
Abstract
Introduction
Innovation
It has been accepted that reactive oxygen species (ROS) and cyclooxygenase (COX)-derived prostanoids are important mediators of vascular alterations in hypertension. Our findings demonstrate that a specific feed-forward relationship between ROS and COX-2/TP receptors serves as a pathological link between Angiotensin II, vascular dysfunction, and hypertension. We suggest that by modulating ROS and COX pathways, antioxidants targeting NAD(P)H Oxidase and/or mitochondria or TP receptor antagonists could be promising therapeutic options with potential beneficial effects to reverse the alterations in vascular function associated to hypertension.
In addition to ROS, vasoconstrictor prostanoids from the constitutive (cyclooxygenase [COX]-1) or inducible (COX-2) COX are mediators responsible of the endothelial dysfunction in hypertension (13). We and others have previously described increased COX-2 expression and participation of COX-2-derived prostanoids on vasoconstrictor- and endothelium-dependent vasodilator responses of vessels from different models of hypertension and from hypertensive patients (1, 2, 16, 20, 28, 32, 34 –36, 38). The increased COX-2 expression and activity found in hypertension were normalized by treatment with an Ang II type 1 receptor antagonist (4), suggesting the participation of Ang II in these effects. In support of this, in vitro studies demonstrate that Ang II induces COX-2 expression and prostanoid production in different vascular cell types (4, 5, 15, 24, 36). In vivo, Ang II infusion induces vascular COX-1 upregulation (21, 30), and either COX-2 downregulation (30) or upregulation (9, 21, 36) in different vessels has been described.
There seems to be a close, yet not totally understood, interaction or feed-forward mechanisms between ROS and COX-derived products. ROS can activate COX expression and/or activity (14, 17, 22), and antioxidant treatments abolish the role of COX on endothelium-dependent contractile responses (37). Additionally, COX activity is also involved in the generation of ROS in vessels from spontaneously hypertensive rats (SHRs) (18, 27). After Ang II stimulation, both positive and negative unidirectional relationships between ROS and COX-2/COX-1 expression or activity have been found (4, 5, 30, 36). In addition, a mutual relationship between ROS and COX activities has been found in liver cells (25). However, studies addressing this relationship at vascular level and the role of mitochondrial oxidative stress are lacking. The aim of the present study was to evaluate the contribution of ROS derived from NAD(P)H Oxidase and mitochondria on the participation of COX-derived products on vasoconstrictor responses in conductance and resistance arteries from the Ang II-induced hypertensive model. We also evaluated whether COX-2-derived products can modulate ROS production and their effects on vascular function. Our findings demonstrate that there is a circuitous relationship between COX-2 and ROS products in hypertension, which in turn participates in the alterations in vascular function associated to this pathology.
Results
ROS and COX-2 blockade decreases blood pressure and cardiac and vascular hypertrophy in Ang II-infused mice
Systemic infusion of Ang II increased systolic blood pressure (SBP), induced left ventricular hypertrophy, reduced aortic lumen diameter, and increased aortic media thickness (Fig. 1A and Supplementary Fig. S1; Supplementary Data are available online at
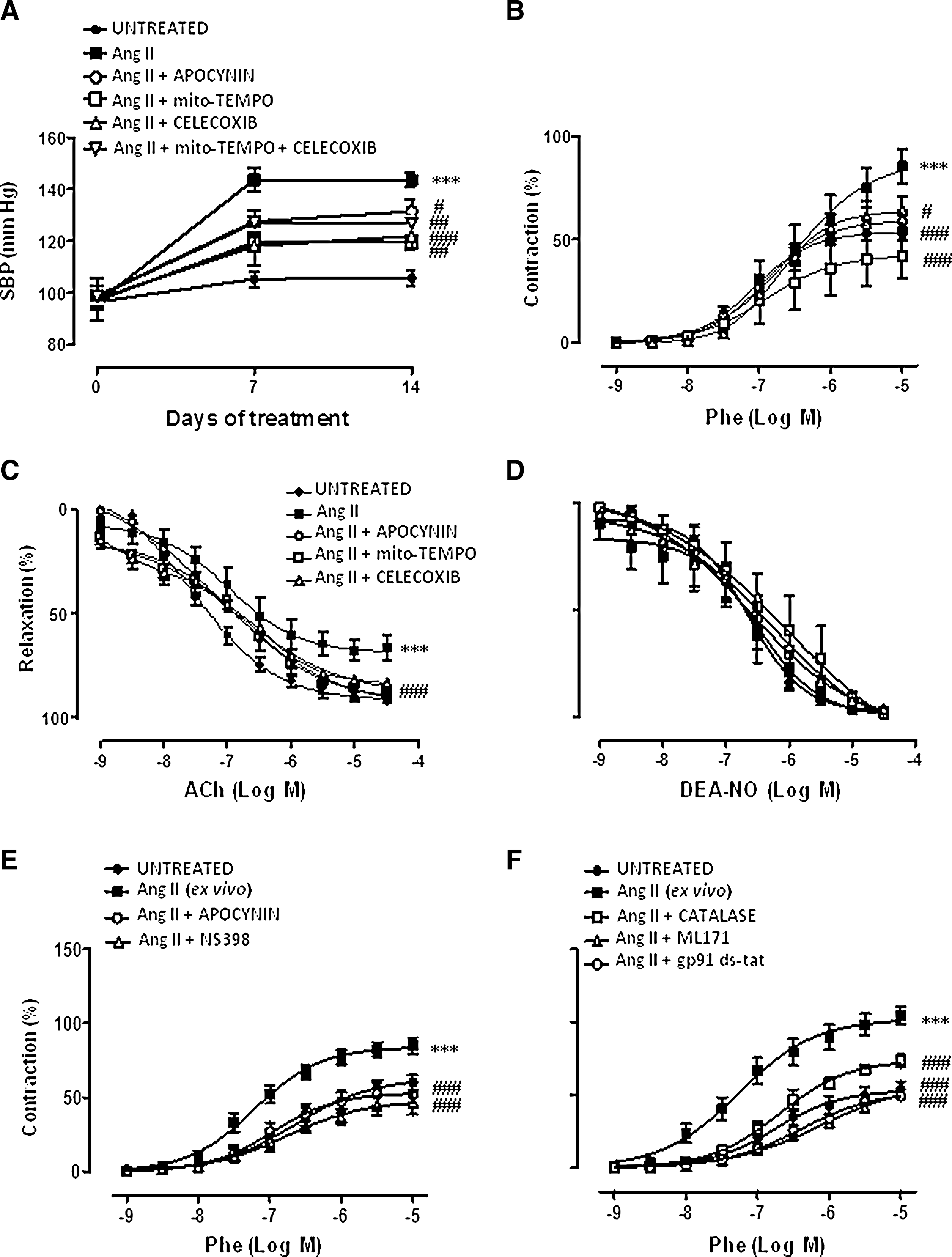
In a previous study, Dikalova et al. (11) demonstrated that Mito-TEMPO decreased established hypertension induced by Ang II. We therefore evaluated the effects of apocynin and Celecoxib when administered 7 days after Ang II infusion; at this time, mice are hypertensive (Supplementary Fig. S2A). Both Celecoxib and apocynin decreased SBP, but not left ventricular hypertrophy (Supplementary Fig. S2A, B). In addition, in a model of established hypertension, Celecoxib, but not the superoxide dismutase (SOD) mimetic tempol or Mito-TEMPO, treatment reduced SBP in SHRs (control: 219.9±7.6 mm Hg; Celecoxib: 184.8±5.5 mm Hg, p<0.01; tempol: 209.7±2.7 mm Hg; Mito-TEMPO: 212.9±5.4 mm Hg, n=4–10). However, neither Celecoxib nor tempol did affect SBP in Wistar Kyoto (WKY) (control: 151.9±3.2 mm Hg; Celecoxib: 148.7±3 mm Hg; tempol: 146.9±2 mm Hg, n=4–7).
