
Abstract
Introduction
When the widespread biological actions of H2S were first being realized, initial estimates of dissolved H2S levels ranged from 20 to 100 μM, but it should be noted that H2S detection is problematic, and these values are likely to be overestimates. Indeed, more recent measurements have downsized these initial values by orders of magnitude [reviewed by Li et al. (29)]. Nevertheless, its biological importance, along with that of NO and CO, has continued to grow apace, and in time all of these gasotransmitters could well be regarded as ubiquitously influential. This is highlighted by the growing number of pathological conditions under which H2S metabolism is altered. Certainly, the list of signaling pathways and effectors regulated by H2S continues to grow, and one major group of target proteins regulated by H2S appears to be ion channels (29). This in itself implies that H2S is influential in membrane excitability as well as any other transmembrane process that might depend on electrochemical gradients.
In this article, we discuss the regulation of known ion channel targets of H2S. While the first ion channel identified as being regulated by H2S was the N-methyl-
ATP-Sensitive K+ Channels
One of the best characterized ion channels identified as a target for regulation by H2S is the vascular ATP-sensitive K+ (KATP) channel (79). This channel, the pore of which is composed of four inwardly rectifying (KIR 6.X) channel subunits, associated with four sulfonylurea receptors (SURs) (Fig. 1), which confer unique pharmacological as well as other properties on the channel, is widely distributed (12). Its activation by H2S, shown schematically in Figure 1, was an exciting breakthrough, since it could account simply for the vasodilatory actions of H2S: KATP activation leads to hyperpolarization of vascular smooth muscle cells, causing vasodilation by reducing voltage-gated Ca2+ influx. Subsequent studies indicated that in addition to this action on KATP channels in vascular smooth muscle, there was a significant endothelial component to the vasodilation caused by H2S; this arose from the ability of endothelium-derived NO to stimulate both the activity and expression of CSE (Fig. 1), enhancing H2S production (78). Since this observation, evidence has accumulated that H2S activation of KATP channels (which are found in the mitochondrial as well as the plasmalemmal membrane) contributes to myocardial protection against ischemia/reperfusion (I/R) injury, control of insulin secretion from pancreatic β cell-derived cell lines, and regulation of inflammation and nociception [reviewed in Ref. (68)]. In addition, activation of neuronal KATP channels by H2S has been suggested to contribute to the protective actions of H2S against glutamate-induced toxicity (24, 25), as discussed in the subsequent section concerning K+ channels and apoptosis.
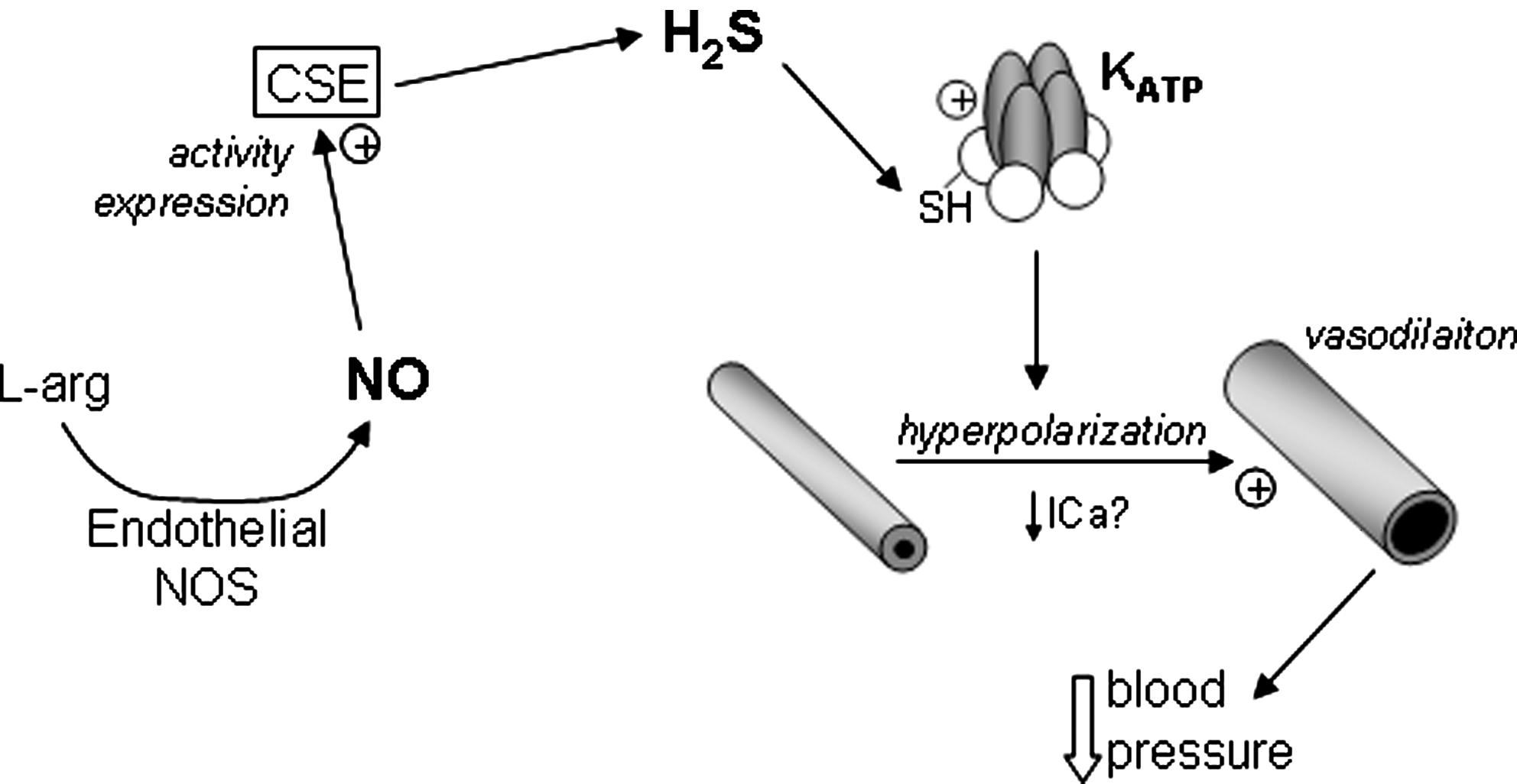
The observation that K+ currents generated by heterologous expression of KIR subunits (in the absence of SUR receptors) were insensitive to H2S strongly suggested that SUR receptors were the sites of modulation by H2S (19). Such a proposal is supported by earlier data indicating that H2S increased the open probability of KATP channels in excised membrane patches independently of ATP levels (73), which argues strongly for a direct mechanism of H2S action. Further insight was gained when it was found that either alkylation or oxidation of key cysteine residues located on the extracellular face of the SUR1 protein prevented regulation by H2S (19). Most recently, Mustafa et al. (38) demonstrated that Kir6.1 is directly sulfydrated by H2S, leading to a reduction of ATP binding (which normally leads to channel closure) and promoting channel binding to phospholipid phosphatidylinositol (4,5)-bisphosphate, which enhances channel activity. This study represents the most detailed investigation published to date of the mechanism by which H2S regulates ion channels; other candidate mechanisms are discussed in detail below.
