
Abstract
The tumor microenvironment is a complex system that involves the interaction between malignant and neighbor stromal cells embedded in a mesh of extracellular matrix (ECM) components. Stromal cells (fibroblasts, endothelial, and inflammatory cells) are co-opted at different stages to help malignant cells invade the surrounding ECM and disseminate. Malignant cells have developed adaptive mechanisms to survive under the extreme conditions of the tumor microenvironment such as restricted oxygen supply (hypoxia), nutrient deprivation, and a prooxidant state among others. These conditions could be eventually used to target drugs that will be activated specifically in this microenvironment. Preclinical studies have shown that modulating cellular/tissue redox state by different gene therapy (GT) approaches was able to control tumor growth. In this review, we describe the most relevant features of the tumor microenvironment, addressing reactive oxygen species-generating sources that promote a prooxidative microenvironment inside the tumor mass. We describe different GT approaches that promote either a decreased or exacerbated prooxidative microenvironment, and those that make use of the differential levels of ROS between cancer and normal cells to achieve tumor growth inhibition. Antioxid. Redox Signal. 19, 854–895.
I. The Tumor Microenvironment
A. Tumor microenvironment components
1. Malignant cells
Malignant cells harbor the genetic alterations that define cancer as a genetic disease. These genetic alterations involve single mutations and amplification or loss of large regions of the genome (6, 327). Dominant gain of function (oncogenes) and recessive loss of function (tumor suppressor genes) are the gatekeeper genes that stand at the root of the initiation of neoplastic growth. The large diversity of mutated genes that exists in cancer cells (6, 327) finally affects intrinsic cellular programs, such as cell cycle checkpoint controls, programmed cell death (PCD), differentiation, metabolism, and cell adhesion, which underlie cancer progression. Cancer exhibits a wide genetic heterogeneity, not only among tumors of different origins but also within the same tumor type. This heterogeneity was historically demonstrated through the expression of tumor-associated antigens and more recently, by advanced genome-sequencing techniques. For instance, cancer cells microdissected from different sections of the same tumor samples showed genetic heterogeneity (120). Thus, subpopulations of cancer cells can be defined as having distinct and complementary capabilities to enhance tumor growth (120).
In recent years, accumulated evidence suggests the presence of a subclass of hierarchical neoplastic cells within tumors, termed as cancer stem cells (CSCs). CSCs were initially identified in hematopoietic malignancies and later in solid tumors, in particular breast, neuroectodermal, pancreatic, and colorectal cancer (120). CSCs are defined operationally by their ability to efficiently seed new tumors upon inoculation in host immunodeficient mice (120). Additionally, CSCs express surface markers typically associated with normal stem cells such as CD44 and CD24 (63). Experimental evidence suggests that CSCs have plastic states governed by microenvironmental conditions, such as hypoxia (37, 186), and hold a key role in the regulation of tumor angiogenesis (36, 37, 246). Moreover, recent reports link the resistance to conventional therapies and the metastatic potential to the CSC population (82).
2. Tumor-associated stromal cells
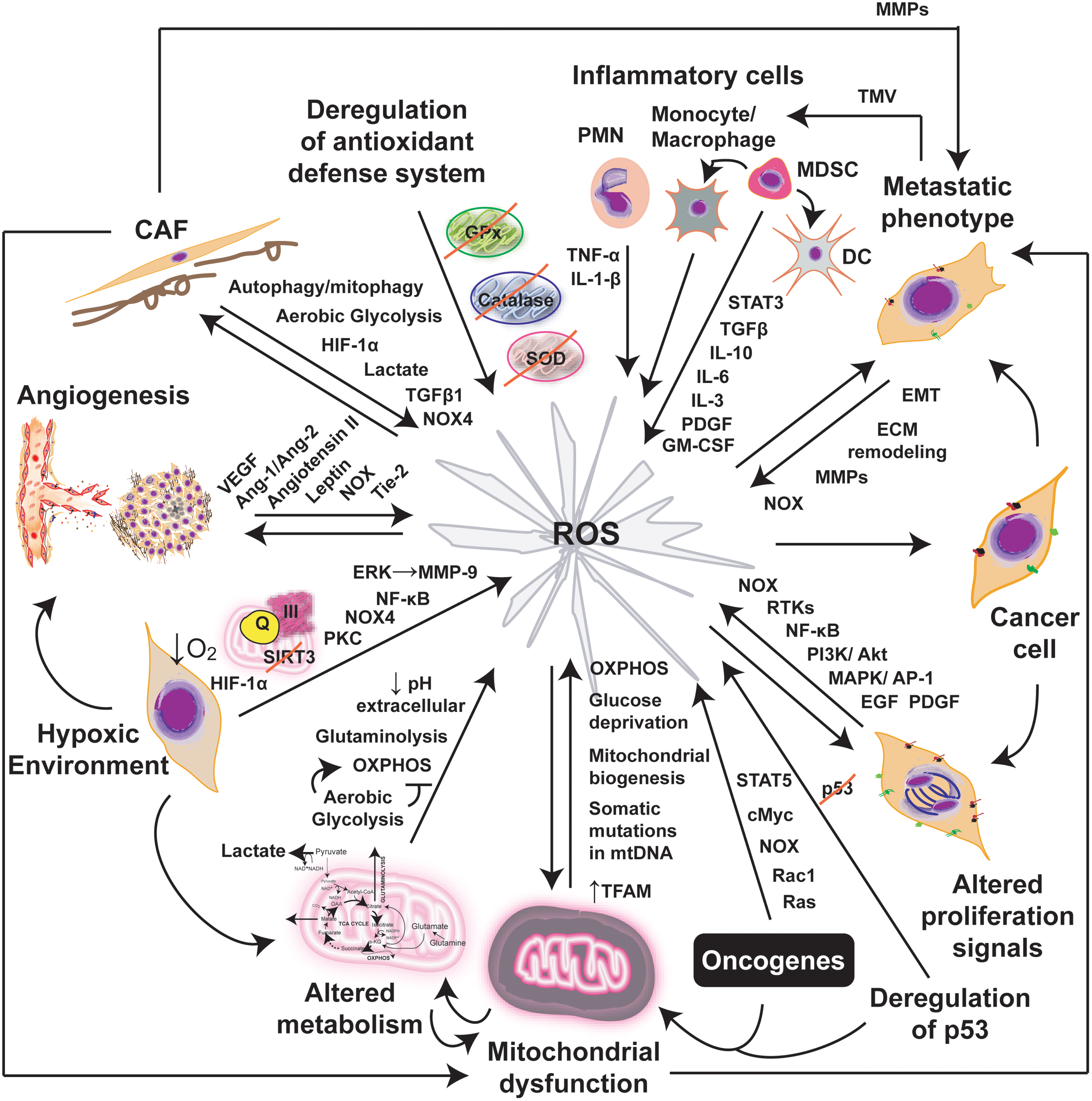
A diverse population of normal or genetically stable cells (currently in discussion) (35) is associated with cancer cells to conform the stromal compartment. Thus, fibroblasts, vasculature-associated cells, immune cells, stem progenitor cells, and other specialized mesenchymal cells support tumor growth and dissemination (Fig. 1A) by inducing different intracellular programs (Fig. 1B), building together the tumor microenvironment. In section II.D, we will discuss how stromal cells may contribute to the generation of a prooxidant tumor microenvironment.
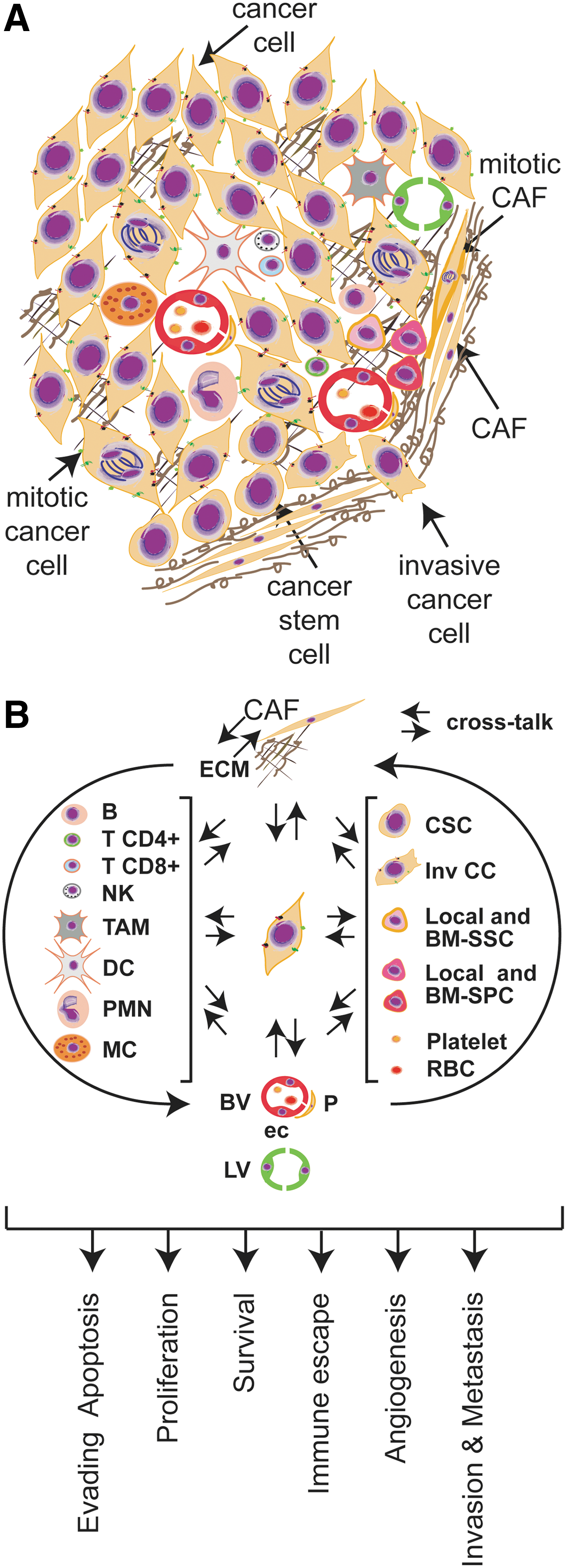
a. Fibroblasts
Fibroblasts constitute the predominant cell type in the stroma of most human tumors and are mainly responsible for secreting ECM components, including collagens, structural proteoglycans, proteolytic enzymes, their inhibitors, and various growth factors (301). Tumor fibroblasts adopt a myofibroblastic phenotype and are called cancer- or tumor-associated fibroblasts (CAFs or TAFs). They typically exhibit a higher proliferative index, as compared with normal fibroblasts, and they often express α-smooth muscle actin, fibroblast activation protein, the membrane glycoprotein Thy-1, desmin protein, S100 calcium-binding protein A4 (S100A4), and others (301, 335, 337). Moreover, they are commonly surrounded by a dense accumulation of fibrillar collagen (301). Although still under controversy, it seems that CAFs originate from diverse sources, such as genetic alteration of normal fibroblasts, from epithelial cells through epithelial–mesenchymal transition (EMT), from endothelial cells (ECs) through endothelial-to-mesenchymal transition, and from bone marrow-derived mesenchymal cells (335, 337). In addition, to synthesize ECM components, CAFs also secrete factors that promote tumorigenesis, including proteinases (203). For instance, matrix metalloproteinases (MMPs) promote ECM degradation facilitating cell migration; chemokines recruit neighbor cells that secrete proangiogenic factors; and growth factors promote malignant cell growth (335, 337). Recently, it was proposed that CAFs and cancer cells orchestrate a tumor–stroma coevolution through the generation of reactive oxygen species (ROS) (discussed in section II.D.1). Thus, CAFs are not by far passive cells within the tumor mass, but rather they are active drivers of tumor progression and metastasis.
b. Tumor vasculature-associated cells
The tumor vasculature-associated cells are one of the major stromal constituents. ECs have a critical role in the formation of new vessels, and marked differences were observed between tumor-associated ECs compared to those present in normal tissues (35, 36, 182). Tumor ECs show an activated phenotype characterized by the ability to degrade the basement membrane and the surrounding ECM. ECs express cell surface receptors for the adhesion to the different ECMs, to circulating leukocytes, and for angiogenic growth factors absent or barely detectable in normal blood vessels (35, 36, 182). Among cell surface receptors produced by ECs are the vascular endothelial growth factor (VEGF) receptors, VE-cadherins, Jagged 1, angiopoietin receptor tie-2, ICAM-1, E-selectin, and Muc-18, which in some cases have been identified as markers of prognostic value (35). Many recent studies revealed the genetic instability of these cells, raising doubts regarding the real efficacy of the myriad of antiangiogenic therapies that assume the genetic stability of tumor ECs (35). Moreover, malignant cells can transfer the genetic material through exosomes and microvesicles to ECs, inducing additional epigenetic changes (36). Other cell types are recruited to the new vessels to support the hydrostatic pressure of blood flow. These mural cells are vascular smooth muscle in larger-caliber vessels (venules, veins, arterioles, and arteries) and pericytes in the capillary context (106). Tumor pericytes present multiple abnormalities, including loss of association with the vessel wall, impaired support of endothelial function, and altered protein expression (106, 224).
c. Inflammatory cells
Inflammatory cells are the most heterogeneous population in the tumor microenvironment (21, 67, 144). Different leukocyte profiles and variations in their state of maturation and/or activation can be found inside the tumor mass. Thus, the evaluation of the tumor immune infiltrate is extremely complex, both in terms of cell type and role (144). Clinical and experimental data indicate that macrophages, mast cells (MCs), neutrophils, eosinophils, dendritic cells (DCs), and T and B lymphocytes are actively recruited within the tumor mass by chemokines produced by neoplastic and tumor-associated stromal cells (Fig. 1B).
Macrophages are the major component of the infiltrate of most tumors and have served as a paradigm for cancer-associated inflammatory response (9, 21, 208, 280). Macrophages differentiate into two distinct types, identified as M1 (or classically activated) and M2 (or alternatively activated) (209). In general, M1 macrophages act as soldiers and fight against tumors, producing high amounts of inflammatory cytokines and activating the antitumor immune response. Instead, M2 cells promote angiogenesis (280, 281), remodeling, and repair of wounded or damaged tissues. Tumor-associated macrophages (TAMs) exhibit an M2 phenotype showing mostly protumoral functions (9, 209). MCs are often abundant within the inflammatory infiltrate and are found in close association with blood vessels. They are co-opted by the malignant cells to promote angiogenesis and lymphangiogenesis and ECM remodeling to facilitate metastatic dissemination (202). In addition, MCs can modulate the immune response by dampening immune rejection or directing immune cell recruitment, depending on local stimuli (202). Eosinophils are also ubiquitous leukocytic infiltrate of solid tumors (180). Although eosinophils tend to accumulate in necrosis or remodeling areas, reports indicate large differences in the amount of infiltrating eosinophils, both among different tumor types and within a given tumor type (180). Neutrophils are commonly found within the tumor microenvironment and were historically considered a means of host defense against cancer; however, their presence in most cases is associated with a poor clinical outcome (117, 128). A recent study by Fridlender et al. suggested that neutrophils may exhibit a unique polarization state (N1 or N2) that dictates their impact within the tumor microenvironment. N1-type neutrophils are capable of killing tumor cells, whereas N2 cells promote tumor growth, and recruitment of either cell type is under the regulation of transforming growth factor-β (TGF-β) (102). Recent evidence from our group has also shown that the matricellular secreted protein acidic and rich in cysteine (SPARC) can induce the recruitment of protumorigenic or antitumorigenic neutrophils and regulate their antitumor cytotoxic capacity (8). Interestingly, SPARC and TGF-β have been shown to transcriptionally regulate one each other expression (192). Natural killers (NKs) are another important inflammatory cell type that greatly infiltrates the tumor microenvironment. NKs are known by their capacity to directly eliminate tumor cells in vitro. However, NKs that infiltrate the tumor in vivo do not appear to make direct contact with malignant cells, but rather reside in the tumor stroma, raising the question whether a lack of direct contact might hamper their capacity to eliminate tumor cells (7).
Among the adaptive immune response, activated CD8+or cytotoxic lymphocytes (CTLs) play a well-defined antitumor role in cancer progression by directly eliminating malignant cells (76). In contrast, the role of CD4+ T helper (TH) in tumor progression is more paradoxical (76). Classically, CD4+ T lymphocyte subsets include TH1 and TH2 lineages. The TH1 lineage can directly and indirectly regulate antitumor programs that restrain cancer development. In contrast, the TH2 lineage alters adaptive immunity by inducing T cell anergy, inhibiting T cell-mediated cytotoxicity, as well as fostering humoral immune responses directed by B cells (76). In addition to the TH1-versus-TH2 paradigm, CD4+-derived lineages have been recently expanded to include a proinflammatory antitumor TH17 response versus CD4+ FoxP3+ T regulatory cells, whose presence often correlates with poor prognosis (76). B lymphocytes and humoral immunity can also modulate solid tumor development, for instance, regulating pathways involved in secretion of anti-inflammatory cytokines (interleukin [IL] 10 and TGF-β), inhibition of CTL activity, perturbation of TH1/TH2 CD4+ T cell lineages, as well as differential recruitment and activation of innate immune cells (76). Antigen-presenting DCs have a crucial role in both the activation of antigen-specific immunity and the maintenance of tolerance, providing a link between innate and adaptive immunity. As it has been extensively reviewed, mechanisms of inadequate DC function result in tumor escape from immune surveillance (199).
3. Extracellular matrix
The ECM acts homeostatically to mediate communication between cells, contributing to survival and differentiation. By this interaction, it provides relevant microenvironmental information, biochemically through soluble and insoluble mediators, and physically through imposition of structural and mechanical constraints (247). Considering their structure and function, the proteins within the ECM can be divided into several classes. The structural ECM proteins consist primarily of the collagen and elastin families that strengthen and organize the matrix. Proteins such as fibronectin, laminin, and tenascin play an adhesive or integral role within the ECM matrix. Other proteins without structural role, called matricellular proteins, are generally involved in the modulation of the adhesive state of cells with implications in malignant dissemination (28). Finally, numerous proteoglycan- and heparan sulfate (HS)-containing proteins form the highly hydrated gel-like mixture that helps to stabilize the matrix within its aqueous environment.
