
Abstract
Classification of EMT
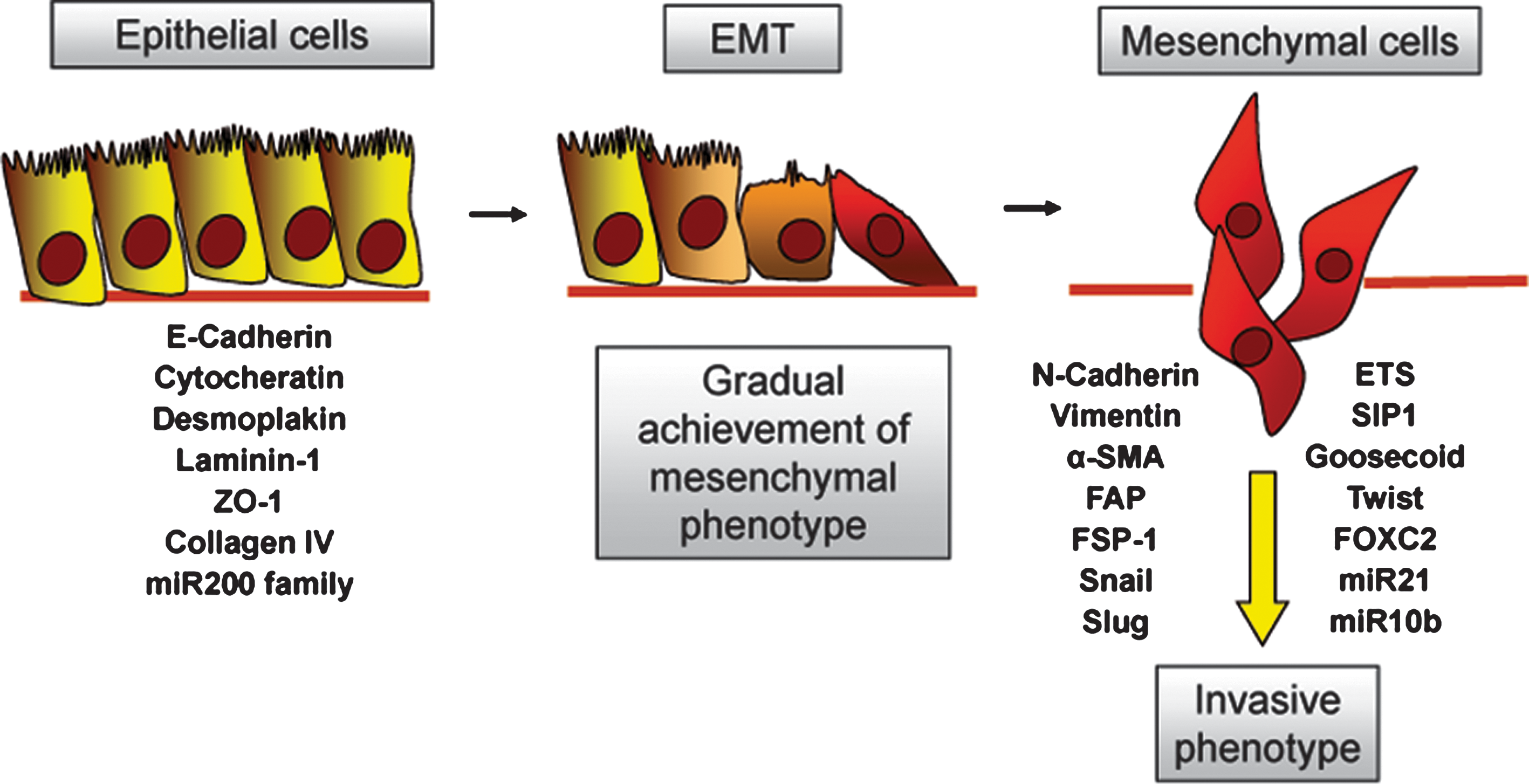
Weinberg and Kalluri proposed a useful classification of EMT into three different subtypes (83). Type I EMT is linked to embryo implantation and development and is aimed to generate the so-called “primary mesenchyme” (i.e., mesenchymal cells endowed with a great phenotypic plasticity) which move away from the original site but subsequently undergo a mesenchymal-to-epithelial transition (MET) to generate secondary epithelia. Type I EMT never causes fibrosis through excessive ECM deposition, or induces an invasive phenotype resulting in systemic spread of cells. Type II EMT is associated with tissue regeneration and healing of wounds. This EMT program is devoted to generation of fibroblasts and other related mesenchymal cells in order to recreate tissues following traumatic events and injury, even if accompanied by inflammation. Tissue fibrosis originates from unstopped ECM deposition after the healing of the wound, due to persistent inflammation, and may lead to severe organ damage (82). Neoplastic cells often undergo type III EMT, after primary carcinogenesis (155). Genetic and epigenetic changes in the primary tumor concur to engage a particular EMT program, very distant from the other two EMTs. Indeed, cancer cells undergoing a type III EMT are the primary source of metastatic cells, endowed with migratory and invasive abilities. Type III EMT is characterized by several intermediate phenotypes between epithelial and mesenchymal ones. Unfortunately, the signals that engage type III EMT in cancer cells are still incompletely clear, but some evidence indicates an involvement of both cell intrinsic and stromal-derived factors.
The term “transition” is indicative of the reversibility of the process. Indeed, EMT may be followed by the reverse process (i.e., MET, rescuing back mesenchymal to epithelial phenotype). Conversely to EMT, MET is yet a relatively unknown process, but it has been called upon for both kidney formation (75) and metastatic colony growth (155). Of note, while type I EMT is independent from inflammation and injuries, both types II and III share their dependence from inflammation and are characterized from their endurance until the provoking spur is removed. Unless the three types of EMT are involved in different biological processes, a common set of molecular elements drives these epigenetic programs.
Role of EMT in Tumor Progression
The process of human tumor pathogenesis consists of multiple steps which can induce or facilitate tumor progression toward metastatic spread. This “long metastatic route” can be categorized in several stages: 1) carcinogenesis; 2) sustained proliferative signaling; 3) generation of hypoxic environment; 4) sustained angiogenesis/lymphangiogenesis; 5) cross-talk with the component of the new microenvironment, including parenchymal, stromal, endothelial, and inflammatory cells; 6) migration through the ECM and invasiveness; 7) intravasation in bloodstream; 8) cell survival in the blood and lymphatic vessels (anoikis resistance, i.e, the ability of cancer cells to survive to lack or improper adhesion to ECM); 9) extravasation from the circulation into the surrounding tissues; 10) preparation of the metastatic niche; and 11) growth of the invading cells in the new microenvironment (Fig. 2).