ROS and COX-2 blockade improves vasoconstrictor- and endothelium-dependent vasodilator responses
Phenylephrine-induced contractile responses were greater in aortic segments from Ang II-infused than control mice. This increase was abolished by apocynin, Mito-TEMPO, or Celecoxib treatments (Fig. 1B). In control mice, these treatments did not have any effect on phenylephrine responses (data not shown). In ex vivo experiments, Ang II (1-μM 1-h incubation) also potentiated phenylephrine responses; this potentiation was reduced by co-incubation with the COX-2 inhibitor NS398 (1 μM), apocynin (0.3 mM), the NOX-2 inhibitor gp91ds-tat (5 μM), the NOX-1 inhibitor ML171 (0.5 μM), or the H2O2 detoxificant enzyme catalase (1000 U/ml) (Fig. 1E, F).
The endothelium-dependent vasodilator responses induced by acetylcholine (ACh) were smaller in arteries from Ang II-infused than control mice. Treatments with apocynin, Mito-TEMPO, or Celecoxib improved the impaired ACh relaxation in Ang II-infused (Fig. 1C) mice, but did not have any effect in control mice (data not shown). However, endothelium-independent relaxation to diethylamine NONOate (DEA-NO) was not modified by Ang-II infusion or by apocynin, Mito-TEMPO, or Celecoxib treatments (Fig. 1D). Celecoxib or apocynin had no effects in phenylephrine responses or in ACh relaxation when administered 7 days after Ang II infusion (Supplementary Fig. S2C, D), suggesting that these treatments prevent, but not reverse, these vascular alterations induced by Ang II.
We have previously described that SHR aorta showed increased contractile responses to phenylephrine and impaired ACh-induced relaxation compared to normotensive WKY rats (2, 3). Neither tempol nor Celecoxib treatment affected phenylephrine responses in SHRs (Supplementary Fig. S3B, D). However, Celecoxib, but not tempol, increased ACh relaxation in SHRs (Supplementary Fig. S3A, C), suggesting that COX-2 is involved in endothelial dysfunction in SHRs, as described (32). In WKY rats, Celecoxib or tempol treatments did not modify phenylephrine or ACh responses (data not shown).
ROS and COX-2 blockade improves the participation of nitric oxide on phenylephrine responses
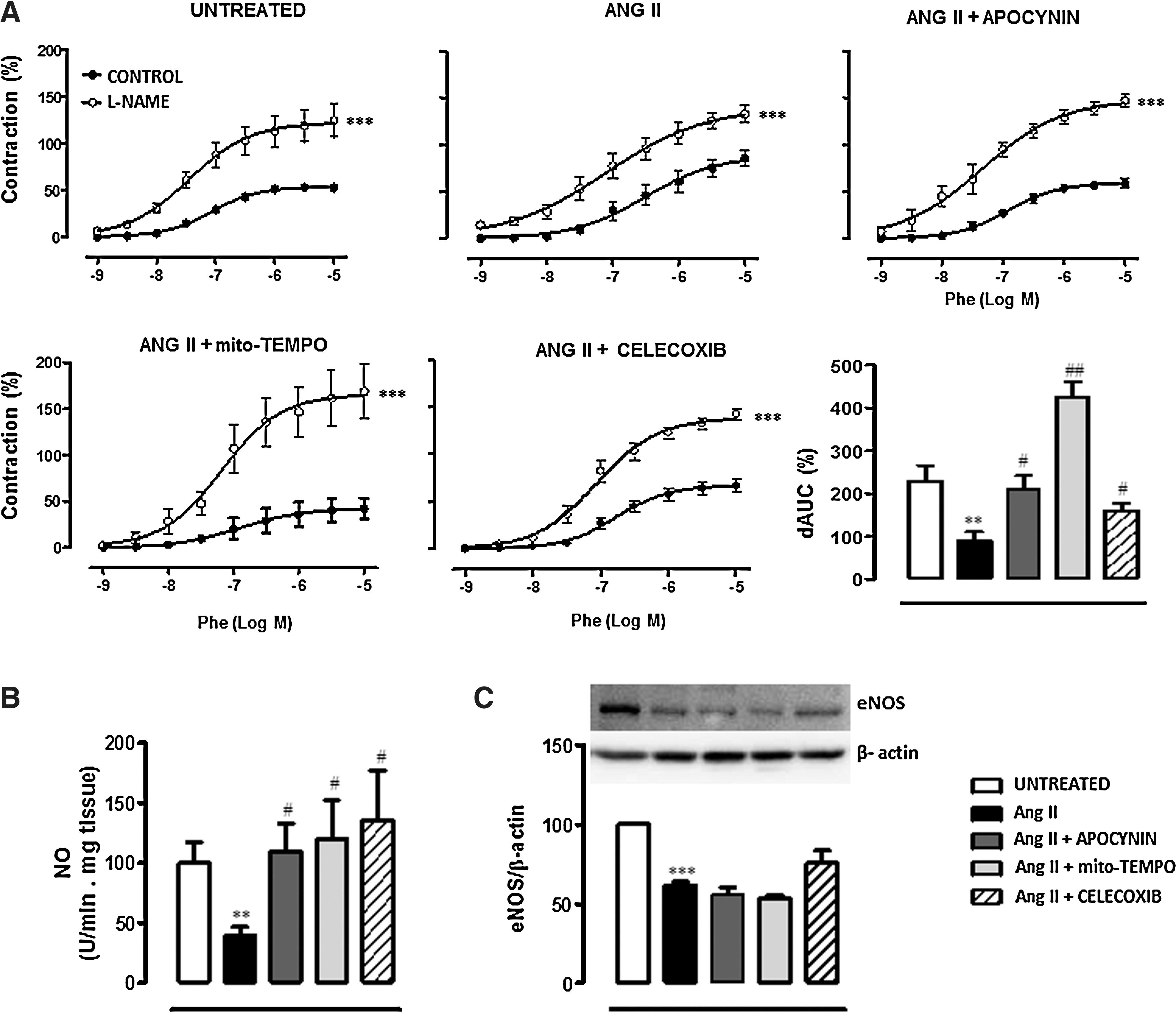
To evaluate whether ROS and COX-2-derived prostanoids might affect the nitric oxide (NO) pathway, we evaluated the effect of the different treatments on the modulation by NO of vasoconstrictor responses, the vascular NO release, and endothelial nitric oxide synthase (eNOS) expression. The NOS inhibitor N-nitro-L-arginine methyl ester (L-NAME, 100 μM) enhanced phenylephrine-induced contraction more in aorta from Ang II-infused mice than control (Fig. 2A). Apocynin, Mito-TEMPO, and Celecoxib treatments increased the effect of L-NAME in the vessel from Ang II-infused mice (Fig. 2A). These results suggest decreased NO production and/or bioavailability after Ang II infusion that is restored by antioxidants and COX-2 inhibitors. In agreement, NO release was reduced in aorta from Ang II-infused mice, and apocynin, Mito-TEMPO, and Celecoxib treatments normalized NO levels (Fig. 2B). Ang II also decreased eNOS protein expression in aorta. However, this decrease was not modified by any treatment (Fig. 2C).