It has long been known that the tumor-derived ECM is biochemically and biomechanically distinct in its properties compared to a normal ECM. Increased ECM deposition with a high content of type I collagen and fibronectin has been observed in tumors, which increases the tumor and the surrounding tissue stiffness compared to normal tissues (252). Other ECM proteins, such as tenascin, decorin, fibromodulin, SPARC, lumican, and osteopontin, have also been shown to be involved in tumor development, modifying both biochemical and biomechanical properties of the tumor ECM (15, 133, 163, 233).
Beyond structural or biomechanical function, the ECM plays a key role in the modulation of malignant processes (68). For instance, increased production of fibroblast-derived fibronectin (153) was observed in metastatic target organs after orthotopic tumor implant, suggesting that tumor-secreted factors can impact in neighbor stromal cells to secrete tumor-supportive ECM proteins.
B. Tumor microenvironmental characteristics
1. Hypoxia
a. General characteristics
Hypoxia is a general term used to describe an oxygenation state that is below the normal O2 levels for a particular tissue. Most mammalian tissues exist at 2%–9% O2 (on average 40 mmHg), and hypoxia and severe hypoxia (or anoxia) are usually defined as ≤2% O2 and ≤0.02% O2, respectively (26). The existence of hypoxic cells within the tumors was suggested very early by Thomlinson and Gray (300) and confirmed in the later decades of the 20th century with the development of precise techniques for measuring O2 levels (26, 50, 332). Tumor hypoxia is generally attributed to chaotic and poorly organized blood vessels within the cancerous tissues (104, 251). O2 diffuse just ∼200 μm, thus malignant cells beyond this diffusion distance from capillary vessels, will shoot angiogenesis-signaling programs (25). Although chronic hypoxia exists in tumor regions beyond the diffusion distance of oxygen, cycling or intermittent hypoxia can also arise from transient fluctuations in tumor perfusion (79, 80, 213, 251). These fluctuations have been attributed to changes in erythrocyte flux, perfusion, and during the development of newer vascular network. Imaging of cycling hypoxia in the tumor mass can provide capabilities to help planning appropriate treatments by taking into consideration the magnitude and frequency of oxygen level fluctuations (213).
Tumor responds to hypoxia not only by promoting angiogenesis or vasculogenesis to offset the oxygen deficit but also by triggering the anabolic switch in central metabolism (31, 33, 78, 332). Furthermore, hypoxia enhances EMT, invasiveness, and metastasis (33, 64, 141, 196), and also has a key role in stem cell regulation (155, 214, 220). Tumor hypoxia has been extensively explored as a cancer target (34, 276, 332). Nowadays, hypoxia and particularly cycling hypoxia are also receiving increased attention (204) because of the significant influence on tumor aggressiveness (314) and resistance to treatment. (80, 213, 332).
b. Molecular control of hypoxia
Fast proliferating cells growing within the tumor limit O2 diffusion from their vascular network and trigger the tumor response to hypoxia (33, 78, 201, 318), by activating broad-action transcription factors named hypoxia-inducible factors (HIFs) (201, 264). HIFs are the master regulators of oxygen homeostasis and play a role in disease pathogenesis as cancer. HIFs are obligate heterodimers composed of an O2-labile α-subunit and a stable β-subunit. HIFα subunits heterodimerize with the stable HIF1β (also known as aryl hydrocarbon translocator) and recognize and bind to hypoxia-response elements in the promoter of hundreds of genes (328) (Fig. 2).
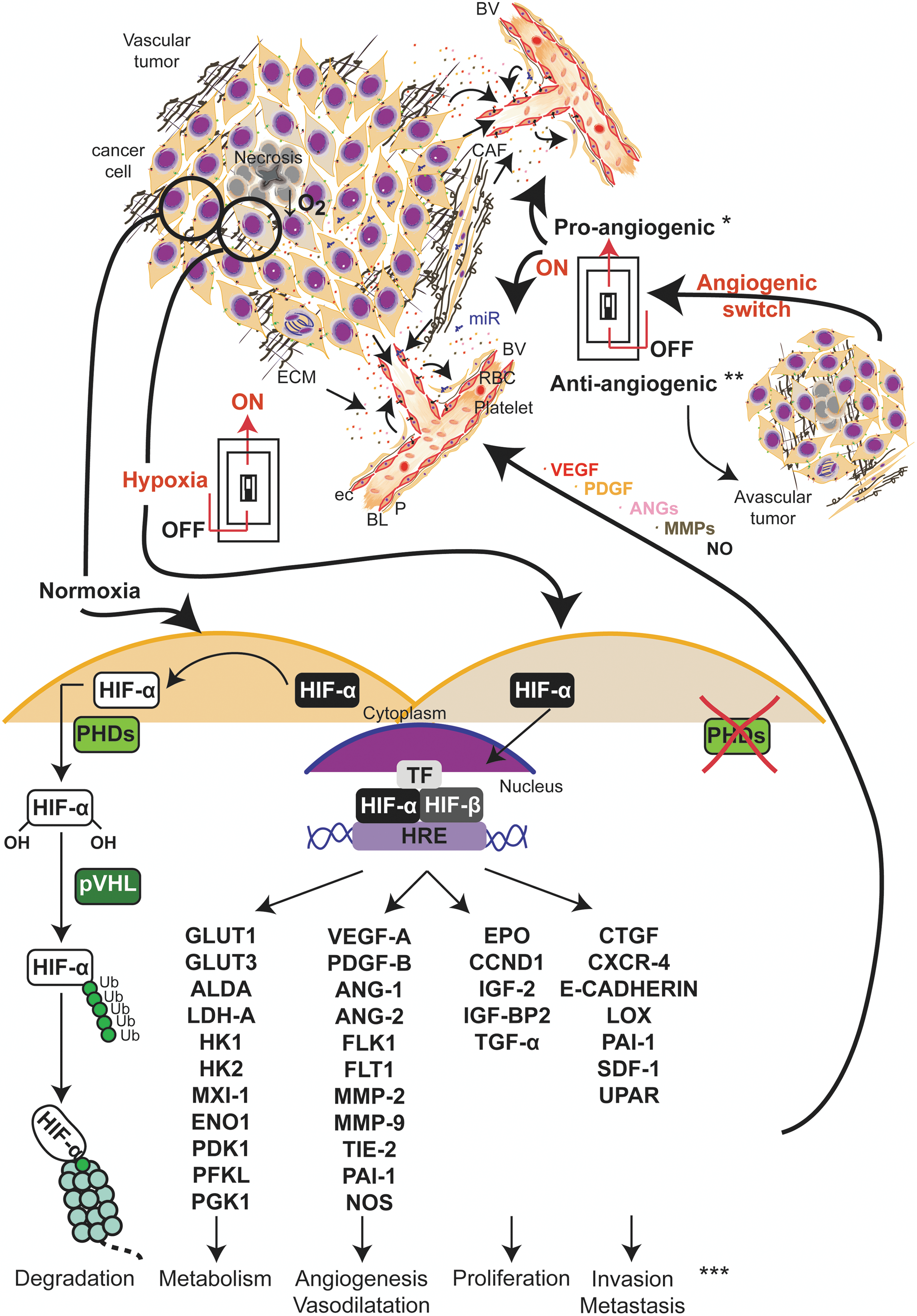
Solid tumors often exhibit high levels of the HIF1α isoform, and this expression correlates with poor clinical prognosis in many cancer types (26, 276, 332). Under normoxia, HIF1α is hydroxylated at conserved proline residues (Pro-402 and Pro-564) by prolyl hydroxylases (PHDs), whose activities are regulated by O2 availability (149, 201) (Fig. 2). Hydroxylated HIF1α is, in turn, recognized and marked by an E3 ubiquitin ligase, von Hippel-Lindau protein, which targets HIF1α for proteosomal degradation (148, 201) (Fig. 2). Under hypoxia, PHD activity is diminished, and HIF1α is stabilized, and migrates to the nucleus. HIF1α dimerizes with HIF1β, and the heterodimer interacts with coactivators CREB-binding protein/p300 and induces transcription of genes that fall into four major categories: glucose transporters and glycolysis; angiogenesis; cell survival and proliferation; and invasion and metastasis (33, 34, 78, 79, 196, 264, 318) (Fig. 2). A list of additional cues modulates the HIF pathways such as oncogenic signals (39, 149, 171, 261, 328, 356), histone deacetylases (149, 328, 356), and microRNAs (miRNAs) (118, 179, 320, 340). ROS also have a key role in the regulation of HIF1 that will be discussed in sections II.C.2 and II.D.4.
2. Tumor angiogenesis
a. General characteristics
The tumor-associated neovasculature emerged as a critical adaptation of the tumor for growing beyond a certain limit and has indeed become a hallmark of cancer (120). Initially, most tumor masses grow avascular, but when the tumor exceeds 2–3 mm3 in volume, a new blood vasculature develops to ensure influx of oxygen and nutrients.
Tumor vessels generally grow from the pre-existing vasculature by a process known as angiogenesis (41, 100). Mobilization of bone marrow endothelial progenitor cells (EPCs) to the tumor and formation of new vessels have also been described (182).
The vessel neoformation in tumors can be triggered by different stimuli, like hypoxia, acidosis, mechanical stress, genetic mutations, or inflammatory processes (41). Pro- and antiangiogenic factors can be secreted by malignant, stromal, and infiltrating bone marrow-derived cells (41). Angiogenesis occurs through a process called angiogenic switch, in which either the secretion of proangiogenic factors is increased, or the production of endogenous antiangiogenic factors is reduced (25) (Fig. 2). The onset of angiogenesis or the angiogenic switch can occur already in premalignant lesions (260) and at any stage of tumor progression (25). In contrast with normal tissues, tumor angiogenesis results from a deregulated balance of pro- and antiangiogenic factors in their temporal and spatial expression (Fig. 2). Thus, tumor vasculature is characterized by an abnormal vascular structure, EC–pericyte interactions, permeability, and blood flow (25, 122, 242). Tumor vessels can grow by different patterns, mostly sprouting and also intussusception, the co-option of existing vessels, and incorporation of bone marrow EPCs. (41, 182). However, the contribution of EPCs to the development of tumor vasculature is controversial mainly because of the lack of a bona fide molecular signature that defines EPCs (331). In addition, certain tumor types are also able to form a vasculature-like system using its own tumor cells through a mimicry process (86, 99).
b. Molecular control
In the past decades, a plethora of pro- and antiangiogenic factors that regulate tumor angiogenesis have been identified (5, 25, 41). Many stimuli, including hypoxia, growth factors, cytokines, and oxidative stress, can increase the expression of VEGF in tumor cells, which is correlated with increased microvessel counts and poor prognosis in many human cancers. VEGF-A is the major regulator of physiological and pathological angiogenesis (5, 25). This factor plays a critical role in tumor angiogenesis, not only through its effect on ECs but also through mobilization of bone marrow-derived EPCs (259). VEGF-A belongs to a gene family that includes placental growth factor (PlGF), VEGF-B, VEGF-C, VEGF-D, VEGF-E, which bind with varying specificities and affinities to VEGF receptors (VEGFRs). This family of receptors is composed by VEGFR 1, 2, and 3 (5). VEGF-A regulates vessel morphogenesis through VEGFR1 and VEGFR2, and proliferation of ECs through VEGFR2. VEGF-B, C, and D contribute to tumor angiogenesis by binding to VEGFR2 and 3. In addition, VEGF-C and VEGF-D were identified as lymphatic endothelial factors, acting mainly via VEGFR3 (5).
Angiopoietins are members of another family of growth factors that play essential roles in modulating the activation status of ECs (5). Angiopoietin-1 (ANG-1) induces the final maturation of blood vessels. The activation of Tie2 receptor by ANG-1 mediates remodeling and stabilization of cell–cell and cell–matrix interactions. Moreover, ANG-1 plays a role in the recruitment of pericytes to the nascent vessels (217). ANG-1 or PlGF can also provide survival signals, and rescue immature blood vessel in the absence of VEGF (25). Platelet-derived growth factor (PDGF) and fibroblast growth factor also stimulate neovascularization in various angiogenesis and animal disease models, supporting their cooperative role in tumor angiogenesis and metastasis (40).
A large number of endogenous antiangiogenic factors have been also functionally characterized. For instance, specific fragments of structural proteins that includes collagen, plasminogen, or ECM glycoproteins (angiostatin, endostatin, tumstatin, and trombospondin-1) or soluble factors like interferon γ and β were characterized. Antiangiogenic factors have been extensively studied during the last decade for their therapeutic value, and more than 40 of them entered clinical trials (5, 25, 41, 272).
3. Tumor metabolism
a. General characteristics
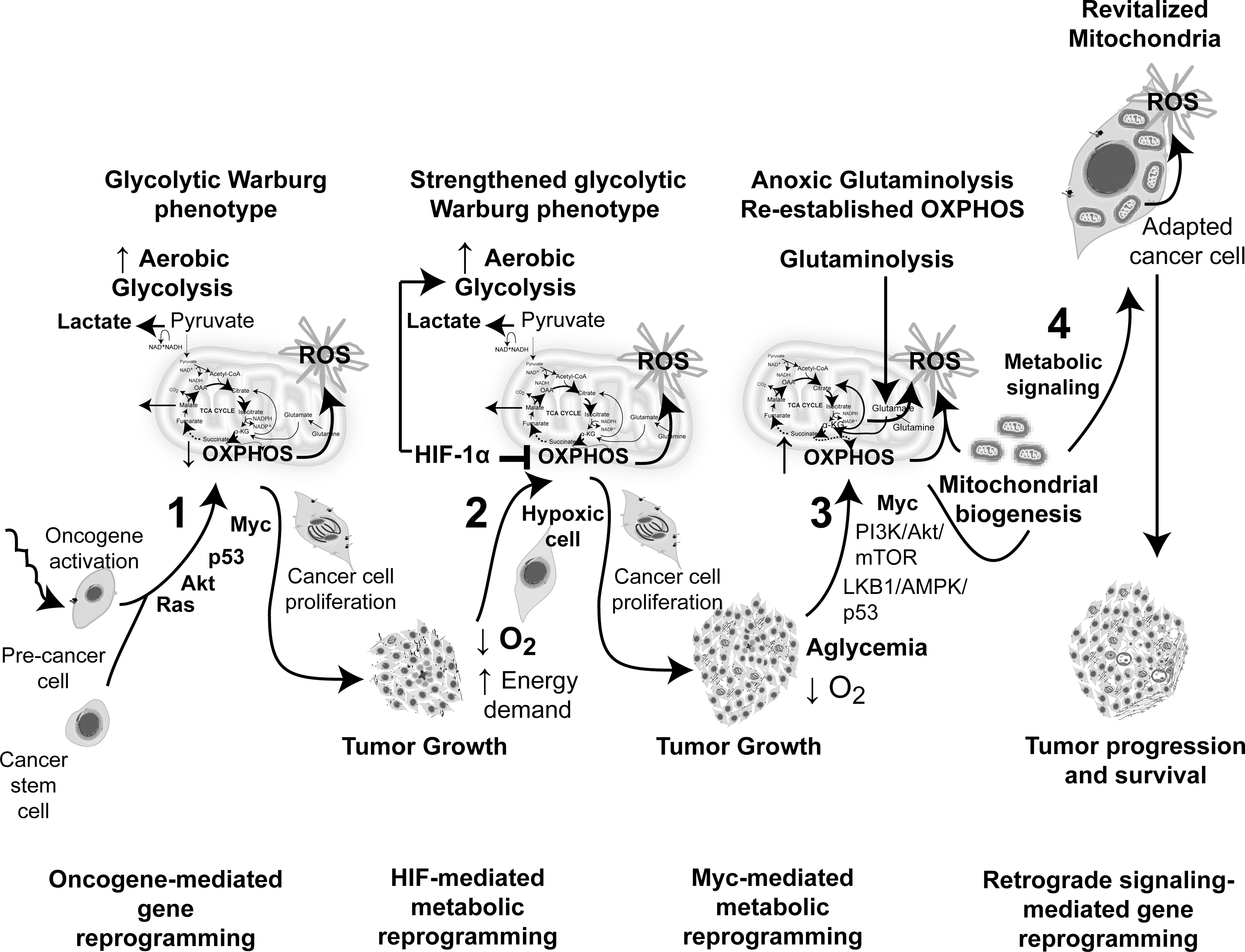
Cancer cells have to reprogram their metabolism to provide the support for the basic needs of proliferating cells: rapid ATP generation and increased biosynthesis of macromolecules (39, 74, 171, 183). The best-characterized metabolic phenotype in cancer cells is the switch of ATP production from oxidative phosphorylation (OXPHOS) to glycolysis, even in the presence of oxygen (39, 74, 168, 171, 183, 323) (Fig. 3). This metabolic switch was identified about 60 years ago by Otto Warburg (168, 323), and is known as the Warburg effect. To compensate the low efficiency of glycolysis in generating ATP, malignant cells increase glucose uptake to abnormally high levels (39), which became the basis for using the glucose analog 2-(18F)-fluoro-2-deoxy-

Although many tumors utilize glycolysis as the principal source of energy, others produce ATP by OXPHOS (105, 146, 222). Recently, it was postulated that waves of gene regulation would suppress and then restore OXPHOS in cancer cells during tumorigenesis (Fig. 7) that can alter metabolic ROS generation (see section II.D.4). It can be hypothesized that the switch between glycolysis and OXPHOS could be an adaptive mechanism of energy production to microenvironmental changes, such as hypoxia, differences in tumor energetics, and biosynthetic requirements.
b. Molecular control
Numerous studies have identified a series of molecular changes responsible for cancer metabolic reprogramming (39, 74, 145, 171, 183). The activation of the PI3K/Akt/mammalian target of rapamycin (mTOR) pathway is a master regulator of aerobic glycolysis and cellular biosynthesis (74), which can be activated through a variety of mechanisms (74). The PI3K/Akt axis increases the glucose and amino acid flux through the plasma membrane and stimulates glycolysis, expression of lipogenic genes, and lipid synthesis (39, 74, 171, 183). Increased levels of Akt stimulate signaling through mTOR kinase that indirectly causes other metabolic changes by activating transcription factors such as HIF1 (discussed below). AMP-activated protein kinase (AMPK) induces opposite effects to Akt, functioning as a potent inhibitor of mTOR. AMPK is a crucial sensor of energy status (it responds to an increased AMP/ATP ratio) and functions as a metabolic checkpoint, regulating the cellular response to energy availability (39). The loss of appropriate AMPK signaling that exhibits many cancer cells contributes to their glycolytic phenotype (39, 183). The tumor suppressor p53 is also an important regulator of metabolism by inhibiting the glycolytic pathway through different mechanisms (39, 254). Thus, the loss of p53, which is frequent in many tumors, may also contribute to the acquisition of the glycolytic phenotype. Under hypoxia condition, tumor cells adapt their metabolism stimulating glucose uptake (31, 78). This response is coordinated by HIF1, which induces energy production by increasing glycolysis and decreasing mitochondrial function. HIF1 can also be activated under normoxic condition by oncogenic signaling activations, including PI3K, or by mutation in tumor suppressors, such as the von Hippel-Lindau gene, succinate dehydrogenase, and fumarate hydratase (39, 171, 261). Thus, the activation of oncogenes and the loss of function of tumor suppressor genes cooperate to enhance the glycolytic metabolic shift.
