
Abstract
The thioredoxin (Trx) system is one of the central antioxidant systems in mammalian cells, maintaining a reducing environment by catalyzing electron flux from nicotinamide adenine dinucleotide phosphate through Trx reductase to Trx, which reduces its target proteins using highly conserved thiol groups. While the importance of protecting cells from the detrimental effects of reactive oxygen species is clear, decades of research in this field revealed that there is a network of redox-sensitive proteins forming redox-dependent signaling pathways that are crucial for fundamental cellular processes, including metabolism, proliferation, differentiation, migration, and apoptosis. Trx participates in signaling pathways interacting with different proteins to control their dynamic regulation of structure and function. In this review, we focus on Trx target proteins that are involved in redox-dependent signaling pathways. Specifically, Trx-dependent reductive enzymes that participate in classical redox reactions and redox-sensitive signaling molecules are discussed in greater detail. The latter are extensively discussed, as ongoing research unveils more and more details about the complex signaling networks of Trx-sensitive signaling molecules such as apoptosis signal-regulating kinase 1, Trx interacting protein, and phosphatase and tensin homolog, thus highlighting the potential direct and indirect impact of their redox-dependent interaction with Trx. Overall, the findings that are described here illustrate the importance and complexity of Trx-dependent, redox-sensitive signaling in the cell. Our increasing understanding of the components and mechanisms of these signaling pathways could lead to the identification of new potential targets for the treatment of diseases, including cancer and diabetes. Antioxid. Redox Signal. 18, 1165–1207.
I. Introduction
A. Redox control and signaling in the cell
ROS are produced by mammalian cells to mediate diverse physiological responses, including cell proliferation, differentiation, and migration. The reductive-oxidative-based reactions that represent the chemical substrates of these signaling pathways are the basis for “redox” signaling which regulates normal as well as maladaptive processes. As the pathways regulating cellular redox biochemistry become better defined, we get a more comprehensive understanding of how cells channel ROS into specific signaling pathways that modulate various cellular outcomes (247).
Redox elements such as redox-sensitive cysteine residues participate in diverse cellular signaling pathways. The organization and coordination of the redox activity of these elements depends on common control nodes or molecular switches such as Trx (132). The Trx system catalyzes electron flux from nicotinamide adenine dinucleotide phosphate (NADPH) through Trx reductase to Trx, which is involved in the redox control of a large number of different signaling pathways through its interaction with a variety of different proteins, some of which are highlighted in this review.
B. Thioredoxin
Trx was first purified and described as being the hydrogen donor for ribonucleotide reductase (RNR) in Escherichia coli in 1964 (161). Sequencing of the bacterial Trx protein revealed the highly conserved prototypical dithiol Cys-Gly-Pro-Cys active site motif that is found in all kingdoms of life from archaea to mammals in this ubiquitous protein (108). Since the 1960s, there have been major advancements in our understanding of Trx biology that are reviewed extensively elsewhere (172). In mammalian cells, there are two isoforms of Trx, the mainly cytosolic Trx1, which can be translocated into the nucleus and secreted out of the cell under certain circumstances, and Trx2, which is the mitochondrial isoform. Unless explicitly stated otherwise, we will refer to Trx as Trx1 in this review. There is also a truncated form of Trx (Trx80) that lacks oxidoreductive properties and is not reduced by Trx reductase (235).
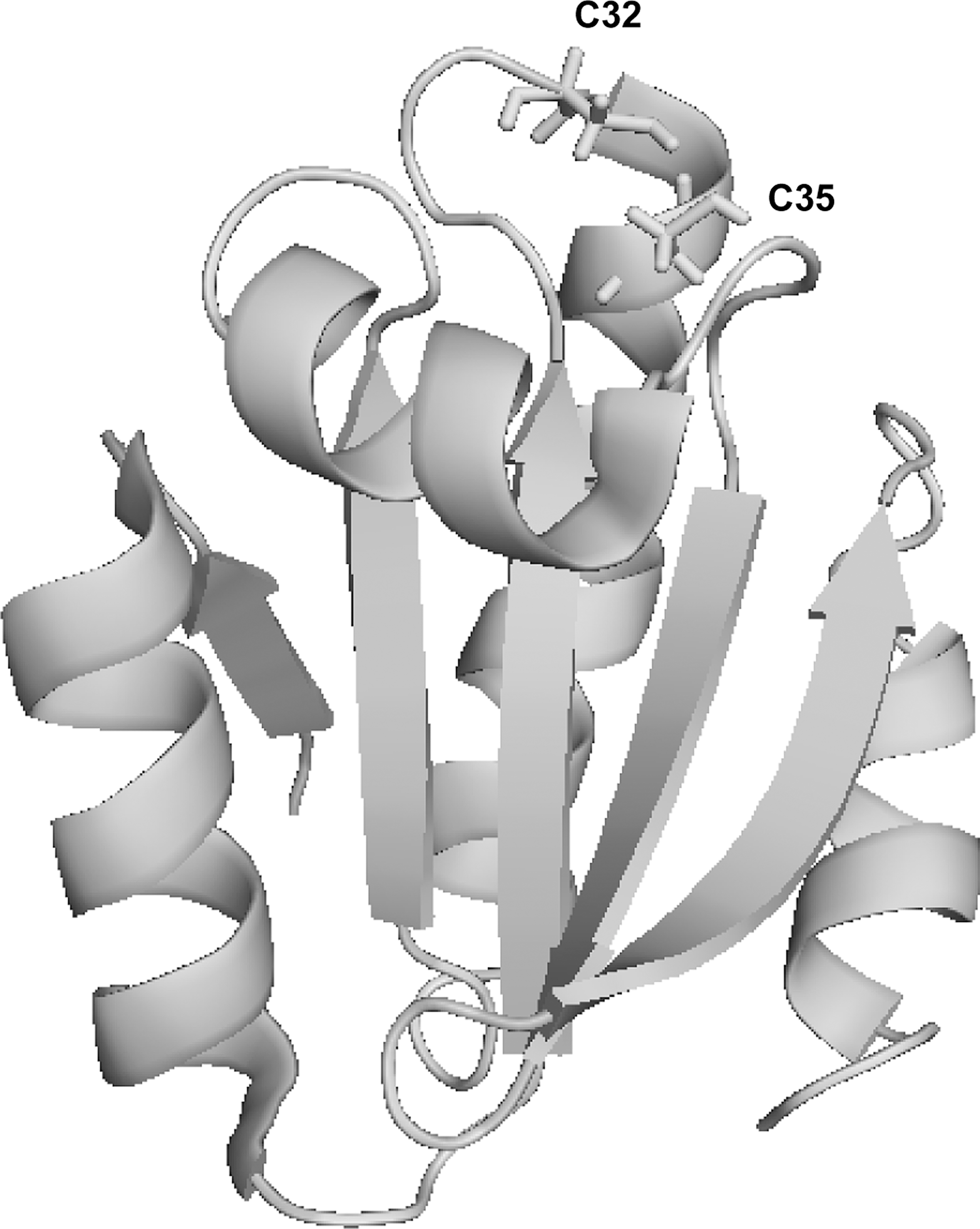
The first description of the three-dimensional structure of bacterial Trx was published in 1975 (111). The crystal structures of many Trxs in both oxidized and reduced states have been resolved (60). There are a number of proteins that share the common Trx motif which has been termed the Trx fold (60, 172). The basic Trx-fold motif consists of four β-beta strands surrounded by three α-helices. Trx itself has an additional α-helix and β-beta strand at the N-terminus (Fig. 1). The main mammalian components of the Trx family of proteins are Trxs, glutaredoxins (Grxs), protein disulfide isomerases, and quiescin-sulfhydryl oxidase, all of which are involved in thiol-disulfide exchange reactions. A thiol-disulfide exchange reaction is a bimolecular nucleophilic substitution reaction that involves the transfer of electrons from Trx to the substrate protein. Trx utilizes its cysteines at position 32 and 35 for this reaction. In the first step of this reaction, the N-terminal cysteine of Trx initiates a nucleophilic attack on the disulfide bond of the substrate protein, resulting in the formation of a mixed disulfide bond between Trx and the substrate protein. The second step is a nucleophilic attack of the C-terminal cysteine of Trx on the intermediate intermolecular disulfide bond, resulting in the formation of a disulfide bond in the oxidized Trx and the breakage of the disulfide bond in the reduced substrate protein (60, 172). Trx is then reduced by Trx reductase, which utilizes NADPH—mainly produced by the pentose phosphate pathway—as an electron donor. (Fig. 2). Other members of the Trx family are glutathione (GSH) transferases, GSH peroxidases, peroxiredoxins (Prxs), chloride intracellular channels, and the copper-ion binding protein Sco1 (234).

In addition to its function as a key regulator in the redox processes associated with oxidative stress, Trx has recently been identified as playing a role in the regulation of nitrosative stress as well (15). Similar to ROS, reactive nitrogen species can generate nitrosative chemistries that are detrimental for certain cellular signaling pathways. However, similar to what we know about the complex and essential redox-dependent regulation of signaling pathways by oxygen and its biochemical derivatives, nitric oxide (NO) has emerged as an essential regulator of numerous cellular functions (10). The morphological correlate of this regulation by NO is the post-translational modification of cysteines through a covalent attachment of NO to their thiol residues, which has been termed “S-nitrosylation.” Similar to other post-translational modifications such as phosphorylation, S-nitrosylation is involved in the regulation of a large number of molecular functions, including enzyme activity, translocation, protein-protein interactions, and protein degradation. The dysregulation of S-nitrosylation is associated with a variety of diseases, including cancer, cardiovascular, and neurodegenerative disorders (10).
Comparable to protein phosphatases, enzymes that catalyze the denitrosylation of proteins are indispensable parts of the S-nitrosylation-regulated pathways. The Trx/Trx reductase system is one of the two enzyme systems that are instrumental for physiological denitrosylation reactions, the other system being the S-nitrosoglutathione reductase system. Similar to thiol disulfide exchange reactions that are catalyzed by Trx, protein denitrosylation by Trx involves the formation of an intermediate mixed disulfide bond between Trx and the substrate protein (14). Alternatively, Trx itself could undergo a transient S-nitrosylation in the process of denitrosylation (290). Either way, denitrosylation of the substrate protein results in the release of nitroxyl (HNO) and the oxidation of Trx, which is reduced and reactivated by Trx reductase.
Through the regulation of redox- and S-nitrosylation-dependent cellular signaling pathways, Trx has an important role in maintaining a physiological environment for the cell, and it interacts with a broad range of different proteins. Since Trx needs to be reduced in order to be able to reduce its substrates, its function depends to a large part on the activity of Trx reductase.
C. Trx reductase
1. Background
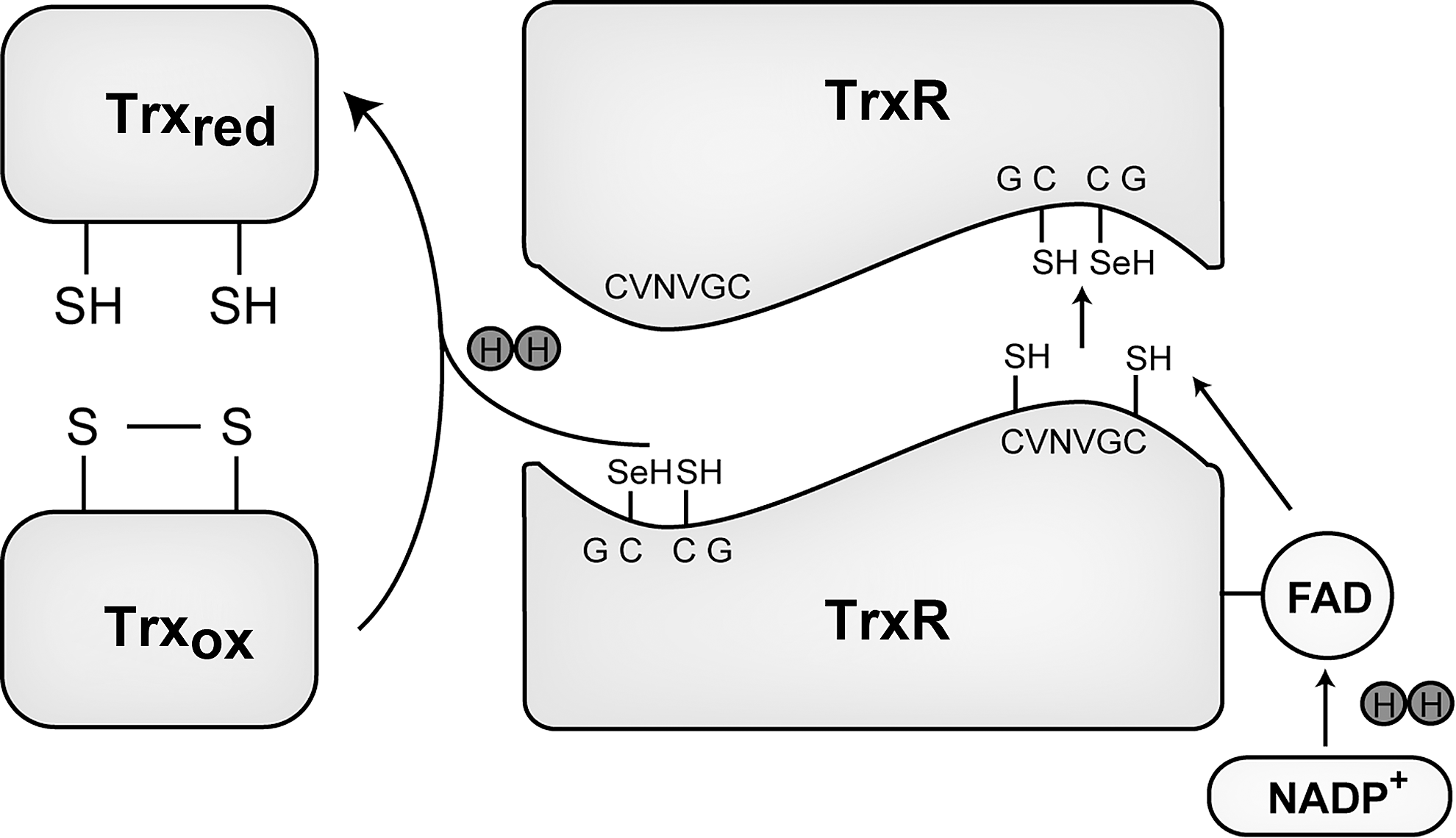
Thioredoxin reductases (TrxRs) are oxidoreductases that are required for the reduction of the active site disulfide in Trx, thus responsible for maintaining the pool of reduced and active Trx. [for review see (13)]. After cloning of human TrxR1 protein, it was found that TrxR is a homodimeric flavoenzyme containing a penultimate C-terminal selenocysteine in its Gly-Cys-SeCys-Gly active site (92, 216). Mammalian TrxR forms antiparallel homodimers with both the subunits necessary for a normal redox reaction during a catalytic cycle (Fig. 3). The first step of the reductive half-reaction of the enzyme involves reduction of the enzyme-bound flavine adenine dinucleotide by NADPH in one subunit. From there, the reducing equivalents are transferred to the Cys-Val-Asn-Val-Gly-Cys active site motif of the same subunit forming a dithiol motif. This dithiol motif is not interacting with the substrate protein; it rather reduces the C-terminal selenenyl sulfide motif of the other subunit of the dimer forming a selenolthiol motif. This reduced selenolthiol motif can, in turn, reduce the substrates of TrxR, including the active site disulfide between positions 32 and 35 of Trx (13). The high reactivity of the active site selenocysteine and its easy accessibility are regarded as the main reasons for the wide substrate specificity, in addition to Trx. Other substrates of TrxR, therefore, include Grx2, protein disulfide isomerase, Trx-like-1, granulysin, and also some nonprotein substrates such as selenite, dehydroascorbate, lipoic acid, ubiquinone, cytochrome C, or the cancer drugs motexafin gadolinium and alloxan (13).
There are three different TrxR isoforms encoded by three separate genes in mammals (13). Human thioredoxin reductase 1 (TXNRD1) is located on chromosome 12q23-q24.1 and contains a core module with 15 exons and several alternative exons in the 5′-region that encode for different splice variants (90). Human TXNRD2 is located on chromosome 22q11.21 and is considered the mitochondrial isoform containing a mitochondrial targeting sequence (192). However, similar to TXNRD1, the TXNRD2 transcript is subject to extensive alternative splicing at the 5′-end, resulting in two other transcripts that do not contain the mitochondrial targeting sequence, thus coding for cytosolic TrxR2 isoforms (293). The human TXNRD3 gene encodes Trx GSH reductase and is located on chromosome 3q21.3. This isoform is predominantly expressed in male germ cells (292).
2. Regulation
While relatively little is known about the transcriptional control of TrxR2 and TrxR3 (13), the TrxR1 promoter was described as having typical characteristics of a housekeeping gene, without a TATA-box with the transcription being driven by Sp1/Sp3 and Oct1 in combination with a nuclear factor (erythroid-derived 2)-like 2 (Nrf2)-regulated antioxidant responsive element (251, 255). The silencing of Nrf2 expression in A549 cells, a lung cancer cell line highly resistant to chemotherapy with very high levels of TrxR1, lowered TrxR1 and increased the sensitivity of these cells to chemotherapy, indicating that TrxR1 and other Nrf2-driven genes contribute to cancer cell resistance to chemotherapy (277). There is also evidence that TrxR1 expression is post-transcriptionally regulated. The 3′-UTR of the human TrxR1 messenger RNA (mRNA) carries adenine/uridine (AU)-rich elements that are thought to mediate rapid mRNA turnover in response to certain cellular signaling events. When the AU-rich elements are removed, TrxR1 mRNA stability is significantly prolonged, suggesting that RNA interference might be involved in the post-transcriptional regulation of TrxR1 expression (89).
TrxR is a selenoprotein, and selenium is required for its expression and activity. Increasing levels of sub-toxic doses of selenium increase TrxR activity, increase TrxR levels, and promote antioxidant defenses (17). Similarly, selenium deficiency in a rat model of selenium depletion significantly decreases TrxR activities in organs such as the liver or kidney (103). In higher doses, selenium acts as a pro-oxidant and is toxic, eventually leading to cell death with reduced TrxR protein and activity levels despite increased transcription (264). In addition to selenium, there are a large number of compounds that increase TrxR expression, including Nrf2 activating factors such as allyl nitrile, acrolein, peroxynitrite, or cadmium. Other inducers of TrxR1 expression include oxidized low-density lipoprotein or estrogen. Of note, TrxR2 expression is significantly down-regulated in skeletal and cardiac muscle of aging rats compared with young ones. These examples illustrate the wide variety of factors that can influence TrxR expression and activity, consistent with its central role in redox signaling (13).
3. Clinical significance
Txnrd1 deletion leads to early embryonic lethality around day 9 with severe growth retardation (25). Txnrd2-knockout (KO) mice die around embryonic day 13 with severe impairment of hematopoiesis, increased apoptosis in liver, and insufficient heart development (61). Both Txnrd1 and Txnrd2 are, therefore, essential for embryonic development, although there are indications for cell- and tissue-type-specific pathways for the different isoforms (13).
Since TrxRs are an integral part of the Trx system, changes in TrxR expression and activity have an immediate impact on Trx activity as well. Thus, the different Trx interacting proteins that are discussed in this review are also indirectly regulated by TrxR. It has been shown that TrxR may be implied in a variety of different human physiological and pathophysiological processes, such as embryonic development, aging, Alzheimer's disease, cancer, hyperoxic lung injury, cataract, skin pigmentation, hemolytic and Fanconi anemia, and even HIV infection (13). The exact role that TrxR plays in these conditions is incompletely defined.
However, since baseline TrxR activity is necessary for cell survival, the potential of TrxR as a drug target in cancer therapy has been extensively investigated. Several electrophilic compounds that interact with the redox-active residues of TrxR and inhibit its activity are currently used in chemotherapeutical regimes to treat cancer, including platinum-containing compounds, arsenicals, nitrosoureas, quinones, and motexafin gadolinium; gold-containing drugs are used in the treatment of rheumatoid arthritis (241, 265). Since treatment with these drugs decrease TrxR activity, they diminish the interaction between Trx and TrxR as well and have an impact on the overall levels of oxidative stress in the cell. While cancer cells are exposed to increased levels of oxidative stress and might benefit from higher TrxR activity, lower-dose selenium could increase baseline TrxR activity and be protective against cancer development as an antioxidant. Therefore, the Selenium and Vitamin E Cancer Prevention Trial (SELECT), a large prospective, randomized, and placebo-controlled trial with more than 35,000 patients, sought to determine whether selenium supplementation conveys protective effects on the development of prostate cancer. In spite of previously published positive epidemiologic and preclinical studies, the SELECT trial showed no effect of selenium supplementation on the prevention of prostate cancer (174). These findings highlight the need for better understanding of the various redox-regulated pathways that are dependent on the Trx-TrxR interaction.
