
Abstract
Adaptation and transformation biology of the mitochondrion to redox status is an emerging domain of physiology and pathophysiology. Mitochondrial adaptations occur in response to accidental changes in cellular energy demand or supply while mitochondrial transformations are a part of greater program of cell metamorphosis. The possible role of mitochondrial adaptations and transformations in pathogenesis remains unexplored, and it has become critical to decipher the stimuli and the underlying molecular pathways. Immediate activation of mitochondrial function was described during acute exercise, respiratory chain injury, Endoplasmic Reticulum stress, genotoxic stress, or environmental toxic insults. Delayed adaptations of mitochondrial form, composition, and functions were evidenced for persistent changes in redox status as observed in endurance training, in fibroblasts grown in presence of respiratory chain inhibitors or in absence of glucose, in the smooth muscle of patients with severe asthma, or in the skeletal muscle of patients with a mitochondrial disease. Besides, mitochondrial transformations were observed in the course of human cell differentiation, during immune response activation, or in cells undergoing carcinogenesis. Little is known on the signals and downstream pathways that govern mitochondrial adaptations and transformations. Few adaptative loops, including redox sensors, kinases, and transcription factors were deciphered, but their implication in physiology and pathology remains elusive. Mitoplasticity could play a protective role against aging, diabetes, cancer, or neurodegenerative diseases. Research on adaptation and transformation could allow the design of innovative therapies, notably in cancer. Antioxid. Redox Signal. 18, 808–849.
I. Introduction
Energy metabolism includes a large number of interconnected biochemical reactions depicted on static metabolic charts (230), but little is known on the biological adaptability of these wide networks. Although the regulatory properties of key composing enzymes, the controlling ones, could permit to predict how the overall bioenergetic network could work, little is known on the adaptations that occur to maintain homeostasy under various physiological conditions. The pioneering bioenergeticists Otto Warburg, Peter Mitchel, and Britton Chance explained how the chemical energy contained in nutrients is converted into a biologically useful form, the adenosine triphosphate (ATP). The transformation of glucose, fatty acids, ketone bodies, and amino acids to ATP necessitates the cooperation of several enzymes forming metabolic networks (e.g., glycolysis and oxidative phosphorylation [OXPHOS]) interconnected by intermediate metabolites (e.g., pyruvate) (Fig. 1). Mitochondria intervene in the ultimate phase of cellular catabolism, following the enzymatic reactions of intermediate metabolism that degrade carbohydrates, fats, and proteins into smaller molecules such as pyruvate, fatty acids, and amino acids, respectively. Mitochondria further transform these energetic elements into nicotinamide adenine dinucleotide (NADH) and/or FADH2, through the Krebs cycle and β-oxidation. Those reduced equivalents are then degraded by the mitochondrial respiratory chain in a global energy-converting process called OXPHOS, where the electrons liberated by the oxidation of NADH and FADH2 are passed along a series of carriers regrouped under the name of respiratory chain or electron transport chain (ETC), and ultimately transferred to molecular oxygen. The ETC is located in the mitochondrial inner membrane (MIM), with an enrichment in the cristae. The ETC consists of four enzyme complexes (complexes I–IV) and two mobile electron carriers (coenzyme Q and cytochrome c). Complex I is the largest enzyme complex of this chain and contains 46 subunits organized in two large domains: the hydrophilic domain and the membrane domain (73, 74, 122). In mammals, Complex II is composed of four subunits, complex III of 11 (126), complex IV of 13, and complex V (the F1F0-ATPsynthase) of 16. Moreover, protein complexes I–IV are organized in a supramolecular assembly, the respirasome, and its structure was recently determined at about 2.2-nm resolution by cryoelectron tomography and subvolume averaging (69). Modeling indicated a loose interaction between the complexes and provides evidence that lipids are gluing them at the interfaces. In presence of energy substrate (NADH or FADH2), the transfer of electrons from complex I (or II) to complex IV mediates the extrusion of protons from the matrix to the intermembrane space, thus generating an electrochemical gradient of protons
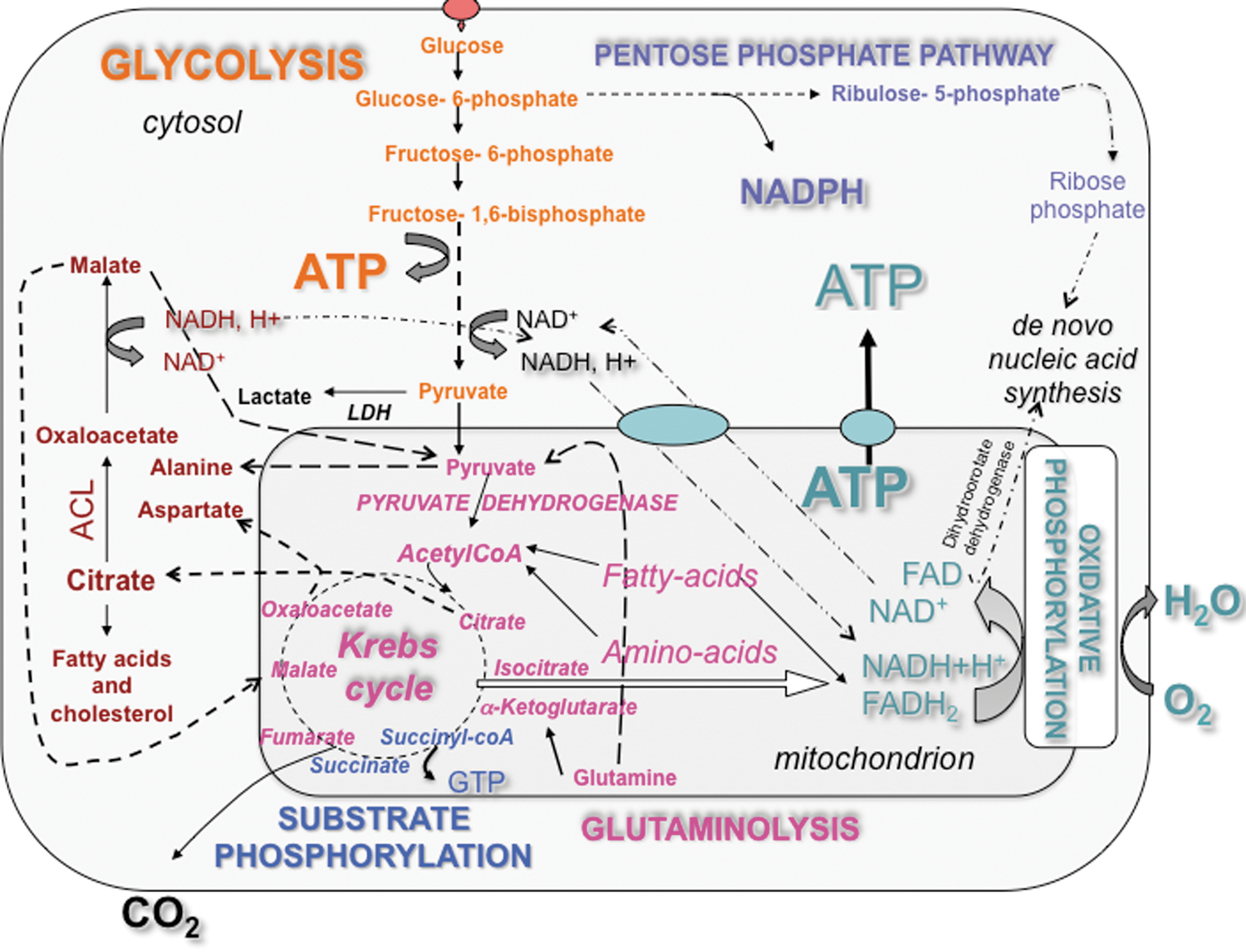

To study the adaptability of energy metabolism also requires the identification of the mechanisms involved in the shift between different metabolic routes. Bioenergetic studies have revealed that metabolic shifts occur when the balance between the energy supply and the energy demand of the cell is challenged, or when the type of energy substrate available is modified (160, 161). Enumeration of the pre-existing metabolic routes and consideration of their thermodynamic and kinetic properties now serve as a scientific basis to predict what metabolic pathways could be favored in a given microenvironment. While in silico modeling of energy metabolism could be helpful to discern between the putative pathways primarily used for energy production, the adaptative capacity of the energy-transducing systems remains underconsidered in biology. In fact, qualitative and quantitative changes in protein expression occur in response to bioenergetic stimuli or mitochondrial stressors, such as hypoxia and aglycemia, so that the metabolic system is not comparable before and after adaptation. Therefore, a better knowledge of the changes that occur in the architecture and the composition of the bioenergetic metabolic systems following a given perturbation, genetic, or environmental is required to propose an accurate model of energy metabolism. The design of therapies targeted against energy metabolism should also consider this adaptability. For instance, cancer cells forced to grow in absence of glucose can derive energy from glutamine as predicted from metabolic charts and demonstrated using carbon-13 isotopes of glucose and glutamine (glutaminolysis), but this shift occurs together with the enhancement of the OXPHOS system when glucose deprivation lasts for more than 72 h (228, 244). This adaptation (discussed in section VI) includes the upregulation of respiratory chain complexes and the morphological remodeling of the mitochondrial network (unpredictable from the charts). Adaptation biology of the mitochondrion is an emerging domain of mitochondrial research that focuses on the study of organelle plasticity. Neuroplasticity refers to the susceptibility to physiological changes of the nervous system, due to changes in behavior, environment, neural processes, or parts of the body other than the nervous system. By analogy, we introduce the term of mitoplasticity to refer to the capacity of the mitochondrion to adapt its shape and functions to the physiological needs. To gain further insight in cell biology and energy homeostasy, it has become critical to decipher the stimuli, the pathways, and the physiology behind mitochondrial adaptation, as this will also allow the design of innovative therapies targeted against the mitochondrion (i.e., bioenergetic therapies). Mitochondrial adaptations occur in skeletal muscle submitted to exercise training, in preadipocytes challenged with respiratory chain pharmacological inhibitors, in the skeletal muscle of patients with a mitochondrial disease or in primary cells undergoing carcinogenesis (Table 1). Adaptations also occur in mammalian cells placed in culture, an artificial phenomenon referred to as the culture shock (241), so that energy metabolism in vitro may not reflect what occurs in the tissues (see section III). So far, little is known on what determines most mitochondrial adaptations: what genes are involved, and what signaling pathways are recruited? The main proteins identified so far as playing a role in cellular bioenergetic remodeling are listed in (Table 2), and their role is mentioned in the text and illustrated in the figures (see Table 1 for details). In the present review, we focused our attention on the importance of these pathways, and we discussed the importance of mitochondrial adaptation for the maintenance of cellular energy homeostasy as well as for tumor survival.
II. Definitions of Mitochondrial Adaptation to Acute or Persistent Redox Change, Mitochondrial Transformation, Metabolic Control, and Adaptative Loops
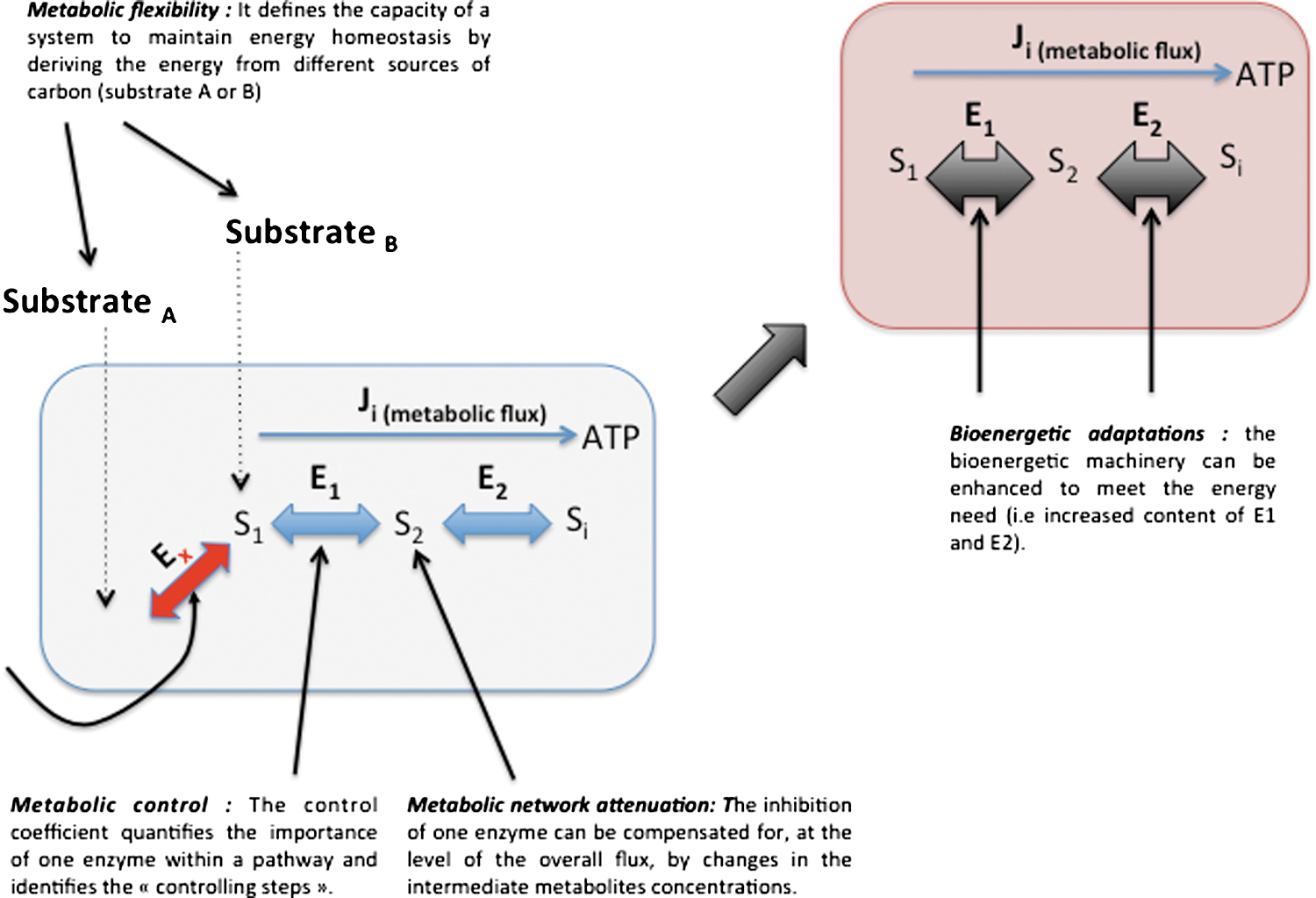
In this part, we define the terms used throughout this review, and we detailed the notions of energy homeostasy, metabolic plasticity, metabolic flexibility, metabolic attenuation, bioenergetic system adaptation, and metabolic control (Fig. 3). The homeostasy of cellular energy production defines a state of cell physiology where the energy needs are adequately balanced by both the energy supply and the activity of the energy-transducing system such as glycolysis and OXPHOS. Such stability is made possible by the existence of mechanisms of adjustment and of adaptation defined below. In the context of energy metabolism, metabolic plasticity means that the metabolic routes involved in energy production are not fixed and can be reassembled according to the biological context. These routes are composed of elementary modes that can be combined to compose dynamic metabolic pathways ultimately used for energy production. The process of shifting from one metabolic mode to another one is known as metabolic remodeling. The term metabolic flexibility applies to a cell system or to an organism and defines its capacity to maintain energy homeostasy by deriving biological energy from different sources of carbon. Such flexibility is predetermined by tissue-specific expression of metabolic enzymes, but adaptations can also occur to enhance metabolic flexibility or to reduce it (i.e., metabolic rigidity; or the dependency on a particular energy substrate). Metabolic control defines the biochemical mechanisms involved in the control of metabolic fluxes. For instance, the control of a metabolic flux by one of the enzymes present in the metabolic pathway can be evaluated by using the metabolic control analysis (MCA). The control coefficient quantifies the importance of one enzyme within a pathway and identifies the so-called controlling steps. In a given pathway, the sum of the control exerted by all the enzymes must be equal to 1. The notion of metabolic network attenuation (MNA) defines a kinetic mechanism of compensation inherent to metabolic networks (16). This mechanism was derived from MCA (111 –113, 131 –133) and takes into consideration the compensatory changes in intermediate metabolite concentrations that reduce the impact of the inhibition of an enzymatic step on the overall flux, within a metabolic pathway working at a steady state. Lastly, bioenergetic system adaptations are observed when metabolic rewiring does not suffice to maintain energy homeostasy, and when the bioenergetic machinery must be enhanced to meet the cellular energy needs (228). Such adaptation requires the synthesis of new proteins, in contrast to metabolic rerouting that utilizes the machinery in place. Adaptative loops or signaling pathways make the link between the biological stimuli and the ultimate modification of the mitochondrion. For instance, we recently showed that the compensatory activation of mitochondrial biogenesis upon ETC inhibition defines a loop that starts with the respiratory chain deficiency and ends up with the stimulation of peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) expression. This loop involves signals (as a decrease in cellular ATP/ADP), messengers (as nitric oxide [NO]), and transcription factors (as cAMP response element-binding protein [CREB]).
III. Mitochondrial Remodeling Pre-Exists in Most Cell Model Systems: Impact of the Culture Shock and the Bioenergetic Shock
Bioenergetics was found on a solid physiological ground, as the first measurements of respiration and OXPHOS by Otto Heinrich Warburg were performed on animal tissues using manometric techniques. Nowadays, most investigators use rapidly growing cell lines as a biological source for mitochondrial studies, and too little concern is made with regard to the relevance of such artificial models for the production of accurate knowledge in bioenergetics. One should question the impact of the placement of primary (noncancer) cells in an artificial culture. How accurate are tumor-derived cell lines, immortalized cell lines, and primary cells in culture to derive relevant bioenergetic knowledge? Do adaptations and remodeling occur in these systems, and to what extent do they compare with the in vivo conditions?
