
Abstract
Introduction
To date, numerous conflicting data regarding the pro- and anti-inflammatory activity of exogenous and endogenous sulfide have been reported. For example, various H2S donors (e.g., sodium hydrosulfide [NaHS]) have been reported to worsen (11, 29, 30) or attenuate (12, 13) inflammation, while CSE inhibitors (e.g., DL-propargylglycine [PAG]) exhibit anti-inflammatory activity in animal models of endotoxemia and sepsis (11, 29, 30). Although the reasons for these conflicting results are likely multifactorial, purity and/or specificity of chemical H2S donors and inhibitors have been questioned. For example, solutions of NaHS appear to include polysulfides and elemental sulfur (2), whereas PAG may affect other pyridoxal 5′-phosphate-dependent enzymes as well (13).
Another critical issue that makes the biological role of H2S controversial is the lack of general consensus about the biologically relevant levels of H2S in tissues and plasma. It is of note that H2S levels of 105-fold concentration range have been reported (8). Effects of exogenous H2S in endotoxemia and sepsis are likely to be affected by the endogenous levels of sulfide and sulfide metabolism.
Innovation
Although H2S has been suggested to have anti-inflammatory as well as pro-inflammatory effects, the role of H2S in endotoxemia is incompletely understood. Moreover, the concentration of the biologically relevant reactive H2S levels in vivo has been the focus of controversy. Using an advanced analytical technique, the current study revealed that LPS challenge decreased plasma sulfide concentration in mice. On the other hand, breathing H2S after LPS challenge restored sulfide levels and markedly increased thiosulfate concentrations in plasma. The increased thiosulfate levels in LPS-challenged mice that breathed H2S appeared to be caused by LPS-induced upregulation of sulfurtransferase rhodanese. Based on these observations, we hypothesized that thiosulfate may be contributing to the beneficial effects of H2S inhalation. For the first time to our knowledge, we demonstrated that administration of STS per se dose-dependently improves survival rate of mice subjected to LPS challenge. Since STS is clinically approved for the treatment of cyanide poisoning, the current observations may have significant translational potential for the treatment of patients with sepsis. Taken together, the current results put forth an innovative hypothesis that breathing H2S exerts anti-inflammatory effects and improves survival during murine endotoxemia, in part by remodeling sulfide metabolism and increasing thiosulfate levels.