4. Tumor acidosis
a. General characteristics
Malignant cells maintain their intracellular pH neutral or alkaline (7.2 to 7.5), but tend to acidify the extracellular microenvironment (pH 5.6 to 6.8) (56). The extracellular acid stress is the consequence of poor blood perfusion, low oxygen availability, increased glucose metabolism, and production of metabolic acids, such as lactic acid (56). Extracellular tumor acidosis facilitates tumor invasion by promoting matrix degradation and death of neighbor normal cells (45, 109) and also promote a reduced immunosurveillance by inhibiting NK and CTL activities (98, 176). In the last 20 years, many studies demonstrated that tumor acidosis is the result of oncogene activation and hypoxia, which promote the shift from OXPHOS to glycolytic metabolism (56).
b. Molecular control
Lines of evidence indicate that the genetic alteration of malignant cells drives intracellular alkalinization and extracellular acidification of the tumor microenvironment. P53 was shown to decrease the activity of glycolytic enzymes, to inhibit glycolysis, by modulating the levels of fructose-2,6-bisphosphate and the expression and activity of proteins involved in mitochondrial respiration (56). P53-deficient cancer cells contribute with to the Warburg effect through aerobic glycolytic compensation, which is accompanied by increased lactic acid production (56). The PI3K/Akt pathway, which is constitutively activated in some cancer types, also increases glycolysis through the induction of HIF-1α expression, leading to acidification of the tumor microenvironment. Other oncogenes such as Ras or c-Myc increase glycolysis and lactic acid production (56). Cytoplasmic alkalinization of cancer cells results from an efficient membrane transport machinery that extrudes H+ and imports HCO3 −, including Na+/H+ exchangers, I−, Cl−/HCO3 − exchangers, Na+/HCO3 − cotransporters, H+/lactate cotransporters, and carbonic anhydrase II, IX, and XII working in a coordinated fashion (241) In addition, malignant cells can induce additional mechanisms for assisting the constitutive pH-regulating systems (241). Hypoxia also promotes acidosis by shifting from OXPHOS to glycolytic metabolism. HIF-1 activates the expression of multiple genes that favor glucose uptake and metabolism, and suppress pyruvate oxidation via TCA and OXPHOS (30). Furthermore, HIF-1 can induce the expression of the H+/monocarboxylate transporter 4, carbonic anhydrase IX, and XII to support cell survival in a hostile microenvironment (57, 241, 291).
II. Redox Characterization of the Tumor Microenvironment
A. ROS and cell sources
ROS encompass a wide range of intermediate oxygen-carrying metabolites with or without unpaired electrons. The species with unpaired electrons or O2-derived free radical include mainly superoxide anion (O2
•−), hydroxyl radical (HO
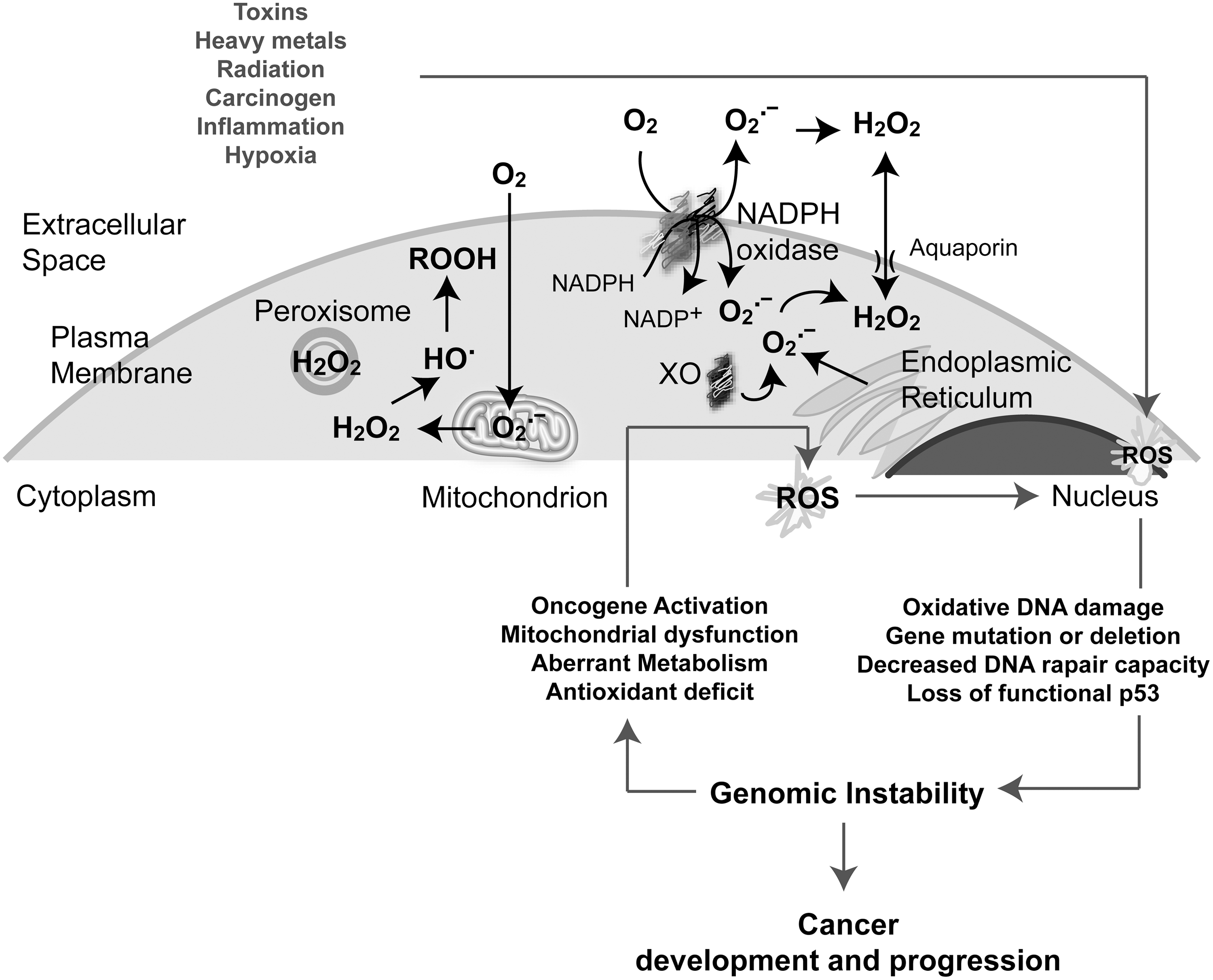
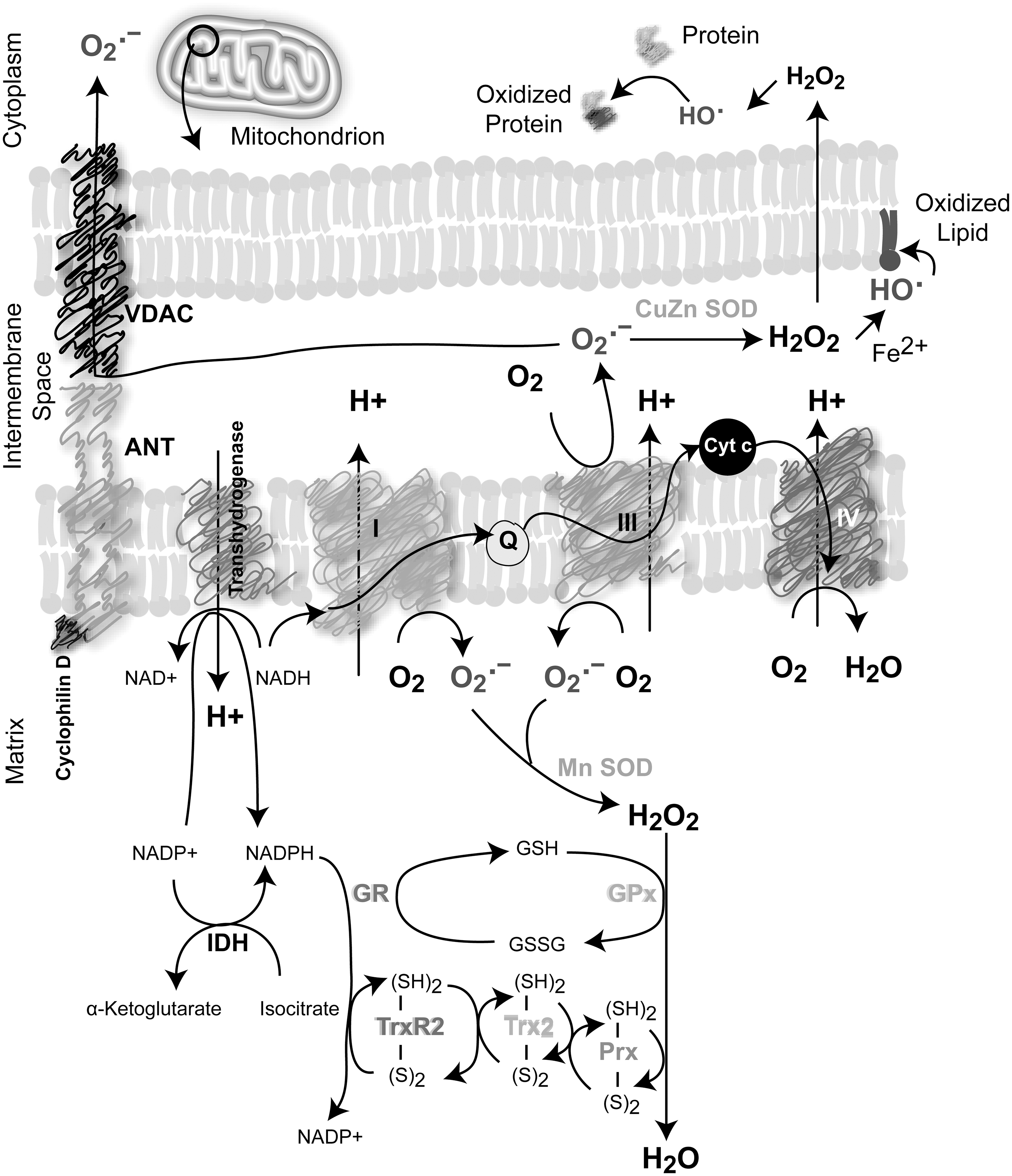
Several cell sources produce ROS under normal physiological conditions, including mitochondrial electron transport chain (mETC), NADPH oxidase (NOX), cytochrome P450, lipooxygenases (LOOX), cyclooxygenases (COX), xanthine oxidases, and peroxisomal enzymes (Fig. 4). In mammalian cells, mitochondria are one of the major sources of cellular ROS produced essentially during respiration through the one-electron reduction of O2 (32, 169, 289). It was estimated that 0.15%–2% of cellular oxygen consumption results in O2 •− in vitro (32, 38, 289). Little is known regarding the regulation of mitochondrial function in vivo in terms of O2 •− production. Five multiprotein complexes compose the respiratory chain embedded in the inner membrane of the mitochondria. Complexes I and II oxidize NADH and FADH2, respectively, and transfer the electrons to ubiquinol, which carries them to complex III, which shuttles the electrons across the inner mitochondrial membrane to cytochrome C. Cytochrome C carries electrons to complex IV, which reduces oxygen to water (Fig. 6). Complexes I and III are the main sites of O2 •− generation; however, complex I produces ROS only inside the matrix, whereas complex III can produce ROS on both sides of the mitochondrial inner membrane (32, 289) (Fig. 6). Superoxide anion is the primary ROS produced by the mitochondria and is converted to H2O2 through the action of superoxide dismutases (SODs). Moreover, significant regulatory effects of NO on mitochondrial respiration have been described as a result of its high-affinity binding to cytochrome oxidase (complex IV). Other components of the mETC can also be inhibited by NO, contributing to an increase in the mitochondrial O2 •− production rate (10, 43).
NOXs are ROS-generating enzymes that produce O2 •− in response to stimuli such as growth factors, cytokines, and calcium present in phagocytic and in various nonphagocytic cell types (24, 174). NOXs are membrane-spanning proteins with NADPH- (or NADH-) and FAD-binding domains in their C termini. Once activated, they produce O2 •− by transferring a single electron from NADPH (or NADH) to FAD, which in turn passes electrons to hemes, and ultimately to molecular O2, forming O2 •− (24, 174) (Fig. 4). At present, seven NOX proteins have been identified: NOX1, NOX2, NOX3, NOX4, NOX5, and dual oxidases DUOX1 and DUOX2 that produce H2O2. These proteins have different tissue distribution and cell-type-specific subcellular localization (24, 174, 353). The prototypical NOX is the phagocyte NOX2, which is a heterodimer formed by the catalytic unit gp91phox and p22phox, which stabilizes gp91phox and enhances O2 •−-producing activity (gp91phox has been renamed NOX2 in the current nomenclature). Rac, a member of small GTPases that are critically involved in cell capacity to adhere and migrate, has been also involved in O2 •− generation by NOX in close association with other cytosolic subunits p40phox, p47phox, and p67phox. NOX2 is highly expressed in neutrophils and macrophages and is usually quiescent, but generates O2 •− at a micromolar-to-millimolar range in response to a challenge from microorganisms or cytokines. In contrast, oxidant production in nonphagocytic cells is low, typically in the nanomolar-to-micromolar range (267). Recent studies revealed some aspects of the functional relationships between the NOX gene family and increased ROS production in tumor cells (151).
Other cellular ROS sources are the endoplasmic reticulum (ER) and several cytosolic enzymes. Particularly, the ER contains cytochromes P450 (CYPs), enzymes that are involved in the metabolism of drugs and other xenobiotics, arachidonic acid, eicosanoids, cholesterol, vitamin D3, and retinoic acid. CYPs catalyze oxidation of substrates by O2, but there are also abortive oxygen reduction that generates O2 •−, which dismutates to H2O2 (347). In addition, the folding of oxidative proteins that occurs in the ER also contributes to ROS generation (306). The xanthine oxidoreductase system, NO synthases (NOS), and COXs are involved in cellular metabolic pathways that contribute additionally to ROS generation (121).
B. Control of cellular redox homeostasis: the antioxidant system
A stringent control of ROS levels is an absolute requirement for cell survival owing to the toxicity of ROS at high levels. Thus, cells have developed a sophisticated intracellular antioxidant defense system to protect themselves from oxidative damage. A complex network of antioxidants includes both enzymatic and nonenzymatic components that regulate ROS cell production, both spatially and temporally (Fig. 5A, B).
