcriptionally by iron in an HIF-dependent manner. Additionally, non-HIF-dependent mechanisms can also transcriptionally regulate such proteins. Indeed, several inflammatory cytokines, including interferon-γ and interleukins 1, 2, and 6, can alter the transcription of mRNAs of key iron metabolism proteins. These include the mRNAs for H-ferritin (379, 381), hepcidin (218, 273), TfR1 (232, 272), and ferroportin (232), both in vitro and in vivo. For example, in human monocytes, combined treatment with the proinflammatory agents, interferon-γ and lipopolysaccharide, decreased TfR1 and ferroportin mRNA and protein levels while increasing DMT1 mRNA and protein levels. The net effect of this treatment was a decrease in Tf-dependent iron uptake and cellular iron export, accompanied by a concomitant increase in NTBI uptake (232). The regulation of iron metabolism by cytokines, particularly in relation to the interleukin-6-dependent upregulation of hepcidin expression, is a key pathogenic mechanism in the anemia of chronic disease (ACD) (122, 411, 412) (see Section VII.A).
In general, the binding of cytokines to their cognate cell surface receptors initiates a signaling cascade that regulates transcription of key genes within the nucleus (Fig. 6B). For example, interleukin-6 increases expression of key genes (e.g., HAMP, which encodes hepcidin) through Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling. Ferritin expression in cultured hepatoma cells is regulated by cytokine signaling (381), and isolated monocytes produce hepcidin in response to interferon-α and interleukin-6 (431). The regulation of ferritin by cytokine is consistent with the behavior of this protein as an acute-phase reactant that is hepatically secreted in response to inflammation (379). In addition, the cytokine-dependent stimulation of ferritin expression in macrophages (197) is important, given their central role in the turnover of effete red blood cells (see Section III.D).
As discussed above, HIF proteins can also regulate the expression of iron metabolism genes. As the HIF hydroxylases that regulate HIFα expression levels at the level of protein degradation (discussed above; see section V.A) require iron [specifically, as iron (II)] for their enzymatic activity (284), the cellular iron status significantly impacts on the activity of these enzymes, and consequently on the expression level of HIFα proteins and HIF-regulated transcription (60, 144, 258). The strict dependence of prolyl hydroxylases on iron is presumed to be largely responsible for the ability of cellular iron levels to regulate HIF-regulated gene expression (60, 258). However, the observation that there are redox-sensitive sites within the prolyl hydroxylases that can be regulated by intracellular antioxidants (e.g., glutathione and/or ascorbate) may indicate that iron-dependent redox regulation of prolyl hydroxylases activity also occurs (280).
B. Overview of systemic iron homeostasis
In the past decade, it has emerged that the keystone regulator of systemic iron homeostasis is hepcidin (121). This small 25-amino-acid peptide is synthesized and secreted by the liver in an iron-dependent manner. Hepcidin is subsequently transported around the body via the circulation, probably bound to α2-macroglobulin (290), and interacts with the iron-efflux protein, ferroportin, in specific cell types (e.g., duodenal enterocytes, splenic macrophages, and iron-storing hepatocytes) (121) (Fig. 9). This interaction decreases ferroportin activity by triggering the degradation of ferroportin within lysosomes (274). This has the effect of decreasing the capacity for affected cells to release iron into the extracellular space, causing a transient increase in the intracellular LIP. This then triggers cellular homeostatic response mechanisms to iron repletion (e.g., decreasing the IRE-binding activity of the IRPs and regulating transcription of the HIF1- and HIF2-target genes). The net response to an increase in hepcidin is a decrease in the amount of iron entering the circulation (Fig. 9). This decreases both dietary iron absorption and TfR1-dependent iron uptake, while increasing iron storage in ferritin by cells (viz. hepatocytes and splenic macrophages). It should be noted however that hepcidin alone is not sufficient to restrict intestinal iron absorption, as mice lacking intestinal H-ferritin show increased body iron stores in spite of increased hepcidin mRNA expression, indicating that H-ferritin plays a limiting role in the hepcidin-dependent regulation of iron efflux from intestinal cells (387).

As will be discussed in more depth in Section VII, hepcidin deficiency induces iron overload, whereas an excess of hepcidin can result in anemia.
1. Models of systemic iron regulation
There are several overlapping and mutually inclusive models of systemic iron regulation, all of which were proposed before the emergence of hepcidin as a keystone regulator of iron homeostasis. It has long been appreciated that, given that no regulated and quantitatively significant pathway for iron excretion exists, systemic iron levels must ultimately be modulated at the level of enteric absorption. This regulation was thought to occur in response to duodenal enterocytes sensing the level of body iron stores and/or the iron requirements of erythropoiesis in the bone marrow. This sensing was presumed to rely on two (not necessarily distinct) regulators: the stores regulator and the erythroid regulator (27, 62, 109), respectively. The stores regulator was thought to be capable of signaling to the duodenal mucosa the levels of body iron stores in iron storage tissues such as the liver, while the erythroid regulator was thought to signal, again to the duodenal mucosa, the iron demand for erythropoiesis. In the first case, a decrease in body iron stores signals for an increase in enteric iron uptake, while in the second, an increase in iron demand for erythropoiesis, relative to iron supply, signals for an increase in enteric iron uptake (62, 109). Conversely, an increase in iron stores or a relative decrease in erythroid iron consumption reduces the signal and consequently decreases enteric iron uptake. In both cases, the regulatory mediators responsible were thought to be soluble plasma components that communicated from the sites of iron storage/utilization to the sites of iron absorption.
