
Abstract
Introduction

Enzymes Involved in GSH Synthesis, Transport, and Recycling
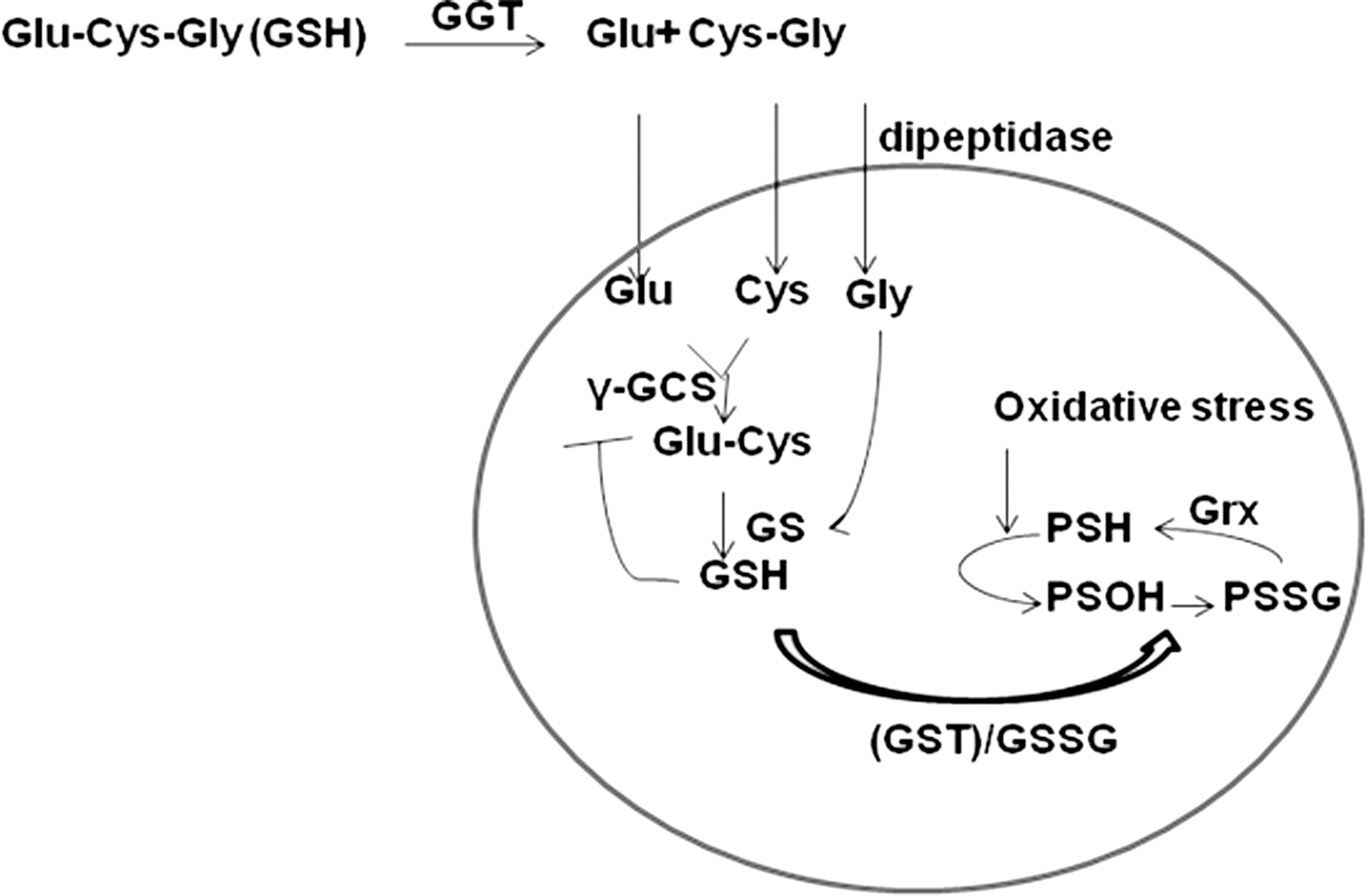
GSH levels in the cells are tightly regulated through control of its rate of synthesis, import, and export (7). Since GSH is negatively charged and cannot cross the plasma membrane, cells have evolved with transporters and enzymes to regulate the GSH levels in the cell (7). For uptake, extracellular GSH is catabolized to cysteinyl-glycine (CG) (64) and glutamate by the enzyme γ-glutamyltransferase (GGT) (7). CG is then cleaved by the plasma membrane-bound dipeptidase to cysteine and glycine (7). Thus produced cysteine, glycine, and glutamate are imported into the cell. Cysteine is then utilized by γ-glutamyl cysteine synthase (γ-GCS) to synthesize γ-glutamylcysteine, and glycine is subsequently added by GSH synthetase (GS) (Fig. 3). The whole cascade is subject to feedback inhibition by GSH. The unusual peptide bond formed between γ-carboxyl group of glutamate and cysteine is not susceptible to cleavage by proteolytic enzymes, hence it is stable in serum and other extracellular environments (7). GSH can be oxidized to the disulfide, GSSG. Furthermore, glutathione peroxidase (GPx) reduces H2O2 while oxidizing GSH to GSSG, which is then recycled by the enzyme glutathione reductase by an NADPH dependent mechanism (5, 7, 14, 63).