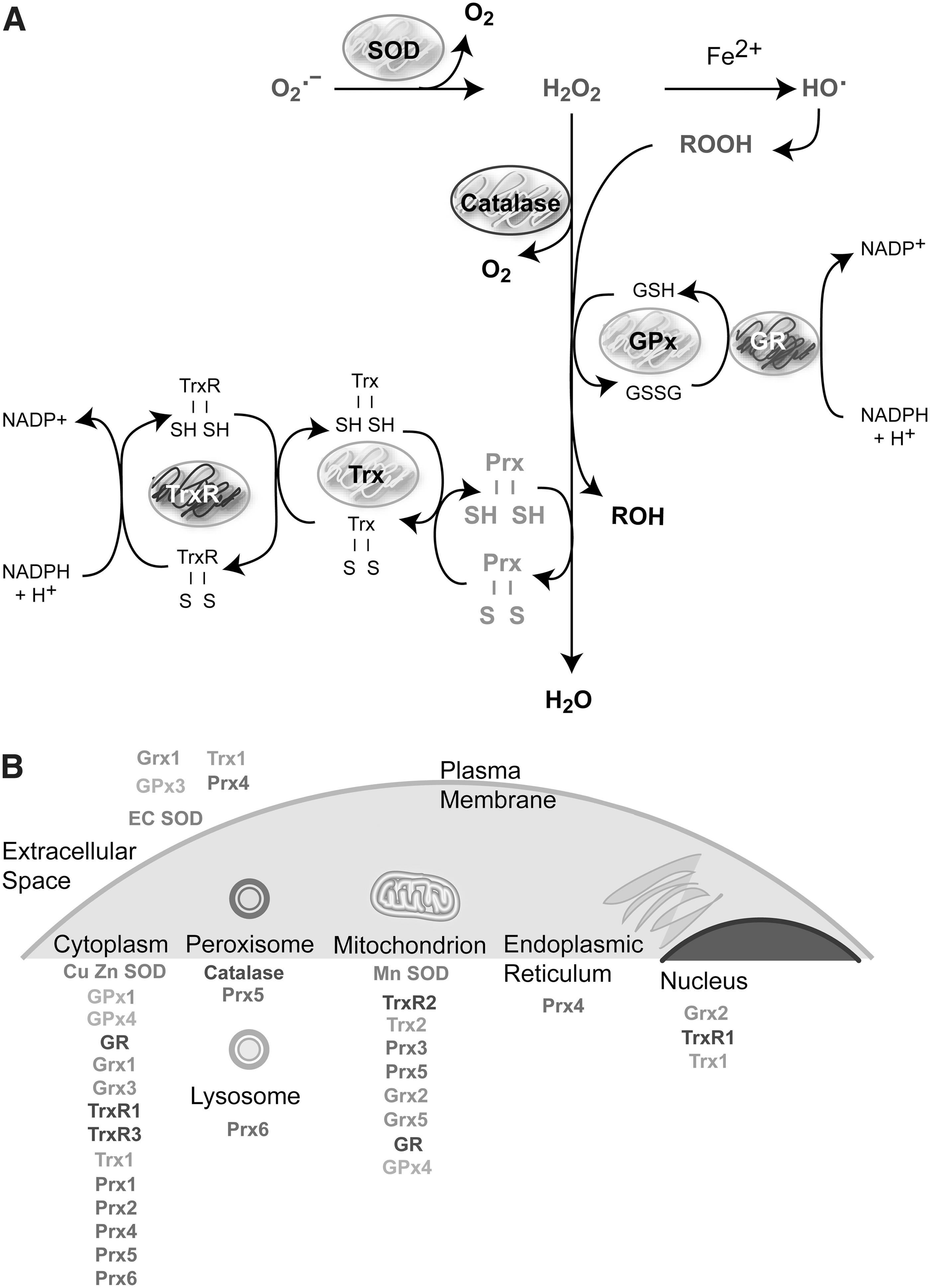
Although O2 •− dismutates spontaneously to H2O2, the SOD family accelerates significantly this reaction (5×105 M −1 s−1 vs. 1.5×109 M −1 s−1). SODs exist in several isoforms: copper–zinc SOD (CuZn-SOD) found in the cytoplasm, nucleus, and plasma membrane; manganese SOD (Mn-SOD) located in the mitochondria; and extracellular SOD (EC-SOD) that maintains the redox status in fluids (187, 313). Several peroxidases convert H2O2 to water and O2. Catalases (CATs) catalyze direct H2O2 decomposition to water in peroxisomes, whereas glutathione peroxidase-1 (GPx-1), located in the cytoplasm and mitochondria, removes peroxide by coupling its reduction to H2O and the oxidation of reduced glutathione (GSH) to oxidized glutathione (GSSG) (302) (Fig. 5A). Other types of GPxs (GPx2, GPx3, GPx4, and snGPx, a specific sperm nucleus enzyme) are mostly specific for GSH as a hydrogen donor, but act not only on H2O2 reduction but also on additional organic peroxides, fatty acid, and cholesterol hydroperoxides (only GPx-4) (239, 349). Intracellular levels of GSH are maintained by the GSH-regenerating system composed by glutathione reductase (GR) and NADPH. Thus, GR catalyzes the reduction of GSSG to GSH coupled to NADPH oxidation (302) (Fig. 5A).
Peroxiredoxins (Prxs) constitute 0.1%–0.8% of the total soluble protein of mammalian cells and are also considered one of the most important cell redox-state-regulating enzymes. Prxs are a family of peroxidases that also reduce H2O2 and alkyl hydroperoxides to water or corresponding alcohol. They are homodimers that contain one or two cysteines at their active site. At least six isoforms of human Prxs (Prx1–6) were located in different subcellular compartments, particularly in the mitochondria (Prxs 3 and 5) (Fig. 5B). Prxs are maintained in the reduced form by the thioredoxin (Trx)/thioredoxin reductase (TrxR) system, which in conjunction with the GSH/GR system maintains the cellular thiol–disulfide redox status in the cell (333).
The Trx system comprises Trx, TrxR, and NADPH. Trxs are small redox-active proteins (about 12 kDa) with a disulfide active site that is reduced to a dithiol by TrxR using NADPH as an electron donor (Fig. 5A). Mammalian Trx and TrxR are expressed as isoforms either in the cytosol and in the nucleus (Trx1 and TrxR1) or in the mitochondria (Trx2 and TrxR2); in addition, there are testis-specific Trx/TrxR systems (Trx3 and TrxR-3) (13, 14, 216, 239). Trx/TrxR plays an important role in the redox regulation of multiple intracellular processes that induce oxidative stress such as DNA synthesis, cell proliferation, and chemotherapeutic drug resistance (13, 14, 216, 239). Unlike Trx that is reduced by its own reductase, glutaredoxins (Grxs) are coupled to GSH/GR. There are four Grx isoforms in humans, Grx1, Grx-3, and Grx5 (primarily cytosolic), and Grx2, displays different splice variants, which are located in the mitochondria and nucleus (216). In addition to antioxidant enzymes, nonenzymatic antioxidants are represented by low-molecular-mass agents such as glutathione (
Transcriptional control of the antioxidant enzyme system is another key point to maintain the cell redox homeostasis. The transcription nuclear factor erythroid 2-related factor 2 (Nrf2) plays a central role in the transcriptional regulation of many antioxidant or detoxifying genes in response to increased levels of different oxidants or electrophiles (156). The kelch-like ECH-associated protein 1 (Keap1) is a cytoplasmic adaptor protein essential for the regulation of the activity of Nrf2. Under normal conditions, Nrf2 is constantly reduced via a Keap1-dependent ubiquitination and proteasomal degradation system (164). In the presence of electrophiles or ROS, Keap1-dependent ubiquitin ligase activity is inactivated by the direct modification of cysteine thiol residues, and subsequently, Nrf2 is stabilized and translocated into the nucleus, where it activates the transcription of various detoxification and antioxidant enzymes genes through its binding to antioxidant-response element (ARE) or electrophile-response elements (294). Nrf2 is tightly involved in glutathione synthesis through its ability to control the expression of glutamate–cysteine ligase catalytic (GCLC) and modifier subunits (GCLM); GCLC and GCLM combine to form a heterodimer that catalyzes the rate-limiting step in GSH biosynthesis. Nrf2 also regulates the production of GR, Trx, TrxR1, peroxiredoxin 1, and sulfiredoxin (124, 156). Thus, the Nrf2-Keap1 system is a key factor for cell protection from oxidative and electrophilic insults that contribute to maintain the redox cellular microenvironment.
C. Altered ROS production in cancer cells
Early studies showed that cancer cells produce large amounts of ROS (292, 303), and a subsequent plethora of accumulating evidence supports the hypothesis that cancer cells are characterized by the production of higher ROS levels compared to their normal counterparts (103, 194, 250). The persistent oxidative stress of cancer cells is caused by an imbalance between ROS generation and the cell's ability to scavenge these species. Chronic oxidative stress in tumor cells is influenced by numerous factors such as genetic alteration of cancer cells, deregulation of antioxidant enzymes, mitochondrial dysfunction, aberrant cancer cell metabolism, alteration in proliferation, and the acquisition of the metastatic phenotype.
Although high levels of ROS generate a chronic oxidative state in the tumor microenvironment that promotes tumor aggressiveness and acquisition of the metastatic phenotypes, the downstream mechanisms that mediate this process are still unclear. Moreover, the antioxidant systems activated intracellularly to scavenge ROS can actively participate in the acquisition of a more aggressive phenotype. The disruption of the xc system (or cystine glutamate antiporter) that maintains an efficient cysteine/cystine redox cycle reduced tumor aggressiveness and the in vivo metastatic capacity of esophageal cancer cells (22, 53). Moreover, human lung carcinomas overexpress Trx and TrxR (44, 286), and lung cancer cells with increased Trx levels exhibited a more aggressive phenotype (44); in addition, human breast cancer patients showing high intratumor expression of Trx exhibited increased resistance to docetaxel neoadjuvant chemotherapy (159). Thus, the upregulation of antioxidant systems can promote tumor progression by increasing aggressiveness and cancer cell resistance to therapy.
1. ROS production due to genetic alterations
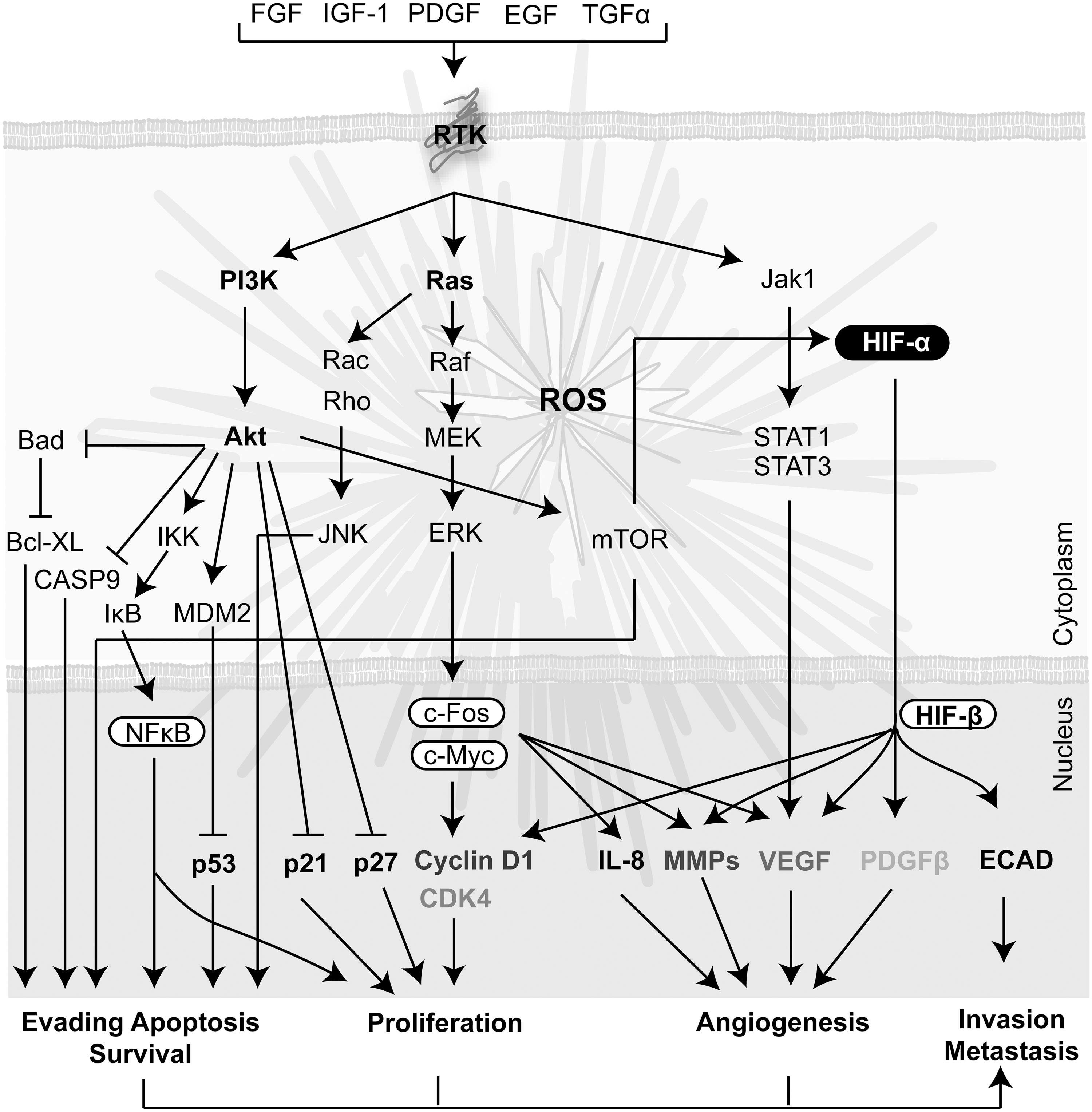
Several lines of evidence suggest that increased ROS production by malignant cells is a consequence of the activation of signaling pathways associated with genetic alterations (325). Ras proto-oncogenes encode membrane-bound GTPases that transduce mitogenic signals from tyrosine kinase receptors (TKR). Ras mutation, which occurs in 15%–30% of human cancer, was early associated with increased oxidant production in transformed fibroblasts (137). Downstream sources of Ras responsible for ROS production are the mitochondria and NOXs. (263, 284, 334). Ras overexpression enhances mitochondrial ROS (mtROS) generation, mitochondrial DNA (mtDNA) synthesis, and biogenesis (263). Increased mtROS are essential for K-Ras-induced cell proliferation and tumorigenic capacity (326). Furthermore, H-Ras induced the transcriptional NOX1 expression through GATA-6 transactivation of the NOX promoter in an MEK-ERK-dependent mechanism (334). NOX1 overexpression has also been linked to prostate and colon cancer (151). Moreover, NOX4 contributes to cell survival of pancreatic cancer cells through a process that involves impaired activities of Akt and its target ASK1 (219). NOX5 has also been also implicated in cell viability in Barrett esophageal adenocarcinoma cells (151)
Deregulation of the activity or expression of transcriptional factors has also been associated with ROS generation in cancer progression. cMyc oncogene overexpression in human cancer was associated with increased intracellular ROS production (325). Recently, it was also reported that cMyc point mutations are associated with ROS production in rat fibroblasts (116). Stat5 is constitutively activated in many human cancers, affecting the expression of genes that control cell proliferation and survival. This aberrant activity induces mitochondrial dysfunction and augmented ROS, leading to DNA damage (97). The transcription factor p53 plays a key role in maintaining redox homeostasis and genome stability. The mechanisms describing the p53 role in modulating oxidative stress and its contribution to tumor development were recently reviewed (173).
Although cancer genetic alterations are directly associated with increased ROS levels that lead to macromolecular damage and increased malignancy, it has been demonstrated that oncogenes can promote tumor aggressiveness through downstream mechanisms that involve ROS-scavenging systems (77, 142). Particularly, the expression of the oncogenic alleles of K-Ras, Braf, and Myc suppressed ROS generation by increasing the basal levels of Nrf2 in murine embryonic fibroblasts and in human pancreatic cancer cells (77). As mentioned before, Nrf2 binding to ARE elements not only triggers antioxidant programs that scavenge intracellular ROS but also induces cell proliferation and tumorigenesis in vivo. It was also demonstrated that Nrf2-null cells display impairment of cell cycle progression accompanied by a reduction in the phosphorylation of Akt, and hence reduced cell survival (268). In coincidence, Nrf2 was shown to upregulate Bcl-2, preventing cellular apoptosis (230). Moreover, constitutive stabilization of Nrf2 has been found in many cancer types to confer resistance to chemo- and radiotherapy, promoting survival of cancer cells under a deleterious environment (177). Nrf2 downstream ROS-scavenging-regulated systems such as heme oxygenase-1, which degrades prooxidant heme into ferrous iron, carbon monoxide, and biliverdin, have a key role in cancer promotion and drug resistance, promoting angiogenesis and metastasis (147). The possibility exists that in addition to scavenging elevated ROS, detoxifying enzymes under Nrf2 control could be acting on additional targets that might promote malignancy. Thus, whether the role of detoxifying systems is only to defend the cell from increased ROS levels or to serve as a downstream mediator of increased aggressiveness warrants further investigation.
2. mtROS produced by malignant cells
As mentioned before, mtROS are directly produced by a leaky transfer of a single electron to molecular oxygen during OXPHOS. O2 •− is rapidly converted by mitochondrial or cytosolic SODs to H2O2, which can diffuse across membranes. The anion superoxide, due to its negative charge, only enters the cytosol through specialized mitochondrial channels, such as the voltage-gated anion channel (VDAC) (279) (Fig. 6). Defects in the mETC (136, 316), prolonged hypoxia and glucose deprivation (4, 287), and the action of some oncogenes on mitochondrial metabolism (152, 263) can lead to increased levels of mtROS production in cancer cells. Elevated mtROS levels in malignant cells can be inhibited by CATs, implying that H2O2 is the predominant ROS overproduced by these cells (316). Somatic mutations in mtDNA occur at a high frequency in many cancer types, as a consequence of the prooxidant tumor environment. These mutations compromise respiratory function, increase ROS production, and promote metastatic dissemination (138). Mitochondria are also implicated in tumorigenesis, favoring oxidative damage-dependent mutagenesis (263). mtROS production might drive the selection of cellular clones capable of supporting an oxidative environment that would promote the amplification of genomic damages and instability. Thus, the greater potential for genetic mutations would lead to further rounds of cell transformation and malignancy (263). The increase in mtROS generation induced by hypoxia and glucose deprivation also contributes with mitochondrial biogenesis (263). The oncoproteins p53, Myc, and Ras were able to increase the levels of the mitochondrial transcription factor A, which promotes mtDNA synthesis and increases mitochondrial numbers in cancer cells, favoring an oxidative steady state (263, 284). Furthermore, Bcl-2, which has been classically associated with protective effect on the outer membrane integrity, was recently associated with an increase in mETC O2 •− generation via complex IV (170).
3. Deregulation of antioxidant mechanisms in cancer cells
Increased ROS levels in malignant cells could arise from the alteration or inactivation of the antioxidant defense system. Low activities of CuZn-SOD, Mn-SOD, CATs, and GPxs have been reported in a variety of transformed and malignant cells compared with their normal counterparts (232, 316). Decreased activity and expression of Mn-SOD were reported in colorectal, prostatic, and pancreatic carcinomas (161). However, Mn-SOD levels were shown to be elevated in mesothelioma, neuroblastoma, melanoma, stomach, ovarian, and breast cancer (161). An imbalance of antioxidant enzymes has been observed in many cancer cell types such as melanoma, lung, prostate, thyroid, and breast cancer, which results in augmented oxidative stress within the tumor microenvironment (316). Particularly, we have demonstrated elevated ROS levels in breast cancer cells in correlation with an increase in the H2O2-generating SOD activity and a decrease of CAT and GPx activity (249). It has also been demonstrated that H2O2 generated in the breast cancer microenvironment may have an oncosuppressor or oncopromoter function depending on EC-SOD enzymatic levels (207).
