
Abstract
Introduction
NO and derived reactive nitrogen species (RNS) are crucial to innate immunity for the control and clearance of pathogens (11), as RNS damage several biological targets, including DNA, lipids, protein metal centers, and amino acid residues of proteins, therefore causing inactivation of key metabolic functions. Hence, to sustain the long-term colonization, H. pylori needs to resist and subvert the effects of the nitrosative stress.
Innovation
On infection, the innate immune system produces nitric oxide (NO), a reactive molecule that helps eradicate pathogens. To date, no system devoted to removing NO has been reported for Helicobacter pylori, a pathogen with a high human health burden. This work provides evidence for the first NO detoxifying enzyme present in H. pylori, namely HP0013. An extensive functional characterization of the gene/protein demonstrates that the enzyme catalyzes the reduction of NO and provides the pathogen with an added defense against inducible nitric oxide synthasemediated host immune attack. Moreover, it is shown that HP0013 constitutes a novel family of NO detoxifying microbial enzymes that is widely spread in prokaryotes.
Regarding RNS defense mechanisms, H. pylori counteracts host NO production by expressing arginase RocF that competes with iNOS for the same substrate,
Results
HP0013 increases resistance of H. pylori to RNS
A mini-Tn3-Km transposon mutant library of H. pylori 26695 (21) was screened for resistance to the NO donor S-nitrosoglutathione (GSNO) using phenotypic disk-diffusion assays. Mutation of the hp0013 gene was shown to yield a strain with higher susceptibility to GSNO (Fig. 1A). H. pylori hp0013 codes for a hypothetical protein that forms a putative operon with the upstream gene hp0012 which encodes the DNA primase DnaG, and the downstream gene hp0014 that encodes a hypothetical protein. However, the hp0014 mutant was equally susceptible to nitrosative stress as the wild-type strain (Fig. 1A).
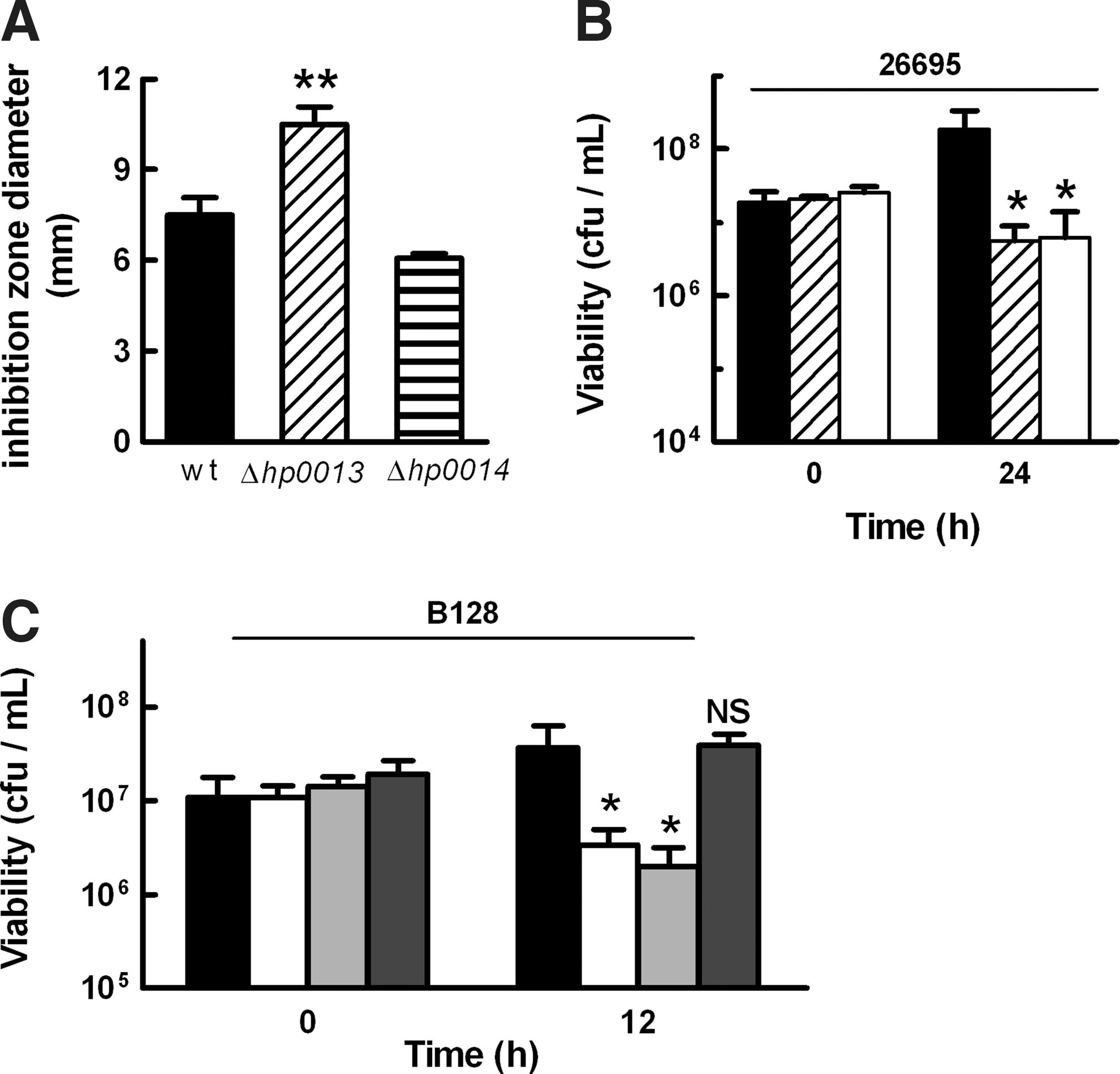
To analyze the role of hp0013, a nonpolar mutant strain was generated in H. pylori 26695 by gene replacement with a kanamycin resistance cassette. The growth behavior of both transposon and nonpolar hp0013 mutants was examined in cultures treated with 200 μM GSNO. Although in the absence of stress the two mutants behaved as the parental strain (Supplementary Fig. S1; Supplementary Data are available online at
To further substantiate the results, the nonpolar hp0013 gene deletion was introduced into another H. pylori strain, namely H. pylori B128, a strain that has the ability to colonize mice and Mongolian gerbils (20). Similar growth assays were performed, and the results revealed that deletion of hp0013 also impairs the resistance of H. pylori B128 to GSNO (Fig. 1C), even though these strains appeared less sensitive than their counterpart H. pylori 26695 derived strains. We further observed that expression of HP0013 from the inducible plasmid pILL2157-HP0013 was sufficient to rescue the phenotype of the H. pylori B128 hp0013 mutant (Fig. 1C).
