
Abstract
Introduction
Innovation
Since the process of neurodegeneration in AD is complex and multifactorial, future therapeutic strategies should consider the use of multifunctional iron chelating drugs, such as M30, which was designed against distinct AD pathological mechanisms. In addition, accumulation and deposition of brain iron is central to various neuropathological processes in AD, including OS, amyloid deposition, and tau phosphorylation. Indeed, in the present study, we have demonstrated the potential neuroprotective effects of M30 on AD-related neuropathology and cognitive deficits in APP/PS1 double Tg mice. The implication of this multifunctional iron chelator/radical scavenger may provide efficient and novel tools against the significant heterogeneity at the molecular level of AD pathology, to make progress in overcoming this disease in the clinic.
As a consequence of these findings, metal chelation is one of the therapeutic strategies for AD (21, 36). Indeed, based on a multimodal drug design paradigm, we deliberately incorporated the propargylamine moiety of the anti-Parkinsonian monoamine oxidase (MAO)-B inhibitor, rasagiline (Azilect®, Teva Inc.) into the antioxidant-iron chelator moiety of an 8-hydroxyquinoline derivative of our potent iron chelator, VK28, thus inheriting some of their neuroprotective properties (58, 64). Among these novel compounds, M30 was the most effective drug, possessing iron chelating potency, radical scavenging, and inhibition of iron-induced membrane lipid peroxidation features (58, 64). In addition, M30 was found to be a potent brain selective inhibitor of both MAO-A and -B activities (18).
A growing body of evidence indicates that an effective therapeutic compound for treating AD has to attenuate Aβ accumulation and tau phosphorylation in the brain, which are recognized as the main hallmarks of AD that contribute to the disease-associated cognitive impairments. Recent positive outcomes for M30 in cell culture studies, regarding aspects of relevance to AD, demonstrated that M30 suppressed the translation of a luciferase reporter mRNA through the 5′UTR sequence (1). This effect may account, at least in part, for the observed downregulatory effect of M30 on membrane-associated holo-APP levels in SH-SY5Y neuroblastoma cells, presumably by chelating intracellular iron pools (2, 4). Furthermore, M30 markedly reduced the levels of amyloidogenic Aβ in the medium of CHO cells, stably transfected with the APP “Swedish” mutation (CHO/ΔNL) (2); and exerted protection against Aβ-induced toxicity in primary cultured neurons (4). Additional studies have reported that M30 enhanced protein kinase B (AKT) and glycogen synthase kinase (GSK)-3β phosphorylation, and attenuated tau phosphorylation in cortical neurons in vitro (3).
In the present study, we extended our experiments to examine the therapeutic effect of the novel multifunctional drug, M30 on AD neurodegenerative pathology, as well as deficits in spatial learning and memory in APP and presenilin (PS1) double-transgenic (Tg) mice. We found that M30 effectively reduced Aβ accumulation and tau phosphorylation, and attenuated memory deficits in APP/PS1 Tg AD mice.
Results
Weekly body weight monitoring showed that vehicle-treated APP/PS1 mice were consistently and significantly lower in body weight gain than the non-Tg mice. Compared to vehicle-treated mice, M30 (1 and 5 mg/kg)-treated Tg mice started to gain more body weight at 6 months of age and this trend was observed for the rest of the experiment. At the end of the experiment, body weights of M30 (1 and 5 mg/kg)-treated APP/PS1 mice were slightly higher than those of vehicle-treated APP/PS1 mice (33.2±1.6 g and 31.8±1.7 g, respectively, vs. 30.6±0.8 g), similar to the non-Tg mice (32.8±1.4 g). Relative brain weights did not significantly differ between the non-Tg and APP/PS1 vehicle-treated mice (1.9±0.11% vs. 1.8±0.09%) and was not significantly affected by M30 treatment (1.8±0.1% and 1.9±0.12% for 1 and 5 mg/kg-treated groups, respectively).
M30 treatment attenuates cognitive deficits of APP/PS1 mice
The major aim of this study was to explore whether the novel multifunctional iron-chelator, M30, exhibits beneficial effects on cognitive impairment and pathological alterations on APP/PS1 Tg mice.
First, we investigated the effect of long-term M30 treatment (1 and 5 mg/kg for 9 months, initiated when the mice were 3 months old) on spatial learning deficits in the Tg APP/PS1 mice. The abilities of the mice to learn and process spatial information were tested by the Morris water maze test, one of the most widely accepted behavioral tests of hippocampus-dependent spatial learning and memory (40). All mice were tested on both the visible and hidden platform versions of the Morris water maze test, as described in Materials and Methods. Figure 1 shows the results of all mice during water maze acquisition training. The visible platform tests showed that the escape latency decreased significantly across the 5 days of visible platform session for all groups. As shown in Figure 1A, despite the significant initial spatial learning impairment exhibited by vehicle-treated APP/PS1 mice, they were able to locate the visible platform proficiently by the fifth day of the visible platform session. In the hidden platform version, vehicle-treated APP/PS1 mice showed impaired acquisition of spatial learning, compared with the non-Tg mice, as indicated by much slower improvements in the escape latency across consecutive trials. M30 treatment ameliorated the performance deficits in APP/PS1 mice during the testing period with the invisible platform, compared with the vehicle-treated group. This was followed by a probe trial performance, showing that M30 treatment not only significantly promoted acquisition phase of place learning, but also significantly improved the memory retention during the probe trial (Fig.1B). Taken together, these data indicate that M30 treatment lead to spatial learning-memory improvement in APP/PS1 mice.
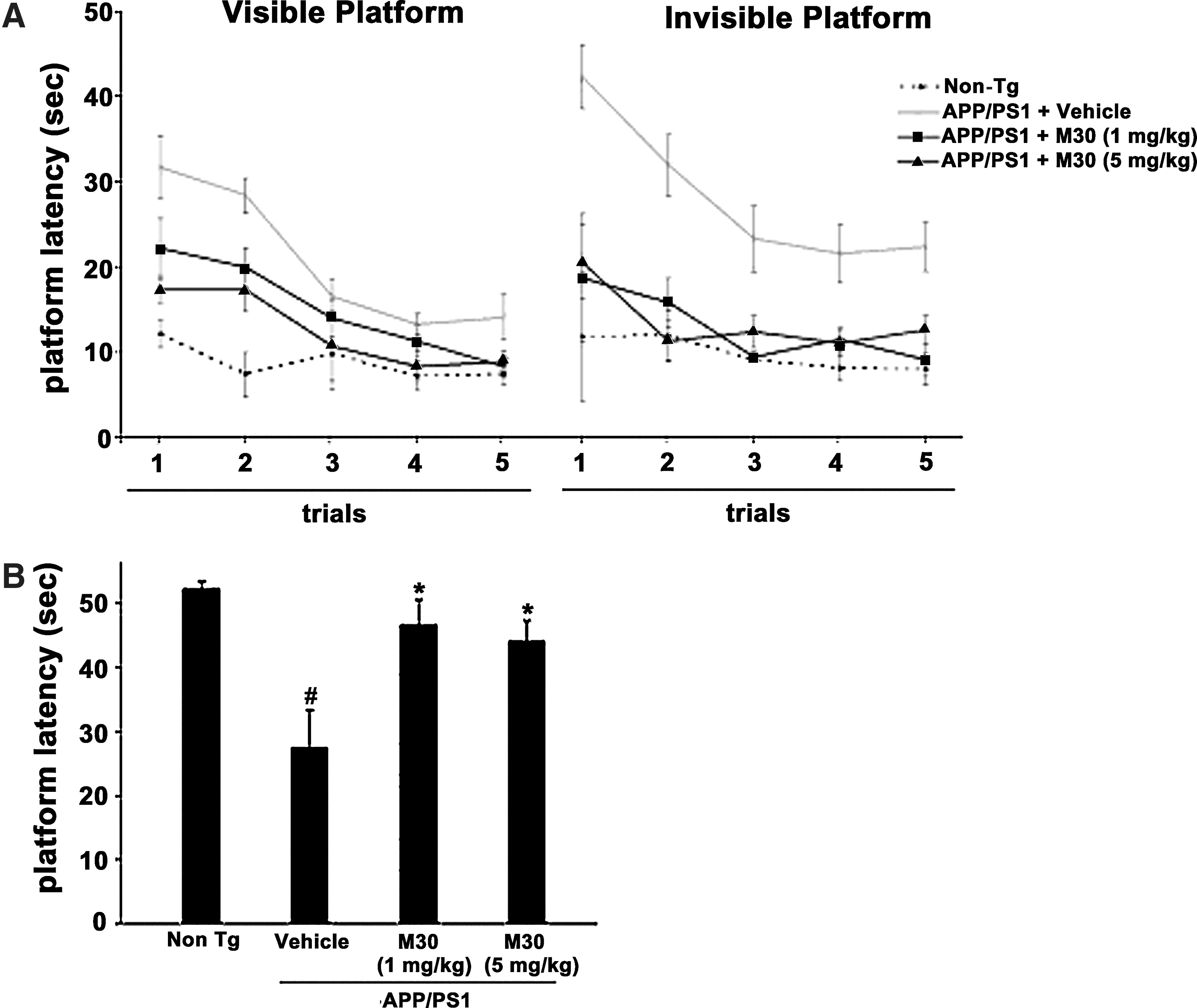
Behavioral testing using the Y-maze test revealed that vehicle-treated APP/PS1 mice were impaired in this task, compared with the non-Tg mice, while APP/PS1 mice given M30 (5 mg/kg) showed significantly higher proportion of spontaneous alternations (Fig. 2A).