4. ROS generated by the aberrant activity of malignant cells
a. Cancer cell metabolism
Although glycolysis dependence was well demonstrated in fast growing tumors, recent studies have revealed the importance of OXPHOS for most of ATP supply that tumors need in crucial steps during malignant progression (146, 222). For instance, glioma, melanoma, colon, lung, cervical, and breast cancer cells are highly dependent on the OXPHOS pathway, from which these malignant cells obtain 70%–90% of cellular ATP (146, 222). Cancer cells can switch from aerobic glycolysis to OXPHOS under limiting glucose supply (146). Recently, an interesting concept of waves of gene regulation was postulated that would either suppress or restore OXPHOS in cancer cells during tumorigenesis (284) in a way that both metabolic pathways might contribute with ROS generation in the tumor microenvironment (Fig. 7). During glycolysis, OXPHOS is diminished, and the consequent slowdown of mETC increases O2 •− generation at complexes I and III. Retardation of electron transport within complex I and H+pumping increased O2 •− formation (85), probably because the generation of longer-lived semiquinone species has higher probability of reacting with oxygen, leading to O2 •−production. The resulting elevated O2 •− formation provides further oxidative stress in a vicious cycle, especially when the damage occurs in both pathways, leading to an even more intensive oxidative damage (85, 138). Frequently, tumor growth requirements exceed the energy supply of nutrients from blood resulting in hypoglycemia or aglycemia (284). Under this condition, energy can be obtained from glutamine by glutaminolysis-related pathways (284). Particularly, the O2-dependent glutaminolysis might elevate mtROS when α-KG and concomitant NADH production exceeds the electron transport rate within complex I (140, 284). Accordingly, it was demonstrated that glucose deprivation in human cancer cells results in a compromised ability to detoxify H2O2 derived from mitochondrial metabolism, due to the diminution of pyruvate and NADPH, which are involved in the cellular detoxification of hydroperoxides (4). Aglycemia and nutrient shortage promote OXPHOS and contribute with mitochondrial biogenesis (263, 284). The increased number of mitochondria in cancer cells would also eventually lead to increased ROS generation. An additional important consequence of cancer cell metabolism is the increased acidosis in tumor microenvironment (discussed in I.B.4). Recently, it was demonstrated that the exposure of cancer cells to extracellular acidosis induced ROS generation, which was abolished by the presence of antioxidants (270). ROS levels increased in the presence of rotenone, an inhibitor of complex I, under acidic conditions, suggesting that mitochondria were the source of oxygen radicals (270).
b. Cancer cell survival and proliferation
It has been demonstrated that ROS levels should rise above a certain threshold to promote cell cycle progression, proliferation, and survival. Particularly, intracellular H2O2 in the range of 0.01–1 μM is associated with cell proliferation (290). Several growth factors such as epithelial growth factor, insulin, and PDGF stimulate their cognate plasma membrane receptors and promote ROS generation and cell proliferation (87). However, malignant cell growth is often independent of mitogenic stimulation (126) due to oncogenic transformation that might arise from high intracellular ROS levels (325, 334). The Ras-ERK, mitogen-activated protein kinase (MAPK), and PI3K/Akt intracellular pathways closely related to cell proliferation and survival are the ones most significantly affected (112, 325, 353) (Fig. 8). ROS may reduce cell dependence on growth factors by lowering the activation threshold of the cognate TKR, or by transactivating the receptors in a ligand-independent fashion (269). Although proliferation of cancer cells is often independent of mitogenic stimulation, stromal cells respond to mitogenic signaling with a concomitant ROS increase, contributing the prooxidant state of the tumor microenvironment. Moreover, ROS levels increase through the cell cycle, and scavenging of ROS by antioxidants leads to late G1 cell cycle arrest (134, 274). Furthermore, mtROS are involved in regulating the activity of kinases that promote cell proliferation, such as ERK1/2 and Akt, and the proapoptotic kinases c-Jun N-terminal kinases (JNK) and p38 MAPK (10). Differential regulation of signaling molecules is mediated by cysteine oxidations, which depend on redox status and steady-state concentration of H2O2 (10, 11). Thus, different levels of H2O2 may lead to opposite responses on cell proliferation, differentiation, arrest, apoptosis, or senescence (10). The NOX enzymes have also been reported to promote malignant cell growth. For instance, NOX4 and NOX5 promote tumor cell survival in pancreatic and lung cancer, respectively (151). Additional studies have demonstrated that low concentration of exogenous ROS, particularly H2O2, induces cell proliferation (290). Thus, ROS generated in the tumor microenvironment could stimulate proliferating signals inducing a positive feedback that leads to a vicious circle of ROS generation. Although ROS are traditionally associated with the induction of cell proliferation, emerging lines of evidence indicate that increased tumor aggressiveness is associated with a reduction in ROS generation (48, 77). Indeed, activation of ROS-scavenging systems, for instance, through Nrf2, and hence a reduction of ROS levels has been shown to be associated with tumor cell aggressiveness in breast cancer (154). The question remains whether only malignant cells that have adapted to oxidative stress by enhancing their endogenous antioxidant capacity to lower ROS levels are prone to disseminate and metastasize (154).
c. Metastatic dissemination
The initial steps of the metastatic process involves an EMT, by which malignant cell lose cell polarity and detach from neighbor cells, augment the interaction with ECM, and migrate toward blood and lymphatic vessels. During this process, the ECM is remodeled by several proteolytic enzymes such as MMPs facilitating cell intravasation into the circulation. ROS generation was associated with several stages of this process (237, 238, 288). Initial studies demonstrated that mtROS generation was the major source to promote cell shape changes and detachment (329). ROS production was associated with loss of cell–cell adhesion and cytoskeleton reorganization (258). ROS produced via the transcriptional activation of NOX have also been linked to the formation of invadopodia and increased cell motility (81). mtROS generation was involved in the regulation of the early focal cell contacts with the ECM, whereas membrane oxidases drive the spreading and actin dynamics of moving cells (293). In addition, a recent study showed higher ROS levels in a cancer-derived metastatic cell line compared with a cell line derived from the primary lesion of the same patient (181). Moreover, it has been recently demonstrated that targeting CATs to mitochondria suppresses the metastatic capacity of breast cancer cells in mouse models (114).
d. Cancer cell death
Cell death occurs by several mechanisms, including necrosis or PCD, in normal and also in cancer cells. However, malignant cells generally induce several mechanisms to resist cell death programs (120). Necrosis has long been recognized as a proinflammatory event in contrast with PCD that is a noninflammatory process. In classic apoptosis (PCDI), early collapse of the cytoskeleton occurs, but organelles are initially preserved. In contrast, PCDII is usually produced by a prolonged autophagy, where some organelles are degraded early with initial preservation of the cytoskeleton (16, 157). Although autophagy can promote cell death, this process is also involved in cell survival, especially under stress condition (353). Autophagy is a catabolic cellular pathway where parts of the cytoplasm and intracellular organelles are sequestered into double-membrane autophagosomes, which are delivered to lysosomes for hydrolytic degradation. This process generates nucleotides, amino acids, and fatty acids, which are recycled for ATP generation and macromolecular synthesis (16). Thus, cancer cells might also support tumor survival by buffering metabolic demands under stress condition, contributing to tumor metabolic autonomy (212, 257).
ROS are involved in the different modalities of cell death. It is well demonstrated that high levels of ROS can induce apoptosis by triggering the opening of the mitochondrial permeability transition pore, a megapore spanning the inner and outer mitochondrial membrane composed by cyclophilin, VDAC, and the adenine nucleotide translocase (61) (Fig. 6). Activation of JNK by ROS can also induce extrinsic or intrinsic apoptosis signaling (61). Excessive ROS production and ATP depletion from uncoupling of OXPHOS promote necrotic cell death. Furthermore, it has been postulated that the switch from apoptosis to necrotic cell death involves not only a decrease in cellular ATP but also a burst in intracellular ROS (16). ROS are also involved in autophagy induction, with protective or destructive consequences. Starvation induces ROS generation, which triggers protective autophagy, contributing to cell survival; however, in some cases, autophagy causes accumulation of ROS and finally cell death. Particularly, it has been demonstrated that during starvation, autophagic cells generate ROS by the selective degradation of CATs that subsequently cause cell death (16). ROS can induce autophagy by the induction of the autophagy-related gene 4 (Atg) and also by disturbances in the mETC. Furthermore, mitochondrial oxidation events, including ROS production and lipid oxidation, play a key role in the induction of autophagy (16, 275).
5. CSCs and redox consideration
The redox status of CSCs in the tumor microenvironment is uncertain (1). Some reports described lower levels of ROS in some CSCs as compared to nontumorigenic cells in human and murine tumors (82, 357). Low levels of ROS seem to be associated with an elevated expression of ROS-scavenging molecules (1). Recent studies suggest that treatment with chemotherapeutic drugs can lead to the emergence of resistant stem cell-like populations expressing stem cell markers such as CD133 and Oct-4; resistance to chemotherapy was associated with reduced ROS levels, augmented activity of ROS-scavenging enzymes, and Nrf2 stabilization (2). Consistent with this, tumor samples obtained from patients that underwent neoadjuvant chemotherapy showed lower ROS levels compared to patient samples that received no therapy (2). In addition, the induction of EMT-like phenomenon in liver CSCs was associated with increased CD13 expression, which plays a role in the reduction of the intracellular ROS level promoting CSC survival (158). Reduced levels of ROS in conjunction with other parameters in CSCs were suggested as a marker of the presence of CSCs in human lung tumors (342). Furthermore, reduced ROS levels in CSCs were closely associated with the induction of radio- and chemoresistance (82). Thus, the possibility exists that the small population of CSCs that was proposed to be present in certain human cancer types might escape antitumor treatment by augmenting scavenging enzymes to reduce ROS levels.
D. ROS generated by the tumor microenvironment
1. ROS generated by CAFs
Recent studies have shown that cancer cells can induce ROS overproduction in CAFs (210, 211). Coculture of human breast cancer cells and immortalized human fibroblasts induced a significant increase in ROS in fibroblasts (210). However, increased ROS levels promoted DNA damage in both cell types. In response, cancer cells induced the expression of antioxidant enzymes (PrxI) and antiapoptotic proteins (Fig. 9). On the other hand, oxidative stress triggered autophagy/mitophagy and aerobic glycolysis in CAFs, mediating the generation of a lactate-rich microenvironment (188). Interestingly, lactate derived from CAFs also induced mitochondrial biosynthesis in breast cancer cells, which can increase ROS generation in the tumor microenvironment (Fig. 9). Recently, it was demonstrated that CAFs in contact with human prostate carcinoma cells mediate a motile and stem-like phenotype through EMT. In response to CAF contact, cancer cells increased oxidative stress through a Rac1b/COX-2 pathway (110). On the other hand, gene expression analysis of primary human prostatic stromal cells induced to undergo fibroblast-to-myofibroblast differentiation with TGF-β1 showed upregulation of NOX4 expression and downregulation of the selenium-containing ROS-scavenging enzymes (273). Thus, CAFs would contribute with the prooxidative tumor microenvironment, promoting genomic instability in cancer cells by enhancing ROS generation with a potential increase in the tumor aggressive behavior (188).

2. ROS generated by inflammatory cells
A plethora of publications exist connecting cancer, inflammation, and ROS, and most of them demonstrate that inflammatory cells induce ROS generation contributing with tumor initiation and progression (9, 21, 72, 280). Macrophages and neutrophils are of the main ROS-generating cells during an inflammatory or immune response. However, this is difficult to reconcile with the evidence that inside the tumor mass, TAMs are polarized into the M2 type, whereas neutrophils are in the N2 state that were not associated with high levels of ROS generation (102, 209). Interestingly, the presence of mutations in the hypoxanthine–guanine phosphoribosyl transferase locus has been associated with the genotoxic capacity of neutrophil-derived ROS, and the grade of mutation also correlated with the levels of neutrophil infiltration (128). Macrophages can be activated by contact with tumor cells (226) or with tumor-derived microvesicles (TMV), which induced ROS generation (20). TMV are small membrane fragments that are released spontaneously by tumor cells during proliferation, migration, activation, and apoptosis. In addition, eosinophils have the most vigorous respiratory burst than any other inflammatory cell type (111) and might also contribute with the prooxidative state of the tumor microenvironment (223).
Proinflammatory cytokines present in the tumor microenvironment also promote ROS generation in phagocytic and nonphagocytic cells through the activation of different signaling pathways. For instance, tumor necrosis factor α (TNF-α) enhanced ROS formation by neutrophils and other cells, whereas IL-1β and TNF-α stimulated the expression of inducible nitric oxide synthase (iNOS) in inflammatory and epithelial cells (96). Furthermore, several damage-associated molecular pattern proteins (DAMPs) passively released by necrotic dying tumor cells can also activate the immune system, inducing an inflammatory response (275). For instance, the chromatin-associated DAMP, high-mobility group box l protein, which is expressed in most tumor cells, (282, 295) stimulates the release of TNF
A very recent finding indicates that tumor cells can acquire metastatic characteristics through the recruitment of myeloid-derived suppressor cells (MDSCs). These cells mediate immune suppression of T cells by ROS-mediated mechanisms (227). In tumor-bearing mice, MDSCs accumulate in the bone marrow, spleen, and peripheral blood and can be found inside primary and metastatic solid tumors, whereas in cancer patients, MDSCs were only found in the circulation (235). MDSCs collected from peripheral blood of patients with stage III head and neck cancer showed fivefold higher ROS levels after phorbol 12-myristate 13-acetate stimulation compared with cells with the same phenotype obtained from healthy volunteers. The increased ROS levels in MDSCs are caused by upregulation of several subunits of NOX under the control of the STAT3 transcription factor (65).
3. ROS generated by ECs and the angiogenic process
Angiogenesis is a process closely related to ROS generation (5, 101, 217, 311, 312). ROS are derived mainly from NOX proteins (101, 311, 312), although mtROS production is also involved (312). Earlier evidence demonstrated that ROS produced by NOX1 trigger the angiogenic switch, allowing vascularization and rapid expansion of the tumor (12). Particularly, VEGF increased two- to threefold intracellular ROS by activation of NOX1 or NOX2, which is essential for their migration and proliferation (101, 107, 311) (Fig. 10A). Ang1 and 2 also increase H2O2 generation, mainly through the activation of NOX2 through the Tie-2 receptor, which is required for EC chemotaxis (311, 312). The functional role of NOX-derived ROS was demonstrated by the observation that the antioxidant or the gp91phox-antisense oligonucleotides significantly block VEGF-induced ROS production and the EC proliferation and migration (101, 107, 312). In addition, the induction of H2O2 generation through the upregulation of Mn-SOD expression was also suggested to be involved in this process (310) (Fig. 10A). The relevance of ROS generation during angiogenesis was also demonstrated in vivo (311, 312). Sponge implant assays in mouse models showed that VEGF-induced angiogenesis was significantly reduced in wild-type mice treated with N-acetyl cysteine (NAC) and in gp91phox−/− mice, suggesting that ROS derived from gp91phox-containing NOX play an important role in angiogenesis in vivo (311, 312). Furthermore, treatment with dietary antioxidants such as food phytochemicals was able to promote an antiangiogenic response (311). In this sense, we were able to inhibit VEGF expression and tumor growth in experimental tumors (134) treated with exogenous CATs (Fig. 10B, C).
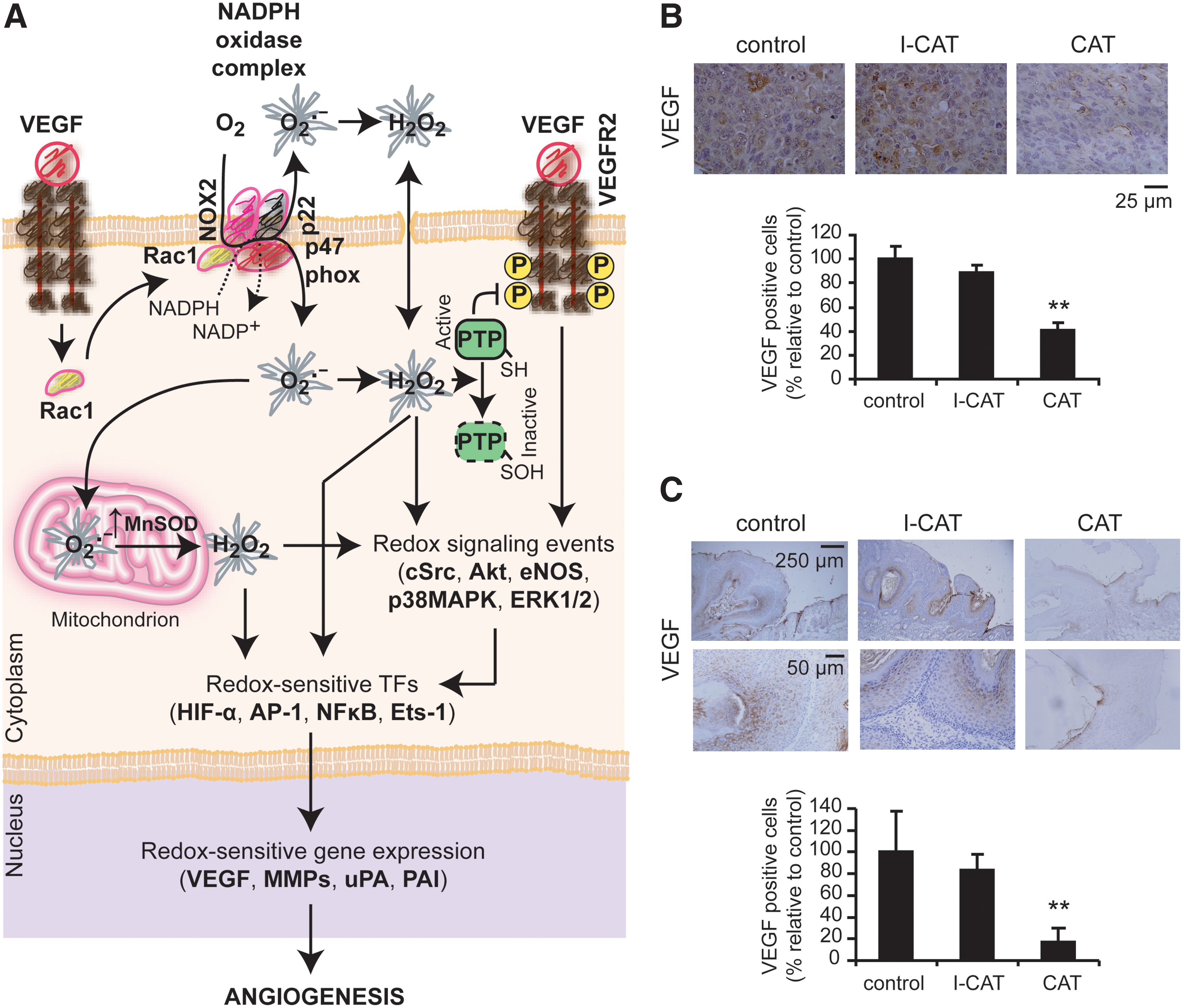
ROS derived from the action of proangiogenic factors activate redox sensor transcription factors such as HIF1α, Ets, AP-1, and NF-κB (311), which in turn promote the transcription of genes involved in the angiogenesis process. MMP activities are essential for ECs to penetrate into the tumor mass. ROS derived from NOX1 inhibited the post-translational modification of the nuclear hormone receptor peroxisome proliferator activated receptor α, promoting the activation of NF-κB and the induction of MMP-9 transcription (107).
