
Abstract
Increased oxidative stress appears to be of fundamental importance in the pathogenesis and development of several disease processes. Indeed, it is well known that reactive oxygen species (ROS) exert critical regulatory functions within the vascular wall, and it is, therefore, plausible that platelets represent a relevant target for their action. Platelet activation cascade (including receptor-mediated tethering to the endothelium, rolling, firm adhesion, aggregation, and thrombus formation) is tightly regulated. In addition to already well-defined platelet regulatory factors, ROS may participate in the regulation of platelet activation. It is already established that enhanced ROS release from the vascular wall can indirectly affect platelet activity by scavenging nitric oxide (NO), thereby decreasing the antiplatelet properties of endothelium. On the other hand, recent data suggest that platelets themselves generate ROS, which may evoke pro-thrombotic responses, triggering many biological processes participating in atherosclerosis initiation, progression, and complication. That oxidative stress may alter platelet function is conceivable when considering that antioxidants play a role in the prevention of cardiovascular disease, although the precise mechanism accounting for changes attributable to antioxidants in atherosclerosis remains unknown. It is possible that the effects of antioxidants may be a consequence of their enhancing or promoting the antiplatelet effects of NO derived from both endothelial cells and platelets. This review focuses on current knowledge regarding ROS-dependent regulation of platelet function in health and disease, and summarizes in vitro and in vivo evidence for their physiological and potential therapeutic relevance. Antioxid. Redox Signal. 17, 1447–1485.
I. Platelet Function in Health
A. Platelets at work in primary hemostasis
Platelets are only apparently very simple cells, being small, anucleated, and originated from cytoplasmic fragmentations of megakaryocytes. Instead, they possess complicated structural features that are related to their functional and biochemical activities (Table 1). Accordingly, platelet activation is a dynamic process that involves cross-talking among multiple cellular systems. Although the primary physiological function of platelets is to prevent blood loss and maintain vascular integrity through the formation of hemostatic thrombi, platelet signaling pathways contribute to their function and involvement in events beyond their classical role in the hemostasis and in its pathological equivalent (thrombosis), such as inflammation and tumor neoangiogenesis.
Among the different types of organelles in the cytoplasm should be mentioned the presence also of mitochondria, electron-dense bodies, glycogen, vesicles, peroxisomes, and glycosomes.
bFGF, basic fibroblast growth factor; EGF, epidermal growth factor; MMPs, matrix metalloproteases; PDGF, platelet-derived growth factor; PF4, platelet factor 4; TGF beta, transforming growth factor-beta; VEGF, vascular endothelial growth factor; VWF, von Willebrand factor.
Under resting, physiological conditions, platelets circulate passively in the flowing blood, rolling along the intact endothelium. Usually, when the prothrombotic subendothelial matrix is exposed, platelets adhere to altered vascular surfaces or exposed subendothelial matrices. After adhesion, they become activated, change shape, secrete the content of their granules, and aggregate with each other to form the first hemostatic monolayer.
Interestingly, despite significant differences in their functions and signaling pathways, several major platelet adhesion receptors share many similarities in their signal transduction mechanisms. Accordingly, the signaling process could be simplified in sequential stages triggered by agonists/platelet receptor interactions and receptor-mediated early platelet activation signaling, followed by intermediate common signaling events and, ultimately, by integrin activation (“inside-out” signaling) and “outside-in” signaling (Figs. 1 and 2) (186). The whole process will be discussed in detail in the course of the review.

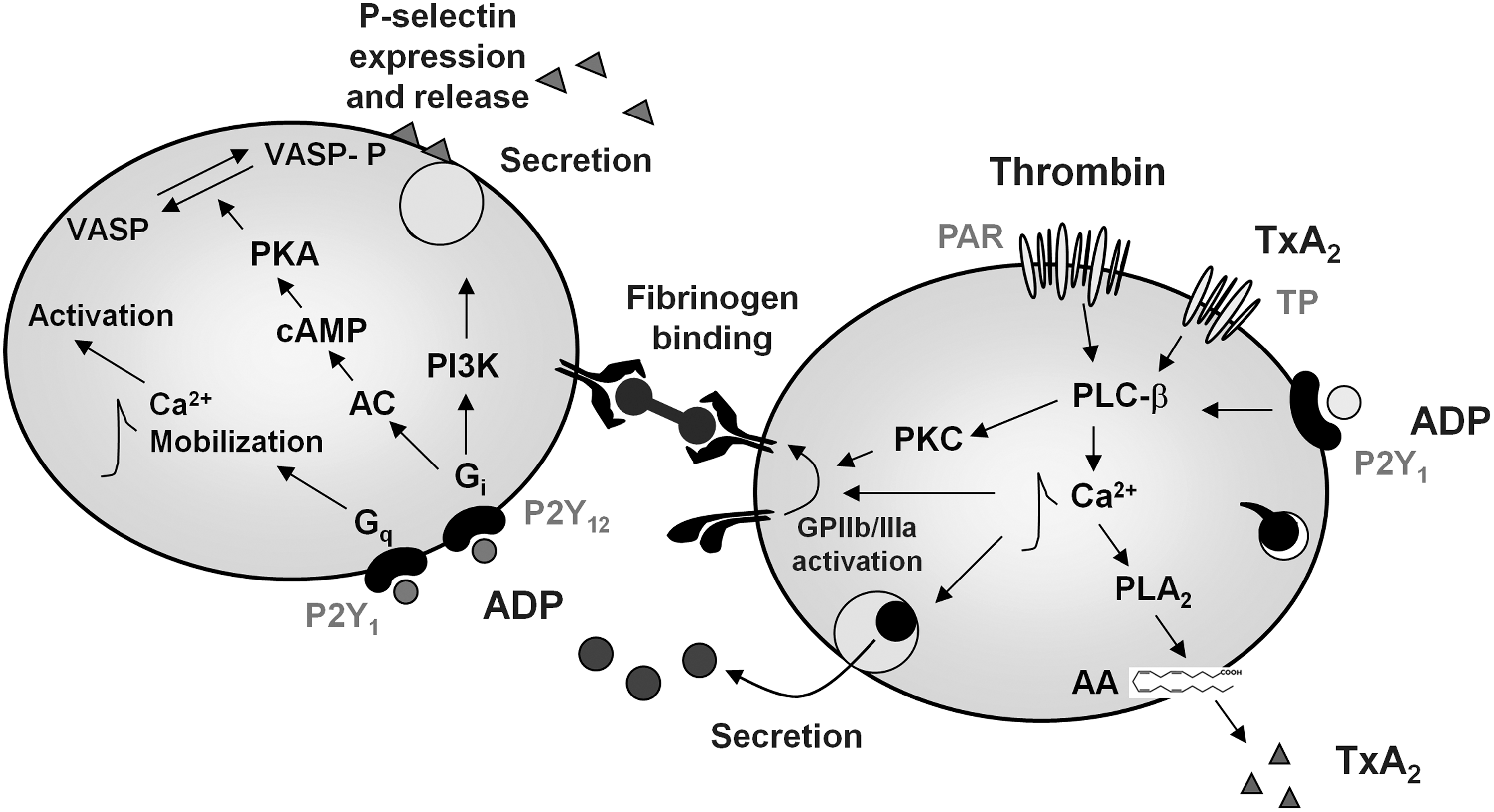
In general lines, an effective primary platelet function requires the combined, synergistic contribution of multiple pathways involving adhesion receptor–ligand interactions, with von–Willebrand factor (VWF)/glycoprotein (GP) Ib (GPIb) interaction playing a pivotal role in initiating the process through which additional platelets are activated and recruited at the site of vascular injury (platelet recruitment) (257), while one or more platelet integrins along with collagen receptor GPVI promote firm adhesion (160) (Fig. 1). These interactions allow the activation of adherent platelets mediated by intracellular pathways that lead to arachidonic acid (AA) metabolism and degranulation. Interactions between integrin adhesion receptors and either counter-receptors on endothelial cells (ECs) or adhesive proteins in the matrix (113, 268) are finely regulated by signals from within the platelet and also trigger platelet responses through mechanisms that propagate information back and forward between the plasma membrane (PM) receptors. This overall process is referred to as integrin signaling (276). The process through which cells regulate ligand binding to integrins is known as “inside-out” signaling or integrin activation, and it is initiated by the binding of one or more agonists to their PM receptors. Once integrins are occupied and clustered by their ligands, they can transmit information to cells (Fig. 1). These “outside-in” signals collaborate with signals originating from growth factor receptors and other PM receptors to regulate a site of anchorage-dependent cellular functions (276).
One of the best studied cases of integrin signaling and the most abundant adhesion molecule present in platelets is GPIIb/IIIa, or integrin αIIbβ3, an integrin receptor present both on the PM and on α-granules, which represents the fibrinogen receptor. It exists as an inactive form in resting platelets, but undergoes conformational changes once the platelets are activated, thus becoming competent to bind soluble plasma fibrinogen (275). αIIbβ3-mediated signaling starts as soon as fibrinogen binds to the integrin. This initial phase of outside-in signaling will contribute toward a further activation of the integrin. αIIbβ3 outside-in signaling results in calcium mobilization, tyrosine phosphorylation of a number of proteins, activation of the phosphoinositide metabolism, and cytoskeleton reorganization (228).
The activating signals triggered by these adhesive interactions initiate a range of platelet biochemical and morphological responses, linked to cytoskeletal remodeling, granule secretion, and generation and release of endogenous soluble agonists, such as adenosine 5′-diphosphate (ADP) and thromboxane A2 (TxA2) (Fig. 2). These processes further help reinforce platelet adhesion and promote the activation of adjacent platelets, recruiting them into the growing hemostatic plug by several feedback amplification loops. The conversion of platelet integrin αIIbβ3 from a “resting” low-affinity state to a high-affinity conformation enables the receptor to firmly bind not only fibrinogen, but also other adhesive ligands, such as VWF and many matrix proteins containing an RGDS (Arg-Gly-Asp-Ser-)-like motif (275). These interactions trigger the series of intracellular events that ultimately cause platelet spreading on the subendothelial matrix, activation of adapter molecules that bind and stabilize the active conformation of integrin αIIbβ3 (67) firm adhesion, full-platelet aggregation, and clot retraction (277).
An increase in intracellular calcium ([Ca2+]i) and the subsequent stimulation of localized Ca2+-dependent signaling processes are critical for the signaling regulated by a complex network of Ca2+ sensors, channels, and binding proteins (Figs. 1 and 2). The whole process of agonist-induced elevation in cytosolic Ca2+ concentrations is essential for platelet activation both in hemostasis and in thrombosis and will be further discussed in a separate paragraph.
As a second messenger, Ca2+ coordinates activation by all agonists under physiological conditions (255). Indeed, an increase in intra-platelet Ca2+ is responsible for a broad range of platelet functional responses, including platelet granule release, integrin αIIbβ3 activation, cytoskeletal remodeling and externalization of phosphatidylserine (PS) on the platelet surface, the latter being a prerequisite for the assembly of coagulation factors (Va, Xa), and the local generation of thrombin, accounting for platelet procoagulant activity (29).
B. Platelet-vessel wall interaction
The vascular endothelium is a multifactorial organ that is able to perceive stimuli (both systemic and local) and modify its functional status to contribute to the homeostasis of the vascular wall (173). Indeed, under physiological conditions, the intimal surface of the healthy endothelium presents both anticoagulant and antithrombogenic properties (165), allowing the exchange of numerous substances between blood and tissues and controlling the vascular tone and trafficking of inflammatory cells toward the vascular bed. Endothelial damage, exposure to certain cytokines or proinflammatory stimuli shifts the balance toward a procoagulant/prothrombotic phenotype of the ECs (173).
Under normal circumstances, platelets circulate without interacting with the intact vessel wall. However, in the event of vessel damage resulting from trauma, endothelial dysfunction, chronic exposure to risk factors, atherosclerosis, or plaque rupture, rapid and complex interactions between circulating platelets and exposed subendothelial structures occur, resulting in the accumulation of platelets at the site of vessel damage that will ultimately lead to the formation of the hemostatic plug (195).
In the initiation phase of primary hemostasis, platelets shift from their physiological resting, inactivated state, roll, adhere, and spread on the exposed extracellular matrix (ECM), which contains a large number of adhesive macromolecules, such as laminin, fibronectin, collagens, and VWF, to form an activated platelet monolayer (307). Depending on the exposed matrix proteins and on the hemodynamic conditions determining the shear stress (defined as the tangential force of the flowing blood on the endothelial surface of the blood vessel), platelet adhesion requires the synergistic function of many platelet receptors that are capable of binding many substrates, including collagens, VWF, and adhesive proteins such as fibronectin, (28), laminin (147), thrombospondin (163), vitronectin (14), and fibulin (121).
The primary tethering of platelets to the damaged subendothelial structures is mediated by the platelet membrane receptor GPIb/IX/V, which interacts with A1 domain of the vessel wall VWF in the exposed subendothelium (307) and by GPVI, which binds to collagen and laminin. VWF is a large adhesive multimeric GP synthesized by ECs and megakaryocytes (261); it is contained in Weibel–Palade bodies of ECs, α-granules of platelets, as well as in the subendothelial matrix (256). In plasma, VWF circulates in a soluble form represented by a series of multimers ranging from 500 to 20,000 kDa with the larger multimers being more hemostatically active. The regulation of VWF multimeric size is regulated through proteolysis by a specific protease ADAMTS13 (92), a plasma metalloprotease that is constitutively active. In addition to its role in mediating platelet-vessel wall interactions, VWF also serves as the carrier molecule for coagulation factor VIII, protecting it from inactivation (320), and acts as a vascular damage sensor by attracting platelets to sites of vessel injury (Fig. 1). Immobilized VWF is sufficient to initiate platelet adhesion under flow, though the kinetics of these interactions varies according to the hydrodynamic conditions (268). A main determinant of the role of VWF on platelet adhesion is shear rate (268), which is a measure of the gradient of flow velocity relative to the distance from the vascular wall. In normal human physiology, shear flow ranges from 50 to 350 s−1 in veins and large arteries, but it considerably increases as the vessels get smaller or under pathological conditions such as stenosed arteries. Indeed, under high shear flow, the VWF-GPIbα interaction is necessary for initial platelet contact with the subendothelium (268), as observed in arterial microvessels, or arterioles (259). Continued platelet recruitment also becomes dependent on VWF-GPIbα as growing thrombi narrow the lumen where blood flows, locally increasing the shear rate up to 20,000–40,000 s−1 (258). The engagement of GPIbα by immobilized VWF elicits typical activation signals such as transient cytoplasmic Ca2+ elevations, protein phosphorylation (phospholipase C [PLC]γ2, ERK-1/2, Syk), TxA2 synthesis, ADP release, and, ultimately, activation of αIIbβ3 (Fig. 1) (98).