While the nature of the erythroid regulator remains uncertain, the BMP6/HJV pathway of hepcidin regulation (see Section VI.C) is a likely candidate for the stores regulator.
C. The hepcidin–ferroportin axis and its regulators
The activity of the hepcidin–ferroportin regulatory axis of systemic iron homeostasis is orchestrated by a host of effectors (151, 190, 348). Although incompletely understood, the emerging signaling pathways and their molecular components, which include BMPs, HFE, TfR2, hemojuvelin (HJV/HFE2), and the recently identified serine protease, matriptase 2 (also known as transmembrane protease, serine 6 [TMPRSS6]), are discussed below.
1. The pivotal role of hepcidin
Hepcidin (also known as LEAP-1) is a small peptide that is predominantly produced by hepatocytes in the liver (198, 288), but also at lower levels by the kidney (200) and possibly by the heart (249). The peptide is initially synthesized as an 84-amino-acid precursor in hepatocytes, which is subsequently enzymatically processed to yield the 25-amino-acid active form, and secreted into plasma. Hepcidin was first identified as a disulfide bond-rich peptide in human blood ultrafiltrate (198) and urine (288), and was initially found to have antimicrobial activity (198, 288). Shortly after its discovery, hepcidin was found to play a crucial role in regulating systemic iron homeostasis (275, 276, 293). The expression of the peptide was found to be regulated by iron, hypoxia, and inflammation (276), and murine hepcidin was found to be overexpressed during iron overload (293). The crucial role played by hepcidin has been demonstrated through genetic studies in which the targeted disruption of the HAMP gene (221), or the indirect loss of hepcidin expression through a targeted disruption of the upstream stimulatory factor 2 (275), leads to severe iron overload in the liver, pancreas, and heart, as well as an increase in plasma iron levels that exceed the Tf-binding capacity of the plasma.
The major molecular event triggered by plasma hepcidin is thought to be the internalization and the lysosomal degradation of ferroportin, which rapidly ensues upon the binding of hepcidin to ferroportin at the plasma membrane (274). In the plasma, hepcidin may circulate bound to the carrier protein, α2-macroglobulin, which also appears to increase the capacity for hepcidin to decrease in ferroportin expression, at least in vitro (290). This post-translational decrease in ferroportin expression ultimately inhibits iron entry into the plasma by downregulating the following iron efflux-dependent processes: (i) iron absorption (i.e., via reduced iron efflux into the portal circulation from duodenal enterocytes), (ii) iron recycling by splenic macrophages (i.e., via reduced iron efflux into the plasma after the phagocytic turnover of effete erythrocytes), and (iii) iron release from iron stored within hepatocytes (Fig. 9). Collectively, these events bring about a unified negative feedback response that decreases the amount of iron entering the plasma, thereby lowering the original effector.
Iron-dependent hepcidin expression is controlled primarily at the transcriptional level. The basal transcription of the HAMP gene requires the liver-enriched transcription factor, CCAAT/enhancer-binding protein-α (C/EBPα) (77). The iron-dependent upregulation of HAMP transcription is additionally dependent on the nuclear-translocatable small mothers of decapentaplegic (SMAD) transcription factors 1, 5 and 8, in conjunction with the mediator SMAD 4 (12), and is controlled by at least two parallel and probably interconnected signaling pathways involving (i) HFE, TfR2, and TfR1, and (ii) BMP (6) and its corresponding cell surface receptors, including the BMP coreceptor, HJV, as well as the serine protease, matriptase-2. The regulation of these two pathways and their possible points of interaction are reviewed below.
2. The HFE/TfR2 pathway
The relative abundance of holo-Tf in the plasma (i.e., typically dictated by the iron saturation level of Tf) is an important mediator of iron-dependent hepcidin expression. In fact, the saturation level of plasma Tf is also a classical indicator of systemic iron levels. As discussed above, Tf saturation is typically in the range of 20%–30% (83). An increase in Tf saturation signals to specific cells (e.g., hepactocytes) to increase their expression of hepcidin. The mechanism by which this activity occurs appears to depend on an intracellular signaling initiated by the interplay between holo-Tf, TfR1, HFE, and TfR2, although the BMP6/HJV pathway may also be involved (see below). TfR2 and HFE may collectively form an iron-sensing and signaling complex, or alternatively, they may signal independently of each other by parallel pathways (400) (see below). The signaling initiated by these proteins may activate the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK)-signaling pathway (45, 297, 304, 400) to upregulate hepcidin expression (Fig. 10). However, it should be noted that the involvement of ERK/MAPK signaling in iron-regulated hepatic hepcidin expression has recently been questioned by results, indicating that the Erk1/2/MAPK pathway was not activated by acute or chronic iron administration in a mouse model (74). Thus, the involvement of the ERK/MAPK pathway in iron-dependent regulation of hepcidin is uncertain. As discussed below, significant recent advances have been made in understanding the aspects of these iron-dependent signaling pathways, although there remain important unanswered questions.