To elucidate the impact of authentic H2S in endotoxin (lipopolysaccharide[LPS])-induced inflammation, we examined the hypothesis that H2S breathing prevents LPS-induced systemic inflammation and organ injury and improves survival in mice. We also sought to determine the plasma levels of H2S and sulfide metabolites using the monobromobimane (MBB)-based HPLC method (20, 27). Here, we report that LPS challenge decreases plasma sulfide levels in mice. Breathing H2S attenuates LPS-induced systemic inflammation and improves survival in mice by restoring sulfide and markedly increasing thiosulfate levels. We further demonstrate that administration of sodium thiosulfate (STS) per se dose-dependently improves survival rate of mice after LPS challenge.
Results
H2S inhalation improves survival rate after LPS challenge
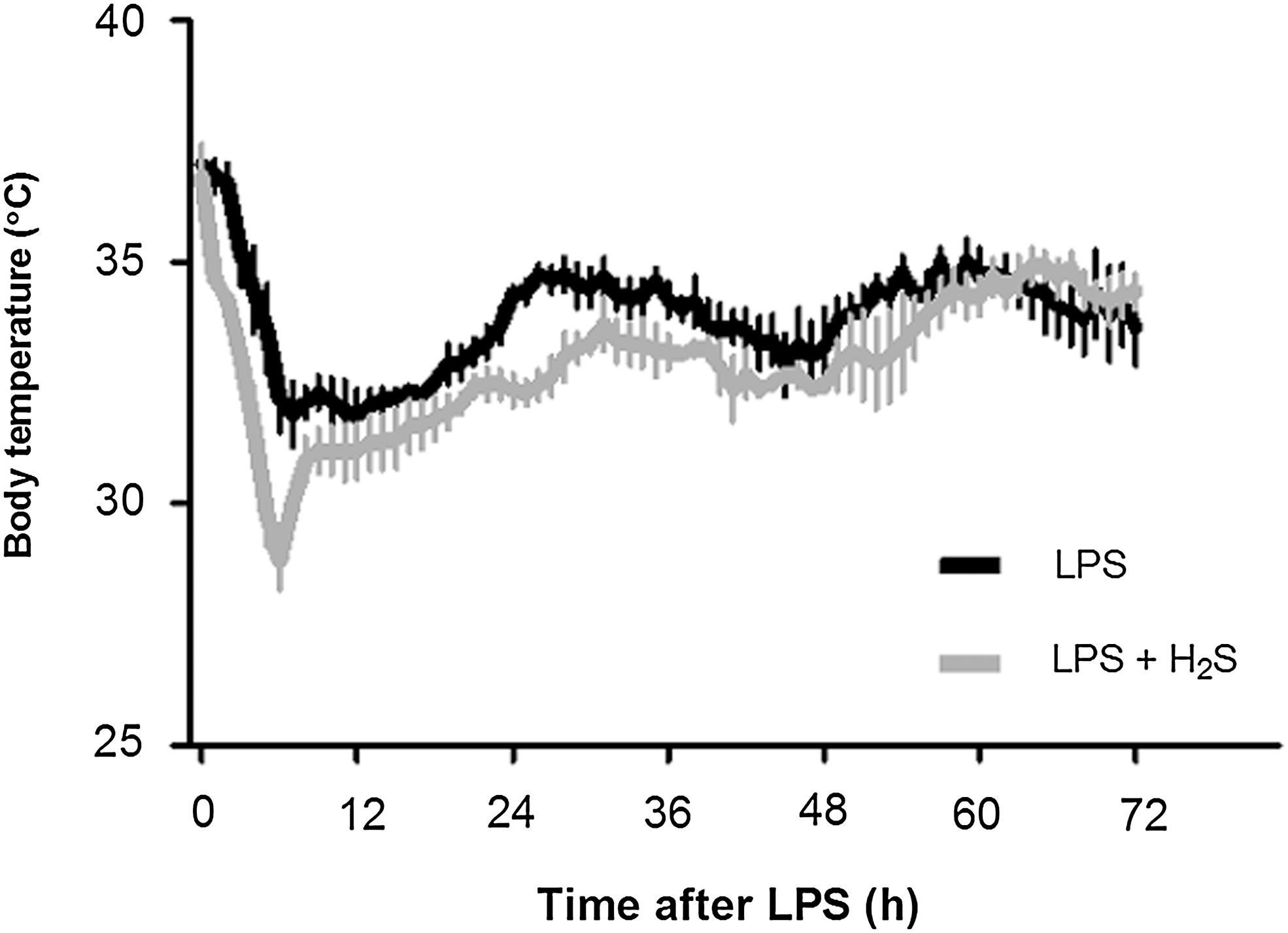
Six out of eight mice died within 72 h after LPS challenge. In contrast, only one out of six mice died that breathed H2S for 6 h after LPS challenge (Fig. 1, p=0.0161 by Log-rank test). No mice died thereafter up to 7 days after LPS challenge (data not shown). LPS challenge decreased body temperature of mice; body temperature reached the lowest points (∼32°C) between 6 h and 12 h and gradually recovered to ∼35°C by 72 h after LPS challenge. While the onset of hypothermia was faster and the lowest temperature (∼29°C) was lower in mice that breathed air supplemented with H2S than in mice that breathed air alone, overall body temperature was comparable for 72 h after LPS challenge between the two groups of mice (p=0.0612, Fig. 2).

H2S breathing attenuates plasma NOx levels
LPS challenge significantly increased plasma nitrite/nitrate (NOx) levels in mice that breathed air, whereas H2S inhalation attenuated LPS-induced rise of plasma NOx levels. These results suggest that H2S breathing suppressed accumulation of NO in mice during endotoxemia (Fig. 3A). Breathing H2S without LPS challenge did not increase NOx levels.
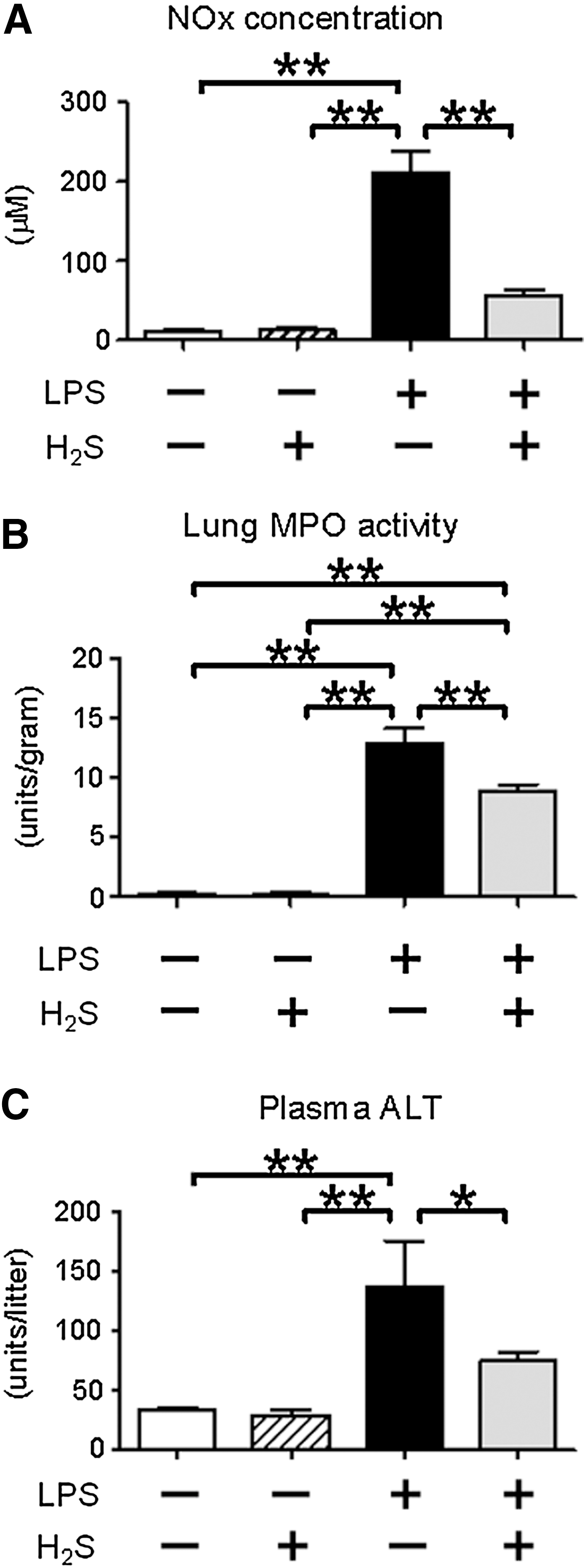
H2S breathing attenuates lung MPO activity
Lung myeloperoxidase (MPO) activity was measured at 6 h after LPS challenge as an index of polymorphonuclear neutrophil infiltration in the lung. LPS challenge markedly increased lung MPO activity. Breathing H2S attenuated LPS-induced increase of lung MPO activity (Fig. 3B). Breathing H2S without LPS challenge did not increase lung MPO activity.
