
Abstract
Introduction
Cellular Sources and Levels of RNOS
RNOS are derived from a variety of cellular sources such as uncoupled NO synthases, xanthine oxidase, NADPH oxidases (NOXs), and complex I and III of the mitochondrial electron transport chain (181). The amount and predominant RNOS formed therein depends on the cell type and activated sources. Accordingly, the major reactive oxygen species (ROS) source in neutrophils is the phagocyte oxidase gp91phox complex or NOX2, which can generate a strong burst of superoxide radicals, for example, 4–20 nM/min/106 cells, that can be converted via myeloperoxidase into hypochlorous acid, a strong bactericidal agent (10, 53, 62, 80). In contrast to this high-capacity system, which is utilized for host defense, endothelial cells possess similar systems consisting of various NOXs, including NOX 1, 2 and 4 (23), and endothelial nitric oxide synthase (eNOS) (170) that release low amounts of RNOS for signaling purposes.
Numerous studies have used exogenously added oxidants to replicate the effects of endogenous RNOS in cell culture models. However, these studies used quantities of oxidants that may not mimic the amounts generated intracellularly. In fact, cytokine-treated human umbilical vein endothelial cells produce submicromolar concentrations of
Redox Signaling Versus Oxidative Stress
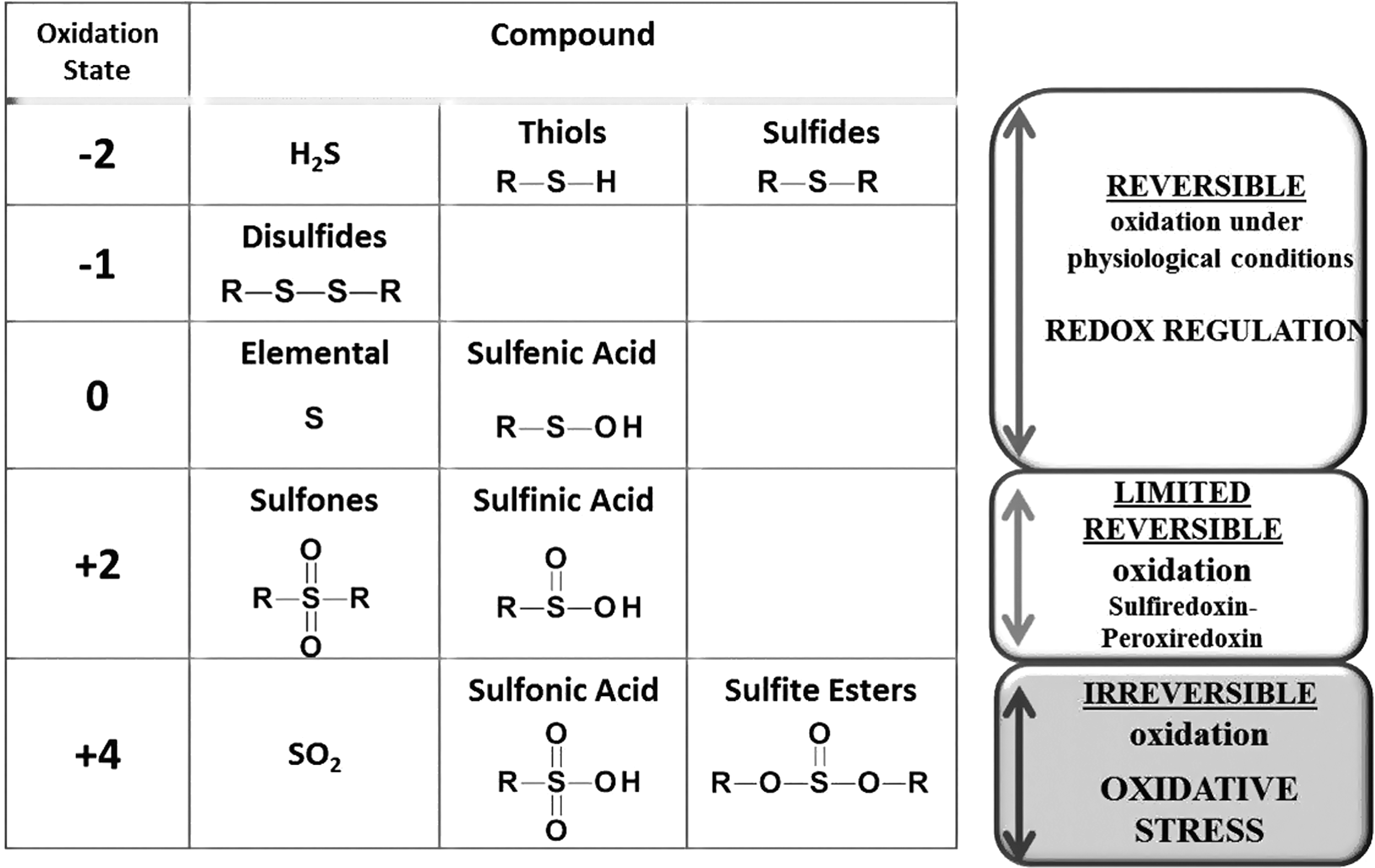
RNOS can lead to either oxidative stress or redox signaling. In oxidative stress, chronic high levels of RNOS lead to irreversible oxidation of a protein that likely functions as a signal for its degradation. OPTMs of this type include hydroxylation of aromatic groups and aliphatic amino acid side chains, nitration of aromatic amino acid residues, chlorination of aromatic groups and primary amino groups, conversion of some amino acid residues to carbonyl derivatives, and hyper-oxidation of thiol groups (150, 151).
In redox signaling, physiological levels of RNOS act on a protein leading to an enzymatically reversible modification, such as S-glutathionylation, that can alter protein function. Classically, a signaling molecule interacts with a protein that then associates with downstream effectors in a local environment and leads to reversible signal propagation through on/off switching (117). In theory, RNOS can activate a multitude of proteins in a spatially diverse manner leading to activation of discontinuous pathways. This might lead to defective signaling, as there is no specificity, localization, or coordinated regulation. Thus, in order to provide specific activation of signaling cascades, endogenous RNOS need to be produced in a sufficient quantity near the affected protein, and result in a reversible modification of either a methionine (→ methionine sulfoxide) or a cysteine residue (→ S-glutathionylation or sulfinylation). Sulfinylation has been shown to be reversed by sulfiredoxin for peroxiredoxin only (134) (Table 1 and Fig. 1). Given these conditions, it is unsurprising that redox signaling targets a very selective number of proteins and should not be expected to be induced by every stimulus.
Glutathione, a Versatile and High Abundant Low-Molecular-Weight Thiol
Glutathione (GSH), the most abundant intracellular small molecule antioxidant in the cell, is an indispensable tri-peptide that participates in various catabolic and anabolic cellular processes (103) (Table 2).
GSH has been thought to mainly act to maintain thiol redox homeostasis; however, recent data suggest a prominent function in iron metabolism as well (90). This article solely focuses on the thiol redox properties of GSH.
Due to its high abundance, water solubility, and strong nucleophilic character after deprotonation, GSH is a central player in the cellular antioxidant system. It is an ideal conjugation partner for detoxification reactions, known as phase II conjugation reactions with xenobiotics. Moreover, GSH enables cellular redox signaling (Fig. 1) by forming stable mixed disulfides at selective cysteine residues within proteins that can be reversed by the cellular reducing system.