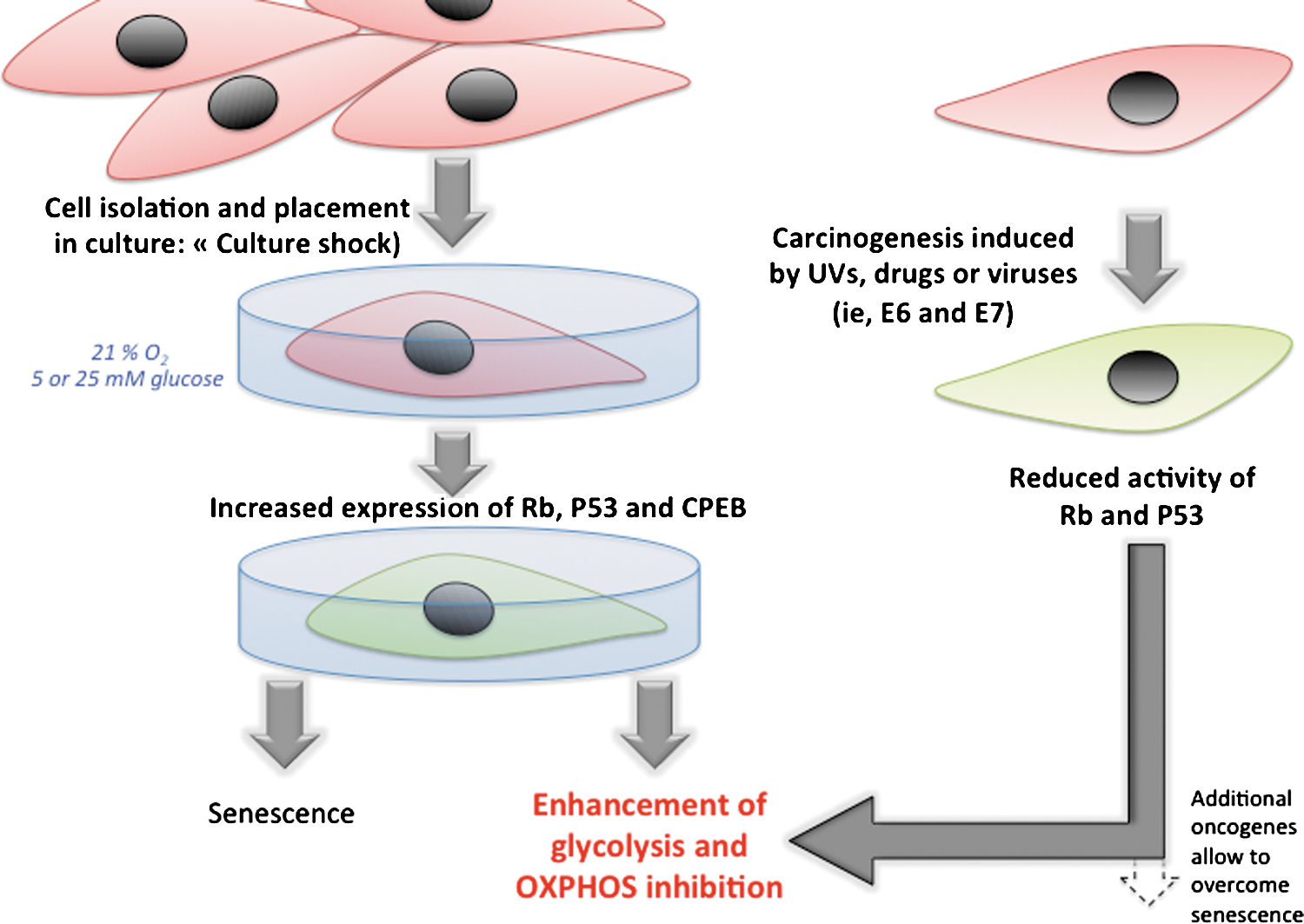
Firstly, the placement of primary mouse cells in culture triggers the so-called culture shock, as the cells immediately start to grow at higher rates as compared to what occurs in the tissue (Fig. 4). It was shown that human primary cells excised from their tissue of origin and placed in cell culture dishes for artificial growth rapidly shift toward a glycolytic phenotype (93, 101). Cell culture is hyperoxic (at the beginning of the culture), and senescence usually results from this culture shock, so that the genetics of senescence has been widely investigated in this context. Under normal physiological conditions, cells are exposed to 3% to 5% oxygen in various tissues, in contrast to the 21% oxygen in vitro (although this parameter could be modulated by researchers in in-vitro conditions for the defined cell culture to simulate the normoxia or hypoxia conditions, respectively). It was shown that genotoxic stress occurs during the culture shock, as mouse fibroblasts isolated from day-14 gestation embryos express P53 protein (p53) in culture conditions, whereas no expression of p53 was detected in these cells in vivo (103). Accordingly, it has been shown that the senescence of MEFs consecutive to the culture shock can be abrogated by maintaining the cells in 3% O2, closer to the original physiological oxygen environment (202). Just after placing the cells in culture dishes, the authors observed an elevated level of negative cell cycle regulators, including the cyclin-dependent kinase (CDK) inhibitors p16INK4a and p21Cip1, the p53 inducer p19ARF, and p53 itself. As proposed by Charles J. Sherr and Ronald A. DePinho (241), we have been conditioned to think that cell culture is a benign process, but nonphysiologic conditions, including disruption of cell–cell contacts, lack of heterotypic interactions between different cell types, the medium-to-cell ratio, persistent Ras activation by mitogens, absence of appropriate survival factors, hyperoxia, and plating on plastic are likely to induce stress responses. Therefore, we should question the occurrence of a bioenergetic shock associated with the culture shock, and further associated with senescence (Fig. 4). While most studies on senescence focused on the changes that occur at the level of cell cycle control, DNA repair, oxidative stress, and apoptosis, little is known on the impact of the culture shock on the mitochondrion. Yet, the two master regulators of cellular senescence activated by the culture shock, namely retinoblastoma protein (Rb) and p53, are also strong modulators of energy metabolism, so that bioenergetic changes are likely to occur. Indeed, one study looked at the status of mitochondrial function during oncogene-induced senescence in cells overexpressing the oncogenic form of Ras (185). The authors found an increase in mitochondrial mass, mitochondrial DNA content, and mitochondrial production of reactive oxygen species (ROS) before the senescent cell cycle arrest. As for the culture shock, these features were dependent on p53 and Rb. By the time the cells entered senescence, dysfunctional mitochondria accumulated around the nucleus; ATP levels decreased; and AMP-activated protein kinase (AMPK) was also activated. Lastly, the sole inhibition of the ETC was sufficient to trigger senescence, suggesting that mitochondrial dysfunction is an effector pathway of oncogene-induced senescence (38, 168). These findings highlight the specificities of cells in culture and raise the question of the relevance of the in vitro findings in bioenergetics with regard to the in vivo situation.
In good complement with senescence research, the field of oncobioenergetics has identified a supplementary role for Rb and p53 in metabolic remodeling. Master regulators of the cell cycle, such as p53, Rb, p21, and p27, and phosphatase and tensin homolog (PTEN) are typically mutated in several cancers and trigger cell cycle progression despite the presence of DNA damage or environmental stress. The overexpression of activated oncogenes can also overcome cell natural defenses by surpassing the capacity of the tumor suppressors. Several oncogenes, tumor suppressors and transcription factors such as hypoxia-inducible factor 1 α (Hif1α), RAS, c-Myc proto-oncogene (C-MYC), SRC, and p53 can influence energy substrate utilization by affecting cellular targets, leading to metabolic changes that favor cancer cell survival, independently of the control of cell proliferation (Fig. 5). These oncogenes usually stimulate aerobic glycolysis, and an increasing number of studies demonstrate that at least some of them can also enhance the OXPHOS machinery. For instance, C-MYC can concurrently drive aerobic glycolysis and/or OXPHOS according to the tumor cell microenvironment, via the expression of glycolytic genes or the activation of mitochondrial oxidation of glutamine (84). The oncogene RAS has been shown to increase OXPHOS activity in early transformed cells (58, 83, 262), and p53 modulates OXPHOS capacity via the regulation of the assembly of cytochrome c oxidase (178). Hence, carcinogenic p53 deficiency results in a decreased level of cyclooxygenase 2 (COX2) and triggers a shift toward anaerobic metabolism. In this case, lactate synthesis is increased, but cellular ATP levels remain stable (297). The p53-inducible isoform of the bisphosphatase domain of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase termed TP53-induced glycolysis and apoptosis regulator (TIGAR), a predominant phosphatase isoform of phosphofructokinase-2 (PFK-2), has also been identified as an important regulator of energy metabolism in tumors (23). In noncancer cells, TIGAR inhibits glycolysis by lowering fructose-2,6-bisphosphate (F-2,6-BP) levels, and upon mutation of p53 as occurs in several cancer cells, such inhibition is released, and glycolysis can proceed (24). Yet, 2-fluoro-2-deoxy-
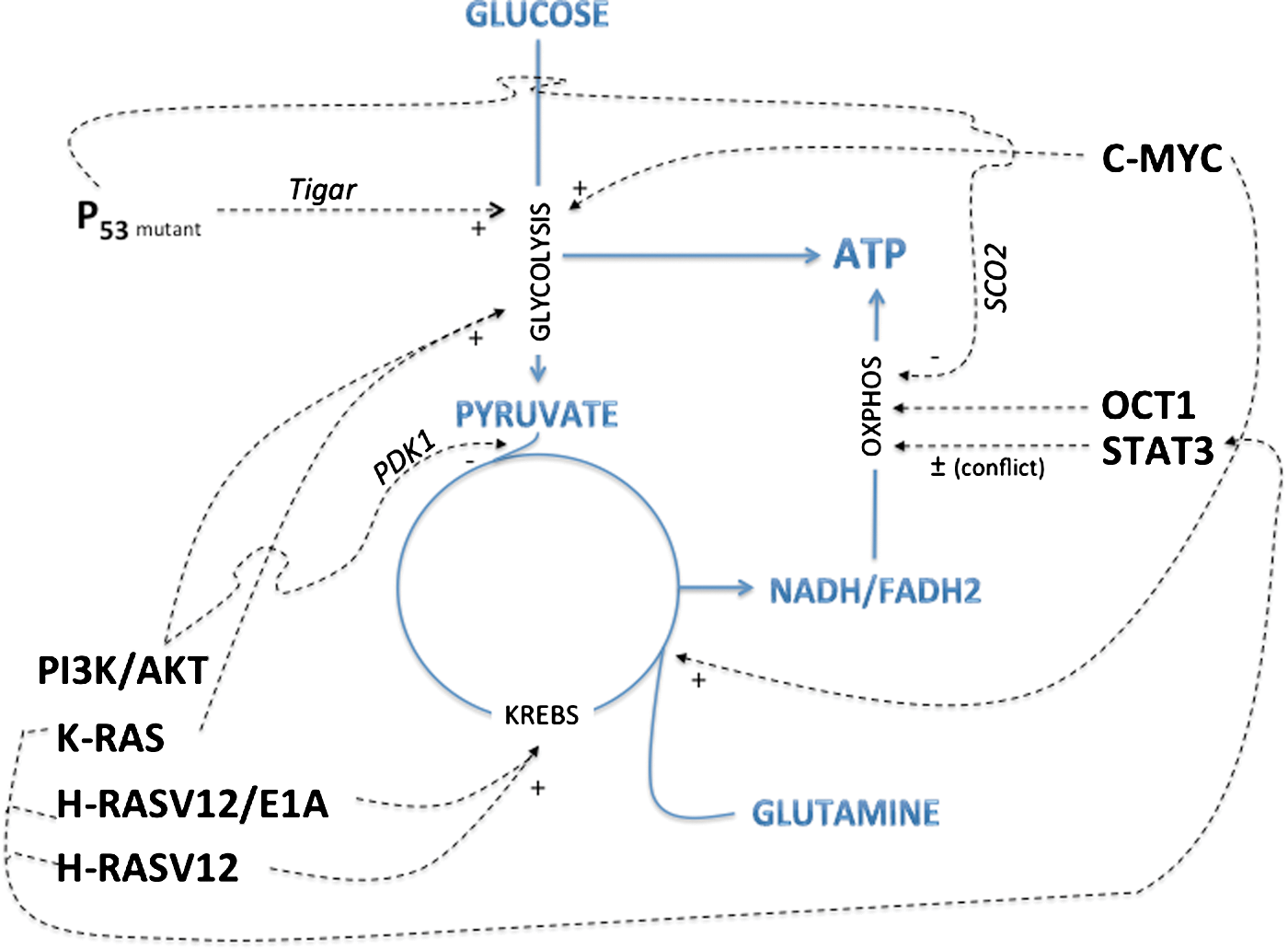
Recent findings revealed that Parkin is a p53 target gene that contributes to the function of p53 in glucose metabolism. Zhang et al. revealed that Parkin deficiency activates glycolysis and reduces mitochondrial respiration, leading to the Warburg effect (296). Restoration of Parkin expression reversed the Warburg effect in cells. Furthermore, in this study, Parkin deficiency sensitized mice to γ-irradiation-induced tumorigenesis, which provides further direct evidence to support a role of Parkin in tumor suppression. p32 sustains mitochondrial OXPHOS by playing a role in the synthesis of mitochondrial-DNA-encoded genes. All the above-listed studies delineate the role of different oncogenes in the regulation of OXPHOS (Fig. 6). Interestingly, while Rb and p53 are obligatory tumor suppressors and loss-of-function mutations in these proteins allow cells to enter the so-called oncogene-induced-senescence, a necessary step in the route to transformation, the completion of the transformation process toward full immortalization requires the bypass of senescence, as obtained with human telomerase reverse transcriptase (h-TERT), small T, and Ras oncogenes (Fig. 6). Those findings were used by Hahn and Weinberg (102) to generate an in vitro model of progressive cell transformation that is now widely used in different laboratories. This bypass is followed by additional changes in energy metabolism, and we proposed a model of metabolic waves to illustrate this phenomenon (247). Among the oncogenes needed to complete cell transformation, h-TERT overexpression improves mitochondrial function and inhibits ROS-mediated apoptosis in cancer cells (123). The authors provided evidence for mitochondrial localization of h-TERT, and a significantly higher activity of cytochrome c oxidase, the rate-limiting enzyme in the mitochondrial ETC, in h-TERT-overexpressing cells. Lastly, besides p53 and Rb, another key player in the bioenergetic changes associated with senescence could be cytoplasmic polyadenylation-element-binding protein (CPEB). The knockdown of CPEB causes skin and lung cells to bypass the M1 crisis stage of senescence. Burns and Richter showed that CPEB knockdown cells have fewer mitochondria than wild-type cells and resemble transformed cells by having reduced respiration and ROS, normal ATP levels, and enhanced rates of glycolysis (32). The mechanisms of CPEB-induced OXPHOS downregulation involve p53, since CPEB knockdown cells showed a 50% decrease in p53 levels (p53 mRNA has an abnormally short poly(A) tail and a reduced translational efficiency). p53 was shown to interact with transcription factor A mitochondrial (TFAM), a critical component of mitochondrial DNA replication, and thereby to participate to mitochondrial biogenesis (201). CPEB could modulate OXPHOS capacity by similar means.

IV. Cellular Energy Homeostasy Relies on the Coordination Between Glycolysis, OXPHOS, and Direct Substrate Phosphorylation: Role of the AMPK
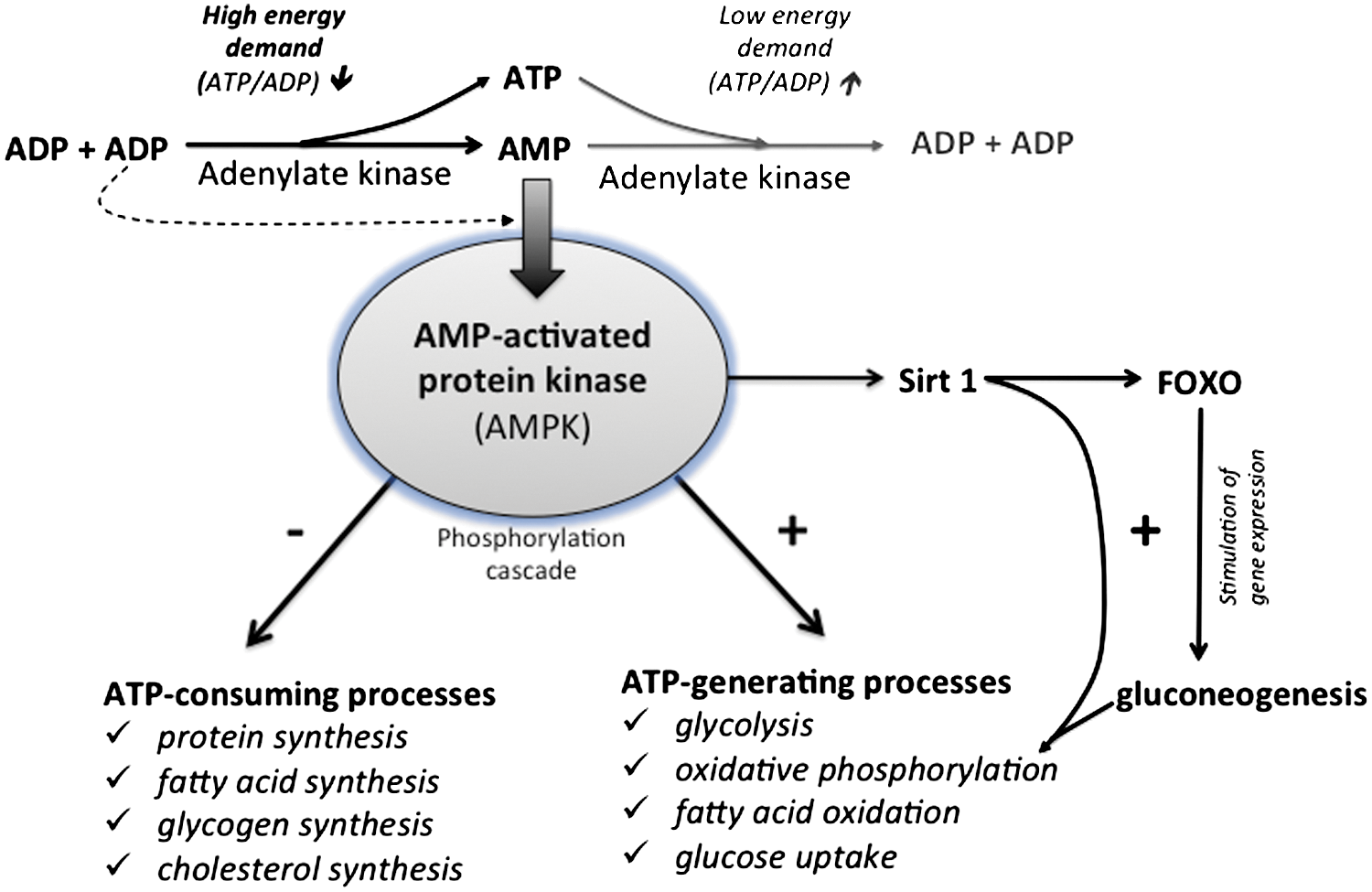
Bioenergetic studies have long used, and still actively utilize, specific chemical inhibitors of the mitochondrial respiratory chain complexes to study the fundamental mechanisms of energy production (44). In the last decade, numerous pharmacological compounds used in clinical practice were also shown to inhibit mitochondrial respiration in vivo and in vitro, so that more priority was given to mitochondrial testing in pharmacological drug development (70). Famous examples are the use of statins to treat hypercholesterolemia, which block coenzyme Q biosynthesis and alter mitochondrial respiration (81), or the use of nucleoside reverse transcriptase inhibitors (NRTIs, such as Azidothymidine), which inhibit mitochondrial DNA replication and further reduce OXPHOS capacity by other mechanisms (22). Lastly, several compounds used for local anesthesia also demonstrated a mitotoxic effect, as widely reported for bupivacaine, which can induce muscle ultrastructural alterations associated with reduction of pain (194). Interestingly, human cells grown in the presence of such mitochondrial stressors revealed the development of metabolic adaptations to promote cell survival (Fig. 7). Typically, ETC inhibition induces an immediate decrease in cellular ATP content (increase in the ADP/ATP ratio), which activates AMPK and inactivates ATP-consuming reactions to preserve ATP during energy stress (109). In particular, AMPK inhibits the synthesis of fatty acid synthesis, triacylglycerol, and cholesterol via the inhibition of the acetyl-CoA carboxylase (ACC), the glycerol phosphate acyl transferase, and the 3-hydroxy-3-methylglutaryl-CoA reductase, respectively. In cells treated with oligomycin and antimycin, the activation of PFK-2 was also observed, with a consecutive increase in F-2,6-BP concentration, and the ultimate stimulation of glycolysis (3). The importance of this AMPK/6-phosphofructo-2-kinase pathway was also demonstrated in neurons where complex IV was inhibited by NO, since neurons that lacked 6-phosphofructo-2-kinase died from ATP depletion (4). In another example, the utilization of oligomycin on OXPHOS-dependent cancer cells (106) induced a 5%–8% ATP drop and a transient AMPK activation during the initial 1–2 h. At 6 h, glycolysis activity was increased along with acetyl-CoA carboxylase phosphorylation, and cellular ATP returned to normal levels. The phosphorylation of acetyl-CoA carboxylase blocks the synthesis of the malonyl-CoA used as a substrate for fatty acid biosynthesis. However, this metabolic shift did not depend on the activation of LKB1 and of AMPK (and possibly PFK2), as the effect was also observed in LKB1-null cells. The authors concluded on an unidentified regulatory mechanism, although this shift could have occurred simply by the rerouting of pyruvate to lactate via the lactate dehydrogenase (LDH). Additional adaptation could occur after the transient AMPK activation, such as enhancement of glycolysis or utilization of alternative energy substrates. Indeed, the molecular determinants of metabolic plasticity include energy sensing and the adjustment of the energy balance. In mammalian cells, energy homeostasy requires a constant coordination between cell activity, nutrient availability, and the regulation of energy transduction processes. This is obtained via a complex signaling system linking energy sensing and nutrient sensing to cellular effectors that include kinases and transcription factors. While the intracellular energy status is mirrored by the respective concentrations of the adenine nucleotides ATP, ADP, and AMP (as well as phosphocreatine/creatine), the nutrient availability is reflected more by the concentration of glucose and amino acids. The S6 kinases 1 and 2 (S6K) and AMPK intervene downstream of these sensors to modulate the growth rate and energy metabolism (2, 267). The AMPKs are activated upon alterations in the cellular AMP/ATP ratio, which is dictated by the balance between energy supply (ATP production) and energy demand (ATP consumption) (Fig. 9). When activated by AMP, the AMPKs initiate a cascade of phosphorylation to switch on the catabolic pathways that produce ATP (glycolysis, OXPHOS via the stimulation of mitochondrial biogenesis), and to switch-off the anabolic pathways that consume ATP (protein synthesis, fatty acid synthesis, and cholesterol synthesis). Yet, AMPK can also be activated by ADP (289), since this enzyme displays significantly tighter binding to ADP than to Mg-ATP, explaining how AMPK is regulated under physiological conditions where the concentration of Mg-ATP is higher than that of ADP and much higher than that of AMP. This property is of critical importance in energy metabolism control (108, 196).

