
Abstract
Hemoglobin (Hb) is the major oxygen (O2)-carrying system of the blood but has many potentially dangerous side effects due to oxidation and reduction reactions of the heme-bound iron and O2. Extracellular Hb, resulting from hemolysis or exogenous infusion, is shown to be an important pathogenic factor in a growing number of diseases. This review briefly outlines the oxidative/reductive toxic reactions of Hb and its metabolites. It also describes physiological protection mechanisms that have evolved against extracellular Hb, with a focus on the most recently discovered: the heme- and radical-binding protein α1-microglobulin (A1M). This protein is found in all vertebrates, including man, and operates by rapidly clearing cytosols and extravascular fluids of heme groups and free radicals released from Hb. Five groups of pathological conditions with high concentrations of extracellular Hb are described: hemolytic anemias and transfusion reactions, the pregnancy complication pre-eclampsia, cerebral intraventricular hemorrhage of premature infants, chronic inflammatory leg ulcers, and infusion of Hb-based O2 carriers as blood substitutes. Finally, possible treatments of these conditions are discussed, giving a special attention to the described protective effects of A1M. Antioxid. Redox Signal. 17, 813–846.
I. Introduction
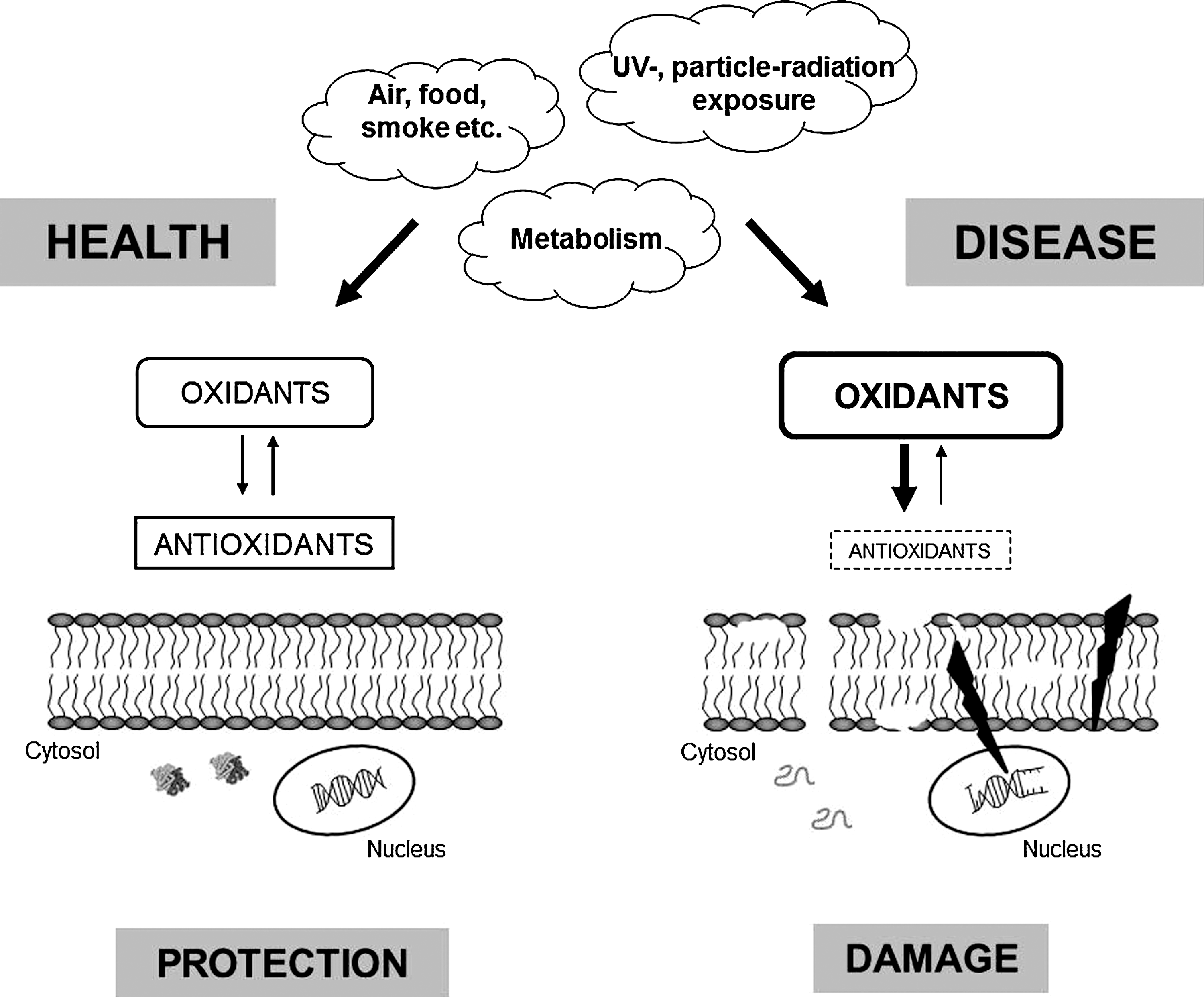
The term “oxidative stress” is used to describe the conditions with an abnormally high production of reductive and oxidative compounds and/or impaired antioxidative tissue defense systems (114). Major mediators of oxidative stress are reactive oxygen species (ROS), including free radicals, which are extremely reactive compounds due to the presence of unpaired electrons. ROS, oxidants, and free radicals react with proteins, DNA, and other molecular components, which can lead to unwanted modifications of the target molecules and loss of their functions. Formation of ROS and free radicals can be induced, for example, by metals and metal-containing compounds and proteins. Since Hb is a metal-containing protein, most of the deleterious reactions of Hb are reductive or oxidative, that is, involves exchange of electrons, and Hb is therefore categorized as a major inducer of oxidative stress. In addition, the Hb-metabolites heme and free iron are also potent inducers of oxidative stress. Therefore, Hb-induced oxidative stress is associated with an extensive cell and tissue damage.
Many pathological situations involve abnormal levels of hemolysis and high levels of extracellular Hb. Below, five groups of such pathological conditions, diseases, and iatrogenic conditions will be discussed. Although they have vastly different etiology, they share many symptoms and clinical sequelae as a consequence of the Hb-induced oxidative damage. (i) Various hemolytic anemias, including inherited RBC dysfunctions and deficiencies, autoimmune tranfusion reactions, and mechanical RBC disruption. (ii) Pre-eclampsia (PE) is a pregnancy disease shown to be associated with elevated levels of fetal hemoglobin (HbF), leaking from fetus to mother. (iii) Intraventricular hemorrhage (IVH) and subarachnoidal bleeding result in locally high concentrations of free Hb in cerebral cavities, and these conditions are associated with severe cerebral damage and dysfunctions. (iv) Chronic leg ulcers are inflammatory conditions that involve dermal and epidermal extravascularization of RBCs and hemolysis. (v) Massive amounts of cell-free Hb in the bloodstream are also found during clinical administration of “artificial blood components”, that is, hemoglobin-based oxygen carriers (HBOC), employed for treatment of blood-loss after trauma, etc.
It is the object of this article to give an overview of the toxic reactions of extracellular Hb focusing on oxidative stress-inducing reactions and of defense mechanisms that protect human tissues against these reactions with a special emphasis on the ubiquitous plasma and tissue protein α1-microglobulin (A1M) (82, 220, 339, 341). We will also describe the five groups of diseases and iatrogenic conditions mentioned above, in which Hb-induced oxidative stress is a major pathological factor. Finally, possible therapies for these types of pathological conditions will be discussed, giving a special attention to the heme-binding and radical-scavenging effects of A1M.
II. Hb, Heme and Oxidative Stress
A. Red blood cells and Hb
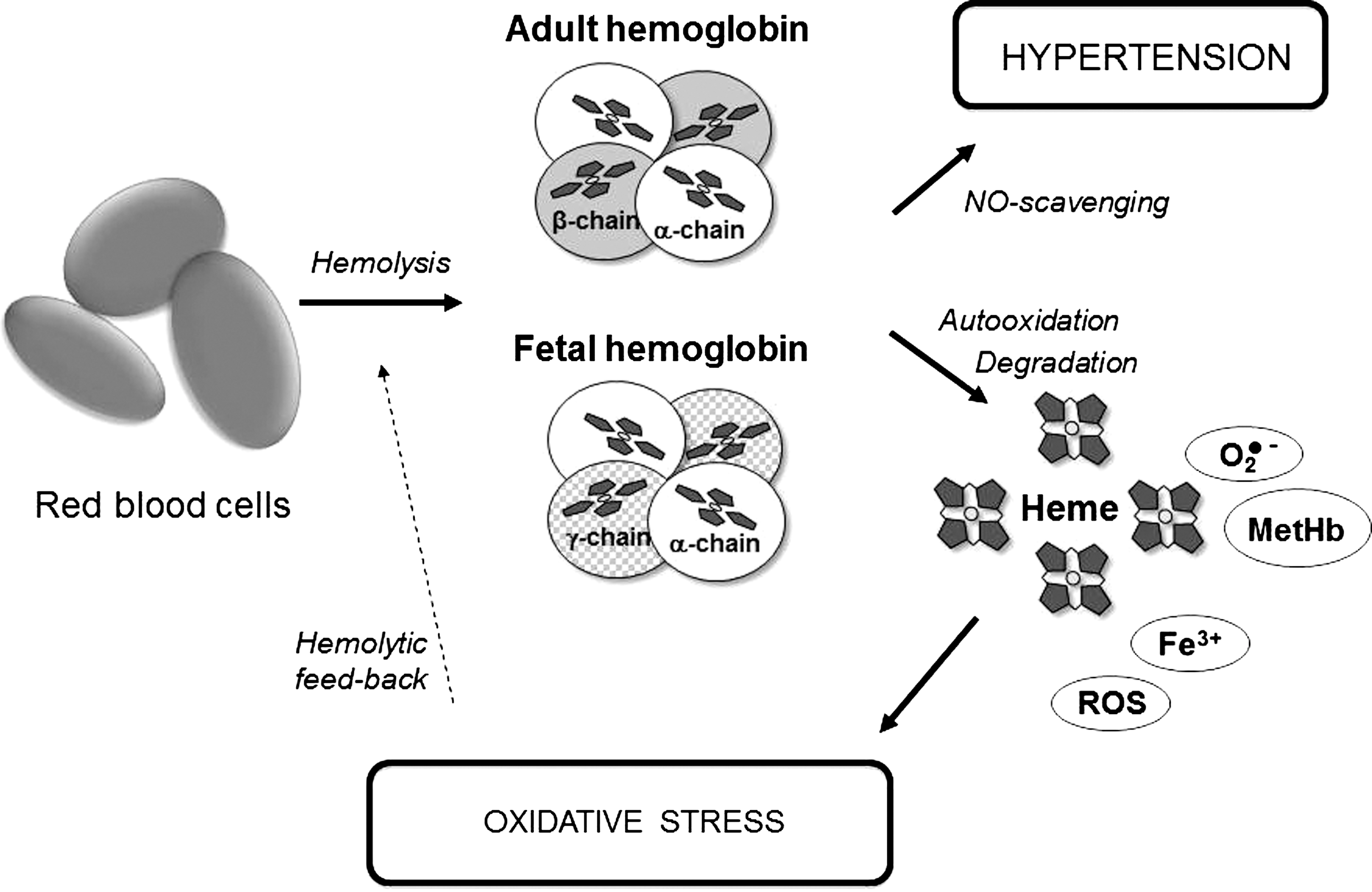
Human RBCs have a lifespan of ∼120 days in circulation, after which senescent RBCs are phagocytized by resident macrophages in the reticuloendothelial system, spleen, and bone marrow (37, 47). The main constituent of RBCs is Hb, and the primary function of this important protein is to bind O2 reversibly and hence to function as an O2-transport protein. The Hb molecule is an assembly of four globular protein subunits where each protein chain arranges into a set of α-helix structural segments connected together in a globin fold arrangement (117, 276). This folding pattern creates a pocket that very efficiently binds a heme group. The heme group consists of an iron atom that is coordinated by four nitrogen atoms of a tetrapyrrole ring (constituting the heme moiety) and a nitrogen of a histidine-imidazole ring, linking the heme to the globular protein structure. This leaves one coordination-site free to reversibly bind gaseous molecules, normally O2, but carbon monoxide (CO) and nitric oxide (NO) can also be bound to the iron (235). In adults, the most common Hb isoform is a tetramer denoted adult hemoglobin (HbA), which consists of two α- and two β-subunits (α2β2) (see Fig. 1). In the fetus, the Hb molecule is made up by two α-chains and two γ-chains (α2γ2, denoted HbF). The γ-chains are gradually replaced by β-chains at birth, and as the infant grows (50), HbF has a higher affinity for O2 and a lower dissociation constant required for sufficient oxygenation of the fetus in utero. In healthy adults, HbF is restricted to RBCs called F-cells, which constitute a limited proportion of the total red cell mass (∼1% of total HbA is HbF).
1. Hb variants
Besides HbA and HbF, there are also a number of Hb variants that are found in different disease conditions. Hb variants occur when genetic changes in the globin genes cause alterations in the amino acid sequences of the globin proteins. These changes may affect the structure of the Hb, its behavior, its production rate, and/or its stability. Thus, more than 1400 hemoglobinopathies, inherited disorders of Hb, and thalassemias, conditions with reduced Hb levels in blood, are caused by different mutations of α-chains and β-chains, respectively, and are listed at the HbVar database (93, 101). Their severity is governed by the number of genes affected. β-chain Hb variants are inherited in an autosomal recessive fashion. Several hundred β-chain Hb variants have been documented, however, only a few are common and clinically significant. A well described β-chain related disease is sickle cell disease. This disease is caused by a substitution of a single amino acid (Glu6Val) in the β-chain, resulting in the HbS variant, which is associated with malformation and destabilization of RBCs (311). HbE (Glu26Lys) is the most common β-chain variant, which manifests in a wide range of clinical symptoms, from asymptomatic to severe thalassemias depending on the genetic background (246, 304). HbC (Glu6Lys) is another common β-chain variant causing aggregation of intracellular Hb resulting in moderate reduction of RBC plasticity in sickle cell HbC disease (298).
B. Extracellular Hb and its intrinsic toxicity
Although Hb is vital for delivery of O2, it needs to be strictly compartmentalized within the RBCs in order to be kept in a functional nontoxic state. The balance between normal physiology and toxicity is largely dependent on the physical and biochemical barriers, presented by RBCs, separating Hb from the extra-RBC environment. Furthermore, RBCs contain a unique setup of enzymatic mechanisms that preserves Hb in this functional nontoxic state (248). However, if these systems are disturbed, for instance, as a consequence of intravascular hemolysis, a number of toxic, potentially dangerous side effects, originating from the rich chemistry associated with the free radical reactivity of the heme group, can occur (see Fig. 1). The key chemistry involved in mediating the intrinsic toxicity of Hb is outlined below. First the ferrous iron (Fe2+) in the oxyHb molecule spontaneously auto-oxidizes, producing the ferric protein (Fe3+, metHb) and the superoxide radical
The superoxide then dismutates to hydrogen peroxide (H2O2), and the ferric protein can react with this peroxide, generating a reactive ferryl iron (Fe4+) and a free radical bound to the protein (R
Both the protein-bound radical (R
Since NO is a highly vasoactive molecule, the binding of NO by Hb affects the vasomotor tone, and hemolysis and elevated extracellular Hb-concentrations are therefore associated with hypertension. This has been described in, for example, sickle cell disease (239) and PE (218) (see also below). Normally, the compartmentalization of Hb in the erythrocyte limits NO scavenging in vivo and subsequent vasoconstriction (162, 302). Interestingly, it has also been proposed that NO may function as an antioxidant, playing a role in the mechanism to detoxify high oxidation states of Hb under oxidative conditions (235).
An important group of oxidant molecules in humans and animals is ROS, which includes, for instance, H2O2, superoxide, and the hydroxyl radical. Superoxide and the hydroxyl radical are also categorized as free radicals, a group of molecules that are highly reactive due to the presence of unpaired electrons in their outer electron shells. All these molecules have the potential to react with proteins, DNA, and other molecular cell and tissue components, leading to unwanted modifications and ultimately loss of function (Fig. 2). The human body is constantly exposed to free radicals and oxidants, both exogenously via the environment (food, air, smoke, irradiation, etc.) and endogenously as by-products of normal metabolism. As these molecules are reactive and potentially dangerous, they need to be balanced by the activity of antioxidants, protective factors that eliminate oxidants or prevent their oxidation reactions (113) (see also below). Thus, it is extremely important that cells maintain a well-controlled redox balance. An imbalance in the redox system will induce oxidative stress, a condition characterized by a surplus of unrestrained oxidants, ROS, and free radicals. Several physiological processes, such as inflammation, ischemia/reperfusion, diabetes, obesity, and kidney insufficiency are associated with oxidative stress. Furthermore, hemolytic diseases involving destruction of erythrocytes in an uncontrolled manner lead to high levels of cell-free Hb, as described above. Additionally, neutrophil activation during infections results in oxidative stress, mainly due to production of superoxide anions and hypochloric acid by the enzymes NADPH oxidase and myeloperoxidase (19, 20, 51, 202, 269).