The mechanism through which the platelets adhere to the damaged subendothelium involves the classical multistep of tethering, followed by rolling and subsequent firm adhesion, and it is mediated by at least two different types of adhesive molecules, selectins and integrins. P-selectin is stored in α-granules of platelets (288) and in Weibel–Palade bodies of ECs (202), from where it is rapidly expressed on the cell surface on activation (Figs. 1 and 2). Indeed, transient platelet interactions with the morphologically intact endothelium have emerged as an important step in the initiation and progression of atherosclerosis (199). Many inflammatory conditions are associated with endothelial activation and release, and the exposure of P-selectin on the surface of the endothelium may promote platelet adhesion by binding to the GPIb complex on platelets. Thus, the GPIb/V/IX complex may mediate platelet interaction with both the ECM and the vascular endothelium (117). In some inflammatory states, platelet adhesion to the endothelium largely depends on platelet leukocyte adhesive interactions, through the mechanisms mediated by P-selectin glycoprotein ligand-1 (PSGL-1) (328). PSGL-1 has been identified on platelets, where it is functional in mediating platelet–endothelium interactions due to its capability to bind P-selectin in a Ca2+-dependent manner (113).
Apart from interacting with ECM proteins and soluble ligands, platelets can directly bind endothelial structures that are usually not accessible, due to the release of antithrombotic mediators such as nitric oxide (NO) and prostacyclin (PGI2) from intact ECs (62), whose antiaggregatory action is exerted through a synergistic enhancement of cyclic adenosine monophosphate (cAMP) and cGMP content, thus preventing platelet aggregation (90) and ultimately limiting the intravascular extent of forming thrombi.
C. Platelet receptors and signaling during platelet adhesion and activation
A number of physiological agonists interact with specific receptors on the platelet surface to induce activatory responses. A wide variety of transmembrane receptors coat the platelet membrane, including many integrins (αIIbβ3, α2β1, α5β1, α6β1, and αVβ3), leucine-rich repeat receptors (GPIb/IX/V, Toll-like receptors [TLRs]), G-protein-coupled seven transmembrane receptors (GPCRs) (protease-activated-receptor [PAR]-1 and PAR-4 thrombin receptors, P2Y1 and P2Y12 ADP receptors, TPa and TPb TxA2 receptors), proteins belonging to the immunoglobulin (Ig) superfamily (GPVI, FcγRIIA), C-type lectin receptors (P-selectin), tyrosine kinase receptors (thrombopoietin receptor, Gas-6, ephrins, and Eph kinases), and others, in common with other vascular cells (CD63, CD36, P-SGL-1, and tumor necrosis factor [TNF] receptor type) (251).
1. Collagen receptors
Among the macromolecular constituents of the ECM, collagen is considered the main character in the process of platelet activation, as it is involved in platelet adhesion through direct and indirect pathways, and it directly triggers both aggregation and coagulant activity (23). Platelet adhesion and aggregation on collagen are integrated processes that involve several platelet agonists acting through a variety of surface receptors, including integrins, Ig-like receptors, and GPCRs. Indeed, platelets express several collagen receptors (64) with different tasks, as different are the collagens present in the vessel wall, of which collagens I and III are considered the most important in supporting platelet adhesion to the damaged vasculature. However, there are two receptors on the platelet surface that bind directly to collagen, the GPVI Ig-superfamily member and the integrin α2β1. An accessory receptor for collagen-dependent platelet adhesion and activation might be represented by GPV, a GP noncovalently linked to platelet GPIb–V–IX complex (292). GPVI is expressed in platelets and mature megakaryocytes, where it associates with the immunoreceptor tyrosine-based activation motif (ITAM)-containing transmembrane adaptor protein Fc receptor γ-chain (119). On the cross-linking of GPVI by its ligand, ITAM is tyrosine phosphorylated by Src family kinases Fyn and Lyn (104) bound to the cytoplasmic domain of GPVI. After binding to phosphorylated ITAM, the tyrosine kinase Syk undergoes autophosphorylation and phosphorylation by Src kinases and initiates a downstream signaling cascade, leading to the activation of a series of adapter and effector proteins (319). One of the main effector enzymes in the GPVI signaling cascade is PLCγ2, which liberates the second messengers 1,2-diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) (76). Binding of IP3 to its receptors triggers the release of calcium from intracellular stores (Ca2+ mobilization), protein phosphorylation, and AA release, which, in turn, causes the aggregation and secretion of TxA2 and ADP (Fig. 1).
The α2β1 integrin, commonly referred to as GPIa/IIa or CD49b/CD29, also plays a role in the adhesion of platelets to collagen for subsequent optimal activation. Similar to what was observed for fibrinogen receptor, integrin αIIbβ3, α2β1 integrin is expressed on resting platelets in a low-affinity state with the extracellular domains folded into a closed conformation. It has been demonstrated that the affinity of α2β1 integrin for soluble collagen increases on cellular stimulation (162), suggesting that α2β1, similar to αIIbβ3, requires an agonist-induced conformational change (via inside-out signaling) to bind to collagen (218). Moreover, recent evidence also suggests that the α2β1-triggered signaling pathway is characterized by the sequential activation of Src and Syk family tyrosine kinases (153), including Rap1b (27) and Rac1 (293), leading to the activation of PLCγ2 and the formation of lamellipodia (153). It is interesting to note that, contrarily to the ability of α2β1 to mediate full-platelet spreading, the engagement of integrin α5β1 by fibronectin (in the presence of a αIIbβ3-receptor antagonist) generates the formation of filopodia but not lamellipodia, eventually causing a stable adhesion of platelets to the ECM (268). In this regard, there is evidence indicating complex and bidirectional cross-talking between αIIbβ3 and α2β1. Indeed, it has been demonstrated that agonist-induced α2β1 activation is regulated by αIIbβ3 outside-in signaling (304) and that α2β1 promotes the activation of αIIbβ3 and induces fibrinogen binding to adherent platelets through a pathway which requires the active form of Rap1b generated downstream of PLC (27).
2. Fibrinogen receptor
A main adhesion molecule involved in platelet aggregation is the membrane protein integrin αIIbβ3. Integrin αIIbβ3 is present at a high density on platelets, both on the PM and on α-granules. After platelet activation and integrin αIIbβ3 conformational changes, receptor-bound fibrinogen acts as a bridge between two αIIbβ3 molecules on adjacent platelets (Fig. 2) (307). This is the final common pathway of platelet aggregation induced by platelet chemical agonists, and in all circulatory conditions of low shear (123), while VWF substitutes for fibrinogen as a bridge molecule between integrin αIIbβ3 for platelet aggregation induced by high shear conditions (123). Although integrin αIIbβ3 is the most widely studied mediator of bridging platelets to each other and stabilizing thrombi, other molecules have been recently proposed as mediating these responses. These include junctional adhesion molecules, signaling lymphocyte activation molecule family proteins, and CD40 ligand (CD40L) (9). Postligand binding events regulated by integrin αIIbβ3 in platelets include the stabilization of large platelet aggregates, platelet spreading, granule secretion, clot retraction, and platelet procoagulant activity (229).
One of the most intensely investigated areas of platelet biology has been defining the mechanisms by which platelet activating signals regulate the adhesive function of integrin αIIbβ3. Soluble agonist receptors coupled to Gαq stimulate PLCβ, triggering the generation of IP3 and DAG. IP3 plays a central role in promoting intracellular Ca2+mobilization, while DAG promotes the activation of specific isoforms of protein kinase C (PKC). A key step in the activation of integrin αIIbβ3 is the Ca2+- and DAG-dependent regulation of the guaninenucleotide exchange factor 1 (CalDAG-GEF1). CalDAG-GEF1 is a potent inducer of the small GTPase molecule Rap1b, which plays a key effector role in inducing integrin αIIbβ3 activation (72). Activated Rap1b-GTP translocates to the PM, where it binds to its effector, Rap1-GTP interacting adapter molecule (RIAM). RIAM unmasks the talin binding site, allowing talin to bind the cytoplasmic tail of integrin β3 subunits, thereby activating integrin αIIbβ3 (294). The activation of integrins by talin is enhanced by focal adhesion protein co-activators, kindlin-2 and kindlin-3. Kindlins bind to the cytoplasmic tail of β-integrin at sites distinct from those bound by talin. The C-terminal region of kindlin-3 aids the conformational activation of integrin αIIbβ3 by connecting it to the cytoskeletal protein actin (300).
The secretory phase of platelet activation amplifies the initial platelet response to ligands (Figs. 1 and 2). The exocytosis of platelet granules, namely α-granules, dense granules, and lysosomal granules, causes the release of messengers that help recruit additional circulating platelets and contributes toward altering the conformation of the αIIbβ3 receptor, finally leading to aggregation (39). In particular, platelet exocytosis of α-granules releases adhesive molecules such as fibrinogen, VWF, and P-selectin; coagulation and fibrinolitic factors; chemokines such as platelet factor 4 (PF4), RANTES (regulated on activation, normal T cell expressed and secreted), and interleukin (IL)-8; growth factors; and adhesion receptors (247). The exocytosis of dense granules releases nucleotides, including ATP and ADP, serotonin, histamine, and divalent cations. Lysosomal granules release mainly proteolytic enzymes (247).
The model of platelet activation (adhesion, shape change, secretion, and aggregation) involves an organized remodeling of the actin cytoskeleton, mediated by the activation of the small GTP-binding proteins Rho, Rac, and Cdc42. These proteins, which differentially regulate the reorganization of the actin cytoskeleton, exert different roles and lead to the formation of different cellular structures. In particular, Rho activation mainly regulates the Ca2+-independent cell spheration and contractility during shape change through stimulation of the Rho-kinase ROCK; Rac1 activation in platelets is Ca2+-dependent (286), is fundamental for the formation of lamellipodia during platelet spreading (201), and is involved in the regulation of secretion and subsequent aggregation in human platelets stimulated with thrombin (221).
3. G protein-coupled receptors for thrombin, ADP, and prostaglandins
a. Thrombin
Thrombin is produced locally at the surface of the platelets activated by tissue factor (TF) and mediates the generation of fibrin from fibrinogen, which contributes to the formation of the hemostatic plug and platelet thrombus growth (39). Platelet response to thrombin is mediated by PARs (Fig. 2). Human platelets express two PAR receptors, PAR-1 and PAR-4, both coupling to activator Gq and G12/13 (69) proteins resulting in the activation of PLC, Ca2+ mobilization, and PKC activation (324). Among the G protein-coupled receptors, PAR-mediated activation occurs through the proteolytic cleavage of the receptor by thrombin and unmasking a specific ligand (68). Thrombin binds to the extracellular domain of PAR-1 and PAR-4, which are then cleaved to form a new amino terminus with its tethered peptide ligand; the tethered ligand activates the receptor and induces transmembrane signaling. Thrombin signaling via either PAR-1 or PAR-4 induces platelet activation, shape change, and granule release; PAR-1-dependent responses are evident at lower thrombin concentrations than those induced by PAR-4 (164). Available data suggest that PAR-4 activation is not necessary for robust responses in human platelets when PAR-1 function is intact (68).
It seems likely that the differences in the kinetics of PAR-1 and PAR-4 signaling may allow thrombin to trigger distinct events, such as specific transcriptional programs, depending on which receptor is activated (142). Thus, the existence of two receptors that signal with distinct kinetics might allow thrombin to elicit distinct responses in cell types which express different receptors or even produce distinct effects in the same cell type, depending on thrombin concentration. Indeed, with regard to calcium signaling, it has been demonstrated that store-operated Ca2+ entry (SOCE) is important at low thrombin concentrations in that it amplifies and sustains Ca2+ signaling in response to low concentrations of thrombin, whereas both SOCE and noncapacitative Ca2+ entry become increasingly important as thrombin concentrations increase (131).
b. Adenosine 5′-diphosphate
ADP plays a pivotal role in platelet involvement both in hemostasis and in thrombosis, exerting its effects through two GPCRs, P2Y1 and P2Y12 (Fig. 2) (115). P2Y1 is coupled to Gαq, which leads to an increase in cytosolic Ca2+ through the stimulation of PLCβ (160), PKC activation, and platelet shape change, whereas P2Y12 is coupled to Gαi, and has the dual function of inhibiting adenylate cyclase while concomitantly activating phosphatidylinositol 3-kinase (PI3-K) (155). PI3K stimulation leads to the activation of AKT and Rap1B (194). For platelet-platelet adhesion, and, thus, thrombus formation, activation of the fibrinogen receptor αIIbβ3 requires the concomitant stimulation of Gαq and Gαi signaling pathways (160).
c. Prostaglandins
Prostaglandins (PGs) are formed by the actions of cyclooxygenases (COX) 1 and 2 on AA and exert their effects through the actions of eight known receptors. PGE2 can bind at least four receptor subtypes (EP1–4 receptors), whereas PGs D2, F2α, I2, and TxA2 each have a single receptor (DP, FP, IP, and thromboxane/prostanoid [TP] receptors, respectively) (217). The binding of PGE2 to the different G protein-coupled receptor subtypes results in diverse and often opposite effects on platelet function (217), depending on the concentration of PGE2 and the conditions used. Indeed, while the interaction with EP3 receptors promotes platelet function, resulting in the elevation of free intracellular Ca2+ levels, stimulation of the EP2 and EP4 receptors inhibits platelet function by increasing intracellular cAMP levels (124); thus, the overall effect is the result of the balance between these opposing actions (230).
TxA2 is a potent platelet stimulator. On exposure to agonist stimulation and Ca2+ enhancement (which implies both sustained elevation of cytosolic Ca2+ coupled to the influx of extracellular Ca2+), PLA2 causes AA hydrolysis from membrane phospholipids and converts it into TxA2 by sequential oxygenation via COX-1 and TxA2 synthase (215). The released TxA2 acts on its receptor by inducing not only platelet aggregation but also smooth muscle cell contraction (118).
Only one gene encodes for TPs, but it can be alternatively spliced in the carboxylterminal tail (C-tail) leading to two variants, TP and -β, that share the first 328 amino acids (65). Human platelets express both variants. TPs are physically associated with Gαq and G12/13. Since platelet Tps do not couple directly to the Gi family members, platelet aggregation induced by TxA2 requires the secretion of ADP to inhibit adenylyl cyclase. Indeed, TxA2 acts as a positive feedback mediator recruiting even more platelets to the primary hemostatic plug, causing ADP release (187) that exerts its effects through P2Y1 and P2Y12 (115).