The targets of AMPKs are numerous genes involved in lipid, carbohydrate, and protein metabolism, but also cell signaling, transcription, and ion transport [for review see (107)]. More recently, it was discovered in mouse skeletal muscle that AMPK further regulates energy metabolism through the activation of the sirtuin SIRT1 (39, 40). This results in the deacetylation and the modulation of the activity of downstream SIRT1 targets that include the PGC1α, and the forkhead box O1 (FOXO1) and O3 (FOXO3a) transcription factors. FOXO are major downstream targets of the PI3K signaling pathway; they control the transcription of several genes involved in cell life and death, cell cycle, differentiation, ROS detoxification, and DNA repair (99); and particularly, the neoglucogenic enzymes glucose-6-phosphatase and phosphoenol pyruvate carboxykinase, which produce glucose in situations of strong energy demand (as occurs during starvation). Accordingly, the AMPK can be activated by cellular stress, exercise, and a wide range of hormones or pharmaceutical agents that affect cellular energy metabolism [e.g., Metformin, thiazolidinediones, adiponectin, leptin, ghrelin/cannabinoids, and resveratrol (295)]. Besides the master control exerted by AMPK and sirtuins, the regulation of cellular energy metabolism is influenced also by calcium and ROS (see section XII).
In addition to glycolysis, human cells can also compensate for defective respiratory chain activity by deriving ATP indirectly from the Krebs cycle (this GTP serves as a phosphoryl group donor for ADP to yield ATP in the reaction catalyzed by cellular kinases). This is known as substrate-level phosphorylation, and an example of this compensatory mechanism was evidenced in cells bearing a genetic defect in the F1F0-ATP synthase complex (NARP/MILS mutations) or in control cells treated with oligomycin (238). In this study, the addition of α-ketoglutarate (αKG) and aspartate allowed to stimulate mitochondrial substrate-level phosphorylation and to rescue both the ATP content and the cell viability (Fig. 8). Moreover, a genetic screen in yeast identified the importance of α-ketoglutarate for the bioenergetic compensation of OXPHOS deficiency. In this study, the authors isolated the oxodicarboxylate carrier 1 (ODC1), an oxodicarboxylate carrier that transports αKG and α-ketoadipate or any other transported tricarboxylic acid cycle intermediate in a counter-exchange through the inner mitochondrial membrane (236). The observed rescue of the respiratory-growth-deficient fmc1 mutant by overexpression of Odc1p was explained by an increase in the flux of tricarboxylic acid cycle intermediates from the cytosol into the mitochondria, leading to an increase in the αKG oxidative decarboxylation, resulting in an increase in mitochondrial substrate-level-dependent ATP synthesis. This mechanism of metabolic bypass of a defective ATP synthase unravels the physiological importance of intramitochondrial substrate-level phosphorylations and could foster the development of therapeutic solutions in pathologies associated with defects in the OXPHOS system. Lastly, several studies showed that respiratory chain inhibition, either inherited or pharmacological, can trigger a compensatory increase in organelle turnover by mechanisms that involve the transcription factor CREB and the transcription coactivator PGC1α. This is further discussed below in section X.
Another important feature of mitoplasticity is the ability of the F1F0-ATP synthase to work in reverse mode when the respiratory chain is no longer able to generate a sufficient protomotive force. This feature also necessitates the reverse mode of ANT functioning, as demonstrated by Chevrollier A and Stepien G (48 –50, 165). In particular, cells lacking mitochondrial DNA (Rho-0 cells) maintain a mitochondrial membrane potential both by a residual proton gradient (up to 45% to 50% of the potential) and by other ion movements such as the glycolytic ATP(4-) to mitochondrial ADP(3-) exchange. The ANT2 gene, encoding isoform 2 of the ANT, is overexpressed in Rho-0 cells, which are strongly dependent on glycolytic ATP synthesis. The reverse mode of OXPHOS is important for the viability of cancer cells, and the expression of ANT2, which is linked to the rate of glycolytic metabolism, is an important indicator of carcinogenesis. The reverse operations of ANT2 and F1F0-ATPase under glycolytic conditions contribute to maintaining the mitochondrial membrane potential, ensuring cell survival and proliferation.
The above-described pathways of mitochondrial adaptation could be involved in carcinogenesis, as a link between mitochondrial stress response and the initiation of cancer was revealed by Gozalvez., in the early 70's with the discovery of rotenone-induced tumors (95). In these visionary studies, the authors found that rotenone was consistently tumorigenic in the albino rats. Four series of 10 animals were given i.p. injections of rotenone at 1.7 μg/gram-weight daily for 42 days. The authors observed an incidence of 80% mammary tumors that appeared from 6 to 11 months after the end of treatment. Electron microscopy of the tumor tissue showed a progression of mitochondrial lesions. Together with the recent implications of the mitochondrial energy proteins succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase (IDH) in cancer susceptibility (56, 57, 96, 140, 191, 291), the above-described experiments with rotenone might suggest that mitochondrial impairment could favor cancer initiation. This hypothesis was tested by the group of Jose Cuezva by evaluating the tumorigenic potential of cancer cells chronically treated with oligomycin, and the results showed that these cells presented with a higher tumorigenicity when implanted into mice (167). Likewise, mutations in mtDNA can be protumorigenic, and both the type of mutation and the mutant load are important to promote tumorigenicity since high levels of mtDNA mutations become anti-tumorigenic [concept on Oncojanus proposed by Guiseppe Gasparre and colleagues (85)]. Also, Martins and colleagues discussed the relevance of mitochondrial stress response in ischemic preconditioning, a situation in which short ischemic episodes protect the brain and the heart against prolonged shortage of oxygen and nutrients (176). They concluded that mild mitochondrial stress can elicit autophagy, a cytoprotective mechanism relying on the digestion of potentially harmful intracellular structures, notably mitochondria. Lastly, recent developments in cell biology have also involved the mitochondrial adaptation pathway in some unexpected phenomena, such as the humoral response. Indeed, it was recently found that modulators of energy metabolism, AICAR and oligomycin, are able to enhance antibody production, but the mechanisms remain to be clarified (6).
V. Mitochondrial Adaptation During Exercise Training, ER Stress, Genotoxic Stress, and Environmental or Biological Insults: Evidence of Immediate and Delayed Loops of Adaptation
Mitoplasticity is involved in a large number of cellular responses to various types of stress. Two types of adaptations were described, the acute response and the delayed one. A good example to illustrate these two types of responses was evidenced by analyses on the adaptation of mitochondria to exercise training. In rodents, acute exercise increases skeletal muscle uncoupling protein (UCP) gene expression and is associated with elevations in serum nonesterified fatty acids (193). In humans, vastus lateralis biopsies obtained 1 h postexercise from untrained subjects revealed that acute exercise induced a significant increase in both UCP-3(L) and UCP-3(S), but not UCP-2 mRNA levels. Moreover, acute exercise upregulates UCP3, whereas endurance training results in the downregulation of UCP3 protein content (235). In addition to this mitochondrial response to acute exercise, another type of adaptation was described in response to endurance training, as shown by the work of Antonio Ascnesao and colleagues (172). These authors revealed that moderate endurance treadmill training ameliorates gastrocnemius mitochondrial bioenergetics and increases the tolerance to the calcium-induced mitochondrial permeability transition-pore (mPTP) opening. Endurance training induced significant increases in state 3 and in the respiratory control ratio both with complex I- and II-linked substrates. Increased carbonyl cyanide 3-chlorophenylhydrazone (CCCP)-induced uncoupled respiration with succinate as a substrate was also observed (p<0.05).
These two types of mitochondrial adaptations (acute and delayed) were also revealed by the work of Gyorgy Szabadkai while investigating the implication of mitochondria during the ER stress response (30). More precisely, these authors analyzed the role of mitochondria in the adaptive unfolded protein response. The objective of this study was to investigate the metabolic events that occur during the adaptive phase of ER stress, before the cell death response. The results showed that during the onset of ER stress, the reticular and mitochondrial networks are redistributed toward the perinuclear area, and their points of connection are increased in a microtubule-dependent fashion. A localized increase in mitochondrial transmembrane potential was observed only in redistributed mitochondria, and the spatial reorganization of these organelles was in good correlation with an increase in ATP levels, oxygen consumption, reductive power, and increased mitochondrial Ca2 + uptake. These data revealed that ER stress induces an early increase in mitochondrial metabolism that depends crucially upon organelle coupling and Ca2 + transfer, which, by enhancing cellular bioenergetics, establishes the metabolic basis for the adaptation to this response. The molecular basis of the bioenergetics coupling between ER stress and mitochondrial ATP production is supported by the existence of a truncated isoform of SERCA (S1T) localized in the mitochondrion-associated membranes and involved in the transfer of Ca2+ to mitochondria. Chami et al. (43) revealed that S1T knockdown prevents ER stress, mitochondrial Ca2+ overload, and subsequent apoptosis.
Mitoplasticity is also involved in the cellular response to toxic compounds, as exemplified by the work of Bourdineaud's group on environmental pollutants (27, 28, 35, 36, 158). To investigate the alteration of mitochondria by methylmercury, Danio rerio was contaminated with food containing 13 μg of MeHg per gram, an environmentally relevant dose. Mitochondria from contaminated zebra fish muscles presented structural abnormalities under electron microscopy observation. In permeabilized muscle fibers, a strong inhibition of both state-3 mitochondrial respiration and functionally isolated maximal cytochrome c oxidase (COX) activity were observed after 49 days of MeHg exposure. A dramatic decrease in the rate of ATP release by skinned muscle fibers using either pyruvate and malate or succinate as respiratory substrates was measured. This suggested that MeHg induced a decoupling of mitochondrial OXPHOS in the skeletal muscle of zebra fish. Then, experiments were performed in mice, as MeHg constitutes a hazard for human health through eating contaminated fish (28). Mice fed with a vegetarian-based diet containing mercury during 2 months showed negatively altered behavior and increased anxiety, associated with modifications of mitochondrial respiratory protein subunit concentrations in the kidney and brain. In kidneys, the COX-IV subunit was increased by a factor of 3, while in the hippocampus, the authors evidenced a 5-fold, 1.4- fold, and a 5-fold increase of the iron protein of the SDH complex, the COX-I subunit, and the COX-IV subunit, respectively. Such upregulation of OXPHOS proteins might indicate the onset of an adaptative response to compensate for MeHg toxicity. To evidence the mitochondrial adaptative response, a timecourse transcriptomic analysis of the effect of methylmercury was performed on the zebra fish [contamination for 25 and 50 days with food containing 13.5 mug/g dry weight (dw) of MeHg (0.6 mug MeHg/fish/day]. Brain mitochondrial respiration was not modified by exposure to MeHg, contrary to what happens in skeletal muscles. A 6-fold increase in the expression of the sdh gene encoding the SDH Fe/S protein subunit was detected in the contaminated brain after 50 days of exposure. After 50 days, the authors also detected the upregulation of glutathione S-transferase genes (gfap and gst), along with a repression of the glutathione peroxidase gene gpx1. These results match well with an MeHg-induced onset of oxidative stress and inflammation. Lastly, several lines of evidence exist on the implication of mitoplasticity in the cellular response to genotoxic stress. In collaboration with the group of Hamid Rezvani, we showed that a primary DNA damage induced by a KO of the DNA repair enzyme XPC in mice triggers a reduction of OXPHOS and a stimulation of glycolysis recapitulating the Warburg effect. Accumulation of unrepaired DNA after XPC silencing increased DNA-dependent protein kinase activity, which subsequently activated AKT1 and NADPH oxidase-1 (NOX1), resulting in ROS production and accumulation of specific deletions in mitochondrial DNA (mtDNA) over time. Those ROS were able to induce the metabolic shift from OXPHOS to predominant glycolysis.
VI. Mitochondrial Structural Adaptation Is Observed in Cancer Cells Confronted to Aglycemia
Already in 1979, Reitzer and colleagues published an article entitled Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells, which demonstrated that OXPHOS was used preferentially to produce ATP in cervical carcinoma cells (220). Griguer and colleagues also identified several glioma cell lines that were highly dependent on the mitochondrial OXPHOS pathway to produce ATP (100). Glioma stem cells (279) also rely mainly on OXPHOS to derive energy. These findings are in apparent contrast with the field of oncobioenergetics (104, 239), which suggests that most oncogenes trigger the enhancement of glycolysis and the reduction of OXPHOS although not exclusively as explained in previous sections (see also below, sections VII and VIII). This apparent contradiction can be resolved when considering that in addition to the oncogenes, other regulators of carcinogenesis participate to the reshaping of energy metabolism (104, 130). In particular, the microenvironment of tumors was designated as a key regulator of the bioenergetic profile of cancer cells, and we give some examples below.
