
Abstract
Introduction
Key Factors of Epigenetic Regulation
As the substrate of transcription, chromatin is subjected to various forms of epigenetic regulation, including histone modifications, DNA methylation, and chromatin remodeling.
Histone modifications
Histones, DNA-wrapping proteins, are subjected to posttranslational modifications including acetylation and methylation by enzymes primarily on their N-terminal tails for regulating chromatin structure. The core of histones H2A, H2B, H3, and H4, which comprise high-order DNA structure units called nucleosomes, can be modified by these posttranslational additions of chemical groups. Acetylation of histones occurs at multiple lysine sites by the balanced actions of histone acetyltransferase (HAT) and histone deacetylase (HDAC), and the resultant acetylated lysine residues can interfere with the positive charge of histones, resulting in weakening the interaction with negatively charged DNAs that can directly influence chromatin structure and fluidity (Fig. 1A). Histone methylation is the modification of histone proteins by the addition of mono- (me1), symmetrical di- (me2s), and asymetrical di- (me2a) methyl groups at arginine residues (Fig. 1B), and mono- (me1), di- (me2), and tri- (me3) methyl groups at lysine residues (Fig. 1C). Unlike acetylation, methylation is highly site-specific, and is maintained by histone methyltransferase (HMT) and histone demethylase (HDM) (Fig. 1B, C). Histone methylation is generally associated with repression of gene expression. For example, trimethylation of lysine 9 and lysine 27 of histone 3 (H3K9 and H3K27) is correlated with inactive regions of chromatin. However, methylation of lysine 4 of histone 3 (H3K4) or arginine on H3 and H4 result in transcriptional activation.
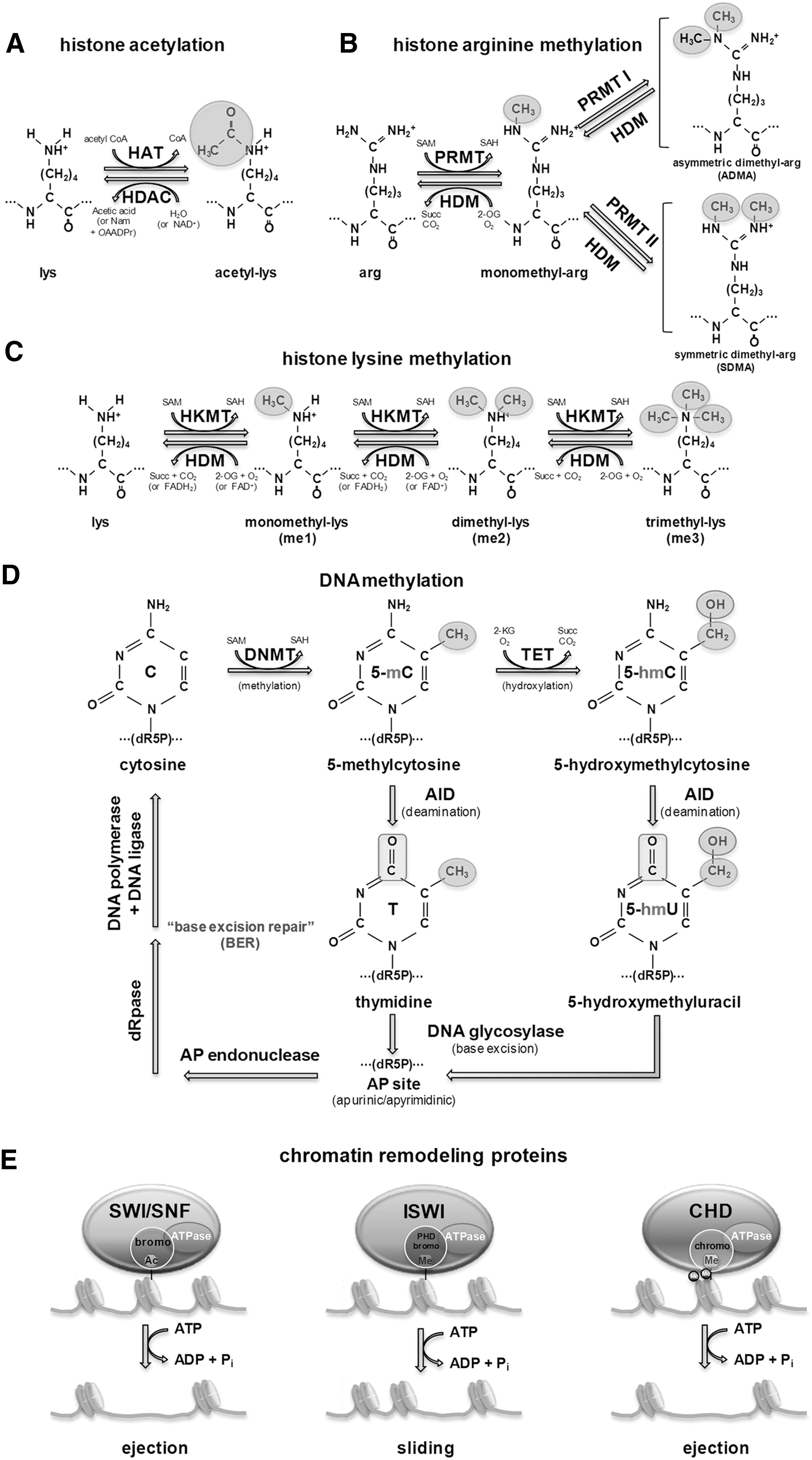
By regulating chromatin structure, epigenetic modifications play an essential role in controlling access to genes and regulatory elements in the genome (20). The differences in epigenetic status between a somatic cell and a PS cell are evident, and dedifferentiation requires global epigenetic reprogramming. For instance, PS cells contain bivalent domains that are characteristic chromatin signatures (10, 11). These are regions enriched for both repressive histone H3 lysine 