III. Protection Mechanisms
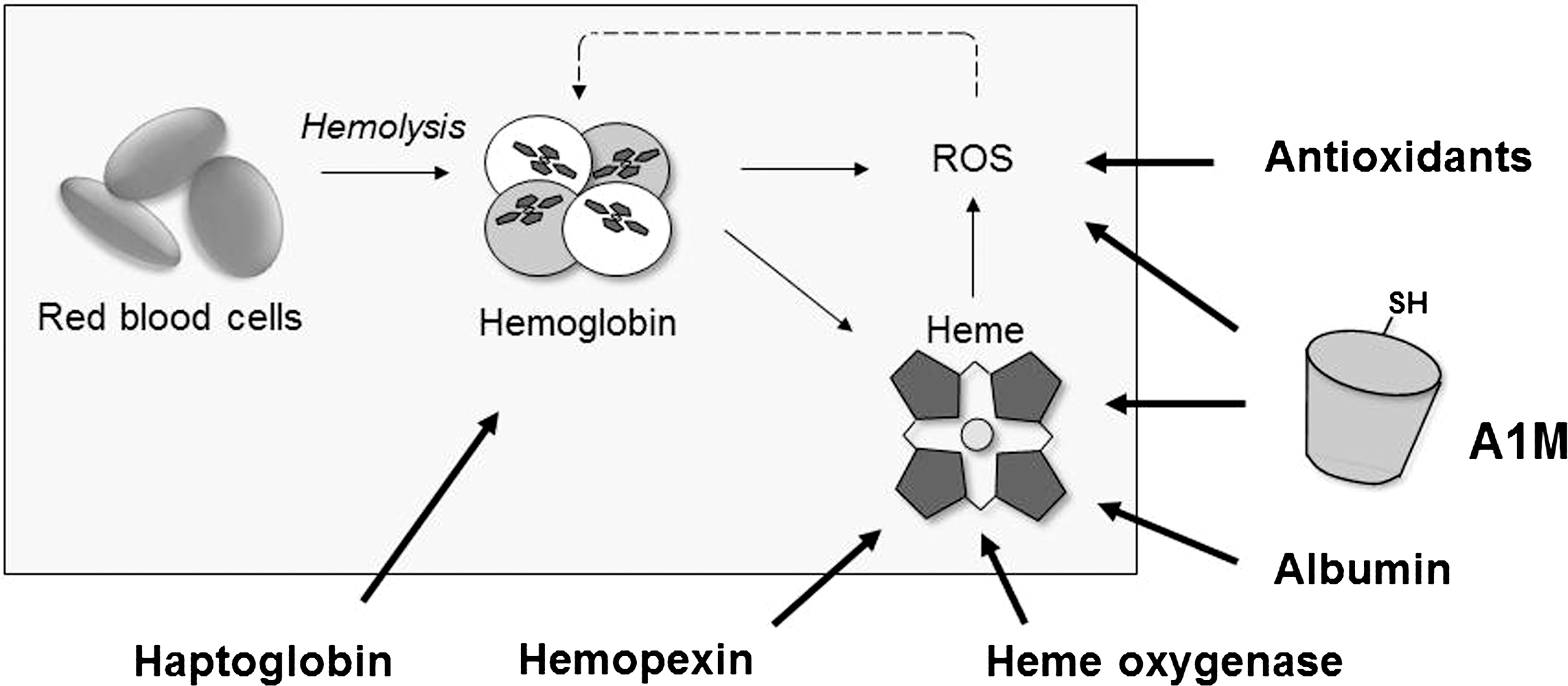
In humans, there are several Hb- and heme-detoxification systems described (Fig. 3). Haptoglobin (Hp), perhaps the most well-investigated Hb-clearance system, binds extracellular Hb in blood (21, 156) and the resulting Hp-Hb complex is cleared from blood by binding to the macrophage receptor CD163 (146). Excess oxyHb that is not cleared by Hp undergoes auto-oxidation reactions resulting in ROS and free heme groups. A complex network of antioxidation mechanisms (such as superoxide dismutase [SOD], glutathione peroxidase [Gpx], and catalase) inhibits and eliminates the oxidative compounds and repairs the oxidative damage caused by them. Free heme in blood is sequestered by albumin and hemopexin (Hpx) (21, 72, 149) and the Hpx-heme complex is cleared from the circulation by the hepatocyte receptor CD91 (127). Intracellularly, heme oxygenase (HO) is the most essential heme catabolic protein converting heme to free iron, biliverdin, and CO (290, 310). The plasma and tissue protein A1M (338) was described to bind and degrade heme and has the ability to reduce metHb (8, 9). Recent reports suggest that a truncated form of A1M is involved in extracellular heme catabolism (8). The heme-binding and putative heme cleavage properties of A1M provide an additional physiologic protection mechanism against extracellular heme (216, 220).
A. Haptoglobin
Several proteins are able to protect against Hb toxicity and one of the most powerful is Hp (235, 318). Hp is synthesized predominantly in the liver by hepatocytes. It is an abundant acute phase protein and its levels in the blood circulation vary greatly within humans, ranging from 0.3 to 3.0 mg/ml. In addition, the synthesis of Hp can be rapidly increased in response to, for example, inflammatory signaling. If free Hb is released into the circulation, it binds immediately to Hp to form an Hp-Hb complex, which subsequently is removed by the CD163 receptor expressed mainly on monocytes, macrophages, and Kupfer cells in the liver (107, 146, 161, 206, 207, 317). The binding between Hp and Hb is among the strongest noncovalent interactions known in biological systems with an estimated Kd of 10−15 and with slow dissociation rates. If Hp becomes depleted as a consequence of hemolysis, Hb accumulates in the kidney and is secreted in the urine. Hp thereby offers an effective protection against kidney damage in the first phases of moderate Hb release.
The Hp monomeric protein consists of two chains; one α-chain and one β-chain, expressed as a single precursor protein (Fig. 4). Two human Hp-gene alleles, Hp 1 and Hp 2, encode two different α-chains, α-1 with Mr≈9 kDa and α-2 with Mr≈16 kDa, whereas the β-chain has an Mr≈33 kDa in both alleles. In mammals, the α-chains and the β-chains are linked together by a disulfide bond, and in most species a disulfide bond also cross-links the α-chains to each other forming a dimer composed of two αβ-subunits. Additionally, Hp can form larger aggregates where the stoichiometry of the polymer is genetically determined and controlled primarily by the α-chain (194, 208, 230). Thus, Hp 1-1 homozygous individuals, having the Hp 1 allele on both chromosomes, form dimers via a single disulfide bond between the α-chains, whereas Hp 1-2 and Hp 2-2 individuals form polymers of higher order because the α-2 chains can form two disulfide bonds each, linking together the polymers. Hp binds Hb αβ-dimers (48). All monomeric forms of Hp and dimeric Hp 1-1, bind the Hb dimers with high affinity. The polymeric forms of Hp can also bind to Hb, though at much lower affinities than monomers and dimers.

Several other functional differences between Hp phenotypes have been reported that could have important biological and clinical consequences. Hp polymorphism, not only linked to different subunit interactions but also to different glycosylation profiles, is associated with the prevalence and clinical evolution of many inflammatory diseases, including infections, atherosclerosis, and autoimmune disorders (70). These differences are explained by an Hp-dependent modulation of oxidative stress (see below) and prostaglandin synthesis. For example, the distribution of highly polymeric Hp proteins in extravascular fluids is restricted by their molecular mass. Consequently, the Hb protective capacity in body fluids is less efficient in Hp 2-2 individuals. Other reported differences among phenotypes include antigenic activities as well as binding to macrophage receptors, including CD163 (206).
The Hb-Hp complex impairs filtration and clearance of Hb dimers by the kidney, and instead directs Hb to CD163 on macrophages for a process of endocytosis and final degradation (138). Within macrophages, the HO enzyme (see below) breaks down the heme group of Hb into bilirubin and CO, which both have been shown to have antioxidant and vasodilatory benefits (206). Therefore, this clearance of Hb indirectly protects against heme-mediated oxidative damage as well as other oxidative enzymatic reactions of Hb, described above, and thus Hp can be viewed as a part of a human antioxidation mechanism (161). Direct binding of Hb to CD163 has also been reported as a secondary clearing mechanism in addition to the Hp-Hb complex formation and subsequent clearance by the macrophages (206). The binding of Hp-Hb to CD163 results in the production of several cytokines, antioxidative genes, and noninflammatory mediators. Particularly, the induction of HO-1 is notable (260).
The literature suggests that O2 binding and redox reactions of Hb are not inhibited by Hp-binding. In addition, upon Hp-Hb complex formation, the Hb moiety retains its NO binding characteristics (23). Likewise, the Hp-Hb complex can still react with H2O2 (46). In spite of this, Hp has been shown to inhibit Hb-induced lipid peroxidation, (112, 194) and it may be speculated that Hp achieves such antioxidation by, for instance, interfering with the diffusion of radical species from the heme center to surrounding tissue components (46, 161). Interestingly, it has been proposed that Hp may also protect the Hb molecule from oxidative attack, including auto-oxidation. In addition to a site-specific protection of amino acids that are prone to oxidation on the surface of the Hb molecule, Hp allows the heme active site to operate unhindered leading to the elimination of oxidants (46). Recent work on the interactions of Hp with Hb has produced a model, which gives a structural basis of the Hp mode of action in protecting Hb against oxidative toxicity. Mutagenesis studies have thus confirmed that a tyrosine residue at the α-subunit of Hb (αTyr42) acts as a conduit for electron transfer to and from the heme, which facilitates the autoreduction of the ferryl Hb (237). In addition, electron paramagnetic resonance (EPR) studies have shown that this electron transfer is also diverted to another Tyr residue on the β-subunit, βTyr145 when Hb is complexed with Hp. Upon addition of H2O2, Hp stabilizes the ferryl-induced free radicals on βTyr145. This radical reactivity may ultimately be directed from Hb to Hp resulting in a safer and more stable redox inactive Hb molecule (7) (Cooper et al., submitted).
It has recently been shown in two animal models, dogs and guinea pigs, that Hp can limit the toxic effects of cell-free Hb after infusion (33). Data obtained from both models showed that the formation of Hp-Hb complexes reduced the hypertensive response during Hb exposure and reduced Hb toxicity in the kidney. Infusion of Hp may thus be explored to control adverse blood pressure responses and tissue toxicity effects associated with hematological disorders and diseases. Indeed, it was suggested that Hp-supplementation may be employed to counter the adverse effects of the high loads of free Hb associated with HBOC therapy (33) (see also below, section IV.E).
B. Hemopexin
Heme scavenging by Hpx (together with high density lipoprotein (HDL), low density lipoprotein (LDL), plasma albumin and A1M) provides protection against oxidative damage, limits access of pathogens to heme, and contributes to iron homeostasis by recycling the heme iron. During the first seconds after heme appearance in plasma, more than 80% binds to HDL and LDL and 20% binds to Hpx and plasma albumin. Heme then transits to Hpx or is slowly removed from HDL and LDL by Hpx (21, 26, 72, 103, 188, 196). Human Hpx consists of a single polypeptide chain containing 439 amino acids residues with six intrachain disulfide bridges (12). The polypeptide chain contains two homologous domains of about 200 residues, each joined by a 20-residue linker (295). The crystal structure of isolated Hpx C-terminal domain (86) showed that these domains have a four-bladed β-propeller fold that is a variant of the larger β-propeller domains found in heterotrimeric G proteins, clathrin, and integrins (151, 274, 292, 295). Such domains appear to be of major importance in cell biology, especially as mediators of protein–protein interactions (273). Each propeller blade comprises a four-stranded antiparallel β-sheet, with the first and fourth blades tied together by disulfide bonds. The heme binds between the two propeller domains. Two histidines coordinate the heme iron, giving a stable bis-histidyl Fe(3+) complex.
A striking feature of the heme-binding site is the preponderance of aromatic and basic residues (224). The construction and location of the heme pocket clearly relates to the requirements of high affinity but reversible heme-binding by Hpx and fits perfectly with the design of other high-affinity binding proteins, such as the bacterial periplasmic binding proteins (232) and iron-binding transferrins (15). Heme-binding and release results from opening and closing of the heme binding pocket, through movement of the two β-propeller domains and/or the interdomain linker peptide (188, 224). Hpx delivers the bound heme upon interaction with its specific receptor, identified as the LDL receptor-related protein CD91, where it is internalized by receptor-mediated endocytosis. The human Hpx-heme receptor is expressed in several cell types, including macrophages, hepatocytes, neurons, and syncytiotrophoblasts. After delivering the heme, the heme group is degraded and intact Hpx can be released back into the bloodstream (21, 72, 188, 295). The Hpx-heme complex removal by a receptor-mediated pathway shows striking similarities to the CD163-mediated Hp-Hb clearance in macrophages (127).
C. Heme oxygenases
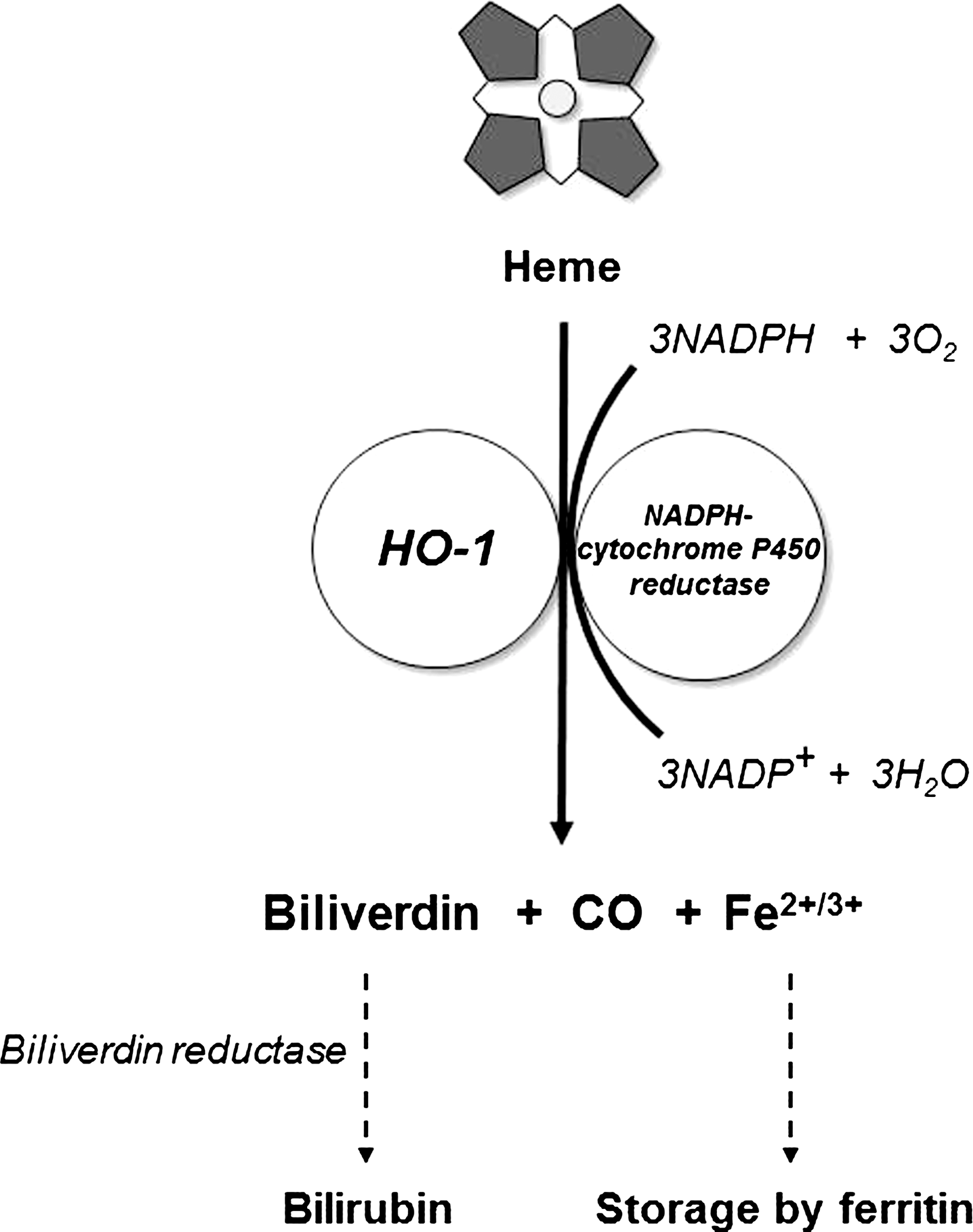
HOs are cytosolic enzymes that participate in heme-detoxification by binding and degrading the free heme group [(290); recently reviewed in Refs. (1, 105, 320)]. Two isoforms of HO have been described: HO-1, the expression of which is induced by a variety of conditions of environmental stress (183, 324), whereas HO-2 is constitutively expressed (184, 296). A third genetic variant, HO-3, has been described but is most likely a nonexpressed pseudogene (120). HO-1 and HO-2 are separate gene products and have different amino acid sequences, structure, and size but have similar mechanisms of heme catalytic activity and specificity.