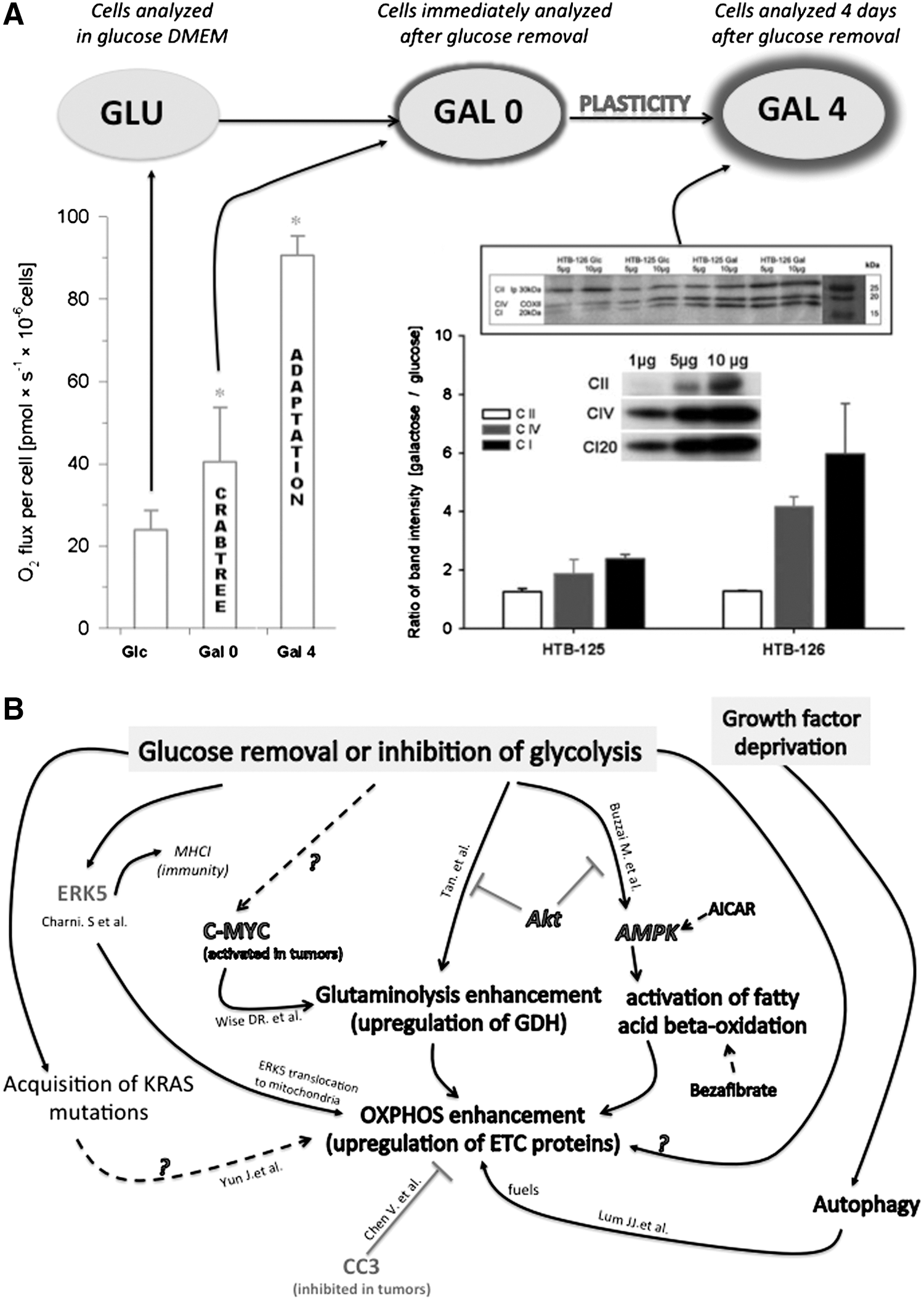
Firstly, glucose deprivation leads to a profound modification of the energy pathways toward oxidative metabolism, with upregulation of respiratory chain proteins and higher branching of the mitochondrial network in osteosarcoma (143B) and hepatocellular carcinoma (HEPG2) (213, 228). Likewise, a subclass of glioma cells that utilizes glycolysis preferentially (i.e., glycolytic gliomas) can switch from aerobic glycolysis to OXPHOS under limiting glucose conditions (12, 29), as observed in cervical cancer cells, breast carcinoma cells (Fig. 8A), hepatoma cells, and pancreatic cancer cells (213, 228, 244). In breast cancer cells, the expression level of the complex IV subunit one was increased by a factor of six after only 4 days of growth in aglycemia, and this phenomenon was associated with a 3-fold increase in respiration, while no significant change was measured in the corresponding noncancer cells (244). In this study, we observed a significant increase (+30%) in the mitochondrial to nucleus areas in HTB-126 cancer cells grown under aglycemic conditions for 4 days. These results suggested a stimulation of mitochondrial biogenesis induced by glucose deprivation, specifically in the cancer cells. In addition to OXPHOS machinery enhancement, glioblastoma cells exposed to glucose withdrawal showed an increase in glutamine metabolism, which was explained by a large increase in glutamate dehydrogenase (GDH) activity (292). This adaptation was linked to the Akt pathway, since inhibition of Akt signaling increased glutamate dehydrogenase GDH activity, whereas overexpression of Akt suppressed it. In that study, GDH activation resulted from the loss of glycolysis, because it could be mimicked with the glycolytic inhibitor 2-deoxyglucose and reversed with a pyruvate analog. In addition to glutaminolysis, the β-oxidation of fatty acids is also activated upon glucose deprivation (33), and the oncogenic kinase Akt blocks these oxidative bypass in presence of glucose (Fig. 8B). Akt is central to bioenergetic remodeling, as it promotes glucose transporter 1 (GLUT1) recruitment to plasma membrane, activates hexokinase association with the mitochondrion, and phosphorylates ATP citrate lyase with downstream activation of fatty acid synthesis. Akt also inhibits fatty acid β-oxidation via the inhibition of carnitine palmitoyltransferase (CPT1A). Of particular importance for mitoplasticity, defects in mitochondrial respiration can lead to Akt activation (206). Pelicano et al. (206) showed that respiration deficiency induced by mitochondrial mutagenesis, pharmacologic inhibition, or hypoxia causes inactivation of PTEN, a negative regulator of Akt.
An intriguing question raised by all these studies concerns the mechanisms involved in the enhancement of OXPHOS observed in HeLa, 143B, HEPG2, HTB126, and other cancer cells shifted from glucose to glutamine–galactose medium (Fig. 8A). So far, the molecular pathways involved in this adaptation remain unresolved. Three possible stimuli should be distinguished: (i) the direct effect of glucose removal, (ii) the effect of prolonged glucose deprivation, and (iii) the secondary effect of mitochondrial reactivation through glutaminolysis. These stimuli activate different signaling pathways that might work synergistically to generate the observed OXPHOS enhancement. Concerning the first possibility, it is known that glucose inhibits OXPHOS in cancer cells, and that removal of this Crabtree effect could liberate OXPHOS and possibly restore a feedback activation of OXPHOS biogenesis (see below, the retrograde response and CREB in section X). We demonstrated the reversibility of the Crabtree effect in breast cancer cells (244). The underlying mechanisms still remain unresolved, and current theories suggest that some intermediates of glucose oxidation could inhibit mitochondrial OXPHOS. Biochemical studies performed in yeast suggest that fructose-1,6-bisphosphate (F-1,6-BP) could play a determining role in this process (61). Hepatoma cells show a 50-fold increase in this metabolite concentration after 5 mM glucose addition to the cells (225). Interestingly, addition of galactose did not change the level of F-1,6-BP in that study, and we also observed the suppression of the Crabtree effect when glucose was replaced by galactose. It is important to notice that galactose is not used as a fuel by cancer cells, but serves for nucleic acid synthesis, while glutamine is consumed by the mitochondria to produce ATP. This phenomenon was demonstrated in HeLa cells or in CHO cells using C13-labeled glucose and glutamine (67, 220). Thus, to compare the bioenergetic status of cancer cells and noncancer cells, it is essential to compare the cells while in the Glc and the Gal 0 conditions, as shown in Figure 9. Moreover, the rapid loss of HTB-126 cancer cell viability induced by various inhibitors of the OXPHOS system was observed only in a glucose-deprived medium (galactose and glutamine are present in this medium), further emphasizing the fact that ATP is derived from OXPHOS in this medium.
The second mechanism by which OXPHOS biogenesis could occur in cancer cells grown in galactose medium is more complex and involves the potency of glucose deprivation to activate a large number of signaling pathways involving specific kinases capable of stimulating OXPHOS biogenesis and intermediate metabolism. Namely, the AMPK, p38 mitogen-activated protein kinase (p38 MAPK), and phosphoinositide 3-kinase/protein kinase B (PI3K/PKB) have been linked to the regulation of glucose uptake elicited by diverse hormonal stimuli in several cell types (53, 82, 134, 255). AMPK is a metabolic-sensing protein that is activated by an increase in the AMP/ATP ratio (see previous section IV). AMPK is activated by allosteric binding of AMP and by phosphorylation by one of the upstream AMPK kinases. Once activated, AMPK regulates ATP levels by suppressing biosynthetic pathways, such as fatty acid and cholesterol biosynthesis, as well as by activating ATP-generating catabolic pathways such as fatty acid oxidation and glycolysis (Fig. 9). Other intracellular signaling systems that may be involved in the metabolic response to glucose deprivation are the stress-activated p38 MAPK (47) and the survival pathway PI3K/PKB (269). As discussed above, the role of Akt in the activation of GDH and of β-oxidation upon inhibition of glycolysis could occur in parallel to OXPHOS upregulation (292) (Fig. 9).
The third mechanism considers the possibility that the higher energy needs of cancer cells for supporting their deregulated growth and extensive biosyntheses stimulate the OXPHOS system when the fuel for energy production switches from glycolytic to oxidative only. Under nonstringent conditions, glucose is the preferential energy substrate of cancer cells, and its consumption is typically increased by the expression of more rapid isoforms of the glycolytic pathway (177). This result could indicate that the strong energy demand of cancer cells dictates the upregulation of whatever energy pathway is used, as determined by the type and availability of the energy substrate. This peculiarity has been observed for other cancer cells [HeLa, 143B (228) and HepG2 (213)] and could designate the underlying pathway of metabolic remodeling as a potential target for anticancer therapy.
The role of PDH also remains to be evaluated in cells grown in absence of glucose. DCA is a pyruvate dehydrogenase kinase (PDK) inhibitor, and typically results in increased pyruvate dehydrogenase activity. The group of Bonnet (26) explained the anticancer effect of DCA by a complex mechanism that involved the resensitization of cancer cells to apoptotic stimuli via the stimulation of mitochondrial respiration and the subsequent (cancer specific) upregulation of a potassium channel (Kv1.5). However, during the adaptation of cancer cells to growth in absence of glucose, PDH reactivation might be required to provide acetyl-coA to the Krebs cycle enzyme αKG dehydrogenase (KDH), since the carbon source and energy source is glutamine, and this amino acid enters the Krebs cycle at the level of KDH. This underestimated role of PDH could explain why we observed a strong upregulation of PDH in HeLa cells forced to grow in absence of glucose (228). Most of the experiments aiming at the evaluation of DCA cancer toxicity were performed in high-glucose media, and in these conditions, DCA induced apoptosis. However, given the potential key role of PDH in absence of glucose, DCA could favor the utilization of glutamine by the Krebs cycle and might benefit the growth of cancer cells in conditions of glucose withdrawal. This hypothesis remains to be evaluated.
Besides glucose, another key player in the definition of a cancer cell's metabolic profile is oxygen tension (p O2). Hypoxia is a common feature of the microenvironment of cancer cells, and tumor oxygenation can be severely compromised as compared to normal tissue (274). Thus, it seems more appropriate to investigate the impact of hypoxia and aglycemia on cancer cells, which typically encounter this type of stress in situ. Vaupel and coworkers observed low values for intratumoral oxygen tension ranging between 3 to 10 mmHg in breast malignant tissue, while noncancer tissue exhibited higher values ∼50 mmHg (276). The Gatenby and Gillies microenvironmental model of carcinogenesis (86, 87) considers that precancer cells are typically found in tissue regions where oxygen and glucose delivery is low. This might have preadapted energy metabolism to a life of uncontrolled growth and deregulated cell death. Gatenby and Gillies further proposed that during tumor growth, angiogenesis leads to the growth of inadequate microvasculature, which results in intermittent oxygen and glucose deprivation in cancer cells along with acidification of the extracellular space (87). Also, Okunieff et al. showed that although glucose supply may be adequate to small murine fibrosarcoma tumors (∼76 mm3), the metabolic baseline of large tumors (∼300 mm3) includes glucose deficit in addition to hypoxia. Although the impact of aglycemia, hypoxia, and acidification on cancer cell metabolic remodeling was analyzed in parcellar studies, their combined interaction has been insufficiently investigated. Different levels of hypoxia are observed in tumors, leading Vaupel and colleagues to propose the Janus face model, whereby metabolic adaptations are thought to occur when oxygen levels decrease below 1%, while more drastic hypoxia (below 0.1%) could trigger the generation of new genetic variants and resistance to apoptosis (273, 275). Another advantage of tumor cell-deviant metabolism is acidification of the extracellular medium, which inhibits the antitumoral immune response (79). In a previous study, we conducted a longitudinal bioenergetic analysis of oxygen and glucose deprivation in human breast carcinoma cells (HTB-126) and breast noncancer cells (HTB-125). We mimicked in vitro the microenvironmental conditions of tumor oxygen limitation (<1% O2; p O2 <7 mmHg or 1 kPa) and glucose deprivation [no glucose; replaced by galactose and glutamine (220, 228)]. We observed that the specific downregulation of OXPHOS triggered by hypoxia in cancer cells did no longer occur when glutamine was the only substrate for growth, that is, when ATP can only be produced by mitochondria. Hence, under conditions of increased mitochondrial energy demand, 1% O2 hypoxia cannot downregulate the OXPHOS system as occurs in the glucose medium. This observation suggests that cancer cells' metabolic apparatus ultimately comprises interactions and coordination between nutrient sensing and oxygen sensing (Fig. 10). In noncancer cells, glucose deprivation did not change the stimulatory effect of hypoxia, and OXPHOS remained intact after 6 days of growth in 1% O2. Of note, stimulation of OXPHOS by glucose deprivation and hypoxia was additive in cancer cells.
Recent studies in the field of immunology have revealed the existence of biochemical pathways involved in the switch of glycolytic to oxidative metabolism associated to the modulation of the immune response. The group of Martin Villalba has shown the role of the ERK5 kinase in the coordinated modulation of energy metabolism and the MHC-I markers of self-immunity. In culture conditions that force respiration in leukemia cells, these authors observed the upregulation of MHC-I transcription and protein levels at the cell surface, whereas opposite changes were observed in the same cells forced to perform fermentation as well as in leukemia cells lacking a functional mitochondrial respiratory chain. Forced respiration triggered the increased expression of the MAPK ERK5, which activated MHC-I gene promoters, and ERK5 accumulation in mitochondria. Such respiration-induced MHC-I upregulation was reversed upon short-hairpin RNA-mediated ERK5 downregulation and by inactive mutants of ERK5. Moreover, short-hairpin RNA for ERK5 leukemia cells did not tolerate forced respiration. This work revealed that the expression of ERK5 and MHC-I is linked to cell energy metabolism.
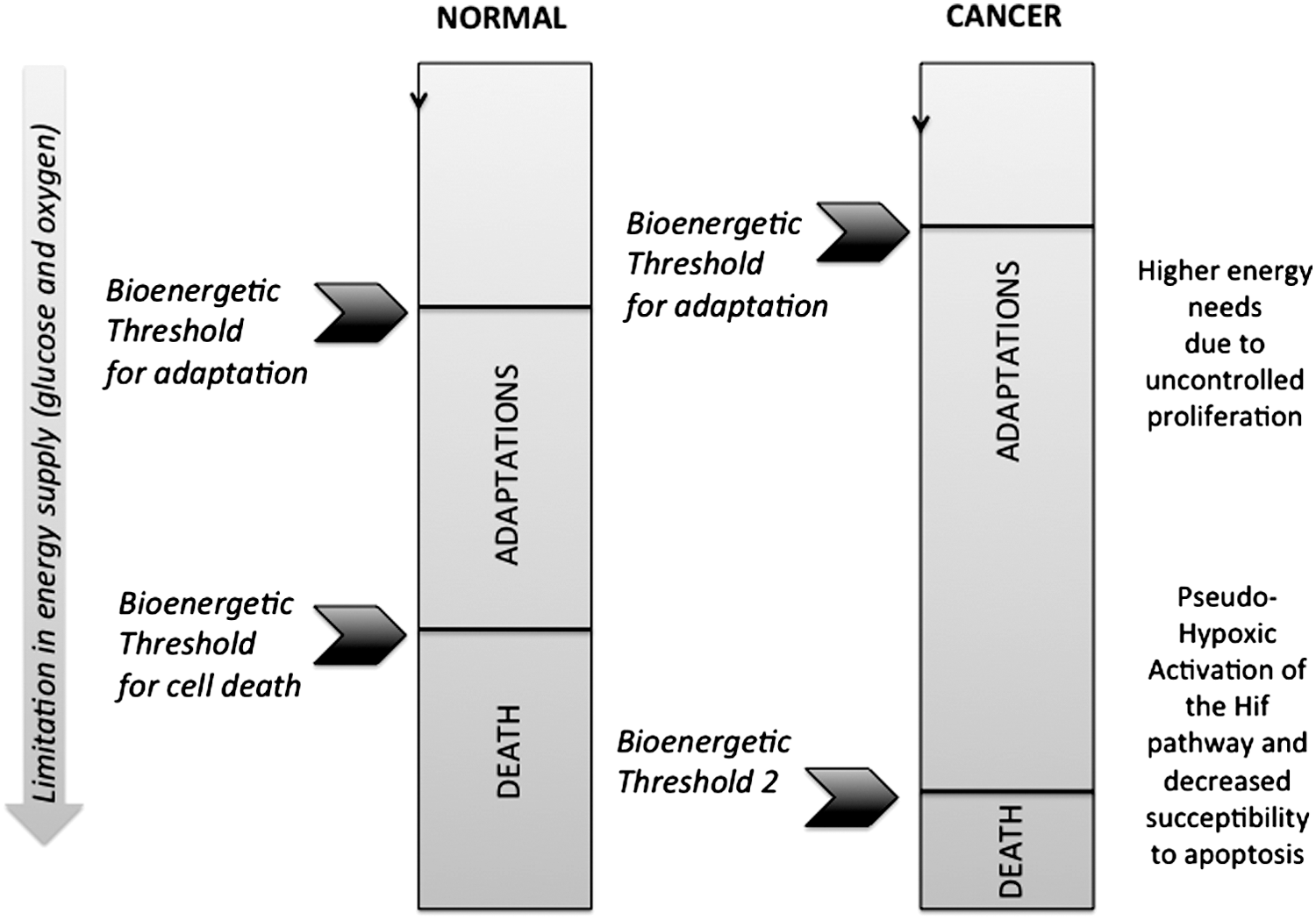
In most of the above-presented studies, mitochondrial adaptation occurred only in cancer cells forced to grow in absence of glucose, while the noncancer cells survived without adaptation. This specificity could be exploited as a therapeutic target, although the determinants remain unclear. We could speculate that the higher energy demand of the cancer cells confronted to their lower OXPHOS capacity (versus their normal counterpart cells; at least for the Warburg type of cancer cells) could trigger the need to upregulate OXPHOS to allow survival in absence of glucose. In our work (129, 228, 244), the noncancer cells possessed a higher contribution of OXPHOS to ATP synthesis, so that upregulation might not be needed in situations of glucose withdrawal.
Then, the laboratory of Glick and Ferrara showed that bone marrow cells proliferating after transplantation increased aerobic glycolysis, but not OXPHOS, whereas T cells proliferating in response to alloantigens during graft-versus-host disease increased both aerobic glycolysis and OXPHOS. Alloreactive T cells also exhibited a hyperpolarized mitochondrial membrane potential, increased superoxide production, and decreased amounts of antioxidants, whereas proliferating bone marrow cells did not. These findings challenged the current paradigm that activated T cells meet their increased demands for ATP through aerobic glycolysis, and identified the possibility that bioenergetic and redox characteristics could be selectively exploited as a therapeutic strategy for immune disorders. Lastly, the group of Ericka Pearce also demonstrated the importance of OXPHOS for the physiology of lymphocytes CD8 T cells, which have a crucial role in immunity. These authors showed that TNF receptor-associated factor 6 (TRAF6) regulates CD8 T(M)-cell development after infection by modulating fatty acid metabolism through OXPHOS. This bioenergetic phenomenon is crucial to maintain the memory function of the immune system.