Ca2+-Activated K+ Channels
Telezhkin and colleagues have demonstrated that H2S (applied as NaHS) inhibited both human recombinant Ca2+-activated K+ (BKCa) channel α subunits (KCNMA1) expressed in HEK293 cells and native BKCa channels expressed in type I cells from the carotid body of the rat (65) (Fig. 2). This channel is widely distributed in most, if not all, tissues, and has well-described roles in the central nervous system and vasculature [reviewed in Ref. (13)]. It is of particular importance to the process of oxygen-sensing by the carotid body, being inhibited by hypoxia and so contributing to type I cell depolarization (47). This in turn leads to voltage-gated Ca2+ entry and transmitter release, which excites afferent sensory nerves and so, ultimately, increases our ventilation in response to acute hypoxia [reviewed in Ref. (26)]. This channel is also sensitive to the gasotransmitter CO, which augments its activity (see Fig. 2) and has been postulated as a mechanism by which the channel may sense hypoxia: in the rat type I cell, the CO-generating enzyme heme oxygenase-2 associates with BKCa channels and regulates their activity through the tonic generation of CO (via degradation of heme, which is an O2-dependent process) (70). It is of interest that both H2S and CO regulate this channel since both gases—and the channel itself—are of influence in the O2-sensing mechanism, which is fundamental to the chemosensory ability of the carotid body. Telezhkin et al. (65) demonstrated that H2S inhibited the channel via a mechanism distinct from its regulation by CO: thus, CO activation was blocked by cyanide, but this had no effect on channel inhibition by H2S. Furthermore, the authors generated a BKCa chimera that lacked both Ca2+ and CO sensitivity, yet remained sensitive to inhibition by H2S.

This study—and its importance for carotid body function—was supported by experiments in which mouse carotid body excitation was observed in response to H2S application. Thus, Li et al. (30) demonstrated the excitatory effects of H2S and, crucially, found that hypoxic excitation of the carotid body was inhibited by suppression of endogenous H2S using two distinct CBS inhibitors (surprisingly, given that type I cells also express CSE, inhibition of this enzyme was found to be without effect). Patch-clamp recordings indicated that H2S mimicked the ability of hypoxia to inhibit BKCa channels in isolated type I cells, and in fact the ability of hypoxia to reduce channel activity was suppressed by a CSE inhibitor, suggesting involvement of H2S in this effect also. An additional study has confirmed the central importance of H2S in carotid body O2 sensing by demonstrating a markedly weakened response to hypoxia in CSE−/− mice (48), although this study did not include direct analysis of ion channel modulation.
Collectively, the above-described studies strongly suggest that inhibition of BKCa channels in type I glomus cells by H2S is of fundamental physiological importance to carotid body function. However, another report has indicated that H2S increases the activity of BKCa channels expressed in the rat pituitary cell line, GH3 (55). The authors proposed that H2S acted in a Ca2+-independent manner to reduce cysteine residues accessible from the cytosolic face of the channel protein. Presently, there is no explanation to account for these opposing reports on BKCa modulation. Possibilities, some offered by the authors (55), include differential effects on splice variants, differing phosphorylation states of the channel protein, involvement of auxiliary (β4) subunits, or the differential presence of unidentified, associated proteins which may regulate channel activity in a H2S-sensitive manner. Interestingly, a most recent report has suggested that H2S can also activate BKCa channels in smooth muscle of small mesenteric arteries (15). Based in part on the ability of the BKCa inhibitor iberiotoxin to occlude the effects of H2S, this study concluded that H2S generated by endothelial CSE caused vasorelaxation not via activation of KATP channels (as described above) but instead by activating BKCa channels, thereby hyperpolarizing and relaxing smooth muscle cells. Although no direct patch-clamp electrophysiological evidence for BKCa activation by H2S was provided, this study supported the idea that in tissues other than the clonal GH3 cell line H2S can activate BKCa channels.
Clearly, further work is required to resolve these opposing effects of H2S on BKCa channels, and indeed to fully characterize the underlying mechanisms in each case. However, it already appears that BKCa represents an influential physiological target for regulation by H2S.