4. Calcium signaling
An increase in [Ca2+]i is a major signal for platelet activation and accompanies activation by all agonists under physiological conditions (255). It occurs through Ca2+ release from intracellular stores and Ca2+ entry through the PM. Ca2+ is initially released from the intracellular stores of the dense tubular system (DTS) in response to the formation of IP3 from PM phosphatidyl inositol-(4,5)-bisphosphate. This initial release from intracellular stores leads to the depletion of the cytosolic stores and triggers the influx of extracellular Ca2+ via store-mediated Ca2+ entry (SOCE) (253). The other major Ca2+ entry mechanism is mediated by the direct receptor-operated calcium channel, P2X1. The increase in [Ca2+]i is very rapid, and it is counteracted by a slower return to lower levels brought about by sarcoplasmic/endoplasmic reticulum Ca2+ ATPase and by plasma membrane calcium ATPase (PMCA), which pump Ca2+ back into the stores or through the PM out of the cell, respectively. PMCA is a highly regulated transporter whose function is to maintain low [Ca2+]i by catalyzing ATP-dependent Ca2+ efflux (46). In human platelets, where a secretion-like coupling mechanism has been demonstrated, Ca2+ entry is proposed to be based on the reversible trafficking of portions of the Ca2+ stores toward the PM to facilitate de novo coupling between the type II IP3R receptor in the store membrane and naturally expressed human canonical transient receptor potential 1 (hTRPC1) in the PM (254).
Ca2+ regulating mechanisms are of great importance for pathological thrombus formation, rendering Ca2+ entry in platelets a promising therapeutic target for both the prevention and treatment of ischemic events. However, since the description of the many proteins involved in Ca2+ homeostasis in platelets is beyond the focus of this review, we refer to more extensive reviews in the field (306).
D. Redox mechanisms
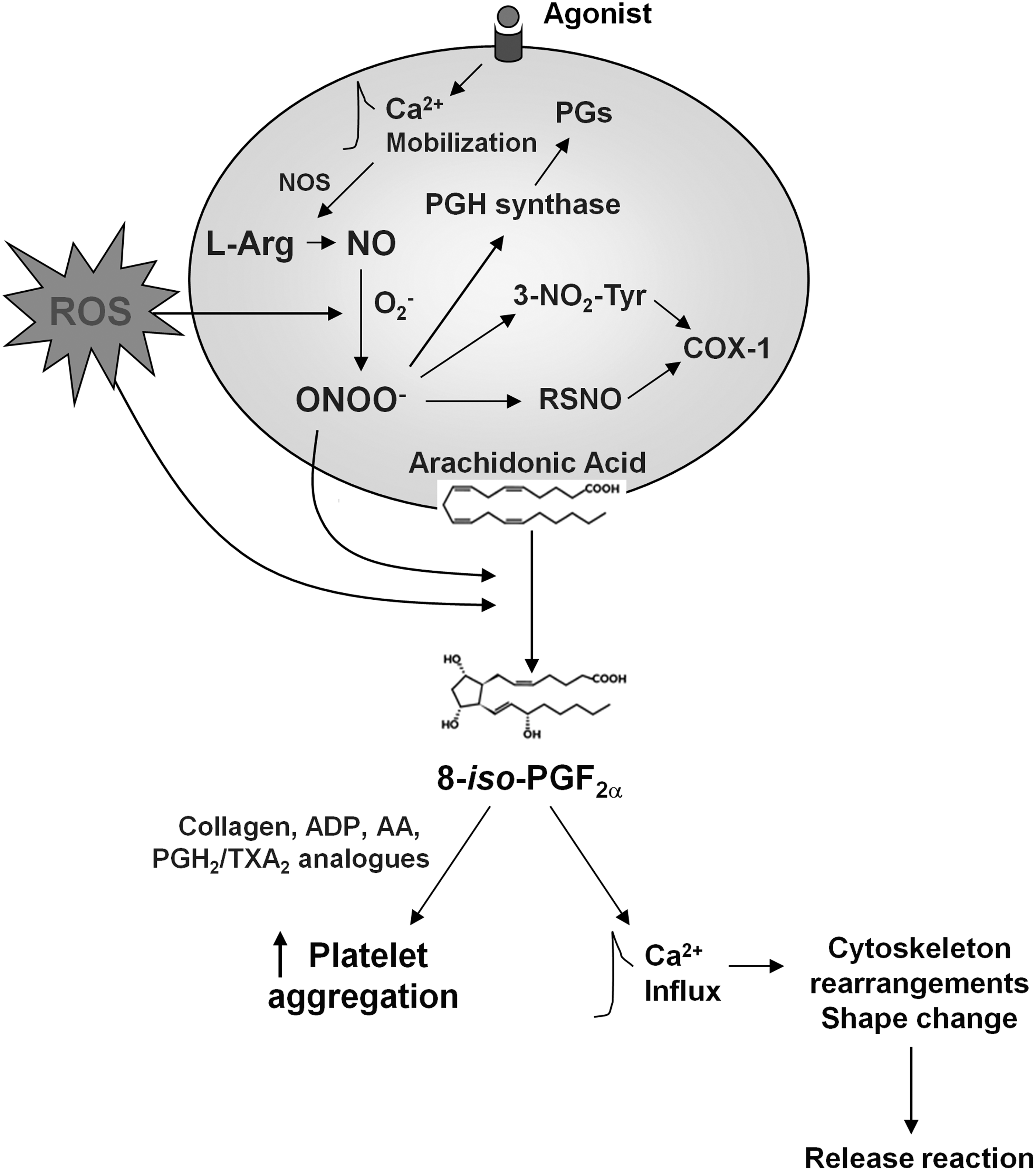
Oxidative stress is thought to be a key event in the initiation and progression of atherosclerotic disease (132). Since platelet function in the development of atherosclerosis is well established, oxidative processes and platelet redox status may have influential effects on the homeostasis of vasculature. On the other hand, oxidant stress and decreased antioxidant levels found in many cardiovascular (CV) diseases are also associated with changes in platelet function. Therefore, platelets represent a prime target for reactive nitrogen and oxygen species (RNOS) produced or released in the vascular lumen (Fig. 3) (177).
1. RNOS generating systems
RNOS include superoxide anion (O2 −), hydrogen peroxide (H2O2), hydroxyl radical (OH·), hypochlorous acid (HOCl), and peroxynitrite (ONOO−) (146). The key modulators of cellular redox regulation are NO and O2 −, which can react with each other, thus causing the formation of ONOO−. The sources of RNOS are a variety of cell types, including vascular smooth muscle cells (VSMCs), ECs, and mononuclear cells. Several enzyme systems contribute to the production of RNOS in vascular tissues, including xanthine oxidase, the nicotinamide adenine dinucleotide (phosphate) (NAD(P)H) oxidases, mitochondrial sources, and NO synthase (NOS) (296).
Vascular ECs are tightly regulated by endothelium-derived NO generated via the catalytic activity of a constitutively expressed NOS, an oxido-reductase found in many cell types, also known as endothelial NO synthase (eNOS) (110). Vascular NO relaxes blood vessels, prevents platelet aggregation and adhesion, limits the oxidation of low-density lipoprotein (LDL) cholesterol, inhibits the proliferation of VSMCs, and decreases the expression of pro-inflammatory genes that advance atherogenesis (109,110). Platelet biosynthesis of NO occurs from
eNOS can be activated in either a Ca2+/calmodulin-dependent or a -independent manner, depending on the stimulus (107, 137), and several kinases (PKA, Akt, calmodulin-dependent kinase II, or AMP-activated protein kinase) appear to be involved in eNOS phosphorylation and activation. Among the kinases, Akt, a Ser/Thr kinase, appears to play a pivotal role in agonist-induced platelet activation and modulation of eNOS (325). In platelets, the β2-adrenoceptor–mediated activation of NOS is adenylyl cyclase-dependent but is not associated with any change in [Ca2+]i (241), suggesting that platelet eNOS is largely Ca2+-independent (107). Indeed, the rate of Ca2+-independent platelet eNOS activity regulation relies on the phosphorylation/dephosphorylation of Ser1177 and/or Thr495 residues. Phosphorylation of Ser1177 residue activates eNOS, while phosphorylation of Thr495 residue, being the negative regulatory site, inhibits the activity of the enzyme (243). There is evidence that persisting oxidative stress will render eNOS dysfunctional such that it no longer produces NO, but O2 − (108).
NO bioavailability in vivo depends mainly on the inactivation induced by superoxide, with the subsequent generation of novel and more RNOS, including ONOO−. ONOO−, besides causing the initiation of lipid peroxidation, nitration of aromatic compounds, and modifications of sulfide residues, was also found to cause Tyr-residue nitration in PGI2 synthase and Mn-superoxide dismutase (SOD) and to participate in providing the peroxide tone for COX (Fig. 3) (16). Indeed, COX-1, also known as PG endoperoxide H2 synthase-1 (PGHS-1), is converted into an active form in which a ferric heme and a tyrosine (Tyr) residue are converted to a ferryl species and a Tyr-radical, respectively. The interaction of this Tyr-radical with the substrate AA forms an arachidonyl radical, which interacts with molecular oxygen to form 15-hydroperoxy-prostaglandin-9,11,-endoperoxide (PGG2) (270). Subsequently, PGG2 is reduced to the 15-hydroxy derivative PGH2, which represents the substrate for many prostanoid synthases, the most relevant of which in human platelet activation is TxA2 synthase (130). The initiation and maintenance of the PGHS catalytic cycle by peroxides was termed “peroxide tone,” requires a steady presence of peroxides or ONOO−, and plays an important role in platelet-derived TxA2 synthesis (270). In conditions of resting equilibrium, in which the uncontrolled formation of TxA2 has to be prevented in order to avoid inappropriate platelet activation, the PGHS-2 isoform is prevalent, thus ensuring a protective release of the antiaggregatory molecule PGI2. Conversely, under pathophysiological circumstances, associated with enhanced oxidant formation, PGI2 formation is inhibited, while TxA2 formation is enhanced (332).
NO bioavailability in vivo also depends on the presence of endogenous NOS inhibitors. These inhibitors take the form of methylated arginine derivatives such as asymmetric dimethylarginine (ADMA) and Nω-monomethyl-
Finally, NO insufficiency can be attributed to limited substrate/cofactor availability as well as interactions with RNOS. A balance of endothelium-derived NO and RNOS modulates endothelial function, while an imbalance between NO and RNOS, so-called oxidative stress, is involved in endothelial dysfunction through the inactivation of NO.
Platelets are capable of producing ROS, such as O2 − and H2O2, during activation (86) (Fig. 3). The first evidence of the release of O2 − and other ROS by platelets was observed by Marcus in 1977 (197).
Increased oxidative stress promotes platelet aggregation, while NO inhibits coagulation and platelet activation. The ROS derived from both platelets or other vascular sources represent modulators of platelet activity to such an extent that it has been shown that the ROS generated by platelets have a direct role in the control of their activity (120).
Agonists that induce platelet activation also activate the platelet isoform of NADPH oxidase through the activation of a gp91phox-dependent enzyme (231). The production of O2 − by platelets that is dependent on NADPH oxidases enhances the recruitment of platelets to a growing thrombus, most likely by inactivating a platelet ectonucleotidase, thereby increasing the bioavailability of ADP. As already stated, the formation of peroxynitrate impairs the antiplatelet activity of NO. O2 − can be converted to H2O2 by SOD; SOD and NO are competitors in O2 −scavenging. H2O2 also serves as a substrate for the production of other detrimental ROS, such as HOCl, generated by enzymatic conversion by neutrophil myeloperoxidase. In addition, H2O2 reacts with ferrous iron (Fe2+) to generate ferric iron (Fe3+) and the OH·. Despite playing an important role in oxidative stress and oxidative biosynthesis, evidence continues to accumulate that H2O2 functions as a signaling agent. Indeed, endothelial-derived H2O2 has proved to be a necessary component of the pathway regulating flow-mediated dilation in human coronary arterioles (192, 208).
In the presence of catalase, H2O2 is degraded to water and oxygen. Glutathione peroxidase (GPX) also exerts an antioxidant enzymatic function, because it catalyzes a reaction that degrades H2O2 by oxidizing reduced glutathione (GSH) to its disulfide form (GSSG) (for an extensive review, please refer reference 86). GSH has proved capable of potentiating platelet aggregation at concentrations (and at a ratio of GSH/GSSG) found in blood in various disease states (102). Intracellular glutathione also plays a role in platelet activation, probably by maintaining the sulfhydryl status of cytoplasmic proteins (101).
It is likely that the source of ROS, the subcellular localization of the ROS generation, and, potentially, the redox-sensitive pathway are dependent on the stimulus. Indeed, recent data indicated that thrombin-induced ROS production through PAR-1, or after membrane depolarization (177), was extracellular, while after collagen stimulation, probably mediated by GPVI, ROS production was intracellular (18). This is consistent with recent reports demonstrating that receptor-stimulated ROS generation is compartmentalized and/or targeted (295).
In patients with hypercholesterolemia, platelet-associated NAD(P)H oxidase produces a thrombogenic phenotype and mediates the arteriolar dysfunction and venular blood cell recruitment (291) [via platelet CD40 and P-selectin (290) and vessel wall CD40/CD40L interactions (105)]. The soluble form of CD40L, sCD40L, mediates the stimulation-induced platelet release of RNOS through the activation of Akt and p38 mitogen-activated protein kinase (MAPK) signaling pathways. These data suggest that sCD40L plays an important role in regulating platelet-dependent inflammatory and thrombotic responses (55). Relative to oxidative stress in platelets, redox-sensitive CD40/CD40L interactions specifically induce the activation of Akt and p38 MAPK (55).
Increased oxidative stress enhances CD40L surface expression in thrombin and GPVI-stimulated platelets, suggesting that platelet-derived CD40L surface expression is redox regulated (18, 111). Accordingly, antioxidants mediate the inhibition of agonist-induced CD40L surface expression (232).
A result of enhanced oxidative stress is the formation of isoprostanes by the peroxidation of AA esterified to membrane phospholipids and that is subsequently released, by PLA2 activity to the blood stream (214, 216). It has been demonstrated that although many isoprostanes are present in very tiny concentrations, their biological effects may add up synergistically to biologically relevant actions (26). Isoprostanes may, thus, link lipid peroxidation seen in CV disorders to enhanced platelet activation (81). One mechanism by which isoprostanes are formed is via redox-generated protein tyrosyl radicals at Tyr385 of the COX, which may further enhance peroxidase activity of COX and thereby aggravate oxidative stress and isoprostane formation (122).