It was clearly demonstrated that exogenous H2O2 added in cultured ECs and vascular smooth muscle cells promotes VEGF expression (217). Thus, ROS generated by the action of proangiogenic factors would promote a positive feedback of ROS generation through VEGF enrichment in the tumor microenvironment contributing with the prooxidative tumor state.
4. ROS generated by the hypoxic microenvironment
Earlier studies have demonstrated the formation of mtROS under hypoxia, and these species were suggested as the real sensor of oxygen deficiencies in cells (162). Since then, many genetic and pharmacological approaches have been employed to inhibit the activity of components of the mETC, preventing hypoxia-mediated stabilization of HIF1α protein (162). These studies have shown that under moderate hypoxia (1.5% O2), mitochondria stimulate ROS generation, which in turn inhibits PHD activity, leading to the stabilization of HIF1α. The pharmacologic and genetic data indicate that the ubiquinone cycle of complex III is the source of ROS generation during hypoxia to stabilize HIF1α protein (162). Recently, it has been described that mitochondrial NAD-dependent deacetylase sirtuin-3 (SIRT3) has tumor suppressor functions via its ability to suppress ROS acting on complex III. The loss of SIRT3 function hyperactivates HIF1α under hypoxia (200). ROS generated at the mitochondrial inner membrane space and in the cytosol under hypoxia were recently visualized using a novel redox-sensitive fluorescent protein (324). Under normoxia, cytosol was the most reduced; the mitochondrial matrix was the most highly oxidized compartment, and the intermembrane space exhibited an intermediate oxidation level. During hypoxia, the mitochondrial matrix underwent a reductive shift, whereas the cytosol and the intermembrane space showed an oxidative change despite the decrease in O2 (324), suggesting that ROS were released from the outer surface of the inner mitochondrial membrane to the intermembrane space and diffuse into the cytosol. However, ROS levels generated during hypoxia appear to be substantially lower than those induced by senescence or apoptosis as assessed using a mitochondrial-targeted redox-sensitive probe (324). NOXs may also contribute to the generation of ROS during hypoxia through protein kinase C activation, suggesting that both mitochondria and cytosolic oxidant systems may contribute to the overall response (81).
Cellular oxidative stress can be increased not only by chronic hypoxia but also by cycling hypoxia (79). During cycling hypoxia, there is a significant increase in ROS generation accompanied by HIF1α stabilization (213), which was demonstrated by oxidative DNA damage and lipid peroxidation measurements in mammary tumors (79) and also by dichlorofluorescein diacetate assays in human glioblastoma cells (129). Furthermore, NOXs seems also to contribute to ROS generation during cycling hypoxia. A recent study demonstrated that NOX4 knockdown or treatment with a NOX inhibitor blocked cycling hypoxia-induced ROS. NOX4-generated ROS are required for cycling hypoxia-induced cell invasiveness through the activation of NF-κB- and ERK-mediated stimulation of MMP-9 (129). Thus, the process of ROS generation from chronic or acute hypoxia might be amplified by cycling hypoxia, contributing to the prooxidative tumor microenvironment. Figure 11 summarizes the contribution of ROS to the prooxidative tumor microenvironment described in this section.
E. In vivo evidence of ROS generation in tumors
Current evidence supports the hypothesis that cancer cells are characterized by enhanced ROS generation, increased ROS accumulation, and deregulation of antioxidant enzymes, thus existing in a state of perpetually elevated stress (119). In vivo evidence of elevated oxidative stress was observed in many solid tumors, including breast cancer, in which H2O2 generated by EC-SOD may exacerbate or decrease the cell proliferation in a tumor microenvironment (207). It has been well established that 8-hydroxy-2′-deoxyguanosine (8-OHdG) is one of the major oxidatively modified DNA base products in vivo. Thus, this lesion has been considered as a biomarker of in vivo oxidative stress. Early reports demonstrated higher levels of 8-OHdG in renal cell carcinoma, mammary invasive ductal carcinoma, colorectal carcinoma, and lung squamous cell carcinoma as compared with their nonmalignant counterpart (139, 166, 303). More recently, this finding was extended to other cancer types (94). High levels of 8-OHdG were also detected in serum, urine, or saliva from cancer patients (19, 47, 172, 341). However, increased 8-OHdG expression was associated with good prognosis in breast cancer, especially related to the ductal type tumors (154). Concomitant with these results, other markers of oxidative stress were described in different types of experimental and human cancer. For instance, high levels of protein oxidation products were found in colorectal cancer patients (47) and in saliva of patients with oral squamous cell carcinoma (19). An increase in lipid peroxidation, measured by the thiobarbituric acid-reactive substances assay, was described in experimental breast cancer (243), as well as in the breast cancer microenvironment, it has been found a peculiar lipid peroxidation and protein oxidative profile (205, 206). Increased levels of lipid peroxidation products were also found in sera of Hodgkin's lymphoma patients as compared with healthy controls (221). Several reports correlated the increase in markers of oxidative stress with a decrease in antioxidant defenses. Bahar et al. reported high levels of oxidative products of DNA and proteins in saliva of patients with oral squamous cell carcinoma as compared with healthy controls, in correlation with reduced values of the total antioxidant capacity and specific antioxidants, that is, peroxidase, glutathione S-transferase, SOD, and uric acid (19). A negative correlation between increased oxidative DNA damage, measured by 8-OHdG, and reduced antioxidant capacity was also reported in tumor samples of patients with glioblastoma multiforme (307). Evidence on the relation between intracellular and extracellular oxidative stress has also been reported. Cancer patients with high levels of generalized oxidative stress markers in their sera also exhibited markers of constitutive oxidative stress within tumors (119).
III. ROS and Cancer Gene Therapeutics
A. Introduction
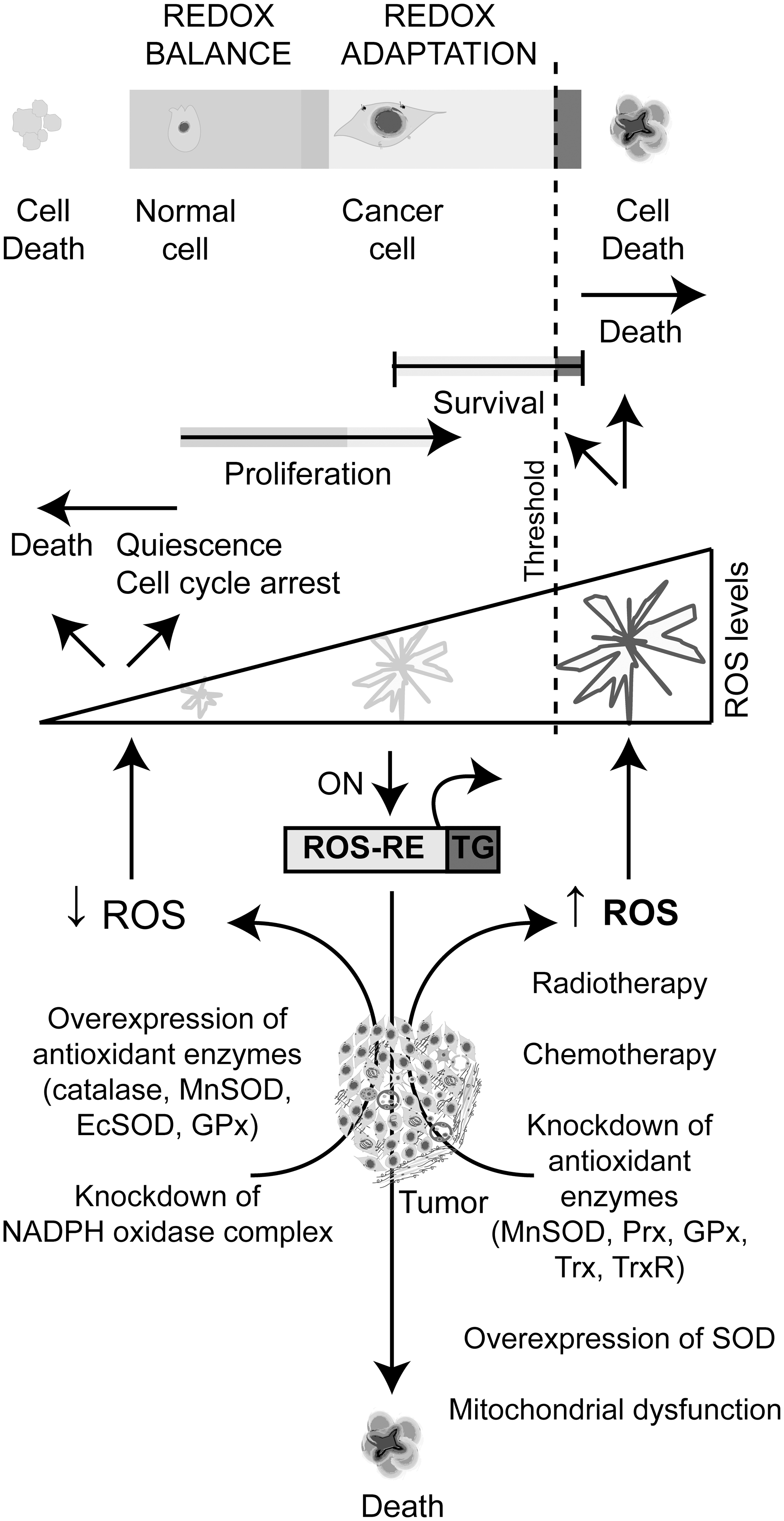
1. Threshold ROS concept for cancer therapy
ROS contribute to several characteristics of the cancer cell phenotype, such as genomic instability and activation of signaling pathways related to survival, proliferation, evasion of apoptosis, angiogenesis, and metastasis. Due to the critical role of ROS in promoting the malignant phenotype, scavenging of ROS has long been accepted as a therapeutic strategy (304, 321). On the other hand, it is widely accepted that augmenting intracellular ROS levels can trigger cancer cell death, and hence it is a plausible tumor suppressor strategy. The mainstay treatments in cancer, such as chemotherapy and radiotherapy, also increase ROS production, which can mediate and induce cancer cell death (236). Thus, increasing the oxidative cells status has also been considered as a conceptual basis to develop different prooxidant cancer therapies (304, 321).
Normal cells maintain redox homeostasis with low basal ROS levels and have a reservoir of antioxidant capacity to tolerate a certain level of exogenous ROS stress. Cancer cells have an increased prooxidant status as compared to normal cells. This prooxidant state generates a redox adaptation response by upregulating the antioxidant capacity to maintain ROS levels below the toxicity threshold (Fig. 12). In consequence, cancer cells have their antioxidant system overloaded and would be more vulnerable to further oxidative stress induced by exogenous ROS-generating agents (167, 304, 321).
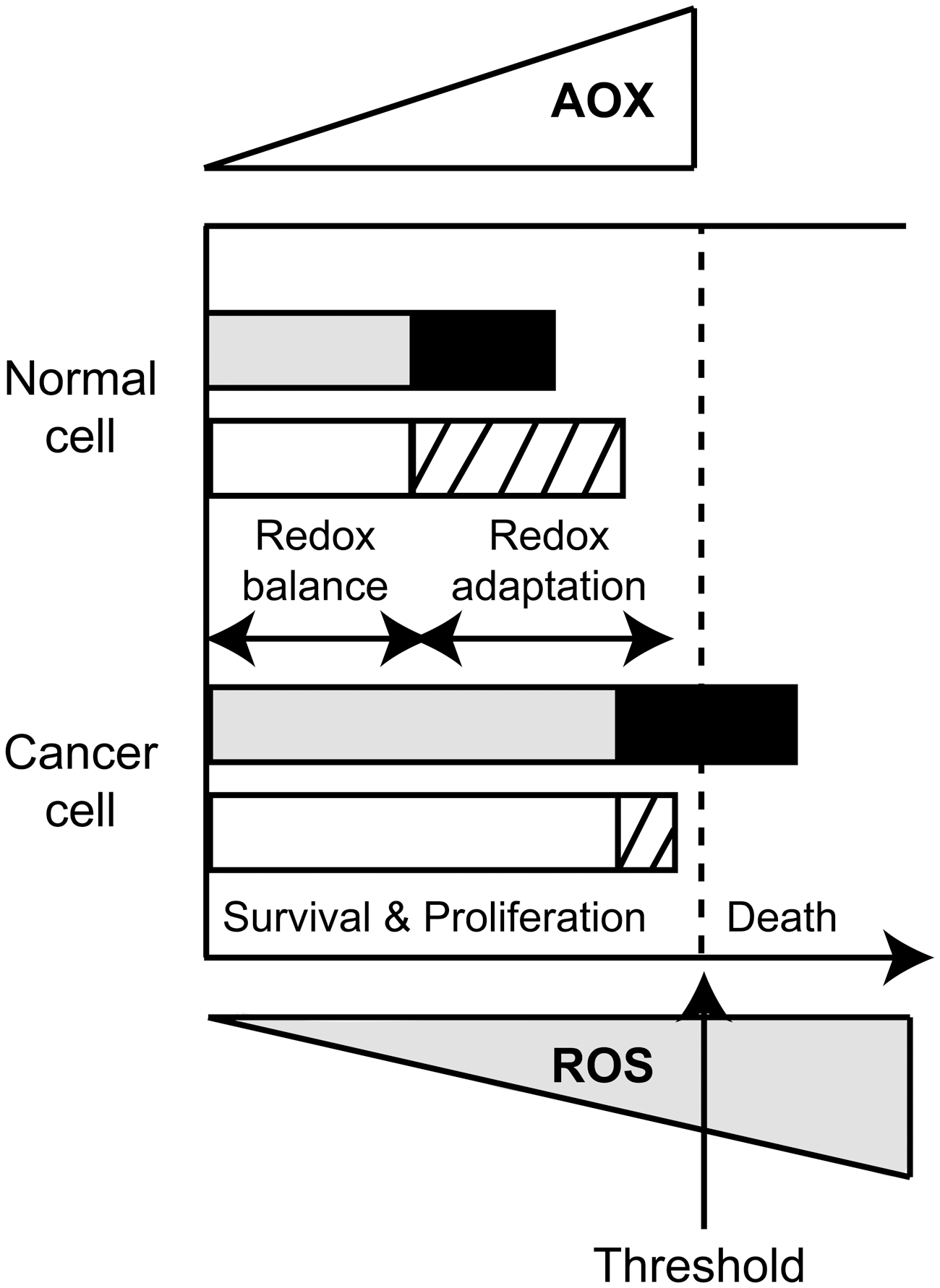
Thus, normal cells have the capabilities to maintain redox homeostasis even when exogenous ROS surpass certain threshold; however, malignant cancer cells appear to lack this capacity and eventually die (Fig. 12). This difference in the ability to dissipate increased ROS levels between normal and malignant cells is the basis for the selectivity of some of ROS-targeted cancer therapeutics. A provocative hypothesis is that metastasizing cells are those capable of overcoming the aggressive ROS microenvironment, by augmenting antioxidant defenses to higher levels than those needed to survive, and thus they would be able to disseminate (237). The CSC appears as the right candidate for that endeavor; however, further investigations are needed (237).
Different therapeutic strategies based on chemicals (95) or genetic drugs (discussed below) appoint to upregulate or downregulate ROS levels in cancer. A more recent approach explored by our group took advantages on the differentially higher ROS levels in malignant tissue. This strategy aims to drive the selective expression of therapeutic gene by ROS-responsive elements that can sense intracellular ROS levels and can activate the downstream gene expression (250). The therapeutic potential of this novel ROS-based approach will be discussed in section III.C.
2. An overview of ROS-based cancer gene therapeutics
Gene therapy (GT) is a relatively new paradigm for treatment of human diseases and is becoming a rationale area for the development of novel agents for cancer treatment. Most clinical trials are still in early phases, and more than 60% of these trials target different cancer types (
ROS-based cancer gene therapeutics focus on scavenging cellular ROS, increasing ROS production or the conditional targeting through ROS-responsive elements (Fig. 13). GT approaches to decrease ROS levels are generally based on the overexpression of antioxidant enzymes. Several approaches included knocking down the activity of some components of the NOX complexes to decrease intracellular ROS production. On the other hand, strategies to increase ROS levels generally involve knocking down specific components of the antioxidant system, or altering specific molecular targets to promote an increase of mtROS. Moreover, in certain circumstances, the overexpression of the antioxidant enzyme SOD increases H2O2 and promotes cell death. We will discuss in detail the different approaches in this section.