K+ channels and apoptosis
In the central nervous system (CNS), H2S is clearly neuroprotective. There are a number of mechanisms that have been proposed to account for its actions in this regard. It has, for example, been shown to increase neuronal levels of reduced glutathione (GSH) in part through increasing cysteine uptake (23). Interestingly, expression of 3MST and CAT in neuronal mitochondria increases resistance to oxidative stresses, implying that mitochondria may be an important source of deleterious reactive oxygen species (ROS) that could lead to neurodegeneration.
Numerous studies have implicated the delayed rectifier channel Kv2.1 as being important in oxidative stress-induced neuronal apoptosis (Fig. 3). Since K+ acts to regulate caspase activation, mitochondrial membrane potential and volume, and (through its abundance) osmolarity and hence cell volume, cellular apoptosis is strongly influenced by intracellular K+ levels (75). K+ loss is a key early stage in apoptosis, and efflux occurs through K+ channels to trigger the apoptotic cascade: cell shrinkage, mitochondrial disruption, cytochrome c release, and caspase activation (75). K+ loss occurs via K+ channels: thus, K+ channel inhibitors can protect against apoptosis triggered by a variety of insults, including oxidative stress (2, 4) and exogenously applied amyloid β peptide of Alzheimer's disease (76). Theoretically, any class of K+ channel could serve to permit K+ loss, but evidence suggests a particularly important role for the delayed rectifier channel Kv2.1 in mediating K+ efflux that leads to neuronal apoptosis: neurons expressing dominant negative Kv2.1 constructs (therefore lacking functional Kv2.1 channels) were protected against experimentally induced apoptosis, and expression of Kv2.1 in Chinese hamster ovary (CHO) cells increased their susceptibility to apoptosis (44). Pro-apoptotic agents cause a rapid increase in the surface expression of Kv2.1 channels (Fig. 3), a process thought to require p38 MAP kinase phosphorylation at Ser-800 in the intracellular C-terminal region of the channel protein and additional phosphorylation of an N-terminal tyrosine (Y124) regulated via Src kinase (51).

An additional feature of neuronal oxidative stress is the up-regulation of the inducible form of the CO-generating enzyme heme oxygenase (HO-1). Since CO provides neuronal protection against stresses such as stroke and excitotoxicity (77), we recently investigated whether CO might regulate Kv2.1 (6). We found that CO specifically protected HEK293 cells overexpressing Kv2.1 against oxidative stress, while the vulnerability of untransfected cells to oxidative stress was unaffected by CO. Furthermore, CO reversibly inhibited the Kv2.1 currents in these Kv2.1-expressing cells. Importantly, in hippocampal neurons, CO selectively inhibited Kv2.1 and reversed the dramatic oxidant-induced increase in K+ current density as well as providing protection against oxidant-induced apoptosis (6). It will be of importance to determine in future experiments whether some of the neuroprotective effects of H2S may involve interruption of Kv2.1 trafficking or activity and hence may be antiapoptotic via this pathway.
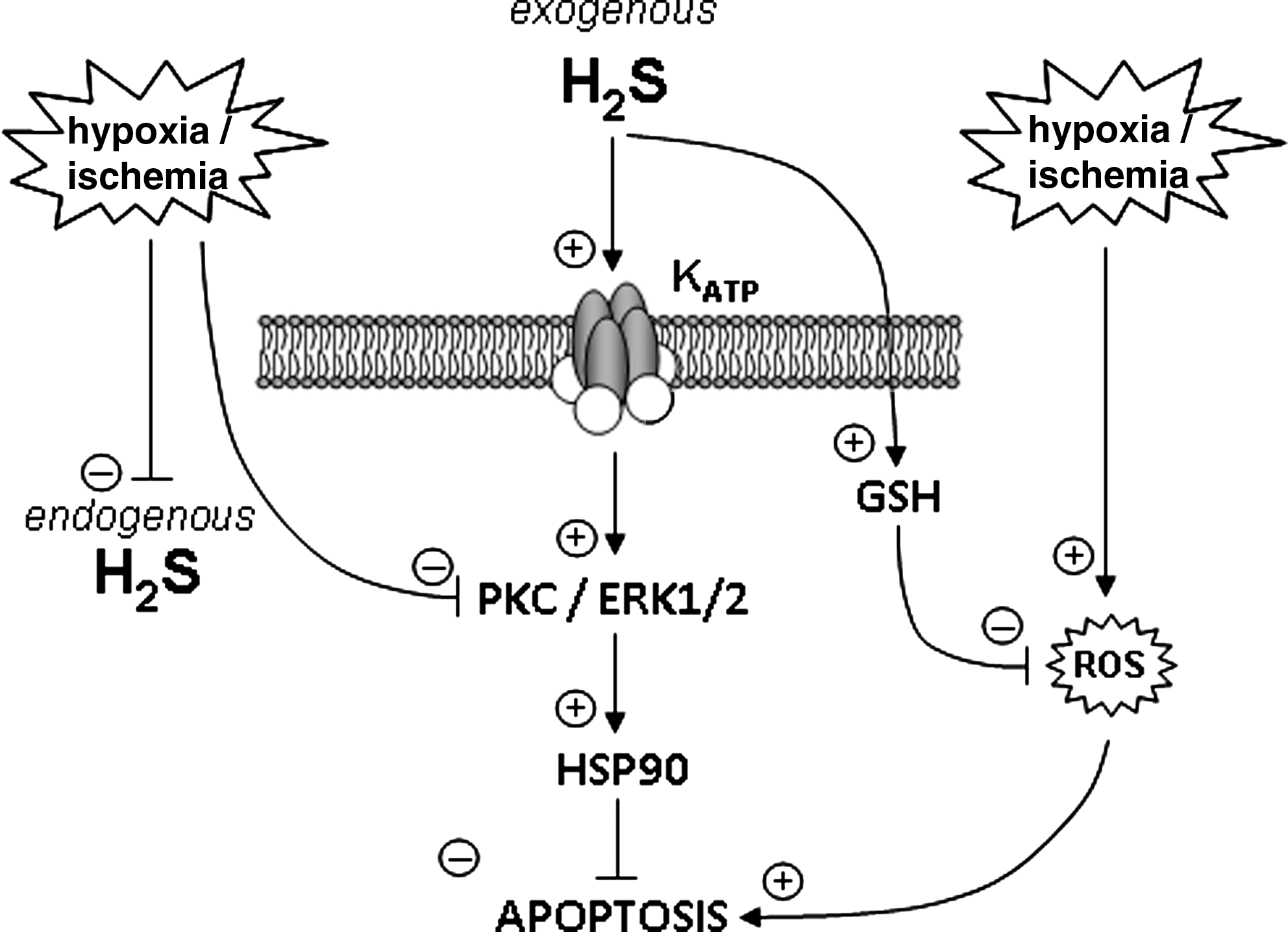
Although a role for Kv2.1 in neuroprotection by H2S remains speculative at present, there is much evidence that KATP channels are involved in this process. For example, Tay et al. (64) demonstrated that prior exposure of neuroblastoma cells to hypoxic/ischemic conditions in vitro provided striking protection (via application of NaHS at 10–100 μM). Protection was prevented by the plasmalemmal KATP channel blocker glibenclamide (a mitochondrial KATP channel blocker was ineffective), and was also dependent on the involvement of protein kinase C/extracellular receptor kinase (ERK1/2) activation, as well as activation of HSP90, which interacts with and prevents activity of various pro-apoptotic proteins (see Fig. 4 for schematic representation). Such an action of H2S is in addition to its known effect to increase reduced GSH levels and so afford protection against oxidative stress (24, 25). This effect may also be of importance in protection against hypoxic insults, since some studies indicate that hypoxia can increase ROS production from mitochondria, thereby causing a seemingly paradoxical oxidative stress (11). It is important to note, however, that convincing evidence for a protective role of H2S in the central nervous system in response to hypoxic/ischemic challenges requires in vivo evidence. Indeed, we need to understand more completely the relationship between oxygen levels and levels of H2S: while some studies suggest that hypoxic/ischemic conditions dramatically reduce endogenous H2S levels (64) (which would remove or reduce protective effects of H2S), other studies argue that tonic production of H2S leads to increased net levels in hypoxia, via H2S accumulation, since its oxidation is limited by available oxygen (43).
T-Type Ca2+ Channels
T-type Ca2+ channels are a unique class of voltage-gated Ca2+ channel (VGCC), distinguished functionally from other VGCCs by their rapid activation and inactivation properties, slow deactivation, smaller single-channel conductances, and their low membrane potential threshold for activation, which is more negative than the threshold for other VGCCs [they were originally termed low-voltage-activated currents (49)]. T-type Ca2+ channels are encoded by three genes (CACNA1G, CACNA1H, and CACNA1I) giving rise to voltage-sensing, pore-forming α1G, α1H, and α1I subunits, now termed Cav3.1, Cav3.2, and Cav3.3, respectively. Also distinguishing them from other VGCCs is their pharmacology and the fact that heterologous expression of these channels gives rise to currents which are remarkably similar to native currents, suggesting that their function is primarily determined by the pore-forming α subunits alone, without major regulation by auxiliary subunits [although their trafficking may be altered (8)]. T-type Ca2+ channels are widely expressed in both central and peripheral nervous systems and serve a wide variety of functions (62). In central neurons T-type Ca2+ channels are responsible for pacemaker activity and low threshold spikes, which in turn give rise to Na+-dependent action potential bursts, and also contribute to rebound bursts of spiking activity following a hyperpolarizing inhibitory postsynaptic potential. This latter property contributes to oscillatory activity, and thus T-type Ca2+ channels have become a focus of study in the treatment of epilepsy [reviewed in Ref. (40)]. They also display a small, but significant, window current (i.e., are tonically active) at membrane potentials close to resting potential, so can contribute to tonic Ca2+ influx (49).
In the periphery T-type Ca2+ channels play an important role in nociception: they are prominently expressed in small nociceptive dorsal root ganglion (DRG) neurons. Cav3.2 (α1H) is the dominant form of T-type Ca2+ channel in these neurons, and although their role in controlling DRG neurons is less well defined than in central neurons, they control burst firing and so influence excitability (40). This implies that they are of central importance to nociception since stimulus intensity correlates with burst frequency, and much information supports this. For example, native DRG T-type currents (and recombinant Cav3.2) are enhanced or inhibited by reducing and oxidizing agents, respectively. Accordingly, hindpaw injections of reducing agents induced thermal and mechanical hyperalgesia, an effect prevented with the T-type inhibitor mibefradil and absent in Cav3.2−/− mice (40, 41). These data clearly suggest that augmentation of T-type Ca2+ currents can be hyperalgesic, while their inhibition may lead to analgesia. Altered function or expression of T-type Ca2+ channels has also been implicated in chronic pain, as studied experimentally in models such as diabetic neuropathy and constriction-induced chronic pain (16, 59). Thus, for example, T-type Ca2+ current amplitudes are enhanced in DRG neurones in experimental diabetic neuropathy (17), and in vivo antisense knockdown of Cav3.2 suppresses hyperalgesia and DRG T-type Ca2+ currents in this model (35).