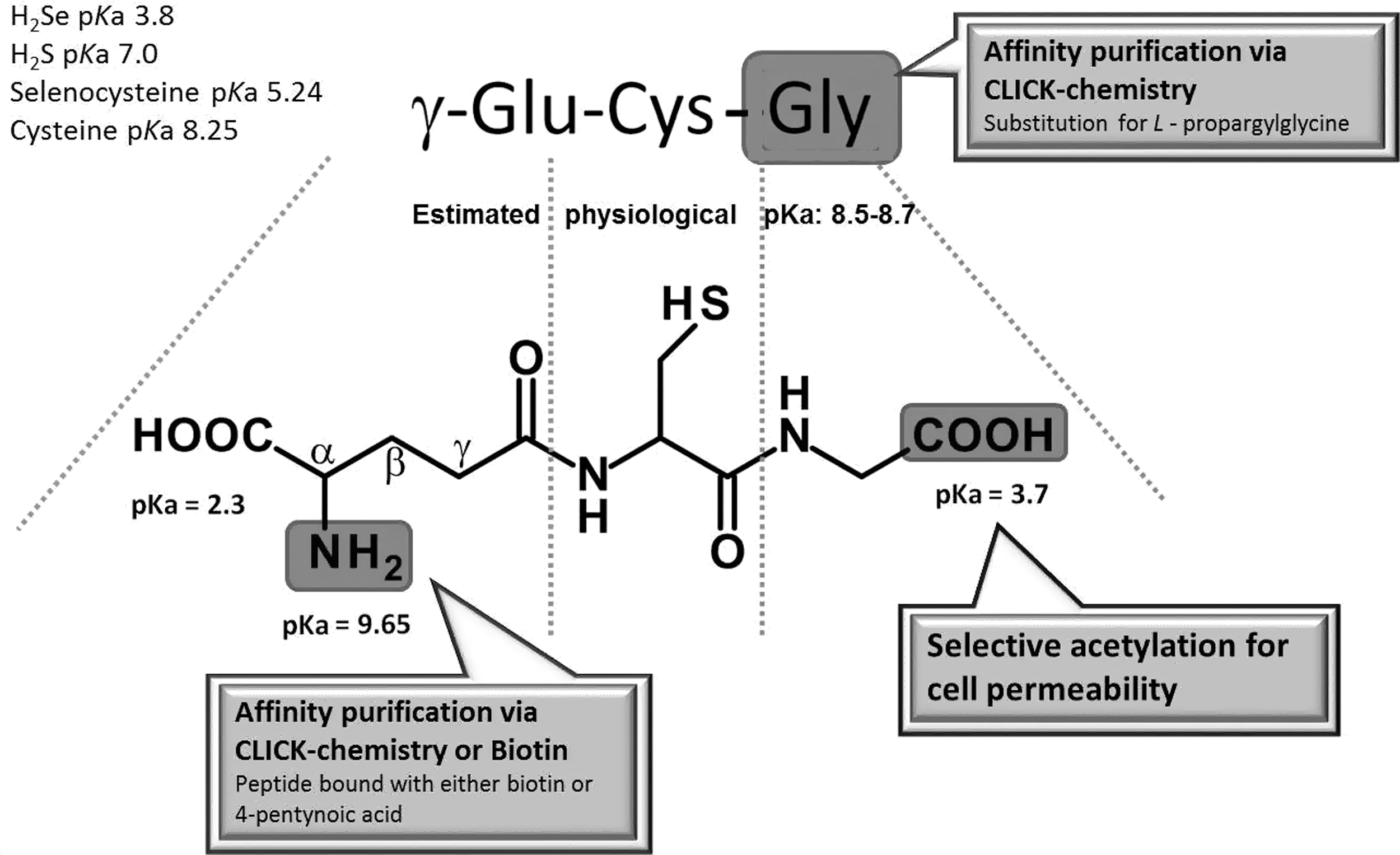
The de novo synthesis of GSH is comprised of two catalytic steps. In the first reaction, a peptide bond between a cysteine and the side chain carboxylic acid (gamma, γ; Fig. 2) of a glutamate is formed by γ-glutamyl cysteine synthase (γ-GCS; alternative name: γ-glutamyl cysteine ligase), which can be inhibited by buthionine sulfoximine to deplete cellular GSH levels. This bond protects GSH from degradation by cellular and serum peptidases. The concluding reaction adds glycine by GSH synthetase. The GSH (γ-glu-cys-gly) produced tightly controls its own intracellular levels by regulating γ-GCS activity through a negative feedback loop. Every cell contains the enzymatic machinery to synthesize GSH in the cytosol keeping concentrations between 1 and 15 mM (redox potential between −220 and −260 mV; GSH:GSSG ratio of 30 to >100:1) depending on the cell type and the extent of rigorous care taken in sample preparation to avoid oxidation of GSH. Therefore, GSH is the major cellular thiol redox buffer constituent and thiol-based antioxidant that helps maintain a reduced intracellular environment. This milieu is necessary to maintain reduced cysteines (thiolate) that are required to catalyze various biochemical reactions including glyceraldehyde 3-phosphate dehydrogenase (115, 136), aldehyde dehydrogenase (169), cysteine proteases (72), and phosphatases (16). In addition, the cellular thiol redox state, commonly expressed by the GSH/GSSG ratio, can control cell proliferation, differentiation, or apoptosis. Therapeutically or experimentally, cellular GSH levels can be replenished with N-acetylcysteine or GSH mono ethylester (5, 6).
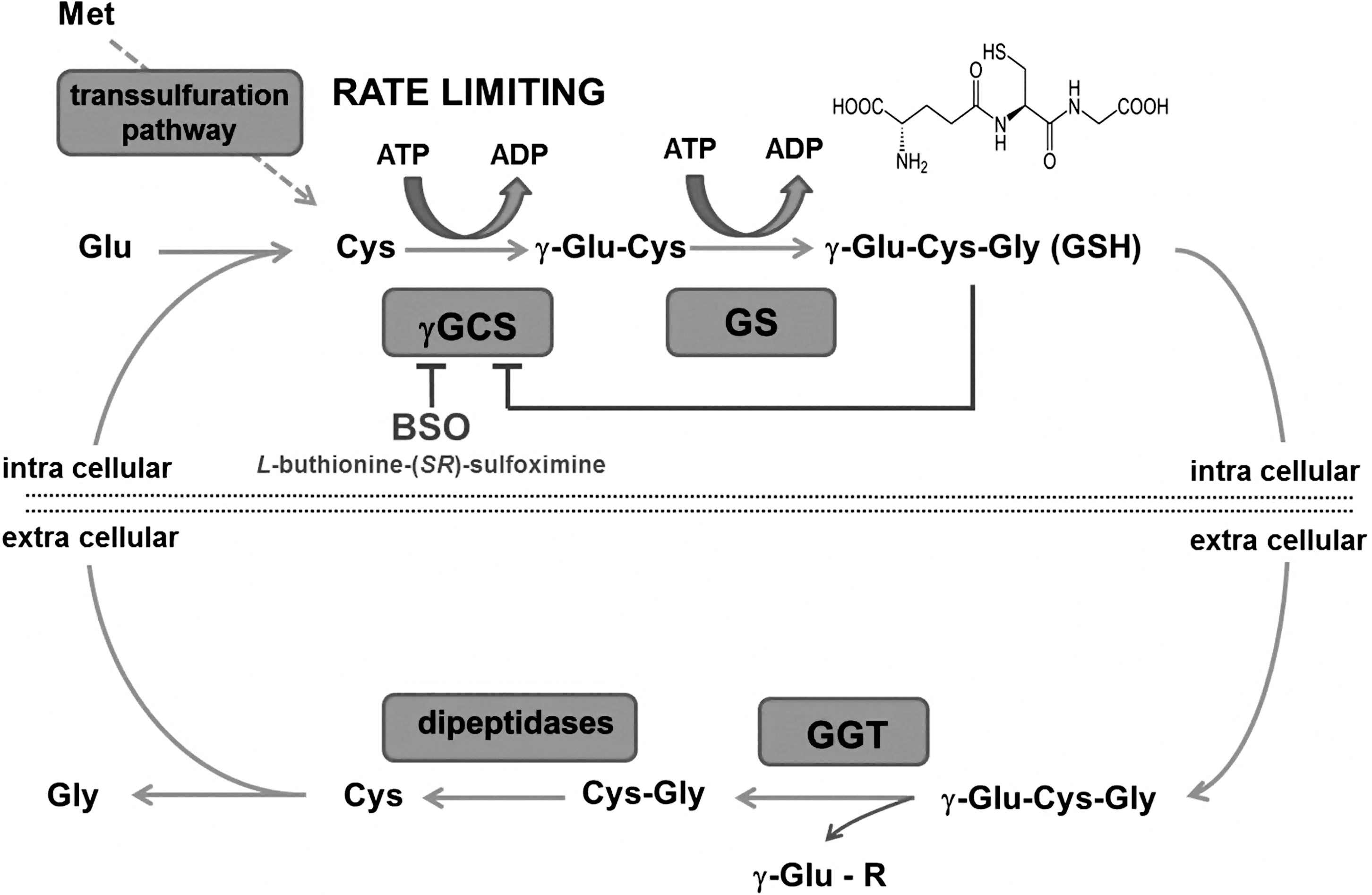
The distribution of GSH is highly compartmentalized with the lowest concentrations in the plasma and interstitial fluid, where it ranges between 10 and 30 μM with a redox potential of about −140 mV. Serum GSH is membrane impermeable, and the γ-glutamine can be removed and conjugated to other amino acids, peptides, or the GSH itself by a specialized cell surface enzyme, γ-glutamyl transferase, which is mainly found on the endothelium of brain capillaries (121) and the epithelium in the kidney, bile, intestine, and pancreas. The resulting cysteinylglycine is cleaved by other cell-surface-localized dipeptidases, and the cysteine can be taken up by surrounding cells and used for de novo GSH synthesis. This mechanism is used by the kidney to recycle GSH and by the brain capillary endothelial cells to promote translocation of GSH precursors across the blood brain barrier (121).