H2S attenuates liver injury after LPS challenge
Plasma levels of alanine aminotransferase (ALT) at 24 h after LPS challenge were measured as a marker of liver injury. LPS challenge markedly increased ALT levels in mice that breathed air, but not in mice that breathed H2S (Fig. 3C). These results suggest that inhaled H2S attenuated liver injury induced by endotoxemia.
H2S breathing prevents systemic inflammation after LPS challenge
Levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and nitric oxide synthase (NOS) 2 mRNA in the liver and lung, and IL-6 and intercellular adhesion molecule (ICAM)-1 in the liver were increased at 6 h after LPS challenge (Fig. 4). H2S inhalation markedly attenuated LPS-induced upregulation of TNF-α, IL-1β, IL-6, NOS2, and ICAM-1 expression in the liver, and IL-1β and NOS2 mRNA expression in the lung. In contrast, LPS-induced upregulation of TNF-α was not attenuated by inhaled H2S in the lung. Breathing H2S without LPS challenge did not alter mRNA levels of these inflammatory mediators in the liver and lung.

H2S breathing attenuates phosphorylation of IκBα after LPS challenge
To elucidate the molecular mechanisms responsible for the attenuated inflammatory cytokine induction by H2S breathing after LPS challenge, we examined phosphorylated IκB levels in the liver that releases the inhibition of NF-κB leading to activation of NF-κB-dependent transcription of cytokines. While phosphorylation of IκBα at serine 32/36 increased in the liver at 1 h after LPS injections, H2S breathing attenuated the phosphorylation of IκBα induced by LPS challenge (Fig. 5A). These results suggest that H2S breathing decreased LPS-induced activation of the NF-κB-dependent signaling.
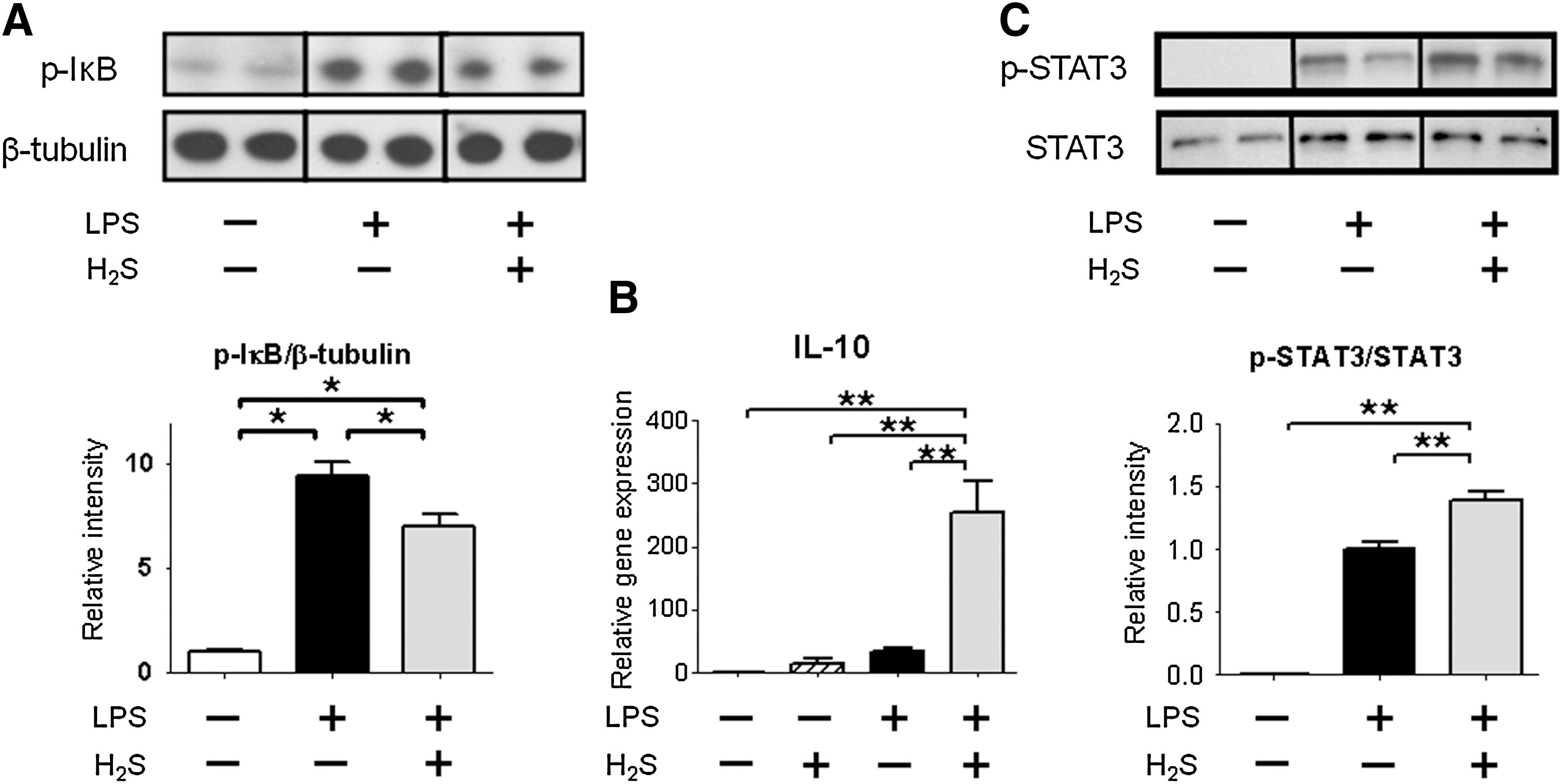
H2S inhalation increases nuclear translocation of phosphorylated STAT3
Although LPS challenge or H2S inhalation alone did not affect IL-10 mRNA levels, IL-10 levels were markedly increased by H2S inhalation after LPS challenge (Fig. 5B). As phosphorylated signal transducer and activator of transcription 3 (STAT3) can translocate into the nucleus and initiate IL-10 gene transcription (18), we examined the extent of phosphorylation of STAT3 at tyrosine 705 in nuclear protein fraction of the liver at 1 h after LPS challenge. LPS significantly increased phosphorylated STAT3 in nuclear extracts in mice that breathed air. The nuclear translocation of phosphorylated STAT3 was further augmented by H2S breathing after LPS challenge (Fig. 5C). These results suggest that anti-inflammatory effects of H2S breathing after LPS challenge may be mediated by STAT3/IL-10-dependent signaling.