Enzymes Involved in the Protein S-Glutathionylation Cycle
S-Glutathionylation
S-Glutathionylation, the covalent modification of reactive protein cysteines with glutathione, has recently emerged as a post-translational modification with cardinal importance in regulation of biological processes. S-Glutathionylation is likely to affect the structure and function of proteins, given the bulky nature of the modification (i.e., addition of three amino acids), and the negative charge contributed by the glutamic acid moiety. One traditionally accepted biochemical scenario for S-glutathionylation represents the reaction of glutathione disulfide (GSSG) with protein sulfhydryls. Indeed, in cells undergoing overt oxidative stress that culminates in full blown cell death, a significant increase in PSSG is proportionally linked to increases in GSSG levels (22). However, under normal physiological conditions, the intracellular milieu favors a fully reduced thiol state, and alternative biochemical processes are more likely to catalyze S-glutathionylation. One plausible alternative scenario is the formation of a sulfenic acid intermediate (hydroxylated cysteine, -SOH) generated after activation of NADPH oxidases, production of ROS by mitochondria, or other processes, which can subsequently react with reduced glutathione (GSH). S-Glutathionylation has been shown to affect proteins in diverse manners. For example, S-glutathionylation of endothelial nitric oxide synthase uncouples the enzyme to produce superoxide instead of nitric oxide (13), suggesting that S-glutathionylation of proteins can alter enzyme function. S-Glutathionylation enhances the activity of enzymes such as SERCA (2) and Ras (1), but inhibits the activity of protein kinase C and inhibitory kappa kinase β (IKKβ) (64, 66). As will be discussed below, S-glutathionylation of the death receptor, Fas, increases the efficiency of binding of Fas ligand to Fas, and promotes formation of the death-inducing signaling complex and apoptosis (5, 35). These diverse outcomes demonstrate that S-glutathionylation evokes target-specific changes in structure and function, in line with the effects of other post-translational modifications. So far, more than 100 proteins have been identified as targets for S-glutathionylation, in response to diverse stresses (87). Despite these recent findings, many of the exact molecular and biochemical determinants that govern S-glutathionylation remain elusive to date, and conclusive evidence of protein S-glutathionylation induced by biological ligands in vivo remains sparse. Recent evidence that the pi class of glutathione S-transferase (GSTP) can catalyze S-glutathionylation (Fig. 2, discussed in the next section) provides exciting new insights into the biochemical events that govern S-glutathionylation in biological settings.
Glutathione S-tranferases (GST)
Classically GSTs (EC 2.5.1.18) are known as detoxifying enzymes that catalyze the conjugation of GSH to various electrophilic molecules (51, 56). A recent body of work has clearly demonstrated the role of GSTs as regulators of molecular pathways that control cell division and apoptosis (3, 27, 82). Based on their primary sequence similarities, substrate specificities, and antibody cross reactivity, GSTs have been classified into seven classes ranging from α to ζ (51, 56). Among them, GSTπ (also known as GSTP) is known to regulate c-Jun N-terminal Kinase (JNK) and its target c-Jun (3, 27, 82). Specifically, GSTP has been demonstrated as an endogenous inhibitor of JNK, and genetic ablation of GSTP, or its pharmacologic inhibition, results in constitutive activation of JNK (82). Because of the cardinal importance of JNK in the causation of apoptosis (80), and the known ability of GSTP to repress JNK, a role of GSTP in the control of apoptosis is therefore easily envisioned. This plausibility is further strengthened by the overexpression of GSTP in many tumors that are known to be resistant to apoptosis (87). GSTs were initially characterized to have significant ligand-binding potential for molecules such as heme, bilirubin, and hypericin; hence they were named as ligandins (1, 48, 49). The interaction between JNK and GSTP is thought to be dependent on the “ligandin” property of GSTP, and does not involve S-glutathionylation catalysis.
GSTP was recently identified as a catalyst of S-glutathionylation reactions (50, 77). GSTP was initially demonstrated to restore the function of oxidized 1 Cys-peroxiredoxin by binding to and S-glutathionylating the sulfenic acid intermediate of 1 Cys peroxiredoxin, a reaction essential for restoration of 1 Cys peroxiredoxin function (50). The role of GSTP as a catalyst of S-glutathionylation addresses potential concerns about spontaneous reaction kinetics between GSH and sulfenic acid intermediates. However, the exact biochemical determinants that govern GSTP-catalyzed S-glutathionylation remain to be fully unraveled. Although it was demonstrated that the sulfenic acid moiety of 1 Cys peroxiredoxin is S-glutathionylated via GSTP (50), it is not clear whether the sulfenic acid moiety is an essential precursor in all GSTP-catalyzed S-glutathionylation reactions, or whether other cysteine oxidations, such as S-nitrosylation (52, 53) may be targets for S-glutationylation (54). It is worthy of mention that GSTP itself can become S-glutathionylated which reduces its activity (75) and may act as a mechanism of negative feedback regulation. Reports published thus far have mainly shown that GSTP is a catalyst of S-glutathionylation following exposure of cells to oxidants (77). Additional efforts will be needed to illuminate the role of GSTP in S-glutathionylation in physiological settings, the protein targets for S-glutathionylation, and whether S-glutathionylation can be catalyzed by other members of the GST family.