COX-2 blockade reduces the increased oxidative stress and its participation on phenylephrine responses
We next evaluated the effects of Ang II infusion in different parameters of vascular oxidative stress, and whether COX-2 blockade might modulate these effects. Figure 3A shows that basal O2·− production was greater in the aortic wall from Ang II-infused than control mice. This was particularly evident in adventitia and media layers. In addition, aortic NAD(P)H Oxidase activity and p22phox, but not p47phox, gene expression were increased in Ang II-infused compared to control mice (Fig. 3B–D). The increased O2·− production, NAD(P)H Oxidase activity, and p22phox gene expression were normalized by apocynin, Mito-TEMPO, and Celecoxib treatments (Fig. 3A–D). Aortic and plasma malondialdehyde (MDA) levels, a marker of lipid peroxidation, were also greater in Ang II-infused than in control mice (Fig. 3E and Supplementary Fig. S4A). All treatments decreased aortic MDA levels (Fig. 3E). However, the increase in plasma MDA was prevented by apocynin and Celecoxib, but not by Mito-TEMPO treatment (Supplementary Fig. S4A), suggesting a greater role of mitochondria in vascular than plasma oxidative stress.
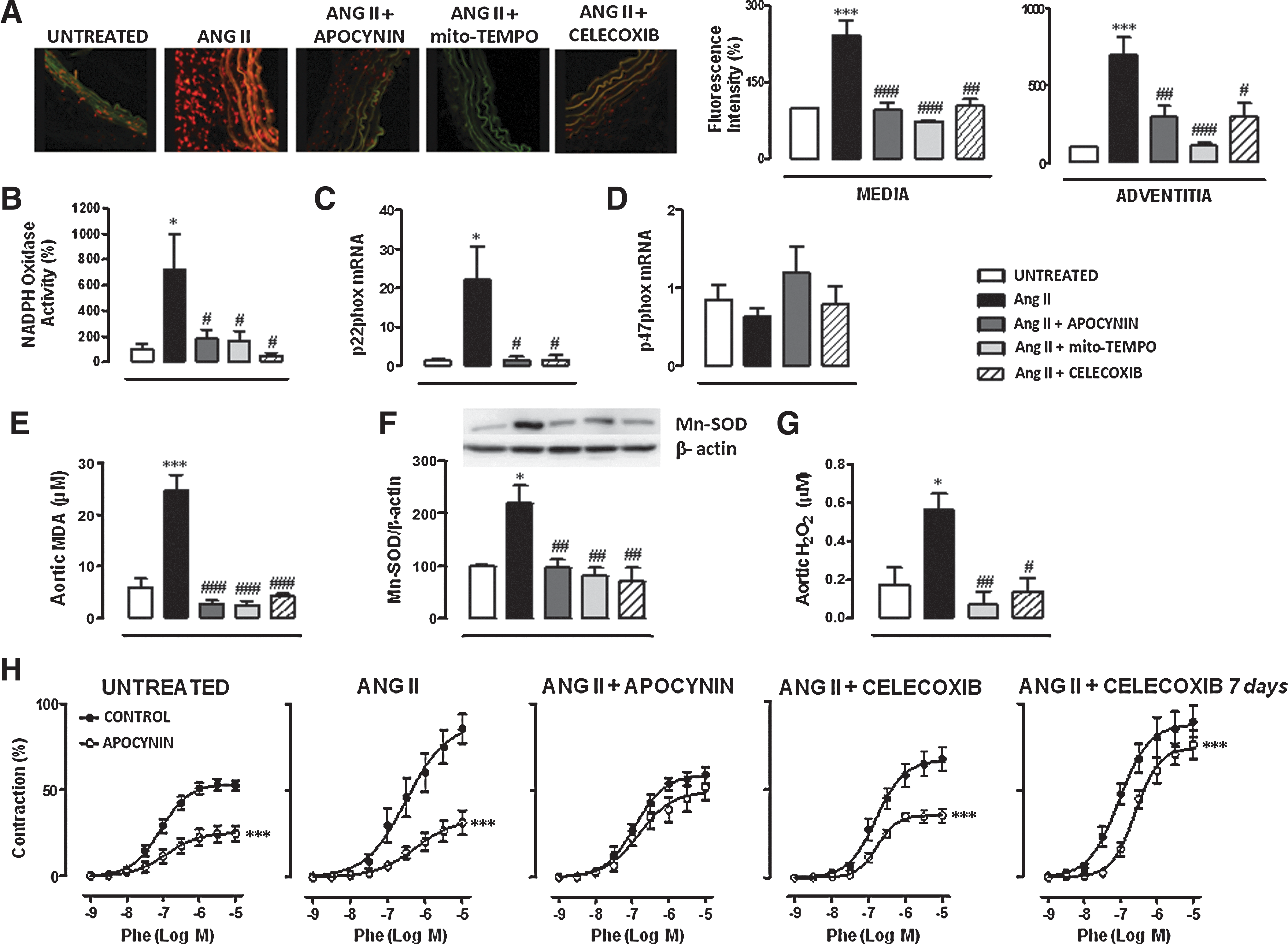
We then evaluated whether alterations in antioxidant mechanisms might be also involved in the Ang II-induced O2·− production. Ang II increased MnSOD, but not Cu/Zn- and EC-SOD protein expression, and this was normalized by apocynin, Mito-TEMPO, and Celecoxib treatments (Fig. 3F and Supplementary Fig. S4B, C). Since MnSOD expression is increased by Ang II treatment, we questioned whether the product of SOD activity, H2O2, was also modified in our experimental paradigm. As shown in Figure 3G, Ang II increased aortic H2O2 production that was normalized by Mito-TEMPO and Celecoxib treatments.
The participation of ROS in vascular contractile responses was evaluated in vitro by preincubation of vessels with apocynin (0.3 mM). As shown in Figure 3H, apocynin inhibited the phenylephrine-induced contraction more in aorta from Ang II-infused than control mice. As expected, in vivo apocynin treatment abolished the in vitro effect of this drug on phenylephrine-induced contraction. In addition, Celecoxib treatment decreased the inhibitory effect of apocynin in Ang II-treated mice (Fig. 3H). When Celecoxib was administered 7 days after Ang II infusion, a decrease in the in vitro effect of apocynin was also observed (Fig. 3H), suggesting that COX-2 blockade not only prevents but also reverses the increased participation of ROS in vascular responses.