Since cysteine is the limiting amino acid in cellular GSH synthesis, methionine or exogenously supplied acetyl-cysteine is utilized. Methionine can be converted via multiple steps in a trans-sulfuration pathway, where serine serves as the backbone, to cysteine.
In the endoplasmic reticulum (ER), GSH is believed to be predominantly in the oxidized form (−150 mV; GSH:GSSG ratio of 3:1) (109, 110), which allows it to function as an oxidizing equivalent for protein disulfide bond formation and proper folding of nascent secretory proteins (73). This environment should, however, also promote S-glutathionylation of ER residing proteins. Bass et al. (17) found that a major fraction of GSH in the ER formed mixed disulfides with proteins. Recently, these findings were reevaluated by Dixon and coworkers (44), who claimed that the redox potential is lower (−176 to −205 mV; GSH:GSSG ratio of 4.7–5.5:1) in maintaining oxidized protein disulfide isomerase (44), which is active and promotes protein disulfide formation. This group also suggested that ER proteins are relatively resistant to protein S-glutathionylation. These issues are most likely due to different isolation procedures, which may have caused artifactual oxidation.
In the nucleus, GSH protects against oxidant- and ionizing radiation-induced DNA damage and maintains nuclear proteins and transcription factors in a functionally reduced state that is required for gene transcription and DNA repair. The nuclear GSH pool seems to be separate from the cytosolic and trafficking of nuclear GSH correlates with cell-cycle progression (43). This concept is further supported by the fact that GSH acts as a hydrogen donor for ribonucleotide reductase (102), an enzyme that catalyzes the formation of deoxyribonucleotides from ribonucleotides required for DNA synthesis and cell proliferation (43). The molecular mechanisms underlying the trafficking and dynamics of nuclear GSH are poorly understood and controversial (147). Nevertheless, various transcription factors including activator protein 1 (AP-1) and nuclear factor κB (NFκB) (54) have been shown to be S-glutathionylated in the cytosol. For transcriptional activity, the factors translocate into the nucleus where deglutathionylation occurs, which enables DNA binding and transcription (54).
Mitochondria import their GSH from the cytosol and contain about 10%–15% of the cellular GSH (58, 59, 76) that slowly equilibrates with the cytosol. Soderhal et al. (147), using an antibody raised against GSH conjugated bovine serum albumin, identified that protein-SSG adducts are unevenly distributed throughout the cell with high abundance at the mitochondria as well as the plasma membrane. GSH in combination with mitochondrial GSH peroxidase (GPx) is an important mitochondrial antioxidant system that protects against H2O2 derived from electron leaks in the mitochondrial respiratory chain (148). In contrast to the cytosol, mitochondria cannot synthesize GSH de novo and require specific carriers and transporters. The 2-oxoglutarate and dicarboxylate carriers may be such systems that can transport cytosolic GSH in exchange for dicarboxylates. However, it is unknown how either oxidized or conjugated GSH exits the mitochondria.
Most of these reported GSH measurements have been performed on crude isolated organelle fractions; thus, the concentrations of contaminants present from the isolation procedure may lead to erroneous results. In addition, the effect of subcellular isolation on GSH:GSSG levels has not been evaluated. Although several reports have used fluorescent GSH probes, these are quite nonspecific and are highly dependent on the cell type, as conjugation may be aided by GSTs (139, 159). Furthermore, antibodies against GSH-glutaraldehyde conjugates can only measure reduced and oxidized GSH (158). Novel technologies, such as targeted reduction-oxidation sensitive green fluorescent protein (45), may be used to determine whether a pathway contains a uniform redox milieu in the various cellular organelles, but care should be used to control for pH variations. Regardless, multiple confirmatory methods should be used for measurements (16), as adequate evaluation of compartmentalized GSH is experimentally challenging.
Difference Between a Thiol and Thiolate
Thiols are defined as compounds that contain a functional group composed of sulfur and a hydrogen atom (R-SH). This group is also named as a mercaptan due to its high affinity for mercury. The primary determinant of thiol reactivity is its pKa value representing the pH at which 50% of the thiol is deprotonated.
This form is referred to as the thiolate anion (R-S−) representing the strongest cellular nucleophile (R-OH<R-SH<<R-S−) that is important in enzyme catalytic activity, substrate recognition, detoxification, and allosteric redox regulation. The lower the pKa value of a thiolate, the higher its reactivity. The pKa of a cysteine can be lowered by deprotonation from neighboring (vicinal) basic residues such as lysine, arginine, or histidine as well as hydrogen bonding with an adjacent hydroxyl group from serine or tyrosine residues. Thiolates are nucleophiles, as they can donate both bonding electrons to an electron-deficient compound, which is called an electrophile. Conversely, electrophiles can be positively charged compounds or carry an atom that has a partial positive charge allowing it to accept an electron pair leading to covalent bond formation.
The thiol group of GSH has low reactivity at physiological pH due to its high-standard pKa-value of 9.3. The actual pKa under intracellular conditions is, however, estimated between 8.5 and 8.7 (64, 124). Therefore, about 90% of GSH is protonated and relatively unreactive under physiological conditions. Nevertheless, the high intracellular concentration enables the GSH to exert some antioxidant function. This ability is enhanced by enzymes such as GSH S-transferases (GST) that can decrease the pKa of GSH to below 6 and generate a highly reactive nucleophile. GSTs coupled with GSH allow for the detoxification of oxidants, lipid peroxidation products, and xenobiotics. The generated GSH conjugates are then excreted from the cell via specialized transport systems (74, 116). Although GSTs are known to catalyze these detoxification reactions, their role in S-glutathionylation of protein cysteines represents a previously uncharacterized function in redox signaling. The three major GST groups, alpha, mu and pi, utilize the ionization of GSH by hydrogen bonding to the active site tyrosine residue of GST (119). These enzymes among others may allow for S-glutathionylation of higher pKa protein cysteines within the cell.
Chemical Mechanisms of S-Glutathionylation
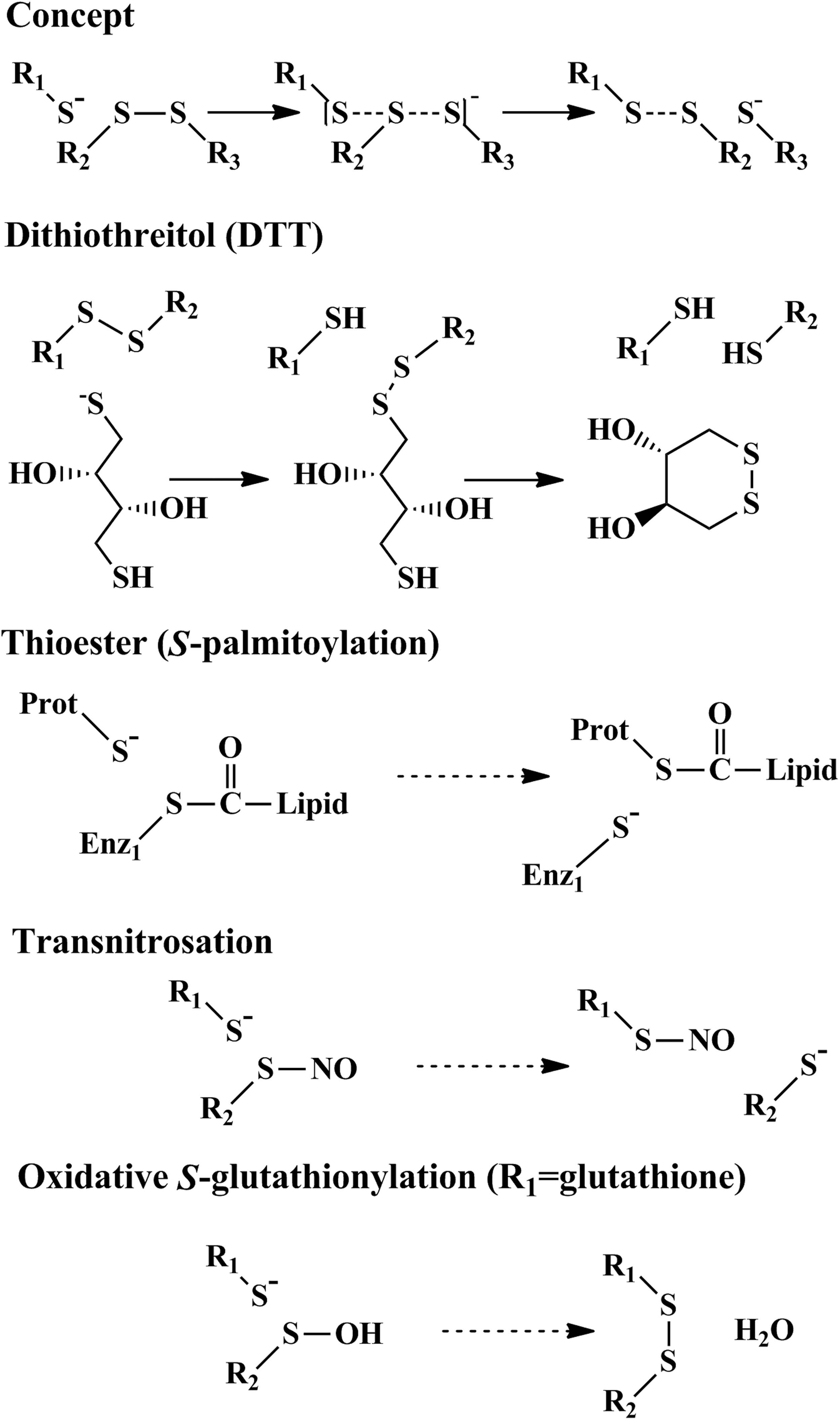
Thiol exchange reaction or shuffling is the principal reaction by which disulfides and mixed disulfides are formed and rearranged in a protein. In this reaction, a thiolate group (R-S−, Fig. 3
ROS-mediated protein S-glutathionylation
Peroxides react and are biologically detoxified through a nucleophilic reaction with thiolates (Fig. 4). Most cellular GSH is protonated, and detoxification by cellular GSH is too slow to be effective (Equation 1). Therefore, the cell has developed a sophisticated enzymatic network to utilize the following reaction for detoxification and redox signaling (Equations 2
–4).