Glutaredoxin
Grx (EC 1.20.4.1) were originally described as thiol transferases which contain a thioredoxin like Cys-Pro-Tyr/Phe-Cys catalytic domain. To date, two mammalian glutaredoxins have been characterized. Grx1 is mostly cytosolic, where as Grx2 has been found in the mitochondria and nucleus (42). De-glutathionylation of proteins by Grxs occurs by a monothiol mechanism that only depends on the N-terminal Cys22 (Cys23 in mouse Grx1), which displays an unusually low pKa (31). Cys22 will become S-glutathionylated and this mixed disulfide is reduced by consumption of GSH. The GSSG formed from this reaction is reduced by GSSG reductase, thereby recycling the enzyme (19, 66). In physiological settings, Grx catalyzes de-glutathionylation of proteins. However, in conditions of overt oxidative stress, Grx can also catalyze PSSG, via a mechanism that involves a thiyl radical (62, 71). Collectively, emerging studies suggest a cooperative role of GSTP and Grx in controlling protein S-glutathionylation; GSTP which catalyzes the forward reaction, and Grx, which catalyzes de-glutathionylation (reverse reaction) (Fig. 3). Modulation of these enzymes in biological settings has the potential to provide new insights into the regulation of protein function via S-glutathionylation.
Recent reports have highlighted how changes in the redox environment might influence activation of the cell death machinery (Figs. 4 and 5). In particular, GSH depletion and S-glutathionylation have been attributed as regulators of apoptosis in response to diverse stimuli that include activation of death receptors, stress, environmental agents, and cytotoxic drugs (22). Subsequent paragraphs of this perspective focus on the apoptotic pathways that are governed by redox-based mechanisms, with a focus on protein S-glutathionylation.
Glutathione (GSH) and its role in apoptosis
Oxidants such as superoxide, hydrogen peroxide, and nitric oxide are generated by a variety of enzymes as a consequence of activation of growth factor receptors, death receptors, environmental agents, and pathogens (7). Protection from oxidative damage is afforded by a number of antioxidant enzymes that include catalase, superoxide dismutase, glutathione peroxidase (29, 40, 69), and peroxiredoxins (46), to name a few, and this repertoire of enzymes is augmented by small molecules such as GSH, ascorbate, tocopherols, and urate, among others (24, 29, 65, 78). GSH is critical for survival, based on findings that GSH synthesis-deficient mice (γ-GCS-/-) die from massive apoptosis (16). GSH depletion in response to oxidants has been widely reported, and linked to cell death (7, 22). Conversely, high intracellular GSH levels have been associated with apoptosis-resistant phenotypes (25). It has been demonstrated that buthionine sulfoximine (BSO)-mediated depletion of GSH does not trigger apoptosis per se, but sensitizes T cells to death receptor-mediated apoptosis (18). Replenishing GSH pools by N-acetyl L-cysteine (NAC) is known to protect against apoptosis (18). The role of GSH depletion and ROS in apoptosis induced by death receptors remains controversial. GSH depletion is known to occur due to physiological death ligands during early stages, resulting in accumulation of ROS which can either be stimulatory or inhibitory to apoptosis dependent on the cell type, stimulus, and experimental context (21 –23, 67). The reader is referred to (7, 22) for a comprehensive review.
Redox regulation of death inducing ligands
Apoptosis is triggered by death receptors such as Fas (CD95), tumor necrosis factor receptor 1 (TNFR1), and death receptors 4 (DR4) and 5 (DR5). These receptors are activated by FasL, TNFα, and TRAIL, respectively (34). Multimers of death receptor ligands such as TNF and FasL are efficient in triggering oligomerization of death receptors in vitro and in vivo, which is important in effective induction of apoptosis (36, 86). Chronic disease conditions are known to be accompanied by an increased oxidative environment, due to enhanced production of oxidants by inflammatory cells, or activated or damaged structural cells (35, 55, 87). To date, the functional link between oxidant production and death receptor activation is not fully elucidated. Oxidant-induced signaling cascades can lead to transcriptional activation of death receptor ligands, such as FasL, which was shown to require activation of c-Jun-N-terminal kinase, and the transcription factor, activator protein-1 (AP-1) (17, 72). It is also possible that direct oxidative modification of death-inducing ligands increases their potency as inducers of cell death. Indeed, one report demonstrated that methionine oxidation of the stalk region of FasL induced oligomerization and increases in death of alveolar epithelial cells in a setting of acute lung injury (35). Since the TNF family of ligands needs to oligomerize to be highly active, this mode of oxidation may be giving rise to potent death ligands in disease settings that are accompanied by increased levels of oxidants. It is not clear to date whether cysteines within death ligands, in particular the cysteines in the stalk region, are involved in intermolecular disulfide linkages and oligomerization of the death receptor ligands.