Both enzymes are anchored to the membrane of the endoplasmic reticulum via a C-terminal hydrophobic domain (192). The enzymes operate in concert with microsomal NADPH-cytochrome P450 reductase to convert heme to biliverdin, CO, and Fe2+, utilizing three molecules each of O2 and NADPH for each molecule of heme (Fig. 5) (209, 257, 291). The products of HO activity have important beneficial physiological activities providing further antioxidation effects besides the mere elimination of heme. Biliverdin is reduced to bilirubin by biliverdin reductase (150), and bilirubin is a powerful physiological antioxidant (279). CO has been reported to have both pro-oxidant and antioxidant effects, mostly as a result of binding to heme-proteins, replacing molecular O2 [reviewed in Ref. (228)]. Indeed, CO had protective effects against lung injury (95, 221). CO also induces vasodilation by binding to the heme-protein, guanylyl cyclase (182). Finally, CO participates in a positive feed-forward loop by inducing an upregulation of HO-1 (145). The release of free Fe2+ iron is closely linked to synthesis of ferritin, an iron-sequestering protein. Thus, HO-1 induction stimulates ferritin expression (81), and inhibition of HO-1 leads to ferritin downregulation (305).
Under resting, nonstressed conditions, HO-2 is the predominating form in all organs except the spleen (36), and the highest activities are found in spleen, testes, and brain (181). The expression of HO-1 is ubiquitously upregulated by a multitude of stimuli related to oxidative stress, including high concentrations of free Hb and heme [(17, 251) reviewed in Refs. (105, 180, 181, 320)]. In fact, elevated levels of HO-1-messenger RNA are commonly used as biomarkers of oxidative stress. In contrast, transcription of the HO-2 gene is only responsive to stimuli from adrenal glucocorticoids (193, 234).
D. Protective heme-binding mechanisms
Plasma albumin binds heme at a protein:heme ratio of 1:1 and 1:2 but with lower affinity than Hpx (Kd≈10−8 M) (171, 247). Since the plasma concentrations of albumin are high (30–50 mg/ml), the amounts of heme that can be bound by albumin are considerable and the albumin binding becomes physiologically important, especially during conditions of heme-overload when the Hpx is depleted (265). This may explain the beneficial effects of albumin infusion on cerebral malaria (185). Albumin partially inhibits the toxic effects of heme (109), but there are no indications of a targeted transport function or specific enzymatic inhibitory actions. Thus, the general belief is that the protective effect of albumin is a result of the protein functioning as a depot of free heme groups.
Free heme in plasma is also bound rapidly, and with high affinity, to the lipoproteins LDL and HDL (196). The heme group catalyzes oxidation of the lipoprotein particle and subsequent endothelial damage and probably contributes to the development of atherosclerosis (27, 55).
The protein A1M binds heme in plasma, extravascular fluids, and cells with a Kd=10−6 M (8, 10, 153). The binding has been shown to yield protection from the toxic effects of heme in various in vitro molecular, cell culture, and ex vivo organ systems. The structure, physiology, and antioxidative properties of A1M are described in some detail below.
As argued in previous publications, the heme group is probably not freely diffusable across membranes (105, 155). Several heme-binding and heme-transporting proteins that mediate the uptake of extracellular heme to the cytosol have been described (233). Two cytosolic proteins, as well as HO-1, have been shown to have protective effects against free heme toxicity, peroxiredoxin I (Prx1 or HBP23) (80, 129, 133, 205) and A1M (220), both of which are upregulated in response to elevated concentrations of free heme (130, 217). Interestingly, intracellular A1M can be derived by local synthesis in most cells but also by uptake of extracellular A1M produced in the liver and transported to tissues via blood (see below).
E. Antioxidation mechanisms
As described above, unsequestered Hb, heme, and iron generate free radicals, mostly ROS. While the Hb, heme- and iron-binding and sequestering mechanisms described above generally operate to prevent the formation of radicals and ROS, a network of antioxidation molecules, acting with many different mechanisms, cooperate to protect cells and tissues against the deleterious reactions of free radicals and ROS once they have been formed. Several high molecular weight enzymatic antioxidants have been described. SOD catalyzes the conversion of O2 •− to H2O2 [reviewed in Ref. (96)]. Three variants of SOD exist in mammals and birds: cytoplasmic Cu/ZnSOD (=SOD1) (191), mitochondrial MnSOD (=SOD2) (312), and extracellular Cu/ZnSOD (=SOD3) (186). Catalase, belonging to the functional group of peroxidases, is a heme-carrying enzyme catalyzing the conversion of two molecules of H2O2 to water plus O2 (61, 104). Catalase is localized to peroxisomes of nucleated cells, (263, 332) where its main function is to eliminate the H2O2 formed in this organelle and to keep cytosolic H2O2 concentrations low. Gpx, the second most abundant peroxidase, is a ubiquitously expressed cytosolic and mitochondrial enzyme (174) catalyzing the reduction of H2O2 by employing glutathione (GSH) as an electron donating substrate. Albumin is generally regarded as an important nonenzymatic antioxidation factor that is present in high concentrations and hence buffers deleterious oxidation reactions in plasma and interstitial fluids.
Several nonenzymatic, low molecular weight radical scavengers operate either by direct reactions with ROS and non-ROS radicals or by acting as cofactors in reactions with the enzymatic antioxidants. The most important is, perhaps, the cytosolic tripeptide GSH. GSH is synthesized in the cytosol and provides reducing equivalents via its free thiol group (187). Thus, the ratio between GSH and its oxidized form, oxidized glutathione (GSSG), constitutes a cytosolic redox buffer, which regulates the balance between antioxidant capacity and beneficial ROS activity (78, 135). Thioredoxin (Trx) is a ubiquitous 12 kDa polypeptide with a Cys-X-X-Cys-motif that acts as an electron reservoir via a disulfide exchange between the two thiol groups (123, 211). High ratios between reduced and oxidized GSH and Trx and thus a high antioxidant capacity are maintained by the regenerating enzymes GSH reductase and Trx reductase, respectively, drawing reducing eqivalents from cytosolic NADH and NADPH (18, 147, 199). The essential nutrients vitamin C (ascorbic acid) and vitamin E (tocopherol) are also generally believed to serve important functions as antioxidants (122, 222). The water-soluble vitamin C is both a reducing agent that is transformed to dehydroascorbic acid when reacting with ROS/radicals, and a cofactor of several enzyme reactions, whereas the lipid-soluble vitamin E is an important lipid peroxidation radical scavenger. In addition, a large number of nutritional low molecular weight radical scavengers with antioxidant properties have been described, such as turmeric curcumin (330) and red wine resveratrol (137).
F. α1-Microglobulin
A1M was discovered in human urine in the early 1970s and was named in the tradition of plasma proteins, reflecting its small size (26 kDa) and its electrophoretic migration slightly behind albumin (82). The protein was characterized by several research groups and is also known under the alternative names, protein HC, “human complex-forming protein, heterogeneous in charge” (289), and α1 microglycoprotein (267). Two striking biochemical properties were described early: a yellow-brown color and a pronounced charge heterogeneity (82, 338).
A physiological role of A1M in heme- and radical-binding was only recently proposed. First, the immunoregulatory (mainly suppressive) properties of A1M were identified during the 1980s and 1990s but did not seem distinct or strong enough to constitute the major function of the molecule in vivo. Then several reports during 2000s showed enzymatic reductase activities as well as heme- and radical-scavenging properties. Recent investigations have described strong protective effects of A1M in molecular, cell, and organ systems, and it has become clear that the protein has a role as an antioxidant, employing several chemical mechanisms. The precise reactions of this protein/enzyme are still largely unknown, however, as is the relationship between its protective and immunoregulatory biological effects.
1. Structure
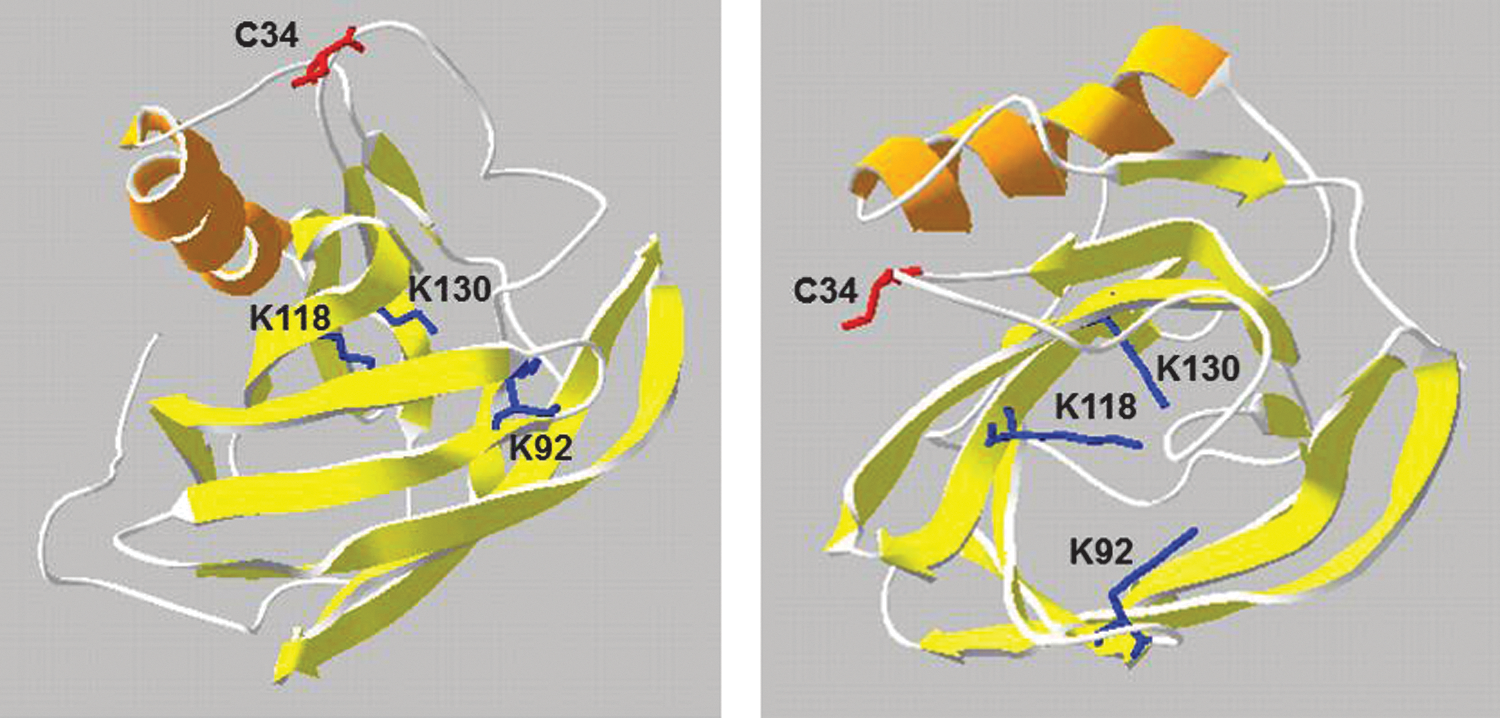
Human A1M has a molecular weight of 26 kDa and consists of a peptide chain with 183 amino acids (82, 140, 283). The peptide is glycosylated with oligosaccharides in three positions, two sialylated complex type, diantennary and triantennary carbohydrates linked to Asn17 and Asn96, and one O-linked oligosaccharide linked to Thr5 (13). The protein is well conserved during evolution, and an A1M homologue has been isolated and sequenced from humans and other mammals, birds, amphibians, and fish (58, 115, 128, 141, 168, 169, 200, 286, 307, 334, 335, 340). No crystal structure has yet been obtained, but a model of the three-dimensional structure was developed based on its membership in the lipocalin protein family (Fig. 6). The lipocalins are a group of 40–50 proteins found in all branches of life (bacteria, plants, fungi, animals) (92, 337) and 12 human genes are known. The lipocalins share a common protein fold: a single polypeptide, 150–190 amino acid residues, forms an antiparallel eight-stranded β-barrel with one closed and one open end. The interior of the barrel often serves as a binding site for small hydrophobic compounds (99). The Cys34 side chain of A1M (Fig. 6), found in all species, was suggested to be located on a flexible loop near the lipocalin pocket and is solvent exposed (306) and involved in complex formation with other proteins, redox reactions, and radical binding (9, 31, 54, 110, 341) (see also below). Covalent modifications of the Lys 69, Lys 92, Lys118, Lys 130, and Cys 34 side chains (Fig. 6) have been found in human A1M isolated from urine, and these “chromophores” have been proposed to cause both the yellow-brown color and the charge heterogeneity of the protein (29, 83). The modifications probably represent the oxidized degradation products of organic radicals covalently trapped by A1M in vivo (see further below).
2. Synthesis
The gene for A1M is called the alpha-1-microglobulin-bikunin precursor gene (AMBP) because it also encodes bikunin, another plasma protein (140) (Fig. 7). Bikunin is a Kunitz-type protease inhibitor (271) and a structural component of extracellular matrix (63). Bikunin is the common subunit of a group of protein/carbohydrate complexes that constitute plasma proteins of the inter-α-inhibitor family (94, 254). Transcription of the AMBP gene produces an mRNA that is translated into a precursor protein consisting of the A1M and bikunin proteins connected by a linker tripeptide (166). The last amino acid of A1M, Arg, plus the linker tripeptide constitute a basic cleavage site, R-V-R-R that conforms to the consensus recognition sequence of furin and other proprotein convertases (PCs) (266). Before secretion, the A1M and bikunin components are therefore separated by proteolytic cleavage by furin or another PC in the late phase of post-translational processing in the Golgi system (39). No physical or functional connection has been found between A1M and bikunin after secretion and the reason for their cosynthesis is not understood. It has also been shown in different expression systems that both A1M and bikunin can be expressed alone (38, 294). In spite of this, the A1M/bikunin genetic construction is conserved in all species where A1M has been found.
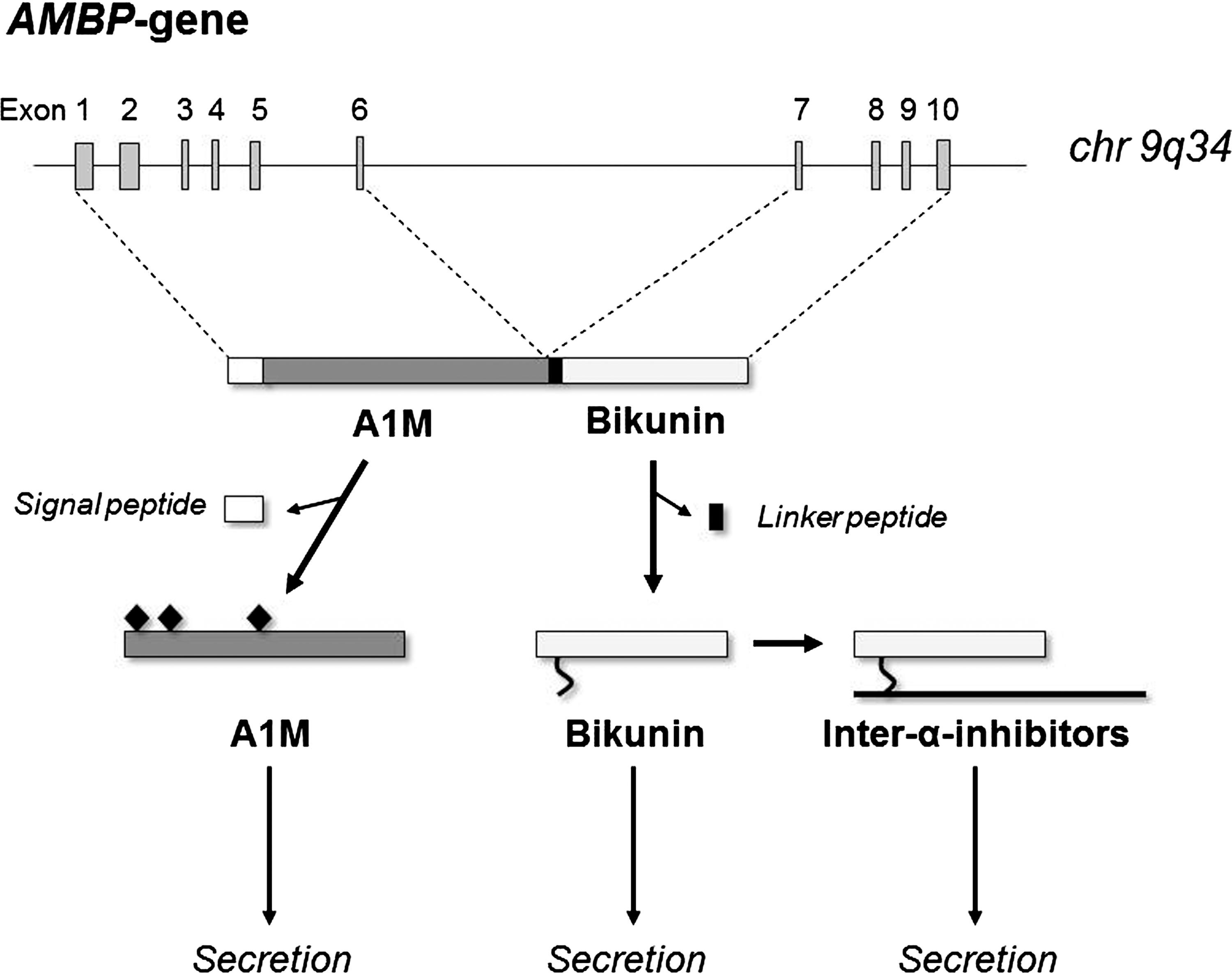
3. Gene
The AMBP gene has been cloned from man (77) and mouse (167). In both species, the gene contains ten exons, of which the six first code for A1M and the four last code for the linker peptide and bikunin (Fig. 7). The AMBP gene has been mapped to the 9q32-33 region in man (76) and to chromosome 4 in mouse (256), in both species in regions where other lipocalin genes have been found to be clustered. The A1M exons are separated from the bikunin exons by intron F, which contains retroposons and other repeated structures, suggesting that it is a recombinatorial hotspot (167). This could have provided the basis for a fusion between an ancestral lipocalin gene (A1M) with an ancestral Kunitz inhibitor gene (bikunin). The pronounced expression of A1M in liver is dependent on a strong liver-specific enhancer located about 2500 nt upstream of the start-codon, and contains eight binding sites for hepatocyte-enriched nuclear factors (249, 250).