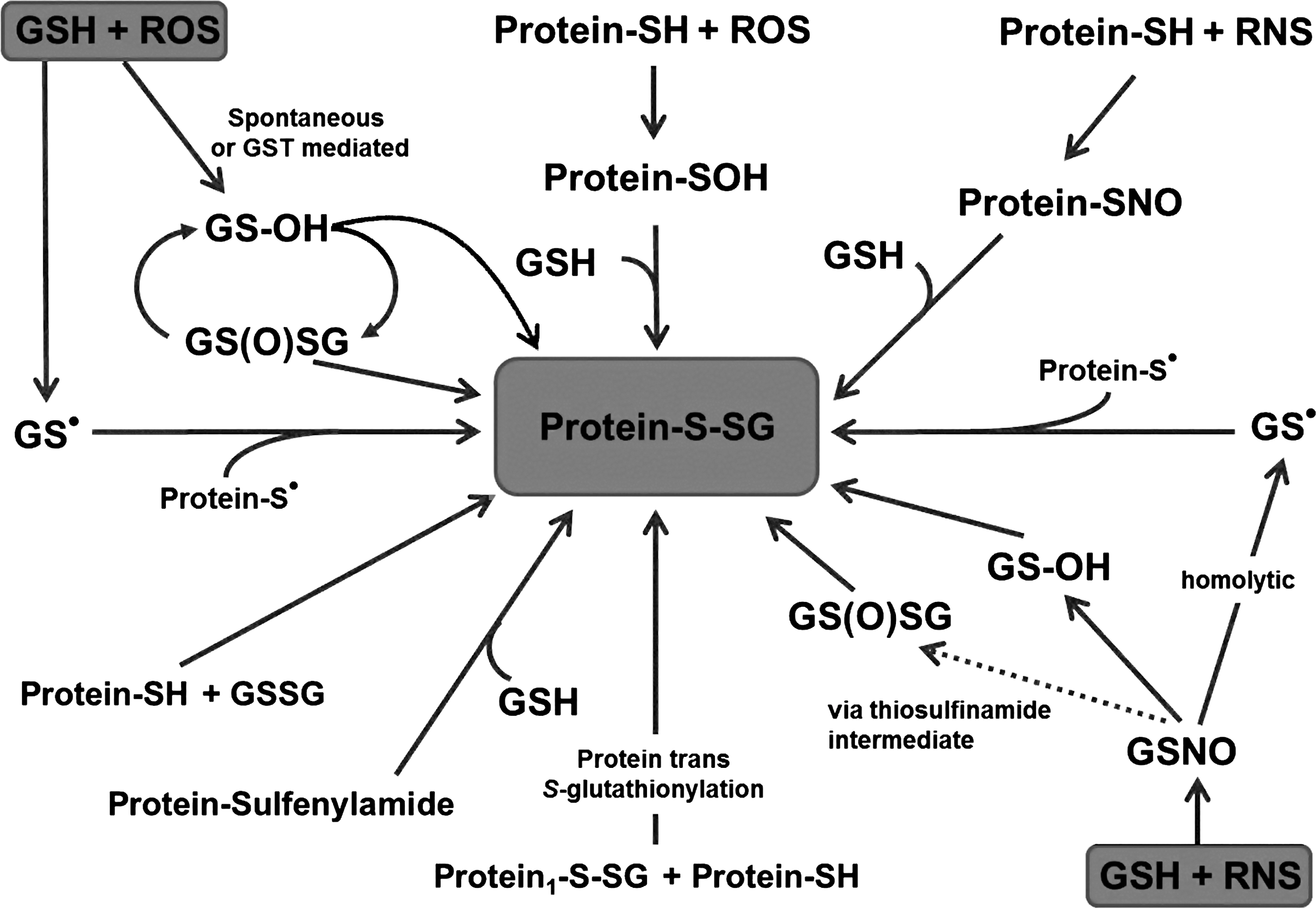
GPx uses selenium at the active site, as the pKa is significantly lower than that of a cysteine, which results in a very reactive nucleophile that efficiently attacks hydrogen peroxide. In this reaction, the selenium becomes oxidized, and needs to be reactivated. This occurs via a GSH, which leads to temporarily glutathionylation of this protein (Fig. 5). In a thiol exchange reaction, a second GSH is utilized to deglutathionylate the enzyme, generating GSSG and the reactivated form of Gpx. Similar reaction schemes that involve a cysteine can be found for the reactivation of several proteins, including peroxiredoxins, with S-glutathionylation being a common intermediate in these reactions. Such a reaction scheme also applies for redox regulation of proteins that contain low pKa cysteines, and, therefore, have a peroxidase-like regulatory enzymatic activity (for example oxy-R sarco/endoplasmic reticulum Ca2+-ATPase [SERCA], and rat sarcoma small GTPase [p21Ras]). In this case, the target protein reacts with the oxidant, and a protein-cysteine (Cys) sulfenic intermediate is formed that reacts with GSH. Second, one can speculate that, in excess, GSTs may mediate and facilitate this process through their peroxidatic activity (catalysis of the decomposition of peroxides), although this enzymatic activity of GST is low. Under these circumstances, activated GSH reacts with the peroxide, and the newly formed GS-sulfenic acid intermediate modifies the thiolate on a target protein. A third possibility is a thioltransferase exchange activity, in which the thiolate may react with GSSG and results in protein S-glutathionylation. A fourth mechanism of chemical S-glutathionylation is described for sulfenic acid, which condensates to GSH disulfide S-oxide (GS(O)SG=anhydride of GSOH). This condensation is a thermodynamically preferred state and is promoted through dimer formation of GSOH by bivalent metals and hydrogen bonding. For this reason, sustained ROS generation will maintain GS(O)SG levels in an autocatalytic cycle. GS(O)SG has been detected in cellular systems, and enzymatic sources of this reaction may include microsomal P450 enzymes and flavin-containing monooxygenases. GS-O-O-SG is suggested as another species that may form Prot-SG adducts (Equations 5–6).
