
Abstract
Introduction
Epigenetic modifications include methylation of DNA, modifications of the proteins that bind to DNA, and the nucleosome positioning along DNA (75). These modifications are essential for normal development and proper maintenance of cell-specific epigenetic phenotype in adult organisms. In normal cells, the genome and epigenome are highly organized and maintained to ensure the proper expression of genetic information.
The physiological template of the genetic information is chromatin that is composed of DNA, RNA, histones, and other chromosomal proteins. The fundamental repeating unit of chromatin is the nucleosome octamer that consists of 146 bp of DNA wrapped around two copies of histones H2A, H2B, H3, and H4 (28). Histones are evolutionary conserved proteins that have a globular carboxy-terminal domain critical to nucleosome formation and flexible amino-terminal tails that protrude from the nucleosome core and contact adjacent nucleosomes to form higher-order of chromatin. At least eight different classes of post-translational modifications, including methylation, acetylation, phosphorylation, ubiquitynation, sumoylation, biotinylation, and ADP-ribosylation can occur on the core histones H2A, H2B, H3, H4, and the H1 family of linker histones (4, 28, 32, 44). These histone marks are essential for organizing chromatin, maintaining genome stability, silencing repetitive DNA elements, regulating cell cycle progression, recognizing DNA damage sites and repair, and for the proper expression of genetic information (7, 8, 53).
Methylation of Histones
Methylation of histones is one of the most studied and intriguing histone modifications that occurs either on lysine or on arginine residues. Currently, there are 24 known histone methylation sites: 17 are on lysine residues, each of which can be mono-, di-, or trimethylated (me1, me2, and me3, respectively) and 7 are on arginine residues, each of which can be mono- or dimethylated (3).
The histone lysine methylation reaction is catalyzed by a family of histone lysine methyltransferases (HMTs or KMTs) that transfer methyl groups from S-adenosyl-L-methionine (SAM) to the lysine ɛ-amino group (Fig. 1). HMTs exhibit remarkable target specificity toward specific lysine residues on a single histone. Depending on the lysine residue and its methylation state, methylation of lysines can lead either to transcriptional activation or to repression. For example, trimethylation of lysine 4 and