In the next behavioral experiment, the effect of M30 on cognitive function was examined by the Hebb-Williams maze test. Figure 2B shows that the total error score of the APP/PS1 vehicle-treated group was higher than that of the non-Tg group. M30-treated APP/PS1 mice showed improved performance in both problems (8 and 12), making significantly fewer total errors, as compared with vehicle-treated APP/PS1 mice. The novel taste neophobia test, which is sensitive to amygdala and hippocampal damage, was used as a measure of anxiety and memory for a novel food. Figure 2D shows that in the vehicle-treated APP/PS1 group, food intake was significantly reduced after the initial exposure, compared to the non-Tg mice. M30 (1 and 5 mg/kg)-treated APP/PS1 mice consumed significantly more novel food during the second encounter, compared with vehicle-treated Tg mice. Nesting behavior studies, followed by analysis of nesting scores, revealed that nesting was impaired in vehicle-treated APP/PS1 mice, in comparison with the non-Tg group. A significantly improved nesting was observed in M30-treated APP/PS1 mice at both given concentrations (1 and 5 mg/kg) (Fig. 2E). No effect was demonstrated for M30 in non-cognitive behavior studies (rotarod task and screen test performance; data not shown).
Effect of M30 on cerebral iron and transferrin receptor expression in APP/PS1 mice
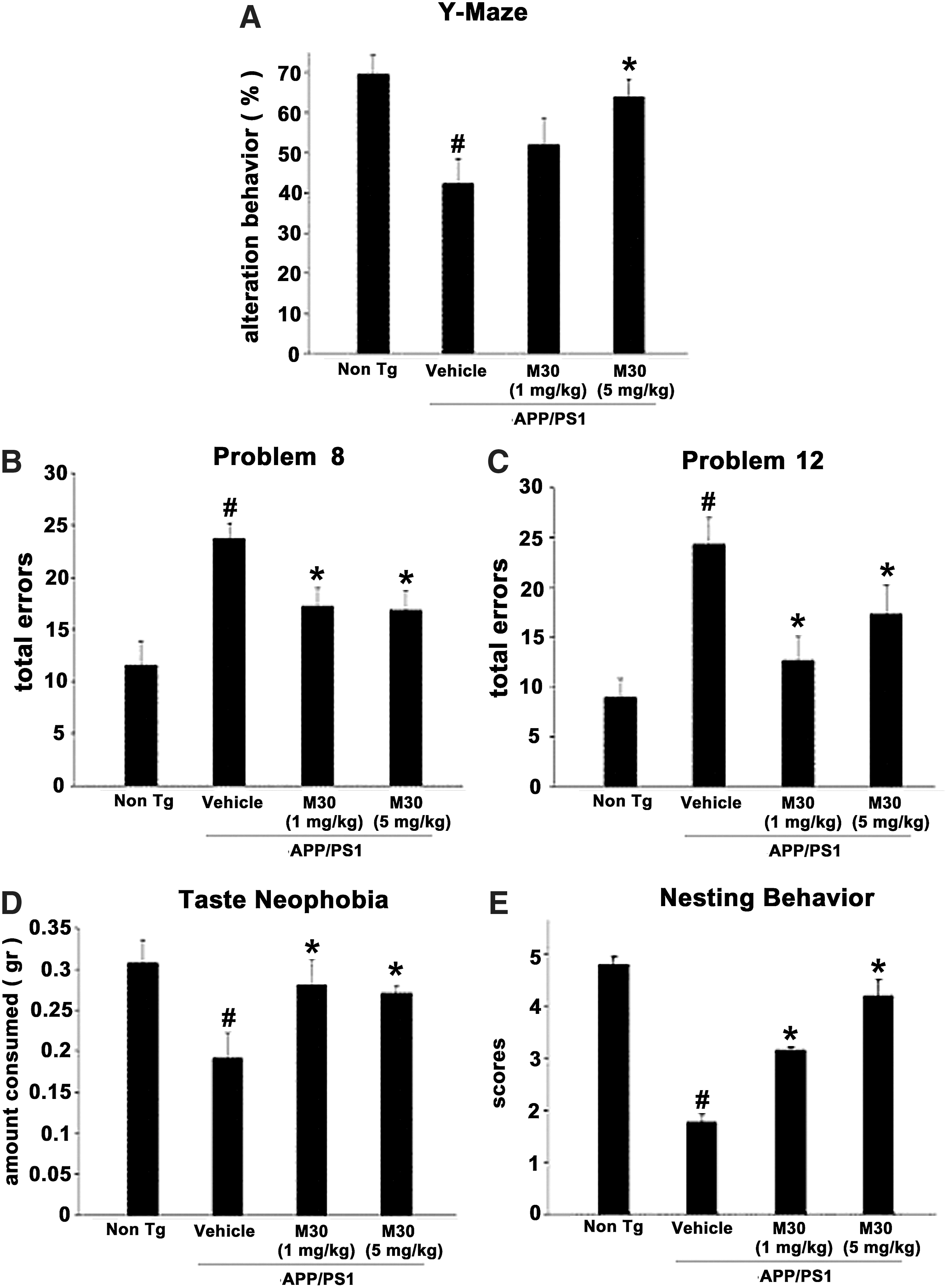
After the behavioral assessment, we studied the effect of M30 on various pathological features of AD, including cerebral iron levels, changes in fibrillar amyloid deposition, and Aβ levels in the brain. Qualitative examination of Perl's-DAB-stained brain sections revealed an increase in iron concentrations in vehicle-treated APP/PS1 mice (n=4), as compared with virtually undetectable levels in vehicle-treated non-Tg mice (n=3), where the iron levels were undetectable. The iron levels were more pronounced in striatum (1.9±0.21) than in cortical and hippocampal areas (0.52±0.21 and 0.29±0.12, respectively), as determined by OD analysis. M30 (1 and 5 mg/kg)-treated APP/PS1 mice showed notably reduced levels of iron staining in all brain regions studied, compared with the vehicle-treated group (Fig. 3A). The cortical and hippocampal iron levels in M30 (1 mg/kg)-treated APP/PS1 mice (n=3) were 0.22±0.06 and 0.16±0.09, respectively, representing a 57% decrease in cortical (p<0.05) and a 44% decrease in hippocampal (p<0.05) iron levels. There was a 24% decrease in striatal iron levels, but this was not found to be statistically significant (0.22±0.14 vs. 0.29±0.1; p=0.1). The cortical, hippocampal and striatal iron levels in M30 (5 mg/kg)-treated APP/PS1 mice (n=3) were 0.18±0.1, 0.14±0.02, and 1.12±0.16, respectively. This represents a 65% decrease in cortical (p<0.05), a 51% decrease in hippocampal (p<0.05), and a 41% decrease in striatal iron levels (p<0.05). Western blot analysis of transferrin receptor (TfR) expression, which is known to be induced by iron chelators through iron regulatory protein (IRP)-mediated mRNA stabilization, revealed that M30 significantly upregulated TfR protein levels in hippocampus and parietal cortex in APP/PS1 mice (Fig. 3B). Previous studies have implicated that, among other protective mechanisms, the neuroprotective effect of propargylamine-containing compounds is ascribed to induction of antioxidant enzymes (42). Here, using real-time RT-PCR, we demonstrated that vehicle-treated APP/PS1 mice exhibited a 71.8±5.26% reduction of hippocampal mRNA expression levels of catalase, as compared with vehicle-treated non-Tg mice (p<0.05). Administration of M30 (5 mg/kg) resulted in a significant increase in mRNA levels of catalase in the hippocampus (2.2±0.39 folds, p<0.05) vs. vehicle-treated APP/PS1 Tg group.