D. Trx target proteins
To maintain a reducing environment, Trx interacts with a large number of different proteins (Fig. 4). Over the past decades of Trx research, numerous redox targets have been identified in different species. With the recent progress in mass spectrometry and other biochemical methodologies, global proteomics studies have become feasible and have been employed to screen for protein-protein interactions. To identify all redox targets in the cell, numerous proteomics studies have been undertaken in different species (194, 325). Recent proteomics studies have investigated the redox as well as the S-nitrosylated target proteomes of Trx (16, 83). An excellent and comprehensive review of the biological and technological aspects of Trx-related proteomics studies including a list of all known mammalian redox and S-nitrosylated targets of Trx is given in Ref. (325).
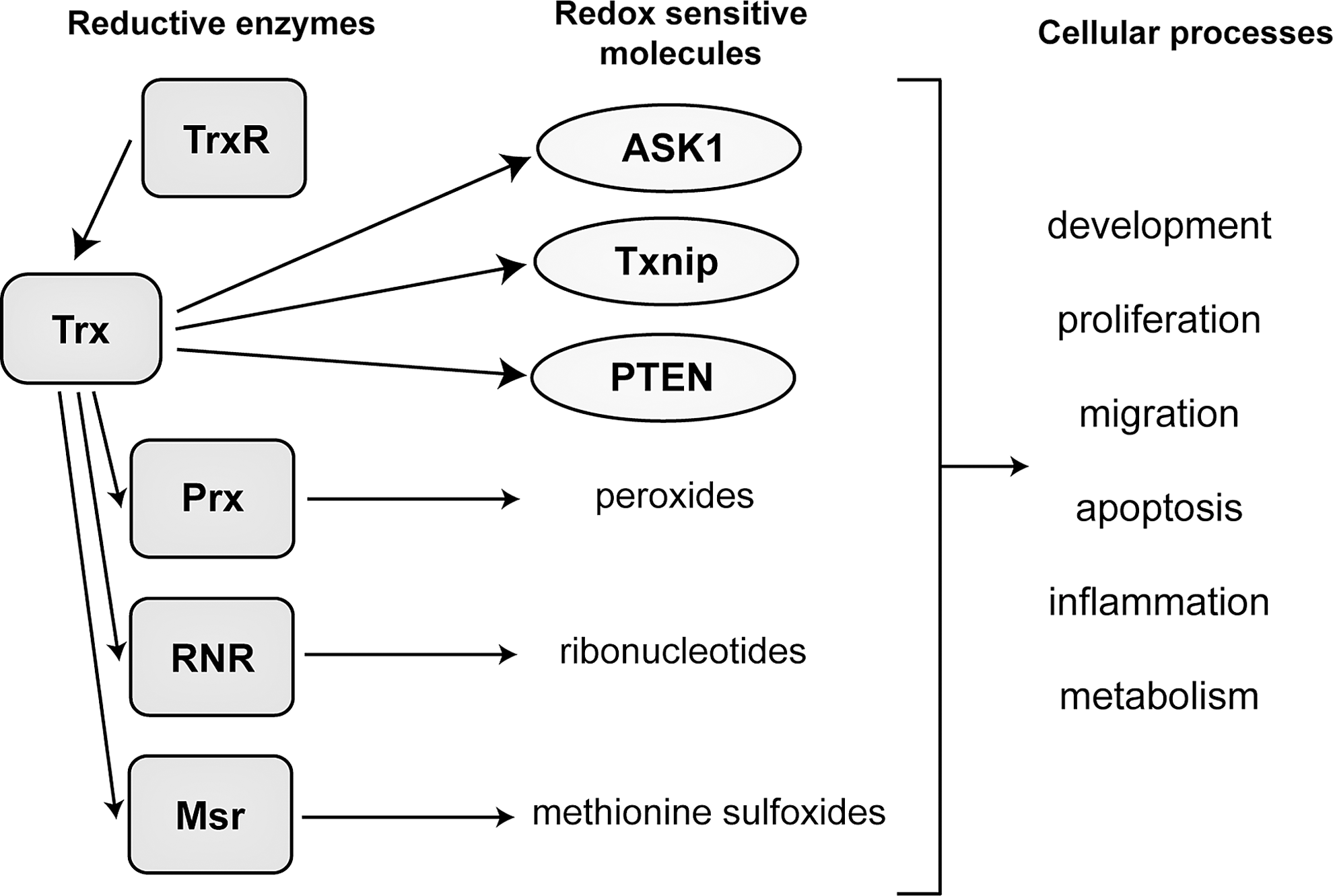
While Trx and its different target proteins are involved in the regulation of many different cellular signaling pathways, the interaction with Trx represents the common thread that links these different proteins to one another. Therefore, there are certain cellular functions that are commonly regulated by this network of Trx and Trx target proteins. Apoptosis is a tightly controlled process that is affected—one way or the other—by all of the proteins that are discussed in this review. The fundamental role of the Trx system, in general, is to provide a reducing environment and to protect the cell from the detrimental effects of oxidative stress that ultimately lead to apoptosis. Any major changes in both Trx activity and activity of reductive enzymes that rely on Trx to regenerate their reductive capabilities will eventually lead to an increased amount of oxidized and nitrosylated proteins with subsequent protein malfunction (121); this is the case for Prx, RNR, and methionine sulfoxide reductase (Msr). In this regard, these reductive enzymes are dependent on Trx. Trx itself is, in turn, dependent on the activity of TrxR to maintain its reducing activity. This leads to the hierarchical nature of the redox chain from TrxR to Trx and other reductive enzymes (Figs. 2 and 4).
However, apart from these general considerations, Trx target proteins are also involved in more specific pathways related to the regulation of apoptosis. Here, apoptosis signal-regulating kinase 1 (ASK1) certainly plays a more central role. A tight control of ASK1 by Trx is a major contributor in regulating apoptosis. Oxidized Trx with an intramolecular disulfide bond between C32 and C35 is inactive and unable to bind to ASK1. Reduced Trx can, however, bind to ASK1 at the N-terminal coiled coil (NCC) domain. On binding, Trx inactivates ASK1 by directing it for ubiquitination and degradation. The inactivation of ASK1 results in diminished c-Jun N-terminal kinase (JNK) and protein 38 (p38) apoptosis signaling cascades, inhibiting apoptosis (119). Interestingly, Prx I is another protein that can bind to ASK1 and inhibit its activation, similar to Trx (147). In contrast, ASK1 activity can be regained in the presence of thioredoxin interacting protein (Txnip). The binding of Txnip inactivates Trx, allowing for the reactivation of ASK1 activity. As a result, the up-regulation of ASK1 signals the cell for apoptosis by activating the JNK and p38 cascades. By inhibiting Trx, Txnip, therefore, affects apoptosis not only through decreased Trx enzyme activity, but also by the disinhibition of ASK1 (134). In addition to the significant contribution of ASK1 in regulating apoptosis, the activity of phosphatase and tensin homolog (PTEN) is also crucial for the proper regulation of programmed cell death. Inactive PTEN is indicated by the presence of an intramolecular disulfide bond between C71 and C124. On reduction of this disulfide bond by either stress or Trx, the catalytic activity of PTEN is regained. Active PTEN is now able to inhibit the Akt signaling pathway. Since phosphorylated Akt directs the cell for growth and proliferation, PTEN stimulates apoptosis by inhibiting the phosphorylation of Akt (286). Identical to the inactivation of Trx by Txnip during ASK1 regulation, the binding of Txnip inactivates Trx, resulting in the down-regulation of PTEN activity. Inactivated PTEN is no longer able to inhibit the phosphorylation and activation of Akt, resulting in activated Akt and thereby promoting cell growth (115). Taken together, apoptosis is achieved by balancing the activities of the upstream signaling molecules, and in the case of the Trx system, many of its pieces are interconnected with each other. Only a combination of all of these signals determines whether the cell will be directed for growth and proliferation or apoptosis. This is a good example for the need to expand our knowledge about the different target proteins of Trx.
For the purpose of this review, we decided to focus on some important target proteins of Trx: Trx-dependent reductive enzymes and Trx-sensitive signaling molecules, with a special focus on the latter. As just mentioned, one of the prototypical functions of Trx as an antioxidant is to function as an electron donor and cofactor for reductive enzymes. These enzymes include Prxs, which are a ubiquitous family of thiol-dependent peroxidases that are responsible in large part for scavenging and reducing peroxides such as hydrogen peroxide (H2O2), RNRs, which are involved in DNA synthesis, and Msrs, which are responsible for the reduction of oxidized sulfur-containing methionines. These reductive enzymes and their interaction with Trx will be discussed in the second part of this review.
Apart from reductive enzymes, Trx also interacts with redox-sensitive signaling molecules. There are a number of transcription factors that are redox regulated and contain redox-sensitive cysteines in their DNA binding domain. These include activator protein 1 (AP-1), NF-κB, protein 21 (p21), protein 53 (p53), hypoxia-inducible transcription factor-1 alpha (HIF-1), the glucocorticoid receptor, the estrogen receptor, PEBP2, EPF, Nrf2, Oct-4, and TFIIIC (172). Trx also interacts with key signaling molecules, including ASK1, Txnip, and PTEN. Although catalyzing redox reactions is the primary purpose of the reductive enzymes that are discussed in the second part of this review, these proteins are essential parts of complex signaling networks, and redox-dependent regulation of these molecules through interaction with Trx is only one of the many regulating factors that are integrated and processed in these networks. To highlight the potential impact that the interaction with Trx could have, even on signaling pathways that are not directly related to the molecular interaction between Trx and the redox-sensitive signaling molecules, the third part of this review will extensively discuss the cellular functions of ASK1, Txnip, and PTEN in a larger context. The focus will be on these proteins, as ongoing research is revealing that they regulate a particularly wide variety of different cellular processes, including metabolism, proliferation, differentiation, migration, and apoptosis.
II. Reductive Enzymes
A. Peroxiredoxins
1. Background
Prxs are a family of ubiquitous peroxidases that reduce peroxides and primarily function as cellular antioxidants [for specific review see (248)]. The first observations of enzymatic activity of Prx were made in Saccharomyces cerevisiae. In 1988, the first yeast Prx protein was purified, and it became apparent that it conveyed the protection of glutamine synthetase against oxidation in the presence of O2, thiol, and Fe3+, suggesting that it had basic antioxidant properties (145). This was further confirmed when it was found that in yeast with the genetic deletion of Prx, growth rate was significantly decreased compared with wild type under aerobic conditions, while there were no such differences under anaerobic conditions. The application of oxidative stress through peroxides led to an even more severe phenotype in the KO strain (144). Interestingly, unlike Trx reductase, Prx does not contain any conventional redox active sites with metals, heme, flavin, or selenocysteine (248). Instead, sequence analysis, and eventually crystallography, revealed two highly conserved cysteine residues at position 47 and 170 in yeast (41). Substituting the N-terminal cysteine with serine (Ser) abolished the antioxidant properties of Prx completely as measured by the preservation of glutamine synthetase activity under oxidative conditions. Further biochemical and structural analyses, including crystallography, showed that Prx forms antiparallel homodimers through an intermolecular disulfide bond between the N-terminal C47 of one monomer and the C-terminal C170 of the other at the essential enzymatic redox center (42, 105). In mammals, the N-terminal cysteine residue is at position 52, and the C-terminal is at position 173. Prx reduces peroxides, such as H2O2, lipid hydroperoxides, and peroxynitrite, using reduced Trx and its dithiol motif at positions 32 and 35 as the immediate hydrogen donor (Fig. 5), thus establishing an enzymatic redox cascade that mediates the flow of electrons from NADPH to TrxR to Trx to Prx to the eventual effector molecule (40).
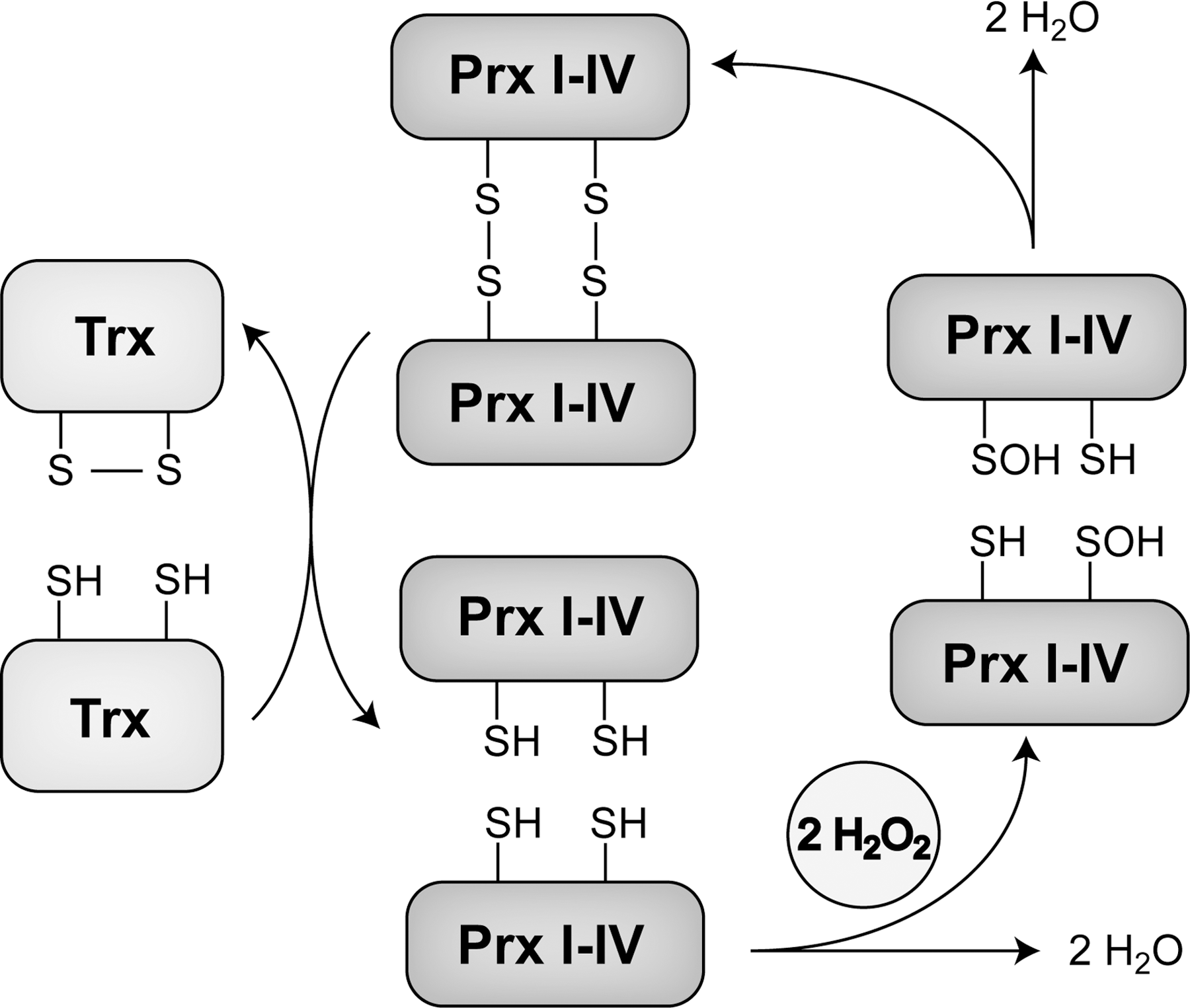
To date, six different mammalian isoforms of Prx have been found, Prx I–VI. These are commonly divided into three subgroups, 2-Cys (Prx I–IV), atypical 2-Cys (Prx V), and 1-Cys (Prx VI) enzymes. All these proteins contain the N-terminal cysteine residue that is selectively and rapidly oxidized by the substrate peroxide (248). Prx I and Prx II are the main isoforms residing in the cytosol and nucleus (242, 271), while Prx III contains an N-terminal mitochondrial targeting sequence (331), and Prx IV can be predominantly found in the endoplasmic reticulum (ER), although containing an N-terminal signal secretion sequence (124). These four isoforms possess both the N- and C-terminal cysteine residues homologous to the C47 and C170 in the yeast protein. The atypical 2-Cys Prx V was first identified in human bronchoalveolar lavage specimens, and it was found to have both an N-terminal mitochondrial and a C-terminal peroxisomal signaling sequence, with detection in the cytosol, mitochondria, and peroxisomes (149). Similar to the 2-Cys Prxs, there are two redox active cysteine residues in the catalytic redox reaction of Prx V, which has an additional cysteine and also forms antiparallel dimers. While the N-terminal cysteine is oxidized by the peroxide substrate in the same way as in the 2-Cys Prxs, the following reaction with the C-terminal cysteine leads to an intramolecular instead of an intermolecular disulfide bond (266). Prx VI is the only known 1-Cys Prx (137). The enzymatic reaction of Prx VI differs from the other Prxs due to the lack of a cysteine residue in proximity to the N-terminal cysteine residue that is oxidized in the first step of the redox reaction; it, therefore, does not establish disulfides. In contrast to the other Prxs, oxidized Prx VI is not reduced by Trx, but uses GSH as an electron donor catalyzed by the π isoform of GSH S-transferase (179).
2. Regulation
Post-translational modifications such as hyperoxidation and phosphorylation regulate the enzymatic activity of Prx (248). The hyperoxidation of Prx occurs at a low rate when exposure to H2O2 leads to further oxidation of the oxidized N-terminal catalytically active cysteine residue, which cannot be reversed by Trx (338). Subsequently, sulfiredoxin (Srx) was identified as the enzyme that was responsible for the reduction of sulfinylated hyperoxidized Prx (20). The hyperoxidation of 2-Cys Prxs leads to structural changes, as sulfinic forms were found to form toroid decamers, whereas the disulfide enzymes form dimers. These structural changes were associated with much higher chaperone activity and lower peroxidase activity, which was reversed on the removal of H2O2 and the reversal to the dimerized state (127). In addition to the switch in enzymatic activity, Prx hyperoxidation also regulates the activity of key transcription factors, for example, leading to the up-regulation of Srx (248). Another way of modulating Prx I activity is through tyrosine (Tyr) phosphorylation at Tyr194 by Tyr kinases of the Src family that are activated by platelet-derived growth factor (PDGF), epidermal growth factor (EGF), or immune receptor signaling. While only a very small part of the total available Prx I is phosphorylated, the phosphorylated molecules are spatially confined to membrane microdomains, called lipid rafts, where certain signaling proteins are concentrated (322). In addition, Prx I and II are also inactivated by phosphorylation through cyclin-dependent kinases (CDKs) (46). It is of interest to note that different post-translational modifications lead to differential regulation of the main cytosolic isoforms Prx I and II (248). Since Prx I is selectively phosphorylated by protein Tyr kinases in a spatially confined area, it functions as a regulator of local H2O2 levels. Prx II is more prone to hyperoxidation in response to global oxidative stress.
3. Clinical significance
Though the primary function of Prxs is the reduction of peroxidases, it has become clear that Prx interacts with a number of different proteins and regulates their function in a redox-sensitive manner (248). One example is PTEN, which will be discussed later in this review. Prx I binds to PTEN and protects it from oxidative inactivation by removing H2O2 produced in response to proliferative signals. Interestingly, Prx I does not require its redox-active cysteine residues to interact with PTEN (34). Since PTEN is considered a tumor suppressor gene, chronic deficiency of Prx I in mice is associated with increased susceptibility for cancer (202). While this clinical outcome of Prx I KO seems to be at least partially independent of its ability to interact with Trx, Prx I KO mice also develop severe hemolytic anemia that is characterized by increased levels of ROS in erythrocytes with protein oxidation and structural abnormalities that lead to increased hemolysis (202). Since most Prxs rely on Trx to be reduced and to be made available for further reductive reactions, it may be speculated that a specific loss of interaction with Trx might yield a similar clinical phenotype. Due to its central role in the regulation of redox status in the cell, Prx deficiency is associated with a lot of different pathologies in diseases that are correlated with increased ROS, such as atherosclerosis, ischemia-reperfusion injury in the heart and liver, and lipopolysaccharide (LPS)-induced inflammation (248).
In addition to its role in diseases associated with increased ROS, autoantibodies against Prxs have been associated with autoimmune diseases such as Kawasaki disease, systemic sclerosis, and systemic vasculitis (84, 125, 139). In these studies, sera from patients with and without autoimmune diseases were screened for autoantibodies against different isoforms of Prx. Patients with Kawasaki disease, systemic sclerosis, and systemic vasculitis have increased levels of autoantibodies against Prx II, Prx I, and Prx II, respectively. In all cases, the presence of Prx autoantibodies also correlated with clinical and laboratory markers of disease progression and severity (84, 125, 139). While these results are certainly interesting, it should be noted that the reported correlations in these studies between antibody titers and clinical parameters were not very high. More studies that investigate the possible implications of Prxs in different autoimmune disease settings on a molecular level are necessary to evaluate the true clinical significance of these findings.