Platelet stimulation leading to various activatory responses, including aggregation and secretion, also considers the involvement of thiol groups or the rearrangement of disulfide bonds. Indeed, it has been suggested that a prerequisite for platelet responses is represented by sulfhydryl groups in platelet surface proteins (101). Sulfhydryl groups that are in proximity to each other such that they undergo reversible dithiol/disulfide conversions are considered vicinal thiols. The mechanism through which protein disulfide isomerase mediates the activation of αIIbβ3 (180) and of other platelet integrins, including α2β1 (179), might be the result of exposure or generation of vicinal thiols both in the αIIb and in the β3 subunits. Platelet redox mechanisms or low-molecular-weight thiols found in the external redox environment possibly regulate the redox-sensitive sites in both integrin subunits. Moreover, it has been suggested that a transplasma membrane oxidoreductase generates thiols from disulfides on the platelet surface (101). Modifications of protein thiols or amino groups might represent an important cause of the lipid peroxidation-mediated inactivation of different membrane-bound enzymes, including protein phosphatases (PTPs). Being the counterpart of protein tyrosine phosphorylation, PTPs are key elements of signaling pathways that regulate the physiological functions of platelets. In particular, PTP activity is essential to avoid inappropriate platelet activation or to bring platelets back to a resting state (185).
2. Antioxidant mechanisms
On the other hand, platelets also express antioxidant enzymes (248). This corroborates the role of ROS in platelet signaling, as these antioxidant enzymes likely not only prevent the cytotoxic effects of ROS but also regulate oxidation-sensitive signaling pathways in platelets (177). Antioxidant status is an important determinant of platelet function: an imbalance between the activities and the intracellular levels of these antioxidants is associated with an increase in ROS production. Antioxidants may exert an indirect inhibition of platelet function by scavenging ROS.
Enzymatic antioxidants include SOD, GPx, and catalase (302), while nonenzymatic antioxidants are represented by ascorbic acid (vitamin C), α-tocopherol (vitamin E), glutathione, carotenoids, flavonoids, and other molecules.
SOD serves as antioxidants by catalyzing the dismutation of superoxide into oxygen and H2O2; GPX reduces lipid hydroperoxides to their corresponding alcohols and reduces free H2O2 to water (108). Hydroperoxides produced by the platelets (PGG2, 12-HpETE, and PLOOH) are metabolized by GPx. Glutathione depletion in platelets leads to attenuated GPX activity, decreased levels of α-tocopherol, and increased lipid peroxidation (43). Catalase promotes the decomposition of H2O2 to water and oxygen.
Vitamin C (l-ascorbate) is considered the most efficacious water-soluble antioxidant in human plasma. This molecule effectively scavenges various RNOS and regenerates α-tocopherol from its radical species (200).
Molecules with vitamin E activity scavenge lipid peroxides. However, α- tocopherol itself becomes, in turn, a radical (the tocopheroxyl radical) with potentially prooxidant activity (108). The platelet inhibitory properties of vitamin E supplementation do not appear to be entirely irrelevant, as supplementation is associated with increased hemorrhagic stroke (2). A study carried out with a mixed tocopherol preparation rich in α-tocopherol demonstrated that such a mixture caused increased NO release, endothelial constitutive NOS activation, and SOD protein content in platelets (191).
Flavonoids are polyphenols (anthocyanins) that display several biologic activities in the CV system by interfering with some of the basic mechanisms involved in the provocation of and in the protection from ischemic CV disease, such as oxidative stress and ROS production, NO formation and activity, and the expression of adhesive molecules by blood cells and the vascular wall (126).
Besides antioxidant enzymes, platelets express scavenger receptors, including CD36. Platelet CD36 is associated with nonreceptor tyrosine kinases of the Src family (151), which have previously shown to be implicated in platelet activation by oxidized LDL (oxLDL) (198). CD36-dependent signaling cascade responsible for oxLDL-dependent activation of platelets includes the Src kinases Fyn and Lyn, the upstream MAP kinase kinase 4, and the MAP kinase JNK2 (56). It has been suggested (233) that platelet CD36 might serve as a sensor of specific oxidized phospholipids generated during oxidative stress. In this review, CD36 engagement might induce an additive activating signal that, in synergism with signals from other receptors, might cause platelet activation by subthreshold concentrations of physiological agonists, thus potentiating members of the platelet activation pathway. Unlike other “co-receptor” ligands, which are primarily localized to the platelet surface during the initial phase of platelet aggregation, CD36 ligands are likely to be presented to the platelet surface before “classical” platelet agonists (233); thus, CD36 may sensitize the platelet for subsequent activation.
Another interesting scavenger receptor present in platelets is the class B scavenger receptor SR-BI, a multiligand receptor of the CD36 superfamily, which seems to be the major receptor on platelets for oxidized high-density lipoprotein (oxHDL) (301). Its major physiologic functions are selective uptake of cholesteryl esters from HDL in steroidogenic tissues and liver (176), stimulation of the bidirectional flux of free cholesterol between cells and lipoproteins, modification of membrane cholesterol distribution, and triggering of signaling events (326).
The findings in animal models have established the key role of hepatic SR-BI expression in the process of reverse cholesterol transport that has proved to be critical in maintaining adequate cholesterol homeostasis and in the prevention of CV disease. If SR-BI plays similar roles in HDL metabolism and protection against coronary artery disease (CAD) in humans as those reported in mice, then it would represent a new target for an antiatherogenic therapeutic approach (184). However, although human SR-BI has the same binding (42) and lipid trading (91) activities as rodent SR-BI, its role in human physiology requires further investigation. A fascinating hypothesis is that LDL binding and cholesteryl ester selective uptake by human SR-BI may contribute to LDL-cholesterol clearance, provided that LDL can bind to SR-BI in the presence of HDL.
To date, the major evidence that SR-BI might be relevant in controlling HDL cholesterol levels and metabolism in humans comes from recent studies of a family with a mutation in the SR-BI gene and increased levels of HDL cholesterol (309). This mutant form of SRBI results in a decreased uptake of cellular cholesteryl esters compared with wild-type SR-BI, indicating that the abnormal HDL cholesterol levels found in these patients might be due to a reduction in the functional activity of this mutant receptor (309).
E. Platelets and the immune system
Studies on platelet postcontact signaling have demonstrated an intense cross-talk among platelets, the coagulation cascade (141), ECs, and leukocytes, thus linking inflammation and atherogenesis (117, 181).
Platelets respond to stimuli by producing a broad array of inflammatory mediators, including CD40L, RANTES, platelet-derived growth factor (PDGF), and macrophage migration inhibitory factor (73). Moreover, it is widely accepted that platelets modify the properties of ECs in a way that facilitates the penetration of lymphocytes and macrophages into the arterial wall, thus favoring the thrombotic complications of atherosclerosis (189). This evidence reveals a synergism between inflammation and thrombosis in the pathobiology of atherothrombosis.
In this setting, the accumulation of activated platelets at sites of vascular lesions participate in the biology of atherosclerosis by producing inflammatory mediators such as CD40L, myeloid-related protein-8/14, and PDGF, as well as directing leukocyte incorporation into plaques through platelet-mediated leukocyte adhesion. CD40 binding to its ligand, CD40L, is likely to contribute to the platelet responses elicited by hypercholesterolemia, as evidenced by studies describing elevated CD40L expression in platelets and increased plasma levels of sCD40L in hypercholesterolemic patients (116, 262). Indeed, the CD40/CD40L dyad has proved a critical component of the mechanisms that underlie the endothelial dysfunction on both the arteriolar and venular sides of the microvasculature during hypercholesterolemia in a recently described model (289) wherein hypercholesterolemia induces microvascular responses with the involvement of T-cell-associated CD40L, arteriolar dysfunction through a pathway involving platelet P-selectin (290), and NAD(P)H oxidase activation (291).
Platelets express components of the bacterial lipopolysaccharide (LPS) receptor-signaling complex, including TLR2, 4, and 9 molecules (8). Platelet TLR4 detects TLR4 ligands in the blood and induces platelet binding to adherent neutrophils, leading to robust neutrophil activation and the formation of neutrophil extracellular traps for bacteria in blood vessels (63). It has been recently demonstrated that LPS mediates platelet activation by interacting with the TLR4-MyD88 receptor-signaling complex and activates the NO/cGMP pathway, thus stimulating platelet granule secretion, leading to the potentiation and amplification of platelet activation and aggregation (330).
Stimulation of the immune TLR2 on the platelet surface leads to P-selectin expression, activation of integrin αIIbβ3, and activation of the PI3K/Akt signaling pathway (32). It is interesting to note that platelet stimulation with the TLR2/1 agonist, Pam3CSK4, causes the release of fibrinogen, which forms a bridge between platelets and immune cells (either neutrophils or monocytes), thus binding them to the site of injury. P-selectin on platelets represents an important adhesion molecule for PSGL-1-bearing immune cells, as it not only mediates the adhesion of activated platelets to leukocytes, resulting in the formation of platelet/leukocyte complexes, but also mediates leukocyte rolling and arrest on surface-adherent platelets (93, 182). Moreover, the cross-linking of PSGL-1 on monocytes by platelet P-selectin induces the upregulation and activation of β1 and β2 integrins and enhances monocyte recruitment to the activated endothelium (74). Due to its dual capability to function as both an adhesion and a signaling molecule, P-selectin on platelets, thus, seems crucial for the recruitment of immune cells. It enables the close contact of platelets with inflammatory blood-borne cells and induces the rolling of these cells, which together with arrest chemokines entails the activation of integrins that are important in mediating firm adhesion (312). Furthermore, platelet P-selectin facilitates the delivery and immobilization of platelet-derived chemokines on activated or atherosclerotic endothelium as culprits for mononuclear cell infiltration.
Due to the presence of proinflammatory mediators and surface receptors predominantly known for their involvement in inflammatory or immune processes, an interesting aspect of platelet behavior in the immune setting is platelet capability that mediates inflammation and clearance of bacteria from the bloodstream (322). There are three basic mechanisms for the mediation of the interaction between pathogens and platelets (Table 2): (i) binding to bacteria of a plasma protein, that is, a ligand for a platelet receptor; (ii) direct bacterial binding to a platelet receptor; and (iii) secretion of bacterial products, that is, toxins, that interact with platelets (70, 171). Some interactions lead to platelet activation, especially those involving cytoskeletal rearrangements and release of granule content, whereas others have no effect on the platelets (70).
CLEC-2, C-type lectin-like receptor 2; CR2, C3d receptor type II; EBV, Epstein Barr Virus; gC1q-R, complement (C1q) multiligand binding protein; HIV, human immunodeficiency virus; TLR, Toll-like receptor.
Many studies investigating bacterial-induced platelet aggregation (Streptococcus sanguinis, Streptococcus gordonii, and Staphylococcus aureus) have elucidated some of the signaling pathways involved in activating platelets, and all reports suggest that the COX pathway is involved (170, 172, 220). Recent findings indicate that Streptococcus pneumoniae-induced platelet aggregation stimulates platelet secretion and synergizes with secreted platelet agonists such as ADP. This may be a mechanism that explains the role of platelets in exacerbating S. pneumoniae-induced sepsis and may contribute to thrombocytopenia (168).
Platelets have also been shown to be capable of killing trypanosomes, though less efficiently than macrophages or neutrophils (278), and there is evidence that platelets are involved in Echinococcus granulosus infectious disease.
F. Coagulation activity of platelets
It is well known that platelets play a dominant role in secondary hemostasis by providing a highly effective catalytic surface for activation of the coagulation cascade (Fig. 4). Indeed, the concept of the “cascade” it is not updated, in that it is now accepted that coagulation occurs in three overlapping stages, in which platelets are always involved: (1) initiation, which occurs on a TF-bearing cell; (2) amplification, in which platelets and cofactors are activated to set the stage for large-scale thrombin generation; and (3) propagation, in which large amounts of thrombin are generated on the platelet surface (148) (Fig. 4).

Indeed, in this new cell-based model of coagulation, the three phases overlap. The initiation of coagulation takes place on TF-bearing cells, such as fibroblasts. If the procoagulant stimulus is sufficiently strong, then enough factors Xa, IXa, and thrombin are formed to trigger the coagulation process. Amplification of the coagulant response occurs and the focus changes from the TF-bearing cells to the platelet surface. The procoagulant stimulus is amplified as platelets adhere, are activated, and accumulate activated cofactors on their surfaces. Finally, in the propagation phase, the active proteases combine with their cofactors on the platelet (148). TF is a protein complexed with phospholipid that triggers the coagulation cascade by activating factor VII and forms a complex with the activated coagulation factor. The TF complex can provide the necessary phospholipid surface for triggering the coagulation process, whereas the surface of the activated platelets in the platelet plug is considered to be of major importance for coagulation in hemostasis. Activated platelets, by binding to coagulation factors, amplify this process and accelerate their catalytic processing by promoting the assembly of the involved enzymatic complexes.
When platelets are activated, negatively charged phospholipids move from the inner leaflet of the membrane bilayer to the outer leaflet. The transbilayer movement of anionic phospholipids is associated with blebbing and release of procoagulant vesicles that are rich in anionic phospholipids. Both activated platelets and the micro-vesicles provide binding sites for enzymes and cofactors of the coagulation system, which then efficiently generate thrombin. During regular platelet activation, besides the surface exposure of aminophospholipids, a particular phenomenon called “microvesiculation” is observed. This represents the formation of small-membrane vesicles containing cytoplasmic material, also called microparticles (MPs). In the vesiculation process, small areas of the surface membrane are shed in a budding process. However, even if the microvesicles are formed in an outside-out configuration, they possess a procoagulant surface that can bind annexin V (75). P-selectin, which in the nonactivated platelet is present in the α-granule membrane, as it is CD40L (145), is also present on the microvesicular surface (75). MPs not only bind the activated platelets, but also fuse with them via PSGL-1, transferring lipids and proteins, including TF, in the PM. The phospholipids on the surface of MPs from platelets and ECs provide a number of binding sites for factors Va, VIII, IXa, and IIa (150).
While moderate and transient rises in [Ca2+]i mediate shape change, integrin αIIbβ3 activation, TxA2 formation, and secretion of granule contents, high and prolonged [Ca2+]i rises are required for the procoagulant response (140). Influx of Ca2+ into the cytoplasm activates a scramblase activity that results in rapid transbilayer phospholipid mixing which leads to a nearly symmetric distribution of phospholipids across the membrane bilayer, thus exposing PS at the outer membrane surface (333). The route of Ca2+ entry is an important regulator of thrombin-induced PS exposure in platelets. Exposed PS provides high-affinity binding sites for key coagulation factors, thereby facilitating the assembly of tenase and prothrombinase complexes, which are responsible for the formation of factor Xa and thrombin, respectively (333), thus ensuring efficient propagation and control of the hemostatic process (Fig. 4).
In one of several regulatory feedback loops, thrombin directly activates platelets via PARs (Fig. 2), thus resulting in a synergistic promotion of platelet adhesion and coagulation (206).
G. Platelet transcription factors
Although platelets are anucleated cell fragments, recent reports show that platelets express transcription factors (TFs) which constitute a new class of platelet-released molecules that could result in a broad range of biological effects.
