
Abstract
Reactive Oxygen Species, Oxidative Stress, and Their Biological Effects

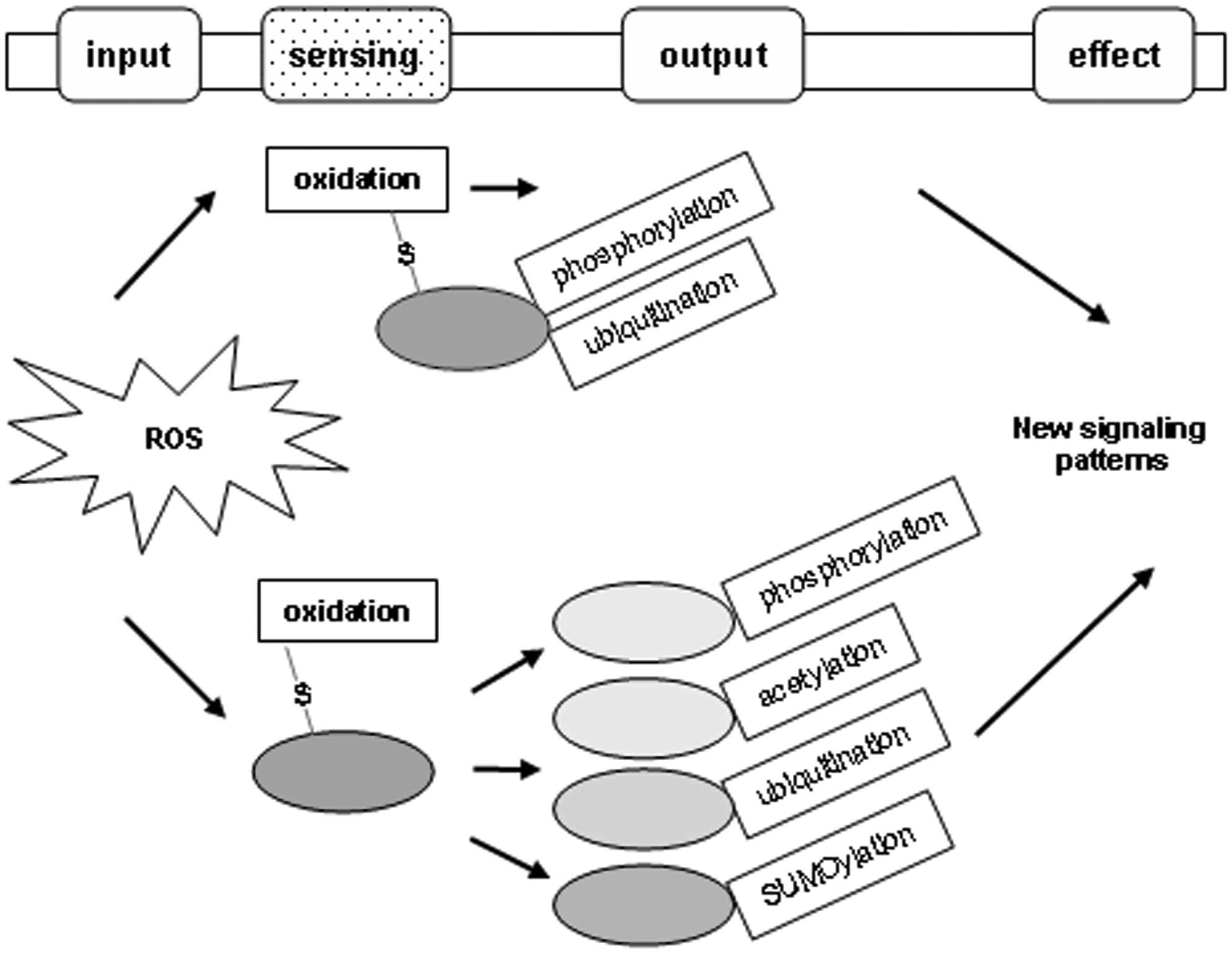
When the basal (low) level of ROS is increased to a modest and nonfatal level, cells can form a new redox balance resulting in a certain type of adaptation, for instance, an adjusted metabolic rate or pathway. When cells experience a high level of ROS, cellular proteins and other macromolecules could be inactivated or even damaged, thus initiating degenerative effects or fatal damage. Under all these circumstances, the functions of cellular proteins are inevitably modulated by changes in the intracellular redox potential; that is, sensing redox changes, cellular proteins modulate themselves in various ways to generate novel signaling patterns in response to mild and severe oxidative stress (Fig. 2).
Redox sensing
The term “redox sensing” has not been clearly defined to date. During the past decade, researchers frequently used “redox sensor” for proteins and small molecules or ions as sensors of cellular redox states. These molecules can sensitively react with ROS and, in turn, regulate protein activity, cell signaling, gene transcription, and various cellular events in response to ROS (28, 41, 44). The molecules, therefore, convert redox changes to protein activity and cellular responses, earning them the label of redox sensors for cells. In this context, redox sensing may mean the way or process in which the redox-sensitive molecules link redox changes to cellular responses.