lysine 36 on histone H3 (H3K4me3 and H3K36me3, respectively) is generally associated with increased transcriptional activity. In contrast, trimethylation of histone H3K9 and, especially, histone H3K27 correlates with transcriptional repression. However, histone lysine methylation is not only relevant to gene activation or silencing, but has broad range implications on chromatin structure, nuclear architecture, cell cycle regulation, and genome stability (10).
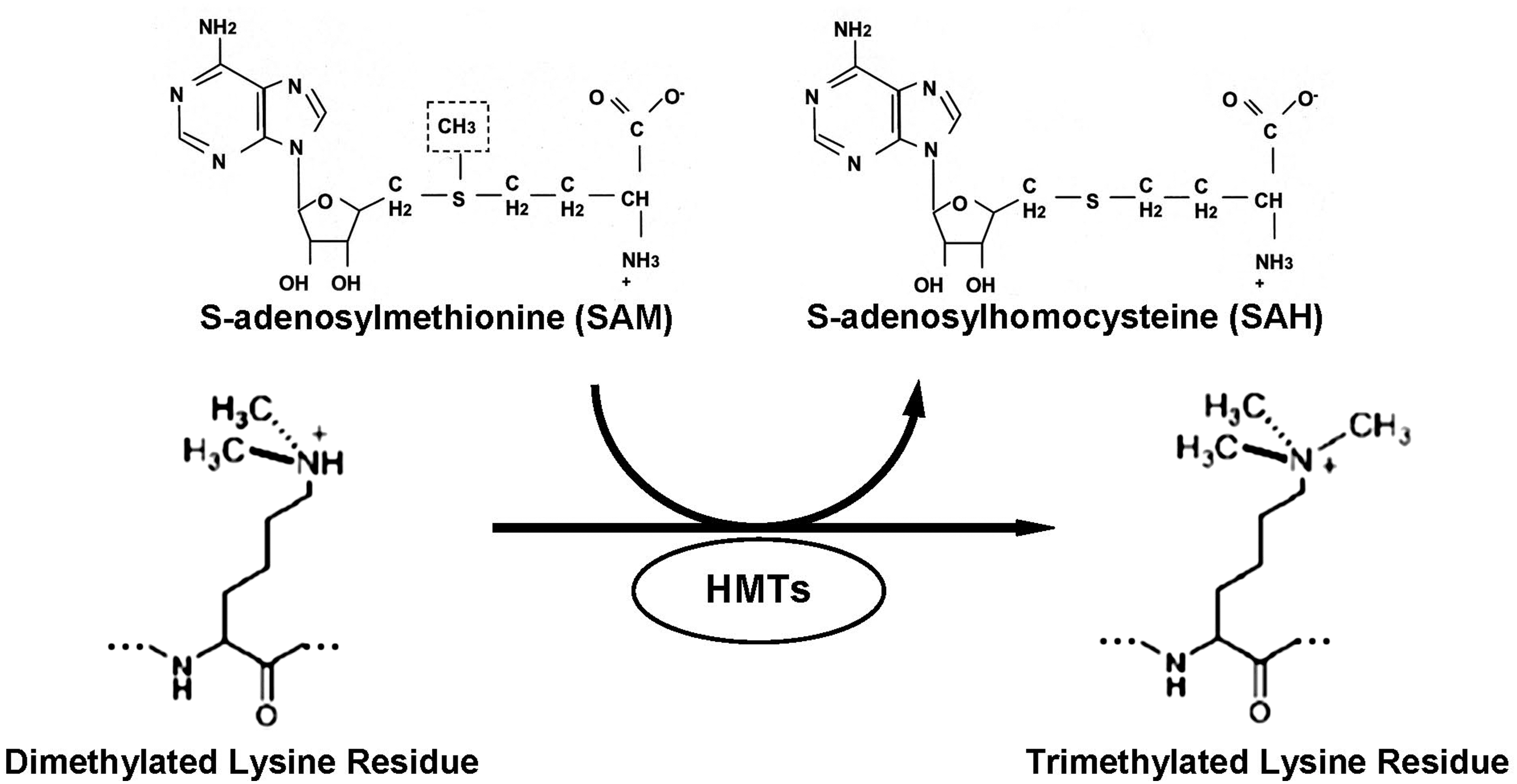
Until the discovery of the first histone demethylase (HDM) lysine-specific demethylase 1 (LSD1) in 2004 by Shi et al. (76), methylation of lysine residues had been regarded as a stable epigenetic mark. LSD1 belongs to the amine oxidase family that specifically demethylates mono- and dimethyl histone H3K4 (Fig. 2). Later, another group of proteins containing the Jumonji C (JmjC) domain that demethylates histone H3K9me3, H3K9me1/2, H3K36 (40), all methylation states of H3K4 (30), and H4K20me1 (49) was discovered (82, 89); however, no specific demethylases for several lysine methylation sites, including lysine 20 on histone H4 (H4K20), have been determined (2).

The mechanisms for maintaining and regulating histone lysine methylation, unlike DNA methylation, are just beginning to be understood; however, a growing body of evidence indicates that the extent of histone lysine methylation depends on the status of intracellular metabolic homeostasis, specifically on the intracellular levels of metabolic coenzymes such as SAM, flavin adenine dinucleotide (FAD), and α-ketoglutarate (12, 17, 79).
SAM Metabolism and Histone Methylation
Methionine, choline, folic acid, and vitamin B12 are essential for the proper functioning of several main interdependent cellular metabolic processes critical for the biosynthesis of SAM, the major methyl donor for cellular methylation reactions, including methylation of histone proteins.
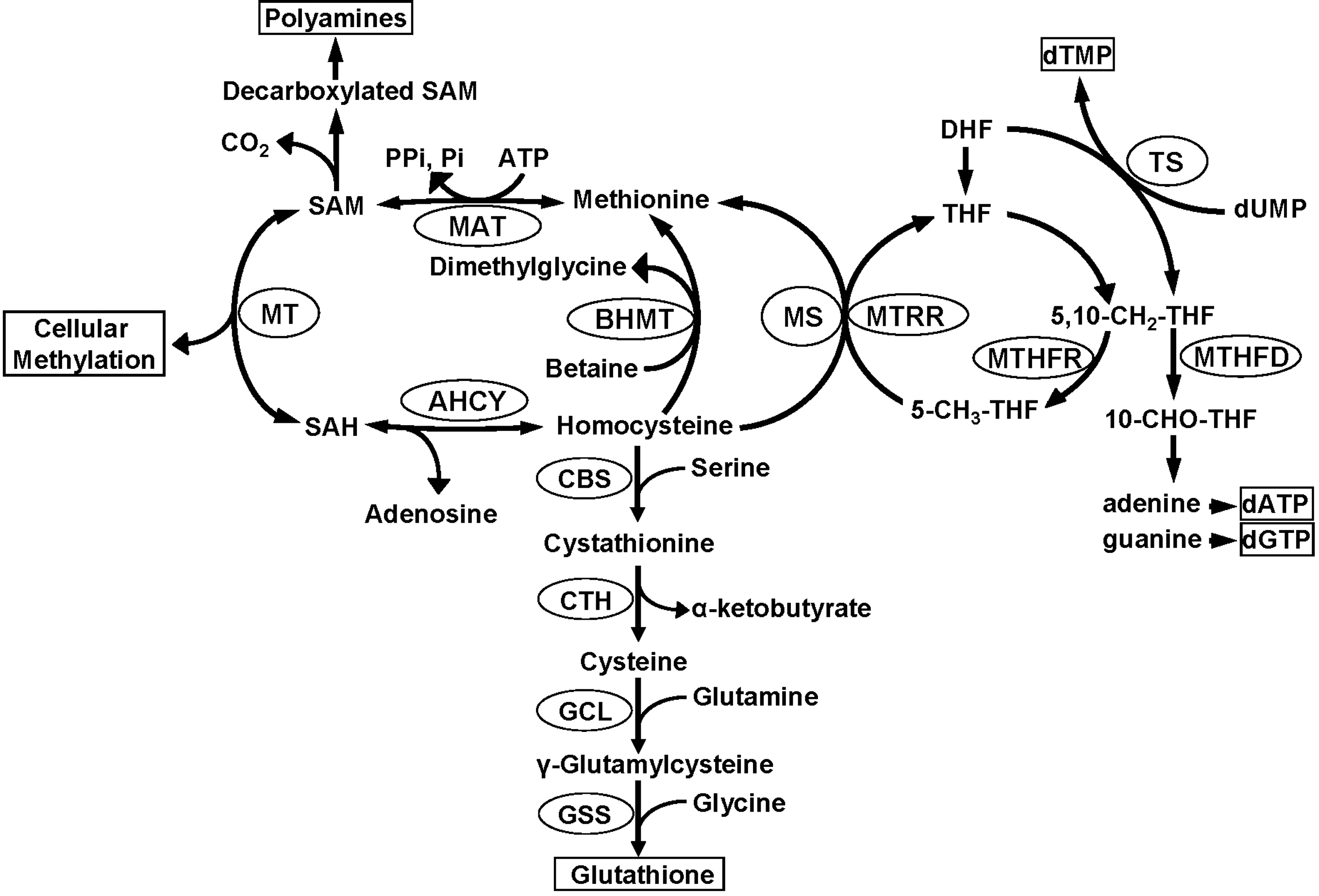
SAM is synthesized by methionine adenosyltransferase that transfers the adenosyl moiety of Mg-ATP to L-methionine (14). SAM is a key component in transmethylation, transsulfuration, and polyamine synthesis pathways (50). In the transmethylation pathway, SAM donates its methyl group to a broad spectrum of substrates, that is, phospholipids, proteins, neurotransmitters, DNA, RNA, and histones, in reactions catalyzed by substrate-specific methyltransferases, including HMTs (65). In the transsulfuration pathway, SAM