Among the first provided evidence, an interesting study reports that platelets express both the estrogen receptor (ER)β and the androgen receptor, suggesting a potential mechanism through which sex hormones might mediate gender differences in the platelet function observed in thrombotic diseases (174). Accordingly, it has been later demonstrated that the interaction of ERβ with 17βestradiol potentiates thrombin-induced platelet aggregation (210). Later studies have shown that platelets express many TFs that, in turn, exert nongenomic functions on platelets and that the activation of these receptors by their ligands inhibits platelet activation. Among the newly discovered TFs is the family of peroxisome proliferator-activated receptor (PPAR)γ (4) and PPARβ/δ (6). PPARγ protein is expressed in human megakaryoblast cell line (Meg-01), in human bone marrow megakaryocytes, and in human platelets, which are themselves targets of selected PPARγ agonists. Indeed, PPARγ agonists dampen platelet release of the key proinflammatory and proatherogenic mediators CD40L, TxB2, and ATP (4). Among the several steps during platelet exocytosis wherein PPARγ could interfere with platelet activation, the authors include Ca2+or PKC signaling pathways, rearrangement of the cytoskeleton during platelet activation, or docking and fusion of granules with the PM.
More recently, it has been demonstrated that human platelets also contain the PPARγ-binding partner retinoid X receptors (RXR), and that PPARγ is released from activated platelets as a functional heterodimer (PPARγ/RXR). Finally, PPARγ/RXR are found to be associated with platelet MPs (244).
As per the nuclear receptor PPARβ, platelets express PPARβ as their precursor megakaryocyte cell line (Meg-01). The activation of PPARβ inhibits platelet aggregation acutely and via a nongenomic mechanism that implies direct binding and repression of PKCα and the consequent inhibition of adenylyl cyclase (which converts ATP to cAMP). This evidence led the authors to hypothesize that some of the antithrombotic properties of PGI2 may be mediated via PPARβ (7).
Finally, platelets express nuclear factor-kappa B (NF-κB), which has proved to be involved in the regulation of the initial stages of platelet activation through the blockade of the ERK-cPLA2-TxA2 pathway, including the cytoskeletal rearrangements that lead to platelet shape change and the active form of the αIIbβ3 integrin (244). Since NF-κB inhibitors decrease P-selectin expression, the authors hypothesized that the nongenomic role of NF-κB in platelets could be associated with the regulation of both hemostatic- and inflammatory-mediated responses (244).
These novel evidences that the enucleated platelets are capable of expressing and releasing active transcription factors which play a great role in hemostasis, immunomodulation, and inflammation suggest the use of TFs both as biomarkers of platelet activation and as targets of antiplatelet intervention.
II. Assessment of Platelet Function
A. Platelet function testing
Tests of platelet function are usually performed in an attempt to measure the different steps of the activation process reached by the platelets of a patient. Possible reasons for measuring platelet function in patients include screening and diagnosis of platelet defects, monitoring antiplatelet therapy, monitoring prohaemostatic therapy, predicting thrombosis, predicting bleeding, and assessing stored platelets (135).
The assessment of platelet function is essential for the diagnosis of congenital/acquired platelet disorders in individuals with pathological bleeding and may be useful for the prediction of surgical hemorrhages. The vast majority of platelet tests focus only on the platelet functions that are directly involved in hemostasis, including adhesion/aggregation, coagulation, and clot retraction.
While hereditary platelet function disorders are very rare, acquired platelet dysfunction is associated with many systemic disorders, such as renal disease, hepatic failure, autoimmune diseases, connective tissue disorders, myeloproliferative disorders, myelodysplastic disorders, malignancy, and CV disease. Clinical features, such as albinism, deafness, nephritis, and susceptibility to infections, may help in the differential diagnosis of the inherited platelet disorders (19).
Drugs represent one of the most common causes of platelet dysfunction. While COX-1 inhibitors (aspirin), P2Y12 antagonists (ticlopidine, clopidogrel, and prasugrel), and integrin αIIbβ3 receptor antagonists (abciximab, eptifibatide, and tirofiban) are well known, and purposely used, platelet-interfering drugs, other widely used agents (e.g., nonsteroidal anti-inflammatory drugs, antibiotics, antihistaminic, serotonin reuptake inhibitors, and volume expanders) have also been described as impairing platelet function (269). Indeed, nowadays, platelet function tests are increasingly being utilized in monitoring the efficacy of antiplatelet therapy.
It has been suggested that the assessment of platelet function may serve for identifying platelet hyperactivity as a potential predictor of an increased thromboembolic risk (298).
A wide variety of tests are available for the measurement of platelet function (Table 3), although, to date, no function test is suitable that addresses all distinct steps of platelet activation or reliably predicts platelet behavior in vivo.
CD40L, CD40 ligand; COX, cyclooxygenase; LTA, light trasmission aggregometry; PRP, platelet rich plasma; Tx, thromboxane; VASP, vasodilator-stimulated phosphoprotein; WB, whole blood; WBA, whole blood aggregometry.
Indeed, in contrast to coagulation defects, where the screening tests (aPTT, activated partial thromboplastin time and PT, prothrombin time) are inexpensive and fully automated, platelet function defects are more difficult to diagnose, because there are no definitive screening tests. No current or future platelet function test is likely to be 100% sensitive to all platelet disorders because of the heterogeneity of platelet defects described so far. The current diagnostic evaluation of a potential platelet defect is heightened on the measurement of platelet aggregation and of granule content/release. These traditional tests, which are complex, costly, time consuming, and prone to operator-specific variables, have been enriched by more easy-to-use point-of-care tests, on fully automated instruments within whole blood, without the requirement of sample processing. These tests were designed to help identifying surgical patients at an increased risk of postoperative bleeding or with resistance to antiplatelet therapy, thus at an increased risk of recurrent thrombotic events.
Thus, a number of platelet function tests and instruments are now available to the clinical laboratory, but most of these tests are, until now, poorly standardized and require expertise and experience to be performed and interpreted (52, 139). Several commercial point-of-care platelet function assays, including the VerifyNow system and the platelet function analyzer (PFA-100), have been introduced for a standardized, user-friendly evaluation of the individual response to antiplatelet therapy. Furthermore, standardization committees (such as the ISTH Platelet Physiology Scientific and Standardization Committee) are trying to produce new platelet function testing guidelines to improve quality assurance and give rise to less variability between laboratories (35). Correlating the results of platelet function tests with clinical outcomes and using the results to guide therapy, however, remain a challenging goal.
1. Bleeding time
The bleeding time (BT) is the oldest in vivo test of platelet function (1) that studies the role played by the vessel wall during the interaction of platelets with endothelium/vascular cells. The BT, developed in 1910 as a clinical bedside test of global platelet function, is still, although less frequently, applied as a classical screening test for primary hemostasis. It measures the time required to stop spontaneous bleeding out of a standardized cut in the skin, on the volar surface of the forearm, using a venostatic pressure (40 mmHg) applied on the upper arm. The normal BT ranges between 2 and 10 min, but severe platelet defects including severe von Willebrand's disease (VWD) can result in a BT > 30 min (157). However, its test procedural variability renders it poorly reproducible and sensitive, and with a high inter-operator coefficient of variation and a subjective end point. Actually, an accurate bleeding history should be regarded as a more valuable screening test (134).
2. Platelet aggregometry
Light transmission aggregometry (LTA), also referred to as turbidometric, spectrophotometric, or optical aggregometry, was developed about fifty years ago and for decades was regarded as the gold standard of platelet function testing (1). LTA measures light transmittance through a platelet milieu before and after the addition of a platelet agonist. The increase in light transmittance is proportional to the platelet aggregates formed.
The principle of LTA consists of exposing platelet-rich plasma (PRP), obtained by the centrifugation of citrated blood to varying doses of platelet agonists such as collagen, ADP, thrombin, or adrenaline (either alone or in combination). On addition of the agonist, the platelets change shape from discs to a more rounded form with pseudopods, resulting in a transient small decrease in light transmission followed by a large increase on the formation of aggregates. The increase in light transmission is classically quantified using optical change technology. This is still the most widely used test for identifying platelet function defects. For instance, VWD by usage of the antibiotic ristocetin as agonist, or Glanzmann's thrombasthenia, when platelets change shape but fail to aggregate in response to all agonists, are easily diagnosed.
Major drawbacks of LTA are represented by the high amount of blood required for PRP preparation, which itself is affected by many preanalytical and analytical variables (51). Apart from the nonphysiologic no/low shear conditions during analysis, the use of PRP is responsible for the fact that LTA does not accurately simulate primary hemostasis and may result in the loss of hyper-/hypoactive platelets, extra time, and technologies experienced in preparation and cell-counting techniques (136).
Thus, whole blood aggregometery using impedance technology, which does not require further blood processing, has been recommended and is sometimes combined with luminometry to simultaneously measure dense granular ATP release. Lumiaggregometry, which measures platelet aggregation and secretion simultaneously, might be preferable to LTA for the diagnostic workup of patients with platelet function disorders (51).
Due to the many disadvantages of BT and LTA, alternative automated technologies have been developed in trying to simulate hemostasis in vitro that might potentially be utilized as point-of-care instruments for assessing bleeding risk, thrombotic risk, and monitoring antiplatelet therapy. However, since they are very poorly standardized between different laboratories, this highlights the need for new guidelines and recommendations on how to accurately perform these tests (135). Finally, large, prospective clinical trials will be required to determine whether these tests are useful for these applications (138).
Here, we briefly describe three of the most frequently used devices: multiple-electrode platelet aggregometry (MEA), PFA-100, and Verify-Now.
a. Multiple-electrode platelet aggregometry
The MEA is a new point-of-care assay, developed for rapid and standardized assessment of platelet function in whole blood (35) in which aggregation takes place on surfaces, thus simulating in vivo conditions. MEA is capable of detecting the degree of platelet inhibition achieved using aspirin, αIIbβ3 receptor antagonists, and different P2Y12 antagonists, including clopidogrel, cangrelor, and the active metabolites of clopidogrel and prasugrel. High on-treatment ADP-induced platelet reactivity assessed with MEA has been shown to be an independent predictor of early stent thrombosis in patients undergoing percutaneous coronary intervention (PCI) who had received a 600-mg clopidogrel loading dose (35).
b. PFA-100
The PFA-100 is a relatively simple bench instrument that provides high shear platelet function within disposable test cartridges (159). This device has been referred to as an “in vitro bleeding time,” in that it consists of an in vitro method for looking at the cessation of blood flow in a high-shear environment, as determined by the closure time (CT) of an aperture by the formation of a platelet plug.
The test is simple to perform, rapid—with maximal CTs of 300 s—and can test relatively small volumes (0.8 mL/cartridge) of whole citrated blood up to 4 h from sampling (133). The test seems reliable, with acceptable variations in normal ranges reported from many laboratories (157), and the awareness that PFA-100 is sensitive to variables that influence platelet function including abnormalities in platelet number, hematocrit (159), and VWF levels.
The ISTH platelet physiology Scientific and Standardization Committee recommends the test as an optional screening test that can potentially be used to detect or exclude severe platelet defects and VWD (139). PFA-100 has high negative predictive value for severe disorders, but it has poor sensitivity to milder platelet function defects, giving false negative results. It has been suggested that PFA-100 CT predicts CV events in aspirin-treated CV patients (71), particularly in patients who appear to be nonresponsive or “resistant” to aspirin therapy. However, residual platelet reactivity assessed by PFA-100 exhibited a limited predictive value for major adverse CV events (MACEs) in patients undergoing elective coronary stent implantation (40). Larger studies are required to assess whether the PFA-100 can reliably predict either thrombotic or bleeding complications in different patient groups.
c. VerifyNow
VerifyNow is a turbidimetric-based optical detection system that measures platelet-induced aggregation as an increase in light transmittance (284). This method uses disposable cartridges containing fibrinogen-coated beads that adhere to stimulated platelets and a platelet activator. The instrument measures the change in light transmission, which represents the rate of platelet/beads adhesion or agglutination, well correlates with conventional platelet aggregometry (284), and has been proposed as a point-of-care test, as it does not require sample processing, time delay, or specialized personnel to perform the test. The test has also been adapted to measure the effectiveness of antiplatelet therapy within specific cartridges (196, 316). Evidence is now accumulating to suggest that the detection of nonresponders to drugs within all the types of cartridge equates with poor clinical outcomes (38, 57, 240, 287), but only the randomized trials in progress might reveal whether monitoring and titrating or changing antiplatelet therapy based on the test result is clinically useful and improves outcomes (35), although preliminary results indicate that, in patients with high on-treatment reactivity, a fixed higher dose of clopidogrel did not reduce CV death, myocardial infarction (MI), and stent thrombosis after PCI compared with a standard dose (238, 239).
3. Flow cytometry
The application of flow cytometry in platelet function analysis is based on the detection of cell surface proteins with fluorescently labeled antibodies. In contrast to the analysis of platelet solutions, whole-blood cytometry allows for fewer artifacts that could occur with artificial platelet (de)activation during sample preparation, but requires access to expensive instrumentation and specialized training to be performed (207). Flow cytometry allows analyzing platelets within whole blood in their circulating state, provided both venipuncture and analysis techniques are well standardized. The most commonly used routine tests are the quantification of GP receptor density (i.e, diagnosis of deficiencies in platelet GPs, for example, Glanzmann thrombasthenia and Bernard Soulier disease), although dense, granular measurements (using mepacrine uptake and release) (313), MP formation, and exposure of anionic phospholipids (procoagulant activity) can be equally performed. Another use of flow cytometry is the detection of platelet autoantibodies in patients with idiopathic thrombocytopenic purpura and drug-induced thrombocytopenias (203).
In conclusion, platelet function testing is increasingly utilized outside of the specialized laboratory. Although this represents advancement, validation, quality control testing, and reliability of these tests will also become an increasingly important issue. Most new platelet function tests have recently and will continue to become available. However, they need to prove to be useful in addition to our existing portfolio of tests.
B. Laboratory markers of platelet activation and their clinical significance
CV disease is a major cause of morbidity and mortality. Numerous risk scores exist that identify healthy individuals at an increased risk of developing an adverse CV event. Markers of CV risk not routinely included in current risk score algorithms, such as platelet activity, may also contribute to enhanced CV risk.
Platelet function can be assessed in vitro by simple platelet properties such as platelet aggregometry, platelet size, and platelet count, in vivo by biochemical immunoassays for plasma markers of platelet activation such as P-selectin, CD40L, or urinary excretion rate of TxA2 enzymatic metabolites. Several prospective studies report significant associations between platelet function and outcomes in patients with established CAD (127, 143). However, since little comparative data on platelet activity and the risk of CV morbidity and mortality in apparently healthy populations are available, it is difficult to determine which parameter of platelet activity has the most promising predictive value for the development of CV disease.