To confirm the role of HP0013, H. pylori 26695 and B128 nonpolar hp0013 mutant strains were exposed to other sources of nitrosative stress. As seen in Figure 2A, B, H. pylori 26695 was less resistant than the B128 wild-type strain to stress induced by dipropylenetriamine (DPTA)-NONOate, an NO donor that releases two molecules of NO with a half-life of 3 h at 37°C. This result was similar to that seen when the strains were tested against GSNO-induced stress. Nevertheless, in both backgrounds, the deletion of hp0013 caused significantly higher NO-dependent killing, with an up to 10-fold decrease of survival relative to the correspondent wild-type strain, an effect that could be reversed with the expression in trans of the HP0013 protein (Fig. 2B).
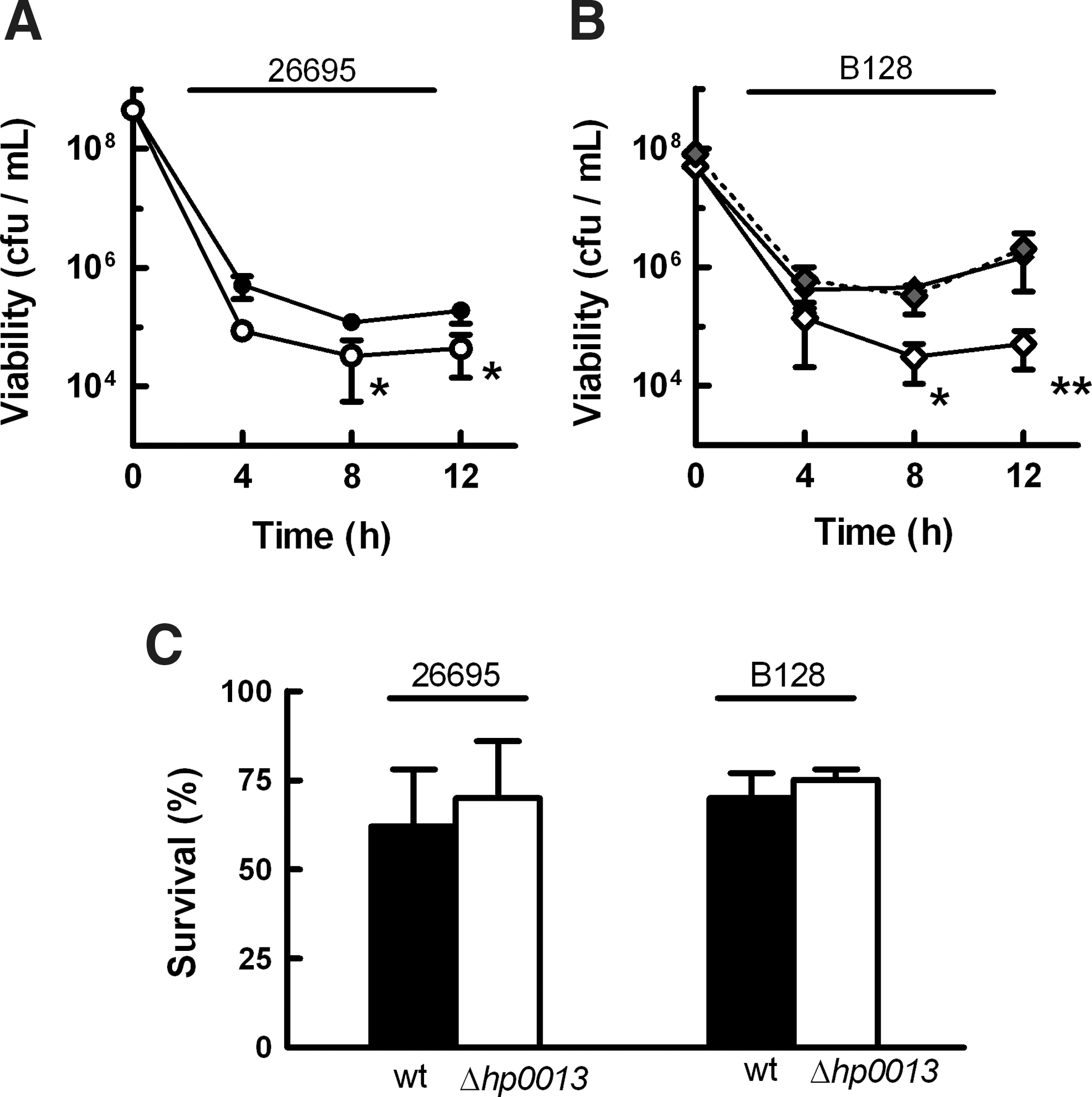
In contrast, on exposure to 200 μM peroxynitrite, wild-type and mutant strains suffered, independently of the time, a small decrease of viability (∼30%) (Fig. 2C), indicating that HP0013 is not involved in the detoxification of the NO congener, peroxynitrite.
Altogether, these results revealed that HP0013 protects the H. pylori species against NO stress.
HP0013 confers NO detoxification ability to H. pylori
Given the phenotypes of the hp0013 mutants, we tested whether HP0013 was involved in the reduction of S-nitrosothiol adducts or in NO detoxification.