is an important precursor of cysteine for glutathione biosynthesis, particularly in the liver. In the synthesis of polyamines, small molecules involved in cell growth regulation and a target for chemotherapy, SAM is decarboxylated and the remaining propylamino moiety is donated to either putrescine to form spermidine and methylthioadenosine, or to spermidine to form spermine and methylthioadenosine.
In the transmethylation pathway, after transfer of the methyl group, SAM is converted to S-adenosyl-L-homocysteine (SAH) within the active site of the methyltransferase enzyme (22). SAH is a potent competitive inhibitor of virtually all cellular methylation reactions, including histone lysine methylation (65), and since most of the cellular methyltransferases bind SAH with greater affinity than SAM, the efficiency of methyltransferase reactions is absolutely dependent upon the prompt removal of SAH. This is effectively achieved by SAH hydrolase that catalyzes conversion of SAH, in a reversible reaction, to homocysteine and adenosine. Since the equilibrium constant of SAH hydrolase favors SAH synthesis, the SAH hydrolysis reaction greatly depends on the efficient removal of homocysteine (Fig. 3). Homocysteine can be removed via remethylation to methionine in a folate/B12-dependent methionine synthase (5-methyltetrahydrofolate-homocysteine methyltransferase) reaction and/or a betaine-homocysteine methyltransferase reaction. Betaine-homocysteine methyltransferase uses betaine, a metabolite of choline, as the methyl donor for homocysteine methylation. A third route of homocysteine removal, especially in the liver, is the irreversible transsulfuration pathway that involves two pyridoxal phosphate-dependent enzymes cystathionine β-synthase and cystathionine γ-lyase (Fig. 3). Consequently, continuous homocysteine recycling or removal is necessary for the methionine cycle, choline synthesis, and folate diversion for nucleotide synthesis; therefore, this is the key interacting point for methionine, choline, folate, and vitamin B12 and B6 metabolic pathways. Additionally, these metabolic pathways are tightly connected with folate-dependent DNA synthesis, since 5,10-methylenetetrahydrofolate is not only a substrate for methylenetetrahydrofolate reductase, but also for thymidylate synthase and methylenetetrahydrofolate dehydrogenase (Fig. 1), key enzymes required for biosynthesis of thymidine monophosphate and purines for DNA synthesis, respectively (19, 78).