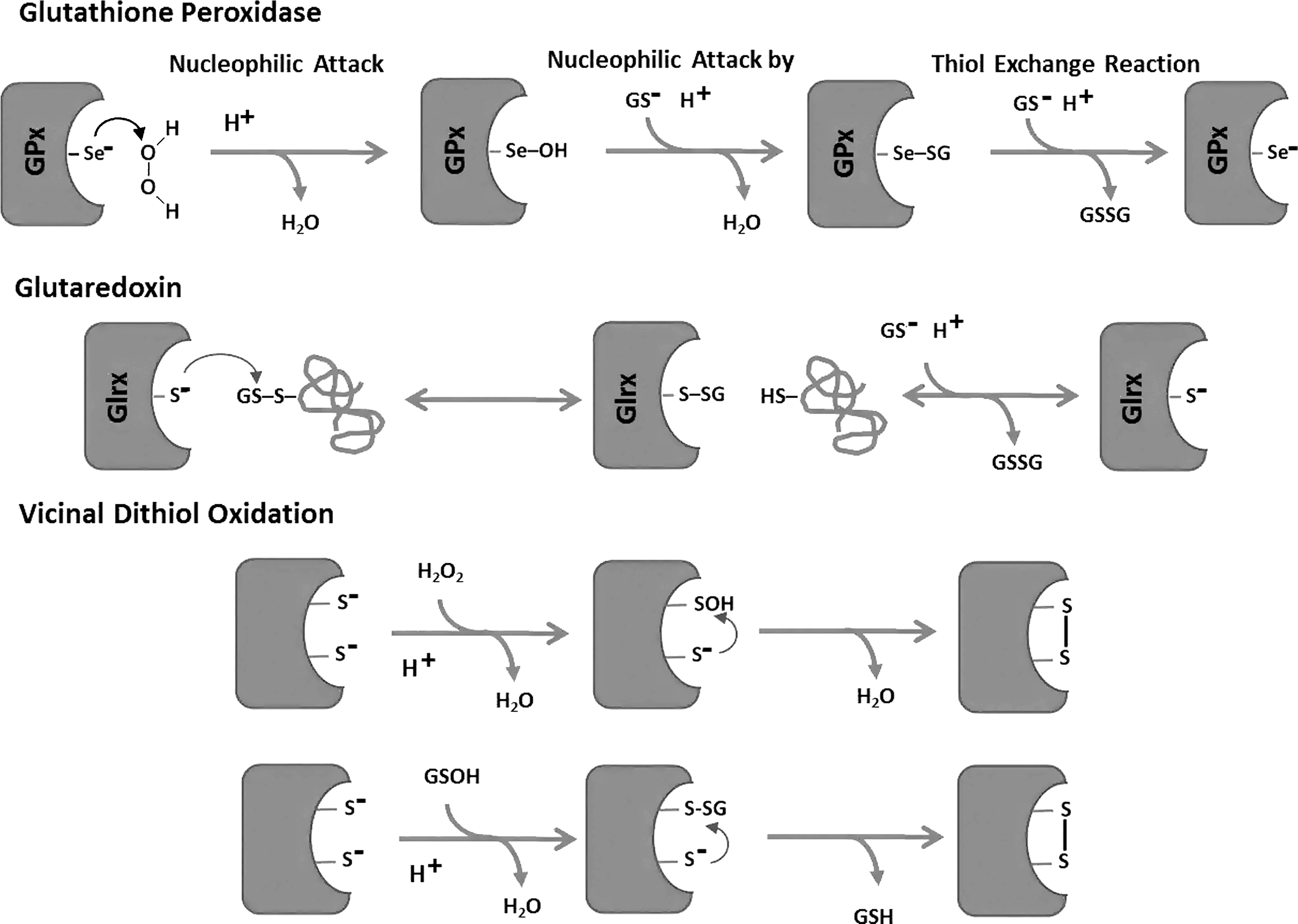
A sixth reaction occurs with the formation of a cyclic sulfenylamide bond, for which protein phosphatase 1B (PTP1B) oxidation acts as a prototype. Under extreme oxidative conditions, the sulfur and the backbone nitrogen of the PTP1B active site cysteine formed a cyclic sulfenylamide bond. This intermediate was fully reversible and prevents further oxidation of the protein. In the presence of GSH, however, the sulfanilamide is reduced, leading to S-glutathionylation of the enzyme. This mechanism is very interesting, as it allows for molecular protection against irreversible oxidation of indispensable active site cysteines during oxidative stress. The role of this mechanism in redox regulation or signaling, however, remains unclear. Given the multiple possibilities of S-glutathionylation, determining the underlying molecular mechanisms is experimentally challenging and likely varies among target proteins, cellular compartments, cell types, and tissue.
Reactive nitrogen species-mediated protein S-glutathionylation
Reactive nitrogen species such as •NO, N2O3 and metal-nitrosyl complexes have been shown to S-nitrosate and to regulate various proteins in vitro and in vivo (Fig. 4). Various S-nitrosated proteins, including SERCA, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and p21Ras, have also been reported to be S-glutathionylated (1, 2, 115, 122). Therefore, S-nitrosation may be a precursor of S-glutathionylation, and several chemical mechanisms have been reported (Equations 7
–9).
A protein can either be directly S-nitrosated or react with GSNO in a transnitrosation reaction, which is thereafter exchanged for a GSH leading to protein S-glutathionylation (Equation 10).
Analogous to these reactions on GSH, similar reaction mechanisms apply to reactive protein cysteines. For detailed chemistry on this topic, please refer Xiong and coworkers (174).
This illustrates that the entire cellular “thiol metabolism” is composed of an elaborate network (Fig. 4) of interconnecting and dynamic pathways, which adds confounding complexity to the study of redox biology.
A complete list of reactions that lead to protein S-glutathionylation as well as candidate enzymes that may mediate these reactions are listed in an excellent review by Gallogly and Mieyal (54) as well as by Xiong et al. (174).
Detection of Protein S-Glutathionylation
Measurement of cellular S-glutathionylation has become more feasible due to the generation of novel labeling antibodies and tagged thiol agents. This section reviews several standard techniques used for detecting these proteins.
Accepted approaches include the chromatographic measurement of released GSH after reduction and the detection of protein-SSG adducts with indirect switch assays (98), biotinylated (153), radiolabeled, or isotope-labeled GSH (83, 84). The former approach solely allows for evaluation of the overall level of S-glutathionylation on cellular proteins that may occur during a pathophysiological setting, but does not allow for the identification of individually modified proteins (126). In contrast, the latter approach can be used to identify specific S-glutathionylated proteins and, when coupled with mass-spectrometry analysis, the exact site of the modification. The stability of the protein-SSG mixed disulfide under nonreducing conditions, as well as its stability in the presence of high intracellular concentrations of GSH, aids in detection of the adducts.
Direct detection under nonreducing conditions is possible with the use of a monoclonal antibody (mAB) raised against GSH covalently bound to target protein(s) (Virogen). With this mAB, it is possible to detect the change in the overall cellular and individual protein S-glutathionylation (67). A major problem with this approach is the sensitivity of the antibody, as large quantities of S-glutathionylated protein may be required to generate an adequate signal for detection. Another direct approach for determining S-glutathionylated proteins is with the use of biotin-tagged GSH ester (153) or its oxidized form, GSH disulfide-ester (22, 47, 178). The tag on the GSH can be used to visualize modified proteins within the cell or on Western blots (Fig. 6). In addition, modified proteins can be purified using an affinity matrix and then identified by Western blotting for specific targets or by using mass spectrometry. Biotinylated GSH esters are commercially available (BioGEE, CLICK-chemistry; Invitrogen) and have been successfully used to identify novel targets. Several indirect techniques also exist for detecting protein S-glutathionylation. This includes the biotin-switch assay (47), where free thiols are alkylated during lysis and then S-glutathionylated cysteines are reduced and labeled with a biotin-linked thiol-reactive agent. By labeling free thiols with a biotin-alkylation agent after samples are treated with a specific reducing agent, such as ascorbate (S-nitrosation), sodium arsenite (sulfenic acid), DTT, or tris(2-carboxyethyl)phosphine (mixed, intra- and inter-protein disulfides), detection of several different forms of reversible protein oxidation is possible (178). A modified version of the biotin-switch method has been developed by Janssen-Heininger for detecting S-glutathionylation in situ. This method utilizes the specific removal of GSH-adducts by recombinant Glrx1 in fixed tissue or cells (4). Another alkylation-dependent technique for analyzing protein oxidation involves using biotinylated iodoacetamide (BIAM) to label free unmodified thiols during cell lysis. The thiols modified by BIAM represent the cellular reduced peptide fraction with a loss in labeling representing an increase in oxidation. Although this method can be used to reliably detect changes in the oxidation state of a protein, it does not provide any information regarding the type of modification. Iodoacetamide does, however, have the distinct advantage that it binds preferentially to reactive thiolates over reduced thiols (156, 176).