The above-described studies shed important light on the mechanism that can account for the hyperalgesic effects of H2S. Thus, for example, the effects of intraplantar injection of NaHS can be abolished by both the oxidizing agent 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) and by known T-type Ca2+ channel inhibitors. Hyperalgesia could also be suppressed by blockade of endogenous H2S production (22). Subsequent studies concluded that H2S could activate or sensitize Cav3.2 channels either in primary afferent or spinal sensory neurons to account for hyperalgesia and in models of chronic pain the associated hyperalgesia and allodynia could be prevented by inhibitors of CSE as well as the T-type channel inhibitor, mibefradil or by antisense knock-down of T-type Ca2+ channels (61). Finally, in a model of H2S-induced colonic pain, the actions of H2S could be mimicked by chelation of Zn2+ (34), which normally tonically blocks Cav3.2 by binding to an extracellular histidine residue (see below) (41).
These behavioral studies strongly indicate that H2S regulation of T-type Ca2+ channels is an important feature of both acute and chronic pain sensation. It is surprising, therefore, that there is no detailed electrophysiological investigation of the modulation of T-type Ca2+ channels by H2S: Kawabata et al. (22) indicated that T-type Ca2+ currents in the NG108-15 hybridoma line were enhanced after 2–3-min exposure to NaHS, and such effects were abolished by either DTNB or the reducing agent dithiothreitol (DTT), but this was only effective at a high concentration of 1.5 mM. There was no evidence presented to indicate that H2S was effective through direct channel modulation (redox or otherwise) or, for example, by altering channel trafficking, which could conceivably be modified within such a timeframe. A schematic summarizing the possible role of H2S in regulating nociception via modulation of T-type channels is shown in Figure 5.

L-type Ca2+ channels
The voltage-gated L-type Ca2+ channel (Cav1.x) has also been shown to be targeted by H2S, but published reports of resulting effects are mixed. In neuronal tissue [both the human neuroblastoma SH-SY5Y (74) and primary cultures of cerebellar granule neurons (10)] evidence has been presented that H2S raises intracellular calcium concentration ([Ca2+]i) via activation of L-type Ca2+ channels. In SH-SY5Y cells, this effect was acute, and rises of [Ca2+]i were observed in response to submillimolar levels of NaHS, peaking after ∼15 min. These rises were inhibited by L-type Ca2+ channel blockers [and also by mibefradil, which preferentially inhibits T-type Ca2+ channels, although SH-SY5Y cells do not express these channels (52)] and also inhibitors of protein kinases A and C. In cerebellar granule neurons, H2S (50–300 μM) also caused rises of [Ca2+]i, which could be inhibited by known L-type Ca2+ channel blockers, but these responses were only reported after much longer periods of exposure (∼1–2 h). Importantly, these rises of [Ca2+]i led to glutamate release and subsequent neurotoxicity, which could be prevented with NMDA receptor antagonists (Fig. 6). This finding contrasts with the previously reported neuroprotective effects of H2S (25), and may also involve the known enhancing effects of H2S on NMDA receptors (1). Despite the general agreement that H2S activates these neuronal L-type Ca2+ channels, conclusions were based on indirect measurements of channel activity (i.e., fluorometric detection of [Ca2+]i) and more direct patch-clamp recordings were lacking.

As was the case in neurons, exposure of hippocampal astrocytes to H2S also raised [Ca2+]i and in some cases induced Ca2+ waves (39), as illustrated in Figure 6. This stimulatory effect of H2S was attributed in part to activation of L-type Ca2+ channels, since effects were reduced in the presence of nifedipine. However, induction and propagation of these Ca2+ waves also involved intracellular Ca2+ stores and alternative (i.e., non-L-type Ca2+ channel dependent) Ca2+ entry pathways, so the relative importance of astrocytic L-type Ca2+ channels—and their regulation by H2S—is not fully understood.
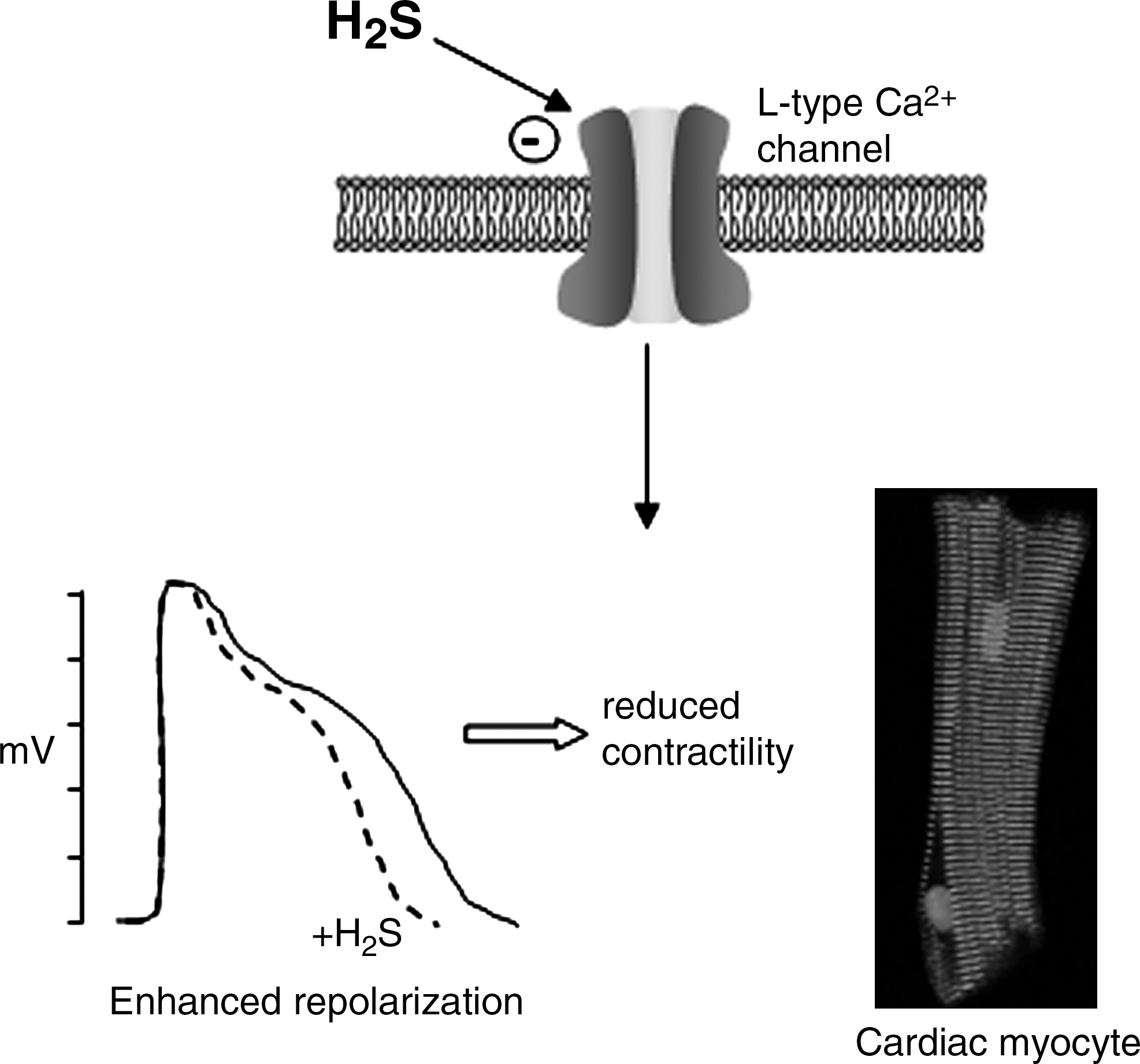