EMT plays several roles in tumor progression and has become prominently implicated as a means by which transformed epithelial cells can acquire a more motile and invasive phenotype (154). Mesenchymal motility strictly depends on ECM proteolysis, through the enhancement in MMPs production, thereby enabling cancer cells to overcome physical barriers and escape from the primary tumor.
Although the major function of EMT in tumors is believed to be the induction of an invasive phenotype, EMT also elicits numerous other features that likely contribute to metastasis formation (Fig. 2).
First, the phenotypic plasticity of cancer cells may enable the formation of functionally distinct subpopulations within a tumor that support its growth in various ways. In this scenario, EMT can convert epithelial carcinoma cells into mesenchymal cells that may well assume the duties of cancer-associated fibroblasts (CAFs) in some tumors, but carry the same genetic abnormalities of cancer cells themselves (84, 137).
Second, EMT is often correlated with tissue inflammation and stromal infiltration. Indeed, pro-inflammatory cytokines, produced both by stromal and cancer cells, exert tumor-modulating effects mainly by recruiting and/or activating CAFs and cancer-associated macrophages (CAMs) (45). CAFs directly induce EMT through secretion of MMPs active on E-cadherin (58). In keeping with the idea that metastasis is a phenomenon reminiscent of the migratory/invasive behavior of inflammatory cells, tumor cells undergoing EMT in response to CAFs exposure share with inflammatory cells the same signals involving activation of cyclooxygenase-2 (COX-2), nuclear factor-κB (NF-κB), and hypoxia inducible factor-1 (HIF-1) (57). CAFs contribute to tumor progression with other key features going beyond their EMT propelling role, as they also recruit endothelial precursor cells from bone marrow, thereby inducing de novo angiogenesis (119), and participate in the preparation of the metastatic site in which the secondary tumor will grow up (44).
Third, EMT is a relevant process for the acquisition of resistance to stresses as treatment with chemotherapeutic drugs or anoikis, namely a particular apoptotic death due to loss or inappropriate cell adhesion to ECM. During EMT several genes, such as SNAI1, Twist, hepatocyte growth factor receptor (HGF-R)/cMet, and NF-κB, are induced and play a crucial role to evade anoikis by constitutively activating specific pro-survival signals (156). SNAI1 inhibits the transcription of the epithelial marker E-cadherin and confers apoptosis resistance by activating survival genes such as the phosphatidyl inositol-3 kinase (PI3K)/Akt pathway (159). In keeping, loss of E-cadherin in mammary tumorigenesis models, grants for anoikis resistance and increased angiogenesis, thus contributing to efficient metastatic spread (41, 118). In addition, cancer cells undergoing EMT are endowed with resistance to both radiation and treatment with chemical agents, in strict correlation with the acquisition of stem-cell features (2, 30, 90). This is related to the inactivation of p53-mediated apoptosis, promoted by SNAI1 and Slug, two major inducers of the EMT program (90). Furthermore, a role of the Hedgehog signaling for EMT-induced chemoresistance has also been proposed (2).
Last, recent research has inter-related the acquisition of cancer stem cell (CSC) traits with the EMT transdifferentiation program. EMT program not only enables cancer cells to disseminate physically from primary tumors, but also can confer to them the self-renewal capability that is crucial to their subsequent clonal expansion at sites of dissemination (135). Intriguingly, Mani et al. initially disclosed that human mammary epithelial cells undergoing EMT are endowed with stem-cell features (103). This connection suggests that the heterotypic signals that trigger an EMT may also be important in creating and maintaining CSCs. In both breast and prostate carcinoma, the generation of CSCs has been shown to be driven by EMT through overexpression of SNAI1 or Twist transcription factors (17, 103). Accordingly, Giannoni et al. reported that CAF exposure promotes EMT in the surrounding prostate carcinoma cells, allowing them to acquire stem cell traits (58).
An apparent contradiction of the association between EMT and metastasis comes from repeated observations that distant metastases derived from primary carcinomas are largely composed of cancer cells showing an epithelial phenotype, closely resembling that of the cancer cells in the primary tumor (26, 135). This discrepancy can be rationalized by the recognition that MET, the reversal of EMTs, likely occurs following micrometastasis growing, due to local selective pressure for the outgrowth of cancer cells with more epithelial features or to the absence of EMT-inducing signals at sites of dissemination (155).
Molecular Mediators and Signaling Pathways Guiding EMT