H2S inhalation increases plasma sulfide and thiosulfate levels after LPS challenge
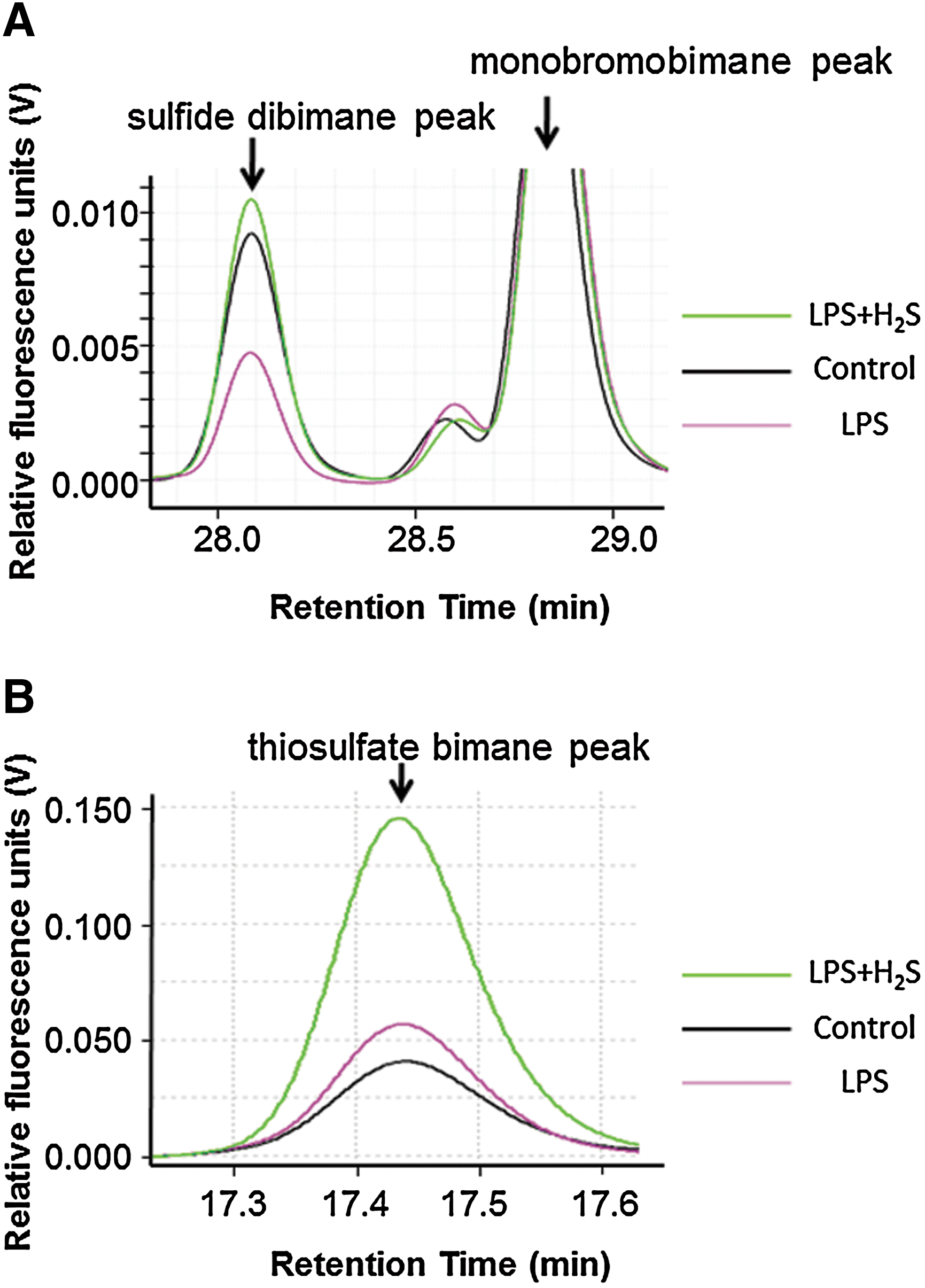
To determine the impact of endotoxemia and H2S breathing on sulfide metabolism, we measured plasma concentrations of sulfide, sulfite, and thiosulfate with HPLC after derivatization with MBB as sulfide dibimane (SDB), sulfite bimane (SB), and thiosulfate bimane (TSB), respectively (Fig. 6). LPS challenge decreased plasma sulfide concentration, whereas 80 ppm of H2S inhalation for 6 h without or with LPS challenge increased plasma sulfide level (Figs. 6A and 7A). H2S inhalation and or LPS challenge did not affect sulfite levels (Fig. 7B). H2S inhalation at 80 ppm per se modestly increased plasma thiosulfate levels, while LPS challenge did not affect thiosulfate levels. H2S breathing after LPS challenge markedly increased plasma thiosulfate levels (Figs. 6B and 7C).