VII. The Rewiring of Pre-Existing Metabolic Routes Accommodates Energy Needs, Anabolic Needs, and ROS Management in Cancer Cells: Role of Hif1α, ChREBP, PDK, and PKM2
Cancer cells are forced to continuously rewire alternative metabolic routes for their energetic and anabolic needs due to the fluctuation in substrate and oxygen levels in the growing tumor. Under conditions of glucose deprivation, mitochondrial activity is increased to sustain ATP production and lipid and protein synthesis. As discussed above, cancer cells can switch from aerobic glycolysis to OXPHOS under limiting glucose conditions. However, when glucose is available in large amounts, mitochondrial respiration is reduced by the Crabtree effect, and the F1F0-ATP synthase activity can even be reversed at the expense of glycolytic ATP. Although this particular functioning of the OXPHOS system (no longer OXPHOS) can deplete ATP and precipitate necrosis, it is limited by the mitochondrial protein inhibitor factor 1 (IF1), an endogenous F1F0-ATPase inhibitor (66). The high energetic needs of tumor cells are often correlated with increasing expression and inhibitory efficacy of IF1. Moreover, IF1 appears to have a role in defining the conformation of the F1F0-ATP synthase and mitochondrial crista structure and function (34, 37). The density of the cristae in IF1 overexpressing HeLa cells delayed the release of cytochrome c from intracristal space and increased survival after staurosporine treatment (257). In this regard, recent experimental evidence has shed some light on a critical impact of mitochondrial morphology on the control of important mitochondrial functions, including apoptosis and OXPHOS (19, 42). In particular, deregulated mitochondrial fusion and fission can now be regarded as important factors in cancer onset and progression, as discussed above. Several studies showed elevated levels of (Δ
The rewiring of energy metabolism in cancer cells was recently shown to modulate the balance between anabolism and catabolism. For instance, it is proposed that pyruvate synthesis occurs primarily by the isoform 2 of pyruvate kinase (PKM2) in cancer cells (180). The tetrameric form of PKM2 channels pyruvate to lactate, and the formation of PKM2 tetramers is stimulated by the glycolytic intermediate F-1,6-BP. The dimeric form of PKM2 retards pyruvate formation and allows the accumulation of upstream glycolytic intermediates, promoting their distribution into the biosynthetic pathways. This regulation of enzyme activity may constitute a molecular switch that diverts glucose metabolites from energy production to anabolic processes in the pentose phosphate pathway (PPP) and the generation of NADPH. Such a switch between glycolysis and PPP was analyzed by Bensaad et al. in the context of cancer and the role of TIGAR in ROS homeostasy (24). Likewise, the work by Vaughn and Deshmukh (272) revealed the importance of glucose utilization through the PPP for neurons and cancer cells to control cytochrome c redox state and thereby modulate apoptosis. In addition, Herrero-Mendez et al. (114) described the role for E3 ubiquitin ligase APC/C-Cdh1 in promoting the degradation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, isoform 3 (PFKFB3), which in turns allows glucose to be consumed through the PPP to avoid oxidative stress (while this work was performed in neurons, it could describe an universal mechanism that regulates this glycolytic/PPP shift). Besides, Smolkova and colleagues reviewed the fate of glutamine in cancer metabolism and evoke two principal pathways of glutamine utilization in cancer cells, one utilizing OXPHOS and another one operating more in hypoxic conditions. The first pathway, dependent on NADPH and independent of respiration, employs only two enzymes of the Krebs cycle that act in a reverse mode and convert αKG to citrate by reductive carboxylation. However, such reductive glutaminolysis cannot support proliferation without ATP produced by glycolysis. Either it can act transiently until the glycolytic ATP pool is consumed, providing anoxic/hypoxic tumors to survive during a critical period until glucose utilization is restored, or it must act in parallel with the oxidative mode. In the reductive carboxylation glutaminolysis, 2-oxoglutarate and NADPH are consumed by isocitrate dehydrogenase isoform 2 (IDH2), followed by the reverse aconitase reaction and citrate efflux (187). This pathway feeds fatty acid synthesis, but does not produce ATP by substrate phosphorylation (via Krebs) or by NADH oxidation (via OXPHOS). Therefore, glycolysis is required to produce ATP in the reductive mode of glutaminolysis (187).
The oxidative mode utilizes the forward-running Krebs cycle truncated after citrate synthase at citrate extrusion. In both modes, citrate is extruded from mitochondria and converted to oxaloacetate and acetyl-CoA by ACL. Acetyl-CoA is then used to produce fatty acids by fatty acid synthase (FASN) and cholesterol for general lipid synthesis. OXPHOS glutaminolysis can compensate for deficiencies in cellular ATP. Both glutaminolytic modes provide pyruvate, lactate, and the NADPH pool, normally supplied by glucose metabolism via the PPP. Increasing anticancer therapeutic approaches target metabolism to selectively kill tumor cells. Glucose catabolism is the pathway the most targeted in this end, but the PPP, TCA cycle, and glutaminolysis are increasingly the focus of research, as the cancer cells are not equally sensitive to agents interfering with glucose metabolism. Lastly, a recent player to consider in the field of energy homeostasy is the carbohydrate-response element-binding protein (ChREBP), the deletion of which alters substrate utilization and produces an energy-deficient liver (31). The KO of this transcription factor in mice induced an increase in PDH activity. This was explained by a diminished PDK activity. In the liver of these mice, the greater pyruvate dehydrogenase complex activity caused a stimulation of lactate and pyruvate oxidation, and it significantly impaired fatty acid oxidation. It remains to be shown whether ChREBP could participate to the maintenance of energy homeostasy in cancer cells, and whether it could play a role during the shift between glucose and galactose media where the expression levels of PDH subunits are strongly increased.
VIII. Cellular and Mitochondrial Adaptations to the Type of Available Energy Substrate: Ras and CC3 Modulate Metabolic Flexibility in Cancer Cells
The impact of various oncogenic mutations on energy metabolism discussed above in section VII could seem paradoxical, as they can disable or, at the opposite, improve the metabolic flexibility, that is, the capacity to derive energy from multiple sources. Yet, nutrient addiction is the Achilles' heel of numerous tumor cells and is often proposed as a rationale for metabolic therapy. Alterations in phosphoinositide 3-kinase (PI3K)/serine–threonine kinase AKT/mammalian target of rapamycin (mTOR) signaling are perhaps the most frequent events observed in solid tumors. Constitutive activation of the oncogenic kinase AKT addicts glioblastoma cells to glucose by interfering with the induction of fatty acid oxidation when glucose is withdrawn (33). Another well-known example of oncogene that causes nutrient addiction is c-Myc, which is frequently deregulated in tumors. Induction of c-Myc expression in MEFs results in the induction of glutamine transporters, glutaminase, and lactate dehydrogenase A (LDH A)(286).
Some transformating mutations can bypass the addiction of cancer cells to glucose (i.e., metabolic rigidity). It has been shown that KRAS or BRAF mutations increase glucose uptake and glycolysis, and at the same time, permit cells to survive in low-glucose conditions. However, the mechanisms that support survival in low-glucose conditions remain controversial, as conflicting results were published recently. For instance, the activation of RAS was shown to confer survival in low glucose via the enhancement of glucose uptake by GLUT1 and the enhancement of glycolysis along with the alteration of respiratory chain activity (10, 59, 294). Conversely, the activation of signal transducer and activator of transcription 3 (STAT3) was shown to stimulate respiratory chain activity in another study (97), while more recently, Ras activation was associated with mitophagy and the reduction of OXPHOS capacity (138). This important issue is further discussed below.
First, Ras oncogenic mutations were associated with mitochondrial protein downregulation mediated by STAT3. The study of Demaria and colleagues (59) evidenced a reduced mitochondrial activity in MEFs with constitutively active STAT3 (Stat3C/C MEFs). The significant downregulation of nuclear-encoded genes involved in mitochondrial function observed in these cells together with their decreased PDH activity led to the reduction of mitochondrial respiration. Mitochondrial ATP production and basal respiratory chain activity were also reduced in these cells, as well as the protein levels of representative components of the ETC, particularly those belonging to complexes IV and V. Lastly, mitochondrial morphology and total area were normal in these cells, suggesting a specific degradation of COX and CV components. Accordingly, the recent study by Kim and colleagues (138) also reported that K-Ras mediates functional loss of mitochondria during cell transformation to overcome an energy deficit resulting from glucose deficiency. In this study, the decrease of respiration was explained by the increased formation of acidic vesicles enclosing mitochondria, during which autophagy-related proteins such as Beclin 1, Atg5, LC3-II, and vacuolar ATPases were induced. The blockade of autophagy recovered respiratory protein expression and respiratory activity. However, in this work, K-Ras (V12) did not induce the expression of GLUT1, the low-Km glucose transporter. Lastly, the interaction between Ras, STAT3, and the mitochondrion was analyzed in immortalized STAT3-deficient mouse embryo fibroblasts (97). In this work, STAT3 was detected in the mitochondria of primary liver tissue, nontransformed MCF10A mammary epithelial cells, and T24 bladder carcinoma cells. In contrast with the two above-cited studies, mitochondrial STAT3 augmented here the ETC activity. Mitochondrial STAT3 also contributed to Ras-dependent cellular transformation by augmenting ETC activity, particularly that of complexes II and V, accompanied somewhat paradoxically by shifted energy production to favor fermentation. In a fourth study, the link between STAT3 and mitochondria was further investigated (282) in noncancer tissues such as the liver and heart. In striking contrast with the study of Demaria et al. where constitutively active STAT3 triggered a reduction of mitochondrial activity in MEFs, the absence of STAT3 in the heart and liver was associated with a significant decrease of complex I and II activities. Moreover, in the study of Wegrzyn et al., state 3 respiration was reduced by 70% in STAT3−/− cells with glutamate as a substrate for complex I and by 50% in cells with succinate as a substrate for complex II. The authors concluded that the most likely mechanism by which STAT3 exerts its actions is not as a transcription factor that regulates nuclear gene expression, but rather through its localization in the mitochondrion. The effects of STAT3 potentially represent a general mechanism by which this protein can influence cell metabolism and tumorigenesis. Already in 1999, Onhami and coworkers showed that the blockade of K-Ras with antisense in pancreatic cancer cells modified (increased) the expression pattern of 11 mitochondrial genes (197). Those included the mitochondrial 16s rRNA gene, cytochrome oxidase subunit 1, NADH dehydrogenase subunit 4, and ATP synthase. Lastly, the group of Weinberg investigated the impact of the K-Ras mutation on mitochondria and proposed that this organelle serves mainly for the production of ROS, which are essential for anchorage-independent growth (283). In this study, human colon cancer cells were able to consume glutamine through respiration, but the contribution of mitochondria to cellular ATP production was not assessed. Interestingly, this study revealed that mice harboring a knockout of TFAM with an activated form of K-Ras had smaller tumors and displayed fewer Ki67-positive cells. Therefore, strategies aiming at the reduction of mitochondrial biogenesis should be tested for their capacity to reduce tumor growth.
In contrast to Ras, CC3/TIP30 is a tumor suppressor with reduced or absent expression in a variety of aggressive tumors. Yet, CC3 could also be involved in the control of metabolic flexibility as Chen and Shtivelman (46) demonstrated that the silencing of CC3 in HeLa cells strongly improves survival and allows superior metabolic adaptation in response to glucose limitation. These authors showed that silenced CC3 increases mitochondrial respiration and expression of mitochondrial proteins of the ETC along while maintaining high levels of c-Myc expression and downstream GLUT1 and PKM2. Moreover, loss of CC3 represses AMPK activation by low glucose. Lastly, metabolic flexibility might also include the capacity of deriving energy from the amino acids obtained by autophagy. In transformed fibroblasts, the Simian Virus 40 Small T (Small T)-carrying cells are more resistant to glucose deprivation-induced cell death than cells lacking ST. ST antigen protects cancer cells from glucose deprivation by maintaining energy homeostasy with activation of AMPK, inhibition of mTOR, and induction of autophagy as an alternate energy source (153).
The above-described changes (sections VI and VII) in mitochondrial energy metabolism raise the question of mitochondrial integrity in cancer cells: Is mitochondria functional or dysfunctional in tumors?
In the 1920s, Otto Warburg evidenced that tumor cells consume large amounts of glucose and convert it mostly to lactic acid despite the presence of oxygen. This led him to hypothesize that cancer arises from the impairment of mitochondrial oxidative metabolism. The aerobic glycolysis of the tumor cell is derived in any case from a disturbance of the respiration. This famous Warburg hypothesis seduced the medical community in that a single metabolic specificity of cancer cells, the high dependency on glycolysis for energy production, could eventually be targeted to cure the disease. Over 90 years of cancer biology research has been conducted with the basis of this predominant Warburg theory. The exclusive respiratory impairment hypothesis is now challenged by an increasing number of bioenergetic studies that already started in the 1950s, when Sidney Weinhouse published experimental evidence in opposition to the Warburg hypothesis. The major argument raised by Weinhouse was that tumors and non-neoplastic tissues show no difference in their ability to convert glucose and fatty acids to carbon dioxide, a process that requires respiration and functional mitochondria. Likewise, Gregg showed that cell respiration increased concomitantly with the increase in glucose consumption in normal and transformed cells, and that HeLa cells presented with a high respiratory rate. It is noteworthy that Warburg himself recognized that respiration was not totally impaired and could even be efficient in a subset of tumors, the frequency of which still remains undetermined. As a rule, the respiration of the tumor cell is small, but in recent years, tumor cells with a large respiration have also been found. Yet, this finding of Warburg remained underestimated so that an accepted consensus on a general mitochondrial impairment in cancer cells had emerged in the 90's and remained highly cited since then. Nevertheless, in the last decade, several groups reported accumulating evidence of functional mitochondria in tumors and demonstrations of a strong dependency of cancer cells survival on mitochondrial oxidative metabolism. Recently, an increase of mitochondrial capacity was even reported in breast cancer (240). This situation was found for other types of cancer as an increased mtDNA content was reported both in chronic lymphocytic leukemias (41) and in lung cancer (121). These findings do not permit to conclude unequivocally on the status of mitochondrial content in cancer and point toward a variability of mitochondrial content and capacity in tumors, rather than a simple general repression. The field of mitochondrial cancer research still lacks of large-scale studies on different types of tumors that could allow to estimate the variability of mitochondrial content and function as well as solicitation in cancer tissues. Alternatively, prospective cohort studies have tried to assess the possible role of mtDNA copy number in blood cells as a risk factor for cancer. The results were extremely variable and suggested again a large disparity between cancer types and even within one type of tumor between different individuals (121, 155, 162, 173).
In addition to biogenesis reduction, the low efficiency of the OXPHOS system observed in the Warburg type of cancer cells could be explained by the alteration of respiratory chain complexes specific activity, possibly caused by cancer-specific protein regulations such as post-translational modifications or ROS-induced alterations. Yet little is known on this particular aspect of OXPHOS dysregulation, and only one study reported a functional alteration of the respiratory chain complex I in cancer (23). Furthermore, abnormalities in the overall morphology of the mitochondrial network and its internal organization have been reported in numerous cancers, including human astrocytomas (27), carcinomas (41), Warthin's tumor (42), xenografted gliomas (39), malignant glioma cells (43), and HeLa cells (44). Most of these studies reported heterogeneous ultrastructural abnormalities, such as organellar swelling with a disorganization and distortion of the cristae and partial or even total cristolysis. Those might underline an activity defect of the OXPHOS system, as such ultrastructural alterations of the mitochondrion are commonly found in mitochondrial diseases. Abnormalities in mitochondrial fusion–fission were also proposed from the analysis of astrocytomal sections by electron microscopy (27). Such structural defects in the mitochondria are frequently seen in cancer cells, although their precise origin and participation in tumor progression are not well understood. An analysis of mitochondrial morphogenesis in cancer cells could help to clarify this question.
Thus, it is clear that mitochondrial dysfunctions exist in some tumors, but this feature is not a hallmark of cancer, since numerous studies have demonstrated that tumor mitochondria can be functional with regard to respiration and ATP synthesis, exhibiting normal respiratory control ratios and normal capabilities for the oxidation of respiratory substrates (71). As soon as in 1976, in a critical analysis of Warburg's theory, Sidney Weinhouse concluded ‘‘no substantial evidence has been found that would indicate a respiratory defect, either in the machinery of electron transport, or in the coupling of respiration with ATP formation, or in the unique presence or absence of mitochondrial enzymes or cofactors involved in electron transport.” In accordance with this, a comprehensive study of mitochondrial function in slowly growing hepatomas showed almost no differences in respiratory parameters as compared to the normal liver mitochondria (205). In some tumors, oxygen consumption was found to be similar, or even higher than in the normal tissue (205). In 1979, Reitzer demonstrated that cancer cells can derive 96% of their ATP from OXPHOS using glutamine as an energy substrate (220). In a study performed on human lung tumors [nonsmall-cell lung carcinoma (NSCLC)], it was found that 70% of the NSCLC presented with a decreased expression of the pyruvate dehydrogenase complex, but 30% of the tumors revealed a higher expression as compared to noncancer tissue (151). They could also explain the difficulty to detect some tumors using PET scan, as OXPHOS tumors would derive energy from glutamine and other oxidative substrate oxidation rather than from glucose. More recently, this distinction was found while comparing the metabolic profile of SiHa human cervix squamous carcinoma cells with WiDr human colorectal adenocarcinoma cells. The first one is of the OXPHOS type and can oxidize lactate, whereas the second one is more glycolytic and generate lactate (151). Other studies have indeed shown that tumor-derived cell lines can use glycolysis or OXPHOS to a variable extent, so that some cancer cells have been classified as glycolytic and other as OXPHOS depending on their utilization of these two systems of energy production. One recent study (283) indicates that a reduction of mitochondrial content in precancer tissues reduces the tumorigenic potential of K-Ras mutations in mice. This strongly suggests that mitochondrial metabolism is required for tumorigenesis, in contrast with the Warburg hypothesis.
IX. Compensation Without Adaptation: Safety Role of the Biochemical Threshold Effect and of Metabolic Attenuation
In most human tissues, mitochondria provide the energy necessary for cell growth and biological activities through OXPHOS. For mammals, the respiratory chain consists of four enzyme complexes (complexes I–IV) and two intermediary substrates (coenzyme Q and cytochrome c) (see the Introduction section for more details). Activity defects in these complexes have been reported in a large variety of pathological situations (281), which generally lead to cellular energy deprivation, redox imbalance, and the excessive production of ROS. However, their repercussion on the fluxes of oxygen consumption or ATP synthesis is highly variable, making the analysis of mitochondrial diseases complicated. One explanation for this observation is the existence of compensatory mechanisms operating at the level of the respiratory chain (227). This compensation can be explained by the interplay of two different fundamental mechanisms: mobilization and attenuation. The former concerns the enzyme complexes themselves and involves the utilization of additional capacity by either molecular recruitment [as demonstrated for the ANT (77)], or direct activation [as demonstrated for the glycogen synthase (232)] to accommodate an increase in flux. The latter concerns each substrate and constitutes a compensatory mechanism inherent to network systems, an original derivation from MCA (113, 132). Sometimes called MNA, it takes into consideration the inhibition of each immediate step within a metabolic pathway working at a steady state that can be reduced at the level of the flux, via compensatory changes in intermediate metabolites' concentrations (Fig. 11). However, the sole kinetic parameters of the respiratory chain do not suffice to determine threshold values; there is also a dependency on additional elements (159). This is the limit of MCA: it cannot predict the extent of maximal respiratory chain complex inhibition above which the global flux can drop, that is, the biochemical threshold effect. Variations in the concentration of intermediate substrates could play an important role in these compensatory mechanisms and in the adaptation of metabolic systems to a variety of situations ranging from the physiological to the pathological. Accordingly, we have previously shown that the magnitude of the biochemical threshold effect is strongly dependent on the concentration of intermediate substrates (reduced form) (159, 179), and the measurement of the cytochrome c redox state in living human cells placed under different metabolic conditions confirmed that variations in intermediate substrate concentration can occur in vivo, in response to perturbations of mitochondrial respiration (119). More recently, we found that the magnitude of metabolic attenuation depends on the concentration of intermediate substrate. By performing cytochrome spectra on mitochondria placed in different energy states, we were able to identify three forms of substrate: the utilized, the mobilizable, and the nonmobilizable. The first corresponds to the substrate engaged during mitochondrial respiration; the second is reduced in case of a perturbation to maintain the energy fluxes at normal values; and the third class is unable to participate in respiration. One should also consider the importance of the redox state of cytochrome c for the regulation of apoptosis. Vaughn and Deshmukh (272) showed that the proapoptotic activity of cytochrome c is influenced by its redox state, and that increases in ROS after an apoptotic insult lead to the oxidation and activation of cytochrome c. Interestingly, the concentration of mobilizable substrates directly correlates with the biochemical threshold value (Fig. 12).