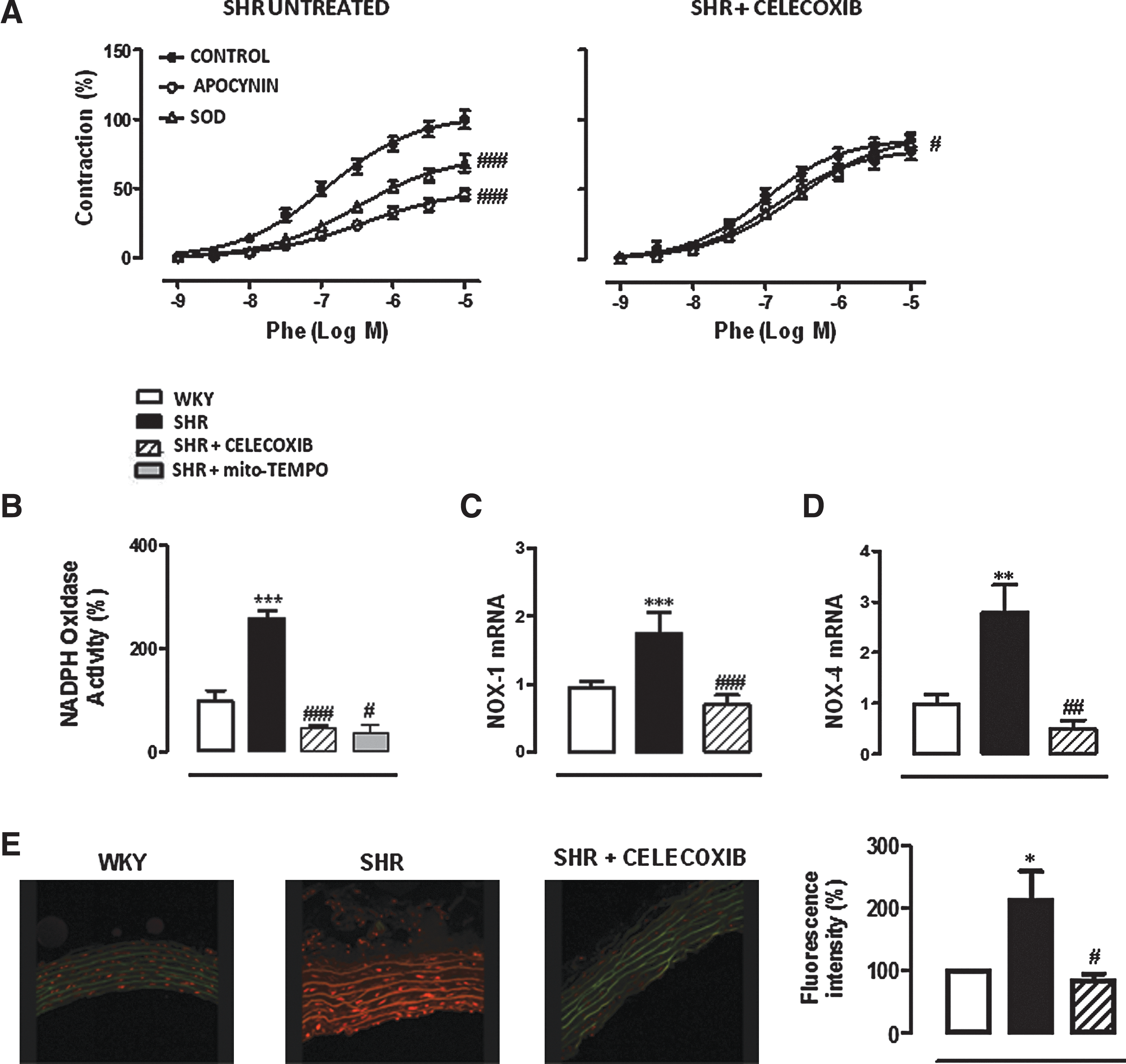
We previously described a greater inhibitory effect of in vitro apocynin and SOD on phenylephrine responses in aorta from SHRs than WKY rats (3). SHR treatment with Celecoxib abolished the inhibitory effects of apocynin and SOD (150 U/mL) on phenylephrine responses (Fig. 4A). In addition, Celecoxib treatment normalized the increased NAD(P)H Oxidase activity (Fig. 4B), NOX-1 and NOX-4 gene expression (Fig. 4C, D) and O2·− production (Fig. 4E), observed in aorta from SHRs. The increase in NAD(P)H Oxidase activity was also diminished by Mito-TEMPO treatment (Fig. 4B).
ROS blockade decreases COX-2 expression and the participation of prostanoids on phenylephrine responses
We then evaluated the effects of Ang II infusion in COX-2 and COX-1 expression and/or activity (evaluated as vascular function) and whether oxidative stress might be modulating COX expression and/or the participation of COX products in vascular responses. Ang II significantly increased aortic COX-2, but not COX-1, expression that was reversed by apocynin, Mito-TEMPO, and Celecoxib treatments (Figs. 5A, B and 6A). The participation of COX-2-derived prostanoids in phenylephrine responses was evaluated by the incubation of vessels with NS398 (1 μM). This drug inhibited phenylephrine responses in aorta from Ang II-infused, but not in control, mice (Fig. 5C). As expected, Celecoxib treatment abolished the inhibitory effect of NS398. In addition, apocynin and Mito-TEMPO treatments