HFE is a transmembrane protein belonging to MHC class I, and is expressed at high levels in the liver (61). Although HFE does not directly interact with iron, HFE can bind TfR1 to form a 2:2 complex at pH 7.5 (413). Both mutational (126, 413) and crystallographic (20) studies have demonstrated that HFE contacts the TfR1 homodimer at a site overlapping the known holo-Tf-binding site in the helical ectodomain of each TfR1 monomer (59). Consequently, HFE competes with holo-Tf for binding to TfR1 (125), and both proteins bind to TfR1 with comparable nanomolar affinities (i.e., 1–5 nM). After an increase in holo-Tf levels, as occurs in iron-overload diseases such as the hereditary hemochromatoses (see section VII.B below), TfR1-bound HFE becomes outcompeted for binding to TfR1 by holo-Tf and eventually dissociates (Fig. 10). Interestingly, the complete dissociation of HFE from TfR1 in cell lysates appears to occur at concentrations of holo-Tf as low as 100 nM (428), which is 100-fold less than the typical physiological concentration of holo-Tf in plasma [i.e., ∼10 μM (263)]. This may suggest that HFE is typically not bound to TfR1 under physiological conditions (see below).
Importantly, HFE can also bind to TfR2 (129), and HFE that is competitively displaced from TfR1 by holo-Tf then becomes available to interact with TfR2, the latter of which is expressed highly in the liver (61). In fact, unlike most cells in which TfR1 expression exceeds that of TfR2, TfR2 expression in liver cells markedly exceeds that of TfR1 (i.e., fourfold to sixfold greater) (61). Moreover, HFE is present at substoichiometric levels relative to both TfR isoforms in the liver, suggesting that HFE is a rate-limiting factor in the formation of the HFE/TfR1 and HFE/TfR2 complexes in hepatocytes (61). These data are consistent with a model that describes TfR1 as sequestering HFE away from interacting with TfR2 at exceedingly low holo-Tf levels, with HFE being released for subsequent interaction with TfR2 at increased holo-Tf levels.
In support of the above model of holo-Tf-regulated HFE-TfR1 and HFE-TfR2 interactions, the interaction between HFE and TfR1 appears to be crucial for the holo-Tf-dependent regulation of hepcidin expression. Mice harboring engineered TfR1 mutations that disrupt HFE binding to TfR1 overexpress hepcidin and consequently develop iron deficiency, whereas those mice harboring mutations that enhance the HFE/TfR1 interaction demonstrate inappropriately low hepcidin levels and develop iron overload (330).
Additionally, the interaction between HFE and TfR2 appears to be important for the holo-Tf -dependent upregulation of hepcidin expression in hepatic cells (123, 129, 400). This interaction requires distinct binding sites from those occurring between HFE and TfR1 (55). Interestingly, the interaction between HFE and TfR2 may not be necessary for these proteins to upregulate hepcidin production. Indeed, as iron overload is more pronounced in TfR2-null mice than in Hfe-null mice, and loss of both Hfe and Tfr2 causes an even more severe iron overload phenotype, Hfe and Tfr2 may in fact signal for hepcidin production by independent, but parallel, pathways involving Erk1/2 and Smad 1/5/8 (400). Moreover, although TfR2 expression is not regulated by the cellular iron status (175, 184), TfR2 expression is regulated by holo-Tf, whereby an increase in holo-Tf protects TfR2 from degradation (54, 175, 312). This degradation occurs within lysosomes (176). Whether this increase in TfR2 stability is due entirely to the direct binding of holo-Tf to TfR2 (54), which occurs with a lower affinity than for TfR1 (184), or whether there is some additional dependence on the interaction of HFE with TfR2, is yet to be resolved.
Overall, HFE appears to transmit information on the elevated holo-Tf status of the plasma, possibly, but not necessarily, by interacting with TfR2 and forming an iron-sensing/signaling complex that then directs the upregulation of hepcidin expression. Conversely, a decrease in holo-Tf levels leads to the increased binding of HFE to TfR1, and a consequent disassembly of the HFE/TfR2 complex. Additionally, as holo-Tf also stabilizes TfR2, while the increases in intracellular iron resulting from its uptake concomitantly downregulates TfR1 via the IRE-IRP system (see above), the extent of binding of HFE to TfR2 is probably enhanced. Notably, a number of recent key findings suggest that HFE in fact acts as a modulator of the BMP/HJV signaling (see below), rather than by activating a separate pathway (74, 183, 321). Indeed, recent data from mouse models of HFE-dependent HH in which Hfe is absent (see also Section VII.B) indicate that the iron-dependent activation of BMP/HJV signaling is defective (73, 183).
The regulation of hepcidin expression by the BMP6/HJV pathway is discussed in the following section.
3. The BMP6/HJV pathway
Emerging data strongly suggest that the BMP/HJV pathway is the central pathway for the iron-dependent modulation of hepatic hepcidin expression. As indicated above, this pathway appears to be responsible for the so-called regulation by iron stores. The BMP/HJV pathway is initiated by the binding of a specific subset of BMPs to their cognate receptors, which are a complex of type I and type II serine threonine kinase receptors (341) and coreceptors (e.g., HJV) at the cell surface of hepatocytes (5, 6, 20). Although it was known that there were four type I BMP receptors (Alk1–4), the identities of the receptors involved in iron-dependent hepcidin expression were unknown. In fact, only recently, two of these receptors, Alk2 and Alk3, were found to be expressed in murine hepatocytes, and their liver-specific deletion caused iron overload (364). Indeed, it appears that Alk3 is responsible for basal hepcidin expression, whereas both Alk2 and Alk3 are responsible for mediating hepcidin expression in response to iron and BMP signaling (364). The activated receptor complex that forms in the plasma membrane can then phosphorylate intracellular SMAD proteins (e.g., SMADs 1, 5, and 8). These phosphorylated SMADs can then form heteromeric complexes with the common mediator, SMAD4 (407), whereby the resulting complex translocates to the nucleus and modulates the transcription of target genes such as HAMP (391) (Fig. 11).