Since the sensors can sense redox changes between reductive and oxidative states, they should have a molecular basis of reversible redox reaction. Cysteine, methionine, and selenocysteine are the only amino acids in proteins that undergo reversible oxidation/reduction under biological conditions, but only Cys and Met are present in most proteins, and Cys is most abundant (16). Once an imbalance of redox state occurs, the cysteine residues of proteins will sense the change and transmit it as a signal to the whole molecule and, further to other molecules in the protein complex or signaling pathway. These cysteine residues of proteins serve as the most essential sensors within the redox-sensitive molecules, and the process can be called redox sensing at a molecular level. Typically, when activation/inactivation of redox-regulated proteins is coupled to oxidation/reduction, then cysteines form the central building block of thiol-based redox switches. A number of such switches have been intensively studied, largely in oxidoreductases, kinases, proteases, and transcription factors, with their enzymatic or transcriptional activities simply coupled to oxidation/reduction of thiols (8). However, if it is true that “thiol-based redox switches” usually confer a simple output, for example, two extremes of an effect, activation/inactivation, to enzymes and transcription factors, then “thiol-based redox sensor” should result in much more diverse and complex effects to a wider spectrum of proteins, including cytoskeleton proteins, membrane or intracellular receptors, membrane channels or transporters, and others (Table 1). Switches can, thus, be viewed as a specific type of sensors.
Oxoform: A, inter/intramolecular disulfide (PrSSPr); B, sulfenic acid (SOH); C, sulfenamide; D, sulfinic acid (SO2H), sulfonic acids (SO3H); ND, not determined.
PTMs, post-translational modifications; TF, transcription factors; Arc, anoxic redox control; ASK1, apoptosis signal-regulating kinase 1; Prx, peroxidase; FoxO, forkhead box class O; PTPs, protein-tyrosine phosphatases; IRP2, iron regulatory protein 2; Trx, thioredoxin; Cys, cysteine; SUMO, small ubiquitin-related modifier; NF-κB, nuclear factor kappa B.
A recent review, formally addressing “redox sensing,” written by Jones, defines the term as “global biological redox control mechanisms.” The author intended to distinguish “redox sensing” from “redox signaling,” emphasizing that “redox sensing” provides a means to globally affect the rates and activities of the redox-signaling systems (16).
We prefer to emphasize the meaning of “sensing redox changes by proteins using their sensory thiols,” because this seems to have more naturally evolved from the concept of “redox sensor.” The process of sensing ought to consist of steps or components to sense the redox changes and to transduce them to the effectors. In this paradigm, the forms of thiol oxidation represent the way sensory thiols sense redox state, and the physicochemical outcomes, that is, conformational changes and diverse chemical modifications of the same or related proteins, function as the transduced outputs. The effectors, hence, are these same or related proteins; the molecular effects are the changes in their abundance, half life, localization, activity, and interaction with other macromolecules.
The Forms of Thiol Oxidation
ROS regulate mammalian proteins through oxidative modifications; this is how proteins sense redox state. Thiol groups of redox-sensitive cysteines are highly susceptible to oxidation by ROS and undergo diverse oxidative modifications, including the formation of sulfenic (SOH), sulfinic (SO2H), and sulfonic (SO3H) acids, disulfide bonds (PrSSPr), and nitrosothiols (SNO) (3). The initial reaction of a cysteine thiolate with ROS yields SOH, which can lead to formation of additional post-translational modifications (PTMs). Sulfenic acids are often considered metastable intermediates prone to further reactions to form stable modifications, such as disulfides with a vicinal cysteine thiol or thiols of other proteins or S-glutathionylation with the small redox-buffering component GSH (7, 34). Both disulfide products can be reduced back to the thiol form by cellular reductive systems (2).
The Physicochemical Outcomes of Redox Sensing
ROS-induced oxidation of cysteine residues of proteins usually triggers protein structural or conformational changes and/or induces other types of modifications to affect signaling, thus enabling cells to obtain a new redox balance adaptive to various stresses.
Conformation
Intra- and intermolecular disulfide bonds affect the global conformational rigidity of a protein. Disulfide bridges have traditionally been thought of as inert covalent linkages between cysteine residues that are formed during protein folding. However, it is now thought that there are two types of disulfide bonds: a structural type and a redox-sensitive regulatory type, the latter of which can directly regulate protein conformation and thereby affect functional consequences (4, 26). For example, depending on the redox state, the serpin plasminogen activator inhibitor type 2 (PAI-2) can exist in either a stable monomeric form or as a loop-sheet polymer, which has an open β-sheet A and can be stabilized by a disulfide bond between Cys-79 and Cys-161. Reduction of this disulfide bond results in closing of the β-sheet A and changing PAI-2 into the stable monomeric form. Here, the stable monomeric and polymerogenic forms of PAI-2 are fully interconvertible, depending on redox state (26).
Zhou et al. recently showed that, in the crystal structure, the angiotensin cleavage site is inaccessibly buried in angiotensinogen; the release of angiotensin requires a change in conformation in angiotensinogen to allow access and complementary binding of rennin (49). The conformational rearrangement that makes this site accessible for proteolysis was revealed in a 4.4Å structure of the complex of human angiotensinogen with rennin. The coordinated changes involved are critically linked by a conserved but labile disulfide bridge that readily undergoes a transition to a more active oxidized form. The oxidative switch of angiotensinogen to its more active sulfhydryl-bridged form promotes hypertension and pre-eclampsia in pregnancy (49).