inhibited the effect of NS398 in Ang II-infused mice (Fig. 5C), suggesting that ROS modulate the COX-2 pathway. When apocynin was administered 7 days after Ang II infusion, a decrease in the in vitro effect of NS398 was also observed (Fig. 5C), suggesting that ROS blockade not only prevents but also reverses the participation of COX-2-derived prostanoids in vascular responses.
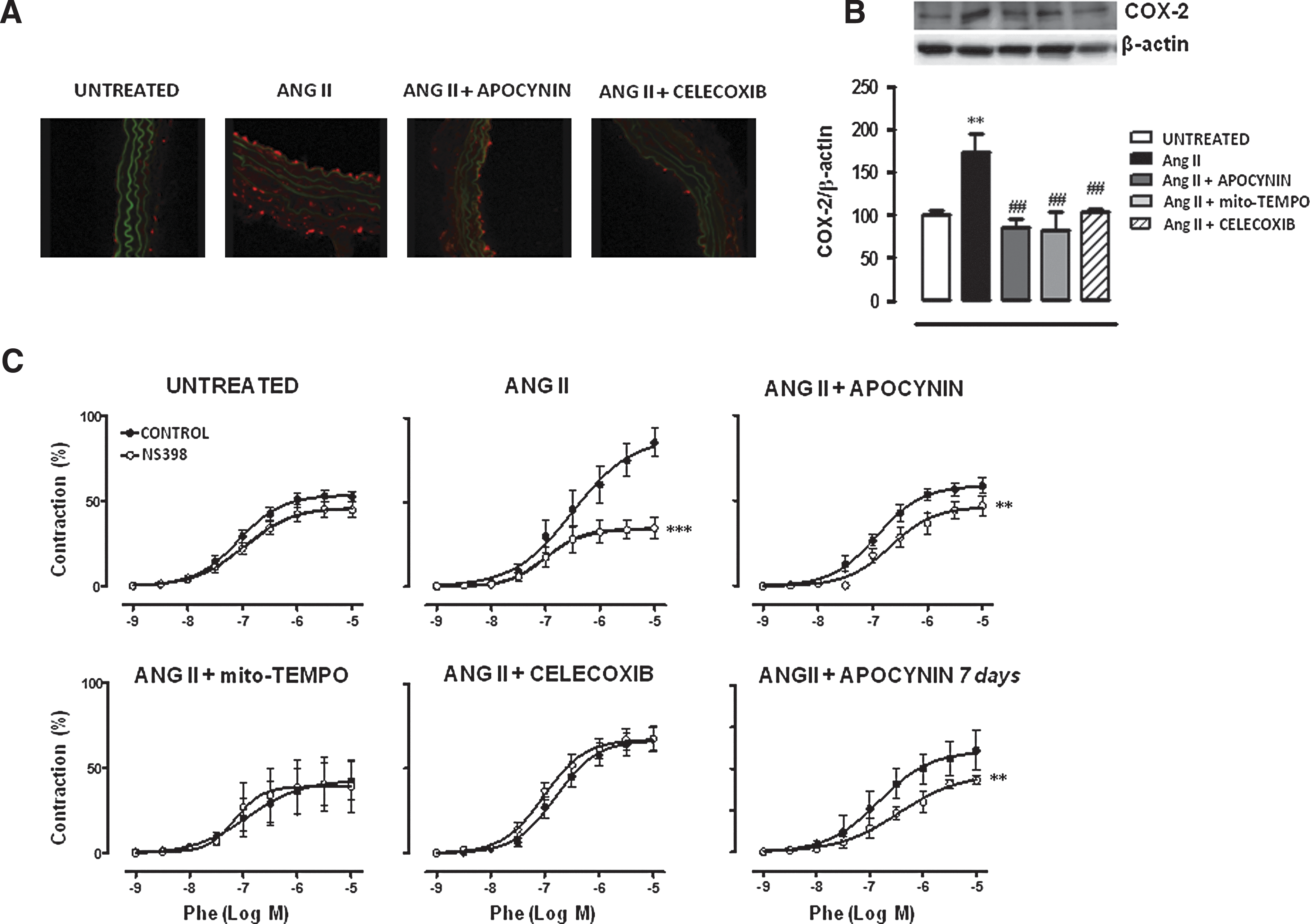
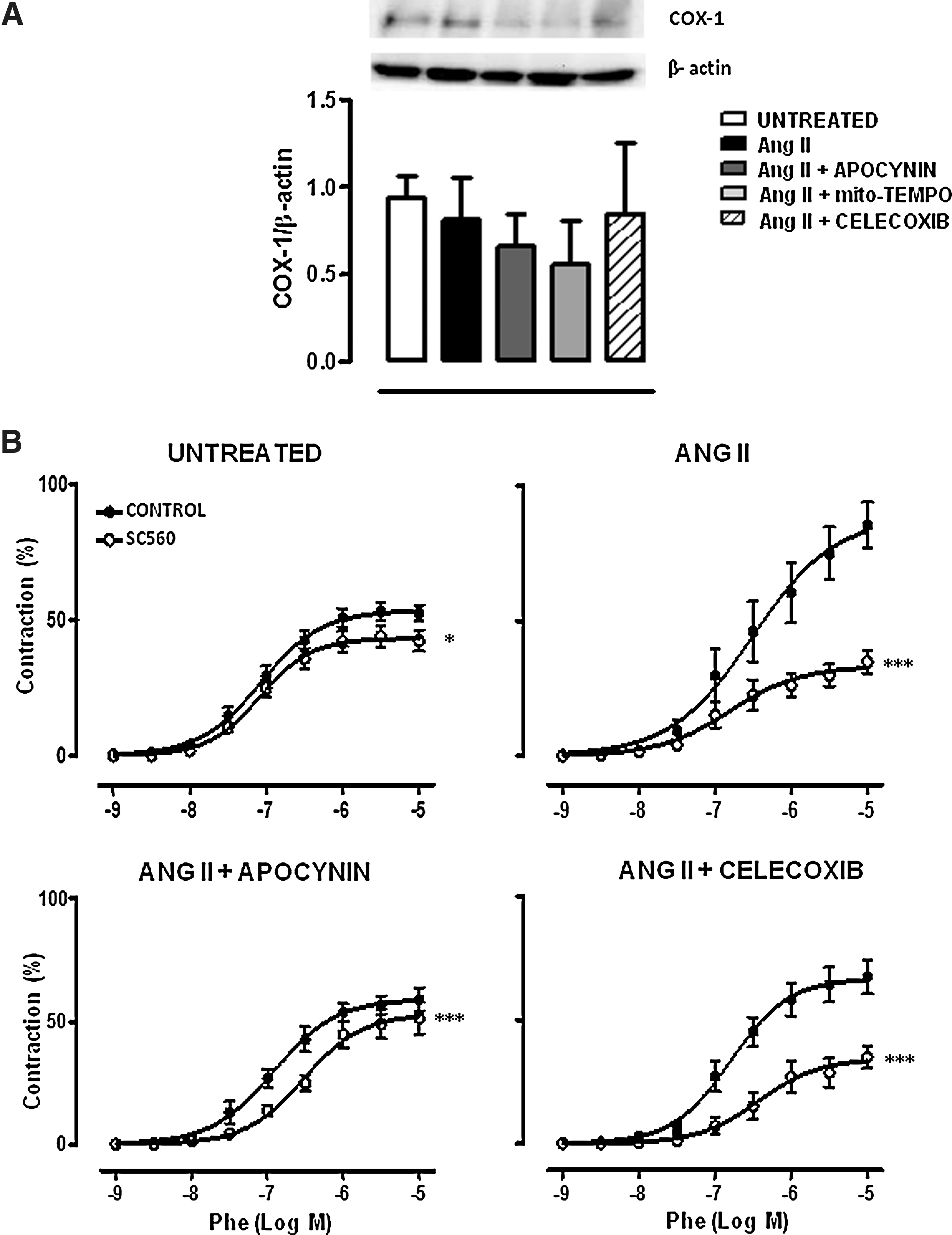
The participation of COX-1-derived prostanoids in phenylephrine responses was evaluated by the incubation with the COX-1 inhibitor SC560 (1 μM). SC560 slightly inhibited the phenylephrine response in control mice. This effect was more pronounced in Ang II-infused mice and was reduced by apocynin, but unaffected by Celecoxib, treatment (Fig. 6B). These results suggest that Ang II also induces COX-1 activity, which is modulated by oxidative stress.
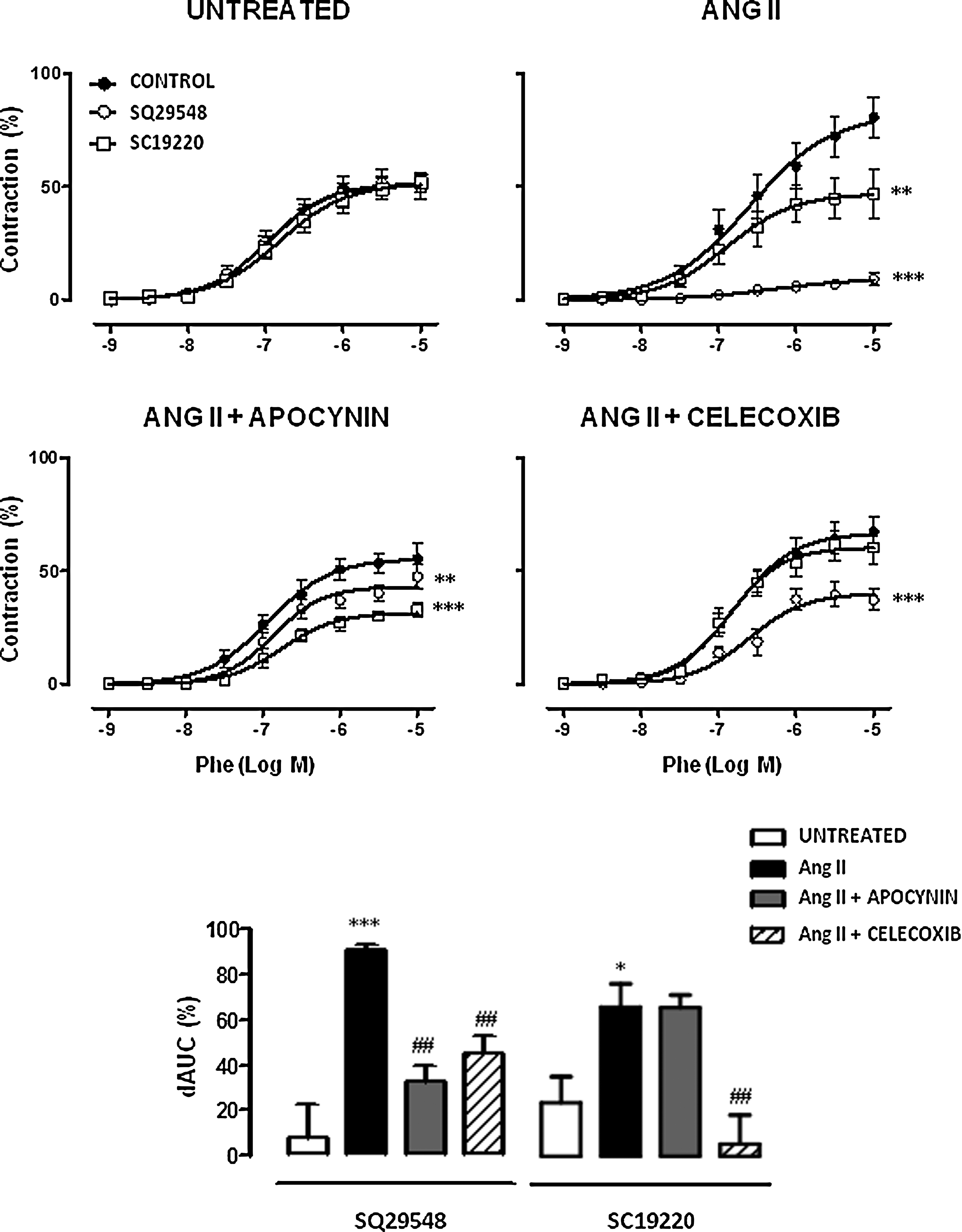
The participation of thromboxane A2 receptor (TP) and EP1 receptors on phenylephrine responses was analyzed using the respective selective antagonists of these receptors SQ29548 and SC19220. Neither SQ29548 (1 μM) nor SC19220 (1 μM) did modify phenylephrine responses in aorta from control mice. However, in Ang II-infused mice, both drugs inhibited phenylephrine responses (Fig. 7). This inhibitory effect of SQ29548 was decreased by apocynin and Celecoxib treatments. However, only Celecoxib reduced the inhibitory effect of SC19220 (Fig. 7).