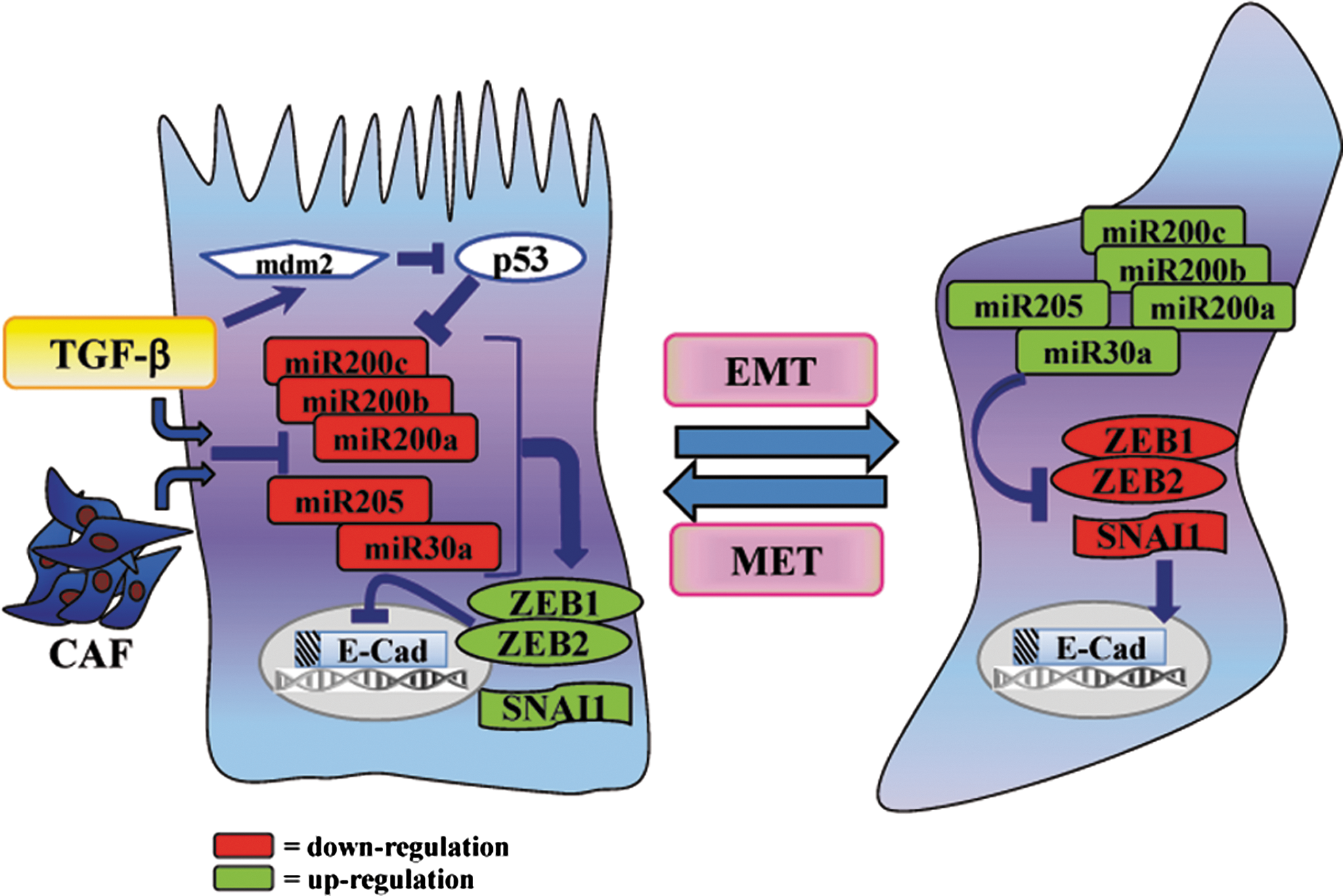
The collection of molecular factors that contribute and cooperate each other to promote EMT is very bulky and continuously growing. The crucial event in EMT is represented by the disassembly of the epithelial structure, for which E-cadherin downregulation is the most relevant step (23, 83, 155). Several transcription factors cooperate for allowing E-cadherin repression, in particular the zinc finger factors SNAI1 and SNAI2 (also indicated as Slug), the two-handed zinc factors ZEB1 and ZEB2 (also designated as SIP1), the basic helix-loop-helix factors Twist, E12/E47, and Twist2, and both the isoforms E2-2A and E2-2B (47, 129, 150). These repressors collaborate with histone deacetylases and histone demethylases (i.e., LSD1), ensuring the maintenance of the silenced state of the E-cadherin gene (99, 113).
Undoubtedly, transforming growth factor-β (TGF-β) is one of the most relevant inducers of EMT and its activity is mediated by both a Smad-dependent signaling pathway and a Smad-independent one. In the first case, the Smad protein machinery plays multiple roles, ranging from the upregulation of SNAI1, SNAI2, Twist, ZEB1, and ZEB2 to the activation of several mesenchymal genes, including α-smooth muscle actin (α-SMA), N-cadherin, fibronectin, and vimentin, all these events contributing to the achievement of a mesenchymal phenotype (23, 107, 167). On the contrary, the non-Smad signaling implicates the involvement of other major effectors of EMT, such as mitogen-activated protein kinase (MAPK), PI3K, and small GTPases of the Rho family, whose contribution to EMT will be discussed later (31).
Soluble growth factors [i.e., epidermal growth factor (EGF), fibroblast growth factor (FGF), HGF, platelet-derived growth factor (PDGF) and insulin-like growth factor-1 (IGF-1)] are another family of crucial inducers of the EMT program, that relies on the recruitment of signaling mediators, such as Ras, PI3K, and Src. Ras activation triggers the MAPK signaling cascade, ultimately leading to activation of several transcription factors, among which SNAI2, Jun, and Fos, which are known and putative repressors of E-cadherin, respectively (36, 130, 160). PI3K is another relevant mediator of EMT (96). Indeed, PI3K promotes the activation of Akt, which in turn, inactivates the glycogen synthase kinase-3β (GSK-3β), whose activity is responsible for the proteosomal degradation of SNAI1 and for the maintenance of the epithelial phenotype (10). GSK-3β is assisted, in its role of modulator of SNAI1 stability, by a recently discovered E3 ubiquitin ligase [i.e., Partner of paired (Ppa)], which also regulates SNAI2, Twist, and ZEB2 proteosomal degradation (94). On the contrary, lysyl oxidase-like 2 (LOXL2), a member of the lysyl oxidase gene family, seems to attenuate the GSK3β-dependent SNAI1 degradation, thus contributing to EMT induction (112, 131). The tyrosine kinase c-Src is another key effector of EMT. Its role in EMT induction depends on its ability to activate the focal adhesion kinase p125FAK, leading to the phosphorylation of both MAPK and the myosin light chain kinase and promoting cell–cell junction disengagement (8).
Recently, three members of the Forkhead box (Fox) transcription factor family, namely FoxQ1, FoxM1, and its splicing variant FoxM1b, are emerging as master regulator of EMT in various human malignancies. FoxQ1 expression is regulated by TGF-β and is able to repress E-cadherin by binding to its promoter (171). FoxM1 contributes to EMT induction and to the acquisition of stem cell traits, through the inhibition of several EMT-related miRNA, among which miR200b is the most relevant one (see below) (12, 139). Otherwise, the splice variant FoxM1b promotes EMT-related changes by activating both Akt and LOXL2 and subsequently upregulating SNAI1, by means of the removal of the GSK-3β-mediated inhibition (125).
An additional crucial event responsible for EMT regulation is linked to β-catenin subcellular localization. While β-catenin sequestration within the cytosol grants for its binding with E-cadherin and the preservation of cell–cell adhesions, during EMT β-catenin translocates to the nucleus, where it associates with TCF/LEF and promotes the transcription of several EMT target genes (63; 151). The Wnt signaling is one of the major regulators of β-catenin localization and relies on the inactivation of GSK-3β, which in turn acts not only by promoting SNAI1 upregulation, but also allowing for the formation of the transcriptional complex β-catenin/TCF/LEF (115).
In addition to soluble factors, some ECM components as collagen type I, III, IV, and V, also contribute to EMT by triggering the activation of the integrin-linked kinase, FAK, Src, Ras/MAPK, and PI3K/Akt (56). Integrin-linked kinase promotes the phosphorylation of Akt and GSK-3β, thus causing the inactivation of GSK-3β and leading to SNAI1 upregulation and β-catenin nuclear translocation (10, 123).