B. Ribonucleotide reductase
1. Background
RNR catalyzes the conversion of ribonucleotides to deoxyribonucleotides (dNTPs) for DNA synthesis. This conversion is based on the direct reduction of ribonucleotides in a reaction initiated by abstraction of the 3′-hydrogen atom of the ribose by a transient thiyl radical of RNR (110, 213). Out of the three known classes of RNR, the mammalian RNR belongs to class Ia. As such, it is an α2β2 tetramer based on the α2 homodimer R1 and the β2 homodimer R2. The R2 subunit harbors a Fe-O-Fe center that generates a stable tyrosyl radical requiring oxygen. Here, the stable tyrosyl radical is stored and is mobilized on substrate binding at the active site of the R1 subunit. The radical is subsequently shuttled to a cysteine residue in the R1 subunit, where it generates the thiyl radical that is necessary for activation of the substrate. The R1 subunit not only contains the catalytic site for the reduction of the ribonucleotide, but also contains the allosteric sites for its regulation. Class Ib, II, and III RNRs differ from class Ia RNR based on their different requirements for cofactors and different radical initiation pathways. While virtually all eukaryotes contain class Ia RNR, Class Ib occurs in aerobic eubacteria requiring oxygen, similar to class Ia RNR. Class II RNRs are microbial enzymes that occur in both aerobic and anaerobic organisms. Class III RNRs depend on anaerobiosis [for review see (213)].
For each cycle of the RNR reaction, reduction of the ribonucleoside is coupled to the formation of a disulfide bond in the active site between cysteine residues at positions 225 and 462 in E. coli. Due to the spatial conformation of the active site, the reduction of this disulfide cannot be achieved by external dithiol-dependent redox active enzymes. Instead, two cysteine residues at position 754 and 759 in the mobile tail of the R1 subunit reduce the active site through a thiol-disulfide exchange reaction (213). To restore the original conformation of the enzyme for the next reaction cycle, an external thiol-dependent reductase system is required to reduce the C-terminal disulfide group. Both Trx and Grx were identified as being dithiol electron donors for RNR in E. coli (109, 161), Trx using its dithiol motif at positions 32 and 35.
2. Regulation
The genes for R1 and R2 are located on different chromosomes and are regulated differently, with R2 being rate limiting for enzyme activity (213). Not surprisingly, RNR activity increases dramatically during the S phase of the cell cycle. During the G1 phase of the cell cycle, the binding of the transcription factor E2F1 represses R2 transcription (63). During mitosis, R2 is degraded via ubiquitination and proteasomal degradation after binding to the Cdh1-anaphase-promoting complex that forms during mitosis (39). In senescent cells, the demand for DNA synthesis is diminished and decreased to the need for DNA repair. Therefore, in resting cells, the R2 gene is not transcribed, but it is also not induced in response to DNA damage (38). Under these conditions, p53-inducible ribonucleotide reductase subunit M2 B (p53R2), an additional mammalian RNR protein, functions as a catalytic partner of the regulatory subunit of R1. Unlike the R2 subunit, the expression of the p53R2 subunit is induced by the DNA damage mediated by the tumor suppressor p53 (300).
It is somewhat surprising that the RNR protein is able to provide a balanced supply of all four deoxynucleotides and is able to rapidly adapt to changes in the demand for dNTPs (213). This is possible through complex allosteric regulation of RNR through the binding of effector molecules to two separate allosteric sites for regulation, one regulating substrate specificity, and the other one regulating the general activity of the RNR. In this system, nucleoside triphosphates are effectors, whereas diphosphates are substrates for class I RNRs. As allosteric regulators, the effectors induce conformational changes of the protein structure, thereby transmitting signals for the required adaptation at the catalytic site (213).
3. Clinical significance
Since RNR is central to the replication of DNA and proliferation of cells, it is a target for cancer therapy (213). Most RNR inhibitors are either radical scavengers acting via destruction of the tyrosyl radical or metal chelators acting via the dinuclear iron center of the R2 subunit. In addition to that, there are a large number of compounds which react with sulfhydryl groups. Since there are a number of cysteine residues that are vital for the enzymatic function of RNR, it is possible that compounds interacting with active RNR thiol functions can act as RNR inhibitors. However, although cancer drugs such as chlorambucil or cisplatin have been assigned RNR inhibitor activity, the clinical relevance of these findings is unclear. The most widely used RNR inhibitor is gemcitabine, which acts as an RNR inhibitor in its biphosphorylated form and as a nucleoside analogon in its triphosphorylated form (110). Since there is a wide variety of cancers that are treated with this drug, a lot of data has been accumulated regarding the characterization, treatment, and outcome of these patients. In an attempt to improve the prognosis of these patients, new approaches for individualized risk stratification and personalized treatment have shifted attention to the R1 subunit of RNR as a possible biomarker with predictive value for patients with different types of cancer, including small-cell lung cancer (133). Several studies have shown that expression levels of the R1 subunit are associated with overall survival in patients with small-cell lung cancer. These findings reveal the potential to improve the treatment of patients by selecting gemcitabine-containing chemotherapy regimens according to expression levels of the R1 subunit or by finding novel approaches to reduce its expression (133). Since most of the data so far has been generated through small retrospective analyses, a larger, prospective, randomized, and controlled trial would be necessary to establish the R1 subunit of RNR as a true prognostic marker for cancer patients.
Since cell proliferation plays a role in the development of atherosclerosis, it could be possible that the inhibition of RNR could have an effect on the development of coronary artery disease. In a single, small study using a combined mechanical and metabolic rabbit injury model of coronary artery disease, treatment with the RNR inhibitors didox and hydroxyurea was associated with decreased atherosclerotic lesion area (87). While this study might be regarded as a hypothesis-generating study, unless new techniques to locally deliver RNR inhibitors to atherosclerotic lesions are developed, the potential severe side effect profile of treatment with these drugs should preclude its clinical use in patients with coronary artery disease in the near future.
It remains to be mentioned that the interaction between RNR and Trx does not seem to be crucial for overall survival of the cell and RNR function in particular, as RNR can be reduced by Grx1 as well as Trx. While Grx1 is the most efficient electron donor for E. coli RNR, it was found that mammalian RNR Trx1 and Grx1 had similar catalytic activities (349). However, since Trx levels are relatively low compared with concentrations of GSH in the mM range in postmitotic cells (178, 181), it is assumed that most R1 subunits are glutathionylated and that subsequent reduction of the C-terminal disulfide of the R1 subunit is primarily catalyzed by Grx. This hypothesis is supported by in vitro studies which showed that there was no change in the dNTP pool after the down-regulation of TrxR in mouse cancer cells (341), and that there is an alternative supply of electrons for RNR to the Trx system, as TrxR-deficient hepatocytes have a normal proliferative potential (249). It is interesting to note that the very first description of Trx was made as a hydrogen donor for enzymatic synthesis of cytidine deoxyribonucleoside diphosphate by RNR in E. coli (161).
C. Methionine sulfoxide reductase
1. Background
Msrs are thiol-dependent antioxidant enzymes that catalyze the reduction of methionine sulfoxide to methionine. The first Msr isoform discovered restored the function of oxidized ribosomal protein L12 in E. coli (32). Msrs can be divided into three groups: MsrA, MsrB, and fRMsr, the latter occurring only in unicellular organisms [for review see (163)]. Oxidized methionine can be found as two diastereomers, methionine-S-sulfoxide and methionine-R-sulfoxide. MsrA is capable of reducing free and protein-based methionine-S-sulfoxide, the only known enzyme in mammalsknown to do so, although it can also reduce sulfoxides as well (270). Since it has a mitochondrial targeting sequence, it was thought to be a mitochondrial protein until a separate promoter and alternatively spliced transcripts were found that lack the mitochondrial targeting sequence, thus leading to cytosolic and nuclear localization of MsrA (143). MsrB has three different isoforms in mammals, MsrB1, MsrB2, and MsrB3 (163). As a common feature, all three enzymes contain Zn in association with two CXXC motifs. MsrBs reduce protein-based methionine-R-sulfoxide, and, to a smaller extent, also free methionine-R-sulfoxide. MsrB1 is the only selenoprotein among this group, and it contains a selenocysteine instead of the catalytic cysteine residue that is present in the other MsrBs. It is located in the cytosol and nucleus and exhibits the highest catalytic activity due to the selenocysteine in its active site (154). MsrB2 contains an N-terminal mitochondrial targeting sequence, making it a predominantly mitochondrial isoform (114). MsrB3 occurs in two isoforms as MsrB3A and MsrB3B via alternative splicing of the first exon. MsrB3A is targeted to the ER by an N-terminal ER targeting peptide and a C-terminal ER retention peptide, whereas MsrB3B is targeted to mitochondria (163).
Since the different Msr isoforms also differ in the number and nature of cysteine residues, their catalytic mechanisms differ (163). MsrA has three conserved cysteines that participate in the catalytic redox reaction of the enzyme. The catalytic cysteine, which is at position 51 in Neisseria meningitidis, generates a sulfenic acid intermediate after attacking the sulfur of methionine-S-sulfoxide. After the formation of a disulfide with one of the other two resolving cysteines and another thiol-disulfide exchange, a disulfide bond between the two resolving cysteines is formed. MsrB1 has a conserved cysteine and a catalytic C-terminal selenocysteine at position 117 in Neisseria meningitidis. The selenocysteine attacks the sulfur of methionine-R-sulfoxide and forms an intermediate selenic acid before it forms a selenenylsulfide with the N-terminal cysteine residue. MsrB2 and MsrB3 contain only one conserved cysteine residue, and, therefore, do not form any disulfide bonds during the redox reaction but only an intermediate sulfenic acid. The reductive capacity of all these enzymes is restored by the reduction of either the disulfide or sulfenic acid by Trx using its dithiol motif at positions 32 and 35 (142).
As antioxidant enzymes, the primary function of Msrs is the reduction of oxidatively damaged proteins. However, Msrs can play regulatory roles in redox-sensitive pathways that are not directly related to oxidative stress. For instance, Ca2+/calmodulin-dependent protein kinase II (CaMKII), which is usually activated by Ca2+/calmodulin, can alternatively be activated by the oxidation of its methionine residue in the regulatory domain. This redox-dependent activation is reversed by MsrA (76). This exemplifies yet another intersection between redox signaling and other signaling pathways.
2. Regulation
Since there are numerous different isoforms of Msr with different subcellular localizations, there is probably a complex transcriptional and post-transcriptional regulatory system in place that controls the expression of this important gene. However, there are not many published reports about the transcriptional regulation of the different isoforms. In retinal tissue of rhesus monkeys, MsrA expression is controlled by two separate promoters (164). While the first promoter controls transcription of the MsrA isoform that is targeted to the mitochondria, the second promoter regulates the transcripts which are targeted to the cytosol and the nucleus. This was later confirmed for the human MSRA gene as well (230). In the same study, all-trans retinoic acid was identified to increase the activity of both promoters, possibly through putative retinoic acid response elements on these promoters.
Since MsrB1 is the only selenoprotein among the Msrs, in contrast to the other Msrs, its expression and activity is dependent on the dietary intake of selenium in mice (214). Interestingly, the activity of MsrB1, but not of MsrA, was diminished in the aging mouse compared with the young one. Overall, there is not much known about the molecular details of the regulation of Msrs, which leaves a lot of room for future studies that could advance our knowledge in this field.
3. Clinical significance
The interaction between Msrs and Trx is important for the regeneration of the redox active catalytic motif of Msr, but there is evidence that other reducing enzymes, such as thionein, can also reduce oxidized Msrs and, therefore, sustain their activity (253). As is often the case with important regulatory pathways, there seems to be at least partial redundancy in the system for the regeneration of Msr (68). Nonetheless, specific disruption of the interaction between Msrs and Trx would probably decrease the enzymatic activity of Msrs. In this context, it is of interest to note that Msrs are thought to be involved in aging, under the hypothesis that ROS accelerate age-dependent cellular changes. It has been shown that MsrA overexpression (OE) in Drosophila extends the lifespan by 70% (250), whereas MsrA deletion in mice shortened the lifespan significantly (196). While these findings are intriguing, more studies are necessary to gain a mechanistic insight into the impact that Msr has on aging (163).
On a slightly related note, reduced methionine sulfoxide repair seems to be critically involved in senile hair graying (323). Graying of hair is associated with increasing H2O2 concentrations, which, among others, is associated with significantly decreased MsrA and MsrB activity, resulting in a functional loss of methionine sulfoxide repair in the entire hair follicle. One of the methionine residues that are affected is the one at position 374 of the active site of tyrosinase, the key enzyme in melanogenesis. A subsequent inhibition of enzyme activity is proposed to lead to gradual loss of hair color (323). This study nicely exemplifies the crucial impact that changes in the function of MsrA and MsrB, in particular, and of enzymes that are involved in redox regulatory pathways, in general, can have on physiological processes which do not seem to be related at first sight, such as senile graying of hair.
III. Trx-Sensitive Signaling Molecules
A. Apoptosis signal-regulating kinase-1
1. Background
a. Mitogen-activated protein kinase signaling cascades
Mitogen-activated protein kinase (MAPK) signaling pathways are Ser/threonine (Thr) kinases that respond to stimuli and stress and regulate cellular responses (Fig. 6). MAPK pathways are evolutionarily conserved in all eukaryotic cells and consist of three, sequentially activated kinase cascades: MAP kinase kinase kinase (MAPKKK), MAP kinase kinase (MAPKK), and MAPK (184). Multiple MAPK signaling cascades that are activated simultaneously or sequentially regulate the activities of the cell. The presence and extent of environmental stress are initially sensed by MAPKKKs. In response, MAPKKKs modify the MAPK signaling cascades. The three major families of MAPK that act in response to various stimuli are extracellular signal-regulated kinase (ERK), JNK, and p38 MAPKs (45, 119). As described next, ASK1 belongs to the MAPKKK family. ASK1 phosphorylates and activates both the JNK and p38 apoptotic pathways (118, 119). All components of the MAPK signaling pathways, including ASK1, regulate stress and control gene transcription, protein synthesis, cell-cycle regulation, differentiation, and apoptosis (160). On sequential activations of MAPKs, the cellular activities are modified in response to environmental changes. Therefore, balancing the signals of multiple signaling pathways that involve pro- and anti-apoptotic factors determines the fate of the cell.

(1) ERK signaling pathway
The ERK family is divided into two classes: (1) the most widely studied members of the family MAPK family, ERK1 and ERK2, which consist of a kinase domain; and (2) the larger members of the family, ERK3, ERK5, ERK7, and ERK8, which consist of both a kinase domain and a C-terminal domain. The C-terminal domain accounts for the higher molecular mass and is involved in the regulation of kinase localization, activation, and up-regulation of certain transcription factors (1, 70, 155).
Generally, ERK1/2 are activated by cell growth and differentiation stimuli to promote cell survival, differentiation, and proliferation (185). Oxidative stress and short-wavelength ultraviolet (UV) light can also lead to ERK1/2 activation through the phosphorylation and activation of various growth factor receptors, including EGF receptor and PDGF receptor (148, 252). ERK1/2 activation promotes the pro-survival pathway of cells on oxidative injury and stimulates keratinocyte proliferation (96, 357). In type 5 adenylyl cyclase (AC5)-deficient mice, the ERK signaling pathway is activated and induces cell protection from oxidative stress and apoptosis, as well as protection from decreased bone density and susceptibility to aging (336). Although ERK1/2 are generally known as pro-survival markers, they can also stimulate the pro-apoptotic pathway. In HeLa cells, the activation of ERK signaling pathway induced cisplatin-induced apoptosis (317). Thus, the ERK family of proteins utilizes both pro-survival and pro-apoptotic signals to regulate cellular activities.
(2) JNK and p38 signaling pathways
In contrast to the ERK1/2 that induce the pro-survival pathways, JNK and p38 are MAPKs that stimulate the pro-apoptotic pathways in response to stress. As just mentioned, ASK1 is a MAPKKK that regulates the JNK and p38 signaling pathways. ASK1 detects environmental stress, including pro-oxidants, such as sodium arsenite and cadmium chloride, UV irradiation, LPS, and inflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor alpha (TNFα) (64, 123, 185, 302). On detection, ASK1 activates the JNK signaling cascade, stimulating apoptosis. Similarly, p38 is activated not only by JNK-activating pro-oxidants, but also by osmotic shock and heat shock (86, 302). The activation of JNK and p38 through ASK1 regulation results in stress-induced apoptosis (45, 185, 329).
In addition to the environmental factors that mediate ASK1-induced apoptosis just mentioned, ROS also stimulates cell death through the activation of the JNK and p38 pathways. In the presence of ROS accumulation, elevated levels of p38α and subsequent decreased tumorigenesis were detected in mouse embryonic fibroblasts (MEFs) (69). It has been demonstrated that PC-12 pheochromocytoma cells derived from rats undergo mitosis in the presence of nerve growth factor (NGF). On removal of NGF, apoptosis occurred in response to the activation of JNK and p38 and the inhibition of ERK (95, 329). Consistent with these findings, inactivation of JNK in animal models showed a significant decrease of brain lesions in ischemia and protection from neuronal cell death (26). Furthermore, the absence or disruption of the JNK gene also decreased glutamate-induced apoptosis of hippocampal neurons and UV-induced apoptosis of primary murine embryonic fibroblasts (309, 337). Thus, the inactivation of the ERK pathway and the activation of JNK and p38 pathways can negatively regulate abnormal cell growth and proliferation by stimulating stress-induced apoptosis (69, 329). As a result, stress-mediated activation of MAPKs can protect against tumorigenesis.
b. Structure and function
Human and mouse ASK1 consist of 1374 and 1380 amino acids, respectively, and both ASK1 molecules possess a Ser/Thr kinase domain that is flanked by regulatory proteins in the N- and C- terminals (305, 306). Crystallization of ASK1 demonstrated that the catalytic domain of ASK1 exhibits the typical structure found in other protein kinases (Fig. 7). However, ASK1 only shares ∼50% of sequence homology with closely related proteins on the phylogenetic tree (28). This suggests that although ASK1 possesses a kinase activity, it is distantly related in sequence to other kinases of a known structure.

In addition to the N- and C-terminals, ASK1 also contains a phosphoserine motif in the C-terminal region to the kinase domain that is recognized by 14-3-3 proteins which bind and regulate the target proteins involved in intracellular signaling. The interaction between ASK1 and the 14-3-3 proteins is specifically dependent on the residue Ser967 (94, 312, 350). Furthermore, the high-resolution structure of ASK1 also revealsthat the catalytic and C-terminal regions of ASK1 are connected by the catalytic adenosine triphosphate (ATP) binding site (28). ASK1 is activated by various types of stress, including ROS, calcium overload, and inflammatory cytokines such as TNFα (101, 208, 298, 306). Even at submicromolar levels of ROS, the growth responses in the cell are modified. Increases in the levels of ROS convert the proliferation signals to apoptosis signals. In the presence of H2O2 in HEK293 cells, elevated levels of ASK1 and subsequent apoptosis were detected (29, 165, 185). ER stress, a result of the accumulation of unfolded and/or misfolded proteins in the lumen of ER, can also activate ASK1 and promote cell death (207, 263). Therefore, the activation of ASK1 that results in apoptosis can be stimulated by both extracellular and intracellular stress.
ASK1 is elevated in various cells undergoing differentiation and proliferation. Constitutive activation of ASK1 and subsequent activation of p38 in keratinocytes induces differentiation. Inhibitors of p38 prevent ASK1-induced differentiation by suppressing the activity of cell differentiation markers (258). Furthermore, constitutive activation of ASK1 up-regulates p38 expression and induces neuronal differentiation and survival of PC12 cells derived from rat pheochromocytoma; these cells exhibit cell growth in serum-starved conditions (297). Interestingly, ASK1 is also involved in hair growth in the presence of active macrophages. In ASK1-deficient mice, hair growth in previously wounded skin is suppressed due to diminished factors that activate macrophages (224). Taken together, these data reveal that ASK1 regulates diverse signaling pathways and the specific effect of ASK1 varies with the cell type.
c. ASK1 signalosome
In nonstressed cells, ASK1 forms a silent homo-oligomer through the C-terminal coiled coil (CCC) domain in a high-molecular-mass complex (>1500 kDa) with other proteins. This complex, designated the ASK1 signalosome, remains in the inactive form until activated by oxidative stress (Fig. 8). Furthermore, this high-molecular-mass signalosome is not formed in an ASK1 mutant lacking the CCC domain, resulting in minimal or completely inhibited ASK1 activity (306).