Previously we found that COX-2 expression, 13,14-dihydro-15-keto-PGF2α levels, and the inhibitory effect of NS398 and SQ29548 on phenylephrine responses were greater in aorta from SHRs than WKY rats (2). Now, we confirmed some of these data, and we observed that COX-1 gene and protein expression were also increased (Fig. 8). SHR treatment with tempol decreased the inhibitory effect of NS398 and SQ29548 on phenylephrine responses (Fig. 8A) and normalized the increased 13,14-dihydro-15-keto-PGF2α levels (Fig. 8D). However, tempol treatment did not modify COX-2 protein expression or mRNA levels (Fig. 8B, C). In WKY rats, tempol did not affect COX-2 protein, 13,14-dihydro-15-keto-PGF2α levels (Fig. 8A, D), or mRNA (data not shown). Interestingly, Mito-TEMPO treatment of SHRs diminished the increased COX-2 and COX-1 gene and protein expression (Fig. 8E–H), suggesting that mitochondrial-specific antioxidants might be more effective than general antioxidants in modulating COX expression in the SHR model.

Effect of mitochondrial ROS and COX-2 blockade in resistance arteries from Ang II-infused mice
To further evaluate whether the observed effects in aorta are vascular bed specific and taking into account the importance of resistance arteries in control and maintenance of blood pressure, we performed an additional set of experiments in resistance arteries. We aimed to evaluate the role of ROS, particularly mitochondrial ROS, and COX-2-derived prostanoids in the alterations induced by Ang II in vascular responses and the possible relationship between these two enzymatic pathways. In mesenteric resistance arteries (MRAs), Ang II infusion did not modify phenylephrine contractile responses, but impaired ACh-induced relaxation (Table 1). Mito-TEMPO, but not Celecoxib, treatment inhibited phenylephrine responses. In addition, both Celecoxib and Mito-TEMPO improved ACh-induced relaxation in MRAs from Ang-II infused animals (Table 1).
p<0.05 versus Ang II.
p<0.05 versus Control.
Data are expressed as mean±SEM of 5–15 animals.
pD2, negative logarithm of the concentration that produces a 50% of maximal response; Emax, maximal response; Ang II, Angiotensin II.
The NOS inhibitor L-NAME increased the vasoconstrictor response induced by phenylephrine in control, but not in Ang II-infused mice (Supplementary Fig. S5A). Mito-TEMPO and Celecoxib treatments restored the increase in the phenylephrine responses induced by L-NAME in MRA from Ang II-infused mice (Supplementary Fig. S5A).
NS398 inhibited phenylephrine responses only in MRA from Ang II-infused mice, and this inhibition was abolished by Mito-TEMPO (Supplementary Fig. S5B). However, SC560 did not affect phenylephrine responses in any experimental group (data not shown). The incubation of the arteries with SOD inhibited the phenylephrine response only in MRA from Ang II-infused mice, and Celecoxib treatment abolished this effect (Supplementary Fig. S5C). These results suggest that the relationship between ROS and COX-2-derived products might be a general vascular mechanism.
Discussion
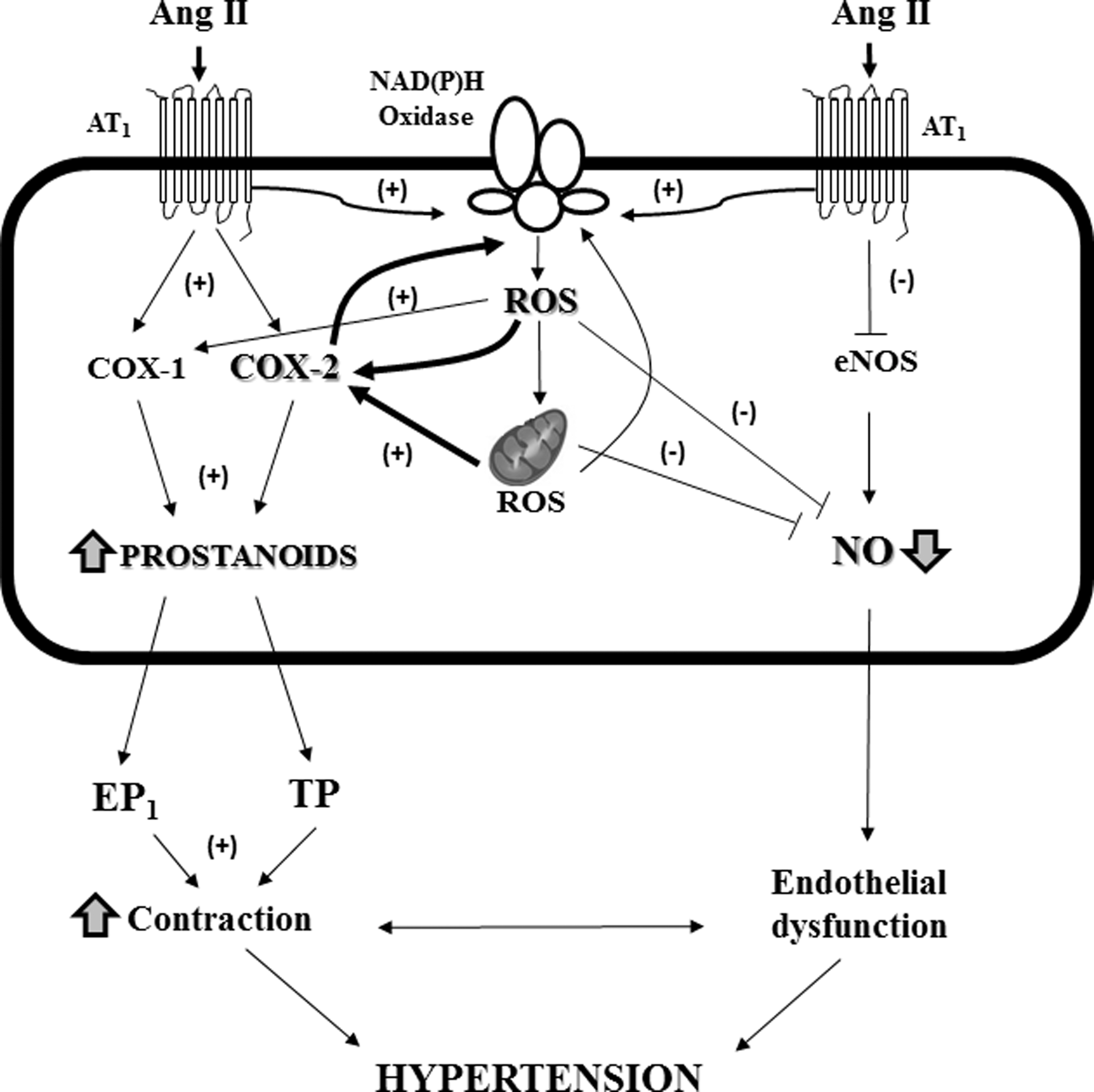
The present study provides evidence that demonstrates that ROS and COX-2-derived prostanoids are important mediators of vascular dysfunction and hypertension development. In addition, we demonstrate that there is a crucial vicious circle between ROS and COX-2-derived prostanoids in this pathology. Both NAD(P)H Oxidase and mitochondrion-derived O2·− participate in the increased COX-2 expression and activity. In addition, COX-2 derivatives modulate O2·− availability by affecting NAD(P)H Oxidase.