The first hypothesis was disproved as H. pylori 26695 and hp0013 mutant cell lysates exhibited similar levels of GSNO reduction activity (Fig. 3A). On the contrary, the rates of anaerobic NO consumption, measured with an NO-electrode, were ∼50% lower in cell extracts of the 26695 hp0013 mutant strain when compared with the wild-type (Fig. 3B).
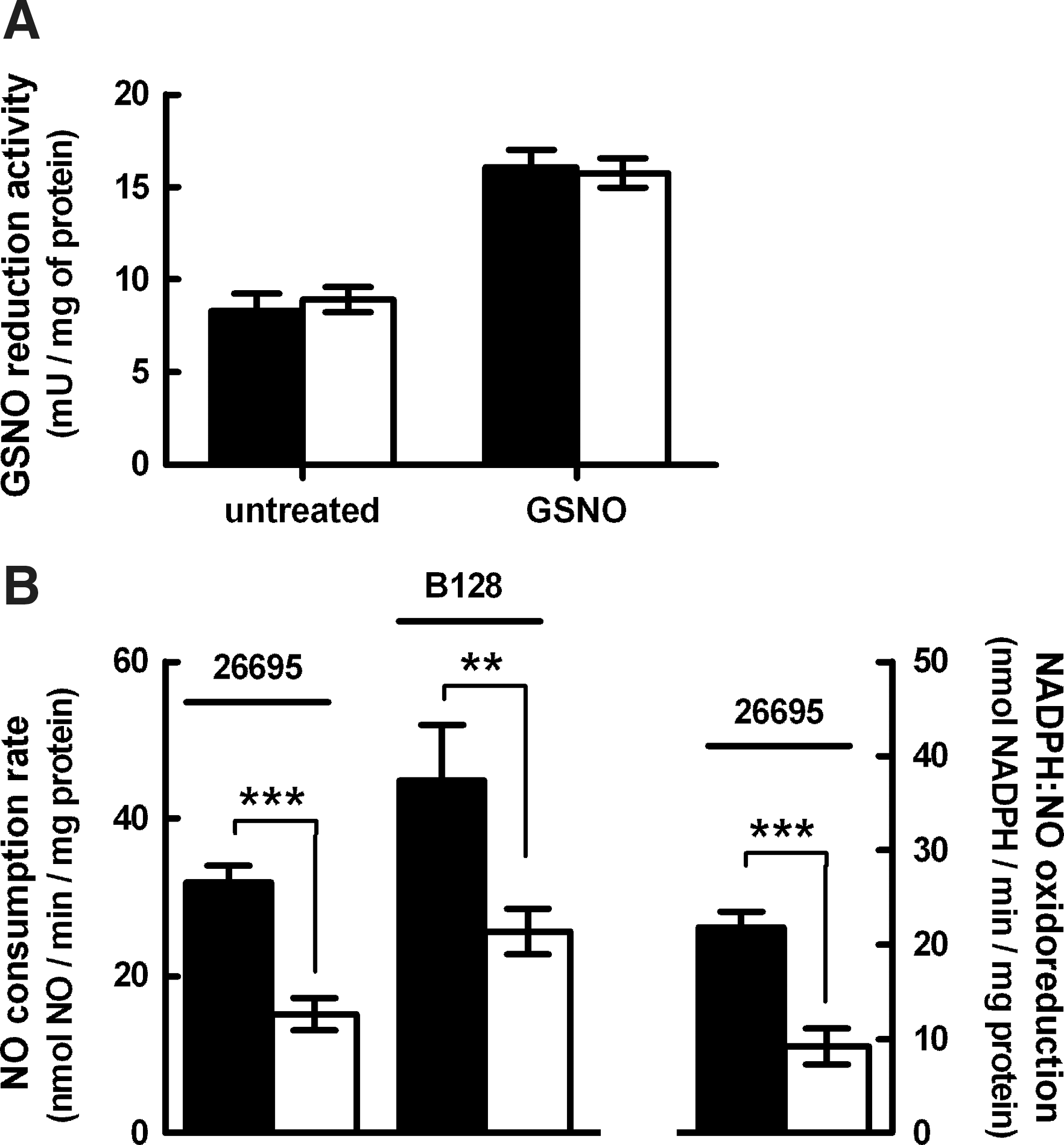
The rate of NAD(P)H oxidation coupled to NO reduction was also assayed in spectrophotometric assays. The cell lysates of H. pylori 26695 wild-type and hp0013 mutant showed rates of NADPH:NO oxidoreduction that were similar to the rates of NO consumption measured amperometrically (Fig. 3B, left), while no activity was detected when using NADH as an electron donor.
We next investigated whether hp0013 deletion also affected the NO reduction ability of H. pylori B128 cells. H. pylori B128 wild-type cell lysates exhibited higher levels of NO consumption than H. pylori 26695. Moreover, the B128 strain mutated in hp0013 had significantly lower activity (Fig. 3B).
We also observed that lysates prepared from H. pylori 26695 cultures pre-exposed for 1 h, to 200 μM GSNO presented NO consumption rates equivalent to untreated cells (Supplementary Fig. S2). In accordance, the hp0013 gene expression was found to be essentially unchanged by the presence of nitrosative stress (data not shown).
Hence, we concluded that HP0013 is involved in the NO detoxification of H. pylori, as the activity of both strains was found to be lowered by ∼50% in the absence of the hp0013 gene.