Effect of M30 on plaque deposition and Aβ levels in APP/PS1 mice
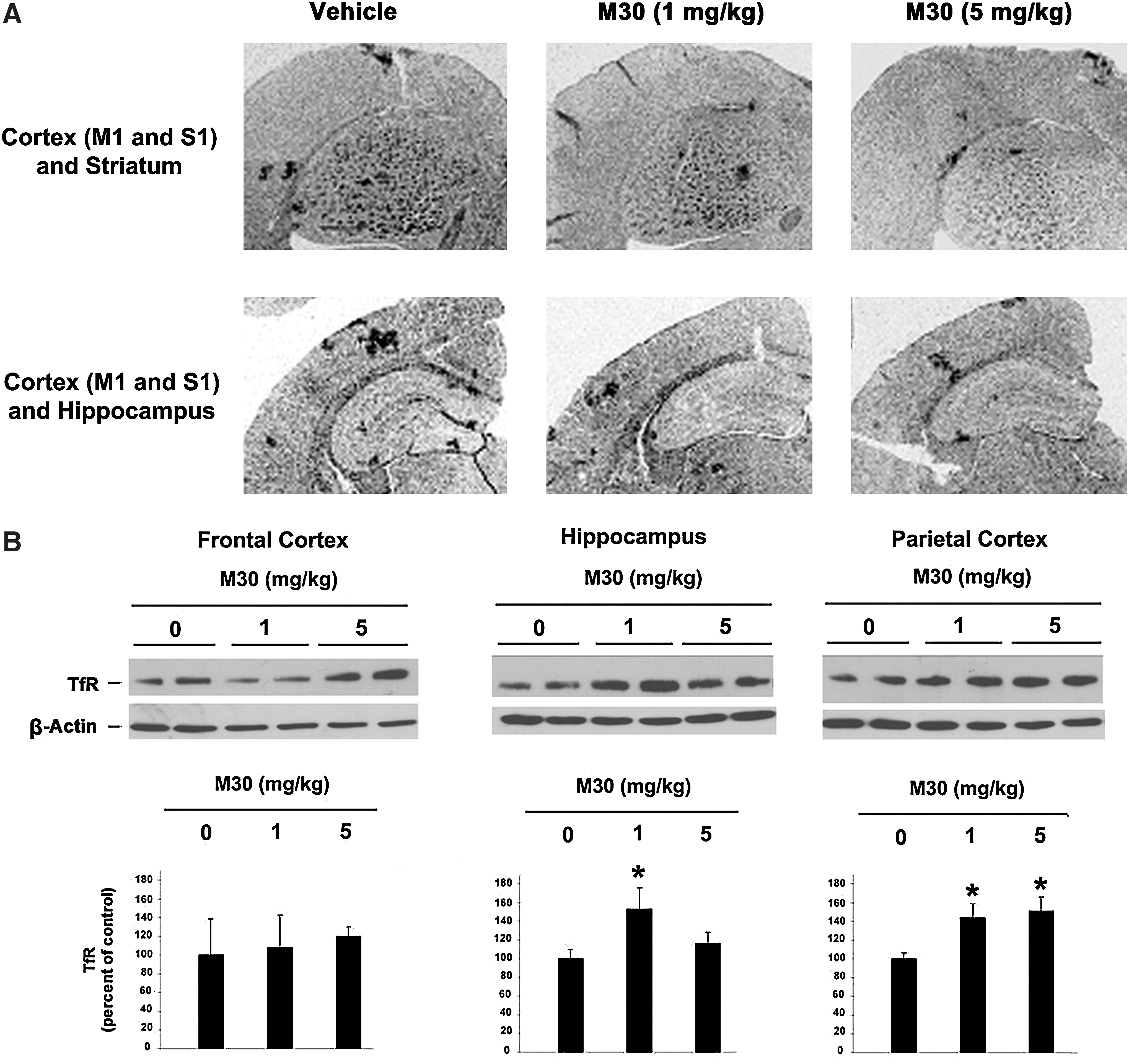
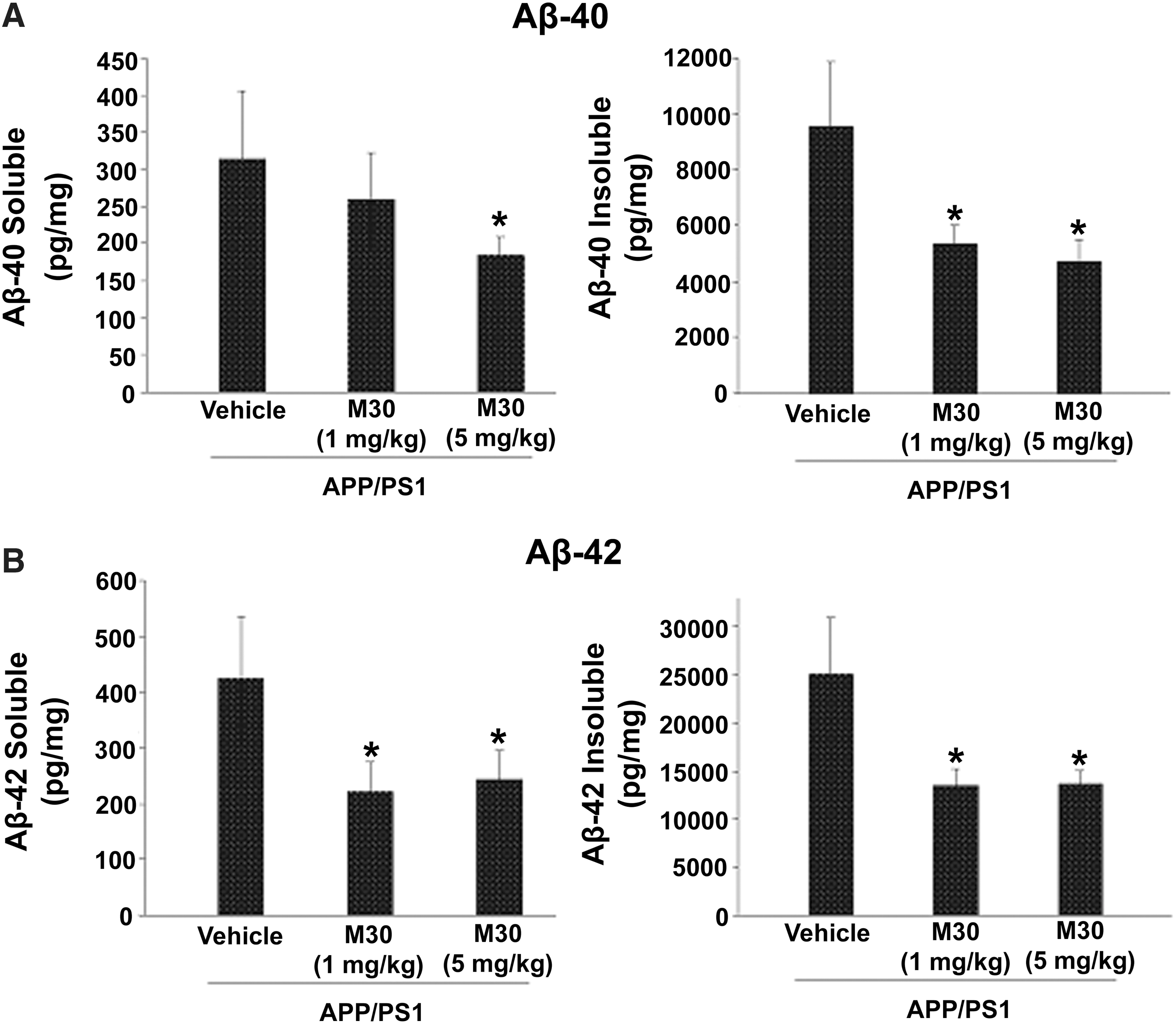
Further biochemical and immunehistochemical studies detected fibrillar amyloid deposits in brain slices by Thioflavin S staining (Fig. 4A and 4B), and total Aβ plaque load, including diffuse and compacted fibrillar plaques, by a specific anti-Aβ-amyloid antibody (6E10, corresponding to amino acids 1–17 of Aβ peptide) (Fig. 4C and 4D). We found that vehicle-treated APP/PS1 mice exhibited high levels of Aβ and fibrillar load, consistent with previous observations (Fig. 4) (10). In contrast, long-term oral administration of M30 resulted in a significant decrease of Thioflavin S-positive plaque deposition (Fig. 4A and 4B) and total Aβ plaque burden (Fig. 4C and 4D) in the frontal cortex, hippocampus, and parietal cortex, compared with vehicle-treated APP/PS1 mice, indicating that M30 was capable to reduce both fibrillar and nonfibrillar/diffused Aβ plaques. Consistent with these findings, high resolution Western blot analysis showed that Aβ levels were reduced in the frontal cortex, hippocampus, and parietal cortex of M30-treated APP/PS1 group (Fig. 5). Next, we examined the effect of M30 on the levels of cerebral Aβ in APP/PS1 mice by a sandwich ELISA. As shown in Figure 6, M30 treatment caused a significant decrease in brain concentrations of Aβ-40 and Aβ-42 in the Tris-buffered saline (TBS)-soluble and guanidine-soluble brain homogenates. This indicates that the reduction in Aβ levels could account for the decrease in Aβ deposition observed in M30-treated APP/PS1 mice.