4. Expression and distribution
Liver, blood plasma, and kidney (Fig. 8) are the major sites of A1M localization (333). The liver is the major site of synthesis in adult tissues (288). The protein is then secreted to the blood, where it exists in free form as well as in disulfide-linked high-molecular-weight complexes with other plasma proteins. The total concentration of A1M in human plasma is ∼2 μM (74), and ∼50% of the total plasma A1M is linked to immunoglobulin A (IgA) via a reduction-resistant disulfide bond, 7% to albumin, and 1% to prothrombin (31, 110, 111). High-molecular-weight forms of A1M have been found in all species examined. In rat serum, A1M can be found covalently linked by a disulfide link to fibronectin (88) and by a reduction resistant bond to the proteinase inhibitor α1-inhibitor-3, a homologue of human α2-macroglobulin (89). A1M complexes were also detected in plaice serum (169). Thus, the complex forming ability of A1M is conserved from fish to man, although the identities of the complex partners are not conserved. Both free A1M and the complexed forms are rapidly equilibrated between the intra- and extra-vascular compartments and their half-lives in blood are only 2–3 min (154, 313). Free monomeric A1M passes almost unhindered through the glomerular membranes out into the primary urine, where most of it is reabsorbed by the proximal tubular cells and catabolized (210). Significant amounts, however, are excreted in the urine, and the A1M-concentration in urine is a sensitive indicator of tubular renal damage (82).
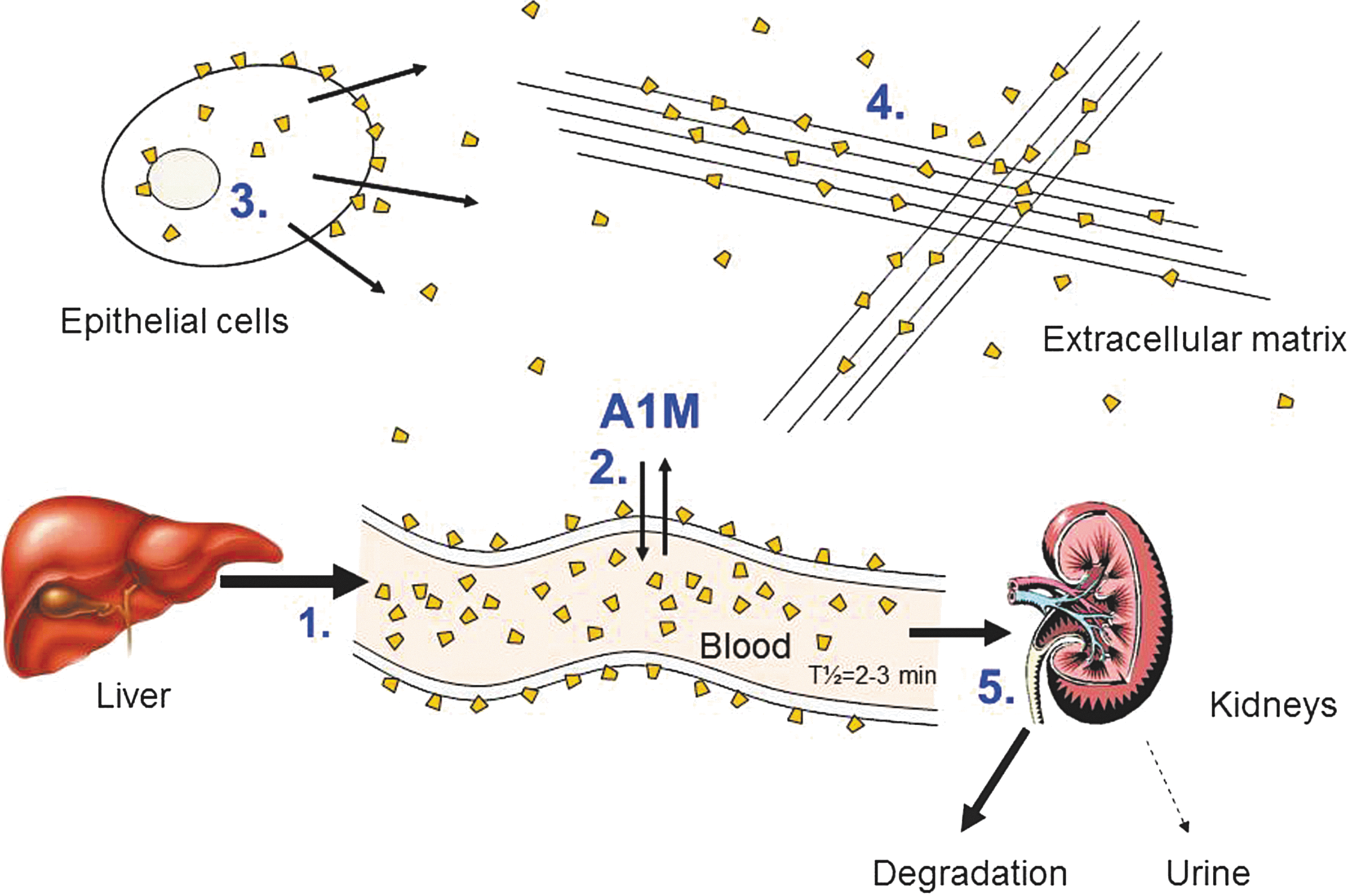
Besides the liver, A1M mRNA has been detected in adult kidney (139, 157), pancreas (30, 132), stomach (286), placenta (218), and blood cells (157, 217). The A1M protein has been identified in the perivascular connective tissue of most organs (30, 212, 213) and is especially abundant in epidermis of skin (10, 34, 216) and epithelium of the gut (35, 154). It is often colocalized with elastin and collagen (34, 212, 213, 216) and binds to collagen in vitro (216, 258). A distribution of A1M at various interfaces between the cells of the body and the external environment (blood/tissue, air/tissue, intestinal lumen/villi), as well as at the interface between maternal blood and fetal tissues in placenta (30, 189), is consistent with a protective role of A1M in vivo (see below).
5. Immunoregulation
A1M inhibits central events of the immune response in vitro. Thus, the antigen-induced cell division and interleukin (IL)-2 production of lymphocytes were inhibited by A1M (178, 179, 314). Inflammatory responses of blood cells were inhibited by A1M; these included migration (177, 178) and chemotaxis (195) of neutrophil granulocytes and the production of free radicals and IL-1β by peripheral lymphocytes/monocytes (258). It is possible that the immunoregulatory effects of A1M are related to its antioxidant and reductase activities, as ROS have been shown to be involved in cell signaling during lymphocyte activation [reviewed in Refs. (78, 321)].
6. Cell receptor binding
A1M has been shown to bind to the surface of various cells, including blood lymphocytes, neutrophils, blood cell lines, and keratinocytes (24, 90, 195, 216, 220, 313, 314). The binding is specific for A1M, saturable and trypsin-sensitive, suggesting the presence of an A1M receptor on these cells. The affinity of A1M for its receptor on T cells and mouse peripheral lymphocytes was estimated to 104–105 M −1 (25, 314) and somewhat higher on the histiocyte cell line U937, 107 M −1 (90). The receptor has not yet been identified, however. Internalization of A1M from medium to the cytosol has been shown in erythroid K562 cells (220), hepatoma HepG2 cells (217), and primary skin keratinocytes (216).
7. Antioxidation mechanisms of A1M
As mentioned above, several reports during the last 10 years have shown enzymatic reductase activities, as well as heme- and radical-binding properties, of A1M. Recent investigations have also described strong protective effects of A1M in molecular, cellular, and organ systems, and it has become clear that the protein has a role as an antioxidant employing several chemical mechanisms. Figure 9 summarizes the reported mechanistic properties of A1M, which may contribute to its possible role as a physiological antioxidant. These include (i) reductase and dehydrogenase activity, (ii) heme-binding and degradation, (iii) covalent radical-scavenging, and (iv) physiological upregulation of the AMBP gene.
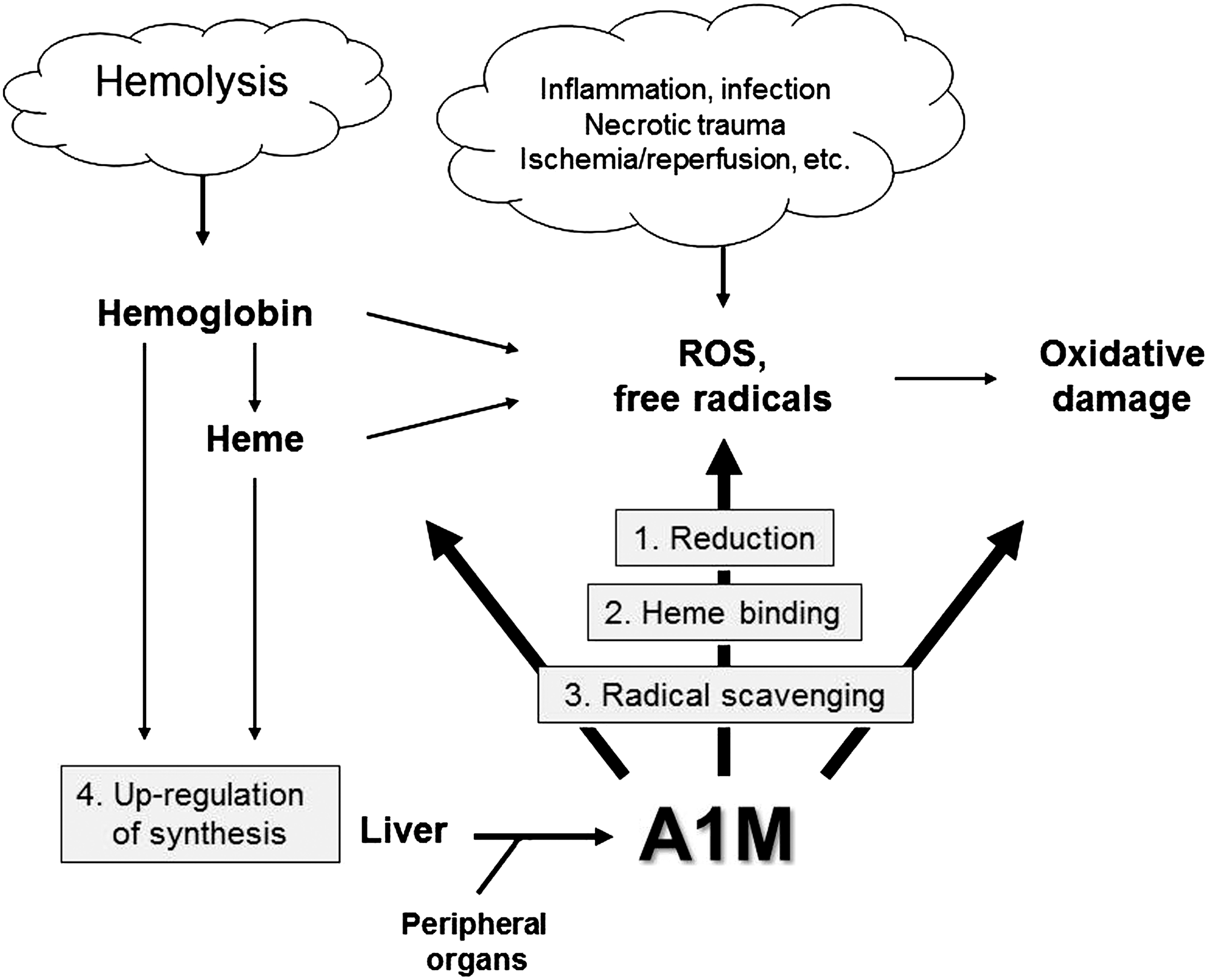
a. Reduction
Enzymatic reductase/dehydrogenase properties were recently described for A1M with a wide spectrum of organic and inorganic substrates (9, 341). The protein was capable of reducing heme-proteins, free iron, and the synthetic compounds nitroblue tetrazolium using the electron donors ascorbate and NADH/NADPH as cofactors (9). Using mutated A1M-variants, it was shown that the reductase and dehydrogenase activities were dependent on the thiol group of Cys34 and the three lysyl residues of K92, K118 and K130 (9). According to the hypothetical structural model of A1M, these four side chains are located in the vicinity of each other at the open end of the lipocalin barrel, Cys34 most likely on a flexible Ω-loop and Lys92, 118 and 130 close to the rim of the pocket (Fig. 6). It was speculated that the role of the three Lys residues in the reaction may be to create a positive electrostatic environment around the Cys 34, thus lowering the pKa of the thiol group and favoring its oxidation (9). This model of the three-dimensional structure of A1M suggests that it may be possible for Cys34 to interact both with the Lys-groups and the electron-accepting substrates. A1M also rapidly reduced the synthetic radical 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS), again in a reaction that was dependent on the free thiol group of C34 (341). Finally, the cell cytosol and thiol groups of cytosolic proteins of the erythroid cell line K562 were reduced by A1M (220).
b. Heme-binding and degradation
A1M binds heme with Kd=10−6 M (153, 216). The heme-binding was shown to occur to A1M in plasma from man, mouse, rat, guinea pig, cow, chicken, and plaice (153), suggesting that this property is evolutionarily conserved. Both free, monomeric A1M and the IgA-A1M complex could bind to the heme group (8, 153). Apart from this noncovalent, relatively weak interaction, a stronger binding was seen, which remained after boiling in SDS, suggestive of a covalent interaction (8, 153). Interestingly, a processed form of A1M, called truncated A1M (t-A1M), lacking the C-terminal tetrapeptide (leucine-isoleucine-proline-arginine; LIPR) of full-length A1M, was generated by a reaction with lysed RBCs or with purified Hb (8). The processed t-A1M also binds heme but was capable of degrading the heme group (8). The t-A1M-heme complex showed a time-dependent spectral rearrangement suggestive of degradation of heme concomitantly with formation of a heterogeneous chromophore associated with the protein (8). The t-A1M form is found in urine, skin, and placenta and is thus formed in vivo (8, 10, 173, 218). Together, these reports suggest that A1M is involved in extracellular heme catabolism by binding to free heme groups, and that a proteolytically activated, heme-degrading form of A1M is induced by free, extracellular Hb. The degradation mechanisms are as yet unknown, but may involve C34-dependent reductase reactions and covalent binding of degradation products to side chains of A1M.