FAD and α-Ketoglutarate and Histone Lysine Methylation
The LSD1 histone demethylase that catalyzes the oxidative demethylation of histones belongs to the family of FAD-dependent amine oxidases. FAD is a redox coenzyme that exists in two different redox states and consists of a riboflavin moiety that is bound to the phosphate group of ADP. In a histone lysine demethylation reaction, FAD oxidizes the methyl group in the methyl-lysine residue to form an imine intermediate that is then hydrolyzed to generate unmodified lysine residue and formaldehyde (Fig. 2A). The reduced FAD (FADH2) is then reoxidized by oxygen to form FAD (23, 29). Additionally, a recent report by Luka et al. (51) has demonstrated that the natural pentaglutamate form of tetrahydrofolate binds with great affinity to the full-length LSD1 indicating that folate directly participates in the enzymatic histone lysine demethylation.
The Jumonji domain-containing HDM (JHDM) belongs to the superfamily of Fe2+-dependent dioxygenases (29). JHDMs use Fe2+ and α-ketoglutarate as coenzymes and in the presence of oxygen convert the methyl group of the methyl-lysine residue to a hydroxymethyl group that is then released as formaldehyde (Fig. 2B). Despite different mechanisms of action of LSD1 and JHDM histone demethylases, efficiency of both of these lysine demethylation reactions heavily depends on the status of cellular metabolic homeostasis, since FAD and α-ketoglutarate, and likewise SAM, are major intermediate metabolites of fundamental metabolic pathways in the cell.
Histone H4 Lysine 20 Methylation in Normal and Pathological States
Among many different types of histone lysine methylation, the methylation of lysine residue 20 on histone H4 is one of the most unique and enigmatic histone modification marks. This lysine residue can undergo mono-, di-, or trimethylation and, depending on the extent of methylation, exhibit different functional characteristics.
Monomethylation of lysine 20 on histone H4 (H4K20me1) has been associated mainly with gene repression (16, 37) and with X-chromosome inactivation (33, 41). However, emerging evidence indicates that this histone lysine methylation mark may also activate gene transcription (5, 38). Specifically, results of a recent study conducted by Li et al. (48) demonstrated that H4K20me1 stimulates the transcription of Wnt genes. Additionally, it is believed that H4K20me1 plays an important role in chromatin organization and is essential for H4K20 di- and trimethylation (55).
Histone H4K20me1 is catalyzed by the Pr-Set 7 (also known as Set 8 or KMT5A) methyltransferase that belongs to the family of suppressor of variegation genes, Su(var)3–9, Enhancer of zeste and Trithorax (SET) domain-containing methyltransferases (Fig. 4). Two other methyltransferases that belong to this family, nuclear receptor-binding SET domain-containing protein 1 (NSD1) and absent small or homeotic-1 protein (Ash1), are capable of monomethylating H4K20 in vitro (6, 66); however, their methylating ability in vivo remains questionable.

Dimethylation of histone H4 lysine 20 (H4K20me2) is the predominant type of histone H4K20 methylation in mammalian cells (57), although it does not play any noticeable role in either the regulation of gene expression or heterochromatin function (57). Several reports have demonstrated that H4K20me2 is uniformly distributed throughout chromatin (37, 57) and plays a crucial role in recognizing DNA damage sites and initiating/allowing DNA repair (56, 68, 69). Specifically, histone H4K20me2 plays a critical role in the recruitment of the checkpoint proteins Crb2 and p53-binding protein 1 (53BP1) to sites of DNA damage (56, 68, 69). Loss of H4K20me2 significantly decreases accumulation of Crb2 and 53BP1 at DNA damage sites and markedly impairs cell survival after genotoxic insults.
Trimethylation of histone H4 lysine 20 (H4K20me3) is one of the most prominent marks of constitutive heterochromatin and mainly affects higher-order chromatin structure (2, 34, 43, 74). H4K20me3, in contrast to H4K20me2, is predominantly located in centromeric, pericentromeric, and telomeric regions (67, 73) and plays a fundamental role in genomic stability by silencing repetitive elements. Besides these main functions of H4K20me3, recent evidence indicates its involvement in regulation of gene expression (87).
Both, di- and trimethylation of H4K20 are catalyzed by Suv4-20h1 and Suv4-20h2 HMTs that induce a transition from a H4K20 monomethylated state to H4K20 di- and trimethylated states (92).