27 trimethylation (H3K27me3) and active histone H3 lysine 4 trimethylation (H3K4me3) (116). It was assumed initially that bivalent domains might be ES-cell specific because they were first identified using chromatin-immunoprecipitation (ChIP) followed by hybridization to microarrays (ChIP-Chip) that featured key developmental regulators. All of these resolved either to a univalent (H3K4me3 only or H3K27me3 only) state or lost both marks in differentiated cells (10). Using ChIP followed by high-throughput sequencing (ChIP-seq) technology, Mikkelsen and colleagues showed that bivalent domains are more generally indicative of genes that remain in a poised or balanced state characterizing their plasticity. Pluripotent cells were found to contain large numbers of bivalent domains (∼2500) compared with multipotent neural progenitor cells (NPCs) (∼200) that still retain multilineage potential but are more restricted than ES cells (109).
DNA methylation
A second component of the epigenetic marks is DNA methylation, which is a stable and heritable mark that is involved in gene silencing, including genomic imprinting and X-chromosome inactivation. DNA methylation patterns are dynamic during early embryonic development and are essential for normal development (141). Overall DNA methylation levels remain stable during ES-cell differentiation, although they are not static at any individual gene level (109). The 5′-promoter regions of many transcriptional units contain clusters of the dinucleotide CpG, which are methylated at transcriptionally silent genes and demethylated upon activation. In differentiated cells, the Oct4, Nanog, and Sox2 promoter regions are highly methylated and in an inactivated state, whereas in ES cells these promoters are unmethylated and in an activated state. During reprogramming, almost complete demethylation of these promoters has been observed (102, 115, 121, 184). Therefore, the loss of DNA methylation at the promoters of pluripotency-related genes appears essential for achieving reprogramming to pluripotency state. Interestingly, loss of DNA methylation at this class of genes seems to be a rather late event in the reprogramming process because cells that have already acquired self-renewing properties still showed high levels of DNA methylation (115).