Redox-based regulation of death receptors
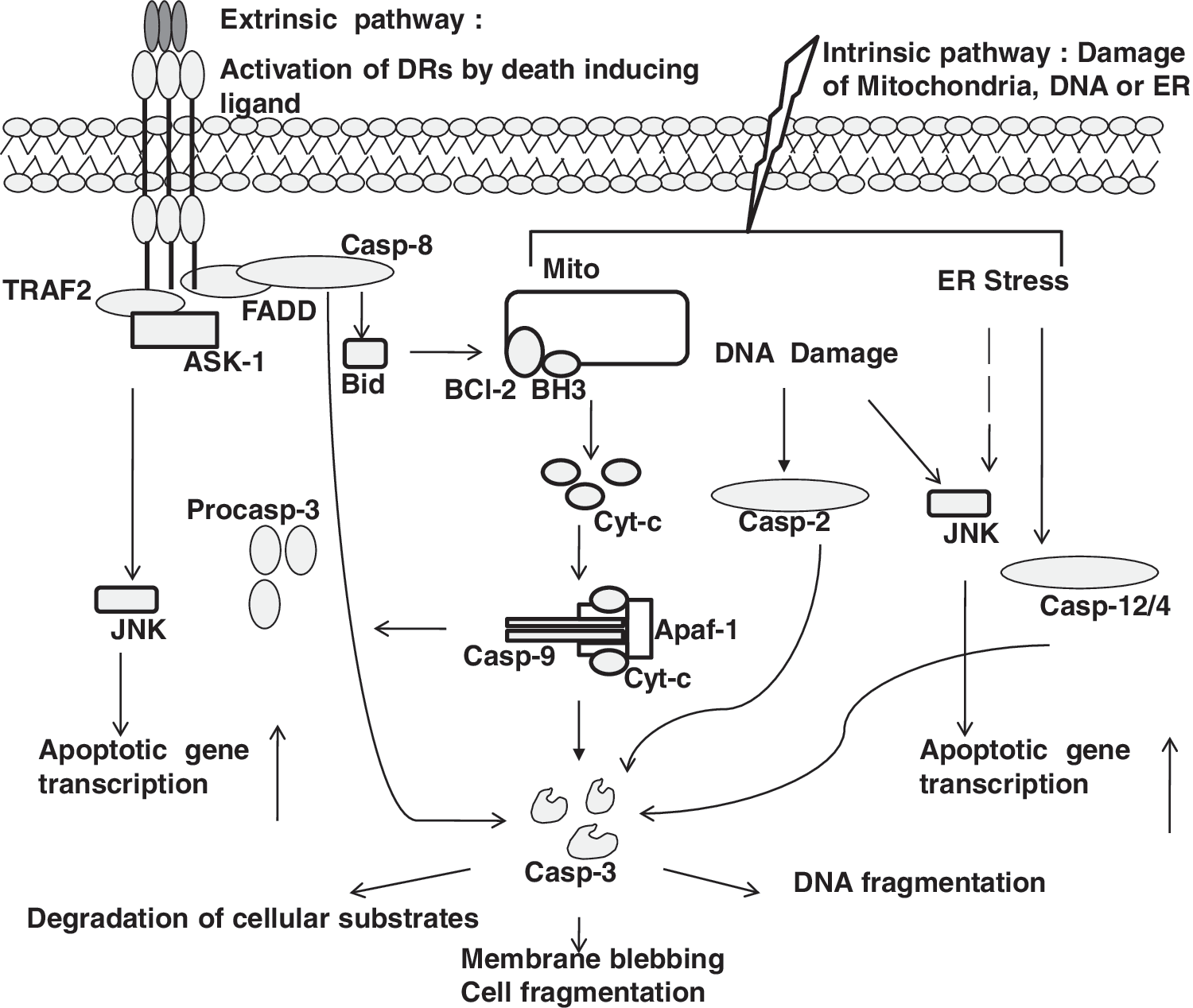
Activation of Fas, DR4, and DR5 leads to the formation of the death-inducing signaling complex (DISC), consisting of Fas-associated death domain (FADD) and procaspase-8/10 (34). Activation of initiator caspase-8/10 further amplifies the apoptotic cascade by activation of executioner caspases-3/6/7 (26). In cell types that have low levels of DISC formation, the progression of apoptosis is amplified by cleavage of the Bcl2 family protein Bid, and its translocation to mitochondria (34). Subsequent release of ROS, along with cytochrome c, activates caspase-9 which enhances the activation of executioner caspases-3/6/7 (34) (Fig. 1). In contrast, TNFR1 signaling results in formation of TNF-induced complex 1 that recruits TNF receptor-associated death domain (TRADD), TNF receptor-associated factor 1/2 (TRAF1/2), but lacks procaspase-8. Translocation of the TRADD/TRAF1/2 complex to the cytosol recruits FADD, caspase-8/10, and leads to apoptosis (8). TNFα-induced recruitment of TRADD and TRAF2 also facilitates pro-apoptotic signaling cascades via the activation of ASK1 and JNK, and subsequent transcriptional activation of pro-apoptotic genes, including FasL (17, 72), as was mentioned earlier. In some cells, TNFα-induced activation of NADPH oxidase mediates activation of the ASK–JNK axis, and this was shown to induce necrosis, without activation of caspases (58).
TNFα-induced apoptosis has been shown to be accompanied by increased protein S-glutathionylation, which could be inhibited by overexpression of Bcl-2 (73). Increases in NADPH oxidase activity may be functionally linked to increases in overall protein S-glutathionylation (PSSG). However, exact molecular details, and protein targets for S-glutathionylation in the TNFR1 receptor pathway remain to be elucidated.