An additional GT strategy is the conditional targeting that provides effective and specific activation of medical products inside the tumor mass. This strategy implies the regulation of viral replication, or alternatively the antitumor activity of a therapeutic gene through a tumor-specific promoter differentially activated in cancer cells. However, the large number and variability of DNA alterations among the different tumors and the genetic heterogeneity among different cell subpopulations of the same tumor cells limit the potential use of a promoter obtained from a tumor-associated gene. Therefore, targeting the tumor mass by taking advantage of a differential microenvironmental characteristic that differentiates cancer and normal tissues is an interesting option. For instance, hypoxia was used for the selective expression in cancer tissues of therapeutic genes driven by hypoxia-response elements (135). In a recent article (250), we proposed the prooxidative microenvironment of malignant tumors as a differential feature of cancer that can be utilized to drive the expression of therapeutic genes through ROS-responsive DNA sequences. As detailed in section II, cancer and stromal cells contribute to ROS generation in the tumor mass, and this characteristic may be considered as another hallmark of cancer microenvironment that could be exploited to develop new cancer gene therapeutics.
B. GT strategies to modulate extra- or intracellular ROS levels
1. Decreasing ROS extra- or intracellular levels by GT strategies
a. Overexpression of the antioxidant enzyme system in cancer cells
ROS favor tumor growth by promoting genetic instability, cell proliferation, angiogenesis, and metastasis. Thus, scavenging ROS by overexpressing antioxidant enzymes is a valuable strategy to suppress tumor growth. The chemical scavenging of ROS and its therapeutic effect have been extensively reviewed recently (195).
The overexpression of H2O2-scavenging enzymes is one of the most explored strategies to inhibit cancer growth. CAT levels are often decreased in a wide variety of tumors and in cancer cell lines as compared to nonmalignant cells (113, 249). The origin of this deficiency in malignant cells remains unclear, but recent data point to the hypermethylation of the CAT promoter, as the explanation for its reduced expression in cancer cells has been presented (218). In initial studies, we demonstrated that stable overexpression of the human CAT gene in breast cancer cells diminished ROS generation, inhibited proliferation, and reverted malignant features (249). Recently, these results were confirmed by showing that the proliferation and migration capacities of breast cancer cells were impaired by CAT overexpression (113). In a recent study, transgenic mice expressing a mitochondrial-directed human CAT gene (mhCAT) were crossed with transgenic mice that develop spontaneously metastatic breast cancer. The progeny expressing mhCAT showed decreased levels of primary tumor invasiveness and suppression of pulmonary metastasis compared to control mice (114). The authors hypothesized that expressing mhCAT inside malignant and stromal cells might increase the antioxidant capacity of the mitochondrial compartment, raising the possibility of using this rational therapeutic approach for treating invasive breast cancer. Scavenging H2O2 by CAT overexpression prolonged cell-doubling time by extending the G0/G1 phase and by modulating the activities of the complexes responsible for the G0/G1-to-S-phase transition, such as cyclin D-CDK4, cyclin E-CDK2, and the CDK inhibitory protein p27(KIP1) (134, 234). In addition, CAT overexpression in breast cancer cells led to sensitization to chemotherapeutic drugs, such as paclitaxel (PTX), etoposide, and arsenic trioxide (ATO), but increases the resistance to the prooxidant effect of ascorbate. However, no effect was observed on the cytotoxic response to 5-fluorouracil, cisplatin, doxorubicin (DOX), or irradiation (113). In contrast, CATs targeted to mitochondria increased the resistance to ionizing radiation in vitro and in vivo (91).
Overexpression of GPx, another H2O2-scavenging enzyme, has also been explored as a gene strategy. Low levels of antioxidant enzymes, including Mn-SOD and glutathione-dependent enzymes, have been reported in pancreatic cancer (191). Delivery of GPx cDNA by an adenovirus vector (Ad-GPx) to pancreatic tumor xenografts slowed tumor growth approximately by 50%. The combination of Ad-GPx with an adenovirus expressing Mn-SOD cDNA suppressed tumor growth almost completely (191). In addition, the expression of phospholipid glutathione peroxidase (PhGPx) is also diminished in pancreatic cancer cells. The overexpression of both the mitochondrial PhGPx form (L-form) and the nonmitochondrial PhGPx form (S-form) by a recombinant adenovirus enhanced tumor growth inhibition in vitro and in vivo (190). SODs show a significant variability in human cancer (161). Many human tumors express high levels of SODs, and this has been associated with aggressive tumor characteristics; meanwhile, other studies have found low SOD activity in the same or different cancer types. Overexpression of SODs in different cancer models will be analyzed in the next sections.
While most data support the view that tumor growth can be inhibited by decreasing ROS levels, large-scale randomized clinical trials searching for the effect of antioxidants in cancer prevention showed inconsistent conclusions regarding their potential ability as tumor suppressors (299). Moreover, supplemental antioxidant administration combined with chemotherapy or radiation therapy might have a bystander effect promoting tumor protection and reduced survival (178).
b. Overexpression of the antioxidant enzyme system in the tumor stroma
A novel approach of cancer gene therapeutics is to suppress ROS generation in the tumor stroma. Recently, Lisanti and colleagues (305) have demonstrated that the loss of caveolin-1 (Cav-1) in human CAFs dramatically promotes human breast cancer cell growth. The loss of Cav-1 expression, the principal component of caveolae, is one of the most relevant stromal biomarkers associated with a poor clinical outcome in breast cancer patients (210). Cav-1 is a potent inhibitor of NOS, and the loss of Cav-1 triggers NO production, leading to mitochondrial dysfunction and an increase of ROS generation in CAFs (210). Interestingly, the breast cancer-promoting effect of Cav-1 knocking down in human fibroblasts was significantly reduced after the stable expression of Mn-SOD (305). Thus, Mn-SOD may also function as a stromal tumor suppressor gene, by lowering oxidative stress in the tumor microenvironment.
The importance of EC-SOD as a potential therapeutic target has also been demonstrated. EC-SOD overexpression by adenoviral vectors attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion (297). Heparanase activity has been widely implicated as an important regulator of proliferation, invasion, and metastasis, and it is involved in the progression of various human cancers, including breast cancer. This enzyme acts both at the cell surface and within the ECM-degrading polymeric HS molecules. Proteolytic HS-derived products act as signaling molecules that can promote cancer growth, angiogenesis, and invasion. ROS are involved in HS degradation, and the overexpression of the human full-length wild-type EC-SOD gene and particularly the EC-SOD gene with a deletion in the heparin-binding domain (ECSODΔHBD) inhibited the in vitro growth and invasion of two aggressive breast cancer cell lines (297). The relative rates of O2 •− formation were significantly diminished in a conditioned medium containing either the full-length EC-SOD or EC-SODΔHBD. The inhibitory effects of either form of EC-SOD were greatly enhanced with the addition of heparin or HS mimetics. Thus, the scavenging of ROS in tumor stroma is an interesting strategy that could be explored as cancer ROS-based cancer gene therapeutics. NOXs are the major ROS generating in endothelial stromal cells and might be an interesting target to inhibit tumor angiogenesis through the suppression of ROS generation.
c. Knocking down NOXs
ROS generated by NOXs have been shown to contribute to tumorigenesis in many types of cancer (151), and the enforced suppression of its activity has been explored in recent studies. For instance, enforced suppression of Rac-1 activity, a key component in the regulation of NOXs, decreased O2 •− levels production and promoted the in vitro and in vivo growth inhibition of human pancreatic cells, suggesting that inhibition of Rac1 may be a potential therapeutic option (88). Hemangioendotheliomas are classified as EC tumors, which are the most common soft tissue tumors in infants. Suppression of NOX4 by stable transfection of small interfering RNA (siRNA) markedly diminished H2O2 accumulation in the nuclear compartment and inhibited hemangioendothelioma formation in a murine model (115). In addition, enforced NOX4 knockdown in human glioblastoma by siRNA inhibited ROS tumor growth and invasion (129).
Although chemical inhibitors of NOXs demonstrated certain efficacy in cancer clinical trials (225), adverse effects on the immune innate defense might restrict their potential use. In this scenario, targeting NOXs at the tissue of interest using a GT approach could provide an alternative strategy avoiding harmful secondary effects.
Another intensive area of research is the use of antioxidants for cancer prevention. However, some data suggest that these compounds may also have toxic effects (42). Thus, targeted vehicles to delivery of genes that will interfere with ROS production could provide an alternative mechanism to avoid adverse effects on normal organs. However, it should be considered that antioxidant treatment would reduce tumor cytotoxicity after radiotherapy or chemotherapy, two of the mainstay cancer treatments largely dependent on ROS to induce cytotoxicity.
d. Knocking down other ROS cell sources
Knocking down the expression of other ROS sources has been also explored; however, the effects on proliferation or tumorigenesis seem to be unclear. For instance, knocking down the expression of myeloperoxidase (MPO) inhibited ROS generation and abrogated the effect of parthenoline (PTL), an antileukemic agent. This study suggested that MPO seems to play a crucial role in determining the susceptibility of leukemic cells to PTL-induced apoptosis (160). Proline oxidase (POX) is a mitochondrial enzyme that oxidizes proline and generates ROS as a byproduct. Although POX is decreased in tumors, it was demonstrated that its expression is induced by serum-oxidized low-density lipoprotein (OxLDL), which has been associated with increased cancer risk (244). Knocked down POX via siRNA reduced the viability of cancer cells treated with OxLDL and decreased ROS generation and autophagy (346). On the other hand, COX-2 is associated with ROS generation and plays a very important role in carcinogenesis. It is overexpressed in many malignant tumors, and higher levels are associated with poor prognosis. Knocking down the expression of COX-2 by siRNA in esophageal carcinoma cells suppressed proliferation and tumorigenesis in nude mice; although the authors did not report ROS levels, it is tempting to speculate that ROS could be involved in mediating this process (351).
2. Increasing intracellular levels of ROS by GT strategies in cancer cells
a. Knocking down the antioxidant enzyme system
An explored strategy to increase ROS levels is interfering with cellular antioxidant enzyme systems. Thus, SODs, Prx, GPx, and the Trx system have emerged as important targets for anticancer gene therapies aiming to increase intracellular oxidative stress levels. One of the most common strategies for knocking down the expression of antioxidants enzymes is the use of iRNA.
(1) Superoxide dismutases
Early studies demonstrated that Mn-SOD-antisense RNA promoted γ-ray-induced apoptosis of oral squamous cell carcinoma by regulating the expression of Bcl-2 family proteins (308). Recently, Mn-SOD expression knocked down by miRNA administration promoted the radiosensitization of nasopharyngeal carcinoma cells (256). In addition, suppression of Mn-SOD activity in conjunction with other mitochondrial enzymes such as GPx and TrxR2 was also explored using miRNA generated from the 3′ arm of miRNA precursors (miRNA*) (339). Increased miR-17* levels through a Tet-on-based conditional expression system in prostate cancer cells markedly suppressed cell growth in vitro and in vivo. This effect involved the inhibition of mitochondrial antioxidant enzymes, suggesting that the miR-17* function in oxidative systems is opposite to its already-described oncogenic function (339).
Additional studies reported that knocking down Mn-SOD by siRNA in ovarian cancer increased ROS levels, which sensitized cancer cells to apoptosis induced by DOX and PTX, leading to a preferential killing of cancer cells (344). This effect was restricted to the activation of intrinsic apoptotic pathways through ERK1/2 and upregulation of caspase-9, which contributed to this synergistic effect (344). Treatment of cells with antioxidants such as GSH or NAC abolished the effects of Mn-SOD siRNA on DOX- and PTX-induced apoptosis. However, an additional report showed an opposite effect in human ovarian cancer cells in response to the suppression of Mn-SOD expression by siRNA (130). Mn-SOD is generally overexpressed in ovarian cancer tissues and positively correlates with bad prognosis in ovarian cancer patients (130). Suppression of Mn-SOD expression by siRNA induced 70% increase of O2 •− in ovarian cancer cells, leading to the stimulation of cell proliferation in vitro and more aggressive tumor growth in vivo (130). Docosahexaenoic acid (DHA) is an n-3 polyunsaturated acid with recognized anticancer properties through the induction of lipid peroxidation. Enforced downregulation of CuZn-SOD with siRNA induced an increase in the ROS level, enhancing cell sensitization to DHA in a resistant cervical cancer cell line (34).
(2) Peroxiredoxins
Several studies using gene-knockout approaches propose that Prxs function as tumor suppressor genes. Homozygous loss of Prx1 increased cancer incidence approximately fivefold, and the loss of Prx2 in thymocytes results in thymic hyperplasia (229). Prx levels were induced by radiation, suggesting Prxs as potential target for radiotherapy in cancer (349). Indeed, Prx1 is elevated in several human cancer cells and tissues (349), and suppression of Prx1 expression by GT strategies increased ROS levels (51, 52), tumor growth inhibition, and radiosensitization in lung, intestinal and rectal cancer (51, 52, 348). Prx1 knockdown significantly increased radiosensitivity, as indicated by a lower capacity to scavenge ROS that leads to a more extensive DNA damage through a mechanism that involves P53 (51, 52). In addition, mouse embryo fibroblasts derived from Prx1-deficient mice showed increased cisplatin-induced apoptosis compared with wild-type cells (198). Prx2 is also associated with radioresistance, and its downregulation sensitizes cells to radiation (349). However, silencing the expression of Prx2 by siRNA only partially reversed the resistance to ionizing radiation in a radioresistant breast cancer cell line, suggesting that Prx2 is not the sole factor responsible for the resistant phenotype (322). Prx3 is a specific mitochondrial H2O2 scavenger enzyme, and transient knocking down of Prx3 by siRNA in breast cancer cells induced the inhibition of cell proliferation (59). High levels of Prx3 are present in radioresistant cancer cells, suggesting that Prx3 plays an important role counteracting the ROS generation induced by ionizing radiation (71). Leukemia cells express high levels of Prx3, and its siRNA-induced depletion led to a significant accumulation of ROS and apoptosis induction in the presence of ATO (317). In addition, knocking down of both human Prx3 and Prx5 in neuroblastoma cells drives cell apoptosis induced by 1-methyl-4-phenylpyridinium ion, a complex I inhibitor (71). Prx4 is overexpressed in head and neck squamous cell carcinoma, and knocking down its expression increased ROS levels, apoptosis, and radiosensitivity (240). Accordingly, overexpression of Prx4 decreased intracellular ROS production and enhanced radioresistance (240). Prdx6, a cytoplasmic protein elevated in certain cancers, is highly expressed in the liver and transcriptionally regulated by various oxidative stressors. The cancerous Hepa1-6 hepatoma cell line is significantly more resistant to peroxide-induced cytotoxicity and exhibited an approximately twofold increased expression of Prdx6 compared to the noncancerous counterpart. Suppression of Prdx6 by siRNA increased the susceptibility to peroxide-induced cell death (319). It can be concluded that Prxs contribute to the radioresistance of cancer cells, and silencing the expression of Prxs by GT strategies might provide a novel approach to enhance the effectiveness of radiotherapy.
(3) Glutathione peroxidases
Although GPxs are widely believed to prevent carcinogenesis because of their ability to balance oxidative stress (302), these enzymes are overexpressed in many cancers, and the suppression of their expression enhances cancer cell death. DNA microarray analysis revealed that GPx2 is commonly upregulated in experimental breast carcinomas induced by chemical carcinogens (228). Suppression of GPx2 expression by siRNA resulted in significant growth inhibition in both rat and human mammary carcinoma cell lines expressing wild-type p53 (228). In colon cancer cell lines, knocking down GPx2 by siRNA resulted in a decreased capacity of colony formation in soft agar and reduced tumor growth in nude mice, but enhanced cell migration (23). GPx2 is associated with the inhibition of malignant characteristics of tumor cells by counteracting COX-2 expression, but is required for the growth of transformed cells, and therefore may facilitate tumor cell growth (23). Knocking down of GPx3 was shown to be associated with chemical sensitization. Indeed, GPx3 was highly expressed in ovarian clear cell adenocarcinoma, and suppression of its expression by siRNA increased cisplatin sensitivity (271). In addition, knocking down of GPx4 by siRNA significantly enhanced the cytotoxic effect of DHA in a human ovarian cancer cell line. This cytotoxic effect was reverted by pretreatment with vitamin E, suggesting that this sensitization is due to changes in the ability of the cells to handle oxidative stress (83).
(4) The Trx system
The Trx system (Trx, NADPH, and TrxR) is an important regulator of cellular redox status (255). Although chemical inhibitors of the Trx/TrxR system like motexafin gadolinium are under evaluation in several clinical trials for various tumor types (195, 304, 321), genetic strategies to knockdown this system have been also explored. Early studies demonstrated that stable transfection of mouse lung carcinoma cells with siRNA that specifically targets TrxR1 inhibited anchorage-independent cell growth properties and induced a dramatic reduction in tumor progression and metastasis in vivo in mouse models (49). Moreover, knocking down TrxR1 in mouse transformed fibroblasts resulted in defective progression in their S phase and decreased expression of DNA polymerase (345). However, in human lung cancer, no observable differences in cell viability, morphology, or phenotype were observed between control and siRNA-TrxR1- or miRNA-TrxR1-transduced cells (93, 248). Nevertheless, TrxR1-knocked-down cells exhibited increased ROS levels and a differential sensitivity to the cytotoxic effects of selenocompounds (248), 1-chloro-2,4-dinitrobenzene or menadione (93), which decreased intracellular GSH levels. This increased cytotoxic effects of selenocompounds were mediated by cellular stress, autophagy, and apoptosis (127).