It is well known that Ang II increases vascular NAD(P)H Oxidase activity and O2·− production (6, 12, 29). In addition, mitochondria are becoming as an important source of O2·− in response to Ang II due, in part, to NAD(P)H Oxidase activation (10, 11). It is also evident that Ang II induces COX-2 expression and prostanoid production in vascular cell types such as endothelial cells (36), vascular smooth muscle cells (4, 24), and adventitial fibroblasts (5, 15) as well as in whole vessels (36). Oxidative stress has been also suggested to induce COX activity (13, 17, 30) or upregulate COX-2 expression (4, 14, 16, 38), and this is particularly increased in hypertension (16). In addition, we and other authors have previously demonstrated that COX-derived products induce O2·− production in SHR arteries (18, 27). Recently, a reciprocal regulation of NAD(P)H Oxidase and the COX-2 pathway has been described in liver cells (25). However, to our knowledge, there are no studies demonstrating a circuitous relationship between these two pathways in the vascular wall.
Effects of ROS and COX-2 blockade in blood pressure and vascular function and structure
In the present study, we observed that treatment of Ang II-infused mice with the selective COX-2 inhibitor Celecoxib, the mitochondrion-targeted SOD2 mimetic Mito-TEMPO, and the NAD(P)H Oxidase inhibitor apocynin prevented, in part, hypertension development. In addition, Celecoxib and Mito-TEMPO partially prevented cardiac and vascular hypertrophy, supporting a role of COX-2-derived products and ROS in hypertension and cardiac and vascular remodeling. This is in agreement with previous reports (11, 28, 33), although no modification of blood pressure after COX-2 inhibition has also been found (31). The combination of Mito-TEMPO and Celecoxib did not further decrease blood pressure; however, it normalized the increased cardiac hypertrophy, suggesting that COX-2 and ROS pathways might be interconnected to participate in the development of hypertension, although they might act through different intracellular mechanisms to modulate cardiac hypertrophy. It has been previously described that Mito-TEMPO, but not tempol, decreased established Ang II-induced hypertension (11). We now observed that apocynin and Celecoxib when administered 7 days after Ang II infusion, decreased SBP, but not left ventricular hypertrophy, suggesting that COX-2 or ROS blockade not only prevents but also reverses hypertension development after Ang II infusion. In agreement, Celecoxib decreased blood pressure in the SHRs, although no effect on blood pressure was observed after Mito-TEMPO and tempol treatments. Higher doses, earlier administration, or longer duration of the treatments might be needed to achieve hypotensive effects of antioxidants in the SHR model.
The exposure to Ang II either in vivo or ex vivo in the organ bath increased phenylephrine responses and impaired ACh relaxation without effect on DEA-NO-induced relaxations in mouse aorta. Other authors have also described endothelial dysfunction after Ang II infusion (21, 30). All chronic treatments abolished the increased phenylephrine-induced contraction and improved ACh responses. In addition, the increased phenylephrine responses after Ang II incubation ex vivo were also normalized by apocynin, the specific NOX-2 inhibitor gp91ds-tat, the specific NOX-1 inhibitor ML171 (19), and the COX-2 inhibitor NS398. These results demonstrate that COX-2, mitochondria, and NAD(P)H Oxidase blockade protects the vascular wall against the damaging effects of Ang II. Our results are in agreement with observations made in this and other models of hypertension where it has been described increased participation of ROS from NAD(P)H Oxidase and mitochondrion- (11, 33) and COX-2-derived prostanoids (1, 2, 21, 32, 34, 36, 38) in vasoconstrictor or endothelium-dependent vasodilator responses.
Reciprocal relationship between ROS and COX-2
It is well known that hypertension is associated with increased vascular ROS production from NAD(P)H Oxidase, among other sources, and/or with altered antioxidant defenses (8, 12). In agreement, we observed in aorta from Ang II-infused mice and/or SHRs, increased vascular oxidative stress as demonstrated by increased ROS production (O2·− and H2O2), MDA levels, NAD(P)H Oxidase activity, NAD(P)H Oxidase subunit expression (NOX-1, NOX-4 and p22phox), and increased participation of ROS in vasoconstrictor responses, as shown by the greater inhibitory effect of apocynin on phenylephrine responses. In addition, we observed increased MnSOD, but not Cu/Zn- or EC-SOD expression, probably as a compensatory mechanism against the increased mitochondrial O2·− production. This upregulation of MnSOD would explain the increased aortic H2O2 production observed after Ang II that might affect vascular function. In fact, catalase incubation abolished the potentiation of phenylephrine responses induced by Ang II.
Prostanoids from COX-1 and COX-2 are also increased in different models of hypertension and are responsible of the endothelial dysfunction observed in this pathology (13). Herein, we observed that arteries from Ang II-infused mice and/or SHRs show increased COX-2 expression and activity and increased participation of COX-2-derived vasoconstrictor prostanoids in contractile responses. Interestingly, COX-1 expression was also increased in SHR aorta, but unaffected in the Ang II-infused mice in spite of the increased participation of COX-1-derived prostanoids in phenylephrine responses observed in this model. Conflicting results on the role of COX-1 or COX-2 in vasodilator and vasoconstrictor responses have been found depending on the hypertension model or the vessel studied (1 –4, 13, 21, 28, 30, 36, 38). It has been proposed that TP receptor activation plays a pivotal role in vascular alterations in hypertension (13). Herein, we confirm the essential role of the TP receptor in the phenylephrine responses in the Ang II-infused model and also suggest the involvement of EP1 receptors in these responses.