BMPs are members of the TGF-β superfamily and were originally identified for their ability to induce bone differentiation (53). While hepcidin expression can be induced by BMPs 2, 4, 5, 6, 7, and 9 (14), BMP6 appears to be the endogenous BMP that regulates hepatic hepcidin expression and systemic iron homeostasis (7, 251). Indeed, BMP6 −/− mice develop severe hepatic iron loading that is associated with reduced hepcidin expression (7, 251). It remains uncertain whether the source of BMP6 is solely the liver, or whether tissues such as the duodenum are also involved. A recent study reported that iron overload in vivo induces BMP6 expression in the liver, but not in the duodenum (182). In contrast, an earlier report showed that the ex vivo treatment of intestinal segments with iron, but not the in vitro treatment of macrophages and hepatocytes with iron, induced BMP6 expression (8). Although the reason for this discrepancy is unclear, it may be that the hepatic production of BMP6 in vivo requires the interplay of other systemic factors. While hepatocytes are probably the major source of BMP6 expression in vivo (182), the duodenal epithelium may contribute to BMP6 production and secretion under certain circumstances.
A crucial component of the BMP6-dependent pathway is HJV (13, 14), which is a GPI-linked membrane protein encoded by the HFE2 gene in humans (427). Loss-of-function mutations in HJV are responsible for Type 2A HH (juvenile hemochromatosis; see Section VII.B) (208, 286). Moreover, Hjv −/− mice have drastically reduced hepcidin expression and develop severe iron overload (158, 277, 305). In adult human tissues, the HJV gene, HFE2, is expressed at the highest levels in skeletal muscle, followed by the heart and liver, and is also expressed highly in the fetal liver (286). In the murine liver, Hfe2 is expressed predominantly by periportal hepatocytes (277). Importantly, the GPI-linked form of HJV appears to function as a coreceptor for BMP6, and facilitates the BMP6-dependent upregulation of hepcidin expression (13). Intriguingly, a soluble form of HJV (sHJV) (228), which can be generated by the furin-mediated cleavage of an endoplasmic reticulum-bound pool of GPI-linked HJV (345), suppresses hepcidin expression probably by competing with membrane-bound HJV for access to BMP6 (228). As furin expression appears to be upregulated by iron deficiency and hypoxia (345), sHJV secretion may function as a soluble systemic signal for iron deficiency that antagonizes hepatic hepcidin production (14). Furthermore, HJV is known to associate with the deleted in the colorectal cancer family member, neogenin (429), a membrane-associated protein that is involved in cell-signaling events. Recent data demonstrating that neogenin-mutant mice display iron overload, reduced BMP signaling, and low hepcidin expression suggest that neogenin can act to antagonize the production/secretion of sHJV (216). In support of this view, an earlier study found that the neogenin/HJV interaction was required for the BMP4-mediated induction of hepcidin expression when HJV is expressed (430). How neogenin and its interaction with HJV are regulated in relation to systemic iron levels remains to be established. Notably, recent data suggest that sHJV derived from skeletal muscle is dispensable for systemic iron homeostasis (56, 128). Conditional knockout mice lacking Hjv in muscle had no detectable derangements or iron metabolism (56, 128), while liver-specific ablation of Hjv caused iron overload (128). Such results indicate that the role of sHJV in regulating hepcidin expression is far from certain.
In addition to sHJV, the type II transmembrane serine protease family member, matriptase-2, acts as a negative regulator of the BMP/HJV pathway, and thus provides a further mechanism for the sensing of iron deficiency. Matriptase-2 (also known as TMPRSS6) was initially identified in 2002 (389). This protein was shown to play a crucial role in systemic iron homeostasis when a screen of mouse mutants generated by N-ethyl-N-nitrosourea exposure identified the microcytic anemic and iron-deficient mask mutant (96). The mask mutant, which expresses a matriptase-2 variant lacking the serine protease domain, has abnormally high hepcidin levels and consequently has reduced dietary iron absorption, which is specifically due to the defect in matriptase-2 (96). A number of mutations in matriptase-2, many of which occur in, or adjacent to, the serine protease domain, are associated with iron-refractory iron-deficiency anemia (IRIDA; see Section VII.A) and abnormally high hepcidin levels in humans (107, 248). Moreover, genetic ablation studies in mice have shown that the protein is an essential regulator of systemic iron homeostasis (116), and further that BMP6 signaling is a prerequisite for the anemia that results from inactivation of matriptase-2 (220). As for the mechanism involved, it has been demonstrated that matriptase-2 inhibits BMP/HJV-dependent hepcidin induction by proteolytically processing and inactivating the GPI-linked form of HJV, but not sHJV (347). Moreover, the endocytosis of matriptase-2 and the direction of the protein into lysosomes appear to negatively regulate the ability of matriptase-2 to downregulate HJV-dependent hepcidin expression, at least in vitro (19).