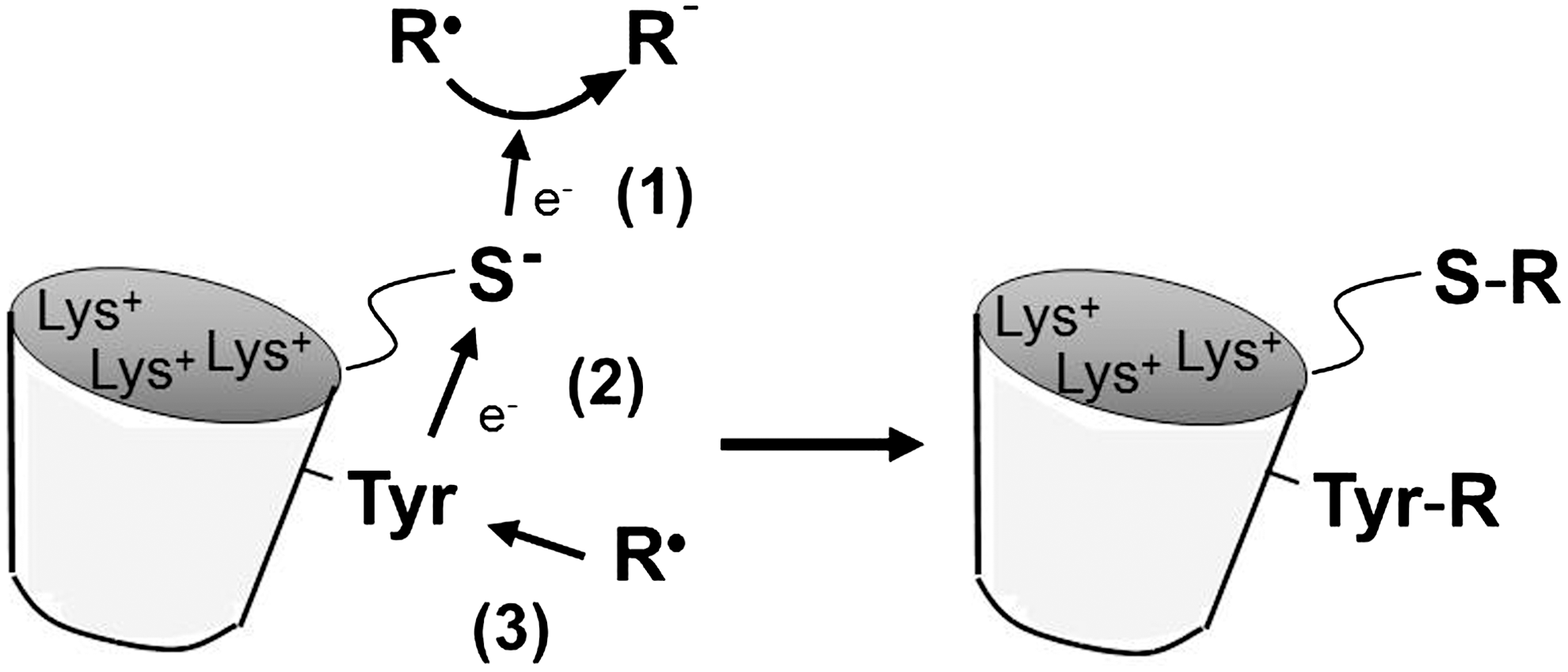
c. Radical scavenging
A1M reacts with organic radicals in a reaction that involves C34-catalyzed reduction and results in covalent trapping of radical metabolites to several amino acid side-chains on A1M (Fig. 10). To study the radical scavenging mechanism, the reaction between A1M and the synthetic free radical ABTS was characterized in detail (341). It was shown that A1M reduced five to six molecules of the ABTS radical in a rapid semicatalytic reaction and covalently trapped three additional ABTS radicals by attaching them onto tyrosyl side chains in a modified, oxidized form giving the protein a purple color. Two of the modified Tyr-residues were identified and localized on the A1M-molecule by mass-spectrometry (Tyr22, Tyr132). A possible reaction scheme involving an intramolecular transfer of electrons between the C34 thiol and the tyrosyl side-chains is shown in Figure 10. Besides these two Tyr-residues, previous reports suggest that several Lys-side-chains may be modified by radical trapping. Lysyl side-chains in urinary A1M from hemodialysis patients were shown to be modified by the kynurenine derivate 3-hydroxy-kynurenin (253). 3-hydroxy-kynurenin is a tryptophan metabolite that has a propensity to form free radicals (131, 215, 303) and is found in elevated concentrations in plasma of, for example, hemodialysis patients (226). Furthermore, although the structures of the modifications could not be identified, the side-chains of Lys69 (336), Lys92, Lys118, and Lys130 were shown to be covalently modified in human A1M isolated from urine (29, 83). The latter three Lys-residues may therefore serve a dual role, both, in activation of the Cys34 thiol by lowering its pKa (9) and as electron donors in the radical-trapping mechanism. In addition, the heme-degradation products strongly attached to A1M, and especially to its t-A1M form described above, could be formed by reactions that are related to this reductase/covalent trapping mechanism. The term “radical sink” was used to describe the radical binding property of A1M (341), referring to the fact that both the radicals and the A1M protein itself are electroneutral after the reactions and therefore do not present any oxidative stress to tissue components.
d. Upregulation of the AMBP gene
The expression of the A1M gene (AMBP) was increased by exposure of hepatocyte-, histiocyte-, and erythrocyte cell lines to oxy- and metHb, heme, and ROS (217). The upregulation, both on the mRNA and protein levels, was reversed by reacting Hb with cyanide, which prevents redox reactions (217). An upregulation of A1M was also seen in primary keratinocytes in response to heme and ROS (216). The latter may be important for the antioxidation defense in the skin, where keratinocytes constitute the major cell type. Recent results (218) show a correlation between the concentrations in plasma and placenta tissue, of A1M and Hb and between A1M and biomarkers of oxidative stress (protein carbonyl groups), respectively, in the human diseases PE (see also below), supporting the notion of an upregulation in vivo of the AMBP gene by elevated concentrations of free Hb and conditions of oxidative stress.
8. Protective effects of A1M
Employing the antioxidation mechanisms described above, A1M can protect cells and tissues from Hb-, heme-, and ROS-induced cell-damage (Fig. 9) (189, 216, 218, 220). A unifying model of the physiological role of A1M is that it functions as a “radical sink,” continuously cleaning tissues from free radicals and oxidants, including free heme and ROS generated by extracellular Hb, heme, and iron and delivering the products to the kidneys for degradation and/or excretion. Below, findings of protective effects of A1M are summarized.
a. Protection against Hb-, heme-, and ROS induced cell- and tissue damage
Several reports describe in vitro protective actions of A1M in cell cultures stressed by exposure to Hb, heme, and ROS. A1M prevented intracellular oxidation and upregulation of HO-1 induced by heme, H2O2, and Fenton reaction-generated hydroxyl radicals in primary keratinocytes (216) and in the erythroid cell line K562 (220). Silencing of the endogeneous A1M expression by addition of siRNA led to an increased cytosol oxidation (220). A1M also inhibited cell lysis of K562 cells caused by heme and cleared the cells from bound heme (220). It was shown that 200 μM heme killed 50% of the cells but adding 2 μM A1M lowered the number of dead cells to 15%, that is, by ∼70% (220). Similar effects were described in heme- and ROS-stressed skin explants (216). It was shown that A1M is localized ubiquitously in the dermal and epidermal layers of skin, and that the A1M gene, which is expressed in keratinocytes, was upregulated after exposure to heme and ROS. Exogeneously added A1M inhibited the heme- and ROS-induced ultrastructural damage, upregulation of antioxidation- and cell cycle regulatory genes, and formation of protein carbonyl groups, a marker of oxidative stress, in skin (216).
b. Protection of irradiated cell cultures
A1M was also shown to inhibit the propagation of cell-death induced by charged particle irradiation of HepG2 cell monolayers (219). The cells were irradiated with a low dose of alpha-particles at a small, restricted area. The directly hit cells were killed and the cells surrounding the irradiation area, not directly hit by the particles (=“bystander” cells), showed a delayed and slowly accumulating necrosis up to 3 days after the irradiation. Furthermore, a significant increase in apoptosis, protein carbonyl groups, and expression of the stress response genes HO-1, p21, and p53 was observed in the whole cell population. Addition of A1M reduced the amount of dead cells by ∼50% in irradiated cells and 100% in bystander cells, and completely inhibited the irradiation-induced apoptosis, formation of carbonyl groups, and upregulation of HO-1, p21 and p53. Irradiation induced an upregulation of endogeneous synthesis and secretion of A1M and an increased uptake of A1M from the medium. This study suggests that A1M protects against bystander cell killing by preventing the propagation of oxidants and ROS produced in the irradiated cells.
c. Protection of ex vivo-perfused placenta
To study Hb-induced tissue damage in a more complex system than cell cultures, the dual placenta perfusion model (262) was employed. In this model, the fetal and maternal placental circulations are perfused separately, and it is possible to study cell- and matrix structure and functions, including the feto-maternal barrier. It was shown that in vitro perfusion of placentas with Hb in the fetal circulation led to a significant increase of the perfusion pressure, feto-maternal leakage of free Hb, morphological damage, and upregulation of genes related to immune response, apoptosis, and oxidative stress (189). Simultaneous addition of A1M to the maternal circulation inhibited the Hb leakage, morphological damage, and gene upregulation. This study suggests that A1M may have therapeutic potential in pregnancy diseases involving placenta malfunction (see below).
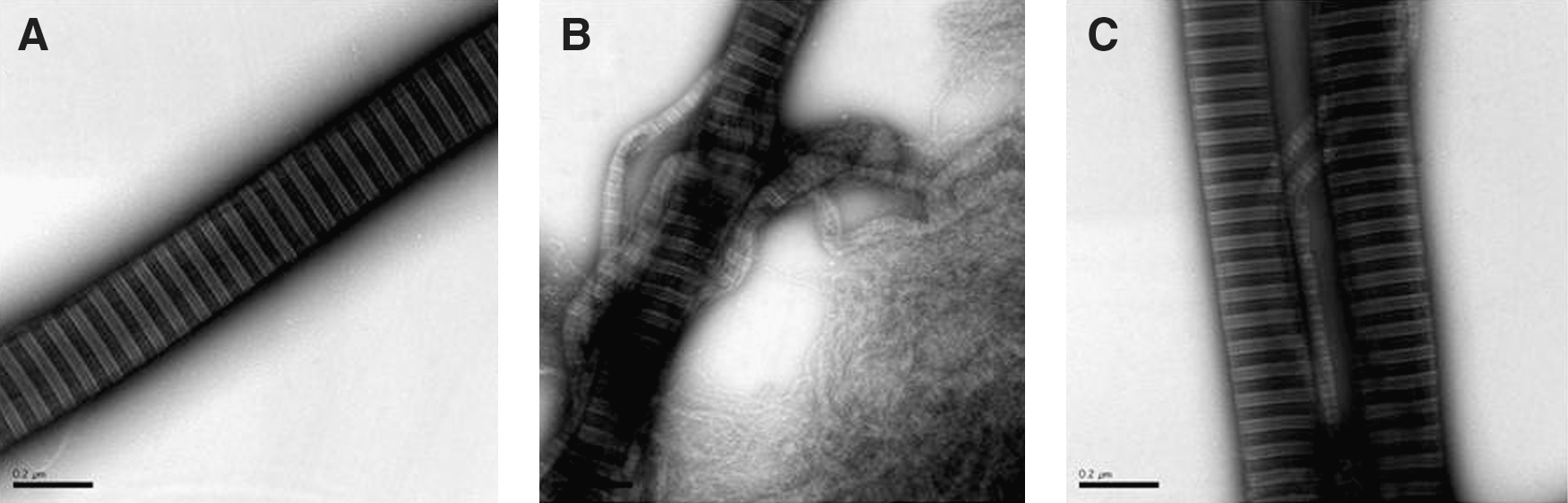
d. Protection of extracellular matrix and collagen fibrils
The skin and placenta studies (189, 216) also demonstrated a protective effect of the protein vis-à-vis extracellular matrix and collagen fibrils. In fact, disruption of collagen fibrils by Hb, heme, and ROS incubation was inhibited by A1M (Fig. 11). The fibrils could also be repaired by addition of A1M after the destruction already had taken place. It was speculated that oxidative lesions of the collagen monomers, induced by reactions with Hb, heme, or ROS, may shift the equilibrium between monomers and fibrils toward the monomers by preventing their interaction, whereas A1M, by removing and/or reducing the oxidative modifications, promotes a shift of the equilibrium back toward fibril formation (216). Interestingly, perfusion of placentas with A1M induced a significant upregulation of extracellular matrix genes, and a remarkable increase was observed in the number of collagen fibrils vis-à-vis Hb perfusion, as monitored by electron microscopy (189).
IV. Pathological Conditions
Hemolysis and elevated concentrations of free, extracellular Hb in blood and extravascular fluids are seen in many different pathological conditions. Five diseases/groups of such conditions will be described in this section. All are associated with a unique set of clinical scenarios and have a variety of underlying causes, but a common denominator is the increased oxidative stress that the extracellular Hb induces, and with a few exceptions hypertension and renal insufficiency, are a common theme. There is still a great need for development of treatment options targeting the oxidative environment that affects patients with hemolytic disease independent of its underlying causes.
A. Hemolytic anemias and transfusion reactions
The clinical definition of hemolysis is a reduction of the lifespan of RBCs to less than the normal range of 100–120 days (227). This premature and uncontrolled destruction of RBCs can lead to symptoms and laboratory signs with different degrees of severity, from none or very mild to fatal reactions. As outlined above, the extracellular Hb released from rupturing RBCs is associated with generation of various ROS, which can induce oxidative damage to matrix molecules, cell membranes, and other tissue components. Thus, in general, one of the key features of the hemolytic state, independent of its origin, is that it puts the individual under heavy oxidative stress (91). In this section, we will briefly outline various clinical situations where hemolysis is associated not only with increased extracellular Hb but also with anemia, that is, loss of total blood Hb, including transfusion-induced hemolysis.
1. Hemolytic anemia
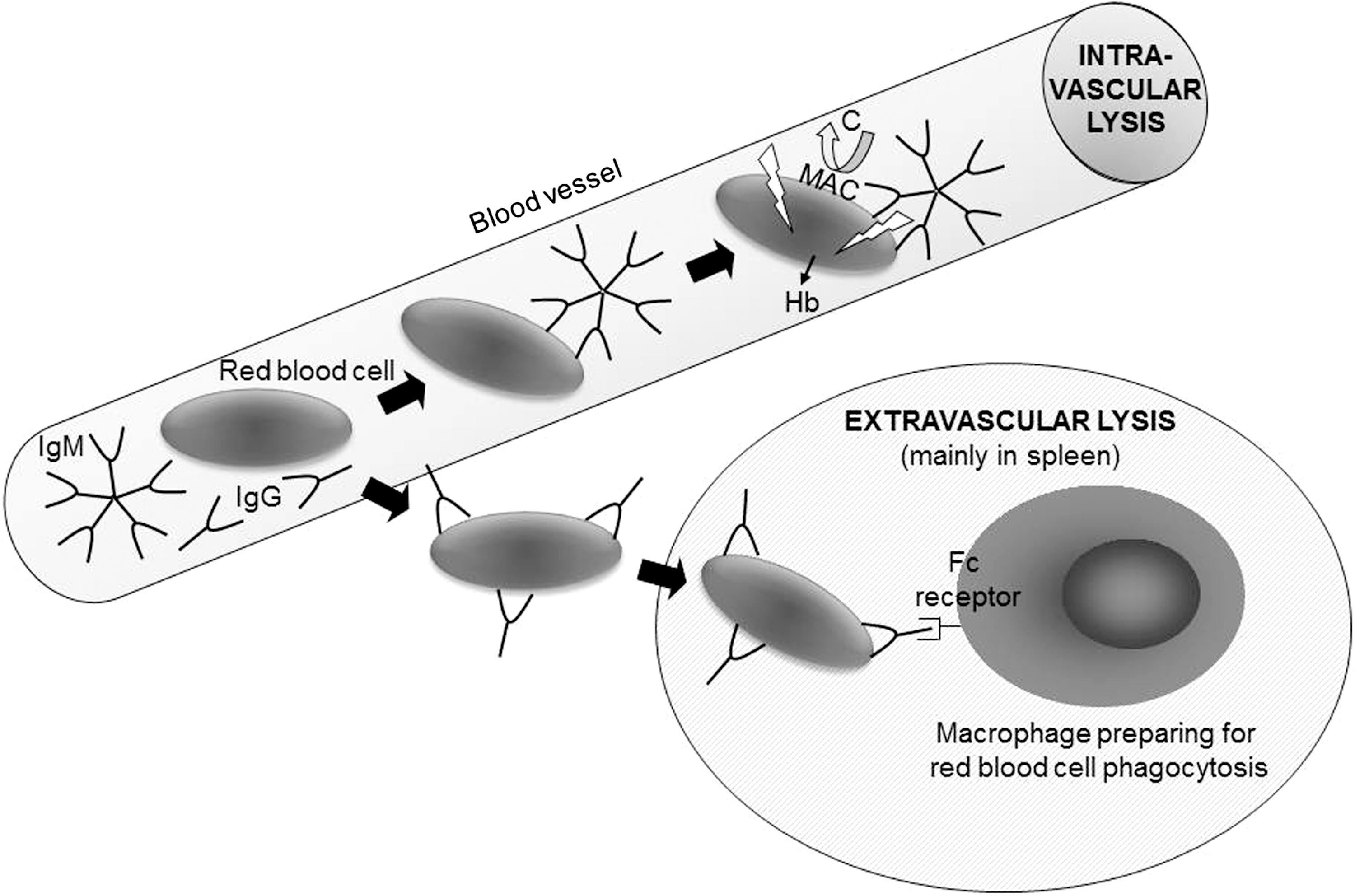
The diagnosis of hemolytic anemia is justified when shortened RBC survival constitutes a major underlying mechanism in explaining a low blood Hb value. The first case of hemolytic anemia was described 140 years ago, and the report includes the hallmark observation of splenomegaly without hepatomegaly and acute attacks of jaundice followed by the passing of reddish brown urine (301). There are many ways to categorize hemolytic disease, but a major distinction is made between hereditary and acquired hemolytic anemias. The former includes various genetic abnormalities of intracellular RBC proteins, including enzymes (287) and structural proteins (73); additionally, alterations in proteins exposed on the extracellular surface of the cell membrane can lead to shortened circulation of RBCs (14, 44). The latter group of acquired hemolytic anemias encompasses many different kinds of cell destruction, including mechanical lysis. This can be observed in population groups as diverse as marathon runners where the RBCs are destroyed due to repeated trauma when passing through the blood vessels in the athletes' feet, and patients whose RBCs are destroyed by nonbiological heart valves or during extracorporeal circulation. Yet another classification targets the location of hemolysis, whether the RBCs are lysed in the blood vessel (intravascular) or outside the circulation (extravascular) (see Fig. 12). Obviously, the mechanical lysis described above occurs intravascularly while spherocytosis, a state in which RBCs do not exist in their normal biconcave discoid shape but are spherical due to errors in the assembly of intracellular structural proteins (73), will mostly result in extravascular destruction and lead to an enlarged spleen. Finally, a very important categorization of autoimmune hemolytic anemias (AIHA) is made on the basis of auto- versus alloimmunity and the nature of the Igs involved in the lytic process. AIHA occurs when antibodies are directed against the individual's own RBCs. From a clinical perspective, this is one of the most common patient groups related to hemolysis in the Western world. AIHA can be categorized as being of the warm or cold type, so named because the autoantibodies in each type react optimally at high or low temperature, respectively. In the warm type of AIHA, the targets of the autoimmune response are protein epitopes (the vast majority of which are Rh-related) on the RBC surface and the antibodies are usually of immunoglobulin G (IgG) type. These antibodies may sometimes activate complement but usually do not. Instead, the main mechanism behind the accelerated RBC destruction in this form is extravascular lysis following Fc receptor recognition of the IgG-coated RBC by immune cells residing in the spleen (Fig. 12). Conversely, the cold type of AIHA is due to immunoglobulin M (IgM) antibodies directed at RBC autoepitopes (mostly carbohydrate structures), and the result is intravascular lysis due to the ability of IgM to activate the complement cascade (Fig. 12). It should be noted that there are cases with mixed type AIHA, where both IgM and IgG mechanisms are at play. There are also other kinds of intravascular hemolysis that are not antibody-mediated but still involve the complement system. For instance, paroxysmal nocturnal hemoglobinuria (PNH) is due to acquired somatic mutations in the PIG-A gene, which normally codes for an enzyme that is necessary for formation of glycosylphosphatidylinositol anchors between proteins and the lipid bilayer of the cell membrane (40). When an abnormal bone marrow progenitor clone is formed where this capacity is lost, the progeny RBCs will lack a whole range of surface proteins, including several blood-group-carrying molecules and most importantly, in this perspective, a selection of complement regulatory proteins. In their absence, the RBC becomes sensitive to complement-induced intravascular lysis and a key symptom is the passing of dark urine when the patient wakes up. In addition, there is a poorly understood relationship between PNH in both leukemia development and the formation of life-threatening clots in the large vessels.