HP0013 protein exhibits NO reduction activity and rescues the NO-deficient phenotype of the Escherichia coli NO reductase mutant strain
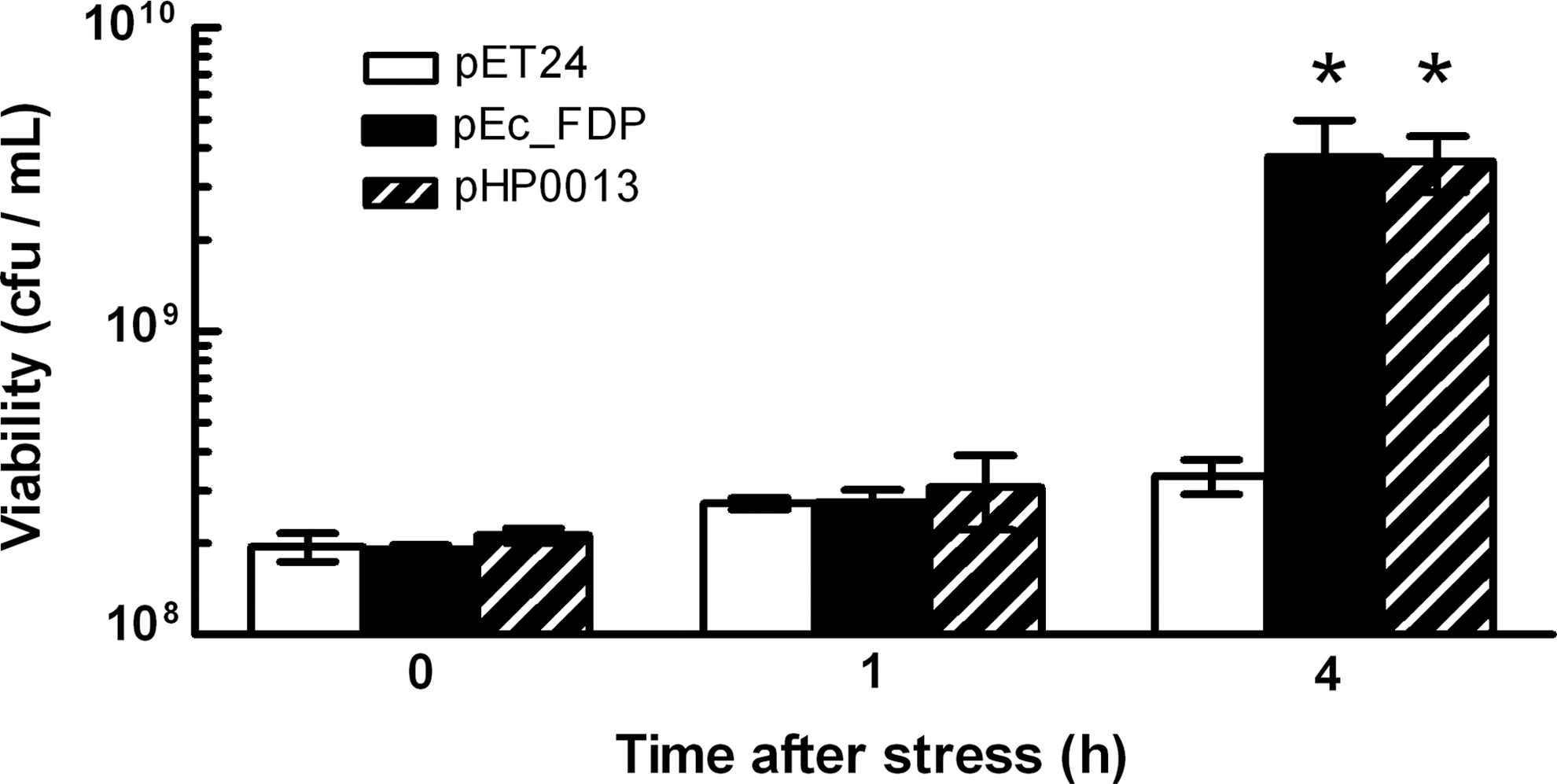
The HP0013 protein was recombinantly produced, isolated, and its enzymatic NO activity was assayed. The purified protein had an NO reduction activity of 0.10±0.03 nmol NO.(s.nmol protein)−1 and a rate of coupled NADPH:NO oxidoreduction of 0.07±0.03 s−1, consistent with a stoichiometry of two molecules of NO reduced to N2O, per molecule of NADPH oxidized, similar to what is generally described for other NO reductases (16). The NO turnover of the recombinant HP0013 protein is lower than that of qNORs and FDPs (10–50 s−1), but within the same range of the NO reductase activity of flavohemoglobins (HMP) (31). Nevertheless, we hypothesize that such a low turnover rate may be due to the instability exhibited by the protein. To further confirm the role of the HP0013 protein as an NO reductase, we tested whether the expression of HP0013 rescued the anaerobic NO-phenotype of Escherichia coli cells deficient in the FDP-type NO reductase. As shown in Figure 4, the E. coli fdp mutant cells carrying the plasmid expressing H. pylori HP0013 were more resistant to NO stress than those carrying the empty vector. Moreover, the expression of HP0013 complemented the NO-phenotype to an extent similar to that achieved on expression of the E. coli's own FDP protein (Fig. 4) from the same vector. These results showed that the NO reductase activity of HP0013 was able to protect the E. coli cells to an extent similar to the one conferred by the E. coli FDP.
HP0013 contributes to the survival of H. pylori in macrophages
Since NO production is a crucial factor of macrophage-dependent eradication of H. pylori (8, 15), we evaluated the killing of H. pylori strains (26695, B128) and their isogenic hp0013 mutants by RAW264.7 macrophages. After 24 h of incubation with RAW264.7 cells, the survival of the two hp0013 mutant strains was ∼50% lower than that of the correspondent parental strains (Fig. 5A, B), that is, the absence of the hp0013 led to a significant increase of RAW264.7 mediated killing of H. pylori. Moreover, expression of the HP0013 protein from an inducible plasmid in the B128 hp0013 mutant strain restored the wild-type phenotype (Fig. 5B), in an expression level-dependent way (Supplementary Fig. S3).