In the present study, besides the demonstration of the role of COX-2-derived products and ROS in vascular responses, we uncover a reciprocal relationship between ROS and COX-2-derived products in aorta from Ang II-infused mice and SHRs. This is based on the following findings: first, Celecoxib treatment normalized the increased NAD(P)H Oxidase activity and expression, ROS production, and their participation on vascular responses in both the Ang II-infused mice and SHRs. In addition, Celecoxib normalized the increased MnSOD expression probably as a consequence of the normalized oxidant status. Celecoxib treatment also decreased COX-2 expression probably due to the observed effects of Celecoxib on blocking oxidative stress. Secondly, Mito-TEMPO and/or apocynin normalized the COX-2 upregulation and the increased participation of COX-1- and COX-2-derived prostanoids in vascular responses induced by Ang II infusion. Furthermore, the activation of TP, but not of EP1, receptors is modulated by oxidative stress. In SHRs, Mito-TEMPO, but not tempol, normalized the increased COX-2 expression, suggesting that specific mitochondrion-targeted antioxidants might be more efficient in modulating COX expression than nonspecific antioxidants. However, tempol was able to diminish the increased PGF2α release and the participation of COX-2-derived prostanoids in vascular responses in SHRs. Interestingly, Mito-TEMPO also decreased COX-1 expression in SHRs. This relationship between ROS and COX-2 is not specific of conductance arteries, since in MRA, a similar ROS-COX modulation was observed. This is based on results demonstrating that Mito-TEMPO and Celecoxib treatments abolished the inhibitory effects of NS398 or SOD on phenylephrine responses, respectively. As described above, several studies have demonstrated a unidirectional relationship between ROS and COX-derived products (4, 13, 14, 16 –18, 27, 38). However, to our knowledge, this is the first study that demonstrates that there is a circuitous relationship between these two pathways in the vascular wall and particularly, the essential role of mitochondria in this relationship.
The fact that Celecoxib abolished the inhibitory effect of NS398, but not of the specific COX-1 inhibitor SC560, on phenylephrine responses suggests that the observed effects on the COX-2 blockade pathway are specific. COX-2 blockade also affects downstream pathways, since the participation of TP and EP1 receptors in vascular responses is reduced. On the other hand, both apocynin and the mitochondrion-targeted antioxidant Mito-TEMPO normalized NAD(P)H oxidase activity and expression, ROS production, and MnSOD expression. This is in agreement with the recent evidence demonstrating that O2·− derived from Ang II-activated NAD(P)H Oxidase induces mitochondrial ROS production, which further activates NAD(P)H Oxidase and increases cellular O2·− production (10, 11).
It is well known that prostanoids and ROS can oppose to the vasodilator effects of NO by counteracting its effects or by reducing its bioavailability. Our results in Ang II-infused mice demonstrate that treatment with Celecoxib, Mito-TEMPO, or apocynin normalized the decreased vascular NO production and also restored the impaired modulation induced by NO of phenylephrine responses, the latter found both in conductance and resistance arteries. Interestingly, any of the treatments improved the decreased eNOS expression induced by Ang II, suggesting that alterations in NO availability are responsible of the altered endothelial function. Alternatively, changes in eNOS activity might also explain the observed differences. In fact, some investigators have demonstrated that antioxidants promoted increased proportion of eNOS expressed as a dimer (23).
In conclusion, our results point to the excess of ROS from NAD(P)H Oxidase and/or mitochondria and the increased vascular COX-2/TP receptor axis acting in concert to decrease NO bioavailability in hypertension, thus inducing vascular dysfunction and hypertension development (Fig. 9).
Material and Methods
Animal models
We used C57BL6 mice infused with Ang II (1.44 mg/Kg/day, 2 weeks, subcutaneously by osmotic minipumps; Alza Corp) alone or in combination with (i) the NAD(P)H Oxidase inhibitor apocynin (1.5 mM in the drinking water); (ii) the mitochondrion-targeted SOD2 mimetic Mito-TEMPO (0.7 mg/Kg/day i.p.); (iii) the COX-2 inhibitor Celecoxib (25 mg/Kg/day i.p.), and (iv) Mito-TEMPO plus Celecoxib. All treatments started 24 h before Ang II infusion. Control animals without treatments or with Celecoxib or apocynin treatments were also studied. In another set of experiments, mice were treated with Mito-TEMPO or with Celecoxib 7 days after Ang II infusion.
Six-month-old male WKY rats and SHRs were used. SHRs were divided into four groups: (i) control, (ii) treated with the SOD mimetic tempol (1 mM, in the drinking water, 18 days), (iii) treated with Mito-TEMPO (0.7 mg/Kg/day i.p., 18 days), and (iv) treated with Celecoxib (25 mg/Kg/day i.p., 3 weeks). Untreated WKY rats or rats treated with tempol or Celecoxib were also used.
In preliminary experiments, we observed that vehicles did not affect SBP or vascular function neither in the Ang II-infused mice nor in the SHRs (data not shown).
Blood pressure was measured by tail-cuff pletysmography. All experimental procedures were approved by the Animal Care and Use Committee of our Institution, according to the guidelines for ethical care of experimental animals of the European Community. The study was conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and the current Spanish and European laws (RD 223/88 MAPA and 609/86).
For blood samples acquisition, left ventricular hypertrophy determination, and tissue preparation, please see the Online supplement.
Histological analysis
Aortas were fixed with 4% paraformaldehyde and embedded in a Tissue Tek OCT medium. Sections from fixed aortas were stained with hematoxylin and eosin.
Reactivity experiments
Reactivity of mouse aorta and first-order branches of the mesenteric artery or rat aorta was studied in a wire myograph or by isometric tension recording, respectively, as described (4, 17).
NO release
NO release was measured with the fluorescent probe DAF-2.
Measurement of O2·− production
The oxidative fluorescent dye dihydroethidium (DHE) was used to evaluate production of O2·− in situ, as described (7).
Measurement of H2O2
H2O2 measurements in aortic segments were performed using the horseradish peroxidase-linked Amplex Red fluorescence assay.
NAD(P)H Oxidase activity
The lucigenin-enhanced chemiluminescence assay was used to determine the NAD(P)H Oxidase activity in total protein aortic homogenates.
Western blot
Aortic homogenates were electrophoretically separated on 7.5% SDS– polyacrylamide gel electrophoresis, as described (4). Western immunoblot was performed with antibodies for eNOS, COX-2, COX-1, MnSOD, Cu/Zn-SOD, and EC-SOD.
Immunofluorescence
COX-2 was localized in frozen transverse sections by immunofluorescence, as described (2).
Measurement of MDA production
Plasma and aortic MDA levels were measured by a modified thiobarbituric acid assay.
Reverse transcription-polymerase chain reaction assay
COX-2, COX-1, β2-microglobulin, NOX-1, NOX-4, p22phox, p47phox, and cyclophilin mRNA levels were determined in aortic homogenates. Total RNA was obtained by using TRIzol. Quantitative polymerase chain reaction was performed using TaqMan Gene Expression Assays or the fluorescent dye SyBRGreen.
Measurement of prostaglandin F2α production
The levels of the metabolite of PGF2α 13,14-dihydro-15-keto-PGF2α were determined using an enzyme immunoassay commercial kit.
Data analysis and statistics
All data are expressed as mean values±standard error of the mean, and n represents the number of animals. Results were analyzed by using an unpaired Student's t-test or a one-way or two-way analysis of variance followed by Bonferroni's post hoc test. A p<0.05 was considered significant.
The detailed methods are included in the Supplementary Materials and Methods section.
Footnotes
Acknowledgments
This study was supported by Ministerio de Ciencia e Innovación (SAF2009-07201 and Red RECAVA, RD06/0014/0011, and RD06/0014/0005), Sociedad Española de Farmacología-Almirall Prodesfarma, and Fundación Mutua Madrileña. AMB is supported through the Ramón y Cajal program (RYC-2010-06473), and SM-R by a fellowship from the Alianza Cuatro Universidades.
We thank the Animal Care Facility of the Faculty of Medicine (UAM) for their help with the animals.
Author Disclosure Statement
The authors declare no competing financial interests
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.