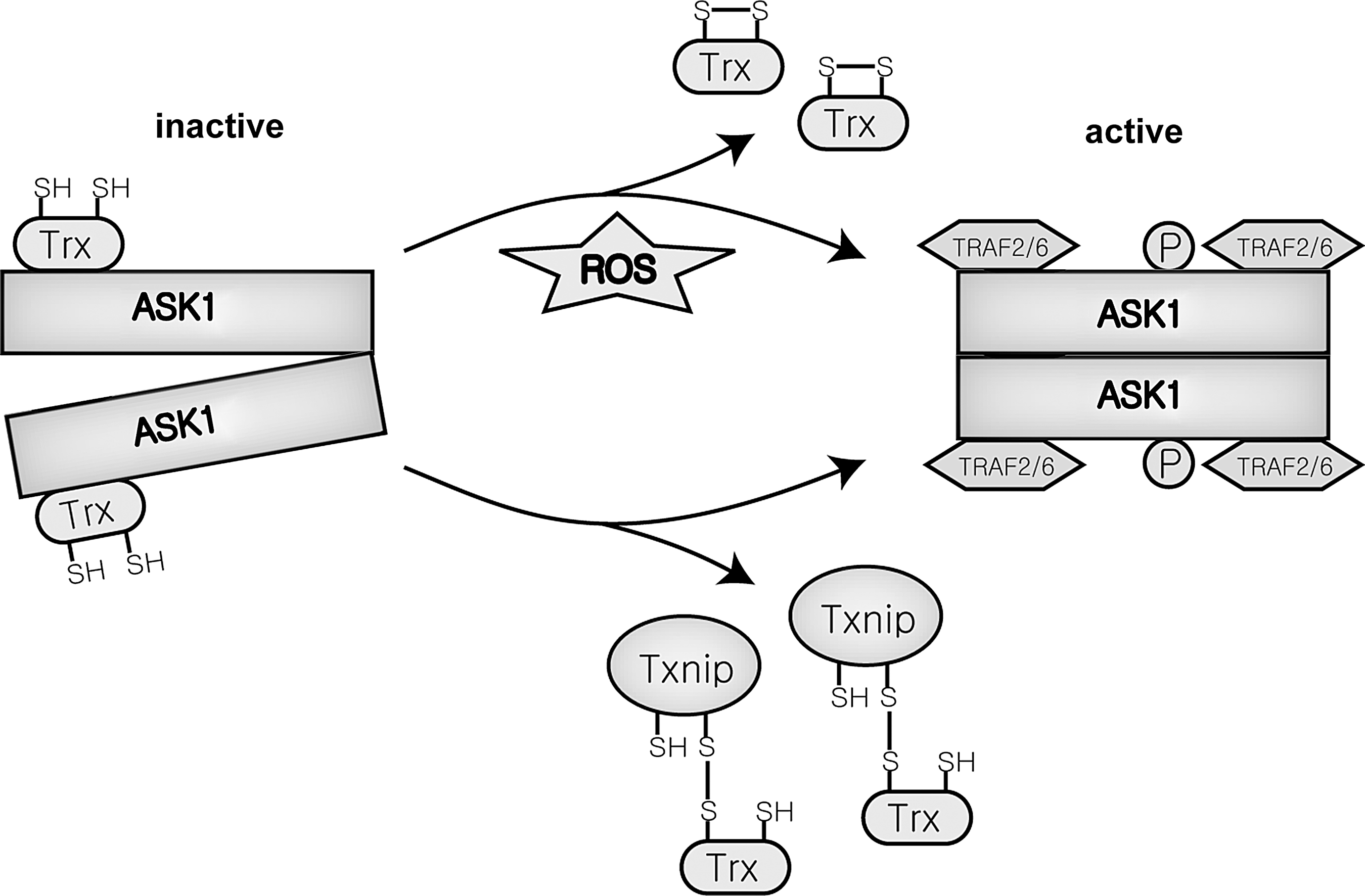
Both positive and negative regulators of ASK1 bind to ASK1 in the ASK1 signalosome. Reduced Trx, a crucial protein for antioxidative defense, binds to ASK1 and becomes a part of the signalosome. Once bound, Trx inactivates ASK1 (175). ASK1 activators, such as tumor necrosis factor receptor-associated factor 2 (TRAF2) and TRAF6, are also recruited to the signalosome for ASK1 activation in the presence of ROS. ASK1 directly binds to the TRAF proteins through the C-terminal region. These TRAF proteins, especially TRAF2, are strong activators of ASK1. TNFα-induced ASK1, JNK, and p38 activation is inhibited in a dominant-negative mutant of TRAF2 (186). Furthermore, H2O2-induced activation of ASK1, JNK, and p38 are strongly suppressed in TRAF2 and TRAF6-deficient MEFs (212). This suggests that both TRAF2 and TRAF6 recruitment is necessary for the activation of JNK and p38 pathways on stress-induced activation of ASK1.
2. Regulation
a. Post-translational regulation
(1) Phosphorylation
The activation of ASK1 occurs through a site-specific phosphorylation of the residues Thr838 of human and Thr845 of mouse ASK1 (305, 306). The residue Thr838 in human ASK1, as well as Thr813 and Thr842, undergoes autophosphorylation during ASK1 regulation. However, mutations at these sites still show active catalytic activity of ASK1. This suggests that the kinase activity of ASK1 is directed independently of its regulation. Other sites such as Ser83, Ser967, and Ser1034 are important for the recruitment of other ASK1 binding partners for regulation (28).
(2) Ubiquitination
The binding partners of ASK1 are crucial for a tight regulation of ASK1. One of these ASK1 binding proteins is a ubiquitin ligase, the C-terminus of heat shock protein 70-interacting protein (CHIP). CHIP binds to ASK1 in the tetratricopeptide repeat domain that is also present in heat shock protein 70. On binding, CHIP induces ubiquitination of ASK1 and directs it for a proteasome-dependent degradation, inhibiting apoptosis (117). In addition to CHIP, β-arrestin1/2 also binds to ASK1 and stimulates proteasome-dependent degradation through the ubiquitination of ASK1 (353). Ubiquitination, therefore, serves as a marker for ASK1 degradation.
(3) S-nitrosylation
In the past, endogenous NO has been shown to regulate the functions of many proteins, including ASK1. In murine fibrosarcoma L929 cells, ASK1 activity is inhibited in the presence of NO by a thiol-redox mechanism, resulting in S-nitrosylation. NO targets C869, one of the four cysteine residues in the kinase domain of ASK1, inhibits the kinase activity of ASK1 (229). Thus, the S-nitrosylation of ASK1 by NO inhibits ASK1 activity.
b. Protein-protein interactions
The regulation of signaling proteins such as ASK1 is crucial for controlling many of its downstream targets that regulate cellular activities. Various inducers and inhibitors of ASK1 either directly or indirectly bind to control ASK1 activity. The structure of ASK1 is important for these interactions. As just discussed, ASK1 contains a phosphoserine motif in the C-terminal region to the kinase domain and the catalytic ATP binding site. Both of these sites can be recognized and bound by regulating molecules such as 14-3-3 proteins and staurosporine. The 14-3-3 proteins bind to ASK1 through the phosphoserine motif found in ASK1. In nonstressed cells, Ser967 is phosphorylated, and ASK1 is bound to a 14-3-3 protein. In the presence of oxidative stress, however, Ser967 is dephosphorylated, and the 14-3-3 protein dissociates from ASK1. The dissociation activates the kinase activity of ASK1 (94, 312, 350). In contrast, staurosporine binds to ASK1 through interacting with the ATP binding site of ASK1. Staurosporine, functioning as a protein kinase inhibitor, inactivates ASK1 (28). Therefore, the activity of ASK1 is tightly controlled by multiple regulatory proteins that form a functional ASK1 signalosome primarily to induce apoptosis. As discussednext, the regulation of MAPK signaling pathways as a result of modified ASK1 activity could be a therapeutic target for various human diseases.
(1) Thioredoxin
Trx, as a regulator of ASK1 activity, binds to ASK1. Two specific cysteine residues of Trx, Cys32 and Cys35, are required for interaction with its binding partners such as ASK1. The oxidized form of Trx consists of an intramolecular disulfide bond between these two cysteines. Oxidized Trx, therefore, is unable to interact with its binding partners such as ASK1. However, the reduced form of Trx in the absence of the disulfide can bind to the NCC domain of ASK1 signalosome in nonstressed cells and inhibit ASK1. The N-terminal domain of ASK1 is essential for the Trx-ASK1 interaction. Thioredoxin-1 (Trx-1) binds to ASK1 at C250, whereas Trx-2 binds at C30 (352). On binding to ASK1, Trx directs ASK1 for ubiquitination and degradation. Thus, Trx acts as a negative regulator of ASK1 activity. Under oxidative stress, Trx is oxidized and dissociated from the ASK1 signalosome. On dissociation of Trx, ASK1 kinase activity is regained, and ASK1-mediated apoptosis in response to stress is induced (175, 176, 254).
In addition to the importance of the oxidation states of Trx, the regulation of ASK1 by Trx is also dependent on nitrosylation (27). Although NO can directly react with ASK1 through S-nitrosylation in the kinase domain, it has been recently shown that Trx is also sensitive to S-nitrosylation. On S-nitrosylation of the reactive thiol groups of Trx, ASK1 activity is increased. Consistent with this finding, S-nitrosylation is inhibited in the presence of antioxidants such as N-acetyl-cysteine and GSH. As a result, the NO-dependent activation of ASK1 is reversed (291). Interestingly, S-nitrosylation of a specific cysteine residue of Trx, C69, results in increased redox activity of Trx. Increased activity of Trx suppresses the activity of ASK1, inhibiting apoptosis (97). Therefore, S-nitrosylation of Trx is essential for a tight control of ASK1 activity.
(2) Trx interacting protein
Txnip binds to reduced Trx and serves as a negative regulator of Trx function. The interaction between Txnip and Trx results in the activation of ASK1. Thus, Txnip acts as a positive regulator of ASK1 activity. OE of Txnip inhibits the interaction of Trx with ASK1, resulting in the declined proliferation and inhibition of JNK suppression by Trx. On heat shock or in the presence of oxidative stress, Txnip expression is significantly elevated, and the interaction between Trx and ASK1 is inhibited; this suppresses cell growth and proliferation as a result of Txnip OE (134). Therefore, by inhibiting Trx activity, Txnip indirectly activates ASK1 and subsequent apoptosis.
(3) Glutaredoxin
Grx, another redox sensing molecule similar in function to Trx, can also regulate ASK1 activity (172, 280). Oxidized Grx is reduced by GSH, and the oxidized GSH is subsequently reduced by GSH reductase. Grx binds to and inhibits ASK1, acting as a negative regulator of ASK1 activity. Previous studies have indicated that glucose deprivation prevents the interaction between Trx or Grx and ASK1, stimulating cell apoptosis through the activation of the stress-induced MAPKs such as JNK (167, 172, 280). This suggests that both Trx and Grx protect cells from death by inhibiting ASK1 in the presence of glucose. Consistent with these findings, Grx2, one of the three Grxs, protects HeLa cells from undergoing apoptosis. Silenced expressions of Grx2 resulted in the significant stimulation of cell death. In contrast, OE of Grx showed the down-regulation of glucose deprivation-induced apoptosis (173, 281). These findings suggest that inhibiting Trx and/or Grx interactions with ASK1 could be useful in directing tumor cells to cell death.
(4) Ser/Thr protein phosphatase 5
Ser/Thr protein phosphatase 5 (PP5) binds to ASK1 and negatively regulates ASK1 activity. In the kinase domain of ASK1, a specific residue (Thr845) is essential for the activation of ASK1. In the presence of oxidative stress (e.g., H2O2), the interaction between PP5 and ASK1 is induced, and PP5 dephosphorylates Thr845 of ASK1, inhibiting ASK1 activity by negative feedback (195). In response to this dephosphorylation, ASK1 activity is suppressed, and cell survival is stimulated.
3. ASK1 in health and diseases
Although the activation of ASK1 leads to apoptosis and inhibition leads to induced cell growth and proliferation, the ASK1 signalosome functions in response to the extent and duration of various types of stress depending on the cell type. The impact of the signaling cascades is determined by the magnitude and duration of exposure to extracellular stimuli and stress. Low or momentary exposure to extracellular stimuli and stress results in cell survival, differentiation, and proliferation, but excess or prolonged exposure leads to apoptosis (69, 185). Thus, tight control of ASK1 activity is crucial in the development and treatment of various types of human diseases such as diabetes.
a. Innate immune response signaling
The innate immune system functions to protect multicellular organisms from invasion of microorganisms that produce infections. These invasions are primarily recognized by Toll-like receptors (TLRs). Expressed in various types of immune cells as well as nonimmune cells, TLRs recognize distinct pathogen-associated molecular patterns exhibited in lipids, proteins, and nucleic acids. So far, 11 TLRs in human and 13 TLRs in mouse have been identified (6, 185). TLR4 protects against gram-negative bacterial infections by recognizing LPS. On recognition, TLR4 signaling activates JNK, p38, and NF-κB (295). Mutations or deletions of the TLR4 gene hinder LPS recognition by TLR4, thereby increasing the susceptibility of infection by gram-negative bacteria (112, 240).
Previous studies have identified the roles of ASK1 in innate immune signaling. LPS recognition by TLR4 stimulates the production of ROS. In response, the TRAF6-ASK1 complex is formed, and the p38 signaling pathway is activated. In ASK1-deficient mice, LPS-induced activation of the p38 is decreased. Furthermore, ASK1 is also required for the LPS-induced stimulation of certain proinflammatory cytokines. LPS-induced inflammatory cytokines, including TNFα and IL-6, are diminished in dendritic cells (DC) and splenocytes derived from ASK1-deficient mice. LPS-induced ROS production also results in the dissociation of Trx from ASK1, forming the TRAF6-ASK1 complex and subsequently activating ASK1 (187). Thus, the activation of p38 stimulated by the formation of TRAF6-ASK1 complex is required for the innate immunity regulated by TLR4.
The ASK1 signalosome has functions similar to inflammasomes, which can induce both inflammatory and apoptotic signals. Inflammasomes are multiprotein complexes that consist of proinflammatory caspases activated by the nucleotide-binding oligomerization domain-like receptor (NLR) family of proteins (182, 294). The capability of producing both apoptotic and inflammatory signals allows the ASK1 signalosome to determine the magnitude of stress and ASK1 activation to produce diverse cellular responses.
b. Cardiac hypertrophy and remodeling
The regulation of ASK1 activity is crucial in different types of receptor-mediated redox signaling in response to ROS, including the angiotensin II (Ang II) signaling pathway involved in cardiac hypertrophy and remodeling (185). In wild-type mice, Ang II induces cardiac hypertrophy and remodeling by producing superoxide. ROS production by Ang II induces the activation of ASK1, JNK, and p38. In ASK1-deficient mice, ASK1 activation in the left ventricle is not detected, and the activation of JNK and p38 is significantly attenuated (126). This suggests that ASK1 plays a key role in Ang II-induced cardiac hypertrophy and remodeling, as well as various cardiac diseases that involve Ang II or other receptor-mediated signaling.
c. Neurodegenerative diseases and ER stress
Alzheimer's disease is a neurodegenerative disorder that is characterized by cerebral neuritic plaques of amyloid-β peptide and neurofibrillary tangles, which eventually leads to the apoptosis of neurons and progressive memory loss. Amyloid-β activates ASK1 through ROS generation in neuronal cells in vitro, inducing ASK1/JNK-mediated apoptosis. Primary neuronal cultures derived from ASK1-deficient mice show that the activation of endogenous JNK by amyloid-β is suppressed (136). Therefore, amyloid-β could be used to target ASK1 activity and possibly decreaseneuronal cell death.
Interestingly, amyloid-β can induce ER stress through ROS-mediated ASK1 activation, thereby stimulating cell death. ER stress has been implicated in various types of neurodegenerative diseases, including Alzheimer's, Parkinson's disease, and polyglutamine diseases. Inositol-requiring enzyme 1 (IRE1), a transmembrane protein that functions under ER stress, stimulates the formation of the TRAF2-ASK1 complex, activating the JNK apoptosis pathway. The aggregation of polyglutamine also induces ER stress and stimulates apoptosis through the IRE1-TRAF2-ASK1 complex formation (120, 201, 207, 313). Furthermore, resistance to ER stress-induced apoptosis was also demonstrated in neurons derived from ASK1-deficient mice (136). These findings suggest that amyloid-β plays a key role in various types of neurodegenerative diseases by regulating ASK1 as a mediator of stress-induced apoptosis.
d. Cancer
MAPK signaling pathways participate in diverse mechanisms of cell regulation, including apoptosis and carcinogenesis. Recently, it has been shown that ASK1-deficient mice are more susceptible for tumorigenesis. On the treatment of wild-type and ASK1-deficient mice with diethylnitrosamine, an inducer of hepatocarcinogenesis, ASK1-deficient mice produce thrice as many tumors as in wild-type mice. Pro-apoptotic markers are also down-regulated in ASK1-deficient mice (200). By suppressing ASK1 activity, cell growth can occur at a higher rate, resulting in tumorigenesis.
Strikingly, however, elevated levels of ASK1 and JNK are detected in gastric cancer tissue specimens. OE of ASK1 up-regulates cyclin D1, a regulator that allows the transition from the G1 to the S phase in the cell cyle, stimulating cell proliferation. However, the levels of ASK1 do not vary between wild-type and tumor cells in colon cancer epithelium (102). This suggests that increased expression of ASK1 is specific to gastric cancer cells. Consistent with the findings of stimulated cell proliferation on the up-regulation of cyclin D1, ASK1 KO mice exhibit fewer and smaller tumors than wild-type mice (102). In addition to the primary function of inducing stress-induced apoptosis, ASK1 can protect cells from death. This suggests that the tight control of ASK1 expression is crucial in altering the downstream signaling pathways which may lead to apoptosis.
e. Diabetes
In cultured human hepatoma (Huh7) cells, ROS production in the mitochondria is stimulated by TNFα, and this significantly activates ASK1 activity. Subsequently, ASK1 activates the JNK and p38 apoptosis signaling pathways. Hyperglycemia, a result of impaired insulin action, increases mitochondrial ROS production. Mitochondrial ROS production may play a role in the development of diabetes as well as diabetic vascular complications (204). Hyperglycemia-induced activation of ASK1 is involved in the senescence of endothelial cells, which leads to accelerated vascular aging and thrombosis in diabetic individuals (340). In conclusion, mitochondrial ROS and high glucose levels activate ASK1 and stimulate endothelial cell senescence, which may provide mechanisms for understanding vascular complications in diabetic individuals.
4. Conclusion
ASK1, as a member of the MAPKKK family that senses oxidative stress, can modify multiple signal transduction pathways. The induction of apoptosis by ASK1 can be used as a molecular target for cells that exhibit abnormal growth, metabolism, and proliferation. Understanding ASK1 may facilitate the development of therapeutic drugs for the treatment of human diseases that result from the dysregulation of ASK1-mediated signaling pathways.
B. Trx interacting protein
1. Background
Txnip was first described as a protein up-regulated in the human promyelocytic leukemia cell line HL-60 treated with 1,25-dihydroxyvitamin D3 in a study identifying genes that are differentially regulated during HL-60 cell differentiation to monocytes (54). It was, therefore, initially known as Vitamin D3 Upregulated Protein 1 (VDUP1) or Trx binding protein 2 (TBP-2). In recent years, a large number of studies were published that collectively point to the involvement of Txnip in diverse biological processes.
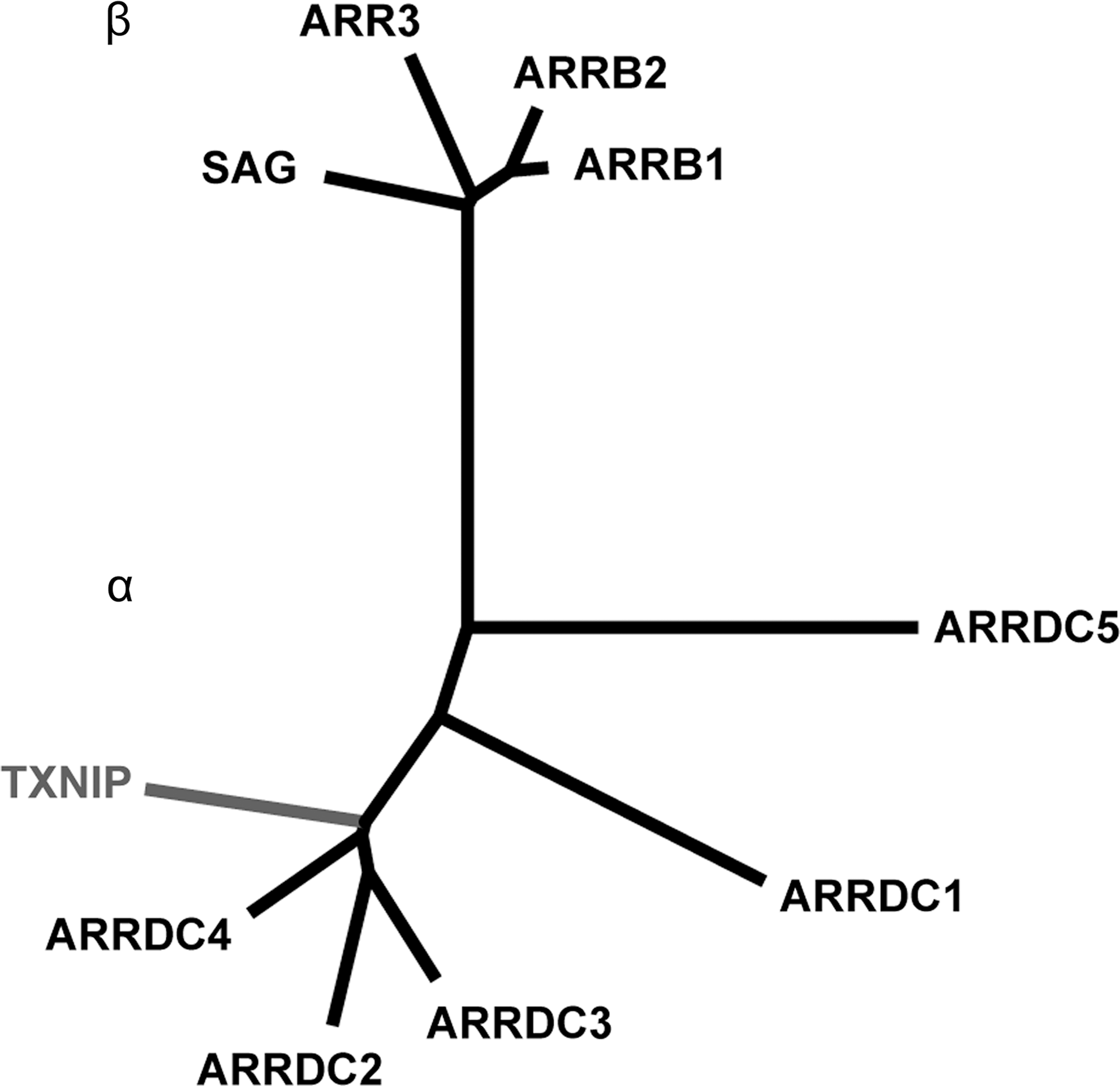
Phylogenetic studies revealed that Txnip is a part of the α-arrestin family of proteins (Fig. 9). The human α-arrestins, which are phylogenetically more ancient than their more prominent β-arrestin counterparts, are a group of six proteins predicted to contain the characteristic two arrestin folds that identify arrestins in general (Fig. 10); Txnip is the only α-arrestin thus far identified that can interact with Trx (9). The β-arrestins are key regulatory proteins that are canonically involved in the desensitization of G protein-coupled seven-transmembrane receptors. In addition, β-arrestins also activate signaling cascades independently of receptor activation, thus functioning as multifunctional adaptor proteins forming scaffolds for numerous intersecting signaling pathways (66). In light of these considerations, it seems plausible to assume that the α-arrestin Txnip could play a central role in the regulation of different biological pathways integrating redox signaling with other metabolic pathways. This assumption would be further corroborated by data about the actual crystal structure of Txnip, as the phylogenetic studies just mentioned were based on genetic sequences and predicted structures of the α-arrestins. So far, attempts to crystallize Txnip have been only partially successful (239). Nevertheless, since its first description in 1994, Txnip has been shown to regulate a surprisingly wide variety of different biological pathways ranging from redox signaling and metabolism to cell-cycle regulation.