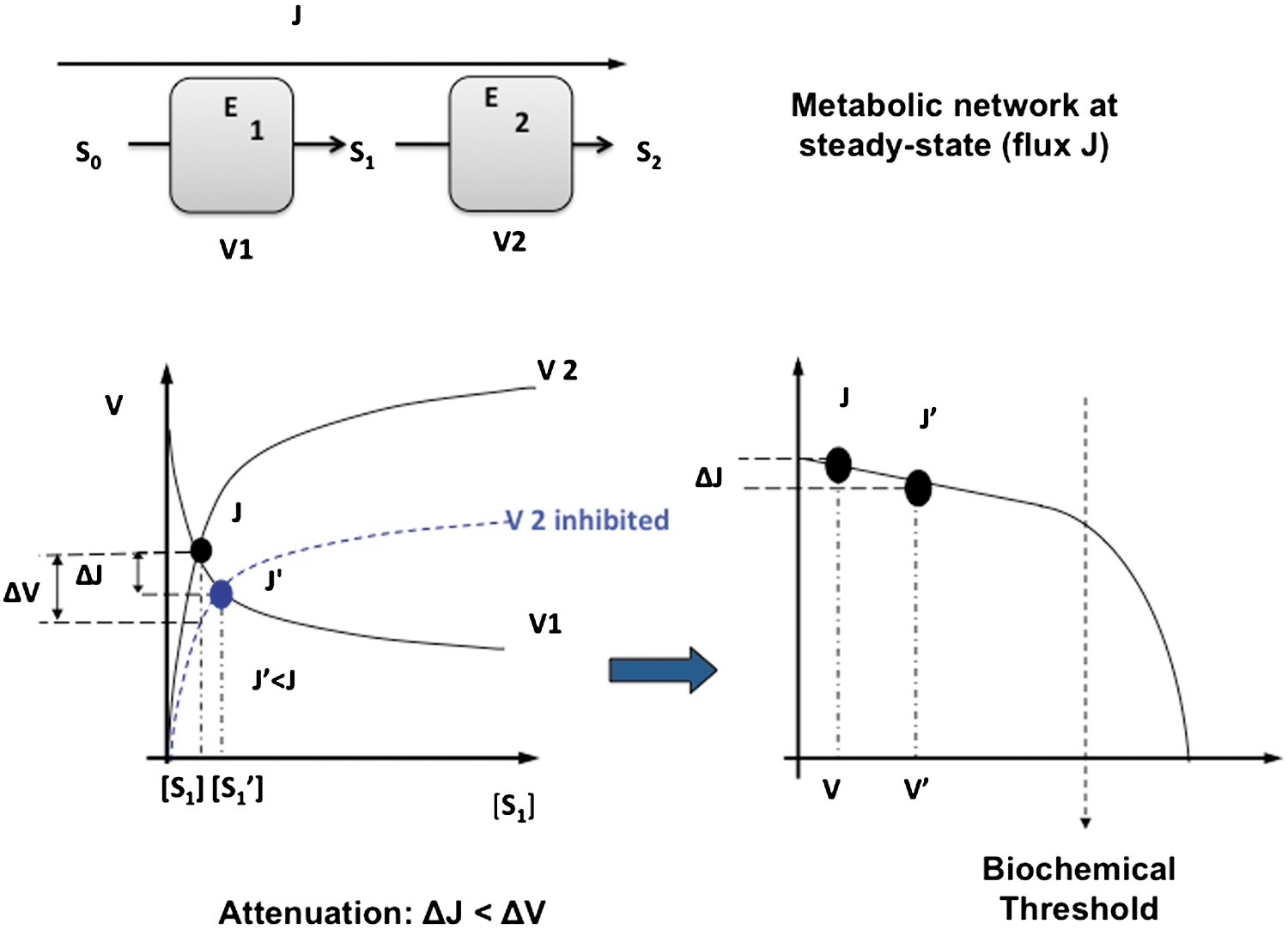
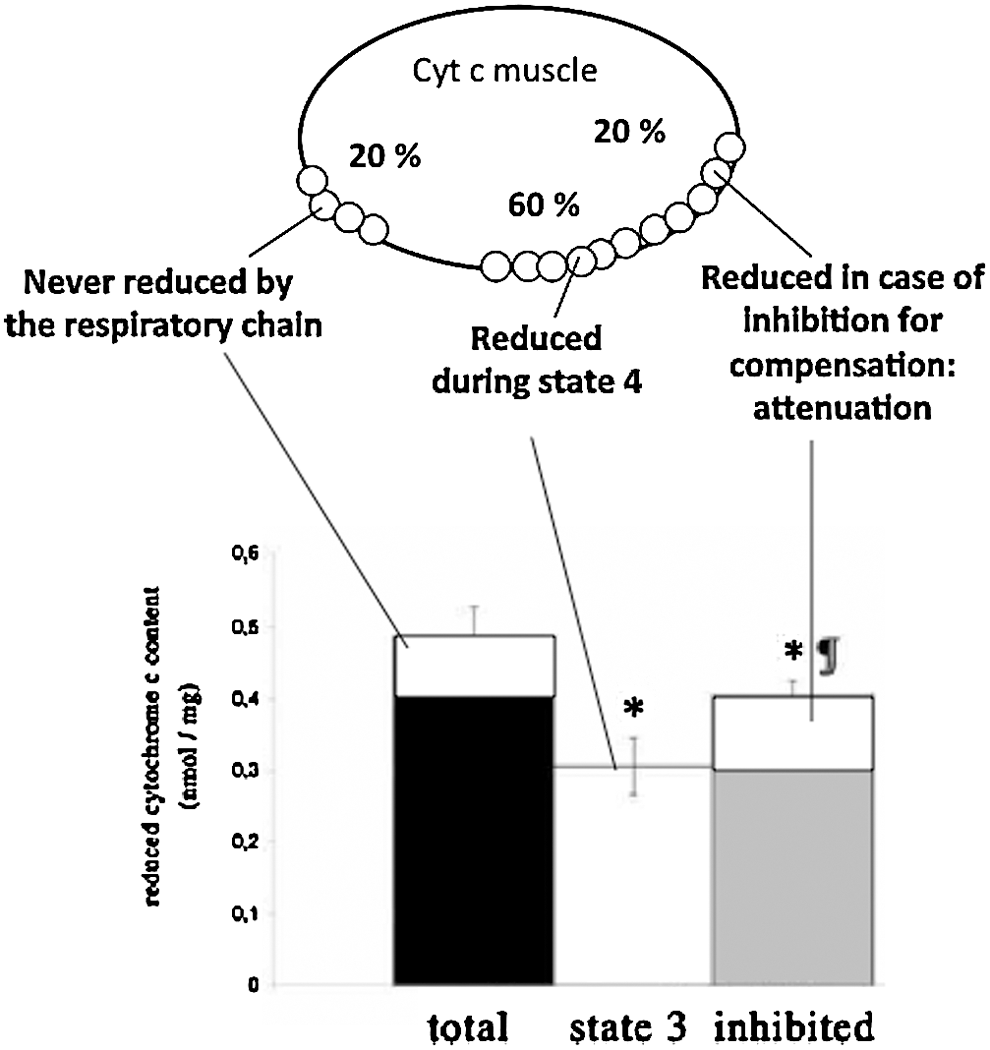
Besides attenuation, the respiratory chain is highly regulated so that energy needs can improve OXPHOS, mainly via post-translational modification of its protein components. For instance, respiratory chain kinetic parameters can be directly modulated by the expression of rapid or slow isoforms of complex IV, the biochemical modulation of complex IV catalytic activity by the electric membrane potential ΔΨ(52, 210), and numerous regulations of all electron transfer chain (ETC) complexes by post-translational modifications, including nitrosylation or phosphorylation. Lastly, the activation of four mitochondrial dehydrogenases (FAD-glycerol 3-phosphate dehydrogenase, pyruvate dehydrogenase phosphatase, NAD-IDH, and oxoglutarate dehydrogenase) by calcium ions is also involved in stimulating OXPHOS in mammalian cells by increasing the amount of NADH supplied to the respiratory chain. Calcium also controls NADH delivery to the respiratory chain as recently shown by the group of F. Gellerich (91). According to this author, a rise of cytosolic Ca2+ concentration causes an up to 5-fold enhancement of OXPHOS rates that is due to an increased substrate supply, while a rise in mitochondrial Ca2+ regulates the oxidation rates of substrates that are present within the mitochondrial matrix (91).
X. Mitochondrial Adaptative Loops in Response to Respiratory Chain Impairment: The AMPK-SIRT-PGC1-FOXO3 Pathway and the CREB Pathway
In mammalian cells, energy homeostasy requires a tight coordination between cell activity, nutrient availability, and the regulation of energy transformation processes. In particular, the oxidative capacity of skeletal muscle fibers is adjusted by changes in the mitochondrial content (163), and a large body of data suggests that in tissues with variable metabolic activity, or during development, mitochondrial biogenesis is tuned to accommodate the energy demand (17, 248). The analysis of mitochondrial three-dimensional structure in living human cells also revealed the importance of mitochondrial fusion and fission, since defective fusion and fission were both associated with respiratory impairment [for review see (15)]. Conversely, little is known on the eventual capacity of mitochondrial fusion to compensate for respiratory chain impairment. Typically, mitochondrial respiration is maintained by the balancing of respiratory chain metabolite concentrations, a phenomenon referred to as metabolic attenuation described just above in section IX (16, 227). Furthermore, in human cells forced to survive under sustained challenging conditions of limited energy supply, energy homeostasy relies on the synthesis of OXPHOS proteins and eventually, the reshaping of the mitochondrial network (228). Accordingly, the observation of electron micrographs obtained from the skeletal muscle of patients with a respiratory chain disorder often reveals an increased mitochondrial content, suggestive of an adaptative compensatory response (55, 98, 105, 127, 166, 186, 243, 264, 287). Similar observations were made in transgenic mouse models of OXPHOS diseases, indicating a possible adjustment of mitochondrial biogenesis to respiratory chain deficiency (287). In these studies, the status of mitochondrial fusion and fission was not investigated, so that it remains unknown whether these processes could contribute to the compensation of respiratory chain defects. Arnould T. and colleagues analyzed extensively the mitochondria to the nucleus communication in cells grown in presence of the respiratory chain complex III inhibitor antimycin A (270), or in conditions of mitochondrial protein synthesis suppression (270), and of mitochondrial DNA depletion (182). All revealed the activation of the CREB consecutive to mitochondrial bioenergetic stress. Cyclic AMP produced inside the mitochondrion also regulates OXPHOS via protein kinase A activation and subsequent COX phosphorylation (1). The group of Semenza further showed that a physiological activator of CREB, the transducer of regulated CREB-binding proteins (TORC1), was able to induce the expression of the peroxisome-proliferator-activated receptor γ-coactivator-1α (PGC1α) and of mitochondrial biogenesis (288). Indeed, PGC1α is the main effector of mitochondrial biogenesis in human skeletal muscle, generally leading to an augmented OXPHOS capacity in response to increased energy demand (39, 120, 154). Yet, the participation of CREB to the mitochondrial adaptation to pathogenic OXPHOS defects remains unknown, and the mechanisms linking the activation of CREB to the downstream induction of mitochondrial biogenesis are also unclear in pathology. The groups of Avadhani and of Arnould revealed that CREB can be activated by calcium in C2C12 myoblasts treated with ethidium bromide or CCCP (25), in mtDNA-depleted lung carcinoma A549 cells (5), in osteosarcoma 143B cells (8), and in mouse immortalized fibroblast L929 cells (182). Arnould and colleagues showed that calcium activates Calcium-/calmodulin-dependent protein kinase type IV (CaMKIV), which in turn activates CREB by protein phosphorylation, leading to mitochondrial biogenesis (8). Recently, we analyzed the adaptative capacity of mitochondrial content and dynamics in muscle biopsies of patients with a complex IV defect, and in skin fibroblasts challenged with complex IV inhibition (21). We observed a biphasic variation of the mitochondrial content upon complex IV inhibition in muscle biopsies and in skin fibroblasts. Adjustment of mitochondrial content for respiratory maintenance was blocked by using a dominant negative form of CREB (CREB-M1) and by L-NAME, a blocker of NO production. Accordingly, cells treated with KCN 6 μM showed higher levels of phospho-CREB, PGC1α mRNA, eNOS mRNA, and mtTFA mRNA. We also observed the increased expression of the fission protein dynamin-related Protein1 (DRP1) during fibroblast adaptation as well as mitochondrial ultrastructural defects indicative of increased fission in patients' muscle micrographs. Accordingly, the expression of a dominant negative form of DRP1 (K38A mutant) reduced the biogenic response in fibroblasts challenged with 6 μM potassium cyanide (KCN). These findings indicate that mitochondrial biogenesis and mitochondrial fission cooperate to promote cellular adaptation to respiratory chain inhibition. The evidenced pathway of mitochondrial adaptation to respiratory chain deficiency provides a safety mechanism against mitochondrial dysfunction.
In addition to Ca2+, other secondary messengers such as ROS, NO, ATP, and NAD+/NADH and proteins such as MIDAS have also been associated with this mitochondrial retrograde response, as recently review by Michel and colleagues (183). The role of ROS in mitochondrial adaptation is discussed below in section XII.
XI. Targeted Mitochondrial Protein Degradation: A Major Component of Mitoplasticity
Evidence indicates that the mitochondrial cellular content can be adjusted to adapt the capacity of energy supply to the physiological demand. Modulation of mitochondrial turnover can be considered as a primary adaptative mechanism of the organelle. The amount of mitochondria at steady state results from the balance between two processes: biogenesis and degradation of the organelle. Mitochondrial biogenesis has been actively investigated in the past decades as extensively reviewed by Hock and Kralli (118) and discussed in sections VI and X. On the other hand, the link between mitochondrial degradation and bioenergetics remains poorly understood, and we summarized here the recent developments in this field. Mitochondrial degradation can occur by two types of mechanisms: (i) degradation of single mitochondrial proteins, or (ii) elimination of whole-mitochondrial fragments. We focused our attention on the connection between these two processes and energy metabolism.
Mitochondrial energy is produced within the inner membrane of the organelle and requires a carefully assembled OXPHOS system. To maintain the efficiency of this complex machinery, a quality control system exists to selectively degrade nonassembled and misfolded OXPHOS complex proteins. The key actors of this system are two AAA–protease complexes, and a third membrane complex formed by several prohibitins. The AAA proteases form a conserved class of ATP-dependent proteases embedded in the MIM, facing the matrix (mAAA–protease) and the inter membrane space (iAAA–protease) (157). In human, the mAAA–protease complex is formed by hetero-oligomeric association of AFG3L2 and Paraplegin. The iAAA–protease consists of an oligomer of ATP-dependent metalloprotease YME1 (YMEL1) (229). The Prohibitin complex is thought to have a chaperone activity. The high-molecular-weight Prohibitin complex is located to the MIM and interacts with the mAAA–protease (250). In the context of mitochondrial bioenergetics, this quality control represents a crucial step, as this system is involved in the elimination of misfolded and dysfunctional mitochondrial proteins (subunits 2 and 3 of cytochrome c oxidase). The quality control of complex I and III and ATP synthase assembly also depends on mAAA and/or prohibitin. Recently, the processing of the dynamin-related GTPase optic atrophy 1 protein (OPA1) has been added to the list of proteins under the quality control of mAAA–proteases and prohibitin complexes (76). The impairment of all above-cited proteases leads to defects in both ATP production and ETC enzymatic activity (9, 188). In human, the loss of mAAA–protease activity induces neurodegenerative diseases. For instance, mutations in the SPG7 gene, which encodes paraplegin, are responsible for an autosomal recessive form of Hereditary Spastic Paraplegia (HSP). Mutation in AFG3L2 leads to dominant hereditary ataxia SCA28, and this disease is associated with complex IV defects and impairment of the respiratory chain (60). Both pathologies show deficits in mitochondrial bioenergetics.
Mitochondrial outer membrane proteins can be eliminated through the ubiquitin proteasome system (UPS) present in the cytosol. In this regard, ubiquination and mitochondrial elimination may also be linked to global degradation of mitochondria through mitophagy (see below). Single protein degradation through UPS involves polyubiquitination of target proteins (115). Polyubiquitination requires the activity of an E3 ligase that recognizes the protein to be eliminated. Recently, several mitochondrial E3 ligases have been identified that participate in the ubiquitination of the machinery involved in mitochondrial morphology (135, 258) and mitochondrially driven apoptosis (18, 124, 259). Interestingly, the mitochondrial transcription factor TFAM undergoes ubiquitinylation in porcine sperma, pointing to the possible relevance of this system for the regulation of mitochondrial energy metabolism (7). Recently, Parkin has been the focus of intense research efforts. This cytosolic E3 ligase can be recruited to the mitochondria by changes in the mitochondrial membrane potential (189, 190). To date, this is the only example directly connecting a bioenergetic parameter with ubiquitination. Unfortunately, it is not known how other key parameters indicative of mitochondrial bioenergetics such as ATP production, respiration rate, or ROS production may affect ubiquitination. Parkin recruitment is mediated by phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1). A recent work by Liu W. and colleagues pointed out that PINK KO flies display reduced ETC activity as well as decreased ATP production. The authors proposed that the importance of the PINK1/Parkin pathway in the regulation of OXPHOS could be linked to the control of mitochondrial dynamics, as bioenergetic deficiency was restored by the expression of DRP1 (164). In mitochondria, Parkin is involved in the ubiquitination of Mitochondrial Assembly Regulatory Factor, Mitofusins 1 and 2, DRP1, and VDAC1, and ubiquitination seems not to be directly involved in the regulation of energy proteins. The exact function of the PINK1/Parkin pathway is still the matter of some debate, as the number of known cytosolic and mitochondrial targets of Parkin does not conform with the dogma of E3 ligases: high selectivity for their substrate. In a recent article, the group of Larsson argued against the exclusive implication of Parkin in the recruitment of damaged mitochondria in neurons of a Parkinson mouse model, as clearance of damaged mitochondria remained unaffected in the absence of Parkin (251). Another study indicated that Parkin could play an ubiquitin ligase-independent function in the transcriptional control of p53 (54). At the boundary between cancer biology and neurodegenerative research, the work of Zhang et al. identified Parkin as a p53 target gene, which is an important mediator of p53 function in regulating energy metabolism (296). In this study, Parkin deficiency activated glycolysis and reduced mitochondrial respiration, leading to the Warburg effect.