H. pylori is known to influence macrophage NO production, for instance, by depleting the substrate of iNOS,
To confirm that the lower viability of hp0013 mutants when infecting macrophages is attributable to their susceptibility to nitrosative stress, assays were repeated in the presence of the iNOS inhibitor NG-monomethyl-
HP0013 contributes to the fitness of H. pylori to colonize iNOS-proficient mice
To further examine the HP0013-dependent NO detoxification activity in vivo, we determined the ability of H. pylori B128 strain and Δhp0013 derived mutant to colonize the stomach of C57BL/6J mice. First, an equal number of mice were orogastrically inoculated with the wild-type and mutant strains. The levels of gastric colonization were evaluated at the peak of infection (15 days) that is characterized mainly by the infiltration of innate immune cells, and at a later time of infection (45 days), when H. pylori's numbers have been drastically reduced by the innate and adaptive immune response (but not eradicated) and an equilibrium between H. pylori and the host has been reached. At these times, mice were sacrificed, and their stomachs were removed to determine the bacterial load. After 15 days, the mean colonization levels of B128 Δhp0013 were comparable to the wild-type B128, but after 45 days, while wild-type H. pylori B128 colonization had reached a steady state, the mutant had been eliminated in seven out of eight mice (Fig. 6A). Hence, the mutant strain exhibited a lower capacity to sustain a chronic infection of mouse stomachs.
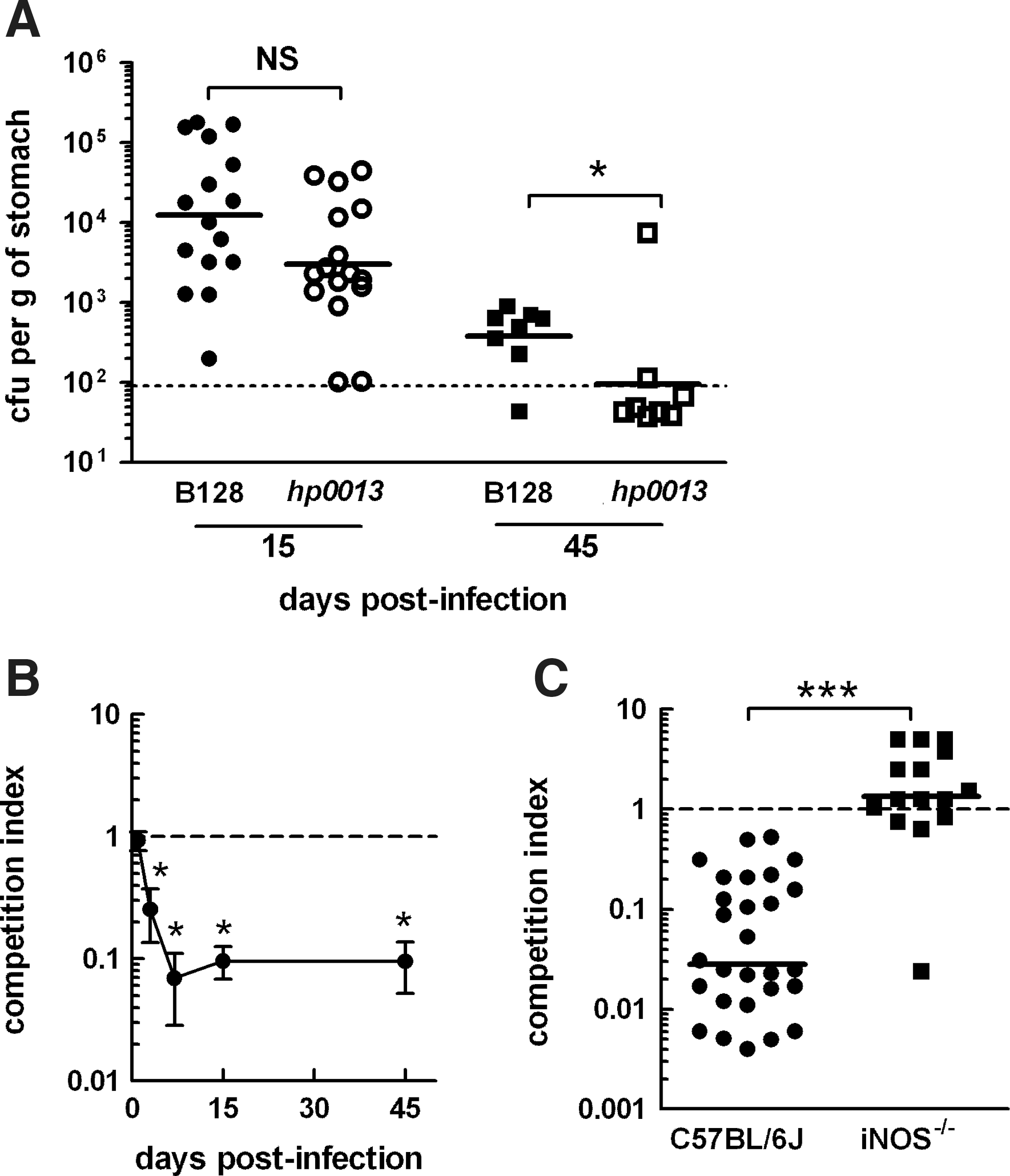
In a second set of experiments, we assessed the colonization efficiency of the H. pylori B128 Δhp0013 mutant during competition with the wild-type strain for the same niche. We inoculated mice with equal proportions of both strains and performed a kinetic analysis of the levels of each strain after 1–45 days. For each mouse, the competition index of the hp0013 mutant versus the wild-type was calculated. The results showed a very clear and rapid displacement of the hp0013 mutant (Fig. 6B), as the competition index started to drastically decline immediately after the first day of infection, and the strain was almost completely eliminated after 7 days. In fact, after 15 days of co-infection, the competition index values determined for 31 C57BL/6J mice were all below 1, with a geometric mean of 0.028, demonstrating that the inactivation of hp0013 renders H. pylori less proficient to colonize the stomach of mice (Fig. 6C). In contrast, when co-infection was performed in iNOS-deficient C57BL/6J mice that lack the capacity to produce NO as an antimicrobial defense, both H. pylori strains showed similar levels of colonization. The geometric mean value of the competition index was determined to be 1.2 (Fig. 6C), which reflects an equal fitness of both H. pylori strains to colonize when no NO is produced by the host immune response. This shows that HP0013 contributes to H. pylori's virulence through the role in NO metabolization.