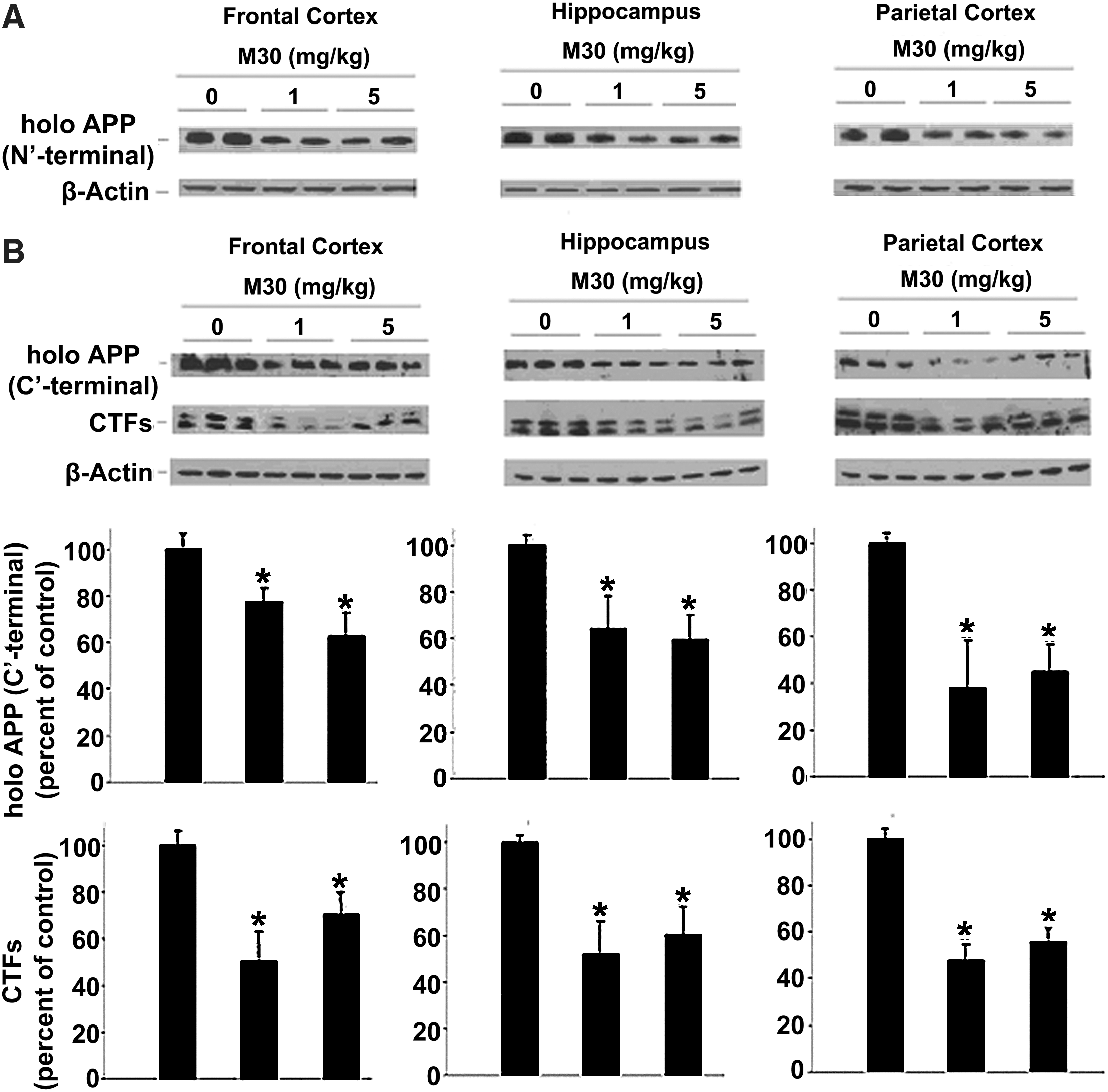
Effect of M30 on levels of APP and APP-C-terminal fragments in APP/PS1 mice
We further assessed the impact of M30 on cerebral levels of the full-length APP and α- and β-C-terminal fragments (CTFs) of APP in APP/PS1 mice. Figure 7A shows that M30 treatment has led to a reduction of holo APP levels in all brain regions assessed (frontal cortex, hippocampus, and parietal cortex), as indicated by Western immunoblotting using the anti-APP antibody 22C11, which recognizes an epitope located between amino acids 60 and 100 in the N-terminal part of the ectodomain of APP. Similarly, immunoblot analysis using an anti- APP C-terminal (676–695) antibody showed that compared with the vehicle-treated group, M30 treatment reduced the levels of holo APP in the frontal cortex, hippocampus, and parietal cortex (Fig. 7B). M30 treatment also caused a significant reduction of the CTFs of APP, produced by α- and β-secretases, C83 and C99, respectively (Fig. 7B). These results complemented the decrease in Aβ levels.
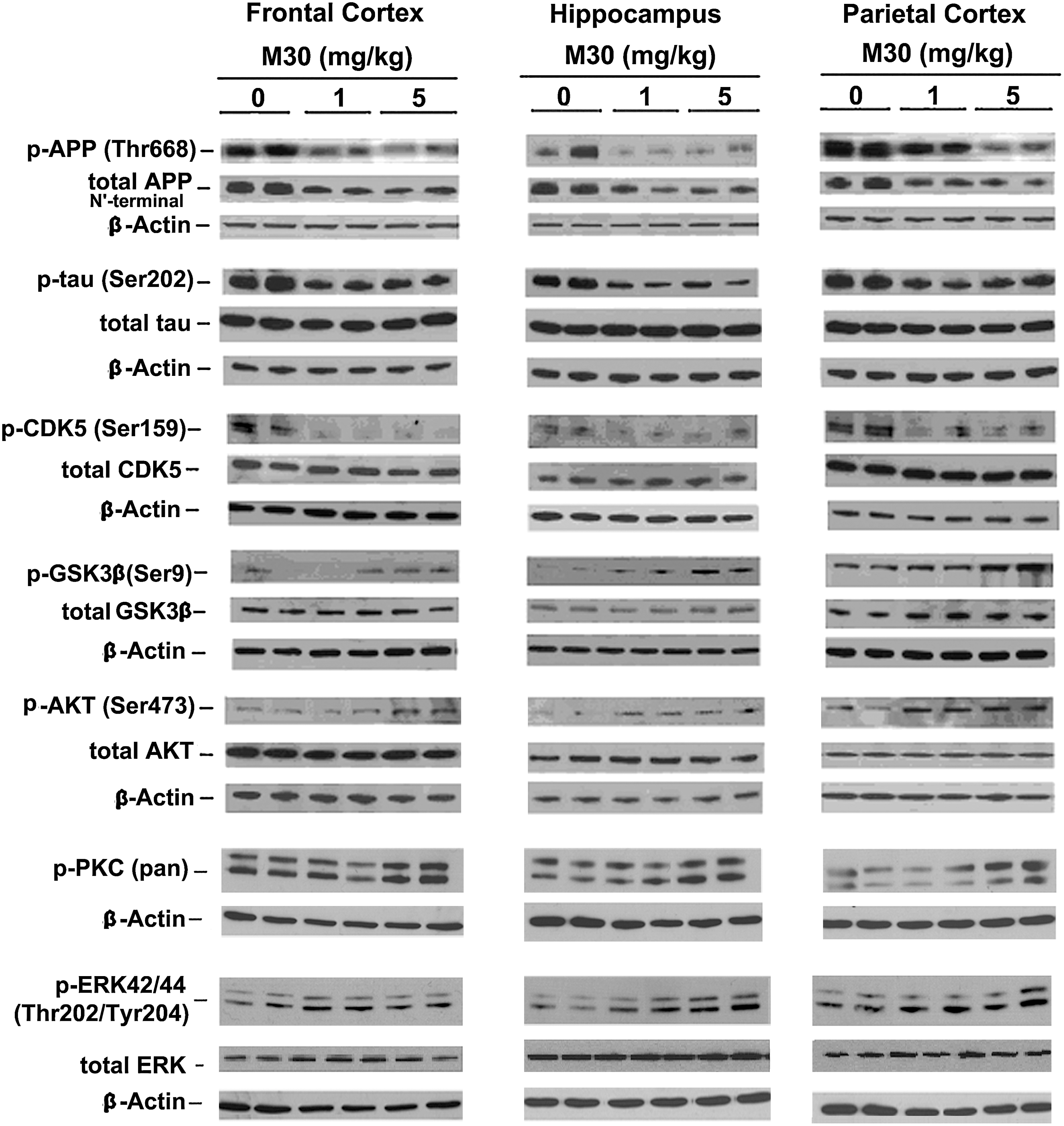
M30 treatment regulated phosphorylation of APP, tau, cyclin-dependent kinase 5, growth-associated protein (GSK)-3β, protein kinase B (AKT), protein kinase C, and mitogen-activated protein kinase/ERK
We explored a possible effect of M30 treatment on phosphorylation levels of APP and tau in the brain of APP/PS1 mice, using specific antibodies against phospho-Thr668 of APP and phospho-tau at Ser202. As shown in Figures 8 and 9, a significant reduction of phospho-APP (Thr668) and phospho-tau (Ser202) was observed in frontal cortex, hippocampus, and parietal cortex of M30-treated APP/PS1 mice, compared with the vehicle-treated group. Given the importance of cyclindependent kinase (CDK)5 and GSK-3β/AKT in the regulation of APP, as well as phosphorylation of tau, we determined the effect of M30 treatment on the phosphorylation levels of these kinases in the brain of APP/PS1 mice. Increased phosphorylation of Ser9 in GSK-3β reflects decreased activity of GSK-3β, whereas phosphorylation of AKT at Ser473 and CDK5 at Ser159 reflects increased activity of AKT and CDK5. Quantification of Western blots revealed that M30 (5 mg/kg) treatment significantly increased the ratio of phospho- GSK-3β (Ser9)/GSK-3β and phospho-AKT (Ser 473)/AKT and decreased phospho-CDK5 (Ser159)/CDK5 ratios, compared with vehicle-treated APP/PS1 mice (Figs. 8 and 9). Since propargylamine containing compounds are known to modulate proteolytic cleavage of APP by regulating mitogen activated protein kinase (MAPK)/ERK and protein kinase C (PKC) pathways (6), the effect of M30 on phosphorylation of MAPK/ERK and PKC was evaluated. M30 (1 and 5 mg/kg) significantly induced phosphorylation of ERK in hippocampus, and M30 (5 mg/kg) resulted in enhanced phophorylation of PKC in hippocampus and parietal cortex. The levels of total GSK-3β, AKT, CDK5 and ERK were unchanged by M30 treatment (Fig.8).