Overall, due to technical limitations, structural illustrations of redox regulation of mammalian protein conformation have been by no means popular, but there are several successful studies revealing the redox-controlled conformational changes of prokaryotic proteins, such as arsenate reductase (1) and MexR (6).
Other types of PTMs
Modification of redox-sensitive cysteines by ROS can be viewed as one type of protein PTM, that is, oxidative modification. It is usually also an initiating modification, followed by other PTMs, that is, phosphorylation, acetylation, ubiquitination, and SUMOylation among others. In some cases, cysteine oxidation of a protein induces other types of PTMs in the same protein. More commonly, according to the biochemical features of the oxidized proteins and their roles in cell signaling, the initial oxidation may have a global impact on the cellular profile of protein PTMs and downstream effects (Fig. 2 and Table 1).
Ubiquitination and SUMOylation After Oxidation of SENP3
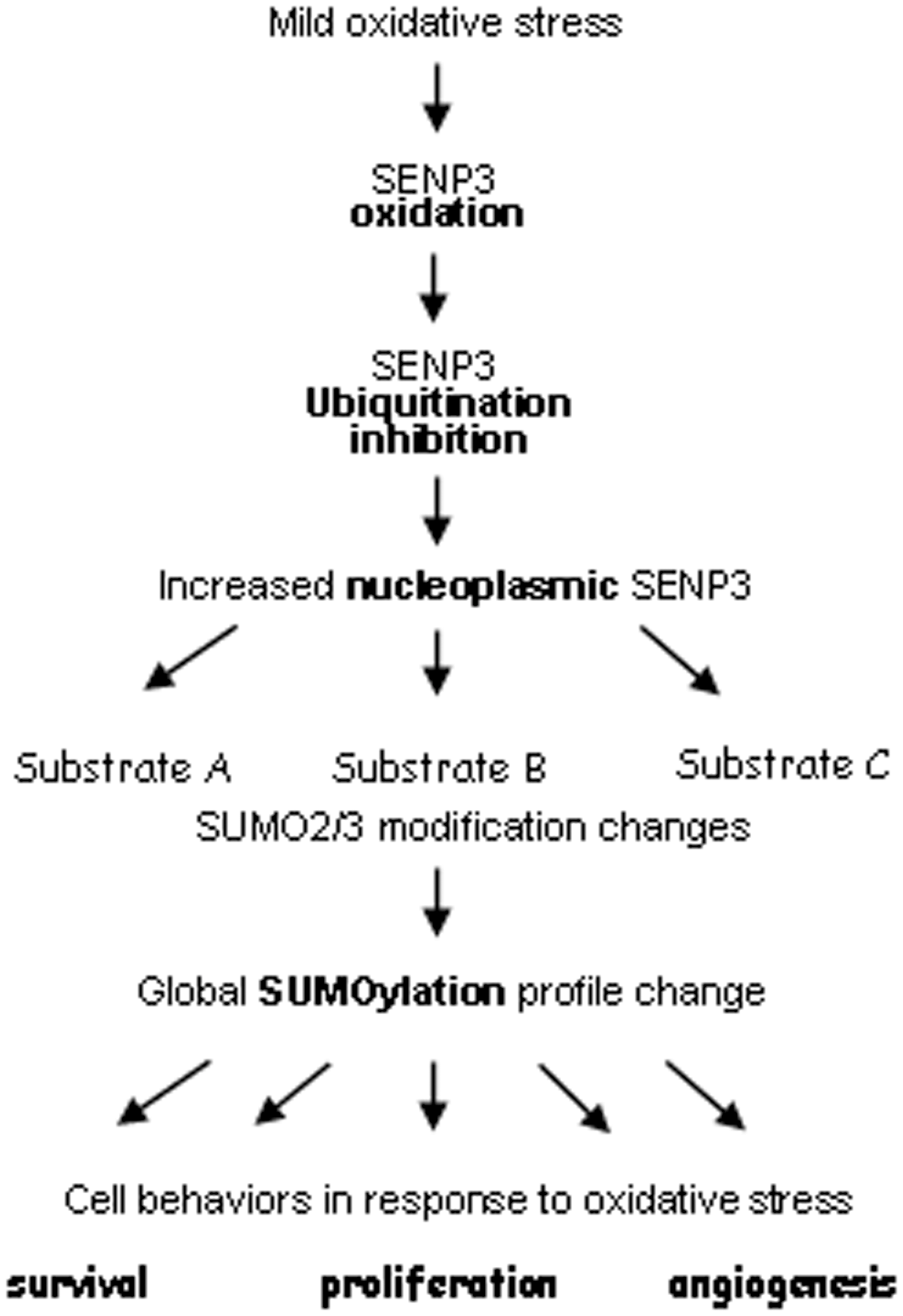
Oxidation of proteins may affect their fate of being degraded by ubiquitin-proteasome pathway. SENP3 is a member of the small ubiquitin-related modifier (SUMO) protease family and deconjugates SUMO2/3 from protein substrates. Our studies indicate that oxidation of SENP3 blocks its ubiquitination and consequent proteasomal degradation, ultimately conferring global regulation of SUMOylation in cells encountering mild oxidative stress.