VII. Diseases of Iron Metabolism
As discussed earlier, in a biological context, iron has two faces: it is an obligate nutrient for almost all cells, being required for the function of many cellular proteins and their iron-containing groups (e.g., heme and ISCs), whereas in excess, iron can become highly toxic by catalyzing the production of ROS (see Section I) that damage cells, tissues, and organs (271). The many diseases of deregulated iron metabolism serve to exemplify this dichotomous nature of iron in human biology, and further provide excellent exemplars of our rapidly increasing knowledge of the mechanisms of cellular and systemic iron metabolism. However, it should be noted that the ensuing discussion will not include the many diseases associated with defects in mitochondrial iron processing, as these have been recently reviewed in this journal (159).
A. Anemias: iron deficiency anemia, ACD, and IRIDA
The most common outcome of systemic iron deficiency, which has been estimated to affect at least 2 billion persons worldwide, is anemia (256, 375). Anemias are characterized by a deficiency in the number of mature erythrocytes in the circulation, which inevitably lowers the oxygen-carrying capacity of the blood, causing tissue hypoxia, and clinical symptoms such as fatigue, weakness, increased cardiac output, as well as increased morbidity and mortality (154). Importantly, iron deficiency is not the only cause of anemia (e.g., see ACD and IRIDA below), but it is clearly a major cause (365). The remaining discussion will deal with several common anemias whose etiologies serve to exemplify the above review of the current state of knowledge on the regulation of systemic iron homeostasis.
1. Iron-deficiency anemia
Iron-deficiency anemia (IDA), which occurs secondary to iron deficiency, is the most common form of nutritional deficiency and anemia (266). IDA is characterized clinically as a hypochromic and microcytic anemia. The increased central pallor and decreased size of the red blood cells reflect defective hemoglobinization that is due to an insufficient supply of iron to the erythropoietic bone marrow. The etiology of IDA is notoriously multifactorial, yet it essentially represents a situation in which body iron requirements are not being met by enteric iron absorption (65). The many environmental causes of this deficit can include insufficient dietary intake of iron, or insufficient intake of other nutritional factors that enhance iron absorption [e.g., ascorbic acid (10, 64, 78, 178)], as well as increased iron requirements during specific physiologic periods (e.g., growth, menstruation, and pregnancy). It is important to note that while deficiencies in nutritional factors such as folate and vitamin B12, which are required for DNA synthesis during cell division (333), may coexist with and complicate the presentation of IDA, deficiencies in these compounds are not regarded as causative of IDA (250). In fact, deficiencies in folate and vitamin B12 can cause a clinically distinct macrocytic anemia that results from megaloblastic transformation within the bone marrow (250, 333).
Pathologic causes of IDA can include body iron losses that are secondary to an underlying medical condition (e.g., gastrointestinal blood loss). While a multitude of causative factors typically converge during the development of IDA, it is generally agreed that the major contributing factor is excessive blood loss (65). From a therapeutic standpoint, when it is apparent that the diet alone is insufficient to restore hematological parameters to acceptable levels, treatments for IDA typically include iron supplementation via oral or parenteral routes (65).
2. Anemia of chronic disease
In the Western world, ACD, now considered as a subtype of the broader anemia of inflammation, is the second most common anemia after IDA (122, 412). In fact, ACD is the most prevalent form of anemia in the hospitalized population, typically occurring in patients with underlying conditions that cause chronic activation of the immune system. Causative underlying conditions include chronic infections, chronic inflammatory diseases, chronic kidney disease, chronic rejection after solid-organ transplantation, and some malignancies. Along with ACD, the anemia of inflammation also includes the closely related anemia of critical illness. The latter occurs in critically ill patients, occurring soon after their admission to an intensive care unit (e.g., after severe acute events such as sepsis, surgery, and major trauma) (76).
Hematologically, ACD is typically characterized as a normochromic, normocytic, and mild anemia (412), and patients with ACD typically have low serum iron, low-to-normal serum Tf saturation, and high-to-normal serum ferritin levels (122, 412). Laboratory evaluations leading to a diagnosis of ACD can include determination of the ratio of a soluble form of TfR1 (sTfR1) to serum ferritin (412), as well as C-reactive protein, the latter of which is indicative of inflammation (122). Intriguingly, sTfR1 is shed from cells (predominantly erythroid precursors) after proteolytic cleavage of the cytoplasmic and transmembrane domains (residues 1–100) of membrane-bound TfR1 (342). Serum sTfR1 levels, which are positively correlated with the iron demand of erythropoiesis and negatively regulated by diferric Tf (82, 161), begin to rise when mild iron deficiency is achieved (351). This is considered to be a consequence of the limitation of iron supply to erythroid progenitors. As sTfR1 levels are not affected by inflammation, whereas serum ferritin levels are, determination of the ratio of sTfR1/log serum ferritin (i.e., the sTfR1 index) helps to differentiate between IDA and ACD, which often occur concurrently (352). For example, in isolated ACD, the sTfR1 index is typically lower (i.e.,<14) than in isolated IDA (i.e.,≥14) (352).