Histone H4 Lysine 20 Methylation in Cancer
The pathogenesis of any given human disease, including cancer, is a complex multifactorial process characterized by many biologically significant and interdependent alterations. One of these changes is a profoundly disturbed pattern of histone modifications, in particular, histone H4K20 methylation. Accumulating evidence clearly indicates that cancer cells are characterized by a global and/or gene-specific decrease in H4K20me3 (24, 46, 72), and an increase in H4K20me1 and H4K20me3 marks at gene promoters (35, 36). Reduced H4K20me3 levels are frequently found in the vast majority of human tumors, including lung, breast, bladder, ovarian, and hematological cancers (20, 24, 46, 72, 80, 83, 84). Based on these findings, loss of histone H4K20me3 is currently regarded as a common hallmark of cancer. Additionally, recent reports also indicate that loss of gene-associated histone H4K20me3 may result in the reactivation of oncogenes. For instance, loss of H4K20me3 at the promoter regions of claudin (CLDN3 and CLDN4) genes results in their derepression which directly correlates with progression of ovarian tumorigenesis (46).
Recognition of the fundamental role of H4K20me3 in cancer suggests that it can be used as a biomarker for the clinical diagnosis and prognosis of cancer. Several reports demonstrated that the extent of histone H4K20me3 is a significant and an independent predictor of lung, breast, and bladder cancer progression (20, 72, 83). For instance, lower level of H4K20me3 in these cancers correlates with a negative cancer prognosis and associates with reduced patient survival.
Histone H4 Lysine 20 Methylation in Carcinogenesis
Carcinogenesis is a complex multifactorial process characterized by a variety of biologically significant and interdependent alterations, among which epigenetic abnormalities, including alterations in histone H4K20 methylation, play one of the key roles. It is becoming increasingly evident that cancer by itself can induce alterations in histone H4K20 methylation; however, distortion of histone H4K20 methylation in normal cells can also predispose them to precancer-specific pathological states and cancer development. In light of this, it has been proposed that changes in histone H4K20 methylation are not only the features of cancer cells, but also an important event in the initiation and progression of cancer.
Histone H4 Lysine 20 Methylation in Genotoxic Carcinogenesis
Carcinogenic agents are considered as genotoxic if they, or products of their metabolism, directly interact with DNA, causing a variety of genetic alterations. However, even within a classical model of genotoxic carcinogenesis, the presence of carcinogen-induced DNA lesions per se is not sufficient for tumor formation and requires much broader alterations in cellular homeostasis. Further, a large spectrum of phenotypic changes observed in preneoplastic and cancer tissues can not be explained by genetic alterations only. Indeed, it is becoming increasingly evident that multiple adverse biological effects, including cancer, observed as a result of exposure to genotoxic agents, are mediated not only via the genotoxic mode of action, but also by epigenetic alterations (11, 52, 62). Further, it is believed that epigenetic alterations may precede and/or provoke genetic alterations (70).
Tamoxifen, a nonsteroidal anti-estrogen that is widely used for treatment of breast cancer in women, is also known as a potent complete hepatocarcinogen in rodents, and exhibits both cancer-initiating (90) and -promoting (18) activities. The mechanism of tamoxifen-induced hepatocarcinogenesis has been attributed to the ability of tamoxifen metabolites, E-alpha-(deoxyguanosin-N(2)-yl)tamoxifen and E-alpha-(deoxyguanosin-N(2)-yl)-N-desmethyltamoxifen, to form stable adducts with DNA (71). However, exposure to tamoxifen, in addition to formation of DNA adducts, also leads to a variety of epigenetic aberrations (81), among which an early and progressive loss of histone H4K20me3 in the livers of tamoxifen-exposed rats is one of the most significant alterations (81).
2-Acetylaminofluorene (2-AAF) is another classic complete genotoxic hepatocarcinogen. Administration of 2-AAF to male rats results in tumor formation without any additional intervention (9). Similar to tamoxifen exposure, 2-AAF-induced rat hepatocarcinogenesis exhibits a progressive decrease in the level of histone H4K20me3 (1). A genome-wide decrease of histone H4K20me3, including loss of the region-specific H4K20 trimethylation in centromeric, pericentromeric, and telomeric regions, occurs in 2-AAF-hepatocarcinogenesis.
Histone H4 Lysine 20 Methylation in Nongenotoxic Carcinogenesis
Nongenotoxic carcinogens are chemicals that induce tumor formation without causing direct DNA damage. Exposure to nongenotoxic carcinogens results in a number of epigenetic alterations, including aberrant methylation of DNA and patterns of histone modifications (62).