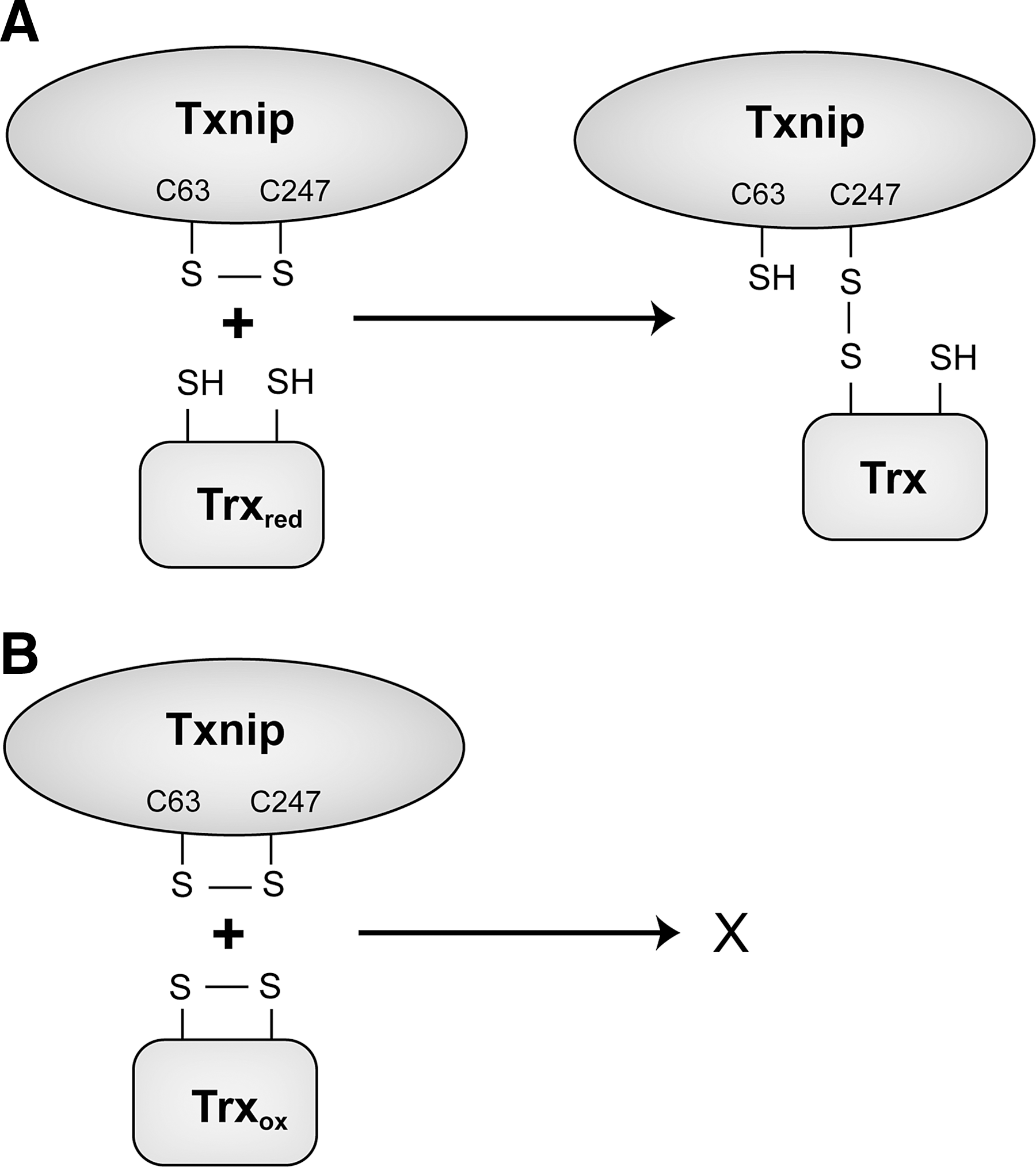
A seminal finding in the field was made when Txnip was identified in a yeast two-hybrid screen for potential binding partners and regulators of Trx (209), which was confirmed by two independent groups shortly after the first description of the interaction (134, 332). Txnip binds to the reduced form of Trx but not the oxidized form (Fig. 11), and also does not bind to a mutant form of Trx that has C32S and C35S mutations at its redox-active catalytic site, indicating an interaction with Trx's redox active site. A C247S mutant of Txnip does not bind to Trx, giving rise to the proposed mechanism of a mixed disulfide by a disulfide exchange reaction between oxidized Txnip and reduced Trx with a proposed disulfide bond between cysteines at position 63 and 247 of Txnip (233). Txnip, therefore, is a negative regulator of Trx and its reducing capacities. It also competes with Prx and ASK1 for an interaction with Trx at its redox active site and is thereby a central regulator of cellular signaling pathways involved in oxidative stress, proliferation, and apoptosis. In addition, Trx also plays an important role for the regulation of nitrosative stress by functioning as a denitrosylase for proteins that were S-nitrosylated (14); protein denitrosylation by Trx is inhibited by Txnip, which is, in turn, repressed by endogenously synthesized NO—the key molecule for S-nitrosylation of protein cysteine residues. NO represses Txnip expression and thereby facilitates Trx-mediated denitrosylation, allowing cells to cope with nitrosative stress (80). These findings revealed Txnip as a feedback regulator of S-nitrosylation and nitrosative stress in addition to its role for oxidative stress.
2. Regulation
a. Transcriptional regulation
The human TXNIP gene is located on chromosome 1q21.1 and includes eight exons and seven introns spanning ∼5 kb. In addition to transcriptional regulation of TXNIP by Vitamin D3 (54), a myriad of stimuli regulate TXNIP expression, including mechanical stress, fluid shear stress, UV light, heat shock, hypoxia, H2O2, NO, nicotinamide adenine dinucleotide (NADH), ATP, glutamine, nicotine, vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), transforming growth factor beta (TGF-β), estradiol, calcium channel blockers, receptor for advanced glycation endproducts (RAGE) activation, insulin, and glucose (48, 65, 104, 134, 135, 162, 237, 261, 267, 268, 311, 319, 333, 348). The control of TXNIP expression by insulin and glucose has generated intense interest, as TXNIP is one of the genes that are most responsive to blood glucose levels and insulin signaling in patients with type 2 diabetes mellitus (228).
Many studies have investigated the transcription factors and TXNIP promoter sequences mediating the effects of stimuli on TXNIP gene expression (Fig. 12). The TXNIP promoter contains a carbohydrate response element (ChoRE) that is responsible for the glucose responsiveness of Txnip expression (190). The heterodimeric transcription factor MondoA:Max-like protein X (Mlx) shuttles from the outer mitochondrial membrane to the nucleus in response to glucose and enzymatic activity of the glycolytic pathway to directly activate the TXNIP promoter through its ChoRE (289). Later on, another ChoRE as well as a CCAAT box and an inverted CCAAT box were identified on the TXNIP promoter (30, 347). The recruitment of MondoA:Mlx to the ChoREs under glucose stimulation is contingent on binding of the trimeric transcription factor nuclear factor Y (NF-Y) to the CCAAT boxes, which results in the synergistic activation of TXNIP transcription by MondoA:Mlx and NF-Y on glucose uptake (347). In addition to the role of MondoA:Mlx in glucose-induced up-regulation of TXNIP, the transcription factor is also involved in regulating TXNIP expression in response to other metabolic parameters such as hypoxia (43), lactic acidosis (53), inhibition of oxidative phosphorylation (346), and adenosine-containing molecules, for example, NADH or ATP (348). Another transcription factor that mediates glucose-induced up-regulation of TXNIP through ChoRE is the MondoA paralog carbohydrate response element-binding protein (ChREBP) (37, 226).

The forkhead box O (FOXO) family of transcription factors is involved in a variety of different cellular functions, including metabolism. A study that investigated the effects of rat synaptic N-methyl-D-aspartate (NMDA) receptor activity on neuronal capacity to deal with oxidative stress showed that FOXO1 and FOXO3a increased Txnip expression and that FOXO1 is associated with the Txnip promoter at an FOXO binding site (227). These findings were validated in human cells, and it was also shown that FOXO1 mediates glucose-induced TXNIP expression via a p38 mitogen-activated protein kinase-dependent pathway, thus revealing an alternative transcriptional control mechanism of Txnip expression in response to glucose (170, 356).
Peroxisome proliferator-activated receptor gamma (PPARγ) is a transcription factor that controls genes in fatty acid and glucose metabolism. After a study showed that PPARγ agonists increased Txnip expression (220), it was later shown that through functional PPARγ response elements in the TXNIP promoter, PPARγ activation regulates TXNIP expression (19). Since Txnip negatively regulates PPARγ activity in adipose tissue, this reciprocal feedback mechanism underscores the role that Txnip plays in cellular metabolism (56).
Apart from their roles in the glucose-dependent regulation of TXNIP expression, the CCAAT box and NF-Y also regulate TXNIP expression by histone deacetylase 1 (HDAC1), a key element in the control of cell proliferation and survival. The HDAC inhibitor suberoylanilide hydroxamic acid (SAHA), an anti-cancer drug, up-regulates TXNIP expression through the inverted CCAAT box in the TXNIP promoter (30). HDAC1 decreases TXNIP expression and is recruited to the TXNIP promoter by forming a protein complex with the RET finger protein (RFP) and NF-Y (140). In addition, other binding sites of transcription factors that are relevant for the regulation of cell proliferation and apoptosis have been identified; AP-1, E-twenty six 1 (ETS1), the glucocorticoid receptor, and Krüppel-like factor 6 (KLF6) increase TXNIP expression (18, 100, 243, 320).
Another mechanism of transcriptional control is epigenetic modification such as the methylation of a regulatory promoter region. During the malignant transformation of human T-cell leukemia virus type I (HTLV-I) infected T cells that results in adult T-cell leukemia, the T-cells undergo a transition from IL-2-dependent to IL-2 independent growth. This transition is accompanied by a loss of Txnip expression due to the DNA methylation at CpG sites in the promoter and exon 1 and histone deacetylation (4). These findings are of special interest considering the role of Txnip itself in cell-cycle regulation and cancer biology.
Other specific factors that regulate TXNIP expression through interactions with its promoter are heat shock factor binding to a heat shock element (146), polycomb repressive complex 2 (354), and heterogeneous nuclear ribonucleoprotein G (276).
b. Post-transcriptional regulation
Post-transcriptional regulation of Txnip mRNA by micro ribonucleic acid (miR)-17-5p may participate in senescence. In human fibroblasts that undergo senescence, the down-regulation of miR-17-5p is associated with increased Txnip expression (356). Another microRNA (miRNA) that is involved in the post-transcriptional regulation of Txnip is miR-373, which promotes breast cancer invasion and metastases. In a proteomics screen of miR-373-regulated genes in a human breast cancer cell line, Txnip protein expression, but not mRNA expression, was found to be down-regulated by miR-373 indicating translational inhibition by miR-373 (334).
c. Post-translational regulation
Recently published data suggest significant regulation of Txnip protein levels at the post-translational level. In rat pancreatic beta cells, cyclic adenosine monophosphate, the second messenger of many G protein-coupled receptors, stimulates proteasomal degradation of Txnip by promoting its ubiquitination (269). Txnip contains two C-terminal PPXY motifs, which are known to interact with WW domains notably present in E3 ubiquitin ligases such as Itch (9). Not surprisingly, Txnip was identified as an adaptor between homologous with E6-associated protein C-terminus (HECT) ubiquitin ligases (WWP1, WWP2 and Itch) and endosomal sorting complex required for transport (ESCRT), a mechanism that is relevant for degradation of plasma membrane proteins or the budding of most membrane enveloped viruses (245). Itch was also identified as being responsible for mediating polyubiquitination and subsequent proteasomal degradation of Txnip through the interaction of Itch's WW domains with Txnip's PPXY motifs (351). The significance of these findings is underscored by their implications for our understanding of Txnip's role in integrating different cellular signaling pathways. A key, and actually eponymous, characteristic of Txnip is its interaction with Trx and its role as a negative regulator of Trx function. A recent study investigates the regulation of adipogenesis by Txnip that counters perception. The dissociation of Trx from Txnip—may be by adipogenic stimulants such as insulin or by mutation of the binding site—targets Txnip for proteasomal degradation and triggers adipogenesis. Mutating the PPXY motifs of Txnip prevented proteasomal degradation and inhibited adipogenesis even when Trx binding was lost (57). In this specific setting, it is not Txnip that regulates Trx, but Trx which regulates Txnip and its intrinsic ability to suppress adipogenesis. Thus, the Txnip-Trx interaction represents a molecular switch at which the cellular redox state modulates metabolic signaling and works as a redox signaling pathway of its own.
3. Txnip in health and disease
a. Development, differentiation and proliferation
Embryonic development as well as tissue regeneration are biological processes that are characterized by orchestrated regulation of cell differentiation and proliferation. The central role that Txnip might play in different regulatory processes is highlighted by the fact which Txnip may play a role in the development and differentiation of a variety of different tissues and cell types, including lung and blood cells.
The first indications that Txnip might be relevant for lung development were found in studies of fetal lung expansion in sheep. Fetal lung development and growth is critically determined by the degree of fetal lung expansion. Txnip levels are decreased in increased ovine fetal lung expansion and raised in decreased fetal lung expansion (284). As observed in other cell types, Txnip expression is inversely correlated with proliferation in the fetal lung. It is positively correlated with SP-B expression, a marker of differentiated type II alveolar epithelial cells, suggesting that Txnip might be involved in expansion-induced lung cell proliferation and alveolar epithelial cell differentiation (79, 284). In later stages of human fetal lung development, Txnip transcription is up-regulated compared with earlier stages (153). Over the first 7 days of murine life, Txnip expression is developmentally decreased. This decrease in Txnip expression is attenuated by exposure of newborn mice to hyperoxia that is thought to be a contributor to development of bronchopulmonary dysplasia for prematurely born infants (303). Thus far, the data regarding the involvement of Txnip in lung development and alveolar epithelial cell differentiation are observational and descriptive. Further evidence from additional studies is needed to confirm the hypothesis that Txnip plays a role in lung development.
Characterization of the Txnip-KO mouse has revealed that Txnip regulates natural killer (NK) cell maturation. While the development of T and B cells is largely intact, there is a profound decrease in the number of NK cells in vivo. Over the course of NK cell maturation in Txnip-KO mice, there is decreased expression of the beta chain of the IL-2 receptor (IL-2RB, CD122), a key marker for NK cell development. Analysis of IL-2RB promoter activity showed that Txnip directly up-regulates IL-2RB gene expression. As a consequence, NK cell-mediated tumor rejection is significantly decreased in Txnip-KO mice (165). Another group confirmed that Txnip-KO mice have a decreased number of NK cells, while Txnip-OE mice have an increase in NK cells. Furthermore, they showed that Txnip-KO mice are resistant to concanavalin A (ConA)-induced hepatitis, while OE of Txnip in vivo leads to increased susceptibility to ConA mediated by NK cell function (222). Since ConA-induced hepatitis reflects aspects of autoimmune and viral hepatitis, these findings highlight the functional relevancy of Txnip's control of NK cell maturation.
Apart from NK cell biology, another immunologic process that has been studied in Txnip-KO mice is T-cell activation by DC. DC derived from Txnip-KO mice were stimulated with LPS, and it was found that under these conditions, Txnip-deficient DC secrete less IL-12, an activator of T- and NK cells, and less IL-6, an activator of the inflammatory acute phase reaction. This leads to decreased DC stimulated T-cell proliferation in vitro, and a delayed hypersensitivity response of Txnip-KO in vivo (279).
Quiescence, activation, and mobilization of hematopoietic stem cells (HSCs) are regulated by a number of different factors. During HSC activation, Txnip is down-regulated, suggesting that it might be important for mobilization of HSC. In Txnip-KO mice, the long-term reconstituting HSC population is decreased and exhausted, and its capacity to repopulate is rapidly lost. Deletion of Txnip results in decreased CXCL12- and osteopontin-mediated interaction between HSCs and the bone marrow, impaired homing, and retention in the osteoblastic niche, leading to increased mobilization of HSCs (129). These findings highlight the importance of Txnip for maintaining and controlling HSC quiescence.
Overall, there is accumulating evidence that Txnip might be involved in the differentiation of many different cell types. Although some of the findings such as Txnip's role in the maturation of NK cells are well documented, the majority of reports that were mentioned in this chapter so far lack a deeper mechanistic insight into the exact regulatory pathways that are affected by Txnip. This is different for Txnip's involvement in the regulation of the cell cycle.
Txnip is highly induced in senescent human fibroblasts, and cell growth is strongly inhibited by OE of Txnip in these cells (131). On the other hand, it was found that in N-methyl-N-nitrosourea (MNU)-induced rat mammary tumors, Txnip expression was significantly down-regulated. The treatment of isolated cells from these tumors with 1,25-dihydroxyvitamin D3 results in increased Txnip expression and attenuated cell growth (339). Higher Txnip expression levels are associated with better prognosis in breast cancer patients as measured by the metastasis-free interval after initial treatment (31). Txnip down-regulation promotes carcinogenesis, and Txnip is down-regulated in many different forms of cancer, including breast, prostate, colorectal, gastric, kidney, liver cancer, squamous cell carcinoma, melanoma, pheochromocytoma, HTLV-1 infected T cells, B-cell and Hodgkin lymphoma, and acute lymphoblastic and chronic lymphocytic leukemia (4, 73, 77, 82, 93, 158, 193, 210, 217, 273, 276, 278, 296, 304, 307).
Txnip OE decreases cell proliferation through the induction of cell cycle arrest at the G0/G1 phase (Fig. 13). Txnip interacts with Fanconi anemia zinc finger, promyelocytic leukemia zinc finger (PLZF), and HDAC1 that form a transcriptional repressor complex. Txnip suppresses the activity of the human cyclin A2 promoter that contains PLZF-response elements (99). Cyclin A2 activates CDK2, which is required for G1/S transition in the cell cycle. Txnip OE increases the protein levels p16Ink4a, an inhibitor of CDK4/6, which is necessary for G1/S cell cycle transition (206). Studies in the Txnip-KO fibroblasts showed that protein levels of p27kip1, another CDK2/4 inhibitor, are decreased compared with wild-type cells, while no transcriptional changes are seen, indicating post-translational regulation of protein levels of p27kip1. The stability of p27kip1 is controlled by Jun activation-domain binding protein 1 (JAB1), which induces translocation of p27kip1 from the nucleus to the cytosol where it is degraded. Txnip interacts with JAB1 and blocks JAB1-mediated translocation of p27kip1 to the cytosol (128).