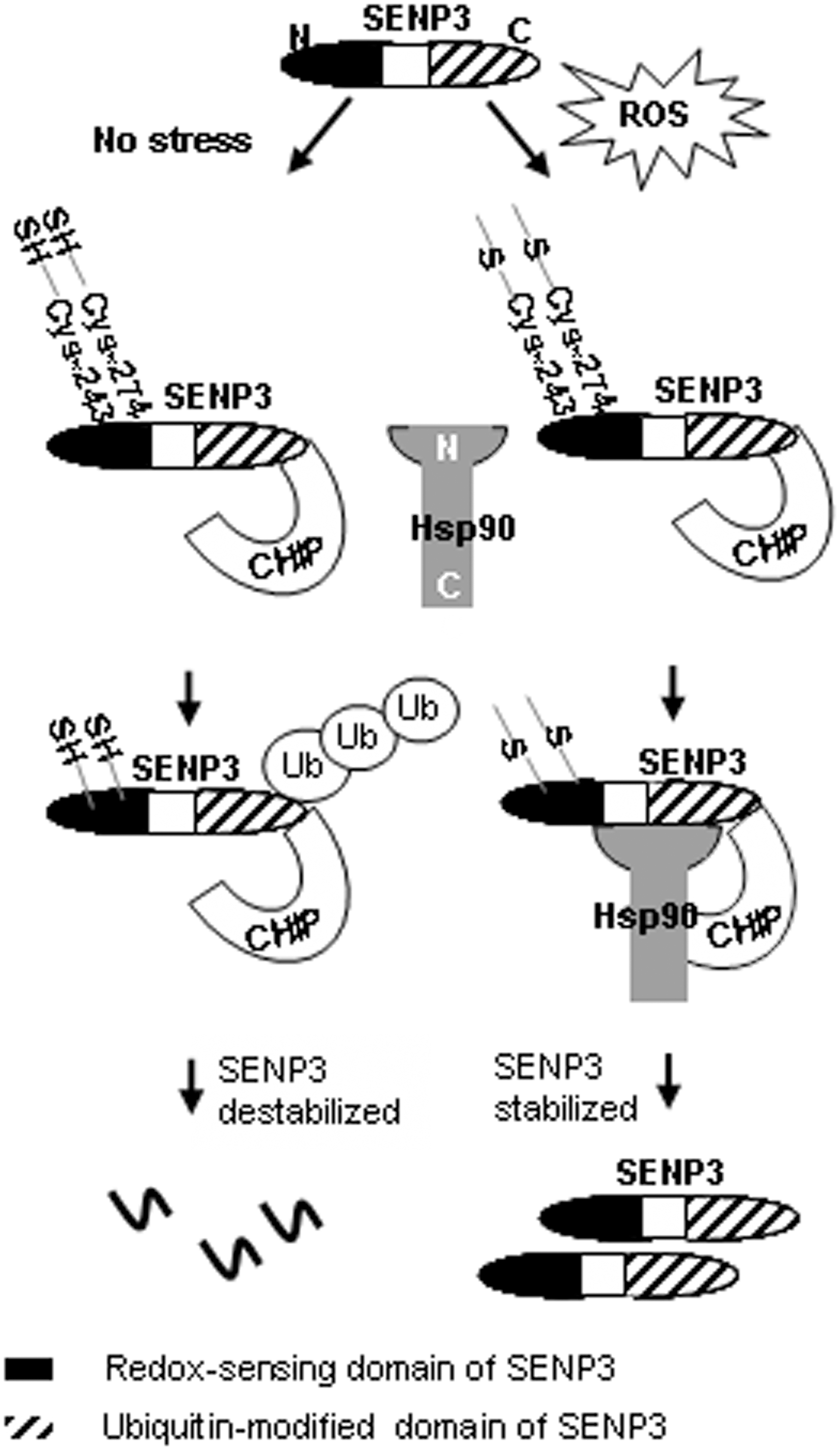
We found that the administration of H2O2 at a series of low doses rapidly increased SENP3 protein levels in various cell lines. This increase in SENP3 protein is due to inhibition of the ubiquitin-proteasome pathway (47). Subsequent study further elucidated that ubiquitination of SENP3 is mediated by Hsc70-interacting protein (CHIP), an ubiquitin E3 ligase, but H2O2 prevents CHIP's activity through recruitment of heat-shock protein 90 (Hsp90). Since CHIP is a cysteine-rich E3 ligase (21), we reasoned that the oxidative effects of H2O2 are likely to disable the catalytic activity of CHIP. However, we eventually confirmed that CHIP is not the target of ROS. Interestingly, an inhibitor of Hsp90, geldanamycin, blocked H2O2-induced SENP3 accumulation. At the same time, we found that Hsp90 transfection, only in combination with H2O2 treatment, decreased SENP3 ubiquitination. This indicates that Hsp90 stabilizes SENP3 protein on H2O2-induced mild oxidative stress by blocking ubiquitin-mediated proteasomal degradation through impairing the E3 ligase function of CHIP (47).