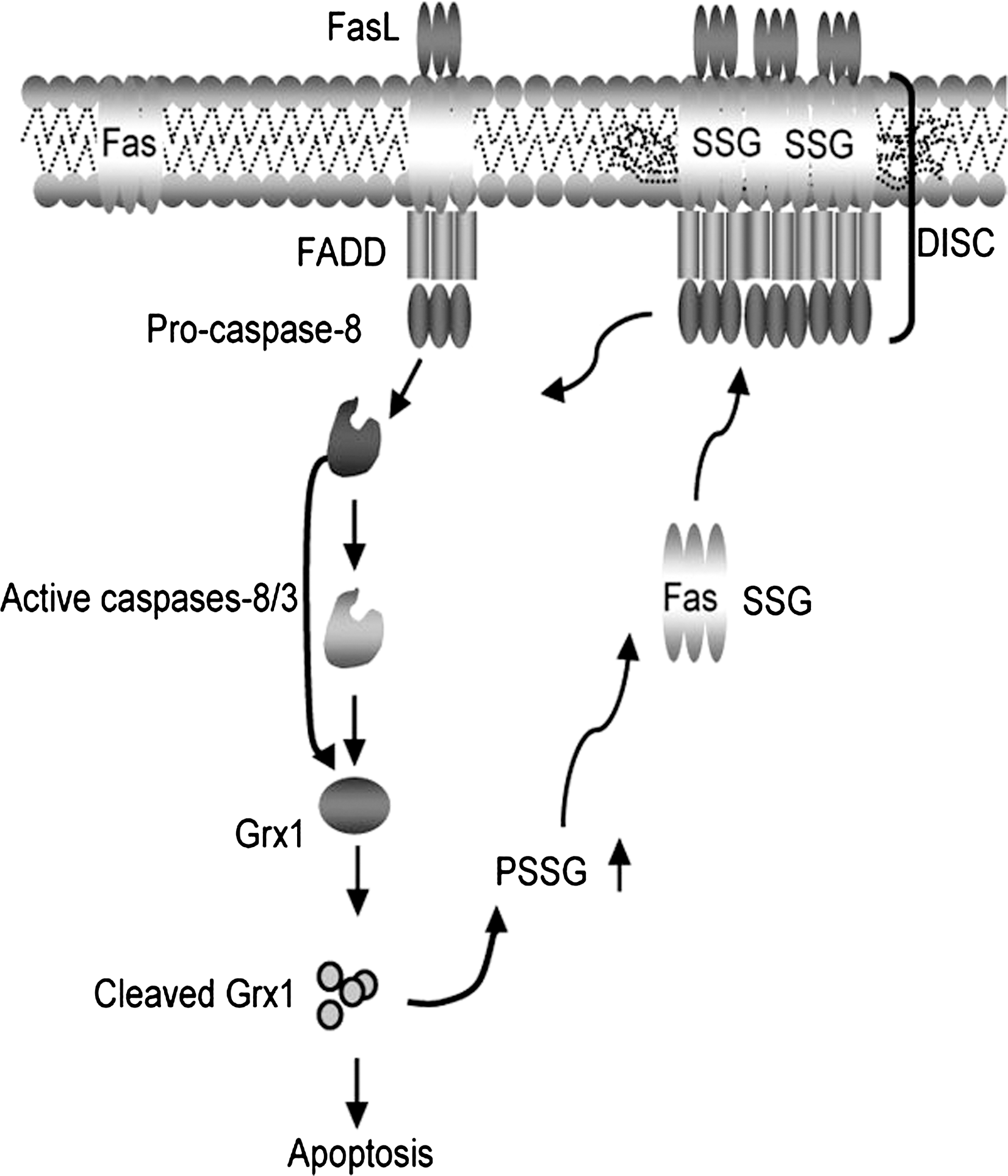
The death receptor Fas, another member of the family of TNFR death receptors, is a transmembrane protein expressed on the surface of a variety of cells. Fas is well studied in cells of the immune system where its primary function is to trigger apoptosis in CD8+ cytotoxic T lymphocytes, and homeostatic proliferation and apoptosis in CD4+ T cells (76). Early work by our laboratory demonstrated that, under conditions of overt stress, nitrotyrosine content of Fas was increased, which facilitated downstream activation of JNK and subsequent apoptosis in epithelial cells (68). Recently, our laboratory has demonstrated that following its ligation, S-glutathionylation of murine Fas receptor, at Cys294 acts as a signal to amplify the strength of apoptotic signal in murine lung epithelial cells, fibroblasts, and CD4+ T cells (5). Specifically, we have shown that S-glutathionylation of Fas occurred in response to stimulation with FasL, in an apparently NADPH oxidase-independent manner. Instead, early activation of caspase-8 and downstream caspase 3 was responsible for degradation of the de-glutathionylating enzyme, Grx1. In association with degradation of Grx1, S-glutathionylation of Fas was increased and sustained. S-glutathionylated Fas (Fas–SSG) was shown to occur in a high molecular weight complex, indicative of its aggregation. Fas–SSG was present in the lipid raft fraction, resulted in more efficient binding of FasL, enhanced formation of the death-inducing signaling complex, and augmented execution of apoptosis. Ablation of Grx1 facilitated Fas–SSG, promoted binding of FasL, enhanced downstream activation of caspases 8 and 3, and augmented apoptosis. Conversely, overexpression of Grx1 attenuated S-glutathionylation of Fas, FasL binding, and DISC formation, and partially protected against FasL-induced apoptosis. Mutation of cysteine 294 to alanine abolished Fas-SSG and resulted in remarkable resistance to FasL-induced apoptosis (Fig. 4). These results demonstrated that protein-S-glutathionylation is a critical regulatory event involved in amplifying Fas-induced pro-apoptotic signaling (5).