Methyl-deficient diets, lacking the major dietary methyl group donors—methionine, choline, folic acid, and vitamin B12, can induce the development of liver cancer in rodents in the absence of any exogenous carcinogens (15, 31, 86). Importantly, this methyl-deficient model of endogenous hepatocarcinogenesis is one of the most relevant models to study the pathogenesis of (i) human liver carcinogenesis (31, 64), since the sequence of pathological and molecular events is remarkably similar to the development of human hepatocellular carcinomas; and (ii) nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (26), the main risk factor for development of hepatocellular carcinomas in developed countries (77, 88).
One of the earliest epigenetic alterations during hepatocarcinogenesis induced by diets lacking methyl donors, in addition to sustained demethylation of genomic DNA (15, 31, 86), is the loss of histone H4K20me3 that is found almost exclusively in the livers of methyl-deficient animals (60). These changes occur before formation of preneoplastic lesions and gradually progress to liver tumors. Importantly, similar effects were observed in mouse studies, where feeding C57BL/6J and DBA/2J mice a methyl-deficient diet resulted in loss of H4K20 trimethylation six weeks after the beginning of diet administration (63).
Peroxisome proliferators, a structurally diverse group of chemicals and therapeutic agents, are one of the most extensively studied classes of nongenotoxic carcinogens. Administration of the model peroxisome proliferator WY-14,643 (4-chloro-6-(2,3-xylidino)-pyrimidynylthioacetic acid) causes profound and sustained loss of histone H4K20me3 in the livers of exposed mice (61).
The mechanistic link between loss of histone H4K20me3 and cancer development is directly related to its localization in chromatin and function in normal cells. The decrease in H4K20me3 may result in organization of a more “relaxed” heterochromatin that markedly impairs genome stability (43, 74). Additionally, it may cause chromosomal abnormalities by the induction of permissive transcriptional activity at the centromere and telomere, resulting in (i) accumulation of small minor satellite transcripts that impair centromeric and subtelomeric architecture, function, and activation; and, (ii) transposition of retroviral mobile repetitive elements that may lead to genomic instability. Any or all of these changes may contribute to initiation and progression of the carcinogenic process. The above described alterations in H4K20 trimethylation are not limited to experimental hepatocarcinogenesis only. Similar changes in H4K20me3 have been found in mammary glands of the ACI rat during estrogen-induced breast