After ubiquitination, the tagged proteins are removed from mitochondria. Karbowski and colleagues have recently proposed that this mechanism requires an outer mitochondrial membrane (OMM) chaperone such as the AAA-ATPase, namely p97 (290). Their findings showed the implication of this ubiquitin system in removing OMM proteins, but little is known regarding the capacity of the UPS system to remove inner membrane proteins. Also, although ATP is crucial for both AAA-ATPase and ubiquitination mechanisms, the connection between these two mechanisms and mitochondrial ATP production has yet to be thoroughly analyzed. Interestingly, the proteins involved in mitochondrial energetic metabolism have never been found to be ubiquitinylated. Almost all cellular important processes display regulation by ubiquitination, but mitochondrial energetic metabolism seems to be an exception. This gap suggests that inner membrane mitochondrial protein turnover and quality control could be compensated for by proteases such as LON, AAA-ATPase, or a global system of degradation such as mitophagy.
Mitochondria can be eliminated by autophagy or by a selective form of autophagy named mitophagy. It involves the sequestering of mitochondria in a newly formed autophagosomal membrane to deliver and degrade the whole mitochondria through fusion with lysosomes. This process is regulated by a growing number of signaling pathways that include the mitochondrial BH3-only proteins (Bad, Bid, Bim, Bik, Nix, and Noxa), the Beclin-1/Bcl-2-Bcl-xL complex, mTOR, AMPK, and HIF-1. These different pathways can activate or suppress mitophagy independently or in concert, according to diverse, but specific, stimuli in response to a need of quality control, a challenged energy status or nutriment deprivation (11, 181). Regulation of mitophagy is therefore highly complex and must be finely balanced, as cell survival or death often depends on the time and degree of activation or suppression of mitophagy. In this chapter, we will discuss how mitophagy interacts with bioenergetics.
Autophagy machinery and membrane dynamics during mitophagy are highly conserved from yeast to mammals. Ubiquitin (Ub)-like protein conjugation systems, including the well-characterized microtubule-associated Protein 1 Light-Chain 3 (Atg8/LC3) and the autophagy gene 12 (Atg12), are integral part of the evolutionary-conserved autophagy machinery. Ub-like conjugation processes are required to initiate autophagy, in which the E1-like enzyme Atg7 covalently attaches Atg12 to Atg5 or Atg3. The Atg12/Atg5 complex allows the conjugation of phosphatidylethanolamine to the soluble Ub-like Atg8/LC3, which mediates formation of the autophagosome membrane. Although the nonselective autophagy and mitophagy pathways generally use the same machinery, neither Atg5 nor Atg7 seems completely essential to mitophagy (192). Atg3 also facilitates Atg8/LC3 lipidation. Atg8/LC3 interacts with adaptor proteins such as p62 and NBR1, which mediate the enrichment of ubiquitinated cargo inside autophagosomes. The conjugation of Atg3 to Atg12 has been recently demonstrated to be important for mitochondrial homeostasy and cell death, independent of starvation signals (217). Interestingly, disruption of the Atg12-Atg3 complex has no effect on nonselective autophagy. Instead, cells display increased mitochondrial mass and enhanced survival in response to activation of mitochondrial apoptosis pathways. Surprisingly, only a moderate decrease in mitophagy is observed. This suggests that the regulation of mitochondrial morphodynamics and mitochondrial homeostasy is closely connected to mitochondrial turnover at the level of mitophagy machinery. Mitophagy may be induced to clear cells of damaged mitochondria as is the case when bioenergetics become substantially impaired (215, 226), and (261) inner membrane potential is decreased (117, 189, 190, 265), or when ROS production is elevated (233, 234) (Fig. 13)
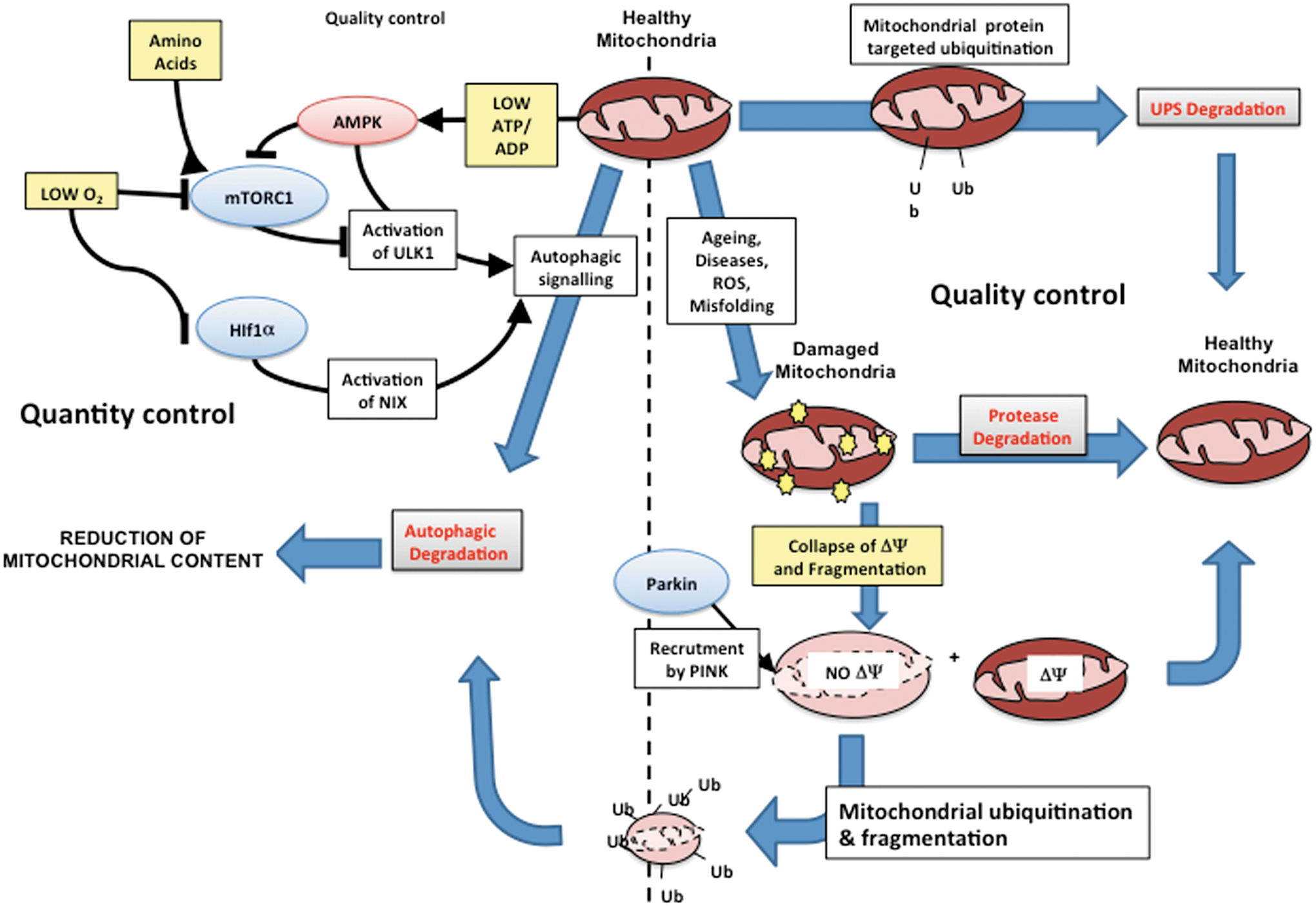
Mitophagy could also act as a metabolic adaptation to oxygen levels and oxygen consumption mediated by HIF1 and Nix/BNIP3 to control overproduction of ROS in cells with impaired ETC by mutation of SDH and FH (125). Moreover, mitochondrial depolarization and mitochondrial fission seem to be critical events needed for elimination of damaged mitochondria through mitophagy. However, membrane depolarization is not always a hallmark of mitophagy induction (137). The best-known pathway in mammalian cells to suppress mitochondrial damage through mitophagy is induced by PINK1 and Parkin (discussed above), where PINK1 is rapidly cleaved and degraded in healthy mitochondria, whereas its proteolysis is inhibited in damaged mitochondria. The activity of stabilized PINK1 then selectively recruits Parkin to damaged and depolarized mitochondria to facilitate mitophagy. Co-overexpression of Parkin and PINK1 produces fragmentation of the normal tubular mitochondrial network into mitochondrial aggregates and/or large perinuclear clusters. As mentioned above, the action of Pink1/Parkin also involves inhibition by ubiquitination of the fusion GTPase mitofusin-1 (MFN1), and this facilitates autophagosomal engulfment of fragmented damaged mitochondria. These studies show the existence of a close relationship between mitophagy, mitochondrial dynamics, and mitochondria bioenergetics. As discussed in the above section, modification of proteins by ubiquitination or other covalently linked tags may mark mitochondria for degradation. Ubiquitination is involved in sorting of vesicular bodies in the cytosol, and it has been shown to tag sperm mitochondria for degradation in the fertilized egg (253).
It appears that mitophagic processes are an essential mechanism to regulate the total content of mitochondria in the cell in addition to maintain a global quality control of the organelle. So far, quality control has been the matter of intensive research, while the role of energy metabolism in the regulation of global autophagy and of mitophagy remains under-investigated. Indeed, AMPK (a serine/threonine kinase) downregulates ATP-consuming pathways and induces pathways that generate ATP when the ATP/ADP ratio falls below a threshold. AMPK is activated in the hearts of starved Atg5 −/− (autophagy-deficient) mice (152). Although autophagy and energy status are interconnected in vivo by the supply of energy substrates, the link is not entirely clear, as Atg5 is not required for mitophagy in lens and erythroid cells, as shown in Atg5−/− neonates.
Autophagy activated by energetic deficit (usually amino acids and glucose) involves inactivation of mTORC1, a suppressor of the autophagic protein, serine–threonine-protein kinase (Ulk1). AMPK, which is activated by the LKB1 complex upon low ATP and high AMP and ADP levels, can stimulate mitophagy by inhibiting mTORC1. The serine–threonine Liver Kinase B1 (LKB1) function is an activator of AMPK. LKB1 is recruited to mitochondria after cell stress, a process that is mediated by interaction with p53 (136). Interestingly, treatment with metformin (activation of AMPK) increases activation of LKB1, but is not known whether this translates to increased LKB1 recruitment to mitochondria. Recently, it was found that AMPK interacts with and phosphorylates Ulk1, and loss of AMPK or Ulk1 results in aberrant accumulation of p62 and defective mitophagy (75). The phosphorylation and direct activation of Ulk1 by activated AMPK bypass the inhibitory activity of mTORC1 on Ulk1. Ulk1 (and Ulk2) in turn negatively regulates AMPK by phosphorylation of all three AMPK subunits. Thus, Ulk1, LKB1/AMPK, and mTORC1 are very likely involved in a regulatory loop able to signal both activation and termination of mitophagy. These findings raise the question of the role played by mitochondrial energetic signaling as a trigger of autophagy and mitophagy. To date, this hypothesis is underestimated compared to the implication in the quality control. As for the Pink/Parkin mitochondrial pathway, it is most probable that mitochondrial energetic could control mitophagy according to the level of the organelle activity. To this aim, AMPK and mTOR seem highly appropriate targets of mitochondria energetic signaling. Mitochondrial content in the cell should be considered as a dynamic process involving both biosynthesis and degradation. As we have noticed, perturbation of one of these processes results into dramatic alteration of energetic homeostasy. It is well established that mitochondrial bioenergetics regulates to a certain extent the cellular mechanism leading to activation of the biogenesis; however, this has not been tested for degradative processes. Recent findings also showed that mitochondrial morphogenesis could play a role during autophagy. Gomes et al. showed that during autophagy, mitochondria elongate, are spared from degradation, and sustain cell viability (94). The mechanism involves the sequestration of DRP1, the mitochondrial fission protein, in the cytosol.
The control of selective autophagy of mitochondria not only is pivotal in cell homeostasy, but may also be central in the prevention, progression, and suppression of cancer. Selective autophagy may be involved in the acquisition of malignancy in response to activated oncogenes. Later, it may also serve to sustain cellular metabolism as tumor grows and oxygen becomes limiting, to delay cell death in a hostile microenvironment. Indeed, several recent studies point to mitophagy as a finely balanced prodeath/prosurvival process in the progression and elimination of cancer (203). Hypoxia is known to induce mitophagy mediated by Nix. Nix mRNA and protein are induced during hypoxia in a p53-dependent manner in nontumorigenic U2OS cells (78). Low oxygen concentration (hypoxia) is typical of solid tumors. Transcriptional upregulation of BNIP3 by Hif1α is indispensable to mitophagy, and this measure is thought to protect cells from the increased ROS production under reduced oxygen availability (13). Both BNIP3 and NIX locate to mitochondria via its C-terminal transmembrane domain, where they act as receptors to directly drive mitophagy by the recruitment of γ-aminobutyric acid receptor-associated protein (GABARAP) and Atg8/LC3-containing autophagic membranes (195). In addition, BNIP3 and NIX might have a more general role in hypoxia-induced selective autophagy by disrupting Bcl-2/Beclin-1 interaction, and thereby activating the Beclin-1/class III PI3K complex (13). Disruption of the complex must be finely balanced, as activation of the Bcl-2 pool is also known to result in apoptosis. In this context, inhibition of mitophagy could help limit tumor growth. During initial cancer development, mitophagy may help suppress cancer by decreasing genotoxic stress. On the contrary, inhibition of selective autophagy under hypoxic conditions should lead to accumulation of stressed mitochondria with an increase in the oxidative burden. It is known that mutations in OXPHOS components such SDH and FH are found in some tumors, although rarely (96). SDH converts succinate to fumarate and is also a functional member of complex II in the ETC. Increases in succinate due to SDH mutation directly inhibit the activity of Hif1α prolylhydroxylases (PHD) as the excess succinate leaks from mitochondria to the cytosol. The inhibition of PHDs leads to stabilization of Hif1α and activation of the hypoxia pathway. In this case, mitophagy could act as a metabolic adaptation to oxygen levels and oxygen consumption mediated by Hif1αand Nix/BNIP3 to control overproduction of ROS in cells with impaired ETC by mutation of SDH and FH (125). This also suggests that a functional ETC coupled to mitophagy inhibition may help in reducing the survival capacity of hypoxic tumors.
When DNA damage occurs, p53 transactivates a number of downstream genes whose products include mitophagy machinery. The p53 is a cell cycle checkpoint protein that functions in the G1 phase of the cell cycle. Downregulation of p53 may contribute to genomic instability and cancer progression. The binding of p53 to DNA is inhibited by ATP and promoted by ADP (199). However, mitochondrial generation of ROS can contribute to p53 induction. Interestingly, as detailed in section III, carcinogenic inhibition of p53 is accompanied by a decrease in COX2 levels and a shift toward glycolysis (297). The control of p53 on gene expression may represent only one of many signaling pathways that regulate mitophagy. Although upregulation of Nix during hypoxia is dependent on p53, p53 inhibition may also promote autophagy after genotoxic stress through activation of LKB1/AMPK and inhibition of mTOR. This may explain why Nix protein expression is absent or downregulated in some lung cancer tissues. Indeed, p53 inhibition efficiently induces reticulophagy and mitophagy (260). As p53 activation leads to mitophagy in a Nix- and hypoxia- dependent manner, the pathways of nonselective autophagy and selective mitophagy that converge on p53 may therefore involve different actors and depend on specific downstream mitochondrial signals. Stabilization of Hif1α combined with expression of p53 is thought to help prevent progression of cancer, and this could be due to the mitophagic clearance of damaged mitochondria. However, cellular apoptosis through overactive mitophagy induced by permanently damaged mitochondria could result in the selection of cells with downregulated or mutant p53. The local increase of a cell population with genomic instability may promote cancer by favoring the accumulation of cells with defective OXPHOS (mutant SDH and FH) and a glycolytic cancer phenotype.
Mitophagy may be inactivated in certain cancers as a result of deletion of Beclin-1 and downregulation of Nix. Beclin-1 (Atg6) is often found haplodeficient in cancer tissues. It is known that Beclin-1 haploinsufficiency and reduction in autophagy lead to increased necrosis in hypoxic tumors. Beclin-1 is inhibited when in complex with Bcl-2 and Bcl-XL, and this suppresses mitochondrial membrane permeabilization (174). Increased expression of Bcl-xL/Bcl-2 in cancer has been shown to confer resistance to a broad range of apoptotic stimuli. It is thought that in Beclin-1-haplodeficient cells, the excess Bcl-2 inhibits mitophagy and also prevents Beclin-1 from activating the mPTP, thereby delaying cell death (174). The BH3-mimetic compound ABT-737, which is able to rescue mitophagy in Nix−/− reticulocytes, has also been used successfully as anticancer in the treatment of breast cancer. ABT-737 works in part by inducing mitophagy through the disruption of the inhibitory interaction between Beclin-1 and Bcl-xL/Bcl-2 (65, 174). Interestingly, ABT-737 only induces mitophagy, but not reticulophagy. The effects of ABT-737 on induction of mitophagy in cancer may be enhanced by the jasmonate vincristine. The synergistic effect on mitophagy of ABT-737 and vincristine results in apoptotic cell death, likely mediated by an important deregulation in mitophagy due to the inhibitory phosphorylation of Bcl-2 induced by vincristine, and the accompanied depolarization of mitochondria. Cancer progression could be promoted by loss of mitochondrial membrane potential signaling Nix to initiate mitophagy in concert with Parkin (63). Parkin promotes mitochondrial ubiquitination and primes mitochondria for mitophagy, although it cannot initiate mitophagy. However, Parkin may also promote recruitment of p62 to mitochondria, and both cooperate to rearrange depolarized mitochondria into perinuclear clusters, which are thought to facilitate their degradation. It is not clear whether p62 and Parkin are essential for mitochondrial degradation, as there are conflicting reports on the role of both proteins (90, 198). Neither p62 expression nor perinuclear clustering is essential for mitophagy, suggesting that there are alternative compensatory pathways for activating mitophagy. However, p62 degradation is accelerated during autophagy activated by hypoxia (216). Degradation of p62 through autophagy is associated with elevated mitochondrial ERK signaling. Chronic activation of the Ras/ERK signaling pathway positively correlates with tumor progression, as elevated ERK signaling desensitizes the mPTP and may protect cancer cells from apoptosis induction through inhibition of mitochondrial depolarization (219). Mitophagy is induced in neuroblastoma cells treated with MPP+, and this is blocked by inhibitors of the MEK/ERK signaling pathway, with ERK2 being localized to mitochondria. Therefore, perturbations in the localization and/or polymerization of p62 and ERK signaling in cancer may have consequences in the execution of mitophagy mediated by membrane depolarization or ETC defects.