In addition to the above-mentioned causes of hemolysis, microbial agents are known to be able to induce hemolysis. This will not be discussed further here but some of the clinically very significant diseases (and agents) involved include malaria (Plasmodium species), hemolytic uremic syndrome (Escherichia coli), dysenteria (Shigella), pneumonia (Mycoplasma), fifth disease (Parvovirus B19), and hemorrhagic fevers (various viruses).
2. Transfusion
When RBCs from a blood donor are transfused to a patient, the donor blood unit could have been stored at 4°C for a maximum of 35–42 days depending on local regulations. Storage of blood units results in storage-time dependent suboptimal function and stability of the cellular elements in the stored blood. Moreover, even if there are regulations to ensure an optimal quality of the blood components transfused, a certain amount of Hb leakage from the cells is unavoidable and expected. A progressive oxidation of cytoskeletal proteins and accumulation of denatured Hb also occurs in the stored RBCs. In addition, a fair percentage of damaged RBCs will be lysed or cleared immediately from the circulation in the recipient upon infusion. Thus, even if the best possible matching of blood groups is ensured by cross-matching, etc., at the blood center, the recipient will suffer an increased load of extracellular Hb following transfusion and consequently, also the negative effects of oxidative stress. Despite this, millions of RBC units are given annually around the globe, and transfusions are considered a prerequisite for many medical procedures, including surgery, safe obstetrics, chemotherapy treatment for cancer, as well as stem cell or organ transplantation. If chronic transfusions are given, the additional risk of iron overload is yet another threat, which is also associated with increased oxidative stress and needs to be treated (40).
The most severe form of iathrogenic hemolysis can be observed, if ABO-incompatible blood is transfused to a patient by mistake. This acute reaction is based on alloimmune RBC destruction and the presence of naturally occurring, pre-existing IgM antibodies against the ABO antigens lacking in the recipient. Even though blood transfusion is a safe treatment, in general, today, ABO incompatibility unfortunately still results in death or injury of patients following transfusion of allogeneic blood (259, 275). However, the consequences of this mismatched transfusion can vary from asymptomatic laboratory signs to death within minutes (165). The reasons why some patients react violently, with disseminated intravascular coagulation and shock, while other patients have no noticable reaction remain obscure. However, it is known that if the incompatibility involves blood groups other than ABO, for instance, Rh, Duffy, or Kell, then the adverse reaction is typically, but not always, milder and slower. Thus, a delayed hemolytic transfusion reaction is much more likely to be due to an anamnestic IgG response to protein epitopes, which will result in opsonisation of the allogeneic RBCs with IgG against the foreign, nonself blood group antigen, and extravascular removal of the targeted cells. A transfusion-unrelated variant of this phenomenon is the hemolytic disease of the fetus and newborn, where maternal IgG against paternal blood group antigens in the offspring passes the placenta and covers the fetal RBCs. This is a potentially lethal disease, since the macrophages of the reticuloendothelial system in the fetus will start phagocytosis of sensitized RBCs, with severe anemia and hepatosplenomegaly. If the Hb levels reach dangerously low values, hydrops fetalis may develop if treatment in the form of intrauterine, antigen-negative RBC units is not started. Hydrops is a critical state in which the fetus accumulates water because of the relative lack of RBCs able to circulate Hb.
3. Hemolysis and kidney function
Depending on what type of hemolysis (as discussed above) is encountered, elevated levels of Hb can be observed in the blood and/or urine. Plasma Hb is filtered through the glomeruli of the kidneys and re-absorbed by tubular cells where it may lead to precipitate formation termed hemosiderin during conditions with Hb-overload, causing oxidative damage and glomerular and tubular necrosis (85, 164, 285). These effects have mainly been reported in intravascular (136, 158, 214) but also in extravascular hemolysis (210). A particularly dramatic scenario is the renal consequences of an acute hemolytic transfusion reaction, after which a possible outcome is that the patient's life is saved, but the massive release of extracellular Hb leads to kidney failure and the need for dialysis and potentially a subsequent kidney transplant.
4. Current treatment of hemolysis
There are various means of treating patients undergoing hemolysis but none of these are specifically aimed at reducing the oxidative stress or inactivating the toxic free Hb molecules. Following massive intravascular lysis associated with ABO-incompatible transfusion, plasmapheresis may be the most efficacious way to remove excessive free Hb from the circulation before it damages the glomeruli permanently. However, because this procedure takes some time to arrange, massive doses of loop diuretics in combination with large volumes of clear fluids are often used to dilute the toxic effects of Hb. In the cold type of AIHA, it is customary to increase the temperature in the patient's room and use warm blankets to prevent hemolysis when the blood circulates to fingers, toes, and nose. Steroids are mainly used in warm type AIHA but the effect can be slow. Monoclonal antibodies against the antibody-producing B lymphocytes are used increasingly (268). A relatively new tool used in PNH treatment is a monoclonal antibody against one of the final components in the complement cascade (176), and it has been suggested that this approach could be applicable in other hemolytic situations as well.
B. Pre-eclampsia
PE has been named the disease of theories (241) and was described as early as 3000 years ago by the ancient Egyptians (277). PE is still one of the dominating obstetric complications that cause perinatal and maternal morbidity and mortality. The worldwide prevalence ranges 3%–7% of pregnancies.
1. Pathophysiology
There is a documented risk of developing PE when there is a maternal and paternal family history of PE (163). Discordance between monozygotic twins indicates that hereditary factors that follow classical Mendelian hereditary traits do not exist (293, 297). Black women are known to have a higher risk of developing PE than Caucasians (116). Several genes are likely to be involved in development of PE (42, 119, 241, 284). Mainly primigravidae are affected indicating that the maternal immune system may play a role. It was recently shown that early and late onset PE, as well as PE with intrauterine growth restriction, (IUGR) all have different inflammatory plasma protein profiles (75). Furthermore, systemic diseases such as diabetes, obesitas, essential hypertension, and renal diseases are all predisposing factors for PE. These data illustrate the complex, multi-factorial nature of PE.
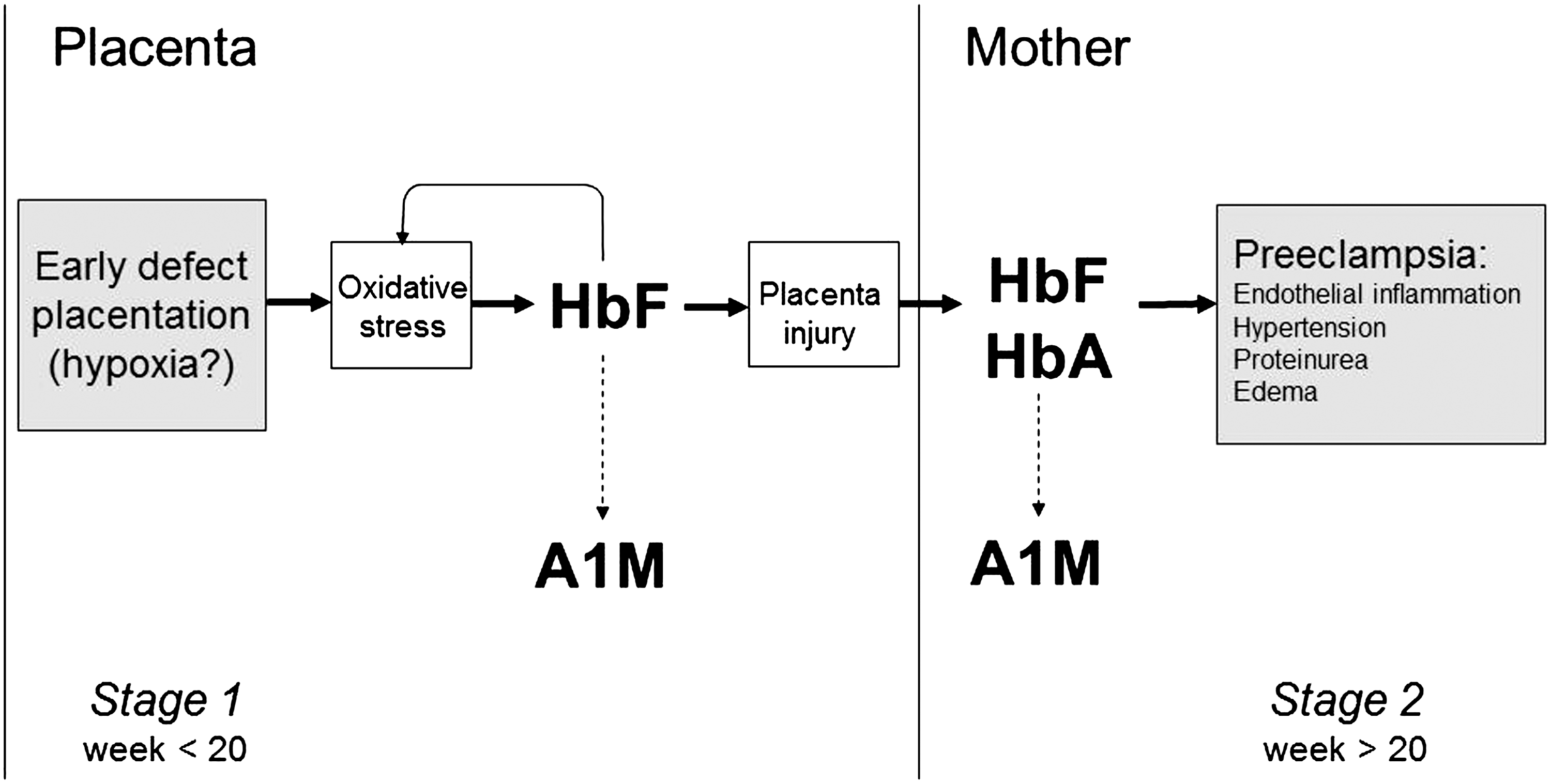
Removal of the placenta is crucial for the resolution of the symptoms (241), a fact that has led to the theory of a placenta-derived factor as a culprit. Although the etiology and pathophysiology are still not known, PE is suggested to evolve in two stages (Fig. 13) (243). Stage one occurs during formation of the placenta, and a defective and shallow invasion of the placental cells, trophoblasts, into the muscle layers of the spiral arteries, has been shown (41). Stage two, the clinical manifestations, that is, hypertension and proteinuria, appear from 20 weeks of gestation onward. Early onset PE (<week 34) is often more severe than late onset PE (>week 34). PE is defined as blood pressure of >140/90 mmHg and proteinuria of >0.03 g/L or a rise of diastolic blood pressure >20 mmHg during pregnancy.
As the disease progresses, angiospasmus in the brain and brain oedema may cause severe epileptic seizures—eclampsia (170). The renal disturbances seen in PE lead to reduced glomerular filtration rate and proteinurea, as discussed above in relation to the hemolytic anemias. Glomerular endotheliosis is characteristic for PE, in fact, it is the most common glomerular disease in the world (278). General endothelial dysfunction and diminished ability to vasodilatation are vascular hallmarks of PE (240). Severe PE also involves pathological activation of the coagulation and fibrinolytic systems that further compromise the blood circulation in general and the utero-placental blood flow specifically (41, 223). The reduced utero-placental blood flow results in IUGR, seen in one out of every four PE cases. The inadequate blood flow also gives rise to a reduced O2 delivery, which results in an ischemia/reperfusion-like situation and oxidative stress. A growing body of evidence suggests that this oxidative stress further aggravates vascular function in the placenta (116, 126, 242, 270).
Decreased placental perfusion, in combination with oxidative stress, causes general endothelial cell damage. An inflammation of the endothelial cells in the vascular system, in kidneys in particular, is a typical histological finding in PE (71, 106, 241, 245, 280). Vasoconstriction and elevated resistance to blood flow follows as a consequence. The increased vascular resistance can be evaluated clinically by using pulsed Doppler techniques on the uterine arteries as a high pulsatility index and/or occurrence of early diastolic notch in the maximum velocity waveform. Patients with persistent bilateral notching in the uterine arteries have a significantly elevated risk of developing PE and IUGR (22, 159).
A severe form of PE, the hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome, is characterized by low platelets, elevated liver enzymes, and hemolysis with high levels of free maternal HbA. Patients with HELLP often present with acute manifestations, possibly due to release of large amounts of free HbA. Oxidative stress is considered a key factor of the HELLP syndrome and significant increased levels of antioxidants in placentas have been shown (100).
2. Hb in PE-placenta
Recently, gene and protein profiling studies have revealed a new potential etiology for PE (Fig. 13). The first stage, caused by a defect formation of the placenta, gives rise to uneven blood perfusion, oxidative stress, production and accumulation of free HbF (56). The mechanisms of the generation of free, extracellular HbF in placenta blood vessels are not known, but it can be speculated that hypoxic conditions induce an upregulation of the HbF genes, and that a microenvironment characterized by insufficient blood flow and oxidative stress promotes hemolysis (218). Interestingly, it was shown that PE is associated with a decreased glucose-6-phosphate dehydrogenase activity and impaired redox regulation in erythrocytes and fetal endothelial cells (5), which may contribute to increased hemolysis in PE. Free HbF further aggravates the oxidative stress, which damages the blood-placenta barrier causing a leakage between the fetal and maternal circulations (189). Recent results show that the free HbF leaks over to the maternal blood stream from the damaged placenta as early as the first trimester (16). Free HbF may also contribute to the general systemic endothelial damage and inflammation that is characteristic of stage two. In addition, maternal plasma levels of HbA are elevated in women with PE at term of the pregnancy as compared to control, and both HbF and HbA are correlated with disease parameters (218). Possible mechanistic explanations of the role of HbF in the early PE-development are, as described above, that the free HbF may generate ROS and free heme groups, which damage the placenta and its barrier function. This may cause a leakage of fetal and placental factors, including HbF itself, from the fetus to the mother, thus linking Stage 1 and Stage 2 of the disease (Fig. 13). In fact, free heme, bilirubin, and biliverdin have been identified among 14 metabolites in a metabolomic signature of PE using first trimester plasma (142).
The hypothesis that Hb may be an important pathological factor in PE by causing placental damage is further supported by a recent report that ex vivo perfusion of placentas with Hb in the fetal circulation led to a significant increase of the perfusion pressure, feto-maternal leakage of free Hb, morphological damage, and upregulation of genes related to immune response, apoptosis, and oxidative stress (57, 189).
Likewise, the elevated concentrations of free maternal plasma HbF and HbA may have a central role in causing the clinical symptoms of the fully developed PE by inducing (i) oxidative stress, (ii) NO-scavenging and vasoconstriction, and (iii) further hemolysis. Indeed, in term pregnancies, elevated biomarkers of oxidative stress and upregulation of the heme-scavenger and antioxidant A1M were found (218), supporting an etiological role of free Hb in PE. Free HbF and A1M were also shown to be useful as prognostic biomarkers for PE as early as in the first trimester, several weeks before clinical manifestations were present (16).
3. Treatment of PE
Only symptomatic treatment of PE is available and includes treatment of the high blood pressure with antihypertensives and prevention of seizures with magnesium sulfate-infusion (79, 264). Several studies have focused on preventing PE by using antioxidative treatment with vitamin C, E, melatonin, and even dark chocolate; however, these treatments have not been able to prevent development of PE (244). Thus, delivery of the fetus, by labor induction or Caesarian section, is the only cure today.
C. IVH in the preterm infant
IVH is a well-characterized hemorrhagic lesion of the brain mainly occurring in premature infants (Fig. 14). The hemorrhage typically initiates in the germinal matrix, a structure located at the head of the caudate nucleus and underneath the ventricular ependyma, which is rich in vasculature and consists of glial and neuronal precursor cells. The hemorrhage can either remain stationary within the germinal matrix or rupture through the ependymal wall resulting in a collection of blood within the cerebral ventricles. Rupture of the germinal matrix vasculature leading to IVH occurs almost without exception during the first 72 h after birth and is easily detected by bed-side ultrasound. Severe cerebral IVH, with more than 50% of the ventricular area filled by blood, occurs in about 15%–20% of very preterm infants (308, 309, 316) and the incidence of IVH has not changed over the last decade (322). Figure 13B–E illustrates severe IVH in a preterm infant as determined by cranial ultrasound.