H. pylori HP0013 protein is widely spread in the microbial world
The NCBI protein database BLAST search of H. pylori 26695 HP0013 retrieved, using as selection criterion an E-value below 10−10 and a span of the query sequence above 60%, ∼200 proteins with sequence identity and similarity ranging from 16% to 99% and 26% to 99%, respectively. This analysis revealed that HP0013 is encoded in all 21, so far available, H. pylori completed genomes. Moreover, the occurrence of HP0013 homologs is restricted to neither the Campylobacterales family nor the ɛ-Proteobacteria class, as they exist in more than 120 distinct species widely distributed through eight bacterial phyla (with a higher predominance in Proteobacteria, Fusobacteria, Firmicutes, and Aquificae) (Fig. 7A). A unifying characteristic of these organisms is their low affinity for oxygen, as they are mainly anaerobes or microaerobes. Around 40%, spread over distant phylogenies, colonize as commensal or pathogenic flora the oro-gastrointestinal tract of humans or animals. These include members of Helicobacter, Campylobacter, Clostridium, Fusobacterium, Brachyspira, and Desulfovibrio genus, as well as of Francisella philomiragia and Wolinella succinogenes.

To the best of our knowledge, no HP0013-like protein has been characterized so far, and the majority of these proteins are annotated as hypothetical or putative argininosuccinate synthetase, as TrmU-like tRNA methyltransferases or ThiI-like proteins involved in thiamine biosynthesis. The annotations are based on sequence similarity confined to the N-terminal region carrying an adenine nucleotide binding motif and a potential Rossman fold motif. However, it was reported that the annotation of ThiI-like proteins is, in most cases, incorrect, as it is based on the presence of these domains, which were shown not to be required for the function of ThiI proteins (2).
The alignment of the amino acid sequences of all retrieved HP0013 homologs revealed a stronger conservation within the first 250 amino acid residues (referring to H. pylori 26695 HP0013), and several highly conserved sequence regions were identified (Fig. 7C and Supplementary Fig. S4).
The first motif (LxSGGLD(S/A)) shares similarity to the P-loop adenosine nucleotide binding motif, which interacts with the phosphate group (36). According to the HP0013 predicted secondary structure, this motif is within a Rossman fold structural motif (Fig. 7C), spanning between the first β-sheet and α-helix structures, a configuration that is typical of NAD(P)/FAD binding motifs (24). Given that the purified recombinant HP0013 was not associated to an FAD group, we propose that this region represents the binding site for a NADPH molecule, which acts as the electron donor for HP0013's catalytic activity.
In the second highly conserved sequence segment (Px2GxGx3CxDC(H/K/R)), in many organisms including all Helicobacter species, the second cysteine residue is followed by a histidine residue. The CXXCH motif is typical of c-type heme containing proteins, which are periplasmic localized (26); however, the predicted cytoplasmic nature of HP0013 suggests that it does not bind a c-type heme.
A previous in silico study aimed at identifying potential disulfide reductases encoded in Campylobacterales genomes reported that the HP0013 protein of H. pylori contains an alternative motif (CXXT) characteristic of the disulfide reductases (23). Our current analysis revealed that this motif is located within one of the conserved regions (Fig. 7C), and that the cysteine residue (numbered 221 in HP0013) is strictly conserved while the threonine residue is highly conserved. This motif is present in the primary sequences of a wide range of diversely functional proteins including those that perform reduction of disulfide bonds and peroxynitrite (23, 34). However, cells of H. pylori 26695 and of the hp0013 mutant showed significant differences neither in the levels of oxidized glutathione (GSSG) reduction activity (144.1±8.9 and 157.9±19.6 mU/mg of protein, respectively [n=3]) nor in the rates of peroxynitrite reduction, as judged by the degree of inhibition of peroxynitrite-mediated oxidation of dihydrorhodamine (respectively, 21.1%±0.5% and 23.8%±1.9% [n=3]).
As previously mentioned, H. pylori lacks any of the known NO detoxifying systems, namely HMP and FDP that, in general, are proposed to act as NO detoxifiers under aerobic and anaerobic conditions, respectively (1, 35). Analysis of the organisms which contain HP0013 homologs showed that ∼20% also lack both HMP and FDP systems (Fig. 7B). The co-occurrence of HP0013-like proteins and FDPs exits in ∼70% of the microbes, while the co-presence with HMPs is lower (15%). Hence, one may speculate a possible association of HP0013 to FDPs and that, similar to FDPs, HP0013 acts predominantly as oxygen-independent NO reductase in organisms growing under low oxygen tension.
Discussion
In this work, we show that H. pylori cells metabolize NO at rates that are within the range of values which were previously reported (32). However, while in this earlier study, NO was assumed to be chemically scavenged, we now reveal that NO consumption is indeed associated to an NADPH-dependent enzymatic activity present in H. pylori cells.
When compared with H. pylori 26695, H. pylori B128 had higher NO consumption rates and, in agreement, increased resistance toward NO. This seems consistent with a different tolerance of several H. pylori strains to nitrosative stress, as H. pylori 26695 and B128 exhibited higher resistance to peroxynitrite when compared with the NCTC 11637 and ATCC 43504 strains (27, 37). GSNO did not induce any morphological changes in H. pylori 26695 and B128 (data not shown), a result that contrasts with previous studies with H. pylori SD14 (9). Although at this stage it is not possible to exclude that these differences are due to the variation of the growth conditions utilized, they may result from the high genetic variability associated to H. pylori (10).