In stark contrast to the effects of H2S on neuronal L-type Ca2+ channels, those recorded in cardiac myocytes were inhibited by H2S (58). This study used whole-cell patch-clamp recordings to measure Ca2+ currents directly, and inhibition by H2S or NaHS was observed at concentrations lower than those required to activate KATP channels. H2S inhibition of the L-type Ca2+ channel current was voltage independent, without effect on channel gating kinetics and independent of activation status (Fig. 7). As might be predicted, these inhibitory effects were associated with a shortening of the cardiac action potential duration and a negative inotropic effect (58). Finally, caffeine-induced Ca2+ transients (arising from mobilization of Ca2+ from intracellular stores via ryanodine receptor activation) were unaffected by H2S, as was also the case also in cultured neurons (10). The authors reported a similar inhibitory effect of H2S on L-type Ca2+ currents in normotensive and spontaneously hypertensive rat strains, and speculated that this important modulatory effect of H2S may contribute not only to a reduction in blood pressure, but also to longer-term protective effects, being potentially antiapoptotic and antihypertrophic. Further studies will resolve the significance of this effect. However, at present it is important to understand why such opposing effects are found on L-type Ca2+ channels in neurons as compared with cardiac myocytes. One suspects that this will only be resolved when we have a more complete understanding of the mechanisms by which H2S regulates ion channels.
Other Ion Channels
A recent study has demonstrated that native (jejunum smooth muscle) and recombinant (Nav1.5) Na+ channels are augmented by H2S (applied as NaHS) with an associated positive shift in steady-state activation and inactivation kinetics (56). These effects were partially reproduced by the reducing agent DTT and prevented by either an oxidant or the alkylating agent N-ethylmaleimide. Although the high (mM) concentrations of NaHS employed in this study suggest that caution should be applied as to the physiological significance of such an effect, the potential influence extends beyond the jejunum, since Nav1.5 is expressed in other tissues, notably the heart, where it gives rise to the upstroke of the cardiac action potential.
Chloride channels represent a widespread, diverse, and relatively understudied group of ion channels that serve numerous roles via permitting Cl− movement not only across the plasma membrane but also across intracellular membranes (42). Indirect evidence, based on the responses to known Cl− channel blockers, suggested that H2S inhibition of Cl− channels in the hippocampal-derived neuronal cell line HT22 contributes to its protective effects against glutamate-induced oxidative stress (24). Similarly, Cl− channels are important in cardiac I/R injury and a recent study demonstrated that Cl− channels isolated from cardiac myocyte mitochondria and inserted into artificial lipid bilayers could be inhibited by H2S and NaHS directly (32). Reduced channel activity was observed when H2S was applied to either face of the channel protein, and was poorly reversible, although no mechanism was proposed for this action. Nevertheless, it could account at least in part for the cardioprotective effects of H2S (27).
Evidence is emerging that H2S can also modulate specific members of the family of cation channels known as transient receptor potential (TRP) channels (45). This channel family is widely distributed and influential in physiological processes as diverse as pain sensation, cardiovascular and renal function, inflammation, and secretion, and as such is currently the focus of intense research. To date, limited studies have suggested that H2S can activate TRPV1 in sensory nerves innervating the airways and so stimulate neurokinin-mediated neurogenic airway inflammation (66). Similarly, TRPA1 in neurons innervating the urinary bladder can be activated by H2S in order to regulate micturition (57). In addition, H2S evoked rises of [Ca2+]i in CHO cells heterologously expressing TRPA1, supporting the notion that H2S may directly activate this channel. Given the diverse and widespread roles of TRP channels throughout the body, their regulation by H2S is likely to account for multiple effects of this gasotransmitter.