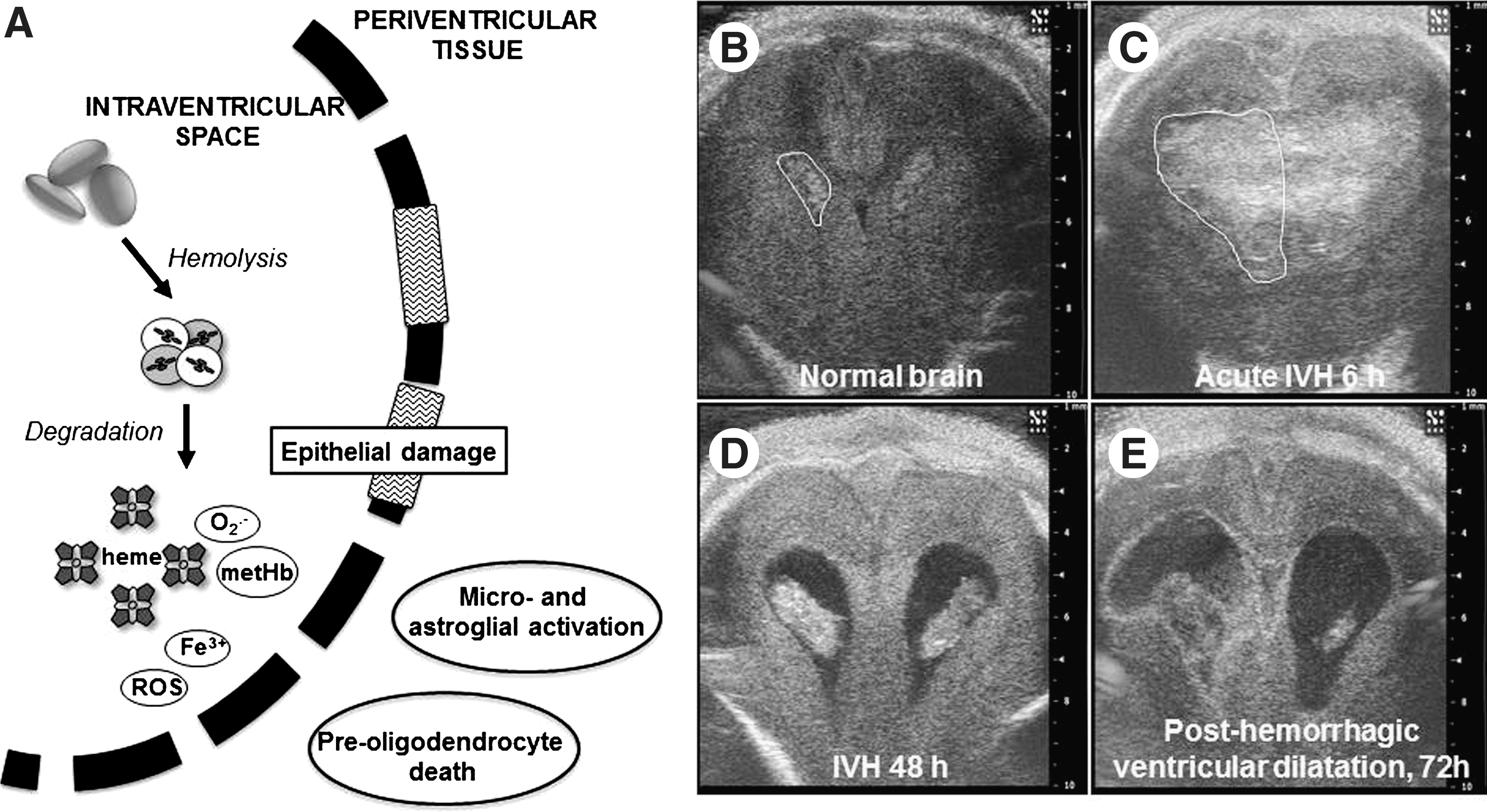
Children with severe IVH have a severe short- and long-term prognosis. The mortality in the neonatal period has been reported to be 20%–50% and of the surviving infants, over 50% develop posthemorrhagic ventricular dilatation leading to hydrocephalus and life-long requirement of a ventriculo-peritoneal shunt. At 2 years of age, 40%–80% of children show signs of severe neurological impairment, mainly cerebral palsy and mental retardation (3, 43, 97, 98, 198).
1. Mechanisms of white matter brain damage in the immature brain
The mechanisms leading to injury of the periventricular white matter following IVH are incompletely understood. The pathology in white matter following IVH is characterized by oligodendroglial cell death and immune cell activation, that is, astrogliosis and microglial activation. The principal cellular target within the white matter of the preterm infant is the developing oligodendrocyte. These early lineages of oligodendrocytes, the preoligodendrocytes, possess an extremely heightened vulnerability to insults associated with increased production of ROS. This is at least partly dependent on a maturational delay in the production of endogenous antioxidants such as SOD, Gpx, and catalase (143). Experimental studies have to date primarily focused on hypoxia/ischemia and inflammation as prime upstream initiators of ROS production. Microglial activation has been shown to play a key role and activation of microglial toll-like receptor (TLR)-2 and TLR-4 by both pathogen-related and endogenous molecules results in generation of ROS and cytokines causing either death or maturational arrest of the preoligodendrocytes. The resulting hypomyelination of corticospinal tracts forms the basis for cerebral palsy detectable during early childhood, most frequently observed as spastic diplegia with a predominant impairment of lower extremity motor function.
2. Free Hb and brain damage
The following will discuss a plausible and potentially treatable mechanism; whereby, IVH causes white matter brain damage (Fig. 14A). Following rupture of germinal matrix vasculature and of the ventricular ependyma, extravasated blood is mixed with intraventricular cerebrospinal fluid (CSF). Ensuing hemolysis of erythrocytes causes release of free Hb. In adults with subarachnoidal hemorrhage (SAH), Hb reaches its peak by day 2 following hemorrhage. Hb, primarily present as oxyHb, will with time be auto-oxidized to metHb. Subsequently metHb may be decomposed into heme and globin. The rates of hemolysis and resulting levels of free oxyHb and metHb in intraventricular CSF have yet to be characterized in preterm infants following IVH. Free Hb is known to be deleterious to the central nervous system and is cytotoxic in neuronal cultures (152, 238). One obvious mechanism is through ROS formation related to free heme release from Hb break-down and subsequently of free iron from the catabolism of heme by HO-1 or the constitutive HO-2. Clinical and experimental studies of adult SAH show convincing signs of immune activation with intracerebral glial activation, macrophage activation, neutrophil recruitment, and increased production of pro-inflammatory cytokines and adhesion molecules. A recent study showed that induced SAH in vivo was associated with upregulation of endothelial TLR-4 and a subsequent in vitro study revealed that exposure to oxyHb activated TLR-4 in vascular smooth muscle cells (323, 331). In summary, available data show that released free Hb and its downstream metabolites exert neurotoxic and pro-inflammatory effects highly relevant for the previously described pathways of white matter brain damage in the preterm infant.
3. Treatment of IVH
There is currently no established therapy to prevent infants from developing neurological disability following detection of severe IVH. Infants who develop posthemorrhagic hydrocephalus receive a life-long ventriculo-peritoneal shunt, which is an efficient means of preventing progressive ventricular distension but does not reduce neurological impairment. Recent data show that an intervention aimed at removing blood clots from the intraventricular space, that is, perfusing the intraventricular compartment via inserted catheters with artificial CSF, can reduce the requirement of life-long ventriculoperitoneal shunt and improve neuro-developmental outcome at early childhood (315). These data support the concept that blood components within the intraventricular space constitute risk factors for development of hydrocephalus and periventricular tissue damage.
D. Chronic leg ulcers
Chronic venous disease and chronic leg ulcers are common in Europe and the United States, and the point prevalence for nonhealing venous ulcers has been estimated to about 0.3% (204). The social and economic impact of these nonhealing wounds constitutes a great burden on both patients and the health care system (203, 204). Although much evidence for a dysregulated inflammatory process in nonhealing venous ulcers has been presented, the exact mechanisms underlying the disease, as well as the relative importance of the so far identified pathogenetic steps, are still not clarified (6). Current evidence, however, indicates that the ulcers are characterized by bacterial colonization, ongoing and unregulated inflammation and excessive proteolysis, chemotactic activity, and endothelial leakage (6).
1. Inflammation
Based on both in vitro experiments and clinical studies, several causative mechanisms underlying the chronic inflammation have been proposed (Fig. 15). The sustained venous pressure of these patients leads to a reduced pressure differential between the arterial and venous systems, and this has been proposed to underlie the fact that these ulcers contain significant amounts of extravasated or “trapped” leukocytes in the connective tissue. Extravasation of leukocytes also occurs in patients with venous disease only, and this in turn, may lead to ulcer formation (319). Extravasation of fibrin leads to the formation of fibrin depositions, possibly trapping, and also inactivating growth factors involved in wound healing in the perivascular areas (87). The high numbers of activated leukocytes lead to release of matrix metalloproteinases (MMPs), which further degrades extracellular matrix components as well as growth factors (6). Furthermore, all chronic ulcers are constantly colonized or infected by various bacteria, such as Pseudomonas aeruginosa, Enterococcus faecalis, Staphylococcus aureus, and Proteus mirabilis, and evidence supports the view that high bacterial loads contribute to nonhealing (68). These findings, together with the observation that keratinocytes display a nonmigratory phenotype in nonhealing venous ulcers suggest a failure of re-epithelialization and thus poor healing.
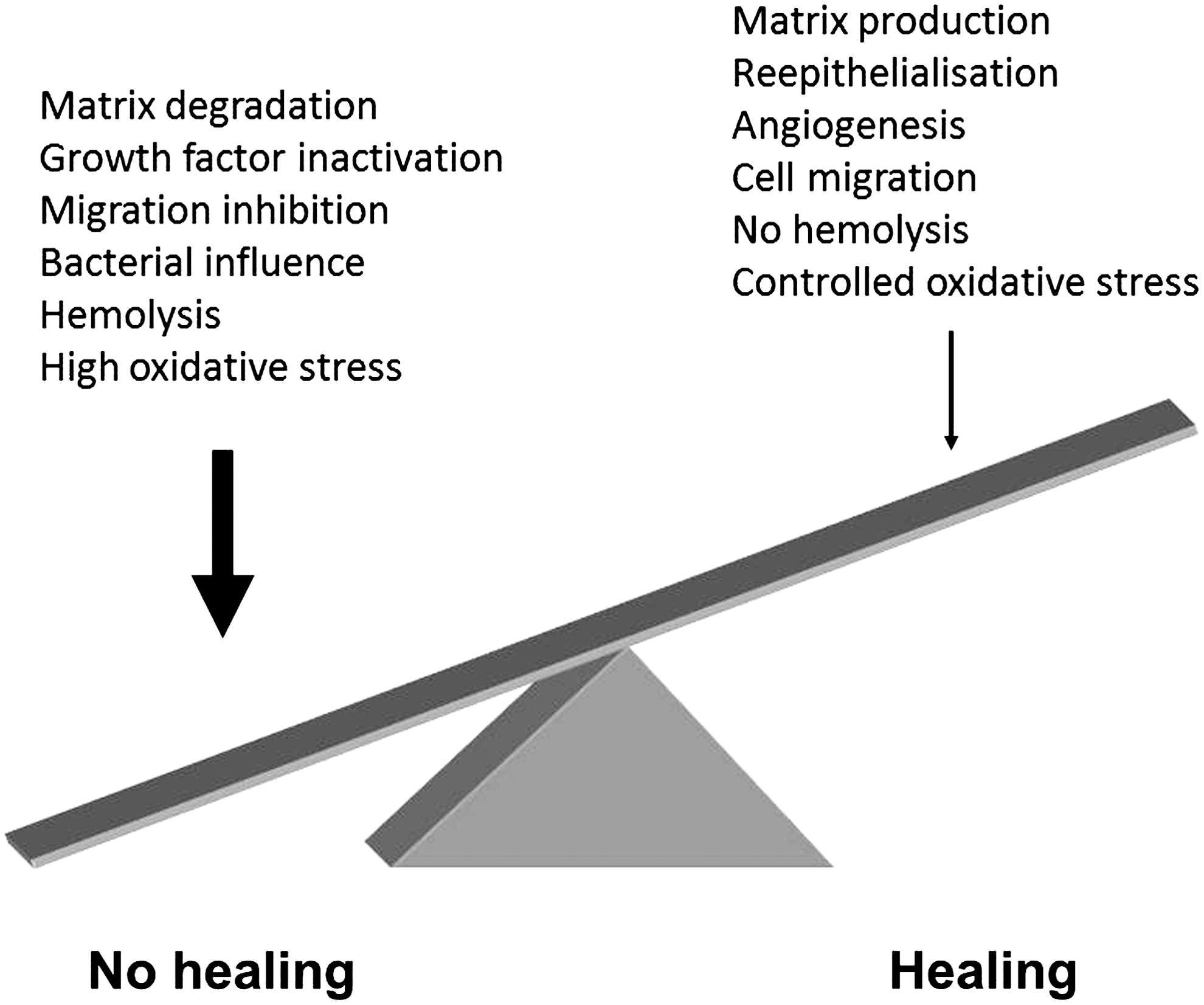
2. Heme, iron, and oxidative stress
Recent studies demonstrate a pivotal role for accumulation of iron in the tissue surrounding venous ulcers, leading to induction of the high inflammation noted in this disease. Iron deposits (Fig. 16) in nonhealing ulcers cause the readily visible brown-colored areas, which often precede and typically are found around nonhealing, venous ulcers. The origin of these increased iron stores is extravasation of erythrocytes due to the venous stasis combined with an excessive capillary leakage. Iron, in free form or complex-bound to protoporphyrin IX in heme, has been implicated as an important pathogenic factor in the initiation of the high inflammation in leg ulcers (2, 10, 310). As described above, the oxidized ferric forms of heme and free iron are strong oxidative agents and the Fe2+ can generate O2-derived free radicals by the Fenton reaction (172, 282). These compounds present a considerable oxidative stress that leads to tissue damage and cell destruction and have been proposed to participate in the pathogenesis of chronic ulcers by initiating local lipid peroxidation of polyunsaturated fatty acids in cell membranes as well as enhancing the synthesis and activation of MMPs (10). Interestingly, heme was identified in significant amounts in fluids from nonhealing ulcers (10). Heme was also found particularly in tissue sections from chronic ulcers as opposed to acute wound sections or normal skin sections (10) (Fig. 16). It thus seems likely that, dependent on venous hypertension in chronic ulcers, heme diffuses from plasma to the extravascular compartments across the endothelium and basement membranes in nonhealing ulcers. Heme may therefore be trapped and accumulated in pericapillary fibrin cuffs of venous ulcers. Thus, excess accumulation of heme causes an increased vasopermeability, increased expression of endothelial adhesion molecules, and infiltration of leukocytes, changes typically characterizing venous ulcers (10). Interestingly, high amounts of A1M were also found in these locals, supporting the view of A1M as a heme scavenger in nonhealing ulcers (10) (Fig. 16). It may be speculated that A1M has a similar effect as HO, promoting iron storage; however, additional studies are required in order to elucidate a role of A1M in iron metabolism in nonhealing ulcers. Unlike HO, A1M functions extracellularly, possibly providing the main source of protection against unsequestered heme in the extracellular space. Interestingly and supporting a role for heme and iron in chronic ulcer pathogenesis, mutations in the human hemochromatosis protein (HFE)-gene seem to be linked to a higher risk of developing ulcers (326 –328).