Independently of the H. pylori background, all hp0013 mutant strains had increased susceptibility to NO, and the NO reduction activity of H. pylori cells was found to be significantly lowered on deletion of the hp0013 gene. Moreover, the purified HP0013 protein showed in vitro activity as NADPH:NO oxidoreductase, and its expression in trans complemented an E. coli strain lacking the FDP-type NO reductase. We further verified that the purified HP0013 protein specifically used NADPH and not NADH as a reducing agent for its catalytic activity. Accordingly, H. pylori cells displayed no consumption of NADH for NO reduction. The preferential usage of NADPH over NADH by H. pylori cells is in line with an earlier work which suggested that H. pylori lacks NADH generating enzymes (12).
HP0013 seems to be dedicated to NO removal, as the deletion of the gene did not influence the peroxynitrite and S-nitrosothiol reduction activities of H. pylori cells. As mentioned in the introduction, H. pylori has been reported to utilize several strategies to survive the damage inflicted by nitrosative stress. However, HP0013 is the first NO detoxifying enzyme, as no genes encoding canonical bacterial NO detoxifiers are found in all the so far available H. pylori genomes.
We further addressed the relevance of HP0013-mediated NO protection in the evasion of host immune response. In vivo studies revealed that deletion of hp0013 in H. pylori significantly reduced the survival of the strains on incubation with macrophages, a phenotype that was shown to be associated to the NO production by macrophages.
Murine colonization assays further demonstrated that the H. pylori B128 hp0013 mutant was also less efficient at sustaining a chronic colonization of mice stomachs. Moreover, in competition assays, in which mice were co-infected with the hp0013 mutant and the wild-type H. pylori B128 strains (1:1), the mutant was much less infectious than the wild-type mice, being eliminated after 7 days of infection. These results contrast somehow with the data from independent infection assays, where the mutant was still found colonizing mice after 15 days of infection. One possible explanation may come from the fact that H. pylori seems to exploit the inflammatory and immune response to its own benefit. Intensifying inflammation creates damages to the epithelial cell barrier and, therefore, conditions to acquire essential nutrients from the sub-mucosa. However, the damage also leads to the infiltration of immune cells (particularly of neutrophils and macrophages that produce NO). Thus, H. pylori has developed a myriad of mechanisms to cope with the immune response, including NO generation by producing an arginase to scavenge the substrate of the iNOS. Hence, it might be postulated that, during the first stages of the infection, H. pylori B128 hp0013 may still deal with a moderate inflammatory response, as the mutant is not completely devoid of NO reductase activity. However, once the adaptive immune response is engaged recruiting an even higher number of neutrophils and macrophages, the threshold of NO is reached, leading to HP0013 eradication. In the competition assays, while the H. pylori B128 wild-type keeps its NO defenses and can sustain a higher inflammatory response, the HP0013 mutant not only has to deal with NO but also has to compete for the access of nutrients with the fitter wild-type strain. Thus, when both strains are co-infecting, the threshold of NO production required to kill a weakened mutant strain is encountered much earlier. In support of this hypothesis, we could attribute the phenotype of the mutant to its lowered tolerance for NO stress, as during competition for a niche in the stomach of iNOS-deficient mice, H. pylori B128 wild-type and hp0013 were equally proficient in colonizing the mouse stomach. Hence, HP0013 was shown to be an important factor for H. pylori to resist macrophages and infect the mouse stomach, constituting an added advantage against NO-dependent immune defenses.
Given that neither the NO consumption activity of H. pylori cells nor the expression of the hp0013 gene were induced by nitrosative stress, HP0013 seems to be a constitutive NO detoxifying protein that may result from the need of H. pylori to survive in an in vivo niche where it is constantly exposed to NO stress.
Finally, phylogenetic analysis revealed that HP0013 homologs have a widespread distribution in microbes (Fig. 6A). The presence of highly conserved residues and motifs in all sequences analyzed further suggests that HP0013-like proteins constitute a family that share a common function in NO detoxification. Although the current knowledge is insufficient to understand the relevance of these conserved regions, their presence in a protein that is able to detoxify NO suggests an unprecedented link between these motifs and NO metabolism. Therefore, we propose to rename HP0013 to NADPH-dependent NO reductase of Helicobacter pylori (NorH).
In conclusion, this work has disclosed a distinct system devoted to the in vivo protection of H. pylori and other prokaryotes, including several pathogens, against NO associated stress.
Materials and Methods
Reagents, strains, plasmids, and growth conditions
GSNO (31), spermine-NONOate, DPTA-NONOate, peroxynitrite (Cayman Chemical), and saturated NO (Gasin) solution (29) were used as NO donors.
All bacterial strains, plasmids, and oligonucleotides used in this study are listed in Table 1. Inactivation of hp0013 in H. pylori 26695 and B128 was done by gene replacement with the nonpolar aphA-3 kanamycin cassette as described in Supplementary Materials and Methods.
FDP, flavodiiron proteins; NO, nitric oxide.
H. pylori was routinely cultivated microaerobically, at 37°C, on blood agar (BA) plates and in brain heart infusion (BHI) liquid medium with 10% (v/v) decomplemented fetal calf serum (Gibco-Invitrogen), supplemented with an antibiotic-antifungic mix (7) and when required with kanamycin (20 μg/ml), chloramphenicol (5 μg/ml). Liquid H. pylori cultures, inoculated from 24 h plates at OD600 ∼0.05, were incubated under microaerobic conditions.
E. coli strains were grown on Luria Bertani medium (LB) and, when required with 30 μg/ml chloramphenicol, 100 μg/ml ampicillin and 30 μg/ml kanamycin.
The susceptibility of H. pylori 26695 and derived mutants to GSNO was analyzed by disk diffusion assays using cell suspensions prepared in BHI (OD600∼2), and spread on HP selective plates (BioGerm Laboratórios), which were incubated at 37°C for 48 h. The disks received 15 μl of GSNO 10 mM. Each strain was analyzed in two independent experiments, performed with duplicate plates.