The scientific literature has recently been enriched with other evidence that highlight the growing variety of signals capable of inducing EMT, among which interleukin-8 (IL-8), macrophage-stimulating protein, and the estrogen receptor (3, 4, 48, 101). In addition, the contribution of several members of the bone morphogenetic proteins, as well as of the Notch signaling and the Hedgehog pathway, has been reported. Bone morphogenetic proteins are members of the TGF-β family that rely on the activation of several Smad members and contributes to SNAI1 upregulation and E-cadherin repression (11). Otherwise, the transmembrane receptors belonging to the Notch family induce E-cadherin downregulation by means of an increased expression of transcriptional repressors, such as SNAI2, ZEB1, or hairy/enhancer-of-split-related or, as recently discovered, by modulating miR-21, miR-200b, and miR-200c (13, 67, 116). Moreover, Gli1/Hedgehog signaling promotes an increase of both SNAI1 and ZEB1, ultimately converging on E-cadherin repression (77, 102).
miRNAs are also emerging as essential components of the cellular signaling circuitry that regulates the EMT program. Indeed, miRNAs are a family of small, highly conserved noncoding RNAs that post-transcriptionally regulate gene expression, by binding to various mRNA targets and inducing either translational repression or mRNA degradation (78). Upon TGF-β stimulation, all the members of the miR-200 family and miR-205 are strongly inhibited, thereby removing their repression on ZEB1 and ZEB2 and ultimately granting for the acquisition of a mesenchymal phenotype, as well as cell invasiveness, stemness, resistance to apoptosis, and chemoresistance (1, 65, 136, 153). Interestingly, a role of miR-205 as a tumor-suppressor in human prostate cancer has been reported, according to its skill to counteract EMT induction, through the repression of ZEB2 and the concurrent upregulation of E-cadherin (53). In keeping, we observed an intriguing role of CAFs in promoting miR-205 downregulation and a concurrent repression of E-cadherin in prostate cancer cells, thereby resulting in cell rearrangements consistent with EMT and stemness (unpublished data). Of note, loss of p53 is emerging as another crucial feature leading to inhibition of miR-200c and EMT induction, accompanied by an increase in stem cell traits (27). In keeping, several mutations in the gene encoding for p53 contribute to EMT activation, by enhancing Twist1 expression (86, 106). Recently, SNAI1 was also found to be downregulated by miR-30a, accounting for the removal of E-cadherin repression and for the maintenance of an epithelial architecture (89). Conversely, miR-155, miR-21, and miR-29a, have been found upregulated in many cancers and contribute to TGF-β-induced EMT (55, 87, 168). In addition, in human breast cancer, high levels of miR-103/107 are associated with metastasis and poor outcome. A key event elicited by miR-103/107 is EMT induction, attained by Dicer inhibition and a subsequent downregulation of miR-200 levels (78, 105). Therefore, this scenario clearly suggests a role of specific miRNAs in EMT induction and of others in the regulation of the reversal phenomenon (i.e., MET) (Fig. 3).
Reactive Oxygen Species and Their Role in Tumor Progression
Reactive oxygen species (ROS) are radicals, ions or molecules that have a single unpaired electron in their outermost shell of electrons and are constantly generated inside cells by dedicated enzyme complexes or as by-products of redox reactions, including those underlying mitochondrial respiration (71, 80, 114). The most well-studied ROS in cancer can be categorized into two groups: 1) free oxygen radicals, such as superoxide (O2 •–), hydroxyl radical (•OH), and nitric oxide (NO•); and 2) nonradical ROS, such as hydrogen peroxide (H2O2).
While generation of NO is largely due to nitric oxide synthases (NOSes), ligand-dependent generation of superoxide anion and of hydrogen peroxide, via superoxide dismutase (SOD) activity, is mediated by the NADPH oxidase (NOX)/dual oxidase (DUOX) family, whose prototypical member is the phagocytic NOX (Phox/NOX2) (92). The small GTPase Rac-1 is a key molecular player of several intracellular ROS sources, including NOX, lipoxygenase (LOX), COX, and mitochondria. Wherever produced, superoxide is dismutated into H2O2, either by the mitochondrial MnSOD or by the cytosolic Cu/ZnSOD) (71, 152).
Species such as hydrogen peroxide, superoxide anion, and nitric oxide act as signaling molecules, and their intracellular concentration is finely modulated and responsive to a wide array of environmental cues (80). These oxidants act by modifying their molecular targets in a reversible fashion, mainly through the formation of cysteine oxidative adducts [nitrosocysteine, SNO, glutathionylcysteine, SSG] or disulfide bonds (S–S). Reversible cysteine oxidation plays a pivotal role in redox signaling cascades, major molecular targets being protein tyrosine kinases, protein tyrosine phosphatases or lipid phosphatases, proteases, signaling adaptors, and transcription factors (80).
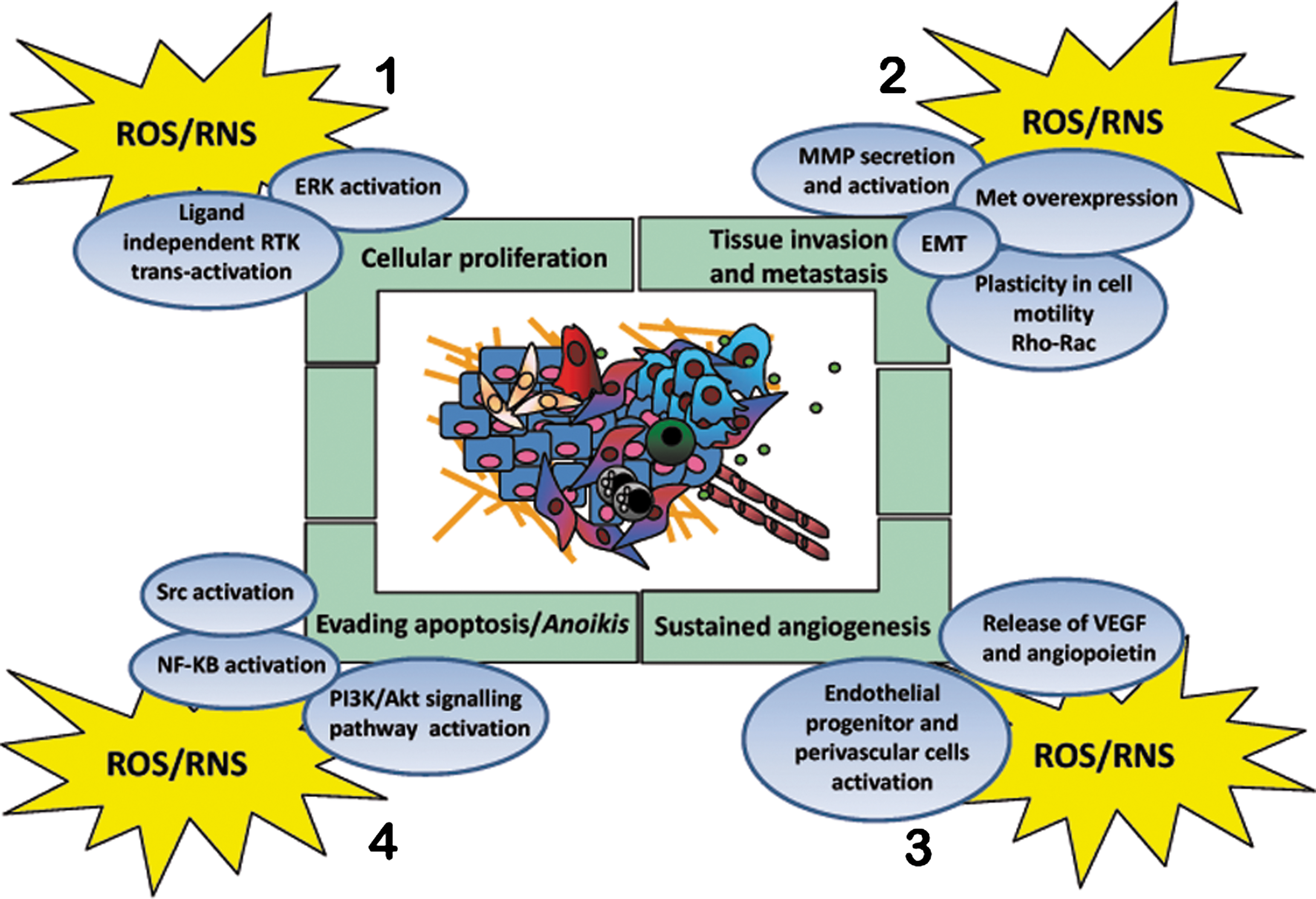