carcinogenesis and were associated with the appearance of putative preneoplastic lesions, such as atypical ductal and alveolar hyperplasia (45). More importantly, a strong decrease in the level of H4K20me3 has been observed in early precursor lesions during human lung carcinogenesis (84). Additionally, recent evidence demonstrating that exposure to different carcinogenic agents, for example, 1,3-butadiene (42) and gamma radiation (58), results in the loss of H4K20 trimethylation and suggests that changes in H4K20 trimethylation may be used for identification and assessment of carcinogenic potential of environmental chemical and physical agents.
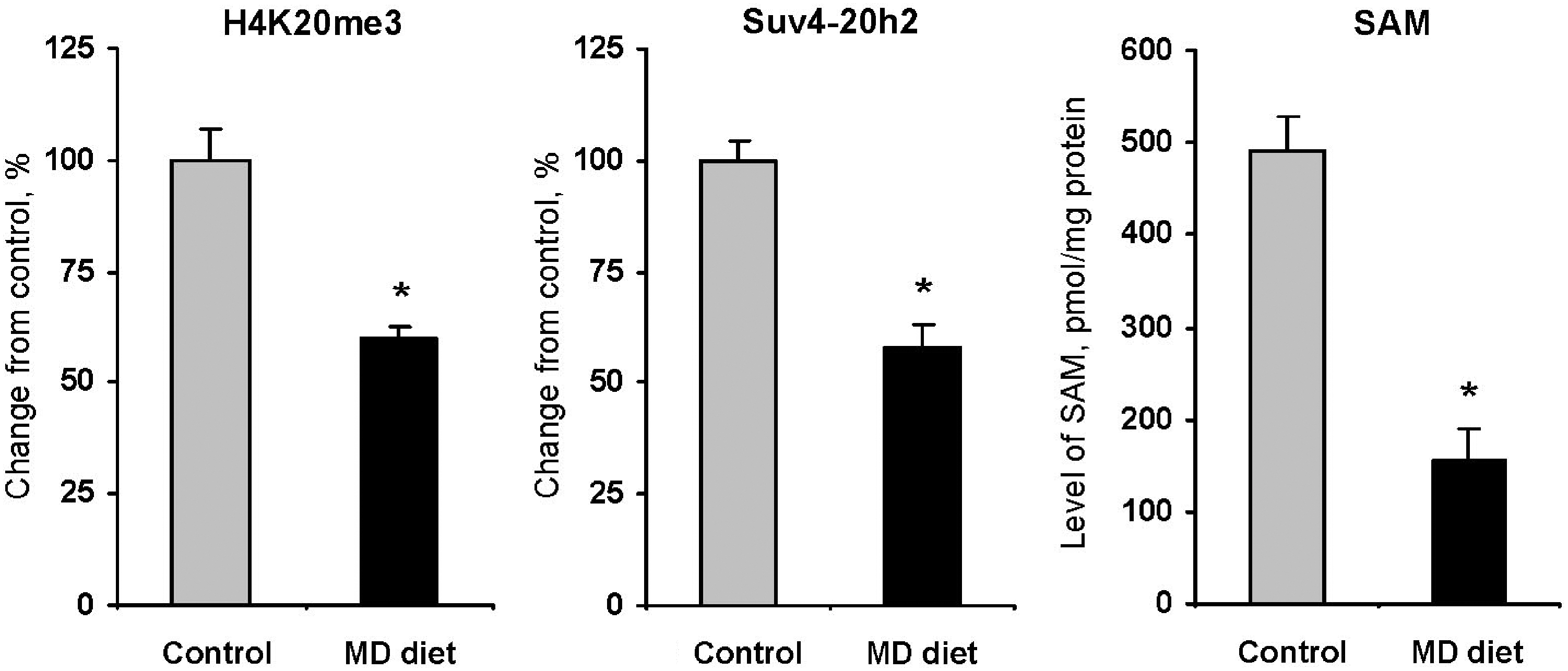
The mechanism associated with the cancer-specific loss of global histone H4K20 trimethylation is largely unknown; however, accumulating evidence indicates that the loss of histone H4K20me3 in cancer is often accompanied by a substantial downregulation of Suv4-20h histone methyltransferases (84). More importantly, these changes occur early during tumor development and progress with cancer advancement. For instance, expression of Suv4-20h2 steadily decreases and significantly correlates with the level of H4K20me3 during hepatocarcinogenesis induced by methyl deficiency (60, 63) and WY-14,643 (61) reaching the lowest level of expression in tumor tissue (59). More importantly, Figure 5 shows that these changes were accompanied by concordant changes in the level of SAM (59) indicating that the Suv4-20h2 HMT may be SAM-dependent. This suggests that regulation of Suv4-20h2 functioning by methyl donor supplementation may be a critical potential target in cancer prevention strategies. This suggestion is supported further by emerging evidence demonstrating that Riz1 (Prdm2) histone methyltransferase is also frequently downregulated in carcinogenesis (39, 93) and the correction of its activity by a methyl-adequate diet substantially decreases tumorigenesis in mice and increases their viability (58).
Glutathione, Oxidative Stress and Histone Lysine Methylation
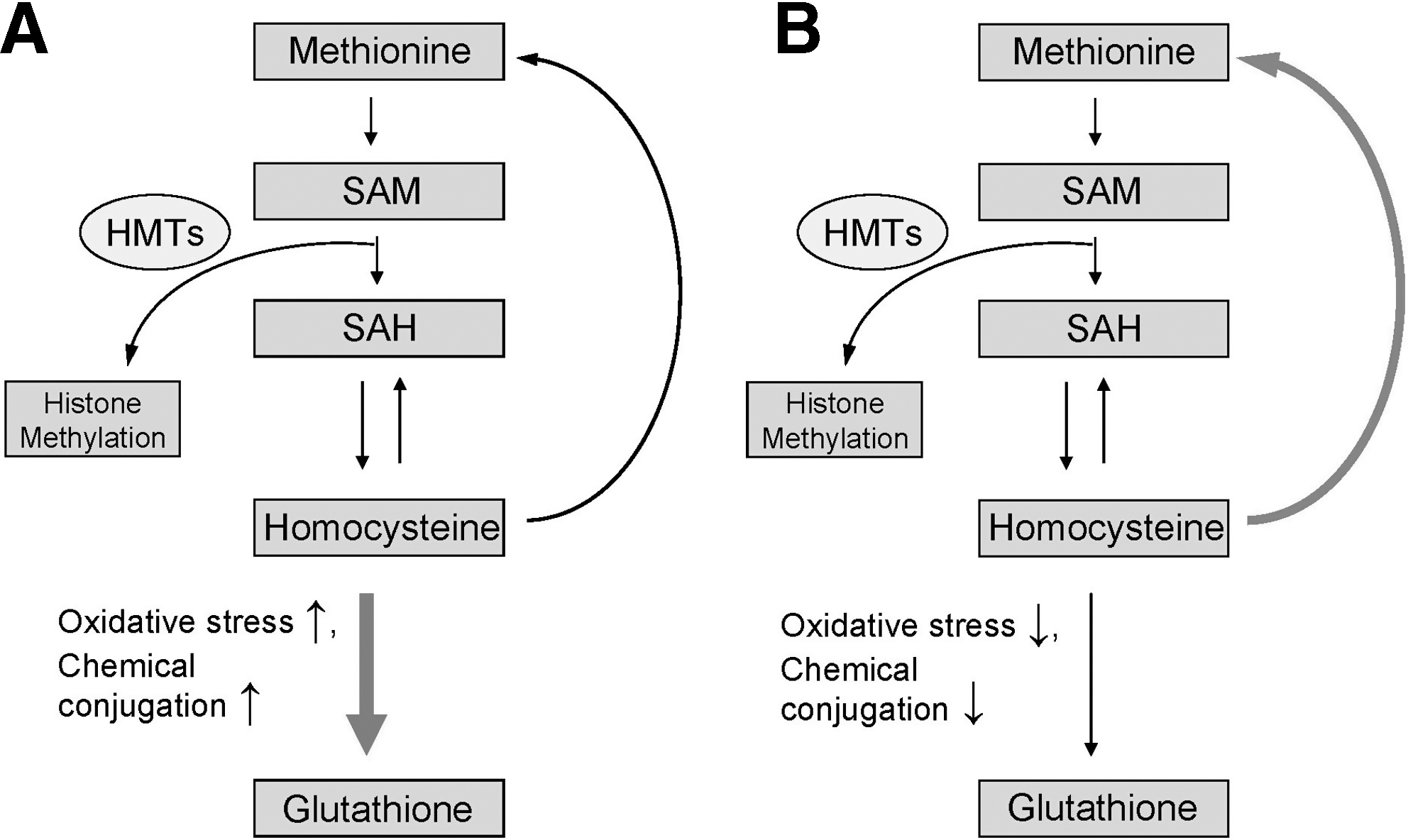
One of the common features of different models of genotoxic and nongenotoxic models of liver carcinogenesis described above is the induction of oxidative stress, a multi-stressor condition that arises when generation of reactive oxygen species exceeds the capacity of the cellular antioxidant defense system to control their production (33). In the liver, during a pro-oxidant state homocysteine is diverted away from the methionine cycle into the transsulfuration pathway (Fig. 3) favoring the glutathione biosynthesis (27, 85). Reduced glutathione (L-gamma-glutamyl-L-cysteinyl-glycine) is the most prevalent low-molecular