The primary pathogenic mechanism of ACD is an increase in circulating levels of inflammatory cytokines (e.g., interleukin-6, interleukin-1, tumor necrosis factor-α, and interferon-γ) that, among other things, signal for increased hepcidin production by hepatocytes. This increase in hepcidin downregulates ferroportin expression in major iron-exporting cells such as macrophages, duodenal enterocytes, and hepatocytes, and ultimately leads to decreased enteric iron absorption and, perhaps more importantly, to increased iron retention within splenic macrophages and hepatocytes (122). While body iron stores may not be significantly reduced, this trapping of iron within macrophages greatly diminishes the body's iron-recycling capacity and is probably the major cause of the restricted iron availability to erythroid precursors (411). The concomitant decrease in circulating iron bound to Tf leads to impaired erythrocyte production as a result of iron restriction. Additional mechanisms involved in the pathogenesis of ACD include (i) decreased erythrocyte lifespan, presumably as a result of an inflammatory cytokine-dependent increase in splenic macrophage activation, followed by premature destruction of aging erythrocytes (122); (ii) impaired erythroid progenitor proliferation, which may involve a cytokine-mediated increase in apoptosis of erythroid progenitors (412); and (iii) a blunted EPO response due to a combination of inflammatory cytokine-dependent downregulation of EPO expression and deregulation of EPO signal transduction (412).
Importantly, it has become clear that the centrality of hepcidin deregulation to the molecular pathogenesis of ACD is paramount. In growing support of this notion, Theurl et al. have recently demonstrated (using an established rat model of ACD) that the blockade of endogenous hepcidin production results in the release of stored iron from splenic macrophages, stimulation of erythropoiesis, and a correction of the anemia (377).
3. Iron-refractory iron-deficiency anemia
In contrast to most patients with IDA, a subpopulation of patients with atypical IDA do not show significantly improved hematologic parameters upon oral iron supplementation, and show delayed and/or incomplete hematologic responses to the parenteral administration of iron (e.g., iron–dextran). This condition has recently been termed IRIDA (OMIM #206200) and is typified by a hypochromic, microcytic anemia, with a very low mean corpuscular erythrocyte volume, low serum iron, and low serum Tf saturation, and variable serum ferritin (107). Instructively, despite being in a state of iron deficiency, patients with IRIDA have abnormally elevated hepcidin levels (107). This contrasts with the observation that urinary hepcidin levels are almost undetectable in patients with typical IDA (187). The increase in hepcidin in IRIDA underpins the iron-refractory phenotype, as it causes inefficient absorption of iron across the duodenal epithelium and impaired release of iron from splenic macrophages.
Although it is unclear whether all cases of IRIDA are genetic, autosomal-recessive IRIDA was recently mapped to chromosome 22q12–13 in a Sardinian kindred in 2008 (248). In the same year, Finberg et al. performed haplotype analysis in five kindreds with IRIDA, supporting the linkage to 22q12–13 (107). These authors also determined that all affected individuals carried mutations in the matriptase-2 gene, TMPRSS6, which is located within 22q12–13. Mutations in TMPRSS6 that are associated with IRIDA have also been identified by others (137, 248). In support of the claim that key defects in matriptase-2 cause IRIDA, the mask mouse phenotype, which is caused by chemically induced mutations in the murine ortholog of human matriptase-2, Tmprss6, is characterized by inappropriately high hepcidin levels and consequent impairment of dietary iron absorption (96). As discussed above, matriptase-2 is a crucial participant in the balancing act of systemic iron homeostasis, as it post-translationally regulates GPI-linked HJV via proteolytic cleavage, thereby attenuating BMP6-dependent upregulation of hepcidin transcription (108, 220, 347). Thus, the loss of matriptase-2 activity in patients with IRIDA impairs their ability to downregulate hepcidin in response to systemic iron deficiency. Importantly, matriptase-2 expression has recently been shown to be upregulated by HIF1α and HIF2α signaling in response to iron deficiency and/or hypoxia (202), suggesting a mechanism by which matriptase-2 responds to iron deficiency. The discovery that key matriptase-2 mutations can cause IRIDA further emphasizes the central role hepcidin plays in regulating systemic iron homeostasis.
B. Iron-overload diseases: the hereditary hemochromatoses and β-thalassemia
Iron-overload diseases can be inherited or acquired, and are typically characterized by the excessive accumulation of iron in various body tissues (e.g., the liver). This accumulation of iron is problematic primarily due to the propensity for inappropriately sequestered iron (i.e., labile iron) to catalyze production of noxious free radicals that lead to cellular, tissue, and organ damage, and eventually to organ failure [(271); see Section I]. For example, in the iron-overload disease, β-thalassemia (see below), plasma malondialdehyde is significantly increased relative to control patients, and α-tocopherol levels are decreased in 32% of patients (402), indicative of increased levels of oxidative stress. Iron overload will usually occur when the iron content of the plasma exceeds the Tf-binding capacity, with a concomitant increase in plasma NTBI (36, 100). However, NTBI may also occur before plasma Tf is fully saturated, and so cannot be regarded as a simple spillover phenomenon (36). In most cases, iron overload results from a genetic defect (i.e., primary iron overload) that impairs the iron-regulated production of hepcidin, or the mechanism of hepcidin action, as is typified by the hereditary hemochromatoses (see below) (21, 83, 271, 295). Additionally, the so-called secondary forms of hemochromatosis occur predominantly as a result of either excessive erythrophagocytosis after multiple transfusions, and/or enhanced dietary iron absorption in the context of an anemia caused by an underlying erythropoietic defect (e.g., hereditary sideroblastic anemias and the hemoglobinopathy, β-thalassemia) (372). In fact, secondary iron overload due to heminoglobinopathic disorders, such as the iron-loading anemias [see β-thalassemia below; other such diseases of secondary iron overload have recently been reviewed (344)], may in fact represent a greater global health burden than hereditary hemochromatoses.