In cancer cells, high levels of ROS can result from increased metabolic activity, mitochondrial dysfunction, peroxisome activity, deregulated cellular receptor signaling, oncogene activity, enhanced activity of COXes, LOXes and thymidine phosphorylase, or depend on cross-talk with infiltrating immune cells (9, 152). Compelling experimental, clinical, and epidemiological evidence indicates that ROS and reactive nitrogen species (RNS) can promote many aspects of tumor development and progression (Fig. 4). The carcinogenic activity of oxidants strongly depends on: 1) their mutagenic potential (initiation), 2) their effects on intracellular signaling pathways controlling cell proliferation and survival (promotion), 3) their impact on cell motility and invasiveness, and 4) their established role in stromal reactions, mandatory for cancer development and dissemination, such as inflammation/repair and angiogenesis.
Endogenous ROS and RNS account for a large fraction of mutagenic DNA damage in mammalian cells, with the hydroxyl radical and peroxinitrite (ONOO−) being the best candidates for formation of 8-oxo-guanine and single/double strand breaks (104).
Tumor-promoting actions of oxidants involve sustained proliferation of carcinogen-initiated cells (25). Low doses of hydrogen peroxide and superoxide stimulate cell proliferation in several cancer cell types (20, 152). Oxidant species may reduce cell dependence on growth factors by lowering the activation thresholds of the cognate receptor tyrosine kinase (RTKs), or by trans-activating receptors in a ligand-independent fashion (141). Since RTKs couple to multiple downstream signaling cascade, many growth-related signaling events triggered by oxidants, such as activation of MAPKs or induction of early responsive genes as Jun, Fos, and Myc, could ultimately reflect, at least in part, the upstream activation of RTK-dependent signaling (121) (Fig. 4).
Clear evidence indicates that tumor progression is closely associated with the hypoxic microenvironment within the primary tumor lesions (5). Cell response to hypoxia, which includes angiogenesis, survival to stress/oxygen deprivation, and metabolic changes, is mediated by the HIF family, reportedly undergoing redox control. Mild hypoxic conditions (1%–3% O2) produce ROS through a deregulation of mitochondrial complex III (85). HIF-1 is stabilized due to a redox-dependent inactivation of their degrading enzymes, called prolyl hydroxylases (PHDs). This redox-dependent stabilization of HIF-1 enables tumor cells to activate a motogenic escape program driven by c-Met (35).
Both ROS and RNS have emerged as critical signaling molecules operating at multiple levels of tumor neo-angiogenesis (52, 88, 124). Several growth factors involved in de novo angiogenesis, such as vascular endothelial growth factor (VEGF) or fibroblast growth factor-2 (FGF-2), undergo redox regulation and ROS/RNS may both act upstream or downstream to their expression. Indeed, the response to hypoxia and to nutrient deprivation is controlled by the redox-sensitive HIF-1 transcription factor, explaining the general pro-angiogenic effects of oxidative stress in cancers. Besides, the vast majority of angiogenic factors, including VEGF or angiopoietins, act through membrane receptors using ROS/RNS as downstream second messengers (108) (Fig. 4).
Although indiscriminate oxidative stress has been extensively linked to apoptosis of cancer cells, several examples exist of the involvement of regulated production of oxidants in survival signaling. ROS generation is indeed firmly linked to activation of the pro-survival pathway driven by PI3K/Akt (32). In addition, survival signaling elicited by growth factor and cytokine signaling is largely mediated by the inflammatory transcription factor NF-κB that has been long known as a factor subdued to redox regulation (120, 143).
It has been shown that most malignancies are resistant to therapies or become resistant during anticancer therapy (140). In particular, it has been suggested that survival signaling pathways involved in the ROS-adaptive response may play a critical role in protecting cells against the damaging and cytotoxic effects of anticancer agents (132). Interestingly, lower ROS levels in breast CSCs are associated with less DNA damage upon ionizing irradiation and with radiosensitization after depletion of ROS scavengers (42).
A strategy used by tumors to avoid anoikis is the employment of oxidative stress. In metastatic prostate cells, the oxidative intracellular milieu, through the oxidation/activation of the c-Src, strongly correlates with anoikis resistance (60). A redox-mediated activation of Src has been recently observed also in several different tumor types overexpressing angiopoietin-like 4 protein (173). In human lung adenocarcinoma cells, Src oxidation/activation compensates for the loss of cell-survival signals caused by disruption of cell–ECM interactions and contributes to anoikis resistance (161) (Fig. 4).
Moreover, accumulating evidence points to the role of ROS as promoters of cell invasion and metastatic spread. Although several of these studies do not directly claim to EMT as the involved phenomenon, many if not all the stimuli eliciting redox-dependent invasion/motility are actually involved in causing EMT. These stimuli include overexpression of RTKs, as EGFR or c-Met/HGFR, hyperactivation of the Rac-1 GTPase, hypoxia, Ras or PTEN mutations (15, 49, 93, 111, 149) (Fig. 4). The specific role of ROS in EMT regulation is the subject of the next section.