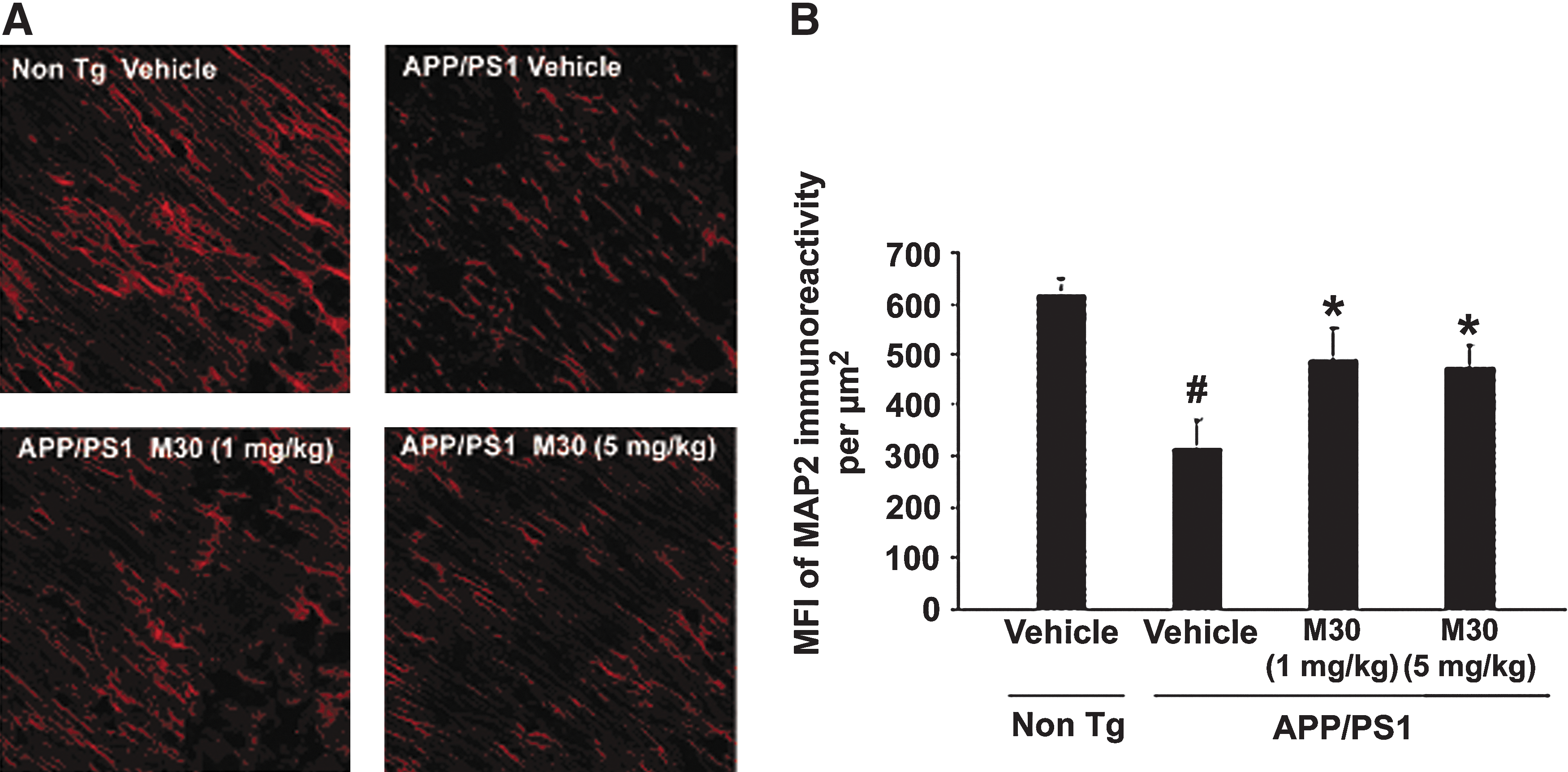
Effect of M30 on microtubule associated protein 2 and brain derived neurotrophic factor protein levels
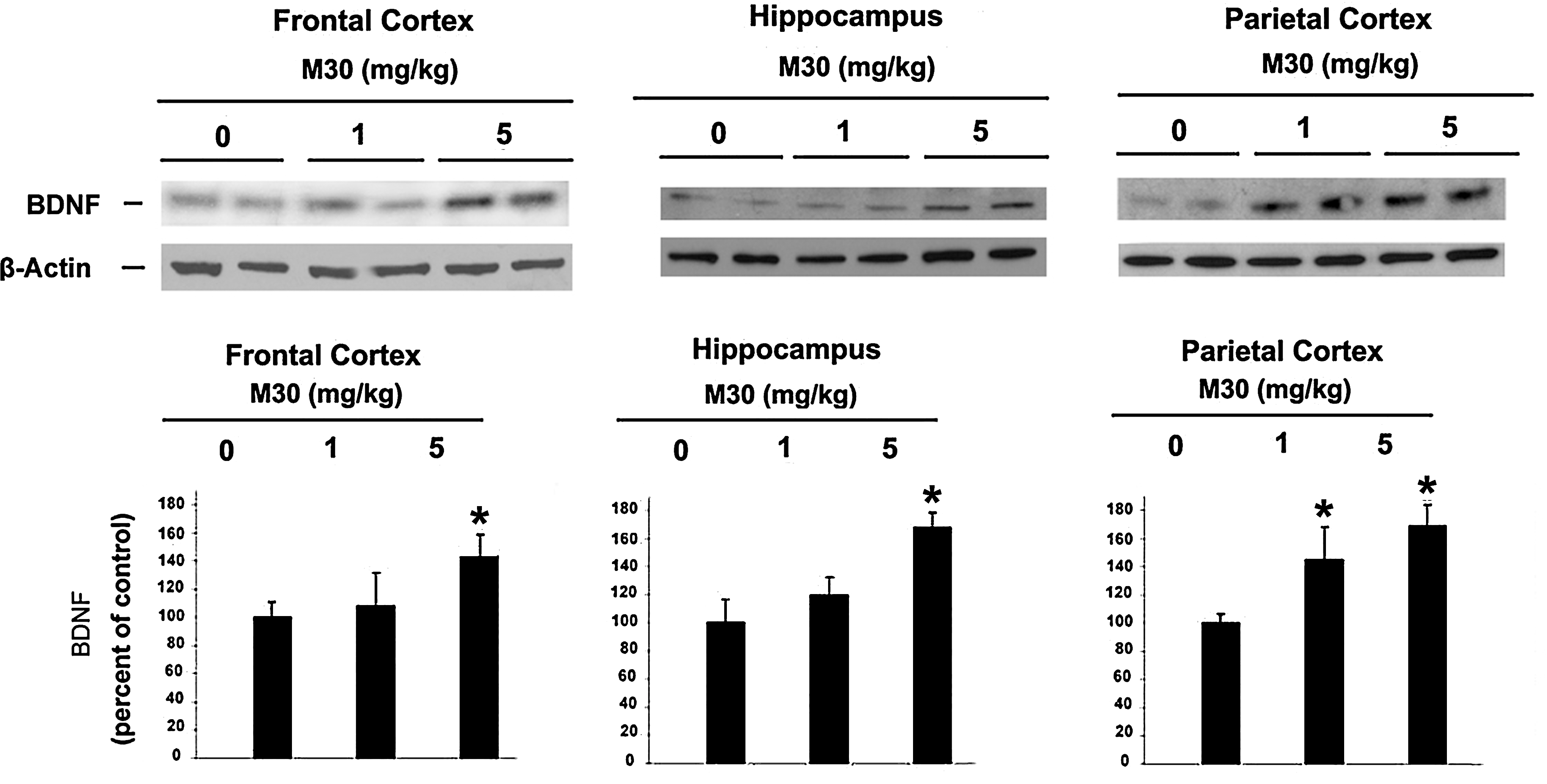
We also sought to determine the effect of M30 on brain levels of microtubule associated protein (MAP)2 [a marker for neuronal cell bodies and dendrites (10)] in the brains of APP/PS1 Tg mice. Since global neocortical neuronal loss is not apparent in this mouse model at this age, and only local neuronal loss in the hippocampal region has been observed (10), we performed immunohistochemical analysis in dentate gyrus, CA1 and CA3 hippocampal areas. Consistent with previous reports on APP/PS1 mouse model of AD (10, 19), we found a marked reduction in the MAP2 immunoreactivity in CA3 hippocampal region (Fig. 10), but only an insignificant decrease in dental gyrus and CA1 (data not shown). However, in M30-treated APP/PS1 mice, a significant preservation of MAP2 expression was observed in CA3 regions, accompanied by a significant improvement of the neuronal fibers integrity and increased neuronal bodies volume, as compared with the vehicle-treated APP/PS1 mice. These data indicate that M30 treatment might decrease the rate of neuronal degeneration in APP/PS1 mouse model. Previous studies found that M30, as well as the propargyl moiety, embedded in the M30 molecule, upregulated the expression of brain neurotrophic factors (e.g., brain derived neurotrophic factor (BDNF) and glial cell derived neurotrophic factor [GDNF]) in various in vitro neurodegenerative models (3, 42), and in vivo (28). In the current study, Western blot analysis revealed that M30 at both concentrations (1 and 5 mg/kg) significantly induced the expression levels of BDNF in parietal cortex, and at 5 mg/kg in the frontal cortex and hippocampus (Fig. 11).