Endotoxin challenge upregulates rhodanese in the liver
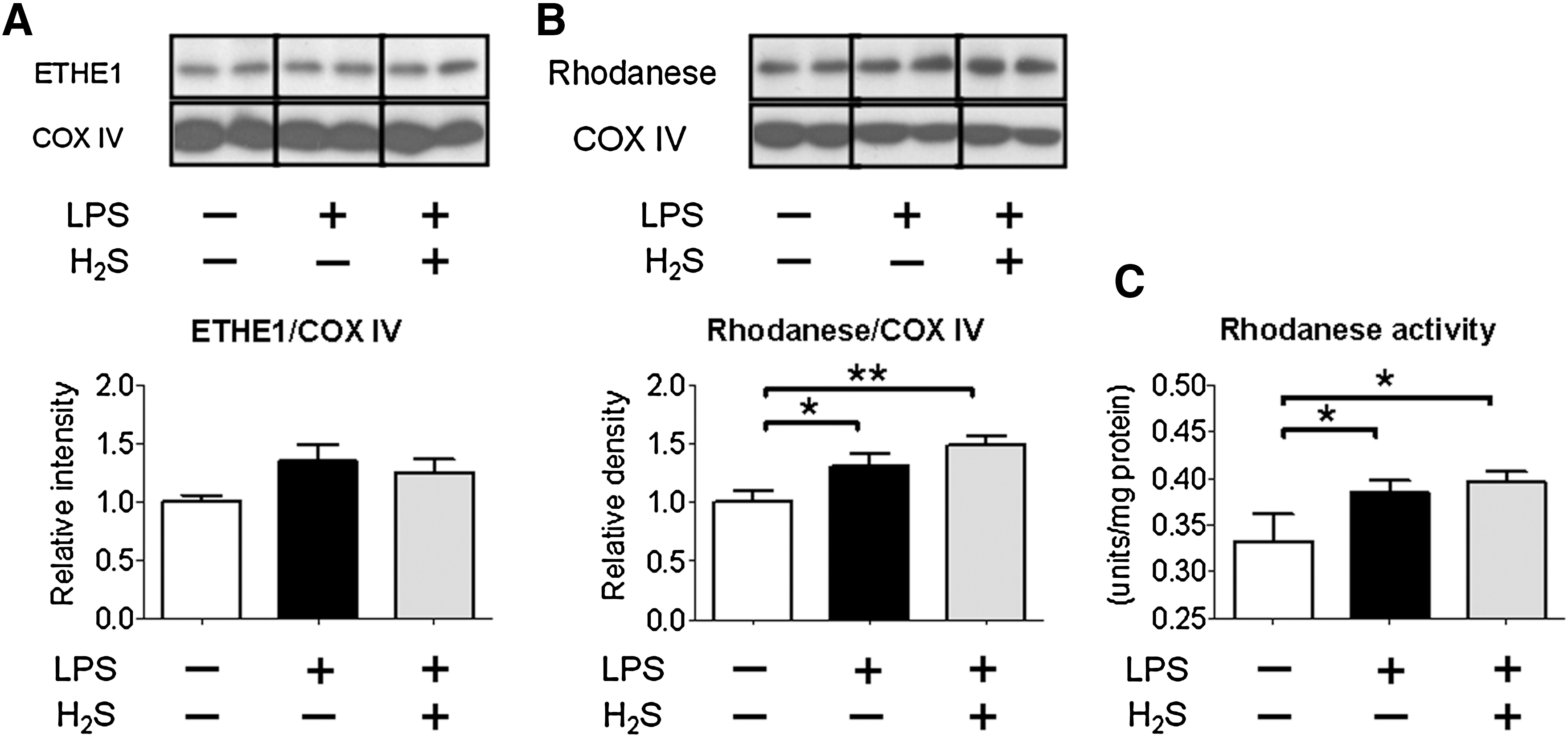
To elucidate the mechanisms responsible for the altered sulfide metabolism after LPS challenge, we examined expression levels of CSE in the liver and lung and ETHE1 and rhodanese in the liver. Although LPS challenge decreased CSE mRNA levels in the liver, protein levels and activity of CSE were not altered by LPS challenge with or without H2S breathing (Fig. 8). CBS and 3MST mRNA levels were not altered by LPS challenge with or without H2S breathing (data not shown). LPS challenge with or without H2S breathing increased levels of rhodanese, but not ETHE1 (Figs. 9A and 9B). In addition, LPS challenge with or without H2S breathing increased rhodanese activity in the liver (Fig. 9C). These results suggest that LPS-induced upregulation of rhodanese contributed to the altered sulfide metabolism after LPS challenge and H2S breathing.