The role of platelet function testing in identifying subjects at risk for CV disease remains elusive as well as the possibility to predict CV risk in apparently healthy subjects seems to be limited. It has been suggested that the use of sub-maximal concentration of agonists in platelet aggregometry might help identify a population with hyperreactive platelet phenotypes (327). Despite several studies evaluating the association between platelet aggregation and incident CV events, overall, the data are largely inconclusive, and most of the studies were unable to demonstrate a significant association between platelet aggregation and CV endpoints (274). It is worth noting that the various studies (many of them performed decades ago) were carried out using different platelet aggregation measurements, with varying agonists, different agonist concentrations, and platelet separation and purification techniques; thus, the results obtained within one laboratory could hardly be compared with others. Since then, a great effort has been made in the standardization of platelet aggregation techniques among different laboratories in order to render efficacius the attempt to define universal cutoff values that identify different degrees of platelet response and their significance.
A few data are available to date for assessing mean platelet volume (MPV) prognostic implication in patients without CAD. MPV is a commonly measured, standardized platelet parameter and has been extensively studied in the context of CV disease with clear associations with other markers of platelet activation. Recently, a Danish study on more than 39,000 participants from the general population showed that a high MPV was associated with an increased risk of MI in subjects with and without antiplatelet therapy (175).
1. Markers of in vivo platelet activation
Since different platelet assays assess different aspects of platelet pathophysiology, the result depends on the clinical condition being investigated and the aspect of platelet activation being measured. Biomarkers of platelet activation may provide clues regarding the extent of basal platelet activation in patients at risk for CV events and may also predict the severity of the thrombotic response in patients with a previous event.
The plasma markers of platelet activation, sCD40L and sP-selectin, are promising prognostic candidates for future CV disease. Both are easily measured with a standardized enzyme-linked immunosorbent assay (ELISA). Although limited, there are some data suggesting a significant association between them and incident CV disease.
a. Soluble P-selectin
P-selectin is an adhesion molecule that is expressed not only on activated platelets but also on the endothelium in the presence of an atherosclerotic plaque. Soluble (s)P-selectin in the plasma is thought to arise predominantly from platelets, with a minimal contribution from ECs, suggesting that sP-selectin is likely to reflect platelet activation (33). Platelet P-selectin can be measured by flow cytometry or, more easily, by ELISA technique as a reliable marker of platelet activation. It has been demonstrated that sP-selectin significantly correlates with all parameters of LTA in response to stimulation with various agonists and proposed that plasma sP-selectin measurement may represent a suitable marker of in vivo platelet activation in atherothrombotic disorders, and a valid alternative to the time-consuming and operator-dependent LTA (106).
A few prospective studies examined whether sP-selectin correlated with future CV events. In the Women's Health Study (249), apparently healthy women who had baseline sP-selectin values in the highest quartile group had a twofold risk of developing a CV event than subjects in the lowest quartile, with an increased 28% risk of CV events for each quartile increase in sP-selectin values. In young men and women followed up for 5 years, sP-selectin levels were significantly associated with carotid intima-media thickness (246).
In acute coronary syndrome (ACS), P-selectin may be useful in risk stratification of patients presenting with chest pain (149). In patients with risk factors for CV, such as diabetes, smoking, and hypertension, raised sP-selectin levels have been proved to be significantly associated (33). In hypercholesterolemic patients, our group first observed higher levels of plasma P-selectin compared with normocholesterolemic subjects (87). Since hypercholesterolemic patients display increased in vivo platelet activation (77), this corrobarates the role of activated platelets in the increase in plasma P-selectin (288).
b. CD40 ligand
The immunomodulator CD40L and its receptor are also present in platelets. CD40L is a ligand for integrin αIIbβ3 that is necessary for the stability of arterial thrombi (9). When activated, platelets strongly express and release CD40L (145) (Fig. 5). CD40L also occurs in a soluble form (sCD40L) that might be biologically active and appears to mostly originate from platelets (234). The CD40/CD40L system was shown to be expressed in vascular cells, including ECs, lymphocytes, and smooth muscle cells. Platelet-derived CD40L can induce a proinflammatory and prothrombotic response in ECs, as evidenced by the generation and release of ROS, adhesion molecules, chemokines, and TF (86, 299). sCD40L significantly correlates with other markers of platelet activation, such as sP-selectin (250), and it is recognized as a useful marker of platelet activation in clinical settings. Limited data are available on the association between sCD40L levels and future CV events in subjects free of CV disease. Among healthy, middle-aged women, those who later developed CV disease had significantly higher baseline levels of sCD40L. Moreover, women with baseline levels above the 95th percentile of the normal distribution were more than thrice as likely to develop a CV adverse event (271). Elevated levels of sCD40L were observed in both stable and unstable angina (UA) (15) and in acute MI (143). Moreover, in patients with UA, the expression of CD40L on T lymphocytes surface and in vitro shedding of sCD40L from these cells were increased (15). Thus, both T lymphocytes and, particularly, platelets represent the major contributor to increased circulating sCD40L in the setting of UA. In patients with ACS, sCD40L identified a subgroup that was at an increased 6-month risk of death or nonfatal MI (263). Accordingly, it has been suggested that the combined assessment of CD40L and troponin-T levels might have a better predictive value of AMI risk. sCD40L seems to have a prognostic role not only in subjects with advanced atherosclerosis but also in the general population (12).

Increased plasma levels of sCD40L have been detected in both type 1 (T1) and type 2 (T2) diabetes mellitus (DM) (265, 308). Moreover, previous studies showed a significantly increased coexpression of CD40 and CD40L in platelets of diabetic patients compared with nondiabetic controls and also a correlation between sCD40L and CD40L expression in platelets (161).
The highly significant correlation between plasma CD40L levels and the urinary excretion rate of 11-dehydro-TxB2, a noninvasive index of in vivo platelet activation, supports the evidence that CD40L is rapidly upregulated during platelet activation (145).
In addition, the positive correlation observed between enhanced sCD40L and 8-isoPGF2α is in line with the report of increased production of endothelial ROS by CD40L (299), suggesting that in T2DM, the release of sCD40L from activated platelets may contribute to increased oxidant stress. Increased lipid peroxidation and persistent platelet activation have previously been reported in patients with T2DM (78, 80). Thus, on the basis of these findings, a possible vicious cycle may be suggested in which inflammatory stimuli involving CD40L upregulation induce increased lipid peroxidation with consequent platelet activation, resulting in further oxidant stress (145).
Although TxA2 enzymatic metabolites have been extensively studied in relation to CV disease and in response to aspirin treatment, no data are available regarding the levels of TxA2 metabolites prospectively used in otherwise healthy subjects as a marker of risk for CV disease (Fig. 6).

c. Phosphorylation of vasodilator-stimulated phosphoprotein
The flow cytometric analysis of vasodilator-stimulated phosphoprotein (VASP) phosphorylation is an assay designed for the measurement of P2Y12 antagonism, thus of clopidogrel efficacy (Fig. 2).
PGE1 binds to its inositol phosphate receptor on the platelet surface and signals through a G stimulatory protein and adenylyl cyclase to convert ATP to cAMP and then, through protein kinase A, converts VASP to phosphorylated VASP (VASP-P). ADP, by binding to its P2Y12 receptor on the platelet surface, inhibits PGE1-induced signaling through adenylyl cyclase. VASP-P is measured by whole blood flow cytometry, using permeabilization and a monoclonal antibody specific for the phosphorylated form of VASP. The advantages of the VASP assay are that it involves a low sample volume of whole blood and that it represents a specific intracellular marker of residual P2Y12 receptor reactivity in patients treated with P2Y12 blockers. The disadvantages of the VASP assay are the requirement of an experienced technician for sample preparation and flow cytometry analysis. High on-treatment reactivity assessed by VASP assay has been associated to an increased CV risk (20). In addition, it has been reported that adjusting the clopidogrel loading dose according to the VASP index significantly improved the clinical outcome after PCI (34).
III. Platelet Function in Disease
A. In vivo redox mechanisms inducing platelet activation
Increased oxidative stress appears to be of fundamental importance in the pathogenesis and development of several disease processes, including atherothrombosis, neurodegeneration, and cancer (81). Various risk factors for atherosclerosis and CV thrombosis, such as hypertension, hypercholesterolemia, hyperglycemia, cigarette smoking, or hyperhomocysteinemia, are associated with ROS-mediated development of endothelial dysfunction (41) (Fig. 7). Several products of glucose, protein, and lipid oxidation are thought to mediate the noxious influences of oxidant stress on cell function (82, 236).
Ex-vivo indices of oxidant stress (e.g., measurement of thiobarbituric acid reactive substances or spectrophotometrical detection of conjugated diene structures) suffer from major limitations in assessing the actual rate of lipid peroxidation in vivo (226).
Isoprostanes are a family of bioactive compounds produced from AA via a free radical-catalyzed mechanism of lipid peroxidation on cell membrane phospholipids or circulating LDL (213). Since 1995, studies have been performed that investigate the formation of F2-isoprostanes in clinical settings putatively associated with oxidant stress. Consistent data support the concept that these compounds are reliable, sensitive, and specific markers of in vivo lipid peroxidation (214). Thus, the measurement of F2-isoprostanes in plasma or urine samples has been extensively employed to assess in vivo lipid peroxidation in several human disease states (226, 235). The measurement of F2-isoprostanes presents several advantages over other biomarkers of oxidative stress in that F2-isoprostanes are specific products of lipid peroxidation, structurally stable and present in detectable amounts in all normal tissues and biological fluids. Isoprostanes are frequently measured in urine, because the method is noninvasive and avoids isoprostanes artifactual ex vivo generation (209).
Among F2-isoprostanes, 8-iso-prostaglandin F2α (8-iso-PGF2α) is the most frequently measured by stable-isotope dilution assay using gas chromatography/mass spectrometry or immunoassay (214, 318). Measurements of the un-metabolized compound can be performed in both plasma and urine samples. Measurable concentrations of the un-metabolized compound are present in peripheral venous blood, and a relatively reproducible fraction is excreted unchanged in human urine, with no detectable circadian variation. Nevertheless, most studies examined its urinary excretion due to the lack of ex vivo artifactual formation resulting from the auto-oxidation of lipids (82, 236).
F2-isoprostanes can modulate both platelet and vascular function (82, 236). In particular, 8-iso-PGF2α is able to modify platelet adhesive reactions and platelet activation induced by low concentrations of agonists (82). Concentrations of 8-iso-PGF2α in the range of 1 nM to 1 M induce a dose-dependent increase in platelet shape change, calcium release from intracellular stores, and inositol phosphate production (212, 237). Moreover, 8-iso-PGF2α causes dose-dependent, irreversible platelet aggregation in the presence of subthreshold concentrations of collagen, ADP, AA, and PGH2/TxA2 analogs, which when acting alone, fail to aggregate platelets (237). The ability of 8-iso-PGF2α to amplify the aggregation response to subthreshold concentrations of platelet agonists might be relevant in settings in which platelet activation and enhanced free radical formation coincide (237).
F2-isoprostane biosynthesis and TxA2-dependent platelet activation are enhanced in association with a variety of CV risk factors.
T1DM at onset represents an interesting paradigm of the interrelationship between immuno-inflammatory reaction, lipid peroxidation, and platelet activation. Indeed, enhanced lipid peroxidation and platelet activation represent early events in the development of T1DM both in children and in adolescents (79). Patients with newly diagnosed diabetes had significantly increased urinary excretion of both 8-iso-PGF2α and 11-dehydro-TxB2 (82), as well as higher plasma levels of a number of inflammatory markers (79). In most of these patients, but not in all, oxidative stress and platelet activation were reduced after 1 year, coincidentally with a fall in the systemic levels of IL-6 and TNF-α. Thus, it appears that biochemical signals of oxidative stress and platelet activation can be appreciated early at the onset of DM, and that their variable intensity is, at least in part, driven by IL-6 production and disease duration. These noninvasive indexes may help in further examining the pathophysiology of T1DM and in monitoring pharmacologic interventions aimed at interfering with disease development and progression.
T2DM is characterized by persistent enhanced TxA2 biosynthesis (78), and evidence has been provided that the metabolic disorder, rather than the attendant vascular disease, is responsible for the persistent platelet activation occurring in this setting (Fig. 8) (84). Furthermore, T2DM is associated with enhanced oxidant stress and increased urinary excretion of 8-iso-PGF2α, with a significant correlation between blood glucose and urinary isoprostane levels, suggesting that lipid peroxidation may be related, at least in part, to determinants of glycemic control (Fig. 8) (80, 89, 266). Accordingly, intensive antidiabetic treatment is associated with a reduction in both urinary 8-iso-PGF2α and 11-dehydro-TxB2 excretion rates, supporting the concept that AA peroxidation that forms bioactive iso-eicosanoids could represent a biochemical link between altered glycemic control, oxidant stress, and platelet activation in this setting (80).

Obesity, and in particular visceral obesity, is associated with an increased CV risk (331). A large body of evidence suggests the presence of a low-grade inflammatory state in this condition, reflecting the release of several inflammatory cytokines from adipose tissue, and the association with increased oxidant stress (82, 211).
We tested the hypothesis that lipid peroxidation and platelet activation are increased in obese women, in the absence of other known CV risk factors, and are modifiable after body weight reduction (85). Obese women had higher levels of lipid peroxidation and platelet activation when compared with age-matched nonobese women. Visceral obesity was associated with a four-fold higher rate of TxA2 metabolite excretion than measured in nonobese women, with a linear relationship between the excretion rates of 8-iso-PGF2α and 11-dehydro-TxB2. Although both hyperinsulinemia and increased leptin production may trigger increased generation of oxygen radicals, possibly contributing to enhanced lipid peroxidation, a multiple regression analysis indicated that C-reactive protein (CRP) levels and the waist/hip ratio predicted urinary 8-iso-PGF2α excretion rate independently of insulin and leptin levels (85). Furthermore, when plasma CRP concentrations were divided into quartiles, the excretion rates of 8-iso-PGF2α and 11-dehydro-TxB2 significantly increased from the first to the fourth quartile (85). To assess the cause-and-effect relationship of these associations, we examined the short-term effects of a weight loss program, by assessing, in the same population of visceral obese women, the changes associated with caloric restriction. Successful weight loss was associated with statistically significant reductions in CRP levels, in urinary 8-iso-PGF2α, and in 11-dehydro-TxB2 excretion rates (85).
Thereafter, data from the Framingham Study confirmed the presence of enhanced oxidative stress in a population of 2828 obese subjects (169). Obesity was a strong independent predictor of systemic oxidative stress, with a positive association between indices of obesity, such as body mass index and waist/hip ratio, and urinary levels of 8-iso-PGF2α (169).