Discussion
In the present study, we evaluated the potential therapeutic effect of the novel brain-permeable multifunctional iron chelating drug, M30 on AD-related neuropathology and cognitive deficits in APP/PS1 double Tg mice, a well established AD mouse model (10, 39, 55, 62). The results show that systemic M30 therapy for 9 months improved cognitive impairment and attenuated both Aβ accumulation and tau phosphorylation in various brain regions of APP/PS1 Tg mice, compared with vehicle-treated Tg mice. This beneficial effect of M30 on cognitive functions may be associated with the inhibitory effect of the drug on the levels of the potentially toxic Aβ and tau phosphorylation, since a clear relationship has been demonstrated between Aβ accumulation and tau hyperphosphorylation and cognitive deficits of these deteriorated mice (48, 60). Indeed, our findings suggest that Aβ plaque burden and Aβ levels were reduced in the frontal cortex, hippocampus, and parietal cortex of M30-treated APP/PS1 mice; quantitative ELISA analysis revealed that M30 caused a significant decrease in brain concentrations of Aβ-40 and Aβ-42, further confirming that M30 lowers Aβ amyloidogenesis in vivo. It appears that this attenuating effect of M30 on cerebral Aβ is associated, at least partly, with the reduction observed following M30 treatment in cerebral levels of its precursors: the full length APP and APP/CTFs, as Aβ (39–43 amino acids) are the product of two sequential endoproteolytic cleavages of the integral membrane protein APP (26). These results are consistent with previously reported in vitro findings showing the regulatory effect of M30 on APP expression/processing, resulting in reduced APP expression levels and Aβ generation in SH-SY5Y and CHO/ΔNL cells (4). It is suggested that the effect may be attributed to the iron-chelating moiety of M30, as mounting evidence has shown that both APP and its proteolytic product, Aβ are regulated by metal homeostasis in the AD brain (7, 11, 32, 47).
Previously, it has been indicated that a direct connection exists between increased APP expression and Aβ levels and AD pathology (50). Indeed, overexpression of mutant human APP gene in Tg mice was found to be necessary for excessive Aβ production that leads to cerebral amyloid deposition and an Alzheimer-like pathology (23). Thus, considering the accumulation of Aβ in the brain as a central pathological event in AD, attenuating Aβ generation in APP/PS1 Tg mice may provide a beneficial effect in the disease.
In regards to the effect of M30 on APP processing pathway, previous in vitro studies in CHO/ΔNL cells demonstrated that the drug increased the levels of the non-amyloidogenic soluble APPα (sAPPα) and αCTF in the medium and cell lysates, respectively (2). This non-amyloidogenic APP processing pathway involves cleavage by α-secretase, which cleaves within the Aβ sequence, resulting in induced sAPPα, thus precluding the formation of Aβ (61). It was reported that PKC and MAPK transduction-dependent pathways modulate the non-amyloidogenic proteolytic cleavage and sAPPα secretion, whereas agents acting on these signaling pathways can induce these processing pathways (5, 20). In accordance, we show in the current study that M30 induced PKC phosphorylation in both hippocampus and parietal cortex and MAPK phosphorylation in the hippocampus. It can be suggested that this effect is related to the propargyl moiety of M30, as structure–activity studies provided evidence that the ability of propargylamine derivatives to regulate APP processing by the α-secretase pathway, via PKC and MAPK signaling, is associated with some intrinsic pharmacological action of the propargyl moiety of these drugs (6). In addition, we demonstrated that M30 treatment reduced cerebral levels of phosphorylated APP695 at Thr668, which has been found to be upregulated in the pathological process of APP/PS1 Tg mice (10). Attenuating pAPP (Thr668) levels may also play a role in APP processing, since it has been suggested that pAPP (Thr668) possesses various regulatory effects on APP metabolism (e.g., induction of β-secretase cleavage and increased Aβ generation) (30).
An additional beneficial effect of the drug, presumably derived from its iron chelating potency, is its inhibitory effect on tau phosphorylation. Indeed, it was reported that iron accumulates in neurofibrillary tangles and can induce aggregation of hyperphosphorylated tau, being regulated mainly by both CDK5 and GSK3β (33).
Additional signaling pathway studies revealed that M30 enhanced brain levels of pAKT and pGSK-3β. Consistent with these data, we have shown that M30 enhanced AKT/GSK-3β phosphorylation pathway in cultured cortical neurons (3) and in vivo in mouse brain (28). The mechanism through which the drug modulates these kinases will require further investigation. Since increased GSK-3 activity has been demonstrated to be linked to spatial learning deficits in AD Tg mice (14, 22), it can be speculated that activation of AKT/GSK-3β pathway also contributes to the improved cognitive abilities observed in M30-treated APP/PS1 mice.
We also observed that M30 treatment attenuated the loss of the immunoreactivity of MAP2, occurred in vehicle-treated APP/PS1 Tg mice. This is in line with recent observations in various neuronal cells (e.g., SH-SY5Y, PC12, and NSC-34), in which M30 promoted neuronal differentiation (including cell body elongation), stimulated neurite outgrowth and upregulated the growth-associated protein (GAP)-43 (2, 29). This effect may be associated with the propargyl moiety of M30, as it has been previously demonstrated that M30, and several other propargyl derivatives significantly upregulated mRNA expression of various growth factors (e.g., BDNF, nerve growth factor (NGF), and GDNF and increased protein levels of BDNF (3, 28, 38, 42, 56). In addition, since it was previously reported that the expression of various neurotrophins, especially BDNF, is impaired in AD and AD-like animal models (17, 41, 52), the observation that BDNF expression levels were enhanced following M30 administration in APP/PS1 Tg mice may further exert the protective effect of the drug.
Accumulated data demonstrated that OS is one of the earliest events in AD progression; it increases Aβ generation and enhances tau phosphorylation. The increased levels of OS in AD brain is reflected by abnormal iron accumulation, which is capable of stimulating free radical formation (e.g., hydroxyl radicals via the Fenton reaction), enhanced lipid peroxidation, increased DNA and protein oxidation and glycation end product, and decreased cytochrome C oxidase (57). In the APP/PS1 Tg mice, it was shown that iron is co-localized with Aβ plaques in the brain (15). Thus, it is reasonable to assume that the neuroprotective action of M30 can be mediated by a reduction of OS, due to its iron-chelating properties (64). Indeed, we observed that iron staining was decreased in the cortex, striatum, and hippocampus after M30 treatment, compared with respective brain regions in the vehicle-treated APP/PS1 Tg group, indicating that the drug may prevent and/or modify the progression of neuronal degeneration by reducing excessive iron and its redox activity. In this context, we also detected a corresponding increase in the cerebral levels of TfR, known to be regulated by iron through IRP-mediated mRNA stabilization (44) and found to be significantly reduced in the hippocampus and temporal and occipital cortex in AD (25).
Noteworthy, an additional beneficial protective property of M30 in AD may be related to the fact that the drug, being a propargylamine derivative, is a potent brain selective MAO inhibitor (18). In agreement, our recent study in aged mice demonstrated that M30 administration caused a significant inhibition of both MAO-A and -B activities in the cerebellum, compared with vehicle-treated aged control mice, associated with a significant positive impact on neuropsychiatry functions and cognitive age-related impairment (27). Given that products of MAO-catalyzed reaction (e.g., aldehydes and H2O2) are compelling inductors of lipid peroxidation, it can be assumed that inhibition of MAO is associated with neuroprotective effects in AD- and age-related disturbances of the homeostasis and generation of free radicals in involution of the nervous system (51). All these beneficial regulatory effects of M30, including iron chelation, antioxidant (catalase) induction, and MAO inhibition may act synergistically with other molecular mechanisms to attenuate AD progression.