Potential Mechanisms
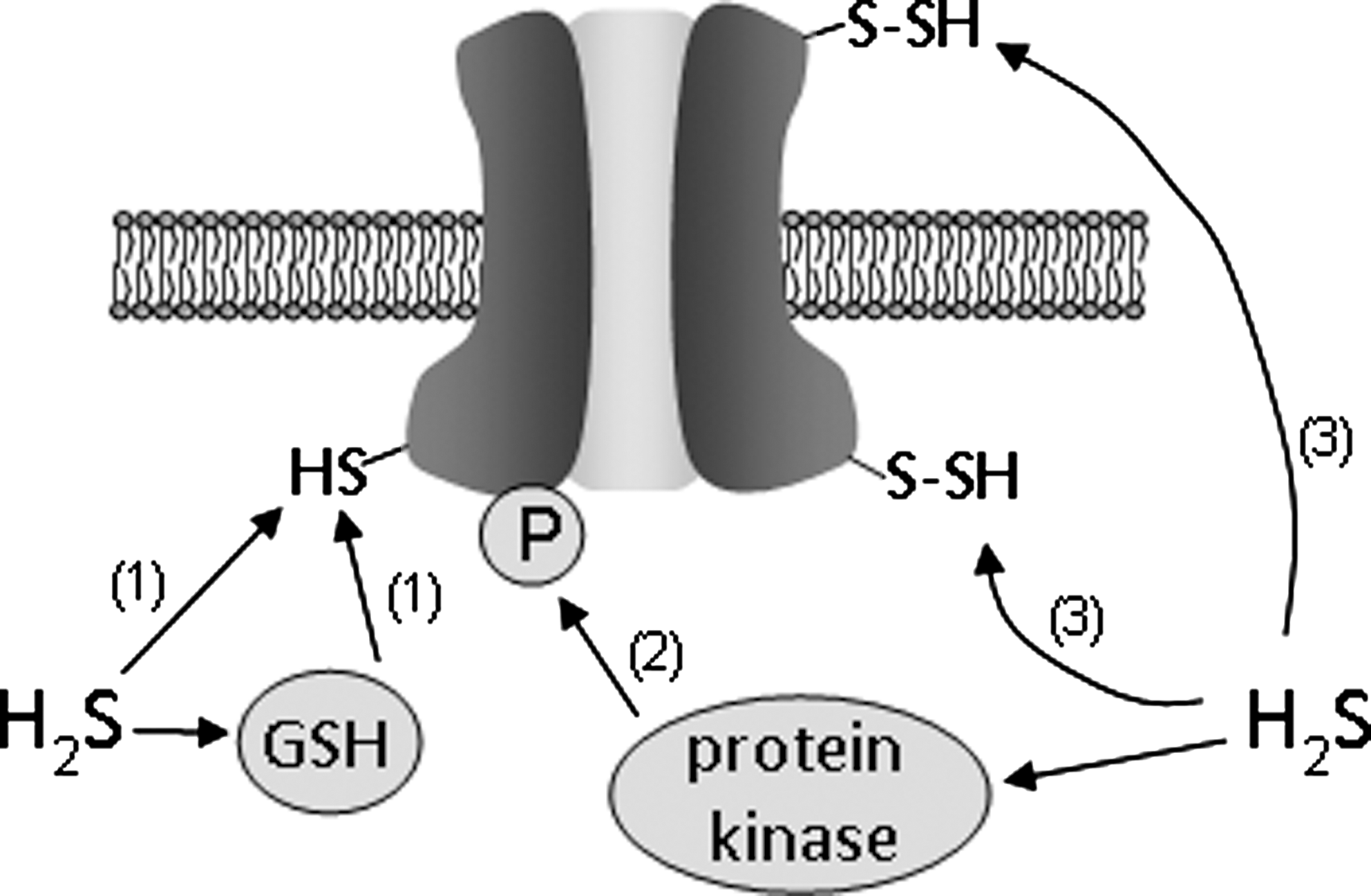
While our appreciation of the widespread and diverse nature of ion channel regulation by H2S is expanding, our insight into possible mechanisms by which such regulation may occur is currently lacking. The most striking candidate mechanisms are illustrated in the schematic of Figure 8 and include the following processes:
Sulfhydration
Snyder and colleagues have recently shown that H2S exerts many of its biological effects through sulfhydration of cysteine thiol groups in target proteins. In this process, –SH groups of cysteine residues are converted to –S-SH groups via addition of sulfur from H2S (37). This post-translational modification appears widespread and analogous to nitrosylation of cysteines (forming –SNO groups) by NO. Indeed, by modifying the biotin switch assay (originally developed to detect NO-mediated nitrosylation of cysteine thiol groups) to permit detection of sulfhydrated proteins, these authors showed a striking loss of physiological protein sulfhydration by comparing proteins from CSE−/− mice with controls, and indicated that it is a remarkably widespread form of protein modification. It remains to be determined whether sulfhydration is a primary mechanism by which H2S regulates ion channels as very little direct information is available. However, likely candidates include the SUR receptor of the KATP channel; these channels are regulated in excised membrane patches (which discounts effects mediated by soluble second messengers) and activation by H2S could be prevented by sulfhydryl alkylation or oxidation (19). Sulfhydration would also seem to be a possible mechanism of regulation of mitochondrial Cl− channels, since these channels were sensitive to H2S when incorporated into lipid bilayers and so H2S could presumably modulate the channels only through a direct effect rather than via involvement of other mediators as discussed below (32). However, sulfhydration is a nonspecific covalent modification with no characterized reversal process (31), so the extent to which it influences specific roles of H2S require further investigation.