Thus, F2-isoprostanes might transduce the effects of oxidant stress associated with complex metabolic disorders into specialized forms of cellular activation. The consistent linear relationship between the excretion rates of 8-iso-PGF2α and 11-dehydro-TxB2 suggests that a low-grade inflammatory state associated with complex metabolic disorders may be the primary trigger of TxA2-dependent platelet activation mediated, at least in part, through enhanced lipid peroxidation. Several feed-forward mechanisms are likely to amplify and sustain the relationship between systemic inflammation and platelet activation (82), including a direct proinflammatory effect of CRP, effects of F2-isoprostanes on inflammatory gene expression, and the synthesis and release of inflammatory cytokines from activated platelets (82, 190).
Oxidative mechanisms have been proved to be implicated in several clinical settings.
Oxidative stress, in terms of 8-iso-PGF2α, has found to be more abundantly excreted into the urine of aspirin low-responders (272), and it has been proposed that residual platelet activity in such patients may be one important cause of recurrent MI.
Platelet-associated NAD(P)H oxidase leads to a thrombogenic phenotype and mediates the arteriolar dysfunction (291) in patients with hypercholesterolemia.
By comparing platelet superoxide production in patients with hypertension alone and in patients with coexistent DM, it has been shown that eNOS can reside in the uncoupled state in patients with hypertension and, to a greater extent, in patients with coexisting hypertension and diabetes, and that this condition significantly contributes to increased superoxide production in these disease states (96).
B. Platelets as predictors of vascular risk
1. Indicators of enhanced platelet turnover (reticulated platelets and MPV)
A higher number of circulating reticulated platelets is considered an indicator of accelerated platelet turnover (97) in that circulating reticulated cells represent young, mRNA rich platelets with enhanced proaggregating and hemostatic potential. Reticulated platelets are larger and more reactive platelets, as platelet size correlates with greater platelet reactivity, measured by aggregation and total release of granular content (297).
MPV is a parameter mirroring in vivo platelet activation (22) and an independent predictor of vascular events (311). Platelets are heterogeneous in size and density. Larger platelets are metabolically and enzimatically more active and have greater prothrombotic potential. Thus, MPV, a standardized platelet parameter, commonly available in the inpatient and outpatient setting, may have prognostic value in patients with previous vascular events. Moreover, MPV is to date recognized as a new independent risk factor for MI, vascular mortality, and restenosis after coronary angioplasty. Thus, MPV is a potentially useful prognostic biomarker for CV risk stratification (60). As compared with other markers of platelet activation, MPV is less expensive, easy to interpret, and can be measured by automated cell counters.
MPV values could reflect global platelet function in that they are associated with markers of platelet activation and with adhesion molecules' expression in platelets. In fact, larger platelets are more reactive and aggregable, contain denser granules, secrete more serotonin and β-thromboglobulin, and produce more TxA2 than smaller platelets.
These factors produce effects on inflammation and endothelial function, promoting cell adhesion and aggregation, vasoconstriction, and, ultimately, inducing thrombosis. Interestingly, large hyperactive platelets might be sequestered in the coronary circulation of CAD patients, as suggested by the finding of lower MPV values within the coronary sinus than in the arterial blood (156).
Larger and more reactive platelets can be found in association with CV risk factors such as DM, obesity, metabolic syndrome, smoking, or hypertension (60) (Fig. 9). Indeed, a higher number of reticulated platelets showing a lower response to antiplatelet therapies including aspirin and clopidogrel can be frequently found in DM (128, 129).

The increase in MPV in diabetics does not seem to be correlated to HbA1c, fasting blood glucose, patient age, and duration of diabetes (144), suggesting that the increase in MPV may be due to the diabetic state per se. The increase in MPV occurs at the onset of the disease and persists for its entire duration.
A recent meta-analysis (60) showed that MPV is associated with MI, mortality after MI, and restenosis after coronary angioplasty, supporting the hypothesis that platelet size may play a role in both the development and the consequences of CV disease. However, it cannot be established whether elevated MPV actually causes CV disease and ACSs, or whether it is merely associated with them. Thus, MPV represents a potentially useful prognostic biomarker in patients with CV disease, although it is likely that an increased MPV pre-dates an acute MI rather than results from it.
2. Biomarkers of platelet activation
a. Platelet-derived MPs
The release of MPs from platelets (platelet-derived MPs [PMPs]) has been associated with the secretion response (206). Therefore, the number of circulating PMPs reflects the platelet activation status in vivo. Indeed, in patients with UA (310), MI (167, 305), cerebrovascular accident (183), and DM (219), PMP subpopulations are increased, as well as in aging and, even further, in peripheral arterial disease (PAD) (305, 329).
b. Tx biosynthesis
TxA2 is a COX product that is synthesized and released by platelets in response to a variety of stimuli, including thrombin, collagen, and ADP. Most of the vascular actions of TxA2 result from the autocrine/paracrine activation of TP expressed on the surface of platelets, endothelial and VSMC (Fig. 10).
Eicosanoids, in general, and TxA2, in particular, are characterized by the episodic nature of their release and the local nature of their actions. These features are ensured by a variable chemical instability (very high for TxA2, due to nonenzymatic hydrolysis to form its stable metabolite TxB2) coupled with a substantial metabolic instability. Thus, extensive enzymatic degradation occurs in the lungs, liver, and kidneys, and it has been suggested to account for the short-lived presence of TxA2 in the human circulation (Fig. 6) (225). Although it has been proposed to measure circulating TxB2 to estimate TxA2 production, TxB2 is not a reliable index of in vivo platelet TxA2 production, as its concentration is readily confounded by ex vivo platelet activation, which occurs readily during sample collection and processing (222). Moreover, the chances of detecting a sudden (mostly unpredictable) and short-lasting change in TxA2 synthesis and release, by one or two plasma sampling, is unreliable. Thus, the measurement of TxB2 in peripheral venous plasma cannot, under any circumstances, reflect changes in TxA2 synthesis and release occurring in vivo (86).
To circumvent this problem, it is necessary to measure a metabolite that cannot be formed in whole blood. TxB2 undergoes enzymatic degradation that generates a series of metabolites. Among them, 11-dehydro-TxB2 has a substantial longer plasma half life, and it is excreted at a higher rate than other metabolites (Fig. 6) (225). Experimental studies reported a linear correlation between levels of exogenously administrated TxB2 and urinary 11-dehydro-TxB2 excretion (61, 78). Thus, it has been hypothesized that urinary 11-dehydro-TxB2 would provide a time-integrated index of endogenous systemic TxA2 biosynthesis, allowing a thorough evaluation of ongoing platelet activation by multiple cell sources (48).
Urinary levels of 11-dehydro-TxB2 have been reported as reflecting TxA2 generation by platelets—and, possibly, other cells—in established vascular disease. Episodic increases in 11-dehydro-TxB2 excretion have been detected in UA, or in transient cerebral ischemic attacks. Thus, TxA2-dependent platelet activation can mediate or amplify, at least in part, the short-term occlusive consequences of acute vascular lesions (e.g., plaque rupture) in the coronary and cerebral circulation. This is likely to reflect a localized (both in time and space) stimulus to platelet activation. Moreover, increased TxA2 biosynthesis has been associated with several clinical settings characterized by increased CV risk, including DM, visceral obesity, hypertension, hypercholesterolemia, heart failure, and PAD (21, 83, 84, 125, 264). In addition, the consistent relationship observed in vivo between the rates of formation of F2-isoprostanes, in vivo indices of lipid peroxidation, and urinary 11-dehydro-TxB2 suggests that a low-grade inflammatory state associated with these metabolic disorders may be the primary trigger of TxA2-dependent platelet activation, partly mediated by enhanced lipid peroxidation (86). Furthermore, the measurement of urinary 11-dehydro-TxB2 levels provides a useful marker that investigates the effect of various cardioprotective interventions, including lipid-lowering therapy, improved glycemic control, weight loss, and antiplatelet agents (86) on platelet activation.
Finally, it has been recently reported that urinary 11-dehydro-TxB2 measurement predicts the future risk of MI or CV death in aspirin-treated patients (99, 100). Nevertheless, whether urinary 11-dehydro-TxB2 levels are independent predictors of future vascular events in untreated patients still remains controversial.
C. Translational research of high platelet reactivity and clinical outcome
Consistent levels of platelet inhibition are required when antiplatelet drugs are used as an adjunct to coronary revascularization. During PCI, atherosclerotic plaque is invariably disrupted, thrombosis occurs, and endothelial healing is delayed. Intensive periprocedural platelet inhibition minimizes morbidity and mortality, whereas failure to provide adequate platelet inhibition may result in stent thrombosis, MI, and death.
1. High (on-treatment) platelet reactivity
High (on-treatment) platelet reactivity (HPR) can be observed in a significant proportion of patients treated with P2Y12 inhibitors (35), the vast majority of the studies having been focused on the antiplatelet action of clopidogrel. Depending on the method used for platelet function testing and the cut-off value chosen to define HPR, the proportion of patients defined as HPR patients varies largely across commonly used assays, from <20% to >50% (13). However, rates close to or more than 50% clearly overestimate the true proportion of patients who are at risk for suffering ischemic events, considering the low rate of stent thrombosis in PCI-treated patients (280). The relation of HPR to a clinical outcome was prospectively shown in large-scale trials and meta-analysis (13, 279) that demonstrated a significant association between the occurrence of stent thrombosis and the presence of HPR. However, only recently, the “Working Group on High (On-treatment) Platelet Reactivity” provided a consensus opinion on the definition (35) of HPR to ADP, establishing specific cut-off values for the most commonly used tests in clinical routine. A crucial point is to assess the stability of the HPR phenotype over time in patients treated with clopidogrel, because platelet function testing may provide variable results measured at different time points in the same patients. Thus, only serial measurements of platelet reactivity may help in guiding tailored antiplatelet therapy (44). Interference with clopidogrel bioactivation may occur for genetic and nongenetic factors.
Apart from drug noncompliance, patients with obesity, DM, older age, or renal insufficiency show higher platelet aggregation values on clopidogrel therapy (280). In patients suffering from acute MI complicated by cardiogenic shock, the poor response to clopidogrel is due to both reduced intestinal absorption of the pro-drug and diminished bioactivation. In addition to clinical conditions, co-medication influences the efficacy of clopidogrel. Proton pump inhibitors interact with the cytochrome P450, calcium channel blockers, and statins interfere with the CYP-dependent bioactivation of clopidogrel. However, since the available data are conflicting, the relevance of this interaction in platelet response and in clinical outcome remains not clearly determined (280). Multiple genetic variants in different genes involved in clopidogrel absorption and bioactivation can be associated with high or low HPR to clopidogrel as well as to bleeding or ischemic events (282).
Several studies have involved the isoenzyme CYP2C19, and the presence of the CYP2C19*2 loss-of-function allelic variant is associated with a reduced response to clopidogrel with a consequent higher risk of stent thrombosis in PCI-treated patients (204, 282).
The CYP2C19*17 gain-of-function allelic variant results in an increased enzymatic function (280). This variant is associated with lower ADP-induced platelet aggregation and increased bleeding risk, especially for homozygous allele carriers, as demonstrated by a genetic substudy of the PLATO trial (315), which showed more bleeding events in patients with this variant treated with clopidogrel.
Other genetic variants have been associated with the clinical outcome of clopidogrel-treated patients, but the results are still conflicting (280), and further studies are needed to better characterize all the genetic variants that are able to influence clopidogrel bioavailability.
2. Variable platelet response to aspirin
Due to the multifactorial nature of atherothrombosis, patients may experience recurrent events (treatment failure) while on aspirin (Fig. 11) as with any other antithrombotic drug. This phenomenon has been inappropriately referred to as “aspirin resistance,” a definition further corroborated by the largely unreliable functional methods used to measure the response to aspirin (252). The vast majority of studies reporting the occurrence of aspirin “resistance” in different clinical settings have relied on a single measurement of platelet function ex vivo using classic LTA or bedside, whole blood assays, all exhibiting less than ideal intrasubject and intersubject variability and limited sensitivity to the effect of aspirin, not directly reflecting its mechanism of action (252). Studies of platelet function have also suggested a lower-than-expected response to aspirin as assessed by PFA-100 or LTA in response to ADP (10).

Recently, a fairly good relationship has been reported between platelet hyperreactivity, as assessed by conventional LTA, and glycemic control in T2DM patients treated with dual antiplatelet agents for coronary stenting (283). In this regard, an analysis from the aspirin-induced platelet effect (ASPECT) study challenged the issue of increasing daily aspirin dosage up to 325 mg to overcome the high platelet reactivity phenotype in diabetics with CAD (94). However, the various methods used to quantitate the anti-platelet effect of aspirin in these studies poorly reflect the biochemical path affected by aspirin, that is, platelet COX-1 activity (267) and variably reflect the aspirin-sensitive TxA2-dependent component of platelet aggregation (86). Whether the measurement of the potential non–COX-1 effects of aspirin might also be important remains unclear (205).
In assessing the effects of a poor response to aspirin treatment, one should be aware that the different platelet function tests correlate poorly among themselves (193). In addition, whether the relative contributions of TxA2-dependent and -independent pathways are constant for any given assay is largely unknown. This is particularly relevant to the current debate on aspirin “resistance,” because the characterization of “resistant” versus “responder” status is typically based on a single determination of platelet function, with the underlying assumption that this determination represents a stable phenotype. In addition, the clinical relevance of aspirin resistance in patients undergoing percutaneous revascularization on dual antiplatelet therapy is still uncertain. Thus, neither routine testing for aspirin resistance nor any change in antiplatelet therapy based on these tests in these patients is recommended.
In contrast, serum TxB2 and urinary 11-dehydro-TxB2 provide reliable information on the maximal biosynthetic capacity of circulating platelets ex vivo and on the actual rate of TxA2 biosynthesis in vivo, respectively (252). Less-than-complete inhibition of platelet TxA2 production and TxA2-dependent platelet activation, as assessed by these methods, are more reliable tools that assess aspirin responsiveness. Residual TxA2 biosynthesis despite low-dose aspirin treatment has been shown to be predictive of vascular events in high-risk patients (99). Thus, elevated urinary 11-dehydro-TxB2 levels identify patients who are relatively resistant to aspirin and who might benefit from alternative antiplatelet therapies or treatments that more effectively block in vivo TxA2 production or activity.
D. Current antiplatelet therapy for atherothrombotic disease
Currently available antiplatelet drugs selectively target key enzymes or receptors involved in platelet activation and aggregation processes (Fig. 12).

Aspirin irreversibly inhibits platelet COX-1, thereby preventing TxA2 biosynthesis (223). The effect of aspirin on platelets is presystemic and begins in the portal circulation. Virtually complete (≥97%) inactivation of platelet COX-1 is required to significantly affect systemic TxA2 biosynthesis (245). A single dose of 160 mg completely abolishes the platelet TxA2 production (223). However, the same effect can be progressively achieved with the chronic administration of a daily dose of 30–100 mg, due to unique pharmacokinetic features of aspirin that allow the cumulative inhibition of platelet COX-1 at low doses while substantially sparing vascular COX-2 (223).