Since cysteines are major targets of oxidative modifications, we aimed to identify the cysteines in SENP3 responsible for sensing ROS and, in turn, regulating SENP3 ubiquitination. The domain responsible for redox sensing was mapped, and all five cysteines in that domain are potentially involved in the ROS regulation of ubiquitination. Strikingly, the C243S and C274S mutants completely abolished SENP3 accumulation in response to H2O2 and Hsp90, indicating that Cys-243 (as well as −274) is required for the stabilization of SENP3. We ultimately confirmed that either Cys-243 or −274 of SENP3 undergoes oxidative modification under mild oxidative stress, and this functions as a signal to recruit Hsp90. The binding of Hsp90 after recognition of oxidized cysteine(s) consequently protects SENP3 from ubiquitination by CHIP (47) (Fig. 3).
SUMOylation has emerged as an indispensable PTM that modulates the functions of a broad spectrum of proteins. Redox regulation of protein SUMOylation has been recently implicated in several investigations (9, 11, 15, 20, 35). A few proteins that have been studied undergo oxidative modification; their functions as regulators of cellular SUMOylation are, thus, modulated under oxidative stress (45, 46), although their own SUMOylation is not changed by ROS.
SUMO2/3 conjugates to substrates in response to cellular oxidative stresses (9, 24). However, we observed that despite the global protein SUMO2/3 conjugation being generally increased by ROS, deconjugation of SUMO2/3 from certain proteins indeed happens under the same conditions, as the global SUMO2/3 conjugation pattern is changed when SENP3 is knocked down by siRNA (11). More impressively, SUMO2/3 conjugation of certain proteins, such as p300 and promyelocytic leukemia (PML), may be dramatically decreased or even eliminated under these conditions, which is mediated by SENP3 (9, 11). SENP3 is rapidly up-regulated by H2O2 at 0.01–100 μM, which is a much lower range than previously reported for SUMO conjugating enzymes (37) or SENP1 (45, 46). It is the most sensitive redox-regulated SUMO protease in the family (9). Our work thereby demonstrates that SENP3 confers a specific SUMOylation profile in cells responsive to mild oxidative stress.
Molecular Effects of Redox Sensing
More often, redox-regulated proteins appear to be central components of nearly every physiological process, thus suggesting that protein redox regulation is the cell's fundamental strategy to fine-tune cellular processes in response to ROS. Oxidative thiol modifications, most often through changes in the conformation and other PTMs of proteins, lead to further changes in their activity, abundance, localization, and interaction with other proteins and with DNA or RNA (Table 1).
We hereby describe three proteins whose abundance, localization, and interaction with DNA are altered after redox sensing.
Redox regulation of stability and distribution of SENP3
The redox-sensitive proteins whose turnover is regulated by the ubiquitin-proteasome system can be stabilized and thereby accumulate under oxidative stress. As just mentioned, our studies showed that after cells had been exposed to H2O2, the amount of endogenous SENP3 rapidly accumulated in a dose-dependent manner via the readjustment of its interaction pattern with CHIP-Hsp90 (47).
Protein localization can be changed by ROS. Our research revealed that SENP3 redistributes between the nucleolus and the nucleoplasm on H2O2 exposure (11). SENP3 is a nucleolar protein (32). Under nonstress conditions, SENP3 is detectable only in nucleoli. However, SENP3 becomes visible in the nucleoplasm when cells are exposed to H2O2, whereas SENP3 in the nucleoli does not decrease at the same time. Strikingly, the proteasome inhibitor MG132 promotes SENP3 accumulation in the nucleoplasm in a pattern similar to that caused by H2O2 (9). This implies that in resting cells, SENP3 is frequently shuttled from the nucleoli to the nucleoplasm where it is degraded. On oxidative stress, this degradation is blocked by inhibition of the ubiquitination of SENP3, which makes it accumulate in the nucleoplasm.
Since SENP3's nucleoplasmic accumulation on H2O2 treatment is coupled with the blockade of ubiquitination after oxidation of specific cysteine(s), we reasoned that the mutants which fail to sense ROS would be unable to localize in the nucleoplasm. As predicted, SENP3 mutant C243/274S is restrained within the nucleoli on H2O2 treatment (unpublished data). Therefore, SENP3's nucleoplasmic distribution on H2O2 treatment can be attributed to blockade of ubiquitination directly after oxidative modification of C243 and/or C274. These findings demonstrate how oxidative modification serves as a switch to control the compartmentalization of protein. It also provides new insight into how a SUMO protease forms its new substrate spectrum along with a change in localization under oxidative stress, as the substrates p300 and PML, which are deSUMOylated under oxidative stress, are nucleoplasmic proteins that are not accessible to SENP3 under normal conditions.
Taken together, SENP3 is an interesting example, in which the ROS-induced oxidation is followed by the inhibition of its own ubiquitination, redistribution, and the consequent changes in global SUMOylation (Fig. 4).
Redox regulation of interaction between caspase-9 and Apaf-1
Intra- or intermolecular disulfide bonds may either modestly or dramatically affect protein–protein interaction, during which ROS play a crucial role (18, 28, 50). Here, we discuss the interaction between caspase-9 and apoptotic protease-activating factor 1 (Apaf-1), which is directly induced by cysteine oxidation on caspase-9, as demonstrated by our work.