DNA methyltransferase (Dnmt)1, Dnmt3a, and Dnmt3b are three enzymes necessary for DNA methylation (14, 53) (Fig. 1D). Dnmt1 maintains DNA methylation at hemi-methylated DNA after DNA replication during cell divisions, whereas Dnmt3a and Dnmt3b are responsible for establishing de novo DNA methylation (28). Lack or mutation of Dnmt1 causes a two-third loss of DNA methylation or embryonic lethality (96). Embryos deficient in Dnmt1 are smaller and seem to have delays in developmental processes (96). Embryos with mutant Dnmt3b seem to proceed normally in their developmental stages initially but show multiple defects in their later stages (126). In mouse embryonic fibroblasts (MEFs), conditional deletion of Dnmt3b leads to partial loss of methylation (38). These findings show that Dnmt1 and Dnmt3b play a significant role in maintenance of proliferating cells and their epigenomic landscape. In contrast, lack of Dnmt3a does not seem to cause obvious developmental defects. A study showed that homozygous Dnmt3a knockout mice developed to term but died within ∼1 month after birth (126). Dnmt3a seems to function as a co-regulator of Dnmt1 and Dnmt3b, and interacts with the N-terminus of histone H3 (67, 127). It has been suggested that it functions as a co-regulator of both Dnmt3a and Dnmt3b and has recently been shown to interact with the N-terminal tail of histone H3 when it lacks methylation at lysine 4 (67, 127).
Chromatin remodeling
Chromatin remodeling enzymes add or remove histone covalent modifications and utilize the energy of ATP hydrolysis to disrupt chromatin structure. This makes DNA/chromatin available to proteins that need to access DNA or histones directly during cellular processes. There are three well-characterized families of ATP-dependent nucleosome remodeling enzymes, SWI/SNF, ISWI, and chromodomain helicase DNA-binding (CHD). These are multi-subunit complexes containing a conserved ATPase domain as the catalytic subunit, with additional components to form large multiprotein-complexes, which promote specific histone posttranslational modifications and incorporation of histone variants (75, 145) (Fig. 1E). The initial creation of the DNA loop or bulge is the result of the translocase activity that has been described to SWI/SNF and ISWI (143, 186, 200). The ATPase of SWI/SNF binds to a specific location on the nucleosome, from which it utilizes its 3′ to 5′ translocase activity to draw DNA into one entry/exit and pump it to the other in a directional wave (144). Binding of the complex to the nucleosome creates significant rearrangement of the DNA with respect to the histone octamer even in the absence of ATP hydrolysis, which may facilitate the creation of the DNA bulge required for ATP-dependent translocation (101). In contrast to SWI/SNF, the smaller ISWI chromatin remodeling complexes make limited contacts with the nucleosome and the extranucleosomal DNA (33, 48, 71, 149). These complexes bind their substrate as a dimeric motor to facilitate the bidirectional and processive translocation of DNA over the nucleosome (15, 47, 133). This is consistent with the role of these remodelers in nucleosome spacing, and their ability to sample DNA linker lengths to position nucleosomes equidistant from either end (48, 165, 195, 199). In this case, ATP hydrolysis by one of the two ATPases results in loosened DNA-histone contacts that may act similarly to the ATP-independent conformational change upon SWI/SNF binding to promote DNA translocation following ATP hydrolysis by the second ATPase (49). ATPases of the CHD family are characterized by N-terminal tandem chromodomains in addition to the conserved DEAD/H-related ATPase domain (132, 173). CHD is targeted to sites of active transcription through PHD-mediated recognition of H3K4me3 (45, 156), and associates with other preinitiation factors to facilitate transcriptional elongation and splicing (157). Despite the similarity between their core enzymatic subunits, their common affinity for nucleosomes, and their common ability to disrupt nucleosomal templates in vitro, there is little functional similarity between members in vivo and as a result, very little predictive power for members of unknown function.
Epigenetic Landscape in Pluripotent Stem Cells
There are unique epigenetic marks in PS cells, ES cells, and iPS cells. It is well known that certain epigenetic marks are critical for maintaining pluripotency, which include histone modifications, DNA methylation of pluripotency genes, and regulation of pluripotency- or differentiation-specific microRNA (miRs) expression. However, other studies still claim that a majority of epigenetic marks are unnecessary for ES cell survival. Although it is possible to maintain PS cells without all of their epigenetic marks, this leads to a decrease in their differentiation potential. Thus, the degree of in vitro epigenetic influence in pluripotency of ES cells needs to be assessed by further studies.
Chromatin structure in PS cells