Recent studies aimed to correlating CSC phenotypes with tumor invasiveness, metastasis, and resistance to therapy, further solicit a critical rethinking of the role of oxidants and redox signaling in malignancy. Recent publications have reported that both normal mammary stem cells and CSCs isolated from human and murine breast tumors have a lower content of ROS compared to their mature progeny, and that in both cases this difference is critical for maintaining stem cell function (42, 122, 133).
Oxidative Stress in Tumor Microenvironment
We now focus our attention on the origin of oxidative stress within tumor environment, the site in which EMT occurs and develops an invasive phenotype, in order to drive metastatic cells to escape the primary site and colonize elsewhere (34). Beside a cell-autonomous process involving genetically transformed cancer cells exposed to intrinsic oxidative stress due to Nox4 or HER2/ERBB2 upregulation (158), the importance of stromal cell types populating the tumoral microenvironment is now well established (16, 157). The environment in which the primary tumor evolves and achieves a metastatic phenotype through EMT is composed by stromal inflammatory cells, as CAMs and CAFs, endothelial precursor cells, and pericytes, usually found at the tumor–host interface of advanced tumors (37, 84). The synergistic activity of these cells facilitates angiogenesis, tissue remodeling by ECM breakdown, thus promoting tumor cell motility. In addition to stromal cells, structural environmental factors have also been reported to affect tumor progression and EMT. These factors include hypoxia, in which oxygen tension may fluctuate from 0.1% to 3%, acidity, and deep change in ECM composition.
Within this complex network of components affecting tumor progression, oxidative stress plays a wide and mandatory role. Indeed, some of the above mentioned components of tumor microenvironment concur to generate an oxidative environment, while some others exploit this pro-oxidant milieu to exert their propelling role for inducing EMT in tumor cells (Fig. 5).

CAMs and hypoxia can be recalled as the main pro-oxidant factors within the tumor microenvironment. It has become clear that chronic inflammation is closely correlated with tumor progression, as cancers are considered ‘‘wounds that do not heal’’ (39). The recruitment of macrophages into several kinds of tumors has been commonly reported and their role in this context is multifaceted, ranging from direct ROS or RNS generation to secretion of pro-inflammatory cytokine enhancing stromal reactivity. Both these activities can affect redox state of the tumor microenvironment, although in different manner. First, the constant production of ROS and RNS within the tumor stroma, due to activation of macrophage NOX-2 and inducible iNOS, could directly promote invasion and metastasis, through recruitment of CAFs (cells that exploit this pro-oxidant environment to differentiate, see below) or activation of MMPs. Besides, CAMs secrete pro-inflammatory cytokines which mainly orchestrate the major inflammatory response (i.e., the NF-κB pathway) in neighbouring stromal and cancer cells, which facilitates both stromal reactivity and metastatic progression of cancer cells through EMT (39, 66). The redox addiction of these cytokine-dependent responses is underscored by the key role played by ROS in the signaling pathways exploited by the master pro-inflammatory cytokines, as TNF-α or interleukins (66, 163).
Hypoxia, a common condition favoring selection of most aggressive and invasive neoplastic cells, has been causally linked to an increase in intracellular/mitochondrial ROS, thereby stressing that hypoxic conditions and ROS can synergize in inducing metastasis outcomes (122, 162). The HIF family members mediate transcriptional responses to changes in oxygen levels (148). Conversely to what expected, inadequate oxygen supply increases mitochondrial ROS rather than diminishing them (85, 148).
Hypoxia elicits the release of superoxide from complex III into the cytosol, where it is converted to hydrogen peroxide to activate oxidant-dependent signaling pathways resulting in the activation of HIF-1. Hypoxia-driven ROS stabilize HIF-1 through oxidation/inactivation of PHDs, enzymes devoted to commit the transcription factor to ubiquitin-mediated degradation in the presence of adequate concentration of oxygen (162). The key role of pro-oxidant environment due to intratumoral hypoxia has been underscored as the main responsible for Myc-mediated tumorigenesis (54), for Met-driven invasive growth of melanoma (35), as well as for EMT commitment (see below).
CAFs are also active propellers of EMT, and their activity greatly depends on oxidative stress (57, 61). CAFs originate by both resident tissue fibroblasts which infiltrate the growing tumors, or by recruitment of circulating mesenchymal stem cells (84, 134). In both cases, these fibroblasts need to be activated through a process called mesenchymal–mesenchymal transition (MMT), which gradually allows them to convert into myofibroblasts (74, 84) or CAFs, contractile cells able to affect tumor progression through secretion of cytokines, metabolites, and ECM deposition. Oxidative stress has dramatic and profound effects on MMT in both neoplastic and fibrotic diseases (6, 19, 24, 158). Indeed, in a tumor-stroma model of skin carcinogenesis, TGF-β1 initiates a ROS-dependent differentiation leading to myofibroblast trans-differentiation. ROS act as modulators of protein kinase C and drive secretion of HGF, interleukin-6, and VEGF, ultimately affecting the invasive ability of skin tumors (24). Moreover, in a model of mouse breast cancer, Toullec (158) indicated oxidative stress again as the driving force for myofibroblast differentiation. In this model, oxidative stress, due to JunD deletion, affects HIF-1 stabilization and has been called upon for activation of fibroblasts into myofibroblasts, cytokine secretion, and tumor spread (158). HIF-1 activation has been involved in MMT of cancer fibroblasts also in another model of genetic-derived oxidative stress, due to deletion of caveolin-1. In this model, myofibroblasts undergo HIF-1-driven mitophagy, overproduction of NO, resulting in tyrosine nitration of the mitochondrial respiratory chain components, as well as a metabolic conversion towards a glycolytic phenotype (127). Last, deregulated redox homeostasis driven by elevated NOX4-derived ROS signaling underlies MMT in the diseased prostate stroma, further indicating both selenium and NOX4 inhibitors as useful tools in preventing stromal reactivity in prostate cancers (145).