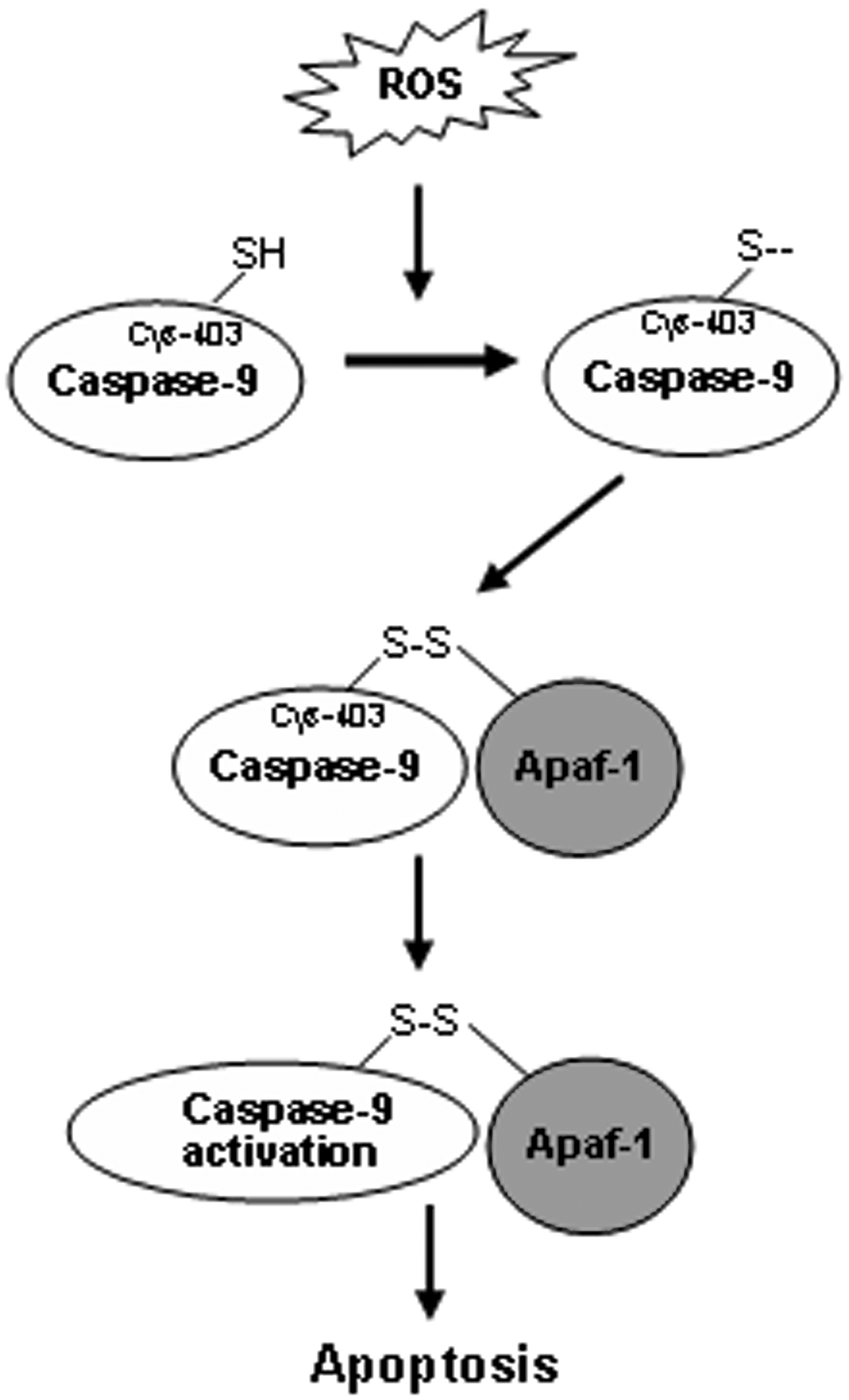
Activation of caspase-9, the initial caspase in the mitochondrial apoptotic cascade, is closely associated with ROS, but it was unclear whether caspase-9 undergoes a direct oxidative modification in its regulation by ROS. All caspases have an active cysteine residue, and the replacement of this cysteine by other amino acids (43) or the oxidation of this cysteine by ROS into another thiol group form will result in their inactivation. Hence, it seems that the enzymatic activity of caspases requires a reducing environment (41).
Caspase-9 activation is initiated by the formation of apoptosomes, a process in which cytochrome c is released from mitochondria into the cytoplasm and binds to Apaf-1 to assemble into an apoptosome. Several plausible proteins that ROS target to promote apoptosis via the mitochondrial pathway have been proposed (19, 27). These findings indicate that ROS may trigger caspase-9 activation by pathways independent of the stimulation of cytochrome c release from mitochondria.