Iron-overload diseases often go hand-in-hand with an increased susceptibility to infections. As bacteria—like all other organisms—need a constant iron supply for proliferation, infections are typically prevented by the low concentrations of soluble iron in the human body (350). Thus, patients with hemochromatosis are highly susceptible to infections with Vibrio vulnificus, a bacterium that usually cannot grow in blood from healthy individuals, but rapidly grows in blood from patients with hemochromatosis (38). In general, iron is essential for bacterial virulence, and during infection, there occurs a battle for iron between the pathogen and host. As the mechanisms of bacterial iron sequestration have recently been reviewed elsewhere (350), we present here only a few brief remarks. Many fungi and bacteria employ small iron chelators, the so-called siderophores, to sequester iron. Many of these siderophores, including the siderophore enterobactin, which is synthesized by a range of gram-negative bacteria, are synthesized by large nonribosomal peptide synthetases (111, 390), whereas others (including the desferrioxamines, which are frequently used in a clinical setting for the treatment of iron overload disorders) are synthesized by widely conserved nonribosomal peptide synthetase-independent pathways (51). The fact that these complex enzymes and pathways are conserved during evolution is indicative of the importance of the siderophores for the microorganisms. Siderophores play a role in the homeostasis of a wide range of metals (328), in the development of bacterial and fungal virulence (143, 322), and in the development of biofilms (322, 393).
Furthermore, several bacteria, including Escherichia coli, Staphylococcus aureus, and Vibrio cholerae, synthesize hemolysins (also referred to as cytolysins) to access heme from the host's erythrocytes (162, 361). Hemolysins are a major virulence factor associated with infection that form pores in the host's erythrocyte membranes to release iron in the form of heme for the pathogen (34).
1. Hereditary hemochromatosis
HH, or primary iron overload, is a collective label for a group of autosomal inherited iron-overload disorders, of which the HFE-dependent form is particularly prevalent in individuals of the Northern European ancestry (416). Essentially, the pathology of HH results from a deregulated and chronic increase in the amount of iron absorbed from the diet, and is typified by excessive iron deposition in the liver, heart, pancreas, as well as the parathyroid and pituitary glands (21). Crucially, HH is genetically heterogeneous and is associated with mutations in at least five genes that regulate the hepcidin–ferroportin axis of systemic iron homeostasis (see Section VI.C). Depending on which genes are involved, HH is classified as type 1 (hemochromatosis protein, HFE dependent); type 2A [HFE2 (HJV) dependent]; type 2B (hepcidin, HAMP dependent); type 3 (TfR2, TfR2 dependent); and type 4 (ferroportin dependent). While the mutations affecting HFE are relatively common, those affecting the other involved genes are very rare (21).
Moreover, it is becoming increasingly apparent that the clinical penetrance of the major HH genotype, type 1 HFEC282Y/C282Y-associated HH (71, 255, 313), is strongly influenced by the presence of genetic modifiers, and probably also by as-yet-uncharacterized epigenetic modifiers.
a. Type 1 HH
Type 1 or HFE-dependent HH (OMIM #235200) is the most common form of HH. Men are predominantly affected, and the most severe effects of the disease do not typically occur until decades of iron loading have passed. The disease is typically autosomal recessive, with ∼85% of patients being homozygous for a C282Y mis-sense mutation in HFE that occurs with a homozygote frequency of ∼5 per 1000 individuals of a Northern European descent, although the clinical penetrance is much lower (21). This mutation prevents the formation of a critical disulfide bridge in the mature HFE protein, with the outcome being that the interaction of HFE with β2-microglobulin (103), and also with TfR1 (102), is disturbed. Additionally, cell surface expression of HFE is impaired, as the protein is retained within the Golgi apparatus (103). Using Hfe knockout mice, which are a widely accepted model of HFE-dependent HH (285), Trinder et al. were able to show that the uptake of radioactive 59Fe from [59Fe]-holo-Tf from the plasma was downregulated in the duodenum of these mice, compared to their wild-type controls, but was not affected in the liver or kidney. Moreover, plasma iron turnover was not affected by the absence of HFE (383). Such results were seen to provide support for a previously proposed model of systemic iron homeostasis in which HFE normally functions to assist in the programming of duodenal crypt cells to extant systemic iron levels. The proposed function of this programming is to regulate the expression of iron transporters such as DMT1 and ferroportin in the derived and differentiated absorptive enterocytes of the villus region (320, 383, 399). However, the observation that the enterocyte-specific ablation of Hfe in mice fails to recapitulate in any way the HH phenotype (397) suggests that the primary pathologic event in HFE-dependent HH is not an HFE-dependent derangement of duodenal crypt programming. More recently, it was shown that only the liver-specific ablation of Hfe in mice is sufficient to disrupt the proper regulation of systemic iron homeostasis (396). As discussed above, the currently accepted view is that HFE in the hepatocytes functions to regulate hepatically derived circulating hepcidin levels, which probably regulates the BMP6/HJV/SMAD pathways (73, 74, 321) (see Section VI.C for further discussion). The derangement of this function alone of HFE in HFE-dependent HH is thought to be primarily responsible for disease pathogenesis.