weight thiol and main nonenzymatic component in the cellular antioxidant defense system (25, 91). Therefore, any event that disturbs the intracellular “redox equilibrium” subsequently leading to alterations in glutathione content may disturb the pattern of chromatin methylation, in general, and histone H4K20 methylation, in particular (Fig. 6A). In addition to its role in the regulation of intracellular redox status, reduced glutathione is necessary for conjugation of chemicals or their metabolites. For that reason, carcinogens that shunt homocysteine from the methionine cycle into the transsulfuration pathway (Fig. 6A) may affect further methylation of histones (47). Additionally, because histone lysine demethylation reactions are redox-sensitive, the intracellular redox homeostasis also plays an important role in their proper functioning. This clearly indicates that accurate maintenance of intracellular glutathione content and “redox equilibrium” by dietary antioxidants and nutrients may avert loss of histone methylation, including histone H4K20 methylation (Fig. 6B) and prevent tumorigenesis.
Concluding Remarks
The above presented evidence demonstrating the loss of H4K20 trimethylation occurs at early preneoplastic stages, persists during later stages of carcinogenic process, and suggests that this may be a “driver” rather than a “passenger” epigenetic event in carcinogenesis and may be used as a biomarker for the molecular diagnosis of cancer. However, the more important question as to whether or not detection of these epigenetic abnormalities can be used as the targets for cancer prevention remains unresolved.
Carcinogenesis, in addition to genetic and epigenetic abnormalities, is also associated with substantial metabolic defects (13, 27). Recently, interest in these cancer-linked metabolic disturbances, in particular to the “Warburg effect,” has been renewed, with special interest regarding influence of metabolic alterations on epigenetic processes (27). Specifically, it has been suggested that the “Warburg effect” substantially influences availability of various metabolites, including SAM, FAD and nicotine adenine dinucleotide, folate, acetyl coenzyme A, and α-ketoglutarate, that are essential for various histone modification reactions and proper maintenance of the epigenome (17, 54, 79). Therefore, timely regulation of the cellular epigenome by providing the appropriate levels of these cofactors for efficient functioning of many intracellular metabolic pathways through dietary intervention may be critical for the prevention of cancer development.
Disclaimer
The views expressed in this article do not necessarily represent those of the U.S. Food and Drug Administration.