Trxs were also explored as possible targets for GT strategies. Trx1 knocked down by siRNA enhanced ROS levels, decreased cell proliferation, and increased the sensitivity to histone deacetylase inhibitors (309) and to ionizing irradiation (75). Additionally, knocking down both Trx1 and CuZn-SOD by siRNA increased the sensitivity to manumycin-induced cell death in glioma cells (84). ROS levels were increased in Trx1 and CuZn-SOD siRNA-transfected cells and were further elevated upon manumycin treatment. This suggests that an elevation in ROS contributes to the manumycin-mediated glioma cell cytotoxicity (84). However, in breast cancer cells, silencing the expression of Trx by siRNA decreased ROS levels and apoptosis in the presence of daunomycin, but Trx overexpression increased ROS levels and apoptosis in the presence of anthracycline drugs (266). It was suggested a novel prooxidant and proapoptotic role of Trx in response to anthracycline drugs by facilitating the redox cycling apoptotic potential of daunomycin (266). The mitochondrial Trx (Trx2) was also downregulated by siRNA in cervical tumor cancer. This resulted in an increased sensitivity to some cationic triphenylmethanes such as brilliant green and gentian violet that were shown to have antitumor and antiangiogenic activity with still unknown mechanisms (352). In addition, knocking down thioredoxin-like 2, a novel Trx-related protein in human breast cancer cells, increased ROS generation, inhibition of cell proliferation, and reduction of tumorigenesis and metastasis upon transplantation into immunodeficient mice (255).
The major problem of radiotherapy or chemotherapy is their intrinsic toxicity. The above-described data support the feasibility of using genetic silencing of antioxidant systems to reduce the dose, and therefore to diminish the systemic toxicity of conventional cancer therapeutics. Mn-SOD and Prxs seem to be interesting targets to manage the toxicity of radiation protocols. Knockdown of the Trx/TrxR system or GPxs seems to be promising targets to induce chemosensitization in several types of human cancer such as breast or ovarian cancer. However, the combination of Trx gene therapeutics and anthracyclines should be carefully studied due to the possible dual role of Trx as an antioxidant or prooxidant (266). The use of selenocompounds in combination with TrxR knockdown may be an interesting approach in cancers expressing high TrxR levels, for example, nonsmall-cell lung cancer. The chemosensitization by GT strategies would represent an opportunity to sensitize cancers that have developed chemoresistance to conventional chemotherapeutic drugs.
b. Overexpression of the antioxidant enzyme SOD
There is a large body of literature linking alteration of antioxidant enzyme systems and cancer. Oberley and Buettner suggested that the normalization of enzyme levels should result in reversion of at least a part of the cancer phenotype (232). They first reported that increased Mn-SOD levels in melanoma cells by cDNA transfection suppressed the malignant phenotype (60). Since then, it has been demonstrated that Mn-SOD-increased expression reverted the malignant phenotype in vitro and in vivo in different cancer models, including human breast carcinoma, lung fibroblasts, viral-transformed WI-38, rat and human glioma, mouse and human fibrosarcoma, human prostatic carcinoma, and human pancreatic cancer cells (231). From these studies, Mn-SOD has been proposed as a tumor suppressor gene through the alteration of the superoxide/H2O2 balance. Indeed, increasing Mn-SOD levels enhanced the conversion of O2 •− to H2O2, which in turn might cause antiproliferative effects. The coadministration of CATs reversed this effect supporting this hypothesis. Moreover, in the breast tumor microenvironment, it has been also demonstrated a switch role of ROS: elevated activity of EC-SOD may generate H2O2 with an oncosuppressor function, whereas CuZn-SOD downregulation may act as an oncopromoter influencing cell proliferation in an ROS-stressed tumor microenvironment (207). Furthermore, in pancreatic cancer, the overexpression of Mn-SOD resulted in high levels of H2O2 and antiproliferative effects, but the overexpression of EC-SOD and CuZn-SOD had even a stronger tumor-suppressive effects (298). On the other hand, in ovarian cancer tissues, Mn-SOD levels were significantly higher than in the normal ovarian epithelium and benign lesions. Suppression of Mn-SOD expression caused ROS accumulation, leading to increased cell proliferation in vitro and enhanced tumor growth in vivo (130).
The administration of a replication-competent recombinant adenovirus expressing the human Mn-SOD gene (ZD55-MnSOD) showed an antitumor effect 1000-fold higher than that observed for the nonreplicative adenovirus (Ad-MnSOD) (355). This vector enhanced Mn-SOD protein levels and increased Mn-SOD activity. Moreover, the combination of ZD55-MnSOD with an adenovirus expressing the TNF-related apoptosis-inducing ligand (ZD55-TRAIL) resulted in a profound tumor growth inhibition and a complete remission of all tumor masses in nude mice. Mn-SOD/TRAIL overexpression enhanced H2O2 levels and decreased tumor cell growth by extending the cell cycle transition time from the G1 to S phase (355). Moreover, the overexpression of Mn-SOD in combination with radiation or certain chemical drugs synergistically increased oxidative cellular stress (92, 330). Recently, a new active recombinant human Mn-SOD (rMn-SOD) was found to exert the same radioprotective effect on normal cells and organisms as any other Mn-SOD, but also radiosensitized malignant cells. Animals exposed to lethal doses of ionizing radiation and daily rMn-SOD injections were protected from radiodamage and were still alive 30 days after the irradiation, whereas control animals exposed to ionizing radiation, in the absence of rMn-SOD, died after 7–8 days from the radiotreatments (29). Thus, a therapeutic approach that might have a radioprotective effect on normal cells warrants further investigation, as radiotherapy is limited mainly by toxicity over normal tissues.
c. Knocking down additional redox-associated cellular genes
In the last years, several studies have demonstrated that knocking down certain cellular targets produces or increases ROS cell generation and cancer cell death. Particularly, specific miRNAs have been explored as therapeutic tools to inhibit tumor growth by mechanisms that include mitochondrial dysfunction. For instance, aberrant expression of miRNA 128 (miR-128) was found to be implicated in different cancers (108). miRNA 128a (miR-128a) is strongly downregulated in medulloblastoma, a malignant primary brain tumor with high incidence in children. Transfection of miR-128 in medulloblastoma cells inhibited cell proliferation by promoting cellular senescence through the targeting of the transcription factor repressor Bmi-1 (315). Mice deficient in Bmi1 had an impaired mitochondrial function, with a marked increase in intracellular ROS and subsequent engagement of the DNA-damage-response pathways (189). miR-128a induced fivefold increase in senescent cells and threefold superoxide (O2 •−) increase when compared to control cells. This effect was reverted by the addition of exogenous CuZn-SOD (315). Moreover, the ectopic expression of hsa-miR-128 induced apoptosis, cell cycle changes, dissipation of mitochondrial membrane potential, and increased ROS generation in HEK293T cells (3). Furthermore, both bioinformatic prediction and experimental results indicate that hsa-miR-128 can target Bcl-2-associated X protein (Bax) (3).Surprisingly, miR-128 seems to downregulate Bax expression and unexpectedly induces HEK293T apoptosis. Another example is the MUC1 oncoprotein, which is aberrantly overexpressed in human carcinomas and hematologic malignancies. Recently, it was demonstrated that miR-1226 interacts with a MUC1 3′- untraslated region and downregulates the endogenous levels of MUC1 (143). miR-1226 induced an increase of ROS cell generation, loss of the mitochondrial transmembrane potential, and a decrease in cell survival. These studies suggest the importance of miRNAs as potential gene therapeutics to increase ROS through enhanced mitochondrial dysfunction, although caution should be taken, since miRNAs can target multiple genes, and the effects on cell survival could be produced by causes other than increased ROS generation.
C. ROS-response elements to drive cancer gene therapeutics
Several groups have shown the therapeutic effect of cytotoxic genes whose activity was driven by promoters corresponding to tumor-associated genes. This strategy named conditional targeting allows the therapeutic gene to be active only in malignant cells without affecting normal cells. Targeting the tumor mass by taking advantage of a defined microenvironmental characteristic that differentiates cancer and normal tissues is a valuable option. For instance, motifs responsive to hypoxia were used for the selective expression of therapeutic genes in the cancer microenvironment (135). We proposed the use of the prooxidative microenvironment of tumors as a feature that distinguishes malignant from normal tissues to direct the expression of therapeutic genes. DNA sequences responsive to ROS are present in the promoters of several redox-regulated genes. Although no sequence consensus has been found among them, different ROS-response motifs were explored to drive therapeutic genes in different models. One of the most explored sequences is the ARE, a cis-acting sequence in the promoters of a number of antioxidants and detoxification enzymes, which are transcriptionally induced under oxidative stress conditions (165). The consensus core ARE sequence (TGAC/GnnnGC) binds basic leucine-zipper (bZIP) transcription factors, most notably Nrf2, which forms heterodimers with other bZIP factors, such as small Maf proteins. As previously mentioned, under basal conditions, Nrf2 is repressed by Keap-1, and during oxidative stress, Nrf2 escapes from the proteasomal degradation machinery, translocates to the nucleus, and activates target genes via binding to ARE sequences (350). This sequence was explored in several models for the treatment of retinopathy of prematurity (253), stroke (54), and cancer (131) directing reporter or therapeutic genes in viral (54, 131) and nonviral vectors (253). This element upregulates the expression of reporter or therapeutic genes in the human embryonic kidney 293T cell induced by H2O2 or diethyl maleate, an electrophilic compound that depletes cellular GSH, thereby producing oxidative stress (131). Moreover, ARE activity was induced by hyperoxia in canine retinal ECs (253). Of note, iRNA-mediated reduction of Nrf2 expression induced ROS generation, suppressed tumor growth, and resulted in increased sensitivity to chemotherapeutic drugs in vitro and in vivo (283)
Another widely studied sequence is the CArG (CC(A/T)6GG) element contained within the promoter of several ROS-response genes. Particularly, the early growth response-1 (Egr-1) promoter contains the CArG elements that were extensively studied as motifs responsive to ionizing radiation. It was well demonstrated that ionizing irradiation activates CArG elements through reactive oxygen intermediates generated by water radiolysis (70). The Egr-1 promoter-incorporated upstream of a cDNA encoding the human TNF-α (hTNF-α) gene was integrated into a replication-defective adenovirus that gave origin to a construct that reached the clinics (TNFerade™) (62). An additional replication-defective adenoviral vector Ad.Egr-TNF.11D was also engineered by ligating several copies of CArG (CC(A/T)6GG) elements in tandem upstream to a cDNA encoding hTNF-α, which was activated by various chemotherapeutic agents that increase intracellular ROS levels (193). Nine tandem copies of the CArG element were also shown to drive the expression of the iNOS gene (66) and an siRNA against urokinase plasminogen activator and urokinase plasminogen activator receptor in cancer models (265). Recently, CArG elements were combined with the low transcriptional activity human telomerase reverse transcriptase (hTERT) promoter (336). This chimeric promoter was better at driving radiation-inducible GT than the hTERT promoter alone and significantly inhibited glioma tumor growth in a xenograft mouse model.
We have recently described a ROS-responsive chimeric promoter that was activated by endogenous ROS cancer cell levels (250). This chimeric promoter was based on a ROS-response motif located in the VEGF gene promoter placed downstream of the aforementioned Egr-1 motif (250). The activity of the chimeric promoter [named E6(40)VE] was largely dependent on variations in intracellular ROS levels, showed a high inducible response to exogenous H2O2 and was differentially activated in cancer or noncancer cell lines (Fig. 14). Transient expression of the thymidine kinase (TK) gene driven by this chimeric promoter, followed by gancyclovir (GCV) administration, inhibited human colorectal cancer and melanoma cell growth in vitro and in vivo (Fig. 15). Moreover, electrotransfer of the TK gene under the control of the E6(40)VE promoter followed by GCV administration exerted a potent therapeutic effect on established tumors. This response was improved when combined with chemotherapeutic drugs such as DOX that by themselves act by increasing ROS levels (250). Thus, the conditional targeting of a cancer tissue by taking advantage of the prooxidant tumor microenvironment might represent a promising strategy of ROS-targeted gene therapeutics not only in cancer but also to tackle other diseases associated with high ROS levels.

IV. Concluding Remarks and Future Perspectives
Throughout this review, we aimed to describe the most relevant features of the tumor microenvironment, addressing ROS-generating sources that promote the generation of a prooxidative microenvironment inside the tumor mass. Genetic therapeutics directed to decrease or exacerbate the prooxidative microenvironment, and even those that take advantage of differential levels of ROS between cancer and normal cells were described. ROS-targeted GT approaches have demonstrated effectiveness in tumor treatment both in vitro and in vivo. In this context, several critical points should be considered as major constraints for developing novel therapeutics based on the oxidative tumor microenvironment if a clinical approach is on the scope. Effectiveness and selectivity are the main goals to develop ROS-targeted cancer gene therapeutics (321).
Potential effectiveness of alternative ROS-manipulating strategies should emerge from understanding the best gene therapeutics for a particular redox characteristic of the tumor. In some cases, enhancing ROS levels appears to be more appropriate, whereas in other cases, scavenging of radical species is more effective. Wang and Yi (321) suggested that cancer cells with a moderate increase in ROS levels are more suitable for ROS-depletion approaches, while those with highly increased ROS levels are suitable for ROS-elevating ones. This stands from the evidence that cancer cells with increased oxidative stress are likely to be more vulnerable to damage by further ROS insults induced by exogenous agents. By contrast, poorly differentiated and highly metastatic tumor cells (147) often do not exhibit an increased ROS accumulation; in this case, ROS-depletion approaches would be the most appropriated ones. In this regard, the characterization of the oxidative profile of a specific tumor is of fundamental importance to decide which strategy is the most appropriate (69). The characterization of this profile that could be made in biopsy or in fluids through the determination of oxidative markers might contribute to predict efficacy and systemic toxicity (46, 132, 343).
Another important issue regarding the effectiveness of redox-directed GT is the capacity of cancer cell adaptation. Under persistent intrinsic oxidative stress, many cancer cells become adapted to such stress and induce an enhancement in endogenous antioxidant capacity, which makes the malignant cells resistant to exogenous stress (175). Adaptive mechanisms not only activate ROS-scavenging systems to cope with the stress but also inhibit apoptosis. Furthermore, metastasizing cells can trigger cellular programs to escape from the high oxidative stress levels observed within the primary tumor (238). Recent reports indicate that transformed cells acquire a cancer genotype by inducing the expression of a set of survival genes such as those of specific antioxidant proteins (262), which are not usually expressed by normal cells. Thus, targeting this set of genes can inhibit growth of rapidly growing and highly invasive cells without a harmful general toxicity, selectively increasing ROS levels and apoptosis of malignant cells (262). Therefore, nononcogene profile would be an interesting approach for new procedures targeting the ROS stress–response pathways in cancer cells.
Tumor heterogeneity is another important issue that should be considered to design a therapeutic strategy. High-resolution imaging methods have already shown that intratumor heterogeneity is a characteristic of the complex tumor microenvironment (73, 185). As hypoxia, glucose or pH, or presumably ROS levels fuel heterogeneity within a tumor. It has been shown in xenograft models of human cancers that the mitochondrial redox state is more oxidized in the core of aggressive tumors, whereas the redox state in the rim is closer to or the same as in normal tissue (338). In this context, compartmentalization of redox states within cancer cells is another topic that should be considered in the design of ROS-targeted GT strategies. It would be of high relevance to establish whether ROS are elevated in the mitochondria or in the nucleus despite the fact that ROS can diffuse from one organelle to the other, especially stable ROS like H2O2. Moreover, heterogeneity of ROS generation within the tumor mass, different cellular sources of ROS generation, and even different compartmentalization of ROS generation could be responsible of some controversies and differences that arose between studies and cancer types. Thus, to avoid treatment failure, adaptive mechanisms, ROS tumor heterogeneity, and even ROS cell compartmentalization should be considered for the rationale design of a ROS-targeted therapeutic strategy. Selectivity of ROS-targeted GT approaches is another critical issue to design novel cancer therapeutics. As described, the threshold concept discriminates normal from malignant cells by their differential capabilities in maintaining redox homeostasis (304, 321). Thus, GT strategies related to ROS levels might involve either the modification of ROS by increasing or decreasing intratumoral concentrations, or the selective expression of therapeutic genes through ROS-responsive motifs by taking advantage of high ROS levels in the tumor microenvironment.
Numerous strategies have been evaluated for detargeting and retargeting adenoviral vectors to tumors. Adenoviral vectors can be retargeted by genetic modification of the capsid proteins (hexon, penton, and fiber), by the incorporation of bispecific fusion proteins or antibodies to detarget adenovirus for their native tropism and retarget the virus to the tumor. Successful retargeting of vectors has been achieved using antiknob antibodies or truncated coxsackie and adenovirus receptor constructs chemically or genetically linked to a variety of targeted ligands or antibodies against cell surface receptors (184, 277). Results from studies using adaptor molecules have shown a 10-fold to 20-fold increase in transgene expression in target tissues in vivo (125). Nonviral vehicles also improve tumor selectivity generally by the attachment of ligand or antibodies on the cell surface used as vehicle (90). On the other hand, the use of specific promoters or conditional targeting to drive the expression of therapeutic genes in viral or nonviral vectors provides additional selectivity to cancer gene therapeutics. Targeting a biochemical alteration in cancer cells might be a feasible approach to achieve therapeutic activity and improve selectivity. For instance, most cancer cells exhibit increased aerobic glycolysis or hypoxia and oxidative stress, which could be important in the development of new anticancer strategies. Another possibility to enhance the therapeutic efficacy is the combination of ROS-response elements with chemotherapeutic agents that enhance intracellular ROS levels such as DOX, bleomycin, and PTX or ROS-generating agents such as gadolinium. Due to the potentially vital roles of stem-like cancer cells in drug resistance and disease recurrence, it is extremely important to examine the redox status in this subpopulation of malignant cell and to devise therapeutically relevant redox modulation strategies. In addition, using motifs capable of sensing and being activated in a ROS-elevated environment could be used as a novel strategy not only in cancer but also in additional diseases. Increased formation of ROS was also shown to be associated with atherosclerotic lesions, diabetes, Parkinson's, and Alzheimer's diseases. Thus, we speculate that this approach might be used for any disease related to high ROS levels, by using the appropriate therapeutic gene.
Footnotes
Acknowledgments
This work was supported by the grants from the National Agency for Promotion of Science and Technology (ANPCyT), Argentina. The authors wish to thank Amigos del Instituto Leloir para la Lucha contra el Cancer (AFULIC) for their continuous support. The authors also gratefully acknowledge Andrea C. Cruz for proofreading the manuscript and advising us on the use of English language.