We investigated the direct effects of ROS and the thiol-oxidizing agent diamide on caspase-9 molecules in an attempt to elucidate the molecular mechanisms by which oxidative stress induces caspase-9 activation in the cytosol, regardless of events originating from the mitochondria. The results demonstrate that H2O2 and the thiol-oxidant diamide can oxidatively modify caspase-9 at C403, which facilitates the interaction of caspase-9 and Apaf-1 through the formation of a disulfide bond and subsequent cleavage of caspase-9, thus ultimately promoting caspase-9 activation (50). Our study demonstrates that oxidative modification of caspase-9 contributes to apoptosome formation under oxidative stress. Unexpectedly, Cys-403, which is not located in the caspase recruitment domain, undergoes thiol oxidation and appears to mediate the formation of a disulfide bond upon oxidative modification. Further, our results indicate that this bond is inter-molecular rather than intramolecular. We, therefore, postulate that this disulfide bond might bridge C403 of caspase-9 with a cysteine either in another caspase-9 molecule or in Apaf-1, or perhaps in other proteins that coexist within the same complex, thus promoting the interaction between caspase-9 and Apaf-1, directly or indirectly (50) (Fig. 5).
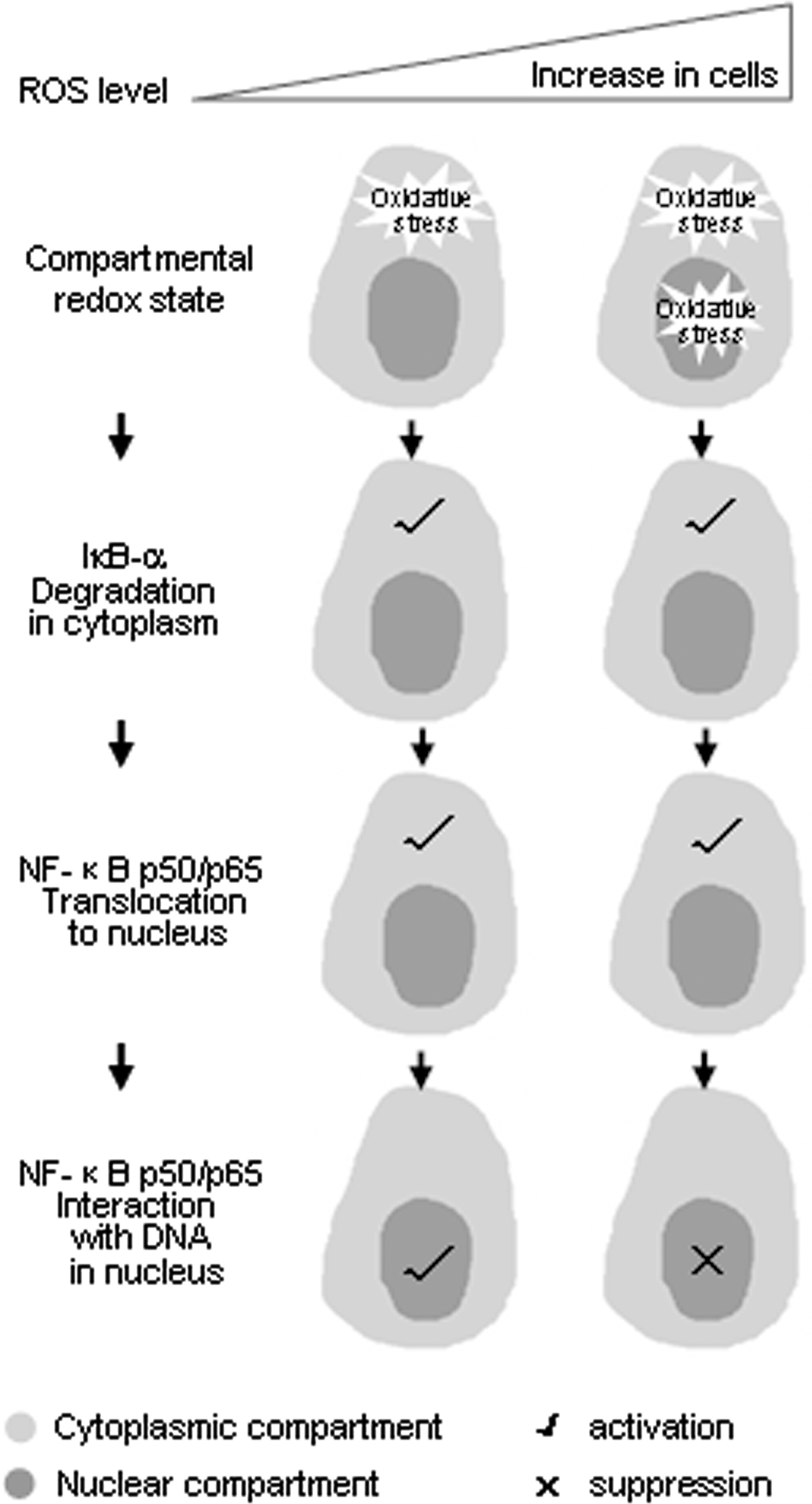
Redox regulation of nuclear factor kappa B–DNA interaction
Protein–DNA interactions play key roles in gene expression, gene replication, and genomic stability (38). They are mediated through the interactions of regulatory proteins with their specific DNA-binding sites at promoters, enhancers, and replication origins in the genome.