b. Type 2A HH
Type 2A or HFE2 (HJV)-dependent HH (OMIM #602390), also known as juvenile hemochromatosis, is an acute form of HH that results in rapid and progressive iron loading of organs and consequent organ failure before 30 years of age (314). It affects men and women equally, and if left untreated, the disease can become fatal with the main cause of death being heart failure (47). The affected gene responsible for this form of HH is HFE2, which was cloned in 2004, with a G320V mutation being present in two-thirds of the patients tested (286). The following year, it was demonstrated that an Hfe2 −/− mouse model recapitulated many of the clinical features of the human disease and was associated with a downregulation of hepatic hepcidin expression and a concomitant increase in duodenal and macrophage ferroportin expression (158). As discussed in Section VI.C, HJV is expressed primarily in skeletal muscle, but also in the heart and liver, and regulates hepcidin expression in a manner involving BMP-dependent signaling. That is, GPI-linked HJV functions as a BMP coreceptor, which is required for BMP6-dependent upregulation of hepcidin, while the soluble form of HJV, which may be released from skeletal muscle stores in a furin- (345) and neogenin-dependent (426) manner, probably antagonizes BMP6-dependent hepcidin expression by the liver. It would appear that the primary biochemical defect resulting in hepcidin deregulation in HJV-dependent HH, as typified by the G320V mutation, is the defective intracellular cleavage and subsequent targeting of the GPI-linked protein to the cell surface (346). Exactly how this leads to a derangement of hepcidin production is unclear, but is probably due in large part to an inability of the intracellularly sequestered protein to facilitate BMP6-dependent signaling (346). Interestingly, results obtained by Silvestri et al. suggest that a decrease in soluble HJV is not likely to be a major contributor to the HJV-dependent HH phenotype (346).
c. Type 2B HH
Some patients with juvenile hemochromatosis do not have mutations in HJV, but instead have mutations in the HAMP gene that encodes hepcidin (21). A variety of mutations have been documented (22), and there may be some interaction with HFE mutations that serves to increase the severity of the disease phenotype. Hepcidin expression is virtually absent in these patients, suggesting that the mutations impair transcription of HAMP.
d. Type 3 HH
A very rare form of HH, in which patients present similarly to HFE-dependent HH, is due to mutations in the gene encoding TfR2. However, the expression of the disease differs significantly from HFE- and HJV-dependent HH, which serves to illustrate the notion that TfR2 is not simply an HFE-binding partner (46). In accordance with this notion, Tfr2 knockout mice have a more severe iron-overload phenotype than Hfe knockout mice, while the Tfr2/Hfe combined knockouts are the most severely affected (400). Interestingly, TfR2 has recently been shown to be a component of the EPO receptor complex in erythroid progenitors, suggesting that it has an extrahepatic function in normal erythropoiesis, and that this may additionally be affected in TfR2-dependent HH (118).
e. Type 4 HH
Mutations that affect the hepcidin receptor, ferroportin, cause an autosomal-dominant form of HH known as type 4 HH or ferroportin disease (84). These forms of HH present differently to the autosomal-recessive forms of HH, and are associated with either (i) loss-of-function mutations that downregulate cell surface expression of ferroportin, thereby causing iron retention within macrophages and a consequent restriction of iron for erythropoiesis (i.e., type 4A); or (ii) mutations that render ferroportin resistant to hepcidin-induced degradation (i.e., type 4B), and are so-called gain of function, as they result in the continued capacity for iron export in the face of normally inhibitory concentrations of hepcidin. The dominant inheritance pattern of this type of HH is likely due the fact that native ferroportin appears to exist as a dimer (86), and that the ferroportin mutant proteins associated with ferroportin disease function in a dominant-negative manner (85).
2. β-thalassemia
β-thalassemia is a secondary iron overload disease, which is characterized by anemia associated with excessive relative α-globin production, ineffective erythropoiesis, and iron overload (410). Genetically, the disease is caused by mutations in the β-globin gene or its promoter that result in reduced or absent production of the β-globin of hemoglobin (410). The relative excess of α-globin chains leads to α-globin precipitation in red blood cell precursors, causing defective erythroid maturation and erythropoiesis, followed by anemia (410). Importantly, the ineffective erythropoiesis causes an increased proliferative drive in the underperforming bone marrow, triggering bone marrow expansion. The latter, by an as-yet-unknown signaling mechanism, and which may correspond to the postulated erythroid regulator (see Section VI.B), causes increased iron absorption by decreasing hepatic hepcidin production (292). Moreover, this iron overload is exacerbated in patients receiving multiple transfusions to combat the anemia (369). The main cause of morbidity and death in β-thalassemia is the cell, tissue, and organ damage caused by oxidative stress resulting from the increase in labile iron, as well as the increased susceptibility to infection that occurs (410) (see above).
VIII. Concluding Remarks
Iron metabolism, including its regulation and role in a range of diseases, has become a major research focus in recent years. In this article, we have reviewed the present understanding of mammalian iron uptake, storage, and homeostasis as well as the role of iron in anemias and iron-overload diseases. For several areas of iron biochemistry not covered in this review, we refer the reader to recent reviews. These areas include iron metabolism in plants (172, 374) and bacteria (35, 374), diseases of mitochondrial iron processing (159), iron in cancer (310), type II diabetes (303), and neurodegenerative diseases (18, 308).
Footnotes
Acknowledgments
We are thankful to the reviewing editors and to Prof. Ann Smith, (University of Missouri-Kansas City, MO) for helpful comments on the manuscript. DJRL acknowledges a Peter Doherty postdoctoral fellowship from the National Health and Medical Research Council of Australia and a Cancer Institute NSW Early Career Fellowship.