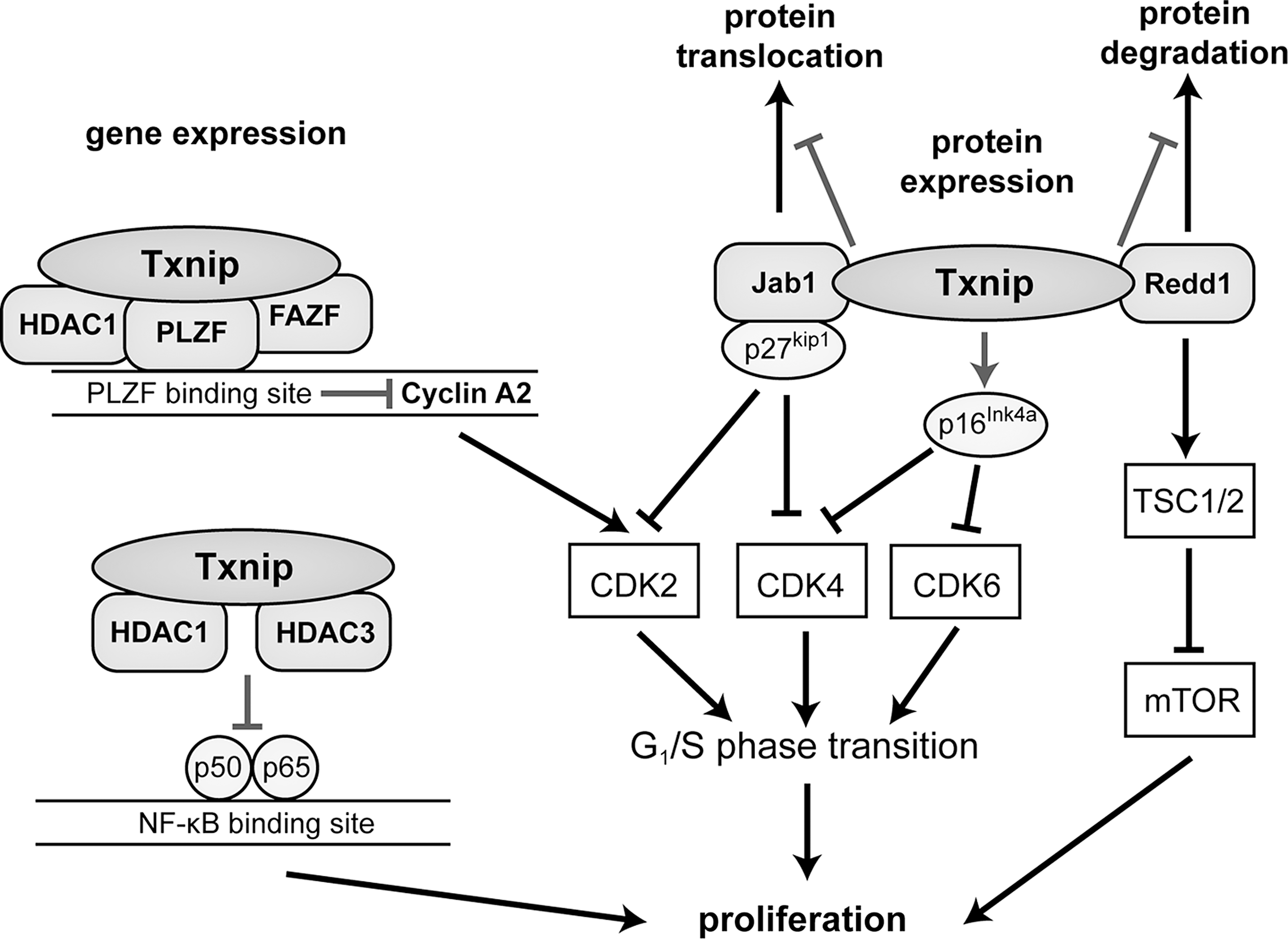
Txnip promotes hepatic carcinogenesis in part through NF-κB activation. OE of Txnip in HEK293 cells resulted in the robust suppression of TNFα induced NF-κB activity. To achieve this, Txnip interacts with HDAC1 and HDAC3, which bind to NF-κB p50 and p65 and lead to transcriptional repression at the NF-κB binding site. Thus, Txnip synergistically suppresses NF-κB activity through a proposed complex with HDAC1 and NF-κB p65 (159). Another key master regulatory protein that is affected by Txnip is the mammalian target of rapamycin (mTOR), which is frequently hyperactivated in cancer and integrates signals from growth factors and nutrients to coordinate cell growth and proliferation. Regulated in development and DNA damage responses 1 (Redd1) protein is a negative regulator of mTOR activity. Txnip interacts with Redd1 and protects it from proteasomal degradation, leading to decreased mTOR activity (130).
Interestingly, not only in cancer but also in the setting of tissue regeneration, Txnip's regulation of the cell cycle may be relevant. In mice that underwent partial hepatectomy, deletion of Txnip led to significantly accelerated liver recovery, accompanied by altered expression of key cell-cycle regulatory proteins, including cyclin D, cycylin E, CDK4, p21, and p27. The induction of growth factors and activation of proliferative signaling pathway components, including ERK1/2, Akt, glycogen synthase kinase 3β (GSK3β), mTOR, and p70S6K, were up-regulated and occurred earlier, indicating that Txnip regulates the cell cycle in the setting of liver regeneration (157).
Collectively, the studies just mentioned show that Txnip is a tumor suppressor which is involved in the key signaling pathways controlling the cell cycle. Evidence from numerous gene expression profiling studies in cancer cells suggested earlier on that Txnip is a critical regulator of proliferation. Since ongoing research continues to reveal the underlying molecular mechanisms, it remains to be seen whether these findings can be translated into the clinical setting of treatment of cancer or to new approaches in regenerative medicine. However, given the fact that since an α-arrestin Txnip is involved in the regulation of numerous fundamental signaling pathways, it will be difficult to predict the possible side-effect profiles of potential therapies.
b. Metabolism
Evidence for the roles of Txnip in lipid metabolism surfaced through attempts to establish an animal model for familial combined hyperlipidemia (FCHL), which is characterized by elevated levels of plasma triglycerides, cholesterol, and apolipoprotein B. Analyses of mouse strains derived from C3H/DiSnA and C57BL/10ScSnA mice identified strain HcB-19 as having dramatically elevated triglyceride, cholesterol and apolipoprotein B levels. The hyperlipidemia gene 1 (Hyplip1) that is responsible for these metabolic disturbances has been mapped to the distal portion of mouse chromosome 3, which is syntenic to human chromosome 1q21-q23. Since this region had been previously shown to contain a gene associated with FCHL, the Hyplip1 gene became a candidate that accounts for FCHL in humans (36). Positional cloning revealed that the Hyplip1 locus is the Txnip gene, where HcB-19 mice have a spontaneous point mutation in exon 2 at amino-acid position 97 from Tyr to a stop codon (24). Although genetic sequencing and association studies in an American, Finnish, and Dutch cohort ultimately did not confirm an association between mutations in the human TXNIP gene and FCHL (62, 225, 314), these studies revealed a crucial role of Txnip in lipid metabolism.
Further characterization of the HcB-19 mouse revealed that elevated levels in triglycerides and free fatty acids in HcB-19 mice were due to increased lipogenesis (71), and that this metabolic profile with increased free fatty acids and ketones was similar in fed as well as fasting HcB-19 mice (274). Since more research groups focused on exploring the functional properties of Txnipin vivo, targeted deletion of the Txnip gene led to the generation of total and conditional Txnip-KO mice by four different laboratories (115, 165, 218, 344). While fed Txnip-KO mice have a similar metabolic phenotype to fasting mice, fasted Txnip-KO mice are actually intolerant to prolonged fasting, being predisposed to death with hepatic and renal dysfunction after 48 h of fasting. The increased levels of pyruvate, lactate, and ketones in these mice suggest a preferred utilization of glucose and decreased Krebs cycle-mediated utilization of fatty acids. This assumption was supported by the observation that fasting-induced death was prevented by supplementation of glucose but not by oleic acid (218). Another study confirmed that Txnip-KO mice have impaired mitochondrial fuel oxidation and increased glycolysis—similar to the Warburg effect that is observed in cancer cells (115). In a recently published study that investigated potential pathways affected by Txnip, a global transcriptomic and proteomic approach revealed the suppression of expression of genes and proteins involved in mitochondrial metabolism. In Txnip-KO hearts subjected to ischemia-reperfusion injury, diminished mitochondrial function was accompanied by enhanced anaerobic glycolysis, which resulted in a net increase of cellular ATP content and a greater recovery of function and decreased infarct size after ischemia-reperfusion injury. Txnip interacts with the PDHE1α-subunit of pyruvate dehydrogenase and inhibits its enzymatic activity, suggesting the redirection of glycolytically derived pyruvate away from mitochondrial toward cytosolic utilization with subsequent anaerobic generation of ATP and lactate (343). Another cellular signaling pathway that is involved in the altered metabolic response to fasting in Txnip-KO mice is decreased in the signaling of the adenine monophosphate-activated protein kinase (AMPK) pathway, which is a major energy sensor in the cells regulating cellular energy homeostasis (11).
In addition to Txnip's role in lipid metabolism, it has also been shown to inhibit adipogenesis (Fig. 14). Txnip-KO mice that were fed a high-fat diet gained significantly more adipose mass than wild-type control mice, and in vitro experiments confirmed that Txnip OE impaired adipocyte differentiation. A potential mechanism for these findings is the regulation of PPARγ expression and activity by Txnip; accordingly, Txnip deletion leads to augmented PPARγ activity (56). Interestingly, Txnip is also involved in controlling whole-body energy homeostasis on the level of the central nervous system, specifically the hypothalamus. Txnip is expressed in nutrient-sensing neurons of the mediobasal hypothalamus and is induced by acute nutrient excess. Down-regulation of Txnip in this area of the brain prevents diet-induced obesity and insulin resistance (22). These studies show that Txnip plays an important role in lipid metabolism on different levels of the mammalian organism.
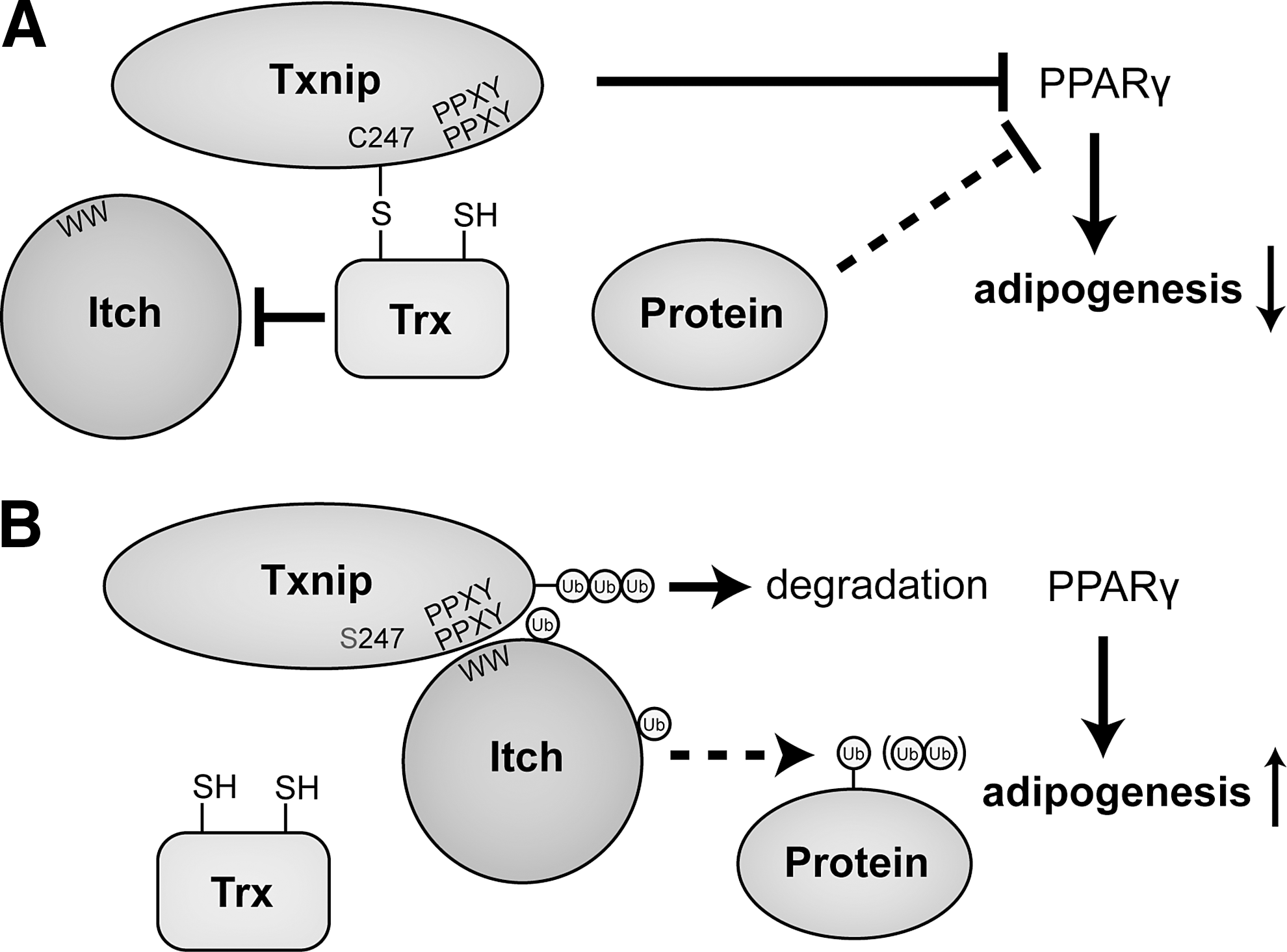
Apart from the changes in lipid metabolism, Txnip also plays a major role in glucose metabolism. The metabolic profile of fed Txnip-KO mice mimicks the fasting state not only with increase in lipogenesis and ketosis but also with marked hypoglycemia (218, 274). Pronounced hypoglycemiais is accompanied by hypoinsulinemia and blunted glucose production after glucagon challenge in Txnip-KO mice, suggesting a defect in hepatic glucose homeostasis. Isolated Txnip-KO hepatocytes have two-fold lower glucose release, while β-hydroxybutyrate release is increased two-fold. OE of Txnip in the KO animals rescues cellular glucose production, but interestingly, OE of the non-Trx-binding C247S Txnip mutant does not (58). The impact that these changes in glucose metabolism through Txnip deletion have on pathologic conditions was shown in a study that examined metabolic adaptation in response to LPS administration. LPS-challenged Txnip-KO mice displayed a predisposition for death without any significant elevation of inflammatory cytokines but with hyperinsulinemia and hypoglycemia. Increased mortality after LPS administration in Txnip-KO mice could be prevented by supplementation of glucose, suggesting that regulation of glucose homeostasis by Txnip might also be an important prognostic factor in pathologies that require metabolic adaptation (219).
The relevancy of these findings for human disease are evident, as Txnip also plays an important role in glucose metabolism in humans. Microarray transcriptional expression profiling showed that TXNIP expression was suppressed by insulin and increased in muscle tissue of patients with diabetes mellitus, consistent with known induction of the TXNIP gene by glucose (228). The question is whether changes in Txnip expression in response to glucose represent adaptive or maladaptive changes that might contribute to the pathogenesis of diabetes. On one hand, increased expression of Txnip leads to increased oxidative stress through reduced Trx activity in vitro and in vivo (262). Since ROS are the key products of different pathogenic biochemical pathways activated by diabetic hyperglycemia, Txnip up-regulation might significantly contribute to diabetic end-organ damage (203). On the other hand, Txnip decreases both basal and insulin-stimulated glucose uptake in insulin-responsive cells (228). This represents a negative feedback mechanism for cells in the setting of hyperglycemia, in which cells may react to increased glucose uptake and metabolism by up-regulating Txnip and inhibiting further glucose uptake. Structure-function analyses of Txnip show that a decrease in glucose uptake is independent of Trx binding, as the C247S mutant form of Txnip that does not bind Trx is still able to inhibit glucose uptake (231). Another member of the α-arrestin family, arrestin domain containing 4 (Arrdc4), has the same effect on glucose uptake, and for both Txnip and Arrdc4, not the C-terminal WW-domain binding PPXY motifs but the arrestin domains themselves are necessary for that metabolic function. These observations suggest again that, while previously thought to function primarily as an inhibitor of Trx, Txnip may actually function primarily as an α-arrestin which is regulated by Trx (231).
Although Txnip-KO leads to hyperlipidemia and increased adipogenesis, insulin sensitivity is actually increased (56, 115, 342). Crossing Txnip-KO mice with ob/ob mice, a mouse model of type 2 diabetes lacking the key metabolic enzyme leptin, leads to improved hyperglycemia and insulin sensitivity (342). As mentioned earlier, deletion of Txnip leads to augmented PPARγ activity, which can explain this metabolic phenotype since PPARγ induces the transcription of a number of genes that regulate glucose homeostasis (56). Increased insulin sensitivity in the Txnip-KO mice is also accompanied by increased Akt signaling, which is a downstream target of insulin signaling leading to enhanced glucose uptake (115, 342). Inactivation of PTEN through oxidation is one way to increase Akt signaling in response to insulin stimulation. Thus, the increased levels of oxidized PTEN found in Txnip-KO mice may link augmented insulin sensitivity and Txnip deficiency (115). Accumulation of NADH indirectly inactivates PTEN by blocking Trx NADPH-dependent reduction and reactivation of PTEN (236). Since mitochondrial respiration is impaired in Txnip deficient cells, the subsequent decreased generation of NADH could explain the increased insulin sensitivity of Txnip-KO mice (115).
In another study that found a surprising role for Txnip, it was shown that Txnip is also involved in inflammasome activation. The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome, composed of the Nod-like receptor protein NLRP3, CARDINAL, the adaptor protein ASC, and caspase-1, is vital for the production of mature IL-1β, and IL-1β contributes to the development of insulin resistance. A yeast two-hybrid screen identified Txnip as a binding partner of NLRP3. Subsequently, it was shown that inflammasome activators induce dissociation of Txnip from Trx in a redox-dependent manner and facilitate binding to and activation of NLRP3. Txnip deficiency impairs NLRP3 inflammasome activation and secretion of IL-1β. Interestingly, Nlrp3-KO mice share parts of the metabolic phenotype observed in Txnip-KO mice, such as improved insulin sensitivity (355). Glucose-induced Txnip up-regulates the expression of IL-1β in human adipose tissue (151). These data show that Txnip functions as a molecular link between oxidative stress and inflammasome activation and highlight that Txnip operates as a redox sensitive regulator in many different relevant cellular pathways, including insulin sensitivity.
Given the implications of Txnip for metabolism, pancreatic beta cells are a logical target of Txnip studies, especially when considering that HcB-19 mice are hyperinsulinemic and markedly hypoglycemic when fasted (116). Transcriptional expression profiling of rat insulinoma INS-1 cells over-expressing Txnip showed that Txnip controls the expression of genes involved in cell apoptosis and survival pathways and insulin secretion, giving rise to the hypothesis that Txnip increases beta cell apoptosis and impairs insulin secretion (191). Glucose induced or forced expression of Txnip in beta cells induced apoptosis in vitro and in vivo (52, 190), which could be inhibited by exanatide, a synthetic version of the glucagon-like peptide-1 analogon exendin 4, which decreases Txnip expression (49). Both HcB-19 and beta-cell-specific Txnip-KO mice have increased pancreatic beta cell mass and are protected against streptozotocin-induced diabetes (51, 183). As a molecular correlate for these findings, in pancreatic beta cells, Txnip shuttles from the nucleus to mitochondria on oxidative stress, a key pathophysiologic factor in diabetes. Through interaction with mitochondrial Trx-2, Txnip decreases binding of ASK1 to Trx-2, thereby allowing for increased ASK1 phosphorylation and activation, resulting in apoptosis (257). It has been further proposed that Txnip deficiency protects against glucose-induced mitochondria-mediated beta cell apoptosis, but not against fatty acid-induced ER stress-mediated apoptosis (50).
Next to peripheral insulin sensitivity and beta cell survival, functional capacity of beta cells is a crucial aspect for maintaining glucose homeostasis. Therefore, it is of interest that in mouse insulinoma MIN6 cells, INS-1 cells and murine pancreatic islets, glucose-stimulated insulin secretion (GSIS) is suppressed with Txnip OE and increased with Txnip deletion, the latter also in ob/ob mice (244, 342). Forced expression of Txnip leads to enhanced expression of uncoupling protein 2 (UCP2) through recruitment of PPARγ coactivator 1α (PGC-1α) to the UCP2 promoter. Since UCP2 is a known negative regulator of GSIS in diabetes, these findings reveal a potential role for Txnip in GSIS (342).