For tests with NO donors, liquid cultures were treated, at OD600 of 0.05–0.1, with 200 μM GSNO and peroxynitrite, and at OD600∼0.5 with 150 μM DPTA-NONOate. The number of viable cells was monitored at intervals of ∼4 h.
E. coli LMS2710 cells (22) were transformed with pET24b, pME2337, and pET24b-HP0013, obtained by NdeI/EcoRI cloning of the hp0013 gene that was amplified with hp13NdeI and hp13EcoRI oligonucleotides. Cells were grown anaerobically in minimal salts medium, and at an OD600∼0.3, 30 μM spermine-NONOate was added and colony forming unit (cfu) was analyzed after 1, 2, and 4 h.
For complementation in H. pylori, the hp0013 gene was extracted NdeI/SacI from pET24b-HP0013 and cloned into pILL2157 and pILL2150, rendering pILL2157-HP0013 and pILL2150-HP0013. Each vector was introduced by tri-parental conjugation into H. pylori B128 hp0013, using E. coli GC7(pRK2013) as mobilizer (19). H. pylori strains were grown and exposed to NO donors as just described, but isopropyl-β-D-thiogalactoside (IPTG) 1 mM was added to the media to induce protein expression.
Production of recombinant HP0013 protein: enzymatic assays in cells and protein samples—phylogenetic analysis
Gene hp0013 was polymerase chain reaction amplified with primers hp13NdeI and hp13XhoI, cloned NdeI/XhoI into pET23a (Novagen) to generate a protein with a C-terminal poly(His) tail fusion, and introduced in E. coli BL21(DE3)Gold. Cells were grown in LB plus 0.4 mM FeSO4. At OD600∼1, IPTG (1 mM) was added, and cultures were grown for 6 h, at 20°C. After cell disruption and centrifugation, the soluble fraction was loaded into a Ni2+-loaded Chelating Sepharose Fast Flow (GE Healthcare) equilibrated with Tris-HCl 20 mM pH 7.5 and 500 mM NaCl, and the protein was eluted with 500 mM imidazole, as confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel.
H. pylori cell lysates from at least three independent cultures were prepared by incubation for 15 min with 0.1 mg/ml lysozyme and 0.02% sodium deoxycholate and used in enzymatic assays. The GSNO reductase activity was determined in cell lysates of H. pylori wt and Δhp0013 mutant after the combined consumption of NADPH and GSNO at 340 nm (ɛ340 [NADPH+GSNO]=7.04 mM −1 cm−1), considering a reaction stoichiometry of one mol of NADPH oxidized per 1 mol of GSNO reduced. Reactions contained 20 mM Tris-HCl pH 7.5, 0.2 mM NADPH, and cell lysates, and were initiated with 0.4 mM GSNO. The NADPH consumption of cell lysates (measured in blank reactions that did not receive GSNO) was subtracted.
NO reduction assays were performed anaerobically, in phosphate-buffered saline with 20 mM glucose, catalase 130 U/ml and glucose oxidase 17 U/ml (Sigma), 0.2 mM NADPH, and 4–6 μM NO and were monitored both amperometrically and spectrophotometrically. The NO consumption was monitored amperometrically using an NO electrode (ISO-NOP) connected to an APOLLO-4000 Free Radical Analyzer (WPI). The NADPH (/NADH) coupled oxidation was monitored spectrophotometrically at 340 nm (ɛ(NAD(P)H)=6.22 mM −1 cm−1), subtracting the NAD(P)H consumption of each lysate in the absence of NO. The specific activity of HP0013 was assayed using 0.5 nmol of purified protein in each reaction. Two batches of purified protein were analyzed in triplicate.
Oxidized glutathione (GSSG) and peroxynitrite reductase activities are described in Supplementary Materials and Methods. Activities (25°C) are defined as unit (μmol substrate consumed/min) per milligram of total protein.
The search for homologs of H. pylori 26695 HP0013 was performed with protein-protein BLAST algorithm at NCBI-BLAST against the nonredundant protein sequences database. The alignment of the retrieved sequences and generation of the unrooted dendrogram (see Supplementary Materials and Methods) were performed using Clustal X 2 (28). Prediction of the secondary structure of HP0013 was done with PSIPRED v3.0 (6), and signal peptide prediction with SignalP 3.0 Server (3).
Macrophage and mouse colonization experiments
For the macrophage killing assays, murine macrophages RAW264.7 (ATCC Tib71) were seeded (5×105 cells per well), in 24-well plates and after 24 h, the medium was changed to Dulbecco's modified Eagle's medium (DMEM) supplemented with 1 mM sodium pyruvate and 0.4 mM
Five-week-old female wild-type C57BL/6J mice (Charles River, France) and iNOS-deficient C57BL/6J(iNOS−/−) mice (Jackson Laboratories) were infected with wild-type H. pylori B128, B128 Δhp0013, and co-infected with a 1:1 mixture of both strains by oral route with feeding needles (2×108 bacteria per mouse). After the indicated time, mice were sacrificed, their stomachs removed, homogenized, and plated to determine the bacterial load as described in Supplementary Materials and Methods. Stomach homogenates from co-infected mice were plated in duplicate, one plate to determine the total bacterial load (wt+mutant), and another with 20 μg/ml kanamycin to measure the survival of the hp0013 mutant.
Statistical analyses
Statistical analyses were performed with GraphPad Prism 5 (GraphPad Software). According to the data, statistical comparisons were assessed using one-way analysis of variance, followed by a Bonferroni multiple comparison test (for more than two conditions), or the Mann–Whitney t test (murine colonization data), with the significance threshold at p<0.05 (95% confidence level).
Footnotes
Acknowledgments
This work was funded by Fundação para a Ciência e Tecnologia (FCT) through projects PTDC/SAU-MII/098086/2008 and PEst-OE/EQB/LA0004/2011. Marta C. Justino is a recipient of the FCT fellowship SFRH/BPD/43172/2008.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.