Redox modulation
Much evidence indicates that H2S can act as an antioxidant, because it has reducing properties (it may break disulfide bonds) and also increases intracellular GSH levels (29). It should also be noted that CSE deficiency might be expected to lead to reduced cysteine and hence reduced GSH levels; this would in theory compromise a cell's antioxidant capabilities. However, while one study does indeed report reduced GSH levels in CSE−/− (associated with marked vulnerability to oxidative injury and myopathy when dietary cysteine was reduced) (14), another, while reporting a similar hyperhomocysteinemia in CSE−/− mice, indicates that they display only slightly lower levels of intracellular GSH, probably due to the minor reduction of circulating
The above-described enhancing effect of H2S on neuronal T-type channels has been attributed to a channel-reducing effect, being inhibited by the oxidant, DTNB (22). Certainly, T-type Ca2+ current amplitudes can be enhanced in nociceptive DRG neurons (which express primarily Cav3.2) by reducing agents (DTT or
Whether or not redox modulation accounts for the actions of H2S on T-type Ca2+ channels (and hence nociception) remains to be determined. However, some interesting and potentially conflicting issues are emerging concerning gasotransmitter regulation of other channel functions and the involvement of ROS. For example, it has been suggested that H2S enhances BKCa activity via cysteine residue reduction (55), yet previous reports indicate that BKCa is enhanced by oxidation (53). Furthermore, it is noteworthy that CO inhibits the cardiac L-type Ca2+ channel, and that this inhibition arises from increased mitochondrial ROS, which regulate the channel via redox control of key cysteine residues (54). It seems unlikely, therefore, that H2S (which exerts a similar inhibitory effect on the channel) acts through channel protein reduction. However, it should be noted that there are numerous conflicting reports of the effects of reducing and oxidizing agents on the cardiac L-type channel [reviewed in Ref. (80)]. An intriguing possible interaction of gasotransmitters arises from these observations, in that CO is able to stimulate ROS production from mitochondria, yet in some cells 3MST is localized to mitochondria where it can scavenge ROS and provide protection against oxidative stress (23).
Involvement of CO/NO production
Interestingly, H2S has been shown to increase nuclear localization of the transcription factor Nrf2, thereby increasing expression of HO-1. Furthermore, exogenous H2S has been shown to increase levels of HO-1 protein and mRNA in pulmonary arteries (50). This enzyme generates CO, which itself is known to modulate ion channels via a number of signaling pathways (46, 69). This raises the possibility that H2S may modulate ion channels (at least chronically, if not acutely) via the generation of CO, adding further complexity to the biological interaction of these gases. Additionally H2S regulates the vascular bioavailability of the third gasotransmitter NO and therefore may mediate some of the reported effects on ion channels through NO-mediated mechanisms (20).
Involvement of kinases
Ion channels are recognized target proteins for phosphorylation, which can have profound effects not only on their activity but also on their localization, since trafficking of ion channels can be directed by phosphorylation. The two kinases most notably associated with H2S are ERK1/2 and p38 MAP kinase. Much evidence indicates that H2S activates ERK1/2 in vascular smooth muscle and other cells (18, 72), and this represents an attractive alternative mechanism by which H2S might regulate T-type Ca2+ channels; in fact, there is a complex association between ERK activation and T-type Ca2+ channel activity: Chen et al. (5) have shown that Ca2+ influx via T-type Ca2+ channels in sensory neurons leads to ERK activation, which may reflect a positive feedback mechanism since ERK1/2 activation is required for neurotrophic factor-mediated up-regulation of neuronal T-type Ca2+ channels (5) and up-regulation in a recombinant expression system (7). However, in cardiac myocytes ERK1/2 appears to mediate down-regulation of T-type Ca2+ channels by estrogen (33). It remains to be determined whether ERK1/2 inhibitors (e.g., U0126) can affect the ability of H2S to modulate T-type Ca2+ channels either acutely or, following chronic exposure, via altered trafficking. Since ERK1/2 can regulate numerous other ion channels [e.g., Kv2.1 (6)] its involvement in their regulation by H2S is worthy of further study.
Reports also indicate that H2S modulates the activity of p38 mitogen-activated protein kinase (MAPK), but there are some conflicts as to its effects and consequences. In vascular smooth muscle H2S activates p38 MAPK, leading to apoptosis (72). Interestingly, p38 MAPK phosphorylation of Kv2.1 is required for its insertion into the plasma membrane as an initial step in the apoptotic response to oxidative stress (51). However, another report suggests that H2S suppresses agonist-induced MAPK activity (9). These issues and further accounts of the regulation of kinases and their downstream targets (including ion channels) will continue to emerge in due course. It is evident from the widespread regulation of ion channels by phosphorylation and the increasing awareness of H2S as a modulator of kinase activity/expression that kinases will be seen to account for many of the signaling effects of H2S on ion channels, as well as other target proteins.
Concluding Remarks
It is clear from the growing number of reports that ion channels are an important and diverse family of target proteins for regulation by H2S. Further research will expand this family, and will hopefully provide greater insight into mechanisms of channel modulation—it will be particularly interesting to determine whether the novel, post-translational modification by sulfhydration can account for any of the effects described herein. Future studies must also take advantage of transgenic models in order to elucidate the tonic, physiological regulation of channel expression and activity by endogenous H2S. The field of H2S biology in general would also benefit enormously from a means by which to detect H2S levels dynamically at the sub-cellular level. Such developments are hopefully near at hand to allow us to explore the regulation of ion channels by H2S more fully, and so provide new opportunities for therapeutic intervention in a wide variety of clinical applications.
Footnotes
Acknowledgments
We are grateful to colleagues for discussions and permitting us to illustrate this review with schematic diagrams based on their data, and apologize to those contributors to the field whose work we could not cite due to space limitations. The authors' own contributions to the field are supported by the Wellcome Trust, the Alzheimer's Society, and the British Heart Foundation.