To definitively identify a type of oxidative modification on a protein, mass spectrometry should be used, which can also provide the exact site of oxidation. This can be accomplished by directly analyzing an alkylated protein using mass spectrometry (MS) to look for peptides that have an additional mass corresponding to S-glutathionylation. Alternatively, an indirect approach can be undertaken using the alkylation techniques just mentioned or isotope coded affinity tags to selectively enrich the pool of proteins with thiol modifications (142), with these proteins then being identified using MS. In addition, with use of stable isotope labeling with amino acids in cell culture, it is feasible to quantitatively measure the percentage change in modified protein between individual samples. The use of MS is limited by the differential optimization required for each individual protein, the quantity of protein needed to adequately identify modified peptides, the use of metal free buffers, and the labor- and resource intensive nature of the procedure.
The Deglutathionylase Glrx: Reversal of S-Glutathionylation
The most well-known and characterized enzyme that removes mixed disulfides from modified proteins is Glrx, which is also known as thioltransferase (Fig. 5). There are two isoforms of Glrx in mammalian cells: Glrx1, which is localized mainly in the cytoplasm, and Glrx2, which is found in either the mitochondria or the nucleus depending on the splice variant. Human Glrx2 exhibits only 34%–36% identity with Glrx1 and has <10% of the specific activity of Glrx1 (57, 101). Glrx1 is a relatively abundant small (12 kD) cytosolic enzyme that was originally discovered in mammals in 1974 by Mannervik's group (8, 48). This thioltransferase was structurally and functionally analogous to Glrx, which Holmgren discovered in a mutant Escherichia coli lacking thioredoxin (48, 69, 70). Although thioltransferase better describes the catalytic function, Glrx has become the common name internationally used. Glrx efficiently catalyzes the reduction of GSH mixed disulfides (R-SSG) in the presence of NADPH and GSH reductase; however, in contrast to thioredoxin, it does not catalyze the reduction of sulfenic acid or intra- and intermolecular disulfides (33). Thus, in vitro, Glrx can act on proteins whose function is influenced by S-glutathionylation. For instance, Glrx restores DNA binding of transcription factors (14, 144), reactivates PTP1B (15, 16) or inhibitory
Physiologic Effects of S-Glutathionylation
S-glutathionylation has been reported to alter protein function by modifying protein-protein interaction, structural conformation, or by adducting an active site cysteine. Since the highest concentrations of oxidants are found in the ER, mitochondria, and at the plasma membrane, these compartments are considered the likely locations in the cell where redox induced signaling occur. As such, we will review the physiological aspects of redox signaling on myocardial contraction, metabolism, cell proliferation/growth, and inflammation.
Cellular calcium homeostasis and regulation of myocardial contraction
Oxidative-stress-induced functional impairment of contractile proteins was one of the earliest described cardiovascular pathophysiological derangements of RNOS (13, 50, 55, 60, 160, 167, 179). An example of this is the actin-actomyosin interaction that occurs at cysteine-374 of actin preventing interaction of the two proteins that leads to diminished force generation (41, 81, 166). Given this, it is unsurprising that a shift in the GSH:GSSG redox potential in skinned cardiac fibers can acutely change contractility up to ∼90% (18, 88). The suggestion that S-glutathionylation by exchange reaction in myocardial sarcomeric proteins reduces contraction may explain why the levels of GSSG are kept low and mostly constant by transporters such as multidrug resistance protein-1 in the cell (173). In addition to myofibrils and the cytoskeletal proteins modulated by thiol exchange, elevated RNOS production at the ER and the plasma cell membrane can modulate cellular contraction by redox signaling and oxidative stress. In the ER/SR, RYR2 (138, 154, 155, 175) and SERCA2 (2, 21, 46, 92, 143, 168) are exquisitely sensitive to thiol modifications leading to functional changes. In addition, plasma membrane bound proteins can also theoretically be regulated by oxidation due to the presence of oxidant enzymes such as NOXs. This is supported by the redox regulation of the plasma membrane bound proteins eNOS and the Na+-K+ ATPase, which are both involved in regulating contractile function, and p21Ras, (36, 153), which regulates cell growth and signaling.
Ryanodine receptor
The cardiac ryanodine receptor (RyR2) is a large ligand-activated intracellular Ca2+ release channel located at the endoplasmic and sarcoplasmic reticulum (SR). In the myocardium, RyR2 plays a crucial role in mediating excitation contraction coupling by increasing intracellular Ca2+ after an action potential. It is regulated by direct phosphorylation at different sites by cAMP dependent kinase (PKA) or calmodulin kinase II (CaMKII), which increase the probability of the RyR2 channel to be open. In addition to phosphorylation, direct oxidation has also been implicated as a regulator of the channel's function. This is not surprising considering that this tetrameric channel contains 364 cysteines with about 84 of them having free thiol groups.
Basal S-glutathionylation of RyR2 was discovered in microsomal fractions enriched in SR vesicles isolated from dog cardiac ventricular muscle (138). This modification was significantly increased after tachycardia and correlated with enhanced oxidant formation and NOX activity. The S-glutathionylation was found to increase RyR2 activity, as the SR had faster Ca2+ release kinetics in the canine model of tachycardia. Furthermore, this finding could be replicated in microsomal fractions where NAPDH addition enhanced both RyR2 S-glutathionylation and Ca2+ release from the SR. In addition to tachycardia, exercise can induce RyR2 S-glutathionylation via NOX activation (137). With both exercise and tachycardia, the administration of the NOX inhibitor, apocynin, prevented RyR2 S-glutathionylation and attenuated Ca2+ release from the SR. However, it is important to consider that apocynin may not be a specific NADPH inhibitor but rather an antioxidant, which could also explain its ability to decrease RyR2 S-glutathionylation in these models (66). The reason for RyR2 S-glutathionylation having a beneficial effect in this model is likely due to improved Ca2+ release during systole that enhances cardiac activity while also decreasing Ca2+ leak during diastole. This would reduce the risk of developing arrhythmogenesis with the additional benefit that a small depletion in SR calcium release during systole could help the SR handle the flood of Ca2+ that occurs during ischemia reperfusion. Therefore, the S-glutathionylation of RyR2 may help explain how tachycardia or exercise can generate preconditioning.
Sarco/endoplasmic reticulum Ca2+-ATPase
In addition to oxidative modification of the RyR2, SERCA has also been described as participating in redox signaling. SERCA2, the dominant isoform in the heart and vasculature, acts as an inward pump that utilizes the energy from ATP to drive the removal of intracellular free Ca2+ into the SR Ca2+ store (165). The ability of this pump to maintain cytosolic free Ca2+ and the quantity available in the SR store during systole makes SERCA2 a major determinant of cardiac contractility and smooth muscle tone. The activity of SERCA2 is regulated by the accessory membrane protein phospholamban (PLN). PLN binds to and inhibits SERCA2 when dephosphorylated. The phosphorylation of PLN by PKA at Ser-16 or CaMKII at Thr-17 dissociates PLN from SERCA and relieves its inhibitory effect on SERCA activity.
In addition to regulation of SERCA2 by accessory proteins, direct S-glutathionylation can modulate the pump's activity. ONOO− increased the binding of SERCA to GSH-sepharose and decreased labeling by the alkylating agent BIAM. These results indicated that RNOS modify SERCA2 by S-glutathionylation, which was demonstrated to be on Cys-674 using mass spectrometry. The GSH adduct on purified SERCA augments its Ca2+ uptake activity. The physiological importance of this redox signaling modification was demonstrated in isolated vessels during relaxations caused by acetylcholine, bradykinin, or •NO. Enhanced SERCA GSH adducts accompanied vessel relaxation that was attenuated by the ONOO− scavenger uric acid. In a rabbit model of atherosclerosis, there was decreased relaxation and SERCA S-glutathionylation in abdominal aorta compared with control animals. The oxidation of SERCA was increased in this disease model, as there was less labeling of this protein with BIAM, and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) MS showed that SERCA was partially hyperoxidized at Cys-674, which also prevented thiol labeling with BIAM. Therefore, the loss in vessel relaxation in the aorta of rabbits with atherosclerosis could be explained by inhibition of S-glutathionylation (2).
In the heart, stimulation of SERCA activity and cardiomyocyte contractility by the thiolating nitroxyl anion generator, Angeli's salt, is also accompanied by S-glutathionylation of and stimulation of SERCA activity (93).