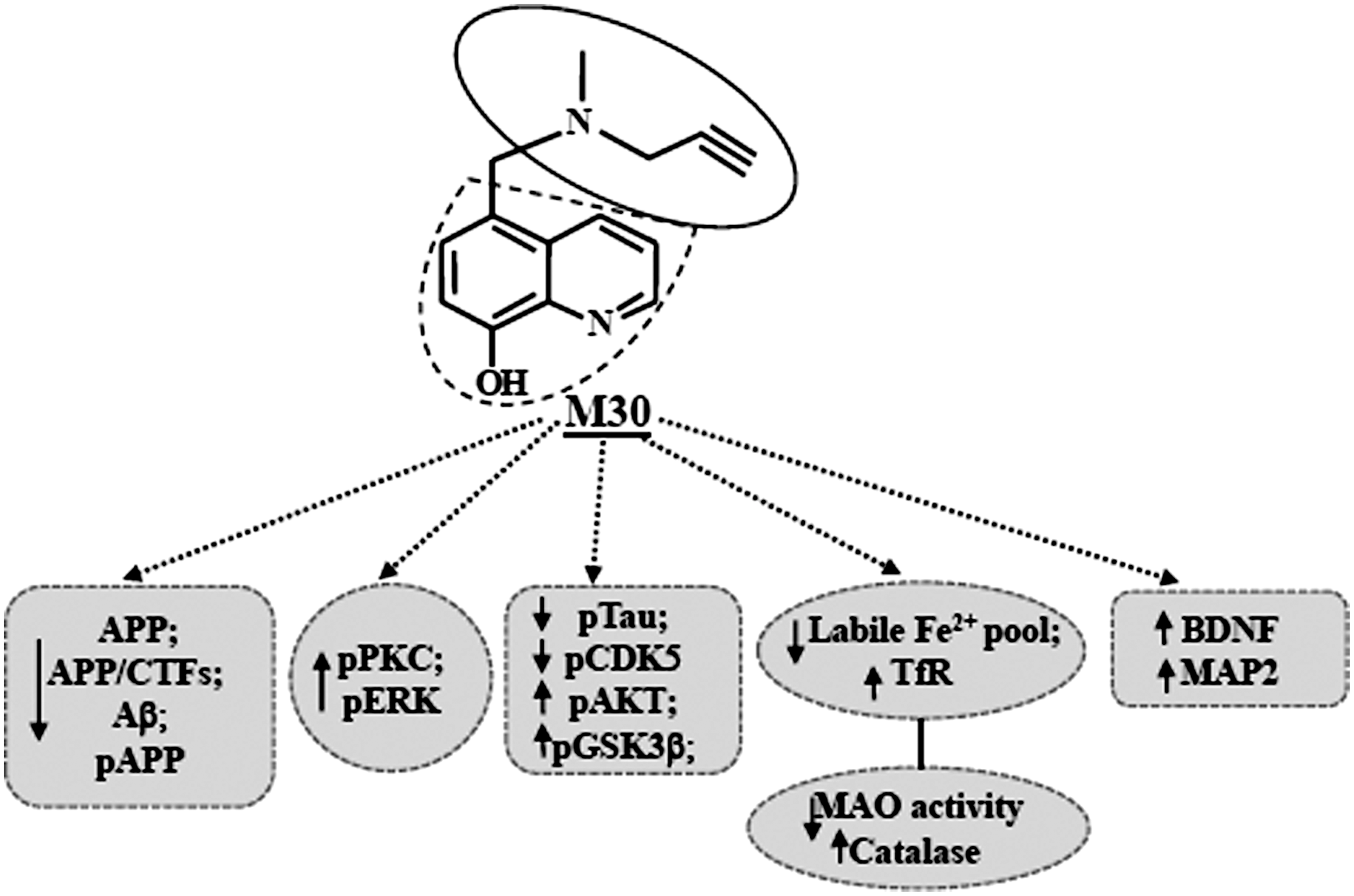
Considering the multiple iron-operating sites in AD and the multietiological character of the disease pathology, novel pharmacological approaches suggest the use of metal-chelating molecules with various active neuroprotective moieties that may simultaneously manipulate multiple desired targets (Fig. 12). Here, we presented data demonstrating that the multi-target brain-permeable iron chelating drug, M30, possesses beneficial effects on several major hallmarks of AD pathology, including improvement of cognitive deficits and reduction of cerebral Aβ plaques deposition and brain Aβ levels in a Tg mouse model of AD. The diverse pharmacological properties and several pathological targets of M30, make this multifunctional compound potentially valuable for therapeutic strategy of AD to delay neurodegeneration.
Materials and Methods
Materials
The following antibodies were used for Western blot and immunohistochemistry: rabbit polyclonal antibodies against phospho- AKT (ser 473), AKT, phospho- GSK-3β (ser 9), GSK-3β, phospho-APP (Thr 668), phospho- CDK5 (Ser159), CDK5, PKC (pan), phospho-p44/42 MAPK/ERK, and MAPK/ERK, and were from Cell Signaling (Beverly, MA). Polyclonal rabbit antibodies against phospho-tau (Ser202), tau and MAP2 were purchased from Upstate (New York). Polyclonal antibody against APP N-terminal antibody (22C11) and monoclonal antibody against Aβ (6E10) were purchased from Chemicon (Temecula, CA). APP C-terminal (amino acids 676–695) antibody β-actin antibody was from Sigma (St Louis, MO). Monoclonal antibody against TfR was purchased from Zymed (San Francisco). Monoclonal rabbit antibody against BDNF was from Epitomics Inc. (Burlingame). Donkey anti-rabbit IgG fluorescein-conjugated antibody was from Jackson ImmunoResearch laboratories Inc. (Baltimore, MD). Mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) was from Vector Laboratories (Peterborough, UK). Electrophoresis reagents and Aβ-40 and Aβ-42 ELISA kits were from Invitrogen (Carlsbad, CA). Other chemicals and reagents were of the highest analytical grade and were purchased from local commercial sources. The novel multifunctional iron chelator, M30 (5-[N-methyl-N-propargylaminomethyl]-8-hydroxyquinoline) (MW 299.3) was synthesized and kindly provided by Varinel Inc. (Philadelphia, PA) (64). M30 possesses solubility and selective iron-chelation properties (compared with zinc and copper) (63, 64). The drug is not cytotoxic, as shown by the genotoxicity assay, performed in several cell lines, inhibition of cytochrome p450 isoenzymes and voltage-dependent potassium channel-blocking test (57).
Animals and treatment
As a model for AD, we used Tg mice expressing mutated human APPswe and presenilin-1 (PS1dE9) genes (24). All procedures were carried out in accordance with the National Institutes of Health Guide for care and Use of Laboratory Animals, and were approved by the Animal Ethics Committee of the Technion, Haifa, Israel. Male APP/PS1 double-Tg mice (B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/J) were obtained from The Jackson Laboratories (Bar Harbor, ME). APP/PS1 Tg and non-Tg mice were randomly assigned into 4 groups: M30 (1 mg/kg)-treated APP/PS1 (n=16); M30 (5 mg/kg)- treated APP/PS1 (n=16); vehicle-treated APP/PS1 (n=16); and vehicle-treated non-Tg mice (n=16). M30 and vehicle were administered by the oral gavage method, four times a week, started at 3 months of age and continued for 9 months. These effective doses of M30 were chosen based on our recent animal studies (18, 65). Mice were weighed once a week started at 3 months of age. The entire brain (without the spinal cord) were dissected and weighed directly after completing all behavioral experiments at 12 months of age. Brain weights are expressed as a percentage of the total body weight.
Behavioral tests
Starting from 10 months of age, the following behavioral analyses were conducted to assess the effect of M30 administration on AD-related behavioral functions.
Morris Water Maze
Spatial learning and memory was assessed with the Morris Water Maze test as previously described (40). Briefly, we used a circular and galvanized water tank (120 cm diameter x 50 cm height) filled to a depth of 25 cm with water and a stainless steel escape platform (10 cm diameter). The surface area of the tank was divided into four equal quadrants. The water was made opaque by addition of milk powder, and its temperature was adjusted to 24°C. Mice were gently released into the water, always facing the tank wall, and given 30 sec to find the platform. On reaching the platform, the mice were allowed to remain on it for 20 sec. If a mouse failed to locate the platform within 60 sec, it was assisted by the experimenter and allowed to stay there for the same period of time (20 sec). The training schedule consisted of 10 consecutive days of testing. During the 5 first days of testing, the mice were training with visible platform which location was changed after each trial for four 30-sec trials per day. During the 5 following days of testing, the mice were trained with hidden platform that was kept at the same (target) quadrant during this period of training for four 30-sec trials per day. To assess memory consolidation, a probe trial was performed 5 days after the 10 day of platform training trials. In this trial, the platform was removed from the tank, and mice were allowed to swim freely. For these tests, time spent in the target quadrant within 90 sec was recorded. The time spent in the target quadrant was taken to indicate the degree of memory consolidation that has taken place after learning. The time spent in the target quadrant was used as a measure of spatial memory. The behavioral testing was performed by an individual blinded to the animal's treatment status.