Aspirin represents a cornerstone of antiplatelet therapy, and its efficacy and safety have been investigated in several populations, ranging from apparently healthy subjects at low risk to patients with established vascular disease or with acute ischemic events (227). The lack of a dose-response relationship in clinical trials evaluating its antithrombotic effects and the dose dependence of its adverse events support the use of the lowest effective dose (50–100 mg daily) as the most appropriate strategy that maximizes its efficacy and minimizes its toxicity (227).
The meta-analysis of the Antiplatelet Trialists' Collaboration (ATC) demonstrated that, in patients with acute or previous CV or cerebrovascular events, aspirin reduced the risk of an MACE (nonfatal MI, nonfatal stroke, or death from vascular causes) by ∼25% (66). Based on this evidence, aspirin is currently recommended for secondary CV prevention (25, 285).
In contrast, results of recent randomized controlled trials and meta-analyses raised questions about the efficacy of aspirin for primary prevention. In particular, the 2009 ATC meta-analysis of primary prevention trials showed that aspirin is of uncertain net value when the reduction in ischemic events is weighed against any increase in major bleeds (17). In addition, the benefit of aspirin in the primary prevention of major CV events in patients with diabetes appears to be limited (88).
It has been proposed that aspirin alone might be not sufficient to prevent ischemic events in patients at a high risk (188). In fact, aspirin inhibits only the COX pathway, leaving the ADP receptor (P2Y12) unaffected. Theoretically, the combined inhibition of the two main amplification pathways of platelet aggregation, ADP, and TxA2 pathways may be superior to the inhibition of either pathway alone in preventing atherothrombosis (30).
The interaction of ADP with one of its platelet receptors can be irreversibly blocked by the active metabolites of thienopyridines—ticlopidine and clopidogrel (224).
Thienopyridines are prodrugs that need to be activated by liver metabolism. This passage critically affects plasma levels of active metabolites. Genetic polymorphisms of the liver enzymes involved in the metabolism of clopidogrel as well as drug-drug interactions are critical determinants of the highly variable circulating levels of its active metabolite (49). This implies that some patients may not have an appropriate platelet-inhibition response to clopidogrel, and this reduced response is associated with an increased rate of subsequent ischemic events (13, 317). Currently, the role of ticlopidine in the antiplatelet armamentarium is limited, at least partially, due to safety concern. On the contrary, clopidogrel, found to be safer than ticlopidine, exhibited a favorable benefit-to-risk profile for CV prevention, both in combination with aspirin and as a single antiplatelet agent (224). The CAPRIE study demonstrated that clopidogrel has a modest but statistically significant benefit compared with aspirin for the prevention of MACE in patients with a recent stroke or MI and in those presenting with symptomatic PAD (3). Thus, according to clinical evidence and cost-effectiveness analyses, clopidogrel should be regarded as a valid alternative to low-dose aspirin in patients with established vascular disease (25).
Combined antiplatelet therapy (low-dose aspirin and clopidogrel) is currently the standard of care for patients with ST-elevation ACS and for those undergoing percutaneous coronary revascularization (25). However, in contrast to the consistent finding of a favorable benefit/risk profile of dual antiplatelet therapy in patients with ACS, the same strategy was not proved successful when compared with single antiplatelet therapy in patients with recent ischemic stroke or transient ischemic attack (95), or in patients with multiple risk factors (but no known vascular disease) (31).
Recently, among patients with a noncardioembolic ischemic stroke, the combination of low-dose aspirin (25 mg) with extended-release dipyridamole—a phosphodiesterase inhibitor that increases intracellular levels of cAMP and cGMP—has been shown to be at least as effective as a single antiplatelet agent (aspirin or clopidogrel) for the prevention of recurrent stroke or MACE (260). Based on these results, it has been proposed that a combined therapy with aspirin and extended-release dipyridamole may offer the best strategy for secondary stroke protection (5, 114).
Integrin αIIbβ3 antagonists (abciximab, eptifibatide, and tirofiban) have been validated as a useful addition to aspirin, particularly in patients with ACS undergoing PCI (178). Interestingly, patients with diabetes seem to benefit more from αIIbβ3 antagonism, thus supporting the hypothesis of a disease-specific antiplatelet treatment due to a platelet hyperreactive phenotype (31).
Despite their proven antithrombotic efficacy, current available antiplatelet agents lack some important features of the ideal antithrombotic agent, such as limited interindividual response variability, and absence of drug-drug interactions or bleeding complications (53). New antiplatelet agents have been developed that overcome these limitations (321). Future clinical research will demonstrate whether improved pharmacokinetics or targeting new platelet activation pathways will translate into better CV outcomes.
E. Novel therapeutic opportunities
Recurrent thrombotic events may continue to occur despite the use of standard dual antiplatelet treatment regimens in high-risk clinical settings. Novel antiplatelet strategies are needed in order to overcome the limitations of current therapies and to provide more effective platelet inhibition. These include action on the thrombin-mediated pathway, through inhibition of the platelet thrombin receptor or the focus of a third generation of P2Y12 antagonists (Fig. 12).
1. PAR-1 antagonists
Thrombin receptors or protease-activated receptors (PARs) are GPCRs differentially expressed by vascular cells, platelets, and other circulating cells (86). Proteolytic cleavage of the receptor by thrombin is irreversible, and is potentially coupled to different G proteins that signal through multiple pathways (11). PAR-1, -3, and -4 are activated by thrombin. Platelets express PAR-1 and -4, and are able to trigger platelet secretion and aggregation (11).
PAR-1-mediated platelet activation is mainly involved in the development of thrombosis. The inhibition of PAR-1 has been shown to reduce arterial thrombosis in a pre-clinical model by reducing thrombin-induced platelet aggregation, without prolonging BT or influencing coagulation parameters (166). PAR-1 antagonism did not inhibit ADP-induced or collagen-induced platelet aggregation, suggesting that PAR-1 antagonism does not affect other platelet signaling pathways.
Vorapaxar (SCH530348) and atopaxar (E5555) are PAR-1 antagonists, which have completed phase II testing, showing overall favorable safety profiles and encouraging clinical outcomes.
Vorapaxar is an orally active, competitive PAR-1 inhibitor. Structurally, it is a synthetic tricyclic 3-phenylpyridine analog of himbacine, a natural product that has been modified as a crystalline salt for drug development and clinical use. In animal models, it seems to completely block the thrombin receptor, inhibiting thrombin-mediated platelet activation without interfering with the cleavage of fibrinogen, which is also thrombin mediated. In addition, Vorapaxar does not affect prothrombin time or activated partial thromboplastin time, suggesting that the potential for bleeding events may not be increased (54). Accordingly, the treatment with vorapaxar either alone or in combination with aspirin plus clopidogrel does not increase BT or surgical blood loss compared with placebo (59). On the basis of these promising results, vorapaxar was advanced into clinical development.
Preclinical studies of atopaxar (E5555), the other orally active PAR-1 antagonist that has completed phase II clinical development, showed interesting antithrombotic effects in animal models (273). In fact, it inhibited neointimal hyperplasia in a rat balloon injury model (158) as well as the production of inflammatory markers: sCD40L, IL-6, and P-selectin (273). Compared with voraxapar, atopaxar has a shorter half life; thus, it may prove useful in surgery settings, in which a rapid suspension of drug effects is needed.
Although extrapolation of these results to humans is highly speculative, it seems that the inhibition of PAR-1 alone may be sufficient for the prevention or treatment of thrombotic events in humans. However, it cannot be excluded that preservation of thrombin signaling through platelet PAR isoforms other than PAR-1 can participate in the maintenance of normal hemostasis. Larger-scale phase III clinical trials will better define the efficacy and safety of PAR-1 antagonists in addition to the standard of care in patients with atherothrombotic disease manifestations.
2. New P2Y12 antagonists
The pharmacopoea of thienopyridines is rapidly expanding. In addition to ticlopidine and clopidogrel, a new third generation of P2Y12 antagonists is in clinical development.
The substantial advantages of prasugrel compared with clopidogrel are represented by the distinct chemical structure, which allows the conversion by esterase to an intermediate metabolite, rather than inactivation, thus resulting in higher plasma levels of the active metabolite, lower inter-individual variability, and faster onset of action (50). Several clinical studies have shown that prasugrel has a greater effectiveness in inhibiting platelet aggregation, as compared with clopidogrel. In fact, it induces 2–3 times more potent platelet inhibition in a 10 times lower dose as compared with clopidogrel or ticlopidine. Even those individuals who had responded poorly to clopidogrel seem to achieve robust platelet inhibition with prasugrel treatment (37). Prasugrel has also been shown to be more effective than clopidogrel in ACS patients undergoing PCI, but with more bleeding (323) and has been approved in the United States and Europe for this kind of patients. A greater net clinical benefit has been observed in patients with diabetes or ST-segment elevation (30).
AZD6140, known as ticagrelor, is a reversible selective P2Y12 antagonist. It acts as an orally active inhibitor of ADP-induced platelet aggregation. In patients with stable atherosclerotic disease, ticagrelor has been shown to inhibit ADP-induced platelet aggregation more rapidly and effectively and with less variability than clopidogrel both after the first dose and after 28 days of therapy (DISPERSE trial) (152). Moreover, in patients with ACS, it seems to be more effective than clopidogrel in preventing MACE, including CV death, nonfatal MI, and stroke. Furthermore, no significant difference in the rates of major bleeding has been found between the patients treated with ticagrelor and those treated with clopidogrel, according to the Thrombolysis in Myocardial Infarction criteria. In particular, ticagrelor was associated with more frequent non–coronary artery bypass graft (CABG)-related bleeding that clopidogrel, but it was safer than clopidogrel in patients undergoing CABG (314). However, an unexpectedly high rate of CABG-related major bleeding has been reported in the two study groups. Regarding safety, dyspnea and bradyarrhythmias occur more frequently with ticagrelor than with clopidogrel (45).
3. TP antagonists
Aspirin-insensitive TP agonists, as well as nucleate sources of TxA2, less affected than platelet TxA2 production by the once-daily dosing, may limit the antiplatelet effect of aspirin (224). In fact, F2-isoprostanes and other structurally related iso-eicosanoids, produced nonenzymatically through a process of oxygen radical-catalyzed lipid peroxidation, represent aspirin-insensitive agonists of the platelet TP. High levels of F2-isoprostanes, such as the compound 8-iso-PGF2α, are associated with lower aspirin effectiveness settings (86).
Thus, a selective TP antagonist could block the interaction of both aspirin-sensitive and aspirin-insensitive agonists with this platelet receptor. Several TP antagonists have been developed since the early 1980s. The most recent one, S-18886 or terutroban, is an orally active, highly specific TP antagonist. However, the PERFORM trial of terutroban versus aspirin, on nearly 19,000 patients with recent stroke, was recently halted on the basis of an interim analysis failing to support the superiority hypothesis. In fact, there was no difference between terutroban and aspirin in the vascular primary endpoint, or any of the secondary or tertiary endpoints. Furthermore, the rate of minor bleeding was slightly increased with terutroban (36). Based on this result, the clinical development of terutroban has been discontinued.
4. Therapeutic use of platelet preparations
Platelets are a source of bioactive factors in the process of wound repair. Indeed, platelets release cytokines and growth factors (e.g., transforming growth factor-β1, PDGF, vascular endothelial growth factor, fibroblast growth factor, epidermal growth factor, and insulin-like growth factor-1) that are involved in the healing process. By virtue of the rich granule content, autologous PRP represents an advanced wound therapy used in acute and chronic wounds. The mechanism of action for PRP is thought to be the molecular and cellular induction of normal wound healing responses similar to that seen during platelet activation, implying the release of granule content. Once the bioactive factors are released at the site of injury, they recruit cells and stimulate their differentiation in order to produce the ECM necessary to replace the tissue (24).
A recent meta-analysis of the literature published over the last 10 years, reviewed randomized, controlled trials and comparative group studies using platelet preparation therapy in cutaneous wounds (47). The comparison with the treatment to standard care or other interventions demonstrated that PRP therapy was significantly favored for complete healing in chronic wounds and for the reduced incidence of infection in acute wounds.
The use of PRP and platelet-poor plasma currently extends to many areas of surgery, from the preparation of preoperative autologous blood component therapy in order to reduce the number of allogeneic blood transfusions (103), to CV and thoracic surgery, neurosurgery, plastic and reconstruction surgery, and dental surgery [see ref. (154) for a review].
In sports medicine, the effects of PRP have been tested in several tendon-related disorders, skeletal muscle traumas, ligament/fascial injuries, and in the treatment of osteoarthritis and cartilaginous injuries [for a review, see ref. (24)].
A very interesting application of PDGF in the healing process is that of MI healing. In an elegant study, Zymek and co-workers (334) demonstrated that the PDGF-mediated signaling pathway plays a crucial role in regulating postinfarction repair by promoting collagen deposition in the infarcted area, modulating fibrosis and angiogenesis, and regulating the maturation of the infarct vasculature. More recently, an intramyocardial injection of autologous platelet gel has proved capable of ameliorating cardiac dysfunction after MI in a rat model (58).
IV. Conclusive Remarks
Multiple redox-sensitive pathways appear to promote agonist-induced platelet function. Overall, there is accumulating evidence supporting a net prothrombotic effect of vascular-derived and platelet-derived ROS in vitro. Several lines of evidence also suggest an oxidative enhancement of thrombus formation in vivo, and the augmented interactions of oxidants with platelets and the release of ROS in several CV disease states further support a causal role of enhanced ROS production in platelet pathophysiology. Taken together, these data point to an important role of platelet derived ROS and the intra-platelet redox state in the regulation of physiological platelet activation and the clinical expression of thrombotic events. The hypothesis that oxidative stress mediates atherothrombosis would implicate a potential for antioxidant therapies to ameliorate and perhaps reverse CV disease. Numerous clinical studies have attempted to validate the salutary effects of antioxidant drugs, or to better characterize the antioxidant effects of existing therapeutic agents. A further examination of the signaling pathways and/or proteins modified by ROS production will help elucidate the mechanism by which ROS generation and oxidative stress regulate hemostasis and coagulation and will facilitate the development of novel therapeutic strategies that target pathologic oxidative mechanisms while sparing homeostatic functions dependent on oxidative signaling.
Footnotes
Acknowledgments
This study was supported by grants from the European Commission (EICOSANOX Integrated Project 005033), the Innovative Medicines Initiative (SUMMIT Consortium). The authors gratefully acknowledge the expert assistance of Silvia De Fulviis, Sara La Barba, Paola Ranalli, and Nicole Santoro.