It is interesting to reflect on the seemingly contrary roles that mitophagy may have in survival or progression of cancer. This may be explained by the multitude of signaling pathways that lead to mitophagy under normal or hypoxic conditions and the normal role and regulation of mitophagy according to the cell type and energetic substrate availability. As mitochondrial activity and energetic substrate utilization are different depending on the cell type, the derived cancer cells are very likely to behave differently with regard to induction or inhibition of mitophagy. Finally, as mitochondria contain several cell death regulators, mitochondrial clearance by selective mitophagy must be kept tightly regulated to avoid caspase activation resulting from mitochondria depolarization and opening of mPTP. Continuous mitochondrial clearance by mitophagy as a function of mitochondrial activity and homeostasy may thus constitute a fundamental checkpoint in cell survival or the execution of cell death.
XII. Role of ROS in Mitochondrial Adaptations in Physiology and Pathology
The study of mitochondrial pathophysiology has revealed that respiratory chain dysfunction can associate with overproduction of ROS at the level of complex I or complex III. Yet, not all ETC dysfunctions lead to ROS overproduction and both the nature of the defect (mutation or pharmacological compound), the complex altered, and the degree of inhibition and the potency of the antioxidant defense determine the extent of ROS production. In particular, the relationships between mitochondrial membrane potential and ROS formation are complex as recently reviewed by the group of Mariusz Wieckowski (252). Besides the variability in the methods to assess ROS production, difference in cell types and cell metabolic state are strong determinants of ROS generation. A study in the rat brain mitochondria revealed that depending on the type of energy substrate utilized for respiration, the relationship between ROS production and ΔΨ was different (280). In a condition of reverse electron transfer from CII to CI, ROS production is large and strongly sensitive to membrane depolarization. In contrast, in presence of complex I substrates alone, little ROS are produced despite the presence of ΔΨ. Therefore, consideration of the link between ETC defect and ROS production should also be taken into account the importance of the type of energy substrate consumed by the mitochondrion. These relationships between ROS and ETC dysfunction were extensively studied by the group of Werner Koopman and Jan Smeitink, notably on fibroblasts obtained from patients with complex I disorder and on cells treated with rotenone (62, 64, 143 –147, 149, 277, 278). These authors showed that cultured fibroblasts of complex-I-deficient patients present with elevated levels of ROS although not systematically, (ii) calcium homeostasy is disturbed; (iii) the mitochondrial network morphology is altered (in proportion to CI defect); and (iv) the matrix viscosity is altered. The link between the changes in ROS levels and the modification of mitochondrial morphology remains unclear, as conflicting studies exist in the literature. On the one hand, mitochondrial fragmentation results from an elevation in ROS concentration (128), while on the other hand, mitochondrial fragmentation is a prerequisite for ROS formation (293). These two hypotheses are not exclusive since similar observations were made for the relationships between mitochondrial energy state and mitochondrial morphogenesis: the link is mutual so that changes in form modulate function, but also the other way round (14). Since ROS are often associated with OXPHOS dysfunction, they could play a role in the initiation of a ROS-specific compensatory adaptative response. We analyzed this possibility in fibroblasts treated with gradual concentrations of potassium cyanide and found that ROS overproduction was only observed at extreme doses of CIV inhibition (21). These elevated levels of hydrogen peroxide were not involved in the bioenergetic adaptative response observed in these cells, although we cannot extrapolate these findings to other types of OXPHOS dysfunction. For instance, it is known that complex I inhibition generates ROS, whereas complex IV inhibition does not or to a lesser extent. As argued by Koopman and colleagues, the activation of ROS signaling depends on the concentration and lifetime of the particular ROS involved (143). It also depends on the activation of ROS-detoxifying mechanisms that are able to modulate both ROS signaling and mitochondrial bioenergetics. In particular, the activation of UCPs by superoxide is able to decrease oxidative stress as well as to reduce mitochondrial protomotive force and subsequent ATP production (72, 256).
Moreover, the adaptative response triggered by ROS might not involve the activation of OXPHOS, but could concern other features of cell adaptation as the stimulation of the antioxidant defenses or the regulation of mitochondrial morphogenesis. An important role of ROS in cell adaptation to ETC deficiency could include the compensatory stimulation of glycolysis via Hif1α (231, 242). The ROS-Hif1α-glycolysis pathway can also be activated by a primary generation of ROS in the cytosol. Indeed, we observed that cells stressed with UV-B irradiation presented a rapid production of cytoplasmic ROS, which were able to downregulate Hif1α expression, but that a delayed mitochondrial ROS generation resulted in its upregulation (222). More recently, we demonstrated that ROS produced by NOX-1 in response to cellular genotoxic stress are sufficient to alter ETC function and to trigger the Warburg effect via Akt-1 (223, 224). While the compensatory activation of glycolysis by ROS was evidenced in cancer cells, the importance of this mechanism in the compensation of bioenergetic ETC deficiencies remains to be analyzed in other type of disorders (as pure mitochondrial diseases). Moreover, the ROS produced by the mitochondrion could participate to cell adaptation via the generation of further ROS production by the p66shc protein. The ROS produced by ETC activate the kinase PKC, which phosphorylates P66shc and triggers the translocation of P66shc to the mitochondrion (212). There, P66shc can directly interact with cytochrome c to generate ROS, and this is further amplified by the p66shc-mediated inhibition of SOD2 expression via the inhibition of FOXO3a (150). The potential role of P66shc in cellular adaptation to low energy state was recently highlighted by the study of P66shc−/− mice confronted to the natural environment where the animals are subjected to food competition and exposed to winter temperatures. (92). Recently, the group of Mariusz Wieckowski investigated the role of p66shc in fibroblasts obtained from patients with a mitochondrial disorder (156). They observed an increase in p66Shc phosphorylation at Ser36 and a decreased level of mitochondrial superoxide dismutase (SOD2) in these cells. Yet, these authors did not analyze the possible adaptative response associated with the vicious cycle of ROS production in OXPHOS disorders, so that the potential role of P66shc in the adaptation to ETC deficiency remains elusive. The role of SOD2 is central to mitochondrial adaptation in response to ETC dysfunction, since two opposite mechanisms regulate SOD2 expression. On the one hand, mitochondrial ROS can activate P66shc and inhibit SOD2 expression as discussed above. On the other hand, Colombo and Moncada (51) showed that mitochondrial ROS generated as a result of the interaction between NO and mitochondrial cytochrome c oxidase activate AMPK-α1 in human umbilical vein endothelial cells at low oxygen concentration and stimulates the expression of genes involved in antioxidant defense, such as manganese superoxide dismutase, catalase, γ-glutamylcysteine synthase, and thioredoxin. These authors showed that PGC1α, CREB, and Foxo3a are involved in this signaling pathway. While this study was performed on endothelial cells, the occurrence of this regulatory pathway could be analyzed in other cell types. Interestingly, we found that CREB was activated in the muscle of patient with a mitochondrial disorder and in fibroblasts treated with cyanide (21), suggesting a central role of this protein in mitochondrial adaptation. The role of ROS in mitochondrial adaptation could also involve the regulation of mitochondrial morphogenesis that impacts organelle functions (19). In particular, elevated oxidative stress can result in the ubiquitination and further proteasomal degradation of Mfn1 and Mfn2 through the PINK1/Parkin pathway (89, 218). A reduction in Mfn2 protein level can displace the equilibrium of mitochondrial dynamics toward fission, and we think that such a change could participate to the preservation of ATP production in the cell. Indeed, in fibroblasts treated with KCN, we observed that during the adaptative response, there was an increase in DRP1 protein content that also triggered the fragmentation of the mitochondrial network. In contrast with the idea that mitochondrial fusion could be protective, we think that mitochondrial fragmentation could allow to isolate defective mitochondrial particles, which could be more easily eliminated by the quality control pathways evidenced by the group of Orina Shirihai (265). This hypothesis is consistent with the observation of fragmented mitochondria in cardiomyocytes, which rely intensively on OXPHOS for ATP production or with the observation of DRP1 increased expression in the muscle of trained individuals (209). Mitochondrial dynamics and motility are linked to organelle biogenesis and recycling, so that the above-described impact of ROS on mitochondrial fragmentation could be linked with the influence of ROS on mitochondrial turnover. According to Speigelman, PGC1α and β are induced when cells are given an oxidative stressor such as H2O2. This results in the activation of mitochondrial biogenesis, but also the stimulation of a PGC1-dependent ROS-scavenging program that includes genes encoding mitochondrial proteins such as SOD2, but also cytoplasmic proteins like catalase and GPX1 (249). The different pathways activated by mitochondrial ROS and potentially involved in mitoplasticity are illustrated in Figure 14.
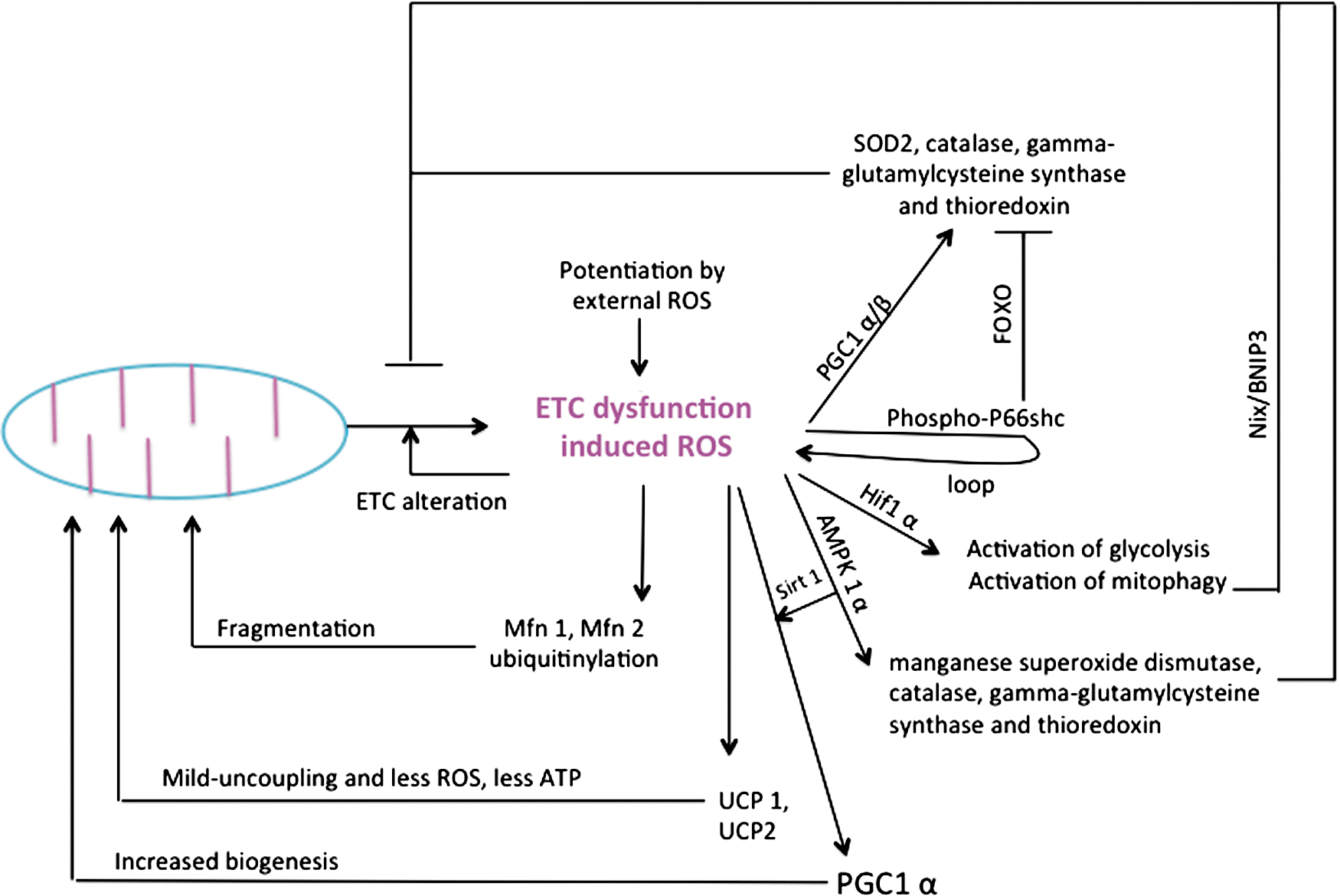
XIII. Conclusion
Altogether, the flexible control of metabolic fluxes, the molecular adaptability of the mitochondrion, and the adjustable turnover of the organelle constitute the fundamental elements of mitochondrial plasticity engaged in mitochondrial bioenergetic homeostasy. The control of mitochondrial respiration and of associated ATP production is tightly adjusted to the steady state of energy production to maintain optimal energy delivery to the cell. A safety margin beyond which changes in respiratory chain activity cannot modulate further the rate of mitochondrial respiration protects the cell from eventual OXPHOS alterations. This so-called biochemical threshold acts as a powerful buffer to provide stability of mitochondrial metabolic fluxes in the mitochondrion. Besides, cancer cells forced to grow in challenging conditions of limited glycolytic substrate delivery develop adaptative mechanisms of molecular enhancement of the OXPHOS components, among which activation of organellar biogenesis and reshaping of the mitochondrial network are the most impressive. Further, cancer cells can acquire the capacity to survive under variable carbon sources, a vital process termed metabolic flexibility, the genetic (oncogenic) bases of which still remain unknown. Ideally, the changes in the bioenergetic profile of cancer cells occur synergistically with the rewiring of elementary metabolic routes to accommodate the tumor-specific anabolism and catabolism. Lastly, a tremendous control of the bioenergetic capacity exists at the level of quality and quantity control of the mitochondrial machinery involved in energy production. The aforementioned mechanism of mitochondrial adaptation to cellular energy needs play a crucial role in the maintenance of cellular energy homeostasy. Mitochondrial adaptation proceeds through a large variety of signaling pathways that necessitate the coordinated action of a priori unrelated transcription factors (Fig. 15). They are also implicated in the etiology of neurodegenerative disorders or in various forms of cancer, so that fundamental research on mitochondrial adaptation could help to discover innovative therapeutic strategies targeted against mitochondrial multiformity.
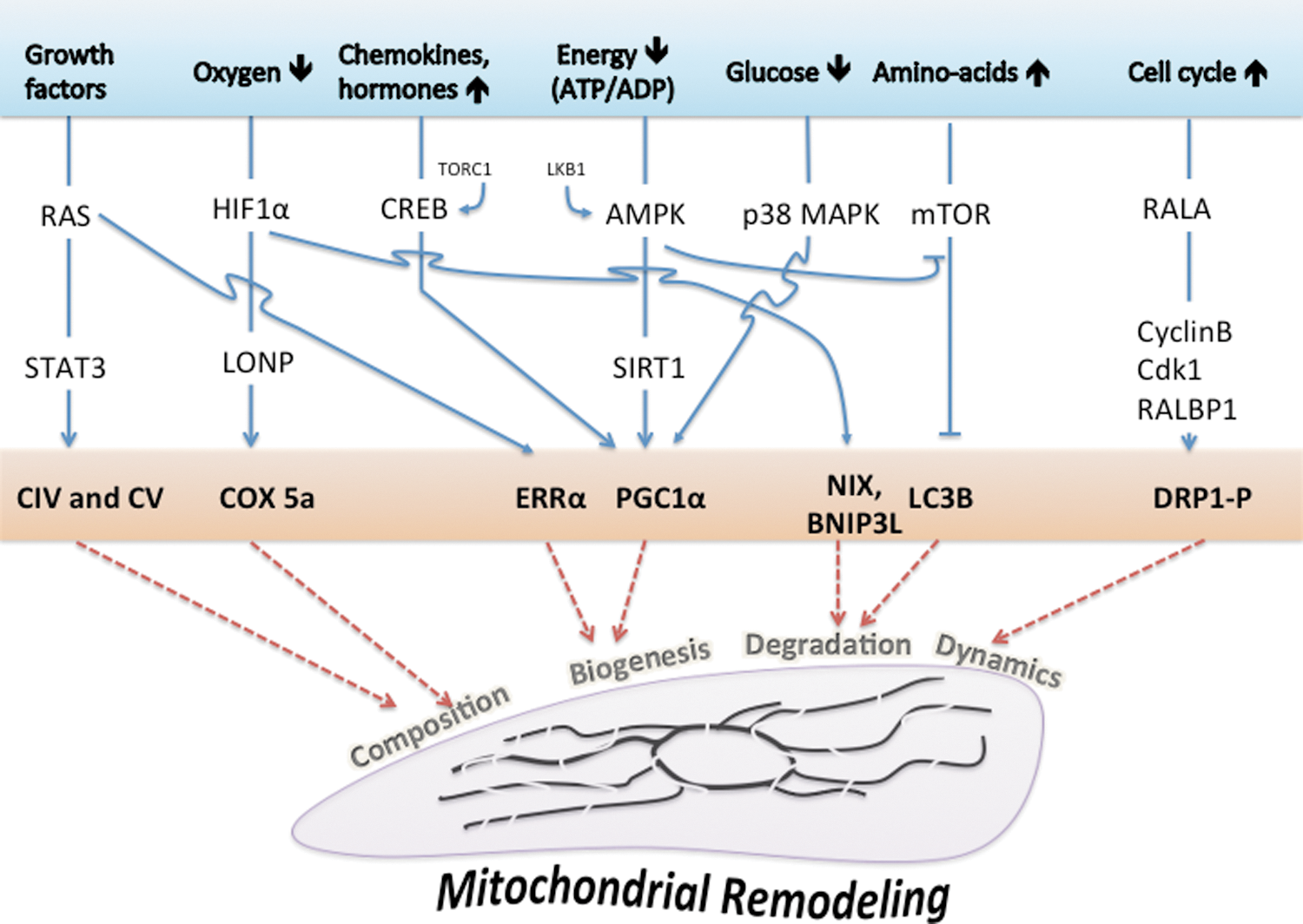
Footnotes
Acknowledgments
The authors wish to thank ANR retour postdoc (G.B., S.M.), Natural Sciences and Engineering Research Council of Canada, AFM, Cancéropôle Grand Sud-Ouest, Université Bordeaux Segalen, MIP society, Région Aquitaine, and Association contre les maladies mitochondriales (Ammi) for financial support.