Endothelial nitric oxide synthase
The eNOS generates nitric oxide (•NO), which has a diverse role in the cardiovascular system that includes the control of blood pressure and smooth muscle tone (39, 152), regulation of platelet aggregation (9), development of arteriosclerosis (129), cytoprotection, and cytotoxicity (52, 82). eNOS is constitutively expressed and is involved in regulating normal cellular function by converting
Na+-K+ ATPase
The heterodimeric Na+-K+ ATPase, which is comprised of an α and β subunit, drives the extracellular extrusion of three Na+ ions in exchange for the influx of two K+ ions by hydrolyzing ATP. The activity of this ion exchanger can be modulated by direct S-glutathionylation of the β1 subunit (49). Paraquat, ONOO−, H2O2, and angiotensin II were all found to enhance the S-glutathionylation of the Na+-K+ ATPase β1 subunit in a concentration-dependent manner. The oxidation of the β1 subunit at Cys-46 was found to prevent its interaction with the α1 subunit, which is usually required for activity. In addition, the mutation of Cys-46 in the β1 subunit to a tryptophan attenuated ONOO− induced S-glutathionylation and inactivation of Na+-K+ ATPase. The potential pathological role S-glutathionylation of the Na+-K+ ATPase was shown using a sheep model of myocardial infarction in which S-glutathionylation of the β1 subunit was enhanced in the peri-infarct zone.
Recently, it has been reported that PKA activation can inactivate the Na+-K+ ATPase through S-glutathionylation (172). The adenylyl cyclase activator forskolin increased β1 subunit S-glutathionylation that could be attenuated by preincubation of cells with SOD. The increase in oxidant formation by forskolin was detected using dihydroethidium-fluorescence and could be attenuated by a PKC-ɛ inhibitory peptide, apocynin and SOD. Furthermore, in myocytes treated with forskolin, there was a decrease in the electrogenic Na+-K+ ATPase consistent with increasing S-glutathionylation that could be attenuated by the PKA inhibitor H-89, SOD, apocynin, gp91ds, the peptide inhibitor for NOX assembly, and U-73122, the phospholipase C (PLC) inhibitor. These findings fit with a signaling cascade whereby PKA activation by forskolin leads to PLC activation that stimulates PKC activity through diacylglycerol formation. The activated PKC then phosphorylates the p47phox subunit of NAPDH oxidase increasing its membrane activity. The oxidants generated by NOX then modify the Na+-K+ ATPase to inactivate it. In contrast to PKA activation, stimulation of β3 adrenergic receptors (β3ARs) increases Na+-K+ ATPase activity. This was demonstrated in patch-clamped cardiac myocytes where the activity of Na+-K+ ATPase was enhanced by the synthetic β3AR agonists, BRL37344 and CL316243, or the natural agonist, norepinephrine. Radicicol, a •NO synthase inhibitor and heat shock protein antagonist, blocked BRL37344's ability to increase the activity of the Na+-K+ ATPase. Furthermore, the guanylyl cyclase inhibitor ODQ also attenuated norepinephrine's ability to stimulate Na+-K+ ATPase activity suggesting cGMP-dependent phosphorylation of Ser69 on phospholemman may be responsible for the observed effect on Na+-K+ ATPase activity, possibly due to a structural change that may expose the S-glutathionylation cysteine in the β1 subunit to Glrx (104). Indeed, BRL37344 and CL316243's ability to increase Na+-K+ ATPase was due to a detectable loss in S-glutathionylation of the β1 subunit. In accordance with these findings, transgenic mice that do not express β3AR have a higher amount of β1 subunit S-glutathionylation compared with wild-type. Furthermore, the importance of β3AR to stimulate Na+-K+ ATPase activity was assessed in a pathological setting using a sheep model of heart failure wherein global damage to the left ventricular (LV) function was simulated by repeated selective coronary micro-embolizations. In the failing heart, BRL37344 increased the LV-end systolic pressure volume relationship (26). This suggests that the β3AR up-regulation in severe heart failure is detrimental, and its function needs to be reassessed.
Proliferation and hypertrophy: p21Ras
Multiple reports demonstrate that RNOS promote cellular hypertrophy and proliferation suggesting that protein oxidation plays a prominent role in regulating cellular growth pathways. One major regulator in growth factor signaling in the vasculature is p21Ras. One of the best characterized models for vascular hypertrophy involves angiotensin II induced RNOS formation through NOX activation (163). These oxidants have the propensity to modify p21Ras, which is known to have 4 exposed redox-sensitive cysteines (142, 182). Adachi et al. have demonstrated that angiotensin II-induced hypertrophy in vascular smooth muscle cells is regulated by S-glutathionylation of p21Ras at Cys-118. The GSH-adduct and the resulting activation of the p21Ras-MEK-ERK pathway were reversed by over-expression of catalase, Glrx1 or dominant negative p47phox. In addition, adenoviral over-expression of a partially redox-insensitive Cys-118S mutant p21Ras decreased angiotensin II-induced p21Ras activity and vascular smooth muscle hypertrophy (1). Campbell and colleagues have reported the importance of a thiyl radical intermediate of p21Ras. The thiyl radical production paralleled a marked increase in the guanine nucleotide dissociation rate, while p21Ras mutated at Cys-118 or chemically blocked thiols were neither subject to free radical induced nucleotide exchange nor produced a detectable thiyl radical signal. Under these circumstances, p21Ras can be S-glutathionylated by formation of a sulfenic acid intermediate after interacting with H2O2 or transnitrosation by GSNO. These reactions would suggest at least two intermediate forms before S-glutathionylation of the protein.
In cardiac tissue, thioredoxin regulates pressure-overload-induced cardiac hypertrophy. Compared with wild-type animals, those without thioredoxin had greater cardiac hypertrophy, while over-expression of the protein resulted in attenuated cardiac size. The mechanism appeared to be due to modulation of p21Ras at Cys-118 as demonstrated by in vitro models of strain and alpha-adrenergic receptor stimulated cardiac hypertrophy in cardiomyocytes (91, 122, 157). The modification of p21Ras likely occurs at Cys-118 and supports a mechanism of p21Ras S-nitrosation, followed by an exchange reaction, which leaves p21Ras S-glutathionylated. Under these circumstances, both thioredoxin, which can catalyze denitrosation reactions (20, 51) and Glrx, at p21Ras-S-NO and p21Ras-S-SG respectively, would be able to interact at the same site, leading to changes in activity. Activation of p21Ras at its redox-sensitive cysteine appeared to be of importance in other cellular systems as well. For example, nitric oxide formation by eNOS induced tumor growth in a p21Ras Cys-118-dependent manner (96) and ONOO− formation in endothelial cells, caused GSH-adduct formation at p21Ras Cys-118 leading to activation of downstream signaling (36, 37). However, the terminal cysteines, especially the palmitoylation sites Cys-181 and Cys-184, can also be targeted by OPTMs, such as S-glutathionylation in vitro (107, 142, 182), or electrophilic lipids such as 15d-PGJ2 in cell culture systems (120, 132) suggesting that altered p21Ras activity and downstream signaling might be affected.
Metabolism
Oxidative stress has been implicated in mediating the induction of cardiac cellular damage from high-fat western diets in animals. Although over-expression of the GPx-1 leads to a diabetic phenotype (112), specific cardiac over-expression of manganese superoxide dismutase (MnSOD) and peroxisomal catalase leads to improvement of contractility in the insulin dependent type I diabetic animal model, OVE26 mice, while catalase over-expression improves contractility in both type I and type II diabetic models (146, 177). Although these two antioxidants scavenge different RNOS in separate compartments, the rescue phenomena are similar suggesting a common mechanism. Since the MnSOD mice have twofold elevated levels of catalase, one can speculate that there are greater numbers of peroxisomes in both that lead to higher beta oxidation flux in the peroxisomes that improves utilization of fatty acids by reducing H2O2 production. In regard to S-glutathionylation, adenoviral Glrx1 over-expression in streptozotocin mice can improve acute cardiac ischemia reperfusion injury leading to smaller infarcts and improved cardiac function modulated by the Akt-FOXO pathway (95). However, the relevance of S-glutathionylation for in vivo metabolism in animals has not been determined. In contrast, much more work has demonstrated a clear role for S-glutathionylation in the metabolism of cultured cells and ex vivo tissues.