Y maze
Spatial working memory was assessed by recording spontaneous alteration behavior in a Y Maze, as previously described (59). All mice were allowed to move freely through the maze during a 8-min session. The number and sequence of arm choices were recorded. The percentage of spontaneous alteration was calculated as the ratio of actual alterations (defined as the arm choices different from previous two choices) to possible (defined as the total number of arm entries minus 2) multiplied by 100.
Hebb-Williams Maze
The Hebb–Williams Maze was constructed, as previously described (53, 54). Each maze consisted of a square box (75×75 cm2) made of black painted wood walls (height 12 cm) and covered with a transparent plastic top. The white floor was divided into 36 black-outlined squares. Start and goal (“reward” chamber in which food was kept) boxes (40×15×12 cm3) were located in two diagonally opposite corners and equipped with sliding doors. Extramaze cues were minimized by conducting the study in an all-black enclosure and by having a dim light as the only source of illumination. 12 h fasted mice were employed in the study. The mice were pre-trained over 5 days in the maze. During the first 2 days of pre-training, mice were placed into the maze (without walls inside) and allowed to explore it freely for 10 min and to have an access to food in the goal box. During the remaining 3 days, four trials per day were performed using simple maze patterns. Following pre-training, all mice were tested on two Hebb-Williams maze-learning problems (problems 12 (easy) and 8 (difficult)) (45), with one problem per day and 6 trials per problem. Initial and repetitive errors were recorded. An initial error was defined as the first entrance with at least the two forepaws into a given error zone of a blind alley on a given trial; repetitive errors were defined as further errors made in the same zone on the same trial.
Novel food neophobia test
The neophobia test was used as a measure of anxiety and memory for the novel food (34, 46). Mice were placed inside a clean, empty tube cage for a 5-min adaptation period. A pre-weighted block of cheese was then placed in the center of the cage. After 15 min, the cheese was removed and weighed, and the mouse was returned to its home cage. Two days later, the procedure was repeated in an identical fashion. For each session, a control block of cheese was placed in an empty cage with no mouse present to control for evaporation of water from the cheese during the 15-min session. All post-session weights were adjusted for the amount of evaporation in the control cheeses block. The amount of cheese consumed by the mouse was calculated as the pre-session weight minus the adjusted post-session weight.
Nest building
APP/PS1 mice and non-Tg controls were housed in single cages containing sawdust for 3 days. On the first day of testing, two pieces of cotton (5×5 cm, Nestlets; Ancare, Bellmore, NY) were introduced in the home cage to permit nesting. The presence and quality of nesting were rated 1 day later on a 5-point scale ranging from 1 to 5 (9, 16), as follows: 1=nestlet not noticeably touched, 2=nestlet partially torn up, 3=mostly shredded but often with no identifiable nest site, 4=an identifiable, but flat nest, and 5=a near perfect nest.
Tissue sample preparation
After the above behavioral studies (at 12 months of age), mice were killed by decapitation, and their brains dissected along the sagittal plane. The right hemisphere brain was placed on an ice-cold plate and dissected into the following regions: frontal cortex, hippocampus, and parietal cortex. The dissected samples were quick-frozen for Western blot analysis. The remaining left hemisphere of each brain was processed for staining or Aβ ELISA analysis.
Immunohistochemistry
Brain hemispheres were post-fixed in 4% (vol/0.1 M PBS vol) paraformaldehyde (48 h, 4°C) and cryoprotected by 30% sucrose (48 h, 4°C). Six series of 40-μm coronal sections were collected in PBS on a freezing-sliding microtome. Immunohistochemical staining was performed as described previously (35) on series 1 (for Aβ analysis) and 3 (for MAP2 analysis). Series 2 and 4 were used for Thioflavin S and Perl's staining. Thioflavin S staining for fibrillar plaques was performed by incubating slides in 1% aqueous solution of Thioflavin S for 10 min, followed by rinsing in 80% and 95% ethanol and then distilled water. For immunostaining with Aβ (6E10) and MAP2 antibodies, sections were first pretreated with PBS containing 10 % donkey serum at 37°C for 1 h to block nonspecific staining. The sections were incubated at 4°C overnight with the primary antibodies (dilution of 1:300 for Aβ, and 1:800 for MAP2). Next, samples were incubated with donkey secondary IgG- Fluorescein- conjugated antibody at RT in the dark for 1 h, and mounted with Vectashield containing DAPI for nuclear staining. Semi-quantitative analysis of MFIs of immunofluorescence of MAP2 and the proportion of the lesion occupied by Thioflavin S and Aβ positive plaques was performed using Laser-Sharp 2000 software. Eight images of slides were obtained per each region of interest. Analysis was performed as previously described (10, 49).
Perl's iron-staining
Iron staining was performed on serial sections using a protocol derived from the standard Perl-diaminobenzidine (DAB) method (43), based on the formation of ferric ferrocyanide (Prussian blue) when ferric ion (Fe3+) released from iron-containing compounds by HCl reacts with potassium ferrocyanide. The ferric ferrocyanide then catalyzes the oxidation of DAB. Sections were rinsed in 0.01 M PBS, followed by 10 min incubation in 0.1% Triton-100/PBS solution. Next, samples were treated in a solution containing equal amounts of 2% potassium ferrocyanide and 2% hydrochloric acid for 20 min in the dark, followed again by 10 min incubation in 0.1% Triton-100/PBS solution. Iron staining was finally intensified using DAB as chromogen for 30 min at room temperature in the dark. After staining, slices were mounted on Superfrost plus slides (Fischer Bioblock Scientific, Ilkirch, France), dehydrated, and coverslipped for microscopic examination. Quantification of iron staining intensities was performed using optical density (OD) analysis (13), which determines levels of iron deposition on the basis of transmitted light in the stained tissue (37). Analyses of OD were automatically performed in different brain regions (M1 and S1 cortical areas, striatum, and hippocampus) using the Bio Rad Radiance 2000 confocal system, supported with Laser-Sharp 2000 software.
Western immunoblotting analyses
For Western blot analyses, brain sections were homogenized in Tris-sucrose buffer pH7.4 (containing a mixture of protease inhibitors, Roche, Inc. and phosphatase inhibitors) and centrifuged at 1000 g for 10 min. Protein content in the supernatants was determined using the Bradford method. Equal amounts of proteins were separated by SDS-PAGE (4%–12% Bis-Tris gels) and blotted on Protran nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). Immunoblot of monomeric Aβ content was performed using 10%–20% Tris-tricine gels (Invitrogen, Camarillo, CA). Membranes were treated with blocking buffer and primary antibodies were incubated with membranes for 20 h at 4°C, followed by incubation with horseradish peroxidase conjugated secondary antibody diluted in the same buffers for 1 h at 25°C. Detection was achieved using Western blotting detection reagent, ECL system (Amersham Pharmacia, Little Chalfont, Buckinghamshire, UK). Quantitation of the results was accomplished by measuring the optical density of the labeled bands from autoradiograms, using the computerized imaging program Bio-1D (Vilber Lourmat Biotechnology, Marne de Vallee, France). The values were normalized to β-actin protein intensity levels.
Quantitative real-time RT-PCR
Real-time RT-PCR was used for detection and quantification of specific catalase gene expression levels. Total RNA extraction, reverse transcription, and quantitative real-time RT-PCR were performed, as previously described (28). The results were analyzed in real-time on the provided program of LightCycler System (Roche Applied Science, USA) and normalized against a reference gene 18S-rRNA in order to correct sample-to-sample variation and compared with control values. The following primer sequence for catalase was used in the study: Forward, 5′-GAG GCAGTGTACTGCAAGTTCC-3′, Reverse, 5′- GGGACAGTTCACAGGTAACTGC-3′.
Measurement of Aβ
To analyze the effect of M30 treatment on cerebral Aβ, cerebral hemispheres were homogenized in 5 volumes (wt/vol) of 1% Triton X-100 in TBS solution pH 7.6, containing protease inhibitors, with a Teflon-glass homogenizer. The homogenates were centrifuged at 100,000 g for 60 min at 4°C, and supernatants were saved as Triton X-100–soluble fractions. The resulting pellets were solubilized by sonication in 5 M guanidine HCl in 50 mM Tris (pH 8.0) with the protease inhibitor mixture, incubated for 2 h at 25 °C, and centrifuged at 13,000 g for 20 min at 4°C. Supernatants were diluted 10-fold to reduce the concentration of guanidine HCl. The amounts of Aβ-40 and Aβ-42 in each fraction were determined using sandwich Aβ-40 and Aβ-42 ELISA kits (Invitrogen), according to the manufacturer's instructions.
Statistical analysis
Differences among means were analyzed using one-way ANOVA; results were expressed as the means±SEM. p values less than 0.05 were considered significant.
Footnotes
Acknowledgments
The authors gratefully acknowledge the support of the Alzheimer's Association (Chicago, USA) and the Technion-Research and Development and Rappaport Family Research Institute, Technion-Israel Institute of Technology (Haifa, Israel).
Disclosure Statement
MBH Youdim is the scientific founder of Varinel Inc. with commercial interest in the M30 drug, and developed the anti-Parkinson's drug rasagiline with the Teva Pharmaceutical Company (Israel), and received royalties