The altered redox environment and enhanced resistance to oxidative stress of tumor cells has been well established (39), and links to death receptor signaling have been reported. In human melanoma cells, increases in intracellular concentrations of superoxide anion (O2 -) attenuated Fas-mediated apoptosis (15) and suggested that excess O2 - may inactivate caspases by oxidative modification of cysteines. In contrast, decreasing intracellular O2 - levels in Fas-resistant bladder tumor and osteosarcoma cells sensitized these cells to FasL-induced death (15). It remains to be determined whether alterations in O2 - correspond to changes in S-glutathionylation, and whether S-glutathionylation of Fas or other downstream members of the pathway, including caspases, was affected. The research area of protein S-glutathionylation of death receptors is just emerging. It is not a leap of speculation to hypothesize that other receptors belonging to the death receptor family can also be modified via S-glutathionylation, given the similar death domain structure, and presence of conserved cysteines in their intracellular death domain.
Redox-based regulation of caspases 8 and 3
Upon FasL binding, Fas is known to interact with FADD and procaspase 8 or 10, in a DISC. These caspases become activated through protein–protein interactions (34). The FADD and procaspase-8 interaction is inhibited by Flice Inhibitory Protein (FLIP) binding to FADD (34, 60). Intriguingly, the ability of FLIP to inhibit the interaction between procaspase-8 and FADD was shown to be enhanced by S-nitrosylation of FLIP (81). The same authors demonstrated that engagement of Fas causes rapid generation of O2 - via activation of NADPH oxidase. It was suggested O2 - scavenged NO, and lowered S-nitrosylation of FLIP. Loss of FLIP S-nitrosylation was associated with its degradation, which in turn facilitated the binding of procaspase-8 to Fas–FADD complex and processing of procaspase-8 to activate caspase-8, resulting in activation of the downstream apoptosis cascade (81). Although the specific cysteine residue involved in S-nitrosylation has not been identified, the nitrosylation of cysteine residues in the vicinity of Lys 192 could be inhibiting ubiquitination and degradation of FLIP.
Cleavage and activation of caspase-3 are known to play central roles in apoptosis. Substantial evidence exists that the activity of caspase-3 is regulated via redox-dependent post-translational modifications. In human umbilical vein endothelial cells (HUVECs), caspase-3 was found to be constitutively S-glutathionylated. Upon TNFα stimulation, caspase-3 was de-glutathionylated by Grx. Small interference RNA-mediated knockdown of Grx significantly inhibited TNFα-induced endothelial cell death due to the attenuated caspase-3 cleavage, concomitant with increased caspase-3 S-glutathionylation. Cysteine-to-serine mutations (C163S, C184S, and C220S) of caspase-3 that were predicted to prevent S-glutathionylation showed increased cleavage compared with wild-type caspase-3 (57). This inverse correlation between caspase-3 S-glutathionylation and cleavage was further confirmed by the observation that S-glutathionylation of caspase-3 in vitro inhibited its cleavage by recombinant caspase-8 (57). These findings demonstrate a novel regulatory role for S-glutathionylation in preventing the activation of caspase-3, and consequently repression of apoptosis.
In addition to S-glutathionylation, caspase-3 also is S-nitrosylated at active site cysteine, C163. Upon ligation of Fas, de-nitrosylation of caspase-3 occurs, leading to active caspase-3 (52), and de-nitrosylation of caspase-3 was catalyzed by thioredoxin 2 (Trx2) which is specifically expressed in mitochondria (9, 37). The exact interplay between caspase S-glutathionylation and caspase S-nitrosylation remains obscure at this time. Given that S-nitrosylation can result in S-glutathionylation (54, 87), it is tempting to speculate that S-glutathionylation of caspase-3 could have been preceded by its S-nitrosylation. This speculation is supported by the observations that protein nitrosothiols represent an activated form of protein thiols that can readily react with GSH and form S-glutathionylated proteins (45, 54, 59). Alternatively, separate pools of S-nitrosylated or S-glutathionylated caspase-3 may exist in distinct subcellular compartments which may confer unique regulation of apoptosis in a context or ligand dependent manner.
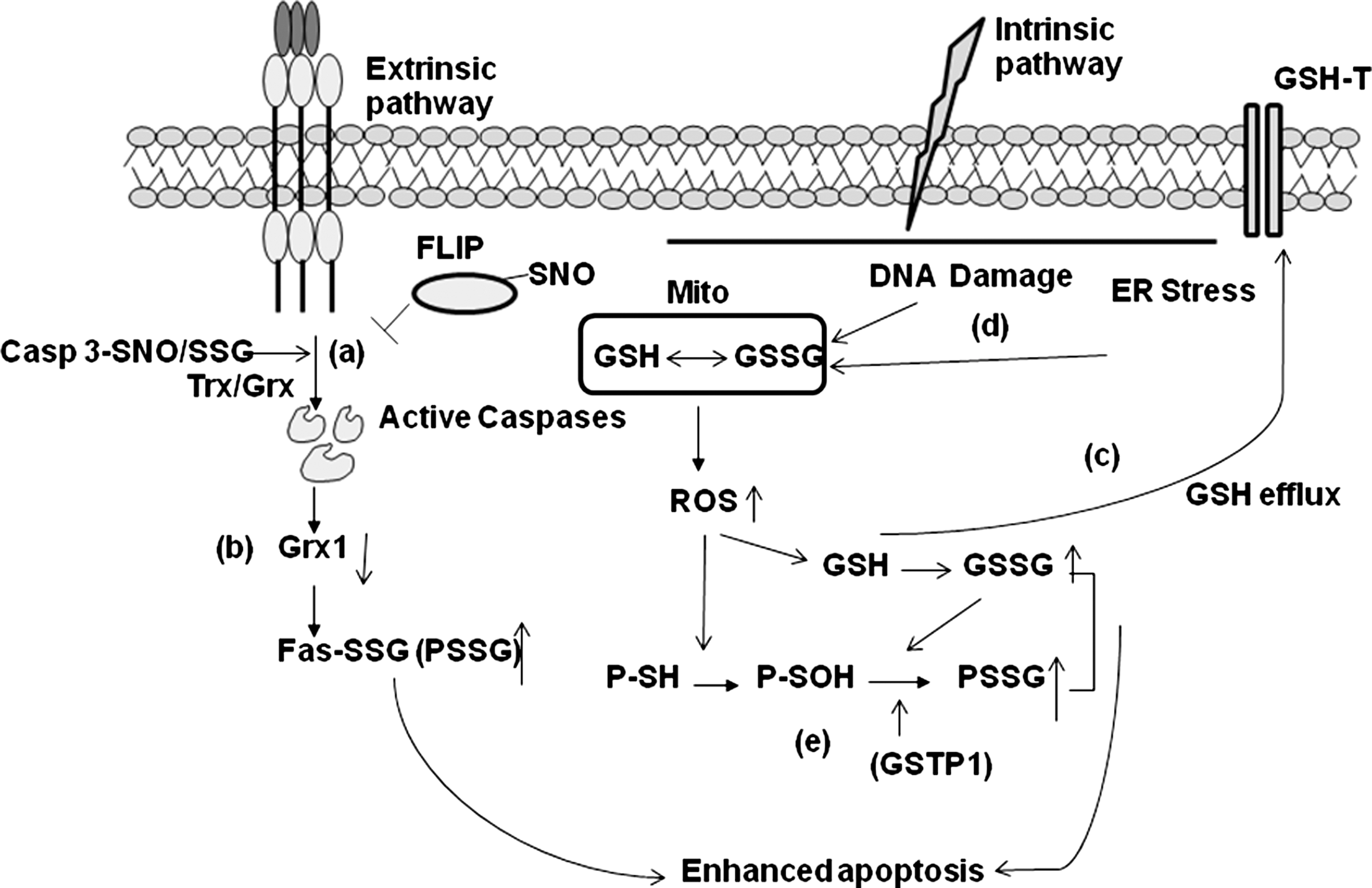
It is apparent that the impact of S-glutathionylation is target dependent: S-glutathionylation enhances the apoptotic function of Fas in a FasL-dependent manner in murine cells (5). It is tempting to speculate that S-glutathionylation of caspase-3 in HUVECs (57) may constitute a regulatory mechanism to control aberrant activation of caspase-3 due to various oxidative and metabolic stresses (11, 79) in the umbilical vein (74, 83). In conditions of overt inflammation, accompanied by release of TNFα, HUVECS induce de-glutathionylation of caspase-3, and subsequently undergo cell death. It remains to be determined whether this scenario also is operative in other cell types or tissues, or in response to different death-inducing ligands. Collectively, the studies described herein highlight that apoptosis is regulated at multiple layers via redox-controlled mechanisms that include S-glutathionylation of multiple proteins in ligand and cell dependent manner (Fig. 5).
S-Glutathionylation, Death Receptors, and Lung Fibrosis
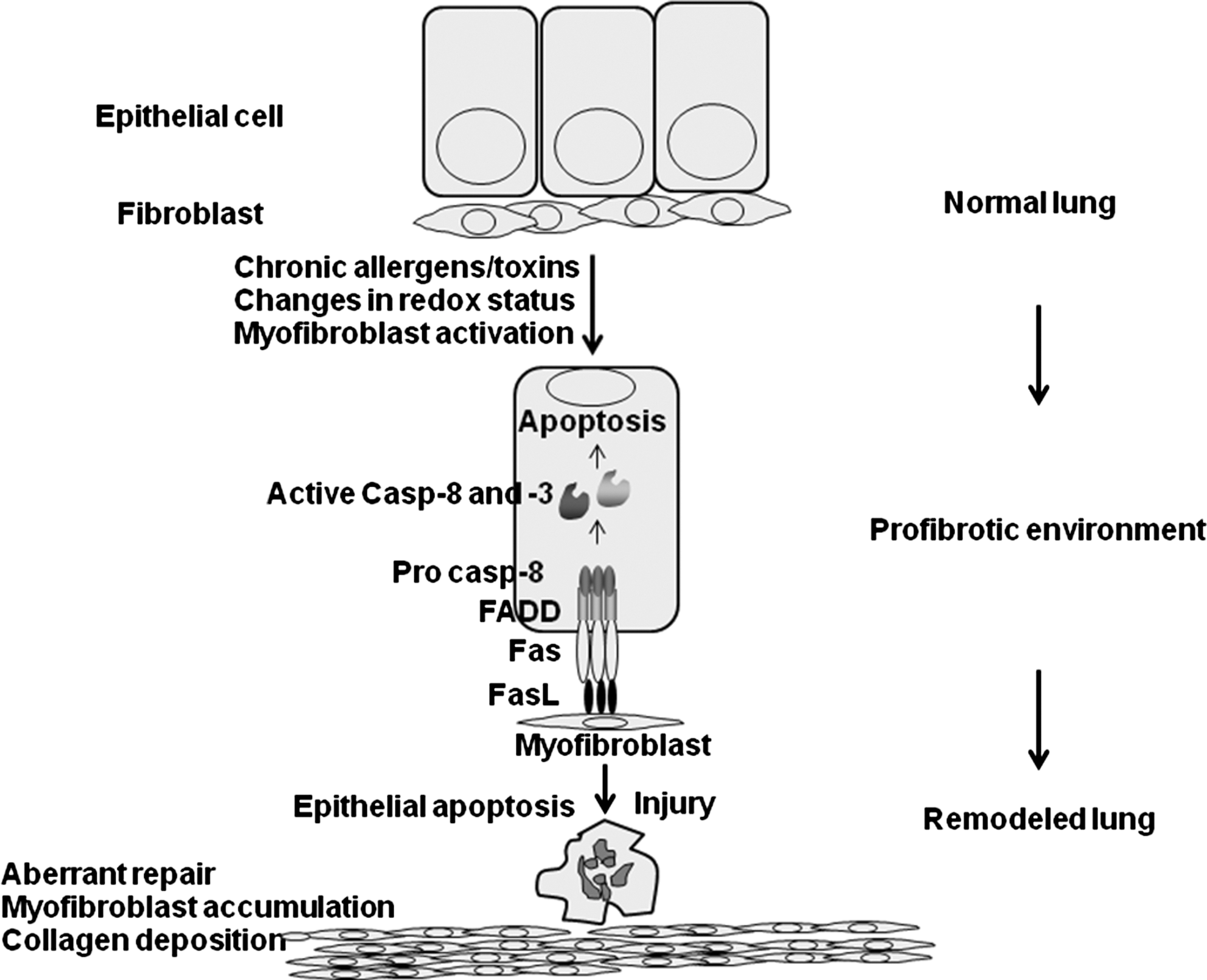
Following injury, impaired re-epithelialization and aberrant myofibroblast activation are believed to be important in pathogenesis of tissue fibrosis. Many inhaled agents, such as mineral dust, cigarette smoke, and microbes, along with radiation and hyperoxia, can cause acute injury and subsequent fibrotic reactions (6). Additionally, these agents are known to cause oxidative stress, apoptosis, and changes in GSH homeostasis (30). Changes in GSH homeostasis have been detected in patients with IPF. Currently, NAC is being evaluated in clinical trials of patients with idiopathic pulmonary fibrosis (17, 41, 44). Signaling via the death receptor Fas is known to play a critical role in the pathogenesis of lung fibrosis (47). Ligation of Fas triggers apoptosis of airway epithelial cells and results in lung fibrosis (32). Similarly, activation of a suicide gene in type II pneumocytes is sufficient to cause fibrotic remodeling of the lung (70). Mice that lack functional Fas (lpr) or FasL (gld) have been shown to be resistant to development of fibrosis and neutralization of Fas similarly has been shown to protect against fibrosis (47). Myofibroblasts derived from patients with idiopathic lung fibrosis were shown to express Fas ligand and were capable of inducing apoptosis of epithelial cells. However, these myofibroblasts themselves appeared to be immuno-privileged and were resistant to Fas-induced killing (28, 47, 85). These findings, coupled to known alterations in GSH homeostasis in the fibrotic lung suggest that altered redox-based regulation of the apoptotic cascade may fuel the pathogenesis of lung fibrosis (Fig. 6). Our recent discovery that S-glutathionylation of Fas promotes its strength as an inducer of apoptosis supports this notion. However additional experiments are needed to determine whether S-glutathionylated Fas occurs in fibrotic lung tissue and whether enzymes in the S-glutathionylation pathway play a role in disease pathogenesis. Such endeavors may yield important new redox-based therapies to combat IPF, a disease for which currently no effective treatment modalities exist.
Conclusion
Redox-dependent post-translational modification of members of the apoptosis pathway were originally considered to occur as by-products or end results of apoptosis, or generated only under scenarios of overt stress. Decades of careful analysis and adoption of new biochemical techniques have spurred the identification of site-specific redox-based post-translational modification of proteins in the apoptosis cascade in settings of physiological ligands and conditions, and has illuminated their functional importance (Fig. 5). Notably, S-glutathionylation has emerged as an important new module in “redox biology,” and identification of GSTP and Grx enzymes that catalyze forward and reverse reactions, respectively, give credence to the potential significance of S-glutathionylation in the apoptosis cascade. Further research will be needed to better determine the context in which S-glutathionylation of the members of the death receptor pathways occur (constitutive or induced), the ligand specificity of S-glutathionylation, and the compartmentalization of these events. Additional investigation into the interplay between S-nitrosylation and S-glutathionylation of apoptosis pathway members is also critically needed to improve our understanding how redox-based control is achieved. The precise link of S-glutathionylation of members of the apoptosis pathway with alterations in redox homeostasis also will need to be elucidated, which includes assessment of the local redox status of GSH, and activities of Grx and GSTP. Such analyses will be important given the emerging importance of these processes in diverse chronic diseases.
Footnotes
Author Disclosure Statement
This work was supported by NHLBI grants HLO7933 and HLO60014 to YJH. VA and YJH have a patent application related to the contents of the manuscript.