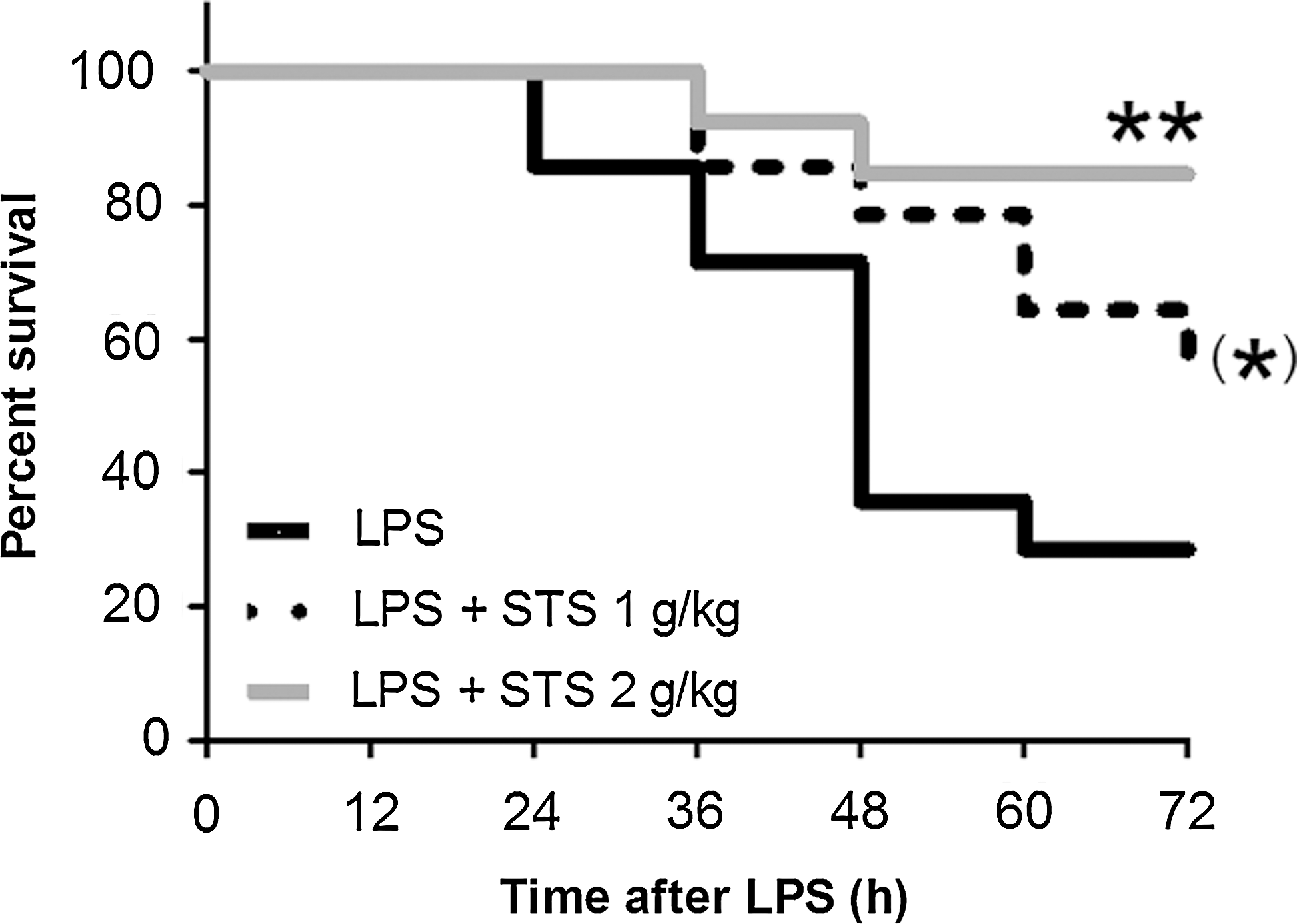
Administration of thiosulfate after LPS challenge improves survival in mice
Since beneficial effects of H2S inhalation after LPS challenge was associated with markedly increased plasma levels of thiosulfate, we hypothesized that thiosulfate per se prevents endotoxin-induced mortality. We found that intraperitoneal administration of STS dose-dependently improved survival of mice subjected to LPS challenge (Fig. 10). These results suggest that beneficial effects of H2S breathing may be mediated in part by increasing thiosulfate levels after LPS challenge.
Discussion
The current study revealed that breathing H2S at 80 ppm for 6 h after LPS challenge markedly improved survival in mice. Inhalation of H2S attenuated LPS-induced systemic nitrosative stress, liver injury, and neutrophil infiltration in the lung. The protective effects of H2S inhalation were associated with inhibition of LPS-induced inflammatory cytokine induction and marked upregulation of anti-inflammatory cytokine IL-10 in the liver. Our study also revealed that plasma sulfide levels were reduced by LPS challenge, while H2S breathing restored sulfide levels and markedly increased thiosulfate levels during endotoxemia. The altered sulfide metabolism after LPS challenge and H2S breathing appeared to be caused by upregulation of rhodanese. Finally, administration of STS dose-dependently improved survival after LPS challenge. These observations suggest that H2S inhalation exhibits potent anti-inflammatory effects leading to improvement of survival during murine endotoxemia in part by remodeling sulfide metabolism in mice.
As in the case of NO, it is likely that H2S dose-dependently exerts wide spectrum of effects during inflammation; low and physiological sulfide concentrations are anti-inflammatory while high sulfide levels are pro-inflammatory. Li and colleagues reported that intraperitoneal administration of NaHS per se at 14 μmol/kg (=784 μg/kg) resulted in marked histological signs of lung inflammation, increased lung and liver MPO activity, and raised plasma TNF-α concentration (11). Therefore, it is no surprise that high dose NaHS (10 mg/kg) aggravates inflammation in septic mice (29, 30). In contrast, our results are consistent with the anti-inflammatory effects of the slow-releasing H2S donor, GYY-4137 [morpholin-4-ium 4 methoxyphenyl (morpholino) phosphinodithioate] (14), in endotoxemic rats and in macrophages incubated with LPS (13, 26). Since NaHS releases large amounts of H2S over a period of few seconds (14, 26), it is likely that tissues are exposed to super-physiological levels of sulfide after systemic administration of NaHS. By demonstrating the beneficial effects of sustained levels of sulfide provided by H2S breathing for 6 h, the current results support the hypothesis that low and physiological levels of sulfide confer anti-inflammatory effects during endotoxemia.
It is well documented that a variety of stressors, including endotoxemia, sepsis, hypoxemia, anesthesia, and surgery, can markedly decrease body temperature of rodents unless they are actively warmed with external heating source (1, 3, 16, 19). In particular, sepsis-induced hypothermia has been suggested to be either protective (10) or detrimental (28). On the other hand, breathing H2S has also been shown to decrease body temperature in mice (25). Since warming mice during sepsis may aggravate or prevent inflammation (10, 28), we chose not to “force” normothermia during endotoxemia with or without H2S breathing in the current study to avoid possible confounding effects of heating. We observed profound hypothermia in mice after LPS challenge without or with H2S breathing, while onset of hypothermia was faster with H2S breathing than with air breathing. Although we cannot exclude the possibility that slightly lower body temperature of mice that breathed H2S, especially during the first several hours after LPS challenge, contributed to the beneficial effects of H2S breathing, it is likely that H2S breathing exerted anti-inflammatory effects independent of its effects on body temperature. Nonetheless, impact of core body temperature on the anti-inflammatory effects of H2S breathing remains to be formally determined.