One of the more active sites of RNOS production is the intermembrane space of the mitochondrion where oxidative phosphorylation occurs. The electron transport chain components of mitochondria undergo S-glutathionylation that alters their activity. Using diamide, Murphy and colleagues have demonstrated that Complex I of the mitochondria can become S-glutathionylated on the 75 kDa subunit of Complex I. Both diamide and H2O2 added to isolated bovine heart Complex I protein leads to modification of predominantly Cys-704 of the 75 kDa subunit causing permanent loss of enzyme activity but unchanged superoxide production (72a). In addition, using LC-MS/MS, Chen and colleagues have demonstrated that an exchange reaction leads to S-glutathionylation of both the 51 kDa and 75 kDa subunits. In this system, the 51 kDa subunit was found to be S-glutathionylated on Cys-206 and Cys-187, while Cys-367 was modified on the 75 kDa subunit. S-Glutathionylated complex I activity actually increases in this model system with minimal loss of
Another protein that has been shown to be affected by oxidative stress that controls metabolism is Sirtuin 1 (Sirt-1). Sirt-1 is an NAD+ dependent deacetylase that can improve insulin sensitivity, glucose tolerance, adiposity, and exercise tolerance. Using purified protein, Zee and colleagues demonstrated that GSNO decreased Sirt-1 activity after resveratrol treatment due to S-glutathionylation of Cys-67, demonstrated by LTQ-Orbitrap MS (180). In addition to this cysteine site being affected, MALDI-TOF-TOF identified Cys-482 as being modified by hydoxynonenal (27). Since Sirt-1 is a nuclear protein, the thiol modifications may be taking place due to a cytoplasmic nuclear shuttle or by the actions of a oxidized intermediate “helper” protein like GAPDH (87).
This concept of helper proteins leads us to a more interesting concept of thiol modifications stemming from exchange reactions from one protein to the next. This possibility has been demonstrated for aldose reductase, which is an aldehyde metabolizing enzyme that has been linked to the development of secondary diabetic complications. In the vasculature, aldose reductase mediates high-glucose-induced smooth muscle cell proliferation and in cardiac tissue, it can modulate the late phase of ischemia reperfusion (130). In vivo, hearts subjected to ischemia reperfusion showed two- to fourfold increases in aldose reductase activity. Using N-ethylmaleimide as an alkylating agent, the pI of aldose reductase was changed in ischemic cardiac tissue suggesting a covalent modification had occurred (78, 79). By MALDI-TOF MS, aldose reductase was S-glutathionylated at Cys-298 and Cys-303. Using electro spray ionization-MS, dimedone binding was noted to occur after H2O2 treatment of purified protein suggesting the formation of a sulfenic acid at these sites. Although it would be reasonable to assume that the sulfenic acid formed would react with GSH, the structure of aldose reductase prevents this binding from occurring and thus a search for a “helper” protein was carried out. Since GSTs modulate protein thiol adducts, GSTπ null mice were used to determine whether aldose reductase could still be S-glutathionylated. The authors noted that no S-glutathionylated protein was formed in these null mice and using isolated protein, they elegantly demonstrated that sulfenic acid formation on aldose reductase was required before GST could transfer GSH to the protein leading to the formation of a mixed disulfide (162, 171). These data suggest that GST or other similar proteins can be used as a carrier of GSH and act as an intermediate in the S-glutathionylation process of target proteins (42). Furthermore, GSTs might also provide localized redox signaling. In addition to GSTs, the sulfhydryl oxidases are known to lead to intramolecular disulfide formation and may be able to cause S-glutathionylation in vitro (54). Whether or not sulfhydryl oxidases, QSox, or ERV1 (Table 1), from the two areas of high oxidizing potential, the ER and inner mitochondrial space respectively, can perform these functions in vivo is, at present, unknown (85, 135).
Inflammation
S-Glutathionylation of AP-1 and NFκB inhibits DNA binding and subsequently alters gene expression that is regulated by these transcription factors (114). NFκB controls transcription of a variety of genes that regulate immune responses and inflammation. The NFκB pathway is inhibited by S-glutathionylation at multiple points including p50 (123), RelA/p65 (128), and IKK-β (133).
Although RNOS have been reported to activate NFκB and are associated with inflammation (140), reduction of intracellular thiols activate NFκB (149) by enhancing subunit binding, while oxidized thiols limit binding and inhibit activity (161). The inhibitory mechanism is thought to be due to S-glutathionylation of the p50 subunit at Cys-62, which has been confirmed by mass spectrometry studies (123). These studies suggest that direct action of RNOS may not be the main mechanism of thiol regulation of NFκB, but rather exchange reactions may be of greater importance (145). In addition, S-glutathionylation of IKK-β, an NFκB activator, at Cys-179 leads to inactivation of the enzyme and subsequently attenuates cytokine-induced NFκB activation in tracheal epithelial cells (86, 133). It is, thus, unsurprising that in vivo Glrx gene deletion results in varied responses to inflammation (3, 35, 133). In addition to affecting transcription factors, the possibility of a thiol modification of an intermediate signaling molecule has been suggested as the S-glutathionylation of tumor necrosis factor (TNF) receptor-associated factor 6 modulates activation of NFκB (28). Glrx-dependent signaling might widely regulate inflammatory pathways. In viral infected cells, interferon regulatory factor 3, an essential transcriptional regulator of the interferon genes, is shown to be S-glutathionylated, and its activation requires deglutathionylation by Glrx (125). A further review regarding S-glutathionylation and inflammatory disease is referred to in the article by Shelton and Mieyal (145).
Animal Models of Glrx Ablation
Glrx is the most specific deglutathionylase or thioltransferase that regulates protein S-glutathionylation. Various mouse models with Glrx gene ablation implicated the involvement of protein S-glutathionylation in inflammatory responses in the lung, cataract formation in the eye lens, and postischemia reperfusion injury of the heart.
Reynaert et al. (133) demonstrated inactivation of IKK-β by S-glutathionylation at Cys-179 and its reversal by Glrx. Glrx knockdown sensitized cells to oxidative inactivation of IKK-β and attenuated TNF-α-induced NFκB activation. Primary tracheal epithelial cells from Glrx −/− mice were shown to reduce NFκB DNA binding and chemokine production in response to LPS (133). Concomitantly, bronchioalveolar lavages of Glrx −/− mice contained fewer activated macrophages, and decreased cytokines levels were measured including interleukin-1β and TNF-α after LPS exposure (3), suggesting that Glrx modulates signaling cascades involved in the pro-inflammatory response.
Conversely, in a model of cigarette smoke induced lung inflammation, Glrx −/− mice showed increased neutrophil infiltration into bronchioalveolar space and pro-inflammatory cytokine release based on enhanced NFκB nuclear translocation (35). These opposing results suggests that in chronic inflammation such as those triggered by smoking, Glrx may protect against the damaging effects of RNOS, whereas after acute inflammatory endotoxin challenge Glrx can amplify pro-inflammatory signaling cascades.
In agreement with this concept, eye lens epithelial cells of Glrx −/− mice showed a higher propensity to oxidative damage induced by UVB radiation, and an increased formation of cataracts was observed (113) in those animals. Similarly, ischemia reperfusion injury in the cardiovascular system was worsened by deletion of Glrx (68, 105). Lastly, it appears that the biologic effects of angiotensin II infusion on cardiac and aortic medial hypertrophy, is inhibited by Glrx deletion. This effect was attributed to attenuation of NOX activity by interference with upstream signaling components (11). A summary of current Glrx −/− animal studies is given Table 3.
Glrx, glutaredoxin; LPS, lipopolysaccharide; NFkB, nuclear factor κB; IKK-β, IκB kinase-β.
Since there have been opposing results and due to the redundancies of cellular thiol-metabolism the need for conditional transgenic and deletion mice will be necessary to better answer questions about ischemia reperfusion, atherosclerosis, angiogenesis, and inflammation in the cardiovascular system.
Conclusions
For more than half a century, the propensity of RNOS to alter protein function has been studied. Oxidants can reversibly oxidize the sulfur found in cysteines and methionine residues, which enable these two amino acids to carry out cellular redox signaling. Protein S-glutathionylation is highly dictated by the thiolate form of protein cysteine residues, participation of RNOS, and presence of GSH or enzymatic catalysts that drive the selectivity of this process. Although scavenging of RNOS has been suggested to worsen cardiovascular outcomes (141), the inhibition of both pathologic oxidative stress and redox signaling may be the cause. Rather than using nonspecific antioxidant scavengers, the restoration of the cellular redox potential may be the strategy of choice, thus still permitting redox signaling. In fact, novel GSH analogs are being studied for adjuvant use in breast cancer therapy (75). The development and utilization of redox potential modulators may be the next phase in the therapy of cardiovascular illness that will improve the pathophysiological processes that are controlled by S-glutathionylation including contraction, cellular growth, metabolism, and inflammation.
Footnotes
Acknowledgments
This work was supported by NIH grants PO1 HL 068758, K08 HL71563, and R37 HL104017 as well as by NHLBI, National Institutes of Health, Department of Health, and Human Services, under Contract No. HHSN268201000031C, and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the awarding offices. This work was also supported by a Sir Henry Wellcome postdoctoral fellowship from The Wellcome Trust (Sponsor Reference 085483/Z/08/Z; to J. R. B.).