It should be also underlined that the different stromal components can synergize in inducing a pro-oxidant environment (Fig. 5). First, we could mention that CAFs and CAMs are often functionally associated in their tumor promoting role and that CAFs can be viewed as conspirators and sustainers of chronic inflammation (50). Indeed, laser capture analysis of prostate and basal cell carcinoma revealed that their stroma contains CAMs inseparable from CAFs (7, 110). In CAFs isolated from the initial hyperplastic stage in multistep skin tumorigenesis, Erez (45) found a pro-inflammatory gene signature, maintained in CAFs from subsequent skin carcinomas, driven by the master regulator of inflammation, the NF-κB transcription factor. Activated CAFs from this pathway promote CAM recruitment, neovascularization, and tumor growth, activities that are abolished when the NF-κB signaling is inhibited (45). The synergy between environmental factors may also be enlarged to hypoxia, which concur to stabilize HIF-1 and hence to myofibroblast differentiation and to matrix deposition, which can compatibly be altered by the redox-dependent transcriptional responses engaged by NF-κB and HIF-1.
Last, it is mandatory to mention senescence as a final factor affecting stromal oxidative stress. DNA damage accumulation occurring in aging has indeed been associated with both deregulation of ROS production and decreased antioxidant defences. Senescent fibroblasts, which constitute an inflammatory environment, secreting several pro-inflammatory cytokines and proteases, the so-called "senescence activated secretory pathway" (SASP), affect the function of surrounding tissues and stimulate tumor growth (38, 40, 91). SASP factors can globally be divided into soluble signaling factors (IL-6, IL1, IL-8), chemokines (CXCL-1, CXCL-2, CCL-8, CCL-13, CCL-2, CCL-7, CCL-26), insulin-like growth factor-1, secreted proteases (MMP-1, -3 and -10), as well as uPA or tissue-type plasminogen activators (tPA), the uPA receptor (uPAR), and the inhibitors of these serine proteases (i.e., the plasminogen activator inhibitors PAI-1 and PAI-2). This SASP turns senescent fibroblasts into pro-inflammatory cells able to promote tumor progression, at least in part by inducing an EMT in nearby epithelial cells, potentially explaining why the incidence of carcinogenesis dramatically increases with advanced age (91).
Redox Control of EMT in Cancer Cells
One of the first studies that established a direct connection between ROS and EMT highlights a direct cross-talk between ROS and TGF-β signaling (142). Upon TGF-β stimulation, there is a significant increase of intracellular ROS, prevented by treatment with inhibitors of both NOX and mitochondrial electron transfer chain. TGF-β-dependent ROS release is then responsible for the phosphorylation of Smad2, p38MAPK, and extracellular signal-regulated kinase 1/2 (ERK1/2), for α-SMA and fibronectin upregulation and for E-cadherin repression (142). All these EMT-related molecular events are well mimicked by H2O2 treatment, with the exception of Smad2 phosphorylation, strictly dependent on ERK1/2 activation (142). TGF-β has been correlated to ROS-dependent EMT induction by another study, dealing with the TGF-β-dependent regulation of ferritin heavy chain (FHC) intracellular levels (172). FHC is an iron storage protein, whose expression is dramatically decreased upon TGF-β stimulation, causing an increase in the intracellular labile iron pool (LIP). LIP increase, in turn, promotes a strong enhancement in ROS intracellular content and a redox-dependent activation of p38MAPK, ultimately leading to the acquisition of a mesenchymal phenotype. ROS elimination, as well as FHC overexpression, clearly abrogates LIP increase and results in EMT suppression, suggesting that TGF-β-induced EMT is dependent on ROS production catalyzed by LIP upregulation (172) (Fig. 6).

The inflammatory cytokine TNF-α secreted by activated macrophages is known to signal through generation of ROS, potent activators of the “master” transcription factor NF-κB (62). It has been reported that during TNF-α stimulation, SNAI1 and vimentin expression is increased, whereas the level of E-cadherin is decreased (43). These events are mediated by the TNF-α-dependent activation of NF-κB, which in turn grants for SNAI1 upregulation, essential for EMT induction (43) (Fig. 6). Recently, several studies further emphasize the role of NF-κB during inflammation-mediated EMT. Wu and colleagues reported that TNF-α, through the activation of the NF-κB pathway, enhances the activity of the COP9 signalosome 2 (CSN2), which, in turn, blocks the ubiquitination and degradation of SNAI1 (164). Altogether, this evidence strongly suggests an involvement of macrophages within the tumor microenvironment, in eliciting EMT activation, although a direct demonstration is still missing.
Accordingly, a recent study from our group further supports the concept that intracellular ROS may regulate EMT through a mechanism involving NF-κB, in strict collaboration with HIF-1 and COX-2. In particular, our data propose the axis COX-2/NF-κB /HIF-1 as a crucial signature for CAFs-mediated EMT in prostate carcinoma cells (57, 58). CAFs may exert their propelling role for EMT by eliciting a pro-oxidant and pro-inflammatory signature in cancer cells. In response to CAF-released MMP-2 and MMP-9, we observed the activation of COX-2 and a consequent COX-2-mediated ROS delivery, responsible for the establishment of a pro-oxidant environment in prostate cancer cells (57). Of note, MMP-dependent COX-2 activation relies on the expression of an alternative splicing variant of the GTPase Rac-1, Rac-1b, although the signaling events underlying the relationship among MMPs, Rac-1b expression, and COX-2 activation need to be clarified. In keeping with a key role of ROS released by COX-2, both antioxidant treatment and COX-2 inhibition or silencing totally impedes engagement of the EMT program (57) (Fig. 6). Besides COX-2, NF-κB, and HIF-1 are also key players of this scenario, since their activation is mandatory for EMT accomplishment, as well as for achievement of stem cell traits (57). Since both HIF-1 and NF-κB are stabilized by a redox-dependent pathway (35, 62, 70), they are major candidates as targets for ROS produced by COX-2 during CAF-induced EMT. Interestingly, a recent report highlights that macrophages induce COX-2 expression in breast cancer cells in a redox-dependent manner, accounting for the enhancement of cancer cell aggressiveness, likely through the involvement of a COX-2-mediated EMT activation (76).
In keeping with the proposed role for HIF-1 as a critical mediator of CAF-induced EMT in normoxic conditions (57), an increasing bulk of literature demonstrates that hypoxia-related activation of HIF-1 acts as an important executor of EMT. Indeed, hypoxic conditions generate and sustain major EMT-triggering pathways for facilitating tumor growth and metastasis, such as TGF-β and Notch signaling pathways (81). Several experimental studies indicate that HIF-1 and TGF-β may cooperate in triggering EMT. On one hand, hypoxia may affect the TGF-β signaling pathway by increasing Smad3 expression (170) or by enhancing TGF-β production (117, 146, 147). On the other hand, TGF-β acts by decreasing HIF-1α-associated PHD2, suggesting that TGF-β enhances HIF-1α protein stability (109). The interplay between hypoxia and Notch signaling has also been recently established. Notch1 has been found to upregulate HIF-1 expression in breast cancer (97). In turn, HIF-1 binds and stabilizes activated Notch, which then translates the hypoxic response into the EMT process (29, 69). In particular, Notch signaling deploys two distinct mechanisms to control SNAI1 expression. First, Notch directly upregulates SNAI1 expression by binding to the SNAI1 promoter (72, 144). Second, Notch enhances HIF-1 recruitment to the LOXL2 promoter and enhances LOXL2 expression, thereby stabilizing SNAI1 (128, 131) (Fig. 6).