Nuclear factor kappa B (NF-κB), composed of two subunits, p50 and RelA (p65), usually exists in the cytoplasm, forming an inactive ternary complex with the inhibitor protein IκB. Following appropriate stimuli, IκB is degraded, NF-κB (p50/RelA) is then released from IκB and translocates into the nucleus, where it binds DNA and activates transcription of target genes. NF-κB has long been known as a redox-sensitive transcription factor. It has been established that ROS often stimulate the NF-κB pathway in the cytoplasm but inhibit NF-κB activity in the nucleus (31).
Studies in the early 1990s demonstrated that oxidation of p50 in its DNA-binding domain prevents its DNA binding. Cys-62 of the p50 subunit of NF-κB is sensitive to oxidation and should be reversed in the nucleus (14). Since p50 is a direct target of oxidative modification, ROS can reduce the transcriptional activity of NF-κB by inhibiting p50 binding to DNA. However, later studies showed that ROS can regulate NF-κB–DNA binding indirectly. The phosphorylation on Ser-276 of RelA, which is thought to depend on ROS, is necessary for the interaction of RelA with CBP/p300 (48).
In the cytosol, ROS have been proposed to both activate and inactivate the I kappa B kinase (IKK) complex (13, 42), to promote activity of NF-κB–inducing kinase (23) or alternative inhibitor of nuclear factor kappa B alpha (IκBα) phosphorylation (39), which may or may not lead to IκBα degradation and p50/RelA translocation (42).
We assume that the differential responsiveness of the components of the NF-κB pathway to ROS or oxidative modification may represent a biphasic redox-sensing mechanism. We have conducted a study to link its compartment-specific redox sensing with its biphasic redox sensing (14).
We investigated the mechanisms by which emodin, a ROS producer, inhibits NF-κB activation in HeLa cells exposed to arsenic trioxide (As2O3). Our results demonstrated that As2O3 exposure induced a mild increase of ROS limited in the cytoplasm and the concurrent slight promotion on tumor necrosis factor-α-caused IκBα degradation, but emodin at high doses or in combination with As2O3, generated excessive ROS, especially in the nucleus, altered subcellular redox equilibrium and, thus, oxidized the redox-sensitive site on NF-κB and prevented its binding to target DNA. This was evidenced by the fact that in the cells expressing C62S NF-κB, its binding and transcriptional activity were no longer inhibited by emodin, in contrast with wild-type NF-κB. Notably, emodin-caused high level of ROS does not block IκBα degradation and does not prevent the consequent p50 translocation, thus indicating that the increased oxidants basically do not suppress, but rather enhance, upstream events in the cytosol. Only in the nucleus is the binding of NF-κB to its target genes apparently blocked, due to oxidative modification of Cys-62 (14).
We, therefore, suggest that when ROS are modestly increased, they activate NF-κB by triggering IκBα degradation and other events in the cytoplasm, which usually lead to prosurvival transcriptional events. However, as ROS become overwhelming, the nuclear environment is shifted from reductive to oxidative, which inhibits activation of NF-κB and other transcriptional factors by preventing them from binding to DNA (Fig. 6). As a consequence, the prosurvival transcriptional activity is abolished. This may explain how the increasing ROS shift survival/death control: they at least shift activation/inactivation control of the NF-κB pathway.
Future Directions
The structural evidence and conformational features of redox sensing by proteins require more studies, particularly in crystallography. Technical optimization remains a challenge for the purposes that oxidized proteins, in particular mammalian proteins, are readily generated, purified, and structurally characterized by X-ray crystallography. Noncrystallographic methods for characterizing redox-dependent conformation changes, especially nuclear magnetic resonance spectroscopy and mass spectrometry, are helpful as well. Moreover, illustration of structural changes during redox sensing by proteins at the fine-tuning level demands the high resolution affordable by state-of-the-art approaches.
Mass spectrometry (MS) is potentially a powerful tool for demonstrating the reversibility and even the kinetics of thiol oxidative modifications. Although detection of various forms of PTMs by MS is becoming popular, skillful application of MS in the detection of protein oxidation, especially metastable forms of oxidation, remains a challenge for most users who aim at revealing mechanistic insights into the redox sensing by proteins.
Organelle-specific ROS capturers and redox-indicating protein probes have been developed during recent years, which allows researchers to precisely visualize spatiotemporal redox changes, even in live cells and animals, thus providing previously unavailable redox information about various cell signaling and biological events (5, 17, 29). However, improved approaches are still required to determine redox states in patients' tissues, and these would also be of value in recognizing oxidative stress-related human diseases, for example, cancer, metabolic syndrome, and neurodegenerative lesions. Moreover, the redox biomarkers for these diseases, if developed, will certainly help in monitoring the clinical progression of the diseases.
Since the increased generation of ROS can occur under both physiological and pathological conditions, opposing cellular responses to increasing ROS are frequently seen, and the underlying mechanisms are still elusive. To understand how proteins mediate bidirectional cellular responses to different levels of ROS, studies on biphasic redox sensing are needed. The clarification of such delicate mechanisms of redox sensing will tremendously enhance our knowledge about why ROS play such complex roles in cells and organisms.
Footnotes
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (91013012, 30971437 to J.Y.), Shanghai Municipal Science and Technology Commission (11ZR1419100 to Y.W.), and Shanghai Municipal Education Commission (J50201, to Yi lab).