The current study showed that inhalation of H2S attenuated LPS-induced phospohorylation of IκB in the liver, supporting the hypothesis that H2S breathing exerts anti-inflammatory effects via inhibition of NF-κB activation. The protective effects of H2S breathing on liver function during endotoxemia were also associated with marked upregulation of IL-10 gene expression and increased nuclear translocation of phosphorylated STAT3 in the liver, consistent with a previous study (13). Taken together, these observations support the hypothesis that STAT3/IL-10 signaling contributes to the anti-inflammatory/pro-survival effects of H2S.
Although several studies reported increased plasma sulfide levels after LPS challenge and peritonitis in rodents and in patients with sepsis (11, 12, 30), these studies used the methylene blue methods to measure sulfide concentrations. The methylene blue method uses harsh acidic conditions under which sulfide leaches from iron–sulfur cluster-containing proteins or is eliminated via desulfuration, leading to overestimation of “free” sulfide levels in biological samples (8, 17). To examine the impact of endotoxemia on biologically available sulfide levels in plasma, we measured plasma sulfide levels with HPLC after derivatization using MBB (20, 27). In contrast to the previous reports that used the methylene blue method, we observed that plasma sulfide levels were decreased at 6h after LPS challenge compared to baseline.
Furthermore, H2S inhalation restored plasma sulfide levels and prevented LPS-induced organ injury and death. These observations are reminiscent of a recent study in which Suzuki and colleagues reported that H2S levels were reduced in diabetic rats and H2S replacement protects vascular endothelium by preserving mitochondrial function (23). It is conceivable that restoration of sulfide levels by H2S inhalation during endotoxemia may prevent endothelial dysfunction via similar mechanisms.
The current results also revealed that H2S inhalation markedly enhanced the LPS-induced increase of plasma thiosulfate levels, whereas plasma sulfite concentrations were not altered by LPS challenge with or without H2S breathing. To define the mechanisms responsible for the altered sulfide metabolite levels after LPS challenge, we examined levels of enzymes responsible for production and or metabolism of sulfide in the liver. LPS challenge decreased CSE mRNA levels without altering protein levels and activity in the liver. It is possible that CSE protein/activity levels are altered at later time points. Protein levels of ETHE1 were not altered by LPS challenge or H2S breathing, consistent with the unaltered levels of sulfite. In contrast, not only protein levels but also activity of rhodanese, that produces thiosulfate from persulfide and sulfite, was increased by LPS challenge with or without H2S breathing. These results suggest that LPS-induced upregulation of rhodanese is responsible for the decreased sulfide levels after LPS challenge. Inhaled H2S after LPS challenge increased plasma thiosulfate levels compared to LPS alone presumably because of synergistic effects of upregulated rhodanese activity and increased sulfide load provided by H2S breathing.
Given the markedly increased levels of thiosulfate in mice that breathed H2S after LPS challenge, we hypothesized that augmented thiosulfate levels may contribute to the beneficial effects of H2S breathing. We found administration of STS dose-dependently improved survival after LPS challenge in mice. These results not only support our hypothesis but also suggest an exciting possibility that administration of STS itself may be beneficial for the treatment of sepsis and systemic inflammation. Thiosulfate is a potent antioxidant and STS has been used for the treatment of cyanide poisoning and calciphylaxis (4). Alternatively, 3MST and rhodanese can convert thiosulfate to H2S that may mediate salutary effects of STS (15). Mechanisms responsible for the beneficial effects of STS during endotoxemia, including the similarities and differences between H2S and STS, remain to be determined in the future studies.
This study has several important limitations. We studied the effects of H2S inhalation started only at the time of LPS challenge. Effects of inhaled H2S started at later time points were not examined. Although H2S and LPS did not alter mRNA levels of CBS and 3MST, we did not examine protein levels and activity of CBS and 3MST. Since 3MST can produce thiosulfate from sulfite and H2S from thiosulfate (15), the role of 3MST in sulfide metabolism during endotoxemia remains to be elucidated.
In conclusion, the current study revealed robust anti-inflammatory effects of H2S breathing that improved survival rate in murine endotoxin shock model. The anti-inflammatory effects of H2S breathing after LPS challenge were mediated at least in part via enhancing STAT3/IL-10 system as well as inhibiting NF-κB-dependent transcription. Our results also demonstrate that plasma sulfide levels were decreased by LPS challenge, whereas H2S breathing restored sulfide levels and markedly increased thiosulfate concentrations. Administration of STS markedly improved survival after LPS challenge. These observations suggest that H2S breathing attenuates inflammation and improves survival after LPS challenge in part by altering sulfide metabolism in mice.
Materials and Methods
Animals and endotoxin challenge model
After approval by the Massachusetts General Hospital Subcommittee on Research Animal Care, all animal experiments were performed in accordance with the guidelines of the National Institutes of Health. Male mice (C57BL/6J, 8–10 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME) and given access to food and water ad libitum in our animal facility until the time of experiments. LPS challenge was performed by administering Escherichia coli 0111:B4 endotoxin (10 mg/kg, Sigma-Aldrich, St. Louis, MO) intraperitoneally. Control mice received saline at the same volume with LPS intraperitoneally.