There is increasing experimental evidence showing that HIF-1 modulates EMT also by regulating the expression and activity of major transcription factors involved in EMT. First, during hypoxia, HIF-1 is able to directly regulate Twist expression (165, 166). In addition, Cannito et al. reported that hypoxia-mediated induction of EMT relies on a biphasic process, involving an early phase dependent on redox-signaling and a late phase, mainly based on HIF-1 and VEGF activity (22). The early ROS generation accounts for the phosphorylation/inactivation of GSK-3β, followed by SNAI1 nuclear translocation and E-cadherin repression, all these events ensuring triggering of EMT. At a later stage, HIF-1 induces a long-lasting activation of Wnt/β-catenin signaling and an increase in migration and invasiveness (22). In keeping with these observations, in hypoxic ovarian cancer cells, the increase in SNAI1 expression correlates with HIF-1α activation, suggesting that hypoxia may act on SNAI1 directly and results in EMT (73). Additionally, HIF-1 may also enhance LOXL2 expression, thereby regulating SNAI1 in an indirect manner (46, 68).
In agreement with the above described redox-regulation of SNAI1 upon exposure to an hypoxic environment (22), another study demonstrates that an oxidative stress elicited by H2O2 administration induces a SNAI1-mediated hypermethylation of the E-cadherin promoter by recruiting histone deacetylase 1 and DNA methyltransferase 1 (98). An alternative strategy for ROS to be involved in EMT regulation has also been proposed. Barnett et al. observed a rise in intracellular oxidants, dependent on SNAI1 activation in prostate carcinoma cells (14). The authors proposed that SNAI1-mediated oxidative burst culminates on ERK1/2 activation, thus granting for EMT accomplishment. According to these data, in addition to the redox-dependent activation of SNAI1, there is also a reverse scenario in which SNAI1 itself has a mandatory role in the induction of an oxidative intracellular environment, mandatory for the EMT program (14).
The role of ROS as crucial mediators of EMT induction is further corroborated by the involvement of MMP-3 (also known as stromelysin) as an EMT inducer in mouse mammary epithelial cells (100, 138). Exposure to MMP-3 is associated with SNAI1 upregulation, loss of E-cadherin, nuclear translocation of β-catenin and, finally, increased motility and invasiveness (100). These events rely on a MMP-3-dependent ROS generation of mitochondrial origin, which in turn is dependent on the expression of Rac-1b (138) (Fig. 6). Accordingly, both SNAI1 upregulation and EMT activation are prevented by antioxidant treatment, while the same phenomena are reproduced by the exposure to H2O2 (138). These results involving MMPs as upstream mediators of the redox-dependent EMT are reinforced by our data, indicating MMP-9 as the driving force in CAF-induced EMT, again a process under redox control (57).
Integrin engagement by ECM components is well known to elicit a LOX-dependent ROS increase (33). This pro-oxidant environment is able to affect several molecular mediators, among which the tyrosine kinase c-Src is one of the major target (59, 61). It is reasonable to suggest that the redox-mediated activation of c-Src may largely participate to execute a downstream signaling pathway, culminating in the acquisition of a mesenchymal phenotype. Accordingly, a ROS-dependent oxidation/activation of c-Src leading to cell–cell disengagement through the Rho/Rho kinase pathway has recently been proposed (79) (Fig. 6).
Many other factors involved in EMT execution are emerging as correlated with ROS production. Aldosterone has been reported to induce EMT in human renal proximal tubular cells, as evidenced by SNAI1 upregulation, E-cadherin inhibition, and α-SMA expression (169). These effects appear mediated by a mitochondrial-dependent ROS generation, responsible for MAPK activation (169). Recent observations by Chang and colleagues also support a pivotal role of ROS in EMT induced by angiotensin II in rat peritoneal mesothelial cells (28). Cell exposure to angiotensin II significantly increases the ROS intracellular content, by means of NOX activity, as confirmed by the effectiveness of diphenylene iodonium to prevent the oxidative imbalance. Moreover, ROS act as essential mediators in promoting plasminogen activator inhibitor-1 and α-SMA upregulation, as well as E-cadherin inhibition, thus supporting a redox-mediated regulation of EMT (28).
Conclusion
In spite of the undoubted role of EMT in embryonic development, organ fibrosis or tumor progression, some key points still need to be clarified by further studies.
1) EMT is a reversible phenomenon, as it may be followed by MET (83, 154), but the stimuli within the metastatic site that induce this reversibility are completely unknown. As EMT is a redox-dependent phenomenon, it is likely that ROS content could also affect MET. If the primary tumor microenvironment has pro-oxidant features, we should assume that the metastatic niche is characterized by low ROS levels, but experimental evidence is still lacking.
2) EMT is clearly associated with selection/induction of cells with stem-like traits (58, 103). Nevertheless, quiescent cells within the stemness niche have been associated with low ROS content (133). The apparent inconsistency between these two concepts needs to be re-discussed, also in the light of the role played by ROS in chemo- and radio-resistance and the acknowledged correlation between resistance to therapy and stemness (42, 95).
3) EMT is not the only example of the enormous plasticity that cancer cells usually undergo. Indeed, mesenchymal-amoeboid motility (MAT) has been reported to be a further escaping strategy adopted by cancer cells when mesenchymal motility is not allowed (by protease or integrin inhibition) (51). Only few indications suggest that also MAT is under redox control (21, 126), thus enlarging the role of ROS in the regulation of whole plasticity of cell motility.
4) An honest view of EMT suggests that, although it acts at multiple levels in tumor progression, it is only one of the steps required by carcinoma cells to successfully spread and grow metastases at distance. This has enormous therapeutic implications for the use of single antioxidants as antimetastatic agents. Indeed, although antioxidant supplements have been used in clinical trials (18, 64), their use may select quiescent stem cells, behaving as dormant metastases. Therefore, studies combining antioxidant therapy with other treatments aimed to target quiescence of CSCs or other kinds of plasticity in cell motility, are highly warranted.
Footnotes
Acknowledgments
This work was supported by the Associazione Italiana Ricerca sul Cancro (AIRC), by Istituto Toscano Tumori (ITT) and Regione Toscana.