Overall, there is ample evidence that Txnip plays a crucial role in the regulation of multiple metabolic pathways, both in peripheral tissues such as muscle and adipose tissue and in pancreatic beta cells that are central for regulation of glucose homeostasis. The striking metabolic phenotype of the Txnip-KO mouse indicates that these regulatory functions of Txnip are highly relevant in vivo. However, there are still lots of open questions regarding the exact molecular mechanisms, particularly of Txnip's regulation of lipogenesis, glucose homeostasis, and insulin sensitivity. Since the rate at which publications in this field have been published recently is rapidly increasing, it can be expected that new and meaningful findings will eventually add to our mechanistic understanding of the involvement of Txnip in these central metabolic pathways.
c. Cardiovascular system
Changes in cellular redox state and lipid, glucose, and energy metabolism usually translate into functional changes for the cardiovascular system as well. In the previous chapter, we have already mentioned the fundamental metabolic changes that are associated with Txnip-KO in hearts which are subjected to ischemia-reperfusion injury (343). Apart from that, Txnip also plays a regulatory role in other areas of cardiovascular research, especially in pathways that are associated with mechanotransduction.
OE of Txnip leads to decreased cardiomyocyte viability and induces apoptosis (48, 319, 330). On the other hand, both in vitro and in vivo experiments showed that OE of Txnip decreases protein synthesis in response to mechanical strain, phenylephrine (an α1-adrenergic receptor agonist), or Ang II, which leads to decreased hypertrophy after aortic constriction (345). To further investigate the role of Txnip for cardiac function in the setting of pressure overload, cardiac-specific Txnip-KO mice have been generated. While KO mice have attenuated cardiac hypertrophy and preserved left ventricular contractile reserve, the beneficial effects are not sustained over time, and Txnip deletion ultimately leads to maladaptive left ventricular remodeling. Interestingly, these effects are not accompanied by global changes in Trx activity or ROS (344). It has been previously shown that nuclear translocation of Txnip is mediated by importin α1 (205). The mechanism through which Txnip regulates cardiac hypertrophy may be related to Txnip-mediated nuclear translocation of Trx. Trx up-regulates DnaJ homolog, subfamily B, member 5 (DnaJb5), a heat shock protein 40, and forms a multi-protein complex with DnaJb5 and class II HDAC4. Both DnaJb5 and HDAC4 form intramolecular disulfide bonds on oxidation in response to ROS-generating hypertrophic stimuli. These disulfide bonds are reduced by Trx, which prevents shuttling of HDAC4 out of the nucleus, thereby maintaining suppression of target transcription factors such as nuclear factor of activated T cell or myocyte enhancer factor 2, both master positive regulators of cardiac hypertrophy (3). These results reveal the role that Txnip plays in the regulation of cardiac hypertrophy.
Txnip's role in vascular biology is shown by its anti-proliferative effects on vascular smooth muscle cells through suppression of Trx activity (260). Moreover, Txnip also plays an important role in endothelial cells. Steady, laminar flow decreases TNF-mediated vascular cell adhesion molecule-1 (VCAM1) expression, which, in turn, decreases adhesion of monocytes, important effectors of atherosclerosis development. The cellular signaling pathway in this process is the ASK1-JNK/p38 pathway that is controlled by Trx and Txnip. Fluid shear stress generated by normal flow decreases Txnip expression leading to inhibited TNF activation of JNK/p38 and VCAM1 expression, establishing Txnip as a regulator of biomechanical signal transduction and inflammation in endothelial cells (333). In a follow-up study, it was shown that Txnip expression was markedly increased in endothelial cells that were exposed to disturbed flow. Endothelial cell-specific Txnip-KO mice exhibited decreased VCAM1 expression and leukocyte adhesion, both important aspects of vascular inflammation and subsequent atherosclerosis. Mechanistically, Txnip is a transcriptional corepressor of KLF2, a transcription factor that regulates a number of anti-inflammatory genes (318). Interestingly, OE of p21Sci/Cip/Waf1 a CDK inihibitor that is up-regulated by laminar shear stress and inhibits endothelial cell proliferation, suppresses disturbed blood flow-mediated up-regulation of Txnip, suggesting a regulatory connection between the two proteins (215).
Another important aspect of vascular biology is angiogenesis, which includes endothelial cell migrationand VEGF-receptor (VEGFR) signaling. Endothelial cell migration triggered by the different angiogenic factors nicotine, VEGF and bFGF is mediated by the down-regulation of Txnip and increased Trx activity (311). Txnip mediates the translocation of Trx to the plasma membrane of endothelial cells subjected to oxidative stress; there, the Txnip- Trx complex promotes Tyr phosphorylation of plasma membrane proteins, including the VEGFR2 (324). Recently, poly-ADP-ribose polymerase 1 was identified as the mediator of Txnip translocation to the cell membrane of endothelial cells (285). These studies suggest that Txnip participates in different aspects of vascular biology which contribute to angiogenesis.
Mechanotransduction—the signaling pathways that translate mechanical stimuli into molecular signaling and cellular responses—is a major area of interest in cardiovascular biology, as these pathways immediately regulate many relevant pathological processes such as cardiac hypertrophy and vascular atherosclerosis. Txnip seems to play an important role as an adaptor protein for translocation of signaling molecules such as Trx to the nucleus or the cell membrane.
d. Other organ systems
The profound impact that Txnip has on fundamental metabolic parameters such as lipogenesis, glucose levels, and insulin sensitivity led a number of research groups to study the potential role that Txnip could possibly play in the development of diabetic end-organ damage and organs which are crucially involved in the pathogenesis of insulin resistance and diabetes mellitus. Here, a brief overview of studies that investigated the role of Txnip in pathologies of the kidney, the eyes, the peripheral nervous system, and the liver is given.
(1) Kidney
A prominent organ that suffers end-organ damage in diabetes is the kidney. Due to the rising prevalence of diabetes in western societies, diabetic nephropathy has become the leading cause of kidney failure and end-stage renal disease. Thickened glomerular basement membranes and progressive accumulation of extracellular matrix proteins in the glomerular mesangium mediated by glomerular mesangial cells represent an important structural pathogenic correlate in diabetic nephropathy. Administration of high concentrations of glucose leads to p38 MAPK-mediated induction of Txnip in mouse and rat mesangial cells, which is suppressed by N-acetyl-cysteine (78, 246). Gene transfer experiments have shown that Txnip OE leads to the induction and accumulation of extracellular matrix proteins, including collagen IV, which is primarily found in basement membranes (55, 150). Knockdown of Txnip leads to decreased 3H-proline incorporation in cultured mesangial cells, suggesting decreased production of collagen (2). As just described, Txnip is a mediator of high glucose-induced apoptosis through increased activation of ASK1. Txnip knockdown in mesangial cells decreased high glucose-mediated apoptosis in mouse mesangial cells (275). Taken together, these studies reveal that Txnip's role in the regulation of metabolism extends to the regulation of pathways important for the development of diabetic nephropathy.
(2) Eye
In economically developed countries, diabetic retinopathy is the leading cause of new cases of blindness among middle-aged people. The activation of RAGE, one of the pathways responsible for diabetic cellular damage, as well as glucose, induces Txnip expression in retinal endothelial cells, accompanied by the up-regulation of inflammatory markers. Knockdown and OE experiments showed that Txnip mediates the regulation of inflammation triggered by RAGE activation and high glucose in a p38 MAPK-NF-κB dependent manner (237). Knockdown of retinal Txnip in vivo abolished diabetes-induced retinal gliosis and ganglion injury, suggesting a role of Txnip in the pathophysiology of diabetic retinopathy (238).
Retinal ganglion cells are another cell type that is affected by not only retinal neurodegenerative disorders, including diabetic retinopathy, but also glaucoma or retinal ischemia. The administration of NMDA, a model that studies retinal ganglion cell death, induces nitrosative stress through peroxynitrite formation. Since Txnip expression is elevated under these circumstances, subsequent ASK1 activation leads to increased apoptosis of these cells (8). Administration of the calcium channel blocker verapamil inhibits Txnip expression and reverses these effects (7). Given that Txnip is an inhibitor of Trx's function as a denitrosylase (80), it can be speculated that in addition to triggering the dissociation of ASK1 from Trx, the modulation of nitrosative stress by Txnip might be relevant for retinal ganglion cell death, although this remains to be directly shown. In a rat model of glaucoma, which features elevated intraocular pressure, Txnip levels were elevated, while Trx levels were decreased. Transgenic OE of Trx decreases the loss of retinal ganglion cells significantly, suggesting that oxidative stress might be involved in the glaucoma-induced degeneration of retinal ganglion cells (35, 197, 198).
(3) Peripheral nervous system
Diabetic polyneuropathy represents a major therapeutic challenge for the treatment of diabetes. Although little is known about the role of Txnip in the peripheral nervous system, one study investigated the role of RAGE and its ligand S100B in Schwann cell migration, which is important for neuron regeneration after peripheral nerve injury. RAGE induces the expression of Txnip in Schwann cells, and Txnip knockdown experiments showed decreased RAGE-induced Schwann cell migration and abolished RAGE-induced expression of fibronectin and IL-1β (259). Further studies are needed to illuminate the potential role that Txnip might have in disease progression or protection of diabetic polyneuropathy.
(4) Liver
In addition to the role of Txnip for hepatic glucose production just described, several studies have investigated the role of Txnip for pathologies of the liver. Gene expression profiling studies first showed that Txnip expression is higher in livers of responders compared with nonresponders to interferon therapy for chronic hepatitis C virus (HCV) infection (98), and that in human hepatoma Huh7 cell clones with higher HCV replication efficiency, Txnip expression was eightfold higher (122). Gene expression profiling of japanese fulminant hepatitis 1 virus (JFH-1)-infected Huh7 cells serving as a model for HCV infection revealed that Txnip expression is increased over the course of infection. Txnip knockdown decreases viral replication as measured by JFH-1 RNA levels by 85% and viral secretion by 90% (21). Since Txnip is an adaptor between HECT ubiquitin ligases and ESCRT (245), and the ESCRT system is required for HCV production (12), it may be speculated that this could be the mechanism through which Txnip is influencing HCV secretion.
While Txnip deficiency is characterized by marked hyperlipidemia and hepatic steatosis, there are no signs of steatohepatitis in the livers of the Txnip-KO mice, even when fed with a methionine and choline-deficient diet, which typically causes nonalcoholic steatohepatitis (NASH). Thus, oxidative stress, DNA damage, neutrophil infiltration, and hepatic fibrosis are attenuated by Txnip deficiency, while gene expression of fibrosis-inducing and inflammatory cytokine-related genes is decreased, indicating that Txnip might be involved in the pathogenesis of NASH (5).
The studies that are mentioned in this chapter show that Txnip, which is one of the genes most strongly up-regulated by glucose, may also play an important role in the pathogenesis of diabetic end-organ damage in kidney, eyes, and the peripheral nervous system. The data that have been published so far are intriguing, but more studies which actually show a direct interaction between Txnip and certain signaling molecules are required.
4. Conclusion
Since the first description of Txnip in 1994 as Vitamin D3 Up-regulated Protein 1, a large number of studies reveals that Txnip is involved in a variety of different signaling pathways. While initially being characterized as a regulator of redox signaling through its interaction with Trx, it is now clear that the cellular and molecular functions of Txnip go beyond classical redox biology and have established Txnip as a potential link between different intersecting signaling pathways. Especially Txnip's role in metabolism, which includes both Trx-dependent and Trx-independent effects, warrants more studies that investigate the molecular mechanisms in greater detail. In light of recent findings regarding other members of the α-arrestin family which suggest that the α-arrestins might function as adaptor molecules for the regulation of different membrane-bound receptors (199, 232), it is conceivable that similar findings will be made for Txnip as well. In general, structure-function analyses of the α-arrestins will be helpful in advancing the field in the future.
C. Phosphatase and tensin homolog
1. Background
a. Phosphatidylinositol 3-kinase signaling pathway
The phosphatidylinositol 3-kinase (PI3K) pathway is found in organisms ranging from yeast to multicellular systems. The members of the PI3K family were classified subsequent to the discovery of vacuolar protein sorting 34 (Vps34), a PI3K found in yeast that is involved in intracellular membrane trafficking. PI3Ks are intracellular lipid kinases that phosphorylate the 3′-hydroxyl group of phosphatidylinositols and phosphoinositides. Using this mechanism of signal transduction, the PI3K family of proteins regulates cell growth, metabolism, proliferation, migration, and apoptosis (75).
PI3Ks are characterized into three classes (classes I, II, and III) by substrate specificity and structure. Class I PI3Ks primarily phosphorylate phosphatidylinositol-4,5-bisphosphate (PIP2) to form phosphatidylinositol-3,4,5-trisphosphate (PIP3), a lipid second messenger that is important for the signal transduction pathway. The activation of class I PI3Ks is stimulated by Tyr kinase-coupled receptors. Class I PI3Ks are further subdivided into two subgroups according to the signaling receptors that are required for activation. Class 1A PI3Ks are activated by Tyr kinase-coupled receptors, whereas class 1B PI3Ks are activated by G-protein-coupled receptors (75, 141).
At present, little is known about the functions of class II and class III PI3Ks. Class II PI3Ks most likely use phosphatidylinositol (PI) and PI-4-phosphate to form PI-3-phosphate. Similar to the method of class I PI3K activation, class II PI3Ks are also activated by signaling from Tyr kinase-coupled receptors, as well as cytokine receptors and integrins. The mechanisms by which class II PI3Ks are activated by these receptors, however, are unclear (75, 141). Furthermore, class II PI3Ks are believed to be involved in membrane trafficking and receptor internalization by binding clathrin in coated pits (85). The ancestor of PI3Ks, Vps34, belongs to class III PI3Ks. Recent studies have demonstrated that mammalian Vps34 may be important for mTOR activity through sensing available amino acids, thereby regulating cell growth. Furthermore, Vps34 may also regulate autophagy in the absence of nutrients (211, 328).
On Tyr kinase-coupled receptors-mediated activation of class I PI3Ks, PIP3 is generated. PIP3 then recruits proteins that contain the pleckstrin homology domain to the plasma membrane and binds to activate its downstream components. The major effector of PIP3 is the Ser/Thr kinase Akt, also known as protein kinase B. Akt binds to PIP3 and is activated by phosphorylation of the residues Thr308 and Ser473 by kinases 3-phosphoinositide-dependent kinase 1 (PDK1) and mammalian target of rapamycin complex 2 (mTORC2) (44, 81). Once activated, Akt controls gene expression by regulating transcription factors such as the forkhead (FOXO) family of transcription factors. FOXO proteins are involved in growth, metabolism, longevity, and tumorigenesis. On Akt-induced phosphorylation, FOXO is unable to activate the downstream pro-apoptotic factors by being fostered in the cytoplasm, stimulating cell growth and proliferation (44, 75).
Akt activates many downstream signaling pathways. In addition to the activation of the transcription factors of Akt, elevated levels of Akt can also activate mTORC1 by dissociating the tuberous sclerosis complex 1 (TSC1)-TSC2 complex, promoting the activity of G protein Rheb. Akt-mediated activation of mTORC1 is involved in ribosome biogenesis, initiation of translation, and nutrient import, thereby promoting cell survival (44, 327). mTORC1 is also activated by a variety of other signaling factors, including mitogens, growth factors, and hormones. By regulating the protein synthesis machinery, the formation of a translational initiation complex is stimulated, and mRNAs are actively translated under nutrient- and energy-sufficient conditions. Furthermore, mTOR is also involved in the translation of HIF-1α. HIF-1α is involved in the expression of angiogenic factors, including VEGF (288). Akt-mediated up-regulation of HIF-1α stimulates angiogenesis. These data support the idea that activation of the Akt signaling pathway results in induced cell growth and proliferation. The role of mTOR in angiogenesis provides a foundation for cells to exhibit altered characteristics that are detected in cancer cells.
In support of promoting the pro-survival pathway, activated Akt also functions to up-regulate other target proteins such as p53 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-КB). Inhibition of the tumor suppressor function of p53 is suppressed by elevated levels of MDM2, which targets p53 for proteasomal degradation. The loss of p53 stimulates cell growth and proliferation. Up-regulation of the transcription factor complex NF-КB also leads to the activation of pro-survival target genes (316). Furthermore, elevated levels of Akt also inactivate GSK3, which controls glucose metabolism and cell cycle, resulting in the induction of insulin-induced stimulation of glycogen and protein synthesis (59). Therefore, the PIP3-induced Akt signaling pathway up-regulates pro-survival factors and protects the cell from death.
b. Structure and function
PTEN, also referred to as mutated in multiple advanced cancers and TGF-β-regulated and epithelial cell-enriched phosphatase (TEP-1), is a tumor suppressor that shares a high sequence identity with protein Tyr phosphatases and cytoskeletal protein tensin (286). Sequence homology with Tyr phosphatases allows for the dephosphorylation activity of PTEN, whereas homology with tensin suggests that PTEN is involved in focal adhesions (168). Human PTEN is located on chromosome 10q23. As discussed next, the disruption of this region is closely associated with the development of tumors. PTEN is composed of two major domains: the N-terminal domain and the C-terminal domain. The N-terminal domain is composed of exons 1–6 that encode the protein and lipid phosphatase activities of PTEN. The C-terminal domain is composed of exons 6–9 that encode the lipid-binding C2 domain, two PEST domains (rich in proline, glutamate, serine, and threonine) and a PDZ domain (Fig. 15). The components of the C-terminal domain are believed to stabilize PTEN and its interactions with other proteins (223).
Among many methods of control, PTEN expression can be regulated through redox signaling. PTEN is oxidized through the formation of a disulfide between Cys124 and Cys71 under oxidative stress. Oxidation results in an inactive form of PTEN that no longer possesses catalytic activity. PTEN remains oxidized until it is reduced by Trx in a redox-dependent manner. On reduction, PTEN becomes active and regains its catalytic activity (166). Activated PTEN can inhibit the Akt pro-survival pathway through its lipid phosphatase activity. Class I PI3Ks phosphorylate PIP2 to generate PIP3, whereas PTEN dephosphorylates PIP3 to regenerate PIP2(44, 75). The reversal of the PI3K activity by PTEN diminishes Akt phosphorylation and activation, inhibiting pro-survival factors. Consistent with this finding, loss of Akt function in Drosophila resulted in induced apoptosis (287). Inactivation of PTEN results in elevated Akt activity and abnormal growth regulation. Thus, the down-regulation of PTEN could potentially be a target for protection against induced cell growth and proliferation.
2. Regulation
a. Transcriptional regulation
Relatively little is known about the transcriptional regulation of PTEN. It has been previously analyzed that ∼80%–85% of individuals with Cowden syndrome (CS), a disease resulting from altered tumor suppressing activity of PTEN, have germline PTEN mutations within the first eight exons of the gene. Splicing alterations can lead to exon skipping, which can modify the function of PTEN (107, 223). In addition to exon splicing alterations, transcriptional activators of PTEN, including p53, early growth response-1, and NF-κB, were found to bind to the consensus binding sites in the PTEN promoter region. Some patients with CS have also been identified as having mutations either in this promoter region or in the translation start sites (223).
PTEN promoter methylation decreases PTEN expression. The association of PTEN promoter methylation and subsequent silencing of protein expression has been studied in various types of cancer (256). About 25%–30% of women with CS develop breast cancer; however, only 5% of the women who developed sporadic breast cancers exhibited mutations to the PTEN gene. On investigation, it became clear that in patients with CS, the incidence of breast cancer was associated with increased PTEN promoter hypermethylation (88). Furthermore, PTEN promoter methylation has been identified to be associated with nonsmall cell lung cancer (NSCLC), endometrial, and gastric carcinoma (138, 256, 283).
b. Post-transcriptional regulation
PTEN expression can also be regulated by miRNAs. Currently, there are many different miRNAs that target PTEN, including miR∼17–92, miR∼19a, miR∼21, and miR-106b∼25. Of these, miR-106b∼25 has been shown to target PTEN. The activity of miR-106b∼25 is accompanied by the function of the mini chromosomal maintenance-7 (MCM7) gene, which is involved in the transformation of various types of cells. Generally, members of the MCM family are necessary for the initiation of DNA replication. It was found that miR-106b∼25 and MCM7 are located in the same genetic locus, and the up-regulated expression of miR-106b∼25 and MCM7 is detected in prostate cancer (188). These data suggest that the amplification of genes located at this locus can contribute to the formation of other tumors as a result of down-regulated PTEN activity.
c. Post-translational regulation
(1) Phosphorylation and protein oxidation
On phosphorylation, PTEN is inactivated and in response, Akt is up-regulated and shows abnormal growth results. It has been previously shown that the C-terminal region of PTEN is necessary for protein stability, and the inhibition of PTEN by phosphorylation of three specific residues (S380, T382, and T383) by protein kinase casein kinase II (308, 315). Similar to PTEN inactivation by phosphorylation, oxidation also inhibits PTEN activity. In the presence of H2O2, diminished PTEN activity is detected in a time- and H2O2 concentration-dependent manner. During the oxidation of PTEN by H2O2, a disulfide is formed in between Cys124 and Cys71 (166). The oxidized PTEN is, thus, inactive and unable to suppress the Akt pro-survival pathway, resulting in stimulated cell growth and proliferation.
(2) Ubiquitination
Nuclear PTEN has been shown to be deubiquitinated and stable, whereas cytoplasmic PTEN has been shown to be polyubiquitinated and degraded. A recent study indicated that NEDD4-1, the E3 ubiquitin ligase of PTEN, mediates the ubiquitination of PTEN. Polyubiquitination of PTEN results in PTEN degradation, whereas monoubiquitination is important for nuclear import and activation of PTEN (311). Thus, ubiquitination of PTEN modifies the cellular function and localization of PTEN.