Survival analysis after endotoxin challenge
Survival after challenge with LPS or saline was studied in age-matched male mice. To avoid dehydration, normal saline (1 ml) was given intraperitoneally to all mice at times 0, 6, and 24 h after LPS challenge. Ambient temperature was controlled at 25°C. In separate groups of mice, changes in body temperature after LPS challenge were recorded with radiotelemetry devices (Data Science International, St. Paul, MN), as described previously (25). To examine the effects of thiosulfate, in separate groups of mice, STS solution was administered intraperitoneally at 1 or 2 g/kg immediately after LPS challenge. Survival rate was compared to concurrent control mice that received LPS alone.
H2S breathing
Immediately after challenge with LPS or saline, mice assigned to H2S breathing were exposed to air containing 80 ppm of H2S (Airgas Inc., Radnor, PA) in custom-made chambers for 6 h, then replaced in their cages to breathe air alone thereafter until the end of experiments. Concentration of H2S was chosen based on dose-ranging studies. H2S concentration, as well as FiO2, was continuously measured using a portable gas monitor (ITX Multi-Gas Monitor, detection limit 1 ppm, Industrial Scientific Corporation, Oakdale, PA).
Measurements of gene expression
Total RNA was extracted from liver and lung tissues of mice at baseline (healthy control) and from mice at 6 h after challenge using the RNAspin Mini kit (GE Healthcare, Piscataway, NJ) and cDNA was synthesized using MMLV-RT (Promega, Madison, WI). TNF-α, IL-1β, IL-6, NOS2, ICAM-1, IL-10, CSE, CBS, 3MST, and 18S ribosomal RNA transcript levels were measured by real-time PCR using a Realplex 2 system (Eppendorf North America, Westbury, NY). The primer sequences are listed in Table 1. Changes in the relative gene expression normalized to levels of 18S rRNA were determined using the relative CT method. The mean value of samples from control wild type mice was set as 1.
CBS, cystathionine β-synthase, CSE, cystathionine γ-lyase; ICAM-1, intercellular adhesion molecule 1, IL-1β, interleukin 1β; IL-6, interleukin 6; IL-10, interleukin 10; 3MST, 3-mercapropyruvate sulfurtransferase. NOS2, nitric oxide synthase 2; TNF- α, tumor necrosis factor α.
Plasma chemistry
Plasma ALT and NOx levels at baseline and 24 h (ALT) or 6 h (NOx) after LPS injections with or without H2S inhalation for 6 h were quantified with an Infinity™ ALT assay regent (Thermo Fisher Scientific Inc., Middletown, VA) or with a Nitrate/Nitrite Fluorometric Assay Kit (Cayman Chemical, Ann Arbor, MI), respectively, according to the manufacturers' instructions.
Myeloperoxidase activity in lung
Polymorphonuclear neutrophil infiltration in the lungs was estimated by measuring MPO activity in lung tissue homogenates as described previously (5).
Protein levels and phosphorylation
Liver tissue was dissected and frozen at 1 or 6 h after LPS injections. Protein levels in liver homogenates were determined using standard immunoblot techniques using primary antibodies (1:10,000, Cell Signaling Technology Inc., Danvers, MA, unless otherwise noted) against phosphorylated IκBα at serine 32/36, STAT3, phosphorylated STAT3 at tyrosine 705, CSE (1:5,000, (7)), ETHE1 (1:5,000, GeneTex, Irvine, CA), rhodanese (1:5,000, GeneTex), β-tubulin, GAPDH, and COX IV. Bound antibody was detected with a horseradish peroxidase-linked antibody directed against rabbit IgG (1:10,000∼1:100,000; Cell Signaling Technology Inc.) or mouse IgG (1:50,000∼1:100,000; Thermo-Pierce, Rockford, IL) and was visualized using chemiluminescence with ECL Advance kit (GE healthcare).
CSE and rhodanese activity in liver
CSE and rhodanese activity in the liver was measured as described previously (7, 24).
Plasma sulfide concentration
Concentrations of free sulfide, sulfite, and thiosulfate in plasma at 6 h after LPS challenge were measured by HPLC after derivatization with excess MBB as stable products SDB, SB, and TSB, respectively, as previously described (20, 27). Briefly, at 6 h after saline or LPS injections, blood was drawn from left ventricle and centrifuged to collect the plasma. 30 μl of plasma was added to 70 μl of 100 mM Tris-HCl buffer (pH 9.5, 0.1 mM diethylenetriamine pentaacetic acid [DTPA]), followed by addition of 50 μl of 10 mM MBB (Sigma-Aldrich). Reaction was stopped by adding 50 μl of 200 mM 5-sulfosalicylic acid after 30 min, and the mixture was centrifuged and the supernatant was analyzed by HPLC with a fluorescence detector (Waters 2475 Multi λ fluorescence detector, Waters, Milford, MA) equipped with Agilent 258 Eclipse XDB-C18 column (Agilent Technologies, Santa Clara, CA) at wavelength of λex=390 nm and λem=475 nm. Typical retention times for SB, TSB. and SDB were around 13.7, 17.4, and 28.1 min, respectively. Standard SDB was synthesized according to the reported method (20). Sodium sulfite and STS stock solutions were diluted to the desired concentrations in Tris-HCl buffer and derivatized with MBB to make SB and TSB standard solutions, respectively. Detection limits of SB, SDB, and TSB were 0.18, 0.029, and 0.84 μM, respectively.
Statistics
All data are presented as means±SE. Data were analyzed by ANOVA using Sigmastat 3.01a (Systat Software Inc., Chicago, IL) and Prism 5 software package (GraphPad Software, La Jolla, CA). Newman-Keuls multiple comparison post hoc test was performed as required. Kaplan-Meier survival analysis was performed using Log-rank test. P values smaller than 0.05 were considered significant.
Footnotes
Acknowledgments
We thank Drs. Takaaki Akaike and Isao Ishii for technical advice.
This work was supported by National Institute of Health grants GM79360 and HL101930 to F. Ichinose.
Author Disclosure
No competing financial interests exist.