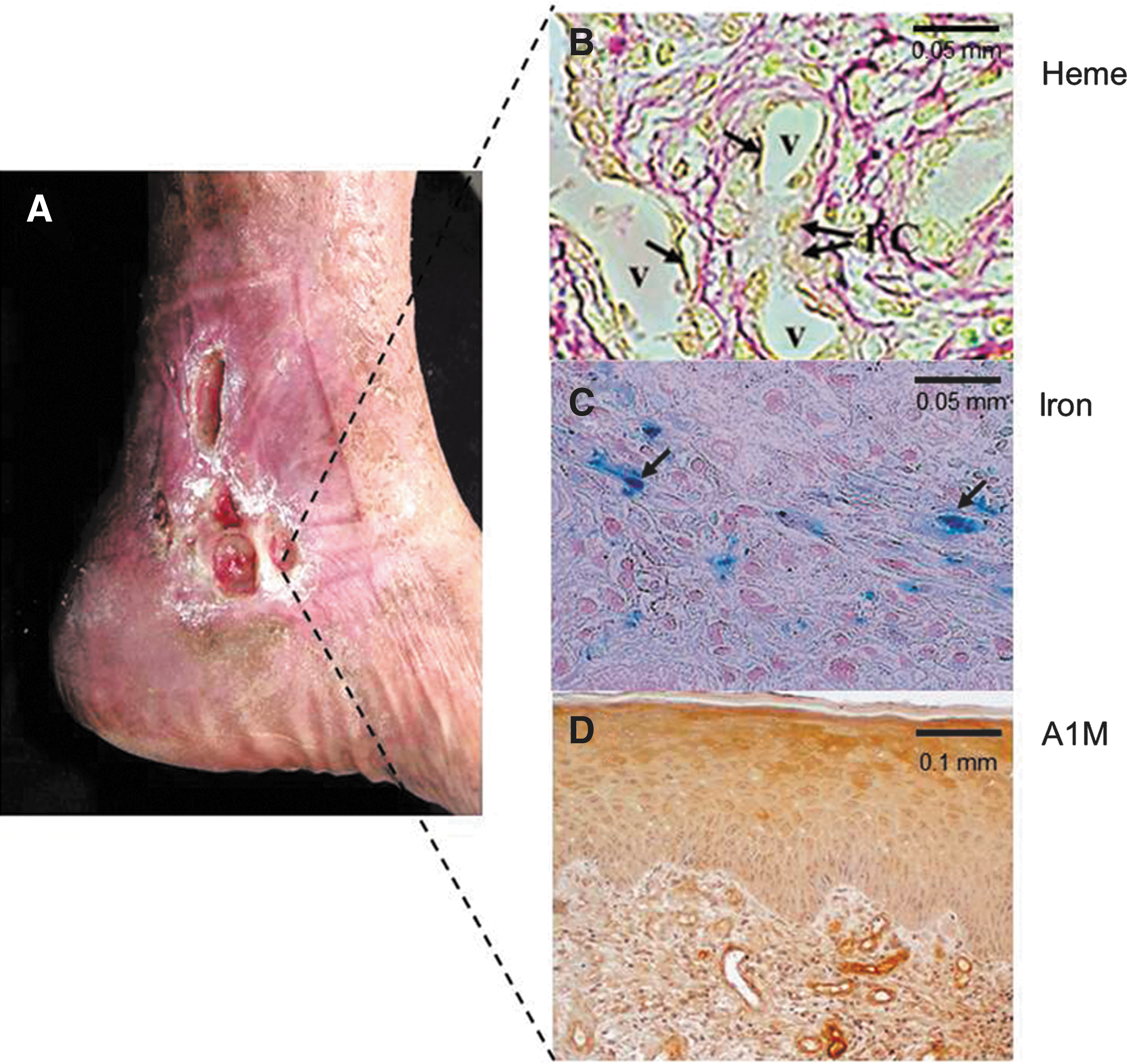
The causes of nonhealing ulcers are summarized in Figure 15. Taken together, the data presented so far support the concept of heme and iron as important mediators of the high inflammation in nonhealing ulcers, with A1M as part of a previously undisclosed endogenous defense mechanism against heme-induced oxidative stress.
E. Blood substitutes and O2 therapeutics
The attempts to develop “artificial blood” or blood substitutes have been ongoing with various intensities for more than 70 years. These research efforts have mostly been focused on the ability of Hbs to carry and deliver O2. Most of the products that have been passed into advanced-phase clinical trials are also derivatives of Hb. The phrase “hemoglobin-based oxygen carrier” (with the acronym “HBOC”) has been applied to these materials, as blood contains many molecules and proteins that provide other functions than O2 transport (49, 60, 62, 325).
There are several important driving forces behind the development of RBC substitutes. For instance, effective trauma care, an increased number of elective surgeries, and the prevalent but small risk of transmission of blood-borne pathogens stimulate this development. In addition, these products can function as organ preservatives to prevent or decrease reperfusion injury to donor organs. Various religious and ethnic groups with concerns regarding the use of human blood products may accept these substances.
The biologic properties of an optimal blood substitute should include a reasonable level of O2 delivery when compared to normal human RBCs. An ideal blood substitute should fulfill several additional requirements, including lack of any antigenicity, readily availability, a long half-life, stability allowing storage at ambient temperature, and elimination of risks to transmit infections. However, to date, no O2-carrying blood substitutes have been approved for use by the U.S. Food and Drug Administration. This fact clearly demonstrates the difficulties that exist in formulating an effective blood substitute. Therefore, most current efforts with these agents do not primarily aim at substituting blood but rather focus on mediating O2 therapeutics in severe clinical settings. HBOCs can thus be viewed as an attractive alternative O2-delivery system, which can partly augment whole blood during a severe blood loss situation or, alternatively, for use when whole blood simply is not available for treatment.
One of the major obstacles in the clinical development of HBOCs has been the uncontrollable and sudden elevation in systematic and pulmonary blood pressure in trauma victims or patients undergoing elective surgery when infused with these products. Furthermore, native free Hb, when infused rapidly, readily splits into dimers followed by its clearance by glomerular filtration. The initial attempts of transfusing Hb-based products thus resulted in renal dysfunction and hypertension. The instability and reactivity of the Hb tetramers have been overcome by altering human (or bovine) Hb by three general approaches: the use of chemical cross-linking reagents that stabilize proteins along with modifications to adjust O2 affinity; exploring reagents specifically designed to increase the size of the Hb product and modifying its reduction to notably NO; protein engineering and expression of modified versions of Hb with desirable properties.
Over the years, several different Hb-based products have been produced. For instance, a so called first generation product is Hemopure, which is a polymerized form of bovine Hb with an O2 affinity that is close to human Hb. It has an intravascular half-life of 8–23 h and a shelf life of 36 months at room temperature. Hemopure is approved in South Africa for the treatment of adult surgical patients who are acutely anemic with the intention of eliminating or reducing the need for RBC transfusions (121). Hb binds NO readily at the same site at which it binds O2 and at the thiol of cysteine (see also above, section II.B). It was apparent at an early phase that the elevating effects on blood pressure was most likely related to strong binding of NO to HBOCs. This inherent property of free Hb would be one of the major causes of vasoconstriction, leading to hypertension. In 2004, the first modified Hb that did not elevate blood pressure was reported. In 2004, the first modified Hb product that did not elevate blood pressure was reported. The Hb molecule was conjugated to polyethylene glycol (PEG) and a safe product could be considered to be a real possibility. PEG-modified Hb, such as MP4, is based on human Hb produced from out-dated blood. In animal models, MP4 has been shown to be effective in cases of hemorrhagic shock. Adverse effects associated with the vasoactive properties of first-generation blood substitutes are not seen with MP4. At relatively low concentrations, MP4 is capable of transporting large amounts of O2 (65).
Besides PEGylation, several lines of developments have been described. For instance, HBOCs have been cross-linked with enzymes in efforts to synthesize compounds that not only perform the function of carrying O2, but also harbor some of the enzyme activity that normal RBCs possess. Polymerized Hb has thus been chemically cross-linked with catalase, SOD, and carbonic anhydrase to form a compound that, in animal models can not only carry O2 but also remove O2 radicals that are responsible for ischemia reperfusion injuries (32). Such conjugates can be prepared by chemical cross-linking or by gene fusion of the corresponding structural genes in order to generate more homogenous preparations (108).
Attempts have also been made to encapsulate Hb within a lipid membrane to create a compound capable of carrying O2 while not being associated with significant vasoconstriction. These liposomes appear to be retained in plasma for a significant period, but they are difficult to produce and can activate the reticuloendothelial system, the complement pathway, and platelets. The ultimate RBC substitute would need to contain an artificial membrane mimicking the native counterpart (252). However, production of such a product would be extremely challenging. Efforts have also been made to use the biopolymer polylactide, which is biodegradable and converted to lactic acid in the body. These artificial RBCs should contain Hb along with SOD, metHb reductase, and catalase (59).
Adverse effects associated with HBOCs include beside hypertension, abdominal pain, skin rash, diarrhea, jaundice, hemoglobinuria, oliguria, fever, stroke, and laboratory anomalies, such as an elevation in lipase levels. Although most of these side effects are transient and clinically asymptomatic, many clinical trials involving these agents have been discontinued or withheld due to the associated adverse effects. In 2008, a meta-analysis of the results of clinical trials of blood substitutes led to the striking conclusions that none are effective and some are not safe. After that report appeared, the U.S. Food and Drug Administration announced that trials of products under consideration would not proceed further (84, 201). With existing documentation of clinical problems, it is clear that new designs and application areas for HBOCs are needed. Ongoing research has improved our understanding of the RBC and its interactions with the surrounding cellular environment. This has facilitated the development of newer products that do not have significant vasoactive properties, as did the first-generation compounds. Several of these recent approaches are promising and involve linking Hb with itself or nonreactive species in order to increase size, emulating the size-expanding effects of PEG (53, 124, 125, 175). For instance, Bucci et al. produced larger materials by combining Hb tetramers under dehydrating conditions to give a “zero-link” product (45), while Zal's group has used protein engineering to express a multi-Hb ensemble that occurs in marine organisms (118). Marden has developed linked Hb tetramers, called octamers and has also attached Hb to nanoparticles as a means to increase size (28). Many of the current polymerized preparations still have a small fraction of free tetrameric Hb in solution. This fact has often been proposed to be a major reason for the observed side effects associated with transfusion. Therefore, novel chemical strategies have been developed to increase the degree of polymerization and minimize free Hb in solution (329). In addition, auto-oxidation and shelf-life are two critical parameters in the design of HBOCs. By storing the product in the carboxy state (carboxy Hb [COHb]) potentially valuable improvements have been observed (300). The use of COHb to release a limited amount of CO may also have potential benefits in ischemic tissues. Small molecules that release CO have been shown to exert vasodilatory and anti-inflammatory properties in a variety of models in the peripheral circulation (197). Transfusion of RBCs containing high COHb was thus shown to be beneficial in limiting microvascular markers of apoptosis after hemorrhagic shock (52).
V. A1M: A Novel Therapeutic Concept
The biochemical and physiological properties of A1M are suggestive of an in vivo protective role of the protein against radicals and oxidative stress. The findings that A1M can bind heme and that Hb induces a proteolytic activation of heme-degradation activities of A1M, suggest that it may have a specific role during hemolytic conditions in protecting tissues exposed to Hb- and heme-induced oxidative attack. Does A1M have properties that allow, and even favor, therapeutic use in clinical situations with a high load of free, extracellular Hb? Below, we speculate on a possible extravascular role of A1M to complement an intravascular role of Hp and Hpx. We will also propose tentative A1M-based therapies in the diseases discussed in this article.
A. Extravascular Hb-protective role of A1M
In a therapeutic situation, what are the possible advantages of A1M compared to the other Hb-protective systems? Available data today points to a specific protective role of Hp and Hpx intravascularly, whereas A1M seems to operate mostly extravascularly. The two former proteins are found in high concentrations in blood and promote clearance of Hb and heme, respectively, from the circulation. However, it should be remembered that the Hp levels fall toward zero very rapidly following the release of free Hb into the plasma, due to scavenger receptors for the Hp-Hb complex, and it is possible that other protective mechanisms are desirable and needed in this situation. The literature suggests that A1M has a role as an antioxidant and heme-scavenger mainly in the extravascular fluids and cytosol rather than in blood. It is secreted to blood from the liver, but its concentration in blood is comparatively low, 1–2 μM, due to a very rapid equilibrium (T1/2=2–3 min) between the intra- and extravascular compartments (see section III.F.4). Likewise, the heme-degrading HO proteins mainly have a cytosolic location, whereas A1M is found both in cytosolic and extracellular locations. Finally, in relation to the vast network of more or less monospecific antioxidants that protect tissues from Hb- and heme-induced oxidative damage/stress, A1M seems to be multispecific, binding, and clearing cytosols and extravascular fluids from ROS as well as small organic radicals.
B. Studies in diseases
1. Chronic leg ulcers
The possible use of A1M in treatment of chronic leg ulcers was suggested by Goldsmith (102). The localization and heme-binding activities of the protein in inflamed tissue and fluid from wounds (10), the heme-binding activities (10), and the in vitro experiments with keratinocytes, skin and collagen fibrils [(216); see also section III.F.8] support this notion. The AMBP gene was also upregulated in keratinocytes and skin by heme- and ROS-exposure, suggesting physiological advantages from increased concentrations of the protein in skin tissue during oxidative stress (216). Furthermore, as described above (section III.F.8.d), several lines of evidence suggest that A1M may have repair-/rebuilding effects on extracellular matrix, promoting increased fibrillation of collagen I and an upregulation of elastin and collagen genes. However, to prove a clinically beneficial effect of A1M in chronic leg wounds or in other oxidative stress-related skin disorders such as during UV irradiation, atopic dermatitis, and psoriasis, in vivo experiments in humans or animals have yet to be performed.
2. Pre-eclampsia
Another target of A1M-based therapy may be PE, which, as described above, is associated with higher than normal plasma and placenta concentrations of HbA and HbF as well as markers of oxidative stress. It was hypothesized that PE manifests as a result of an Hb-dependent oxidative insult and that A1M is involved in the (futile) defense (56, 218). Indeed, elevated serum concentrations of A1M were found in the first trimester of women who later develop PE (16) and in the placenta, urine, and blood from women with diagnosed PE at the term of the pregnancy, and evidence of t-A1M formation in placenta (148, 218, 225). Furthermore, as described above, ex vivo perfusion of placenta showed that the PE-like damage induced by perfusion of Hb was effectively inhibited by A1M. Antioxidative treatment of PE has been suggested and tried before. Several studies have evaluated the use of vitamin C and E in high-risk pregnancies in order to prevent the PE-associated oxidative stress. These studies failed to show a reduction in the rate of adverse maternal or perinatal outcomes related to pregnancy-associated hypertension (231, 244). This does not disprove the oxidative nature of the disease; however, since the scavenging capacity of vitamin C is limited and the oxidized form, dihydroascorbate, which is formed by its oxidation, may also present an oxidative challenge in the tissues during the disease.
3. IVH in preterm infants
Treatment of IVH and/or SAH with A1M may be a possible option if Hb- and heme-induced toxicity and oxidative stress actually are important pathological factors, as described above. The heme- and radical-binding of A1M, the rapid turnover rate, and the fact that it is an endogeneous, nonimmunogenic molecule makes this protein an attractive therapeutic option for these diseases.
4. Hemolytic anemias and transfusion reactions
Hemolytic anemias and adverse reactions following a blood transfusion may also be future targets of A1M-treament, but both scenarios present a much greater challenge than, for instance, chronic leg ulcers or IVH since the whole body is affected. Thus, systemic, rather than local administration has to be considered. Intravascular hemolysis, such as that occurring in autoimmune hemolytic anemia of the cold (IgM-mediated) type, PNH, and paroxysmal cold hemoglobinuria may be diagnosed in which a benefit from systemic A1M administration could be possible since these states are often associated with depletion of Hp as discussed above. Similarly, we know that patients with extravascular hemolysis, for example, diagnoses, including autoimmune hemolytic anemia of the warm (IgG-mediated) type and drug-induced hemolytic anemia, acquire impairment of their kidney functions due to detrimental effects of Hb. It is likely that they also suffer from increased oxidative stress based on the large amounts of extracellular Hb and heme they have to process. It is thus probable that also these patients could benefit from antioxidative/radical scavenging therapy by A1M. Today's storage solutions for RBC concentrates for transfusion do not take into account the problem with oxidative damage during storage. To increase the quality of RBCs for transfusion, one may therefore propose to add A1M before and/or after storage. Thus, A1M could be added separately (i) before the storage period is started (proactive); (ii) as a rejuvenation agent after a certain time period had passed (interactive); or (iii) just prior to issuing the blood unit to the patient/ward (cleaning/therapeutic approach). In analogy with data from other systems, A1M would have the ability to prevent oxidation and, in the latter two cases, also repair oxidative modifications of the blood components. This approach may be especially appealing in the situation of gamma irradiation of blood to prevent graft-versus-host disease, since those units tend to be more damaged with membrane disturbances causing potassium leakage, compared to untreated blood for transfusion.
5. Blood substitutes
As discussed above, many of adverse side effects of treatment with blood substitution products are related to generation of ROS, free radicals, and free heme, and may therefore also be future targets of A1M intervention. For example, A1M may be administered as an addition to the HBOC itself, given to patients as a means of coping with the negative consequences, and oxidative stress induced by free Hb and heme. An interesting alternative could perhaps be to cross-link A1M with Hb or to create a genetically modified fusion protein between Hb and A1M. Another approach would be to encapsulate Hb and A1M together in liposomes. Both ways, A1M would do the job of keeping the Hb molecule in a reduced state and eliminate Hb-generated ROS as soon as they are produced. However, no investigations on in vivo trials with HBOC in combination with A1M have been reported yet.
VI. Conclusion
Toxic effects of extracellular Hb are major components in the pathogenesis of many diseases and iatrogenic situations, including hemolytic anemias and transfusion-induced intravascular hemolysis, PE, intraventricular hemorrhage, chronic inflammatory leg ulcers, and infusion of recombinant Hb. The pathogenesis involves one-electron reactions between oxyHb, its downstream metabolites heme, iron, ROS and free radicals, and exposed tissue components. Many mechanisms have evolved to protect organisms against damage from extracellular Hb. One of these, the ubiquitous protein A1M, is found in all vertebrates, including man and operates by rapidly clearing cytosols and extravascular fluids from heme groups and free radicals released from the oxyHb molecule. A1M has been shown to protect cells and matrix from Hb-, heme-, and ROS-induced damage and is a promising therapeutic alternative in diseases with Hb-induced pathology.
Footnotes
Acknowledgments
Part of the work shown in this article was supported by the Swedish Research Council, governmental ALF research grants to Lund University and Lund University Hospital, the Royal Physiographic Society in Lund, the Foundations of Greta and Johan Kock and Alfred Österlund, and the Blood and Defense Network, Lund University. The authors also wish to acknowledge Mattias Mörgelin and Maria Baumgarten for outstanding electron microscopy work.