(3) Acetylation
PTEN expression is regulated by the direct binding of p300/CBP-associated factor (PCAF), a histone acetyltransferase that regulates gene transcription. The direct association of PCAF with PTEN caused acetylation of PTEN, inhibiting the activity of PTEN. Specifically, PCAF increased the acetylation of two specific lysine residues (Lys125 and Lys128) in PTEN (221). The inactivation of PTEN that suppresses the PI3K signaling pathway results in the inhibition of PTEN-regulated cell-cycle arrest.
d. Localization
In the nucleus, PTEN mutants display centromere breakage and chromosomal translocations. The resulting chromosomal instability is a hallmark of cancer that can stimulate tumor formation and progression. The centromere, a chromosomal domain that is required for accurate chromosomal segregation during mitosis, serves to form a proper assembly of the kinetochore. PTEN was found to be associated with centromer protein C (CENP-C), a centromere-specific binding protein that is essential for forming a functional centromere. PTEN mutants that exhibit centromere breakage result in inaccurate chromosomal segregation. Furthermore, PTEN-deficient cells exhibited spontaneous DNA double-strand breaks (DSBs), resulting in chromosomal translocations. The major mechanism of repairing DSBs is the homologous recombination-directed repair (HDR). HDR is primarily regulated by the members of the DNA repair family, including Rad51. It has been shown that PTEN regulates the expression of Rad51, decreasingspontaneous DSBs and increasing chromosomal stability (272).
Besides its presence and function in the nucleus, the cytoplasmic localization of PTEN has been shown by immunofluorescence (168). In the cytoplasm, PTEN is polyubiquinated and ultimately degraded. A recent study has demonstrated that nuclear PTEN enhances cell death (91). Furthermore, it was found that a specific residue in the C2 domain of PTEN, K289, is necessary for PTEN monoubiquitination that is required for the nuclear import of PTEN (311). Thus, the polyubiquitination of PTEN that sequesters PTEN in the cytoplasm is considered oncogenic in nature. Taken together, these findings indicate that the accumulation of PTEN in the nucleus protects against the growth and proliferation of tumor cells by maintaining chromosomal integrity. Chromosomal instability that occurs in response to PTEN mutations or deletions leads to a higher frequency of genetic alterations in cells, providing a platform for tumorigenesis. Thus, the balance between cytoplasmic and nuclear PTEN may regulate cancer progression.
e. Protein-protein interactions
As just described, the regulation of Akt is significant in determining the fate of the cell. Elevated levels of activated Akt are commonly detected in tumor cells exhibiting characteristics such as increased proliferation and inhibited apoptosis (189). One method of chronic activation of Akt is through increased levels of Trx-1. Trx-1 has many binding partners, including ASK1 and PTEN, that regulate the downstream pathways. OE of Trx-1 results in the activation of Akt and inhibition of apoptosis (282). Similarly, the interaction between reduced Trx and ASK1 stimulates growth and inhibits cell death. In contrast, Trx promotes apoptosis by binding to PTEN. Thus, varying consequences of Trx and its binding partners affect the downstream pathways that lead to controlled cell proliferation and death.
As previously discussed, the disulfide formation between PTENC124 and C71 in the presence of H2O2 is reversible and is primarily reduced by Trx in a redox-dependent manner (166). On reduction of the disulfide, the catalytic activity of PTEN is regained. Therefore, the reversible disulfide formation and inactivation of PTEN provides a powerful mechanism of regulating PTEN activity.
Trx-1 directly binds to the C2 domain of PTEN, inhibiting the lipid phosphatase activity of PTEN. The interaction between Trx and PTEN occurs through the formation of a disulfide bond between Cys32 of Trx-1 and Cys212 of the C2 domain of PTEN (189). In support of this finding, a recent study demonstrated that OE of human Trx-1 (hTrx-1) in Drosophila melanogaster stimulated cell proliferation during the development of the eye. Furthermore, hTrx-1 rescued the small eye phenotype that resulted as a response to the OE of PTEN activity. As expected, hTrx-1 OE leads to an increase in phosphorylation and the activation of Akt (282). Elevated levels of Trx-1 inhibit apoptosis and stimulate cell proliferation and aggressive tumorigenesis. As a result, longevity is decreased. This reveals that increased levels of hTrx-1 could serve to stimulate cell proliferation in human tumors.
PTEN is inactivated through the formation of a mixed disulfide with reduced Trx. However, PTEN activity is also altered in response to Trx regulation by Txnip. On Txnip interaction with Trx, Trx is no longer able to bind and inactivate PTEN. Trx dissociates from PTEN, forming the reduced state and activated PTEN that can inhibit the Akt signaling pathway. A recent study demonstrated that in total Txnip KO mice, elevated levels of Akt, insulin sensitivity, and glycolysis in skeletal muscle and hearts were detected, but not in liver and adipose tissue. Furthermore, the accumulation of oxidized, inactive PTEN in these mice, as well as MEFs derived from the Txnip KO mice, were observed. These MEFs also exhibited faster growth rates and increased dependence on anaerobic glycolysis for energy due to impaired mitochondrial oxidation of fuels. The absence of Txnip resulted in the accumulation of NADH from the impaired mitochondrial respiration that competes with NADPH, inhibiting PTEN reduction and activation by Trx (115). Through the redox mechanism of regulating Trx in the presence of electron transfer cofactors, Txnip indirectly regulates PTEN activity and its downstream signaling components (Fig. 16).
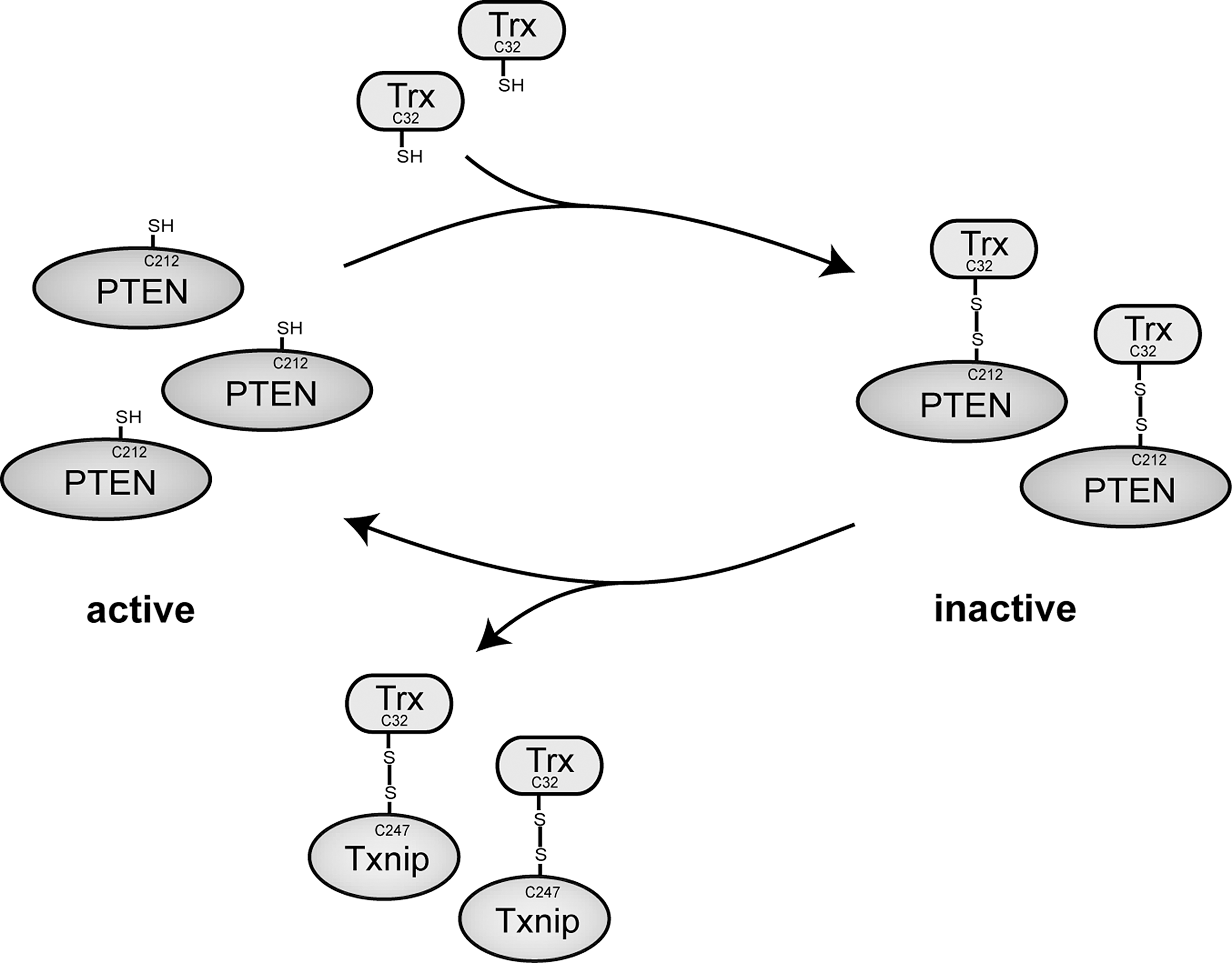
3. PTEN in health and disease
a. PTEN hamartoma tumor syndrome
Previous studies indicate that mutations or deletions of PTEN are frequently detected in certain types of diseases. Inherited mutations of PTEN are most commonly found in clinical syndromes that are collectively known as PTEN hamartoma tumor syndrome (PHTS). PHTS is characterized by neurologic disorders and susceptibility to cancer (44, 106). These syndromes exhibit abnormal cellular growth that often results in benign hamartomas in various organs. Hamartomas are a subtype of benign tumors in which the cells differentiate normally, but are disorganized in architecture. Syndromes that are categorized as PHTS include CS, Lhermitte-Duclos disease (LD), Bannayan-Riley-Ruvalcaba syndrome (BRRS), Proteus syndrome (PS), and Proteus-like syndrome (PLS). In all of the PHTS syndromes, PTEN mutation is detected. The prevalence of PTEN mutation is 80% in CS, 83% in LD, 60% in BRRS, 20% in PS, and 50% in PLS (23, 177, 223).
The prototypic PHTS is CS. CS is defined as an autosomal dominant cancer predisposition syndrome due to its association with increased susceptibility to breast, thyroid, and endometrial cancers (23, 171, 180). As previously discussed, the residue K289 is required for the monoubiquitination of PTEN that allows for nuclear import of PTEN. It was demonstrated that a missense mutation at this site, K289E, is found in individuals with CS. Although PTENK289E did not exhibit significant defects in its catalytic activity, it directed the localization of PTEN to the cytoplasm. Monoubiquitination in PTENK289E was inhibited, thereby inhibiting the intrinsic nuclear import of PTEN. This supports the finding that the residue K289 is essential for the regulation of nuclear import and shuttling of PTEN (311). Taken together, PTENK289E mutant represents a loss of function mutant. Due to the accumulation of cytoplasmic PTEN, the tumor suppressing activity is suppressed, and the susceptibility to disease formation is increased.
b. Embryonic development
PTEN activity is necessary for embryonic development. Silencing mutations in the mouse PTEN gene result in early embryonic lethality. Embryonic stem cells that are derived from PTEN-deficient mice display abnormal embryoid bodies and exhibit altered differentiation of endodermal, mesodermal, and ectodermal derivatives. Furthermore, mice with conditional disruptions in the PTEN gene are more susceptible to the spontaneous development of tumors, such as thyroid and colon tumors (67). Another study showed that certain regions of PTEN-deficient mouse embryos showed increased proliferation (286). These data reveal that PTEN activity is required for normal embryonic development.
c. Cancer
Alterations in the PTEN gene and the mechanisms that promote tumorigenesis have been investigated. Previous studies have demonstrated that the location of the human PTEN gene on chromosome 10q23 raises susceptibility to the development of tumors. It has been postulated that during tumor progression, genetic alterations occur at a high frequency. One alteration that is recurrently detected in multiple tumor types is the loss of heterozygosity (LOH) at chromosome 10q23. Due to the location of the PTEN gene, abnormalities of the PTEN gene are frequently detected in various types of cancers, including brain, breast, and prostate cancer (169). PTEN suppresses tumor cell growth by promoting apoptosis through regenerating PIP2, as well as dephosphorylating phospho-Tyr and phosphor-Ser/Thr-containing substrates (169, 286). Although 10q abnormalities are more frequently detected in advanced tumors, deletions in 10q22–25 are present in various types of cancers, including malignant gliomas, prostate, and breast cancers (33). The tumor suppressor PTEN functions to control cell growth and proliferation by mediating cell death. Therefore, various tumor cells exhibit mutations or homozygous deletions of the PTEN gene.
The tumor suppressing function may be dose dependent, and subtle changes in the tumor suppressor gene expression may change cellular activities. In support of this, a series of hypomorphic PTEN mouse mutants with decreasing PTEN activity (PTENhy/+, PTEN+/−, and PTENhy/−) was created, and the levels of PTEN were measured. This study showed that diminished PTEN expression was accompanied by elevated levels of Akt. Massive prostate hyperplasia and invasive prostate cancer were detected in PTENhy/− mutants. However, these mice did not exhibit an increase in body weight and size. This suggests that the effect of PTEN dose varies according to tissue type. Furthermore, MEFs derived from PTENhy/− mutants exhibited decreased cell growth (310). These findings reveal the importance of PTEN in prostate tumor progression and the significance of subtle changes in the tumor expression gene that lead to the generation of tumor cells. The notion that the effect of the tumor suppressor activity is dose dependent has been studied with a quantitative measurement of another tumor suppressor, the adenomatous polyposis coli (APC) tumor suppressor. APC activity is necessary for suppressing intestinal tumorigenesis. This study demonstrated that LOH of APC resulted in a modest decrease in transcripts and may contribute to attenuated polyposis formation (335).
Although it is clear that the effect of a tumor suppressor varies among different tissue and organ types, it is known that the PTEN deletion alone can cause tumorigenesis in certain tissues. PTEN deletion is found in ∼45% of melanomas, and elevated Akt expression is found in ∼45% (47). However, if PTEN deletion does not trigger tumorigenesis, then PTEN deletion in concert with other genetic alterations contributes to tumor growth (107). Whether it is complete or partial loss of PTEN activity, mutations that alter PTEN function are found in proliferating tumor cells, frequently detected in combination with elevated levels of Akt. At present, inhibitors of the PI3K pathway are being used as therapeutic drugs in an attempt to protect against cancer. PTEN expression controls the pro-survival signaling pathway of Akt and regulates the transformation of normal cells to cells that exhibit abnormal growth characteristics, contributing to tumorigenesis.
(1) Lung cancer
Decreased PTEN activity and the loss of epidermal growth factor receptor (EGFR) in lung cancer result in resistance to gefitinib, an EGFR Tyr kinase inhibitor. Gefitinib prevents various cancer types by inactivating EGFR signaling. The inactivation of EGFR signaling results in diminished Akt phosphorylation as a response to increased expression of PTEN. Although gefitinib has been shown to protect against advanced NSCLC, the addition of gefitinib to chemotherapy did not increase survival in patients with advanced NSCLC (152). Furthermore, increased Akt phosphorylation and activation were detected in subpopulations of an adenocarcinoma cell line that are resistant to gefitinib due to reduced PTEN and loss of EGFR gene mutation (152). From this, we can hypothesize that introducing PTEN activity to cancers that exhibit resistance to gefitinib could be a strategy of cell protection. The re-introduction of PTEN activity in individuals with diseases that result from the loss of PTEN may be a therapeutic target in protection against tumorigenesis.
(2) Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) accounts for ∼6% of all human cancers and more than 600,000 deaths annually worldwide, making it the third most prevalent cause of cancer-related mortality (74). It has been postulated that PTEN inactivation is involved in the development of HCC. PTEN KO studies have demonstrated that heterozygous mice (PTEN+/−) develop neoplasms of the liver. Furthermore, more than 30% of individuals with HCC exhibit a loss of a PTEN allele. These data suggest that PTEN may play a role in liver carcinogenesis. Although contradictory views indicate that PTEN is not involved in HCC development, immunohistochemical analysis demonstrated that PTEN expression was decreased in HCC tissue specimens. Consistent with these findings, the re-introduction of PTEN activity in PTEN-deficient hepatoma cells showed diminished levels of Akt and attenuated tumorigenesis (113).
(3) Prostate cancer
Somatic deletions and mutations of the PTEN gene have been frequently detected in prostate cancer. It has been shown that the loss of PTEN expression in PTEN-deficient mice stimulates tumorigenesis (310). Consistent with the knowledge that PTEN inactivation often results in various types of cancers, PTEN acts as a negative regulator of the PI3K signaling pathway and stimulates prostate cancer formation. Elevated levels of Akt are detected in human prostate cancer cell lines and xenografts lacking PTEN expression (326). As seen earlier, the loss of the tumor suppressor activity of PTEN can lead to cellular transformation and induced tumorigenesis.
(4) Breast cancer
The loss of PTEN activity contributes to the proliferation of tumor cells in individuals with breast cancer. A recent study showed that PTEN deletion is associated with elevated levels of v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ErbB-2) in a transgenic mouse model expressing ErbB-2 under the transcriptional control of its endogenous promoter—ErbB-2 is a receptor tyrosine kinase (RTK) that stimulates cancer. The mammary tumorigenesis was closely associated with elevated levels of ErbB-2. Furthermore, morphologic heterogeneity of tumors was displayed in mice exhibiting PTEN loss and the up-regulation of ErbB-2 compared with mice solely exhibiting upregulation of ErbB-2 (72).
d. Diabetes
Class I PI3Ks play a pivotal role in cell growth and metabolism. The disruption of the PI3K signaling pathways leads to abnormal cell regulation as detected in various types of human malignancies. Mutations of the components in the PI3K pathway often result in human cancers, whereas dysregulation of this signaling pathway may also result in type 2 diabetes. It has been hypothesized that attenuated PI3K signaling downstream of the insulin receptor (IR) contributes to the development of type 2 diabetes (75). Insulin signaling is complex and involves a variety of signaling molecules and proteins in diverse signaling pathways. The two main signaling cascades that are affected by insulin are the PI3K signaling pathway and the MAPK pathway. The PI3K signaling pathway regulates metabolism on insulin signaling, whereas the MAPK pathway regulates cell growth, differentiation, and proliferation. Insulin controls its downstream effectors by stimulating the phosphorylation of IR substrate proteins by the IR, a subfamily of RTKs (301).
On activation in the presence of insulin, IR and insulin-like growth factor-1 receptor phosphorylate substrate proteins that stimulate the generation of the lipid second messenger PIP3. Subsequently, the three known isoforms of Akt are activated by PDK1 and PDK2 (301). Activation of the Akt pathway in response to insulin results in increased glucose uptake into muscle and adipose cells. A recent study showed that PTEN-deficient mice were protected from insulin resistance and diabetes as a result of high-fat feeding in a muscle-specific manner (321). The loss of PTEN function stimulates Akt activity, thereby increasing insulin sensitivity. Consistent with this data, insulin sensitivity and resistance to diabetes were also detected in the adipose tissue of PTEN-deficient mice (156). Therefore, the alterations in glucose metabolism due to PTEN deletion and Akt activation in the absence of cancer development may be used as a target to prevent insulin resistance.
4. Conclusion
Over the past few decades, significant progress has been made in understanding the functions and roles of PTEN in signaling pathways that regulate cell growth, metabolism, proliferation, and apoptosis. Alterations in the PTEN gene and function result in tumorigenesis in both sporadic and inherited tumors. The prevalence of PTEN deficiency in a variety of tumors reveals that the tumor suppressing function of PTEN is crucial in maintaining normal cellular regulation. Thus, methods of treating cancers often target PTEN expression.
Since dysregulation of the PI3K/PTEN/Akt/mTOR pathway has been recurrently detected in many cancers, activators and inhibitors of the downstream components of this signaling pathway have been in use to prevent tumor formation and treat human malignancies. Since a tight regulation of this pathway is critical for cell growth, transformation to malignant cells, angiogenesis, and metastasis in the development of cancer, it is expected that the use of these inhibitors will suppress cell growth, preventing tumorigenesis. Furthermore, targeting these pathways may prevent aging through the stimulation of cellular senescence.
However, the complex network of cellular signaling cannot be easily controlled. Although altered regulation of the PI3K/PTEN/Akt/mTOR pathway can promote the down-regulation of pro-survival markers of tumor cells, the major problem with this mechanism of treatment is that the modified control of this pathway can be detrimental to certain cells. Inhibiting one signaling pathway could modify one or more signaling pathways that could result in altered regulation of cell growth and homeostasis. With many risks at hand, the mechanisms of inactivating the up-regulated PI3K/PTEN/Akt/mTOR pathway in cancers are currently being extensively studied. Down-regulation of this pathway may represent a therapeutic target for cancer and other human diseases.
Footnotes
Acknowledgments
This work was supported by the German Research Foundation (LE 2728/1-1 to S.L.) and the National Institutes of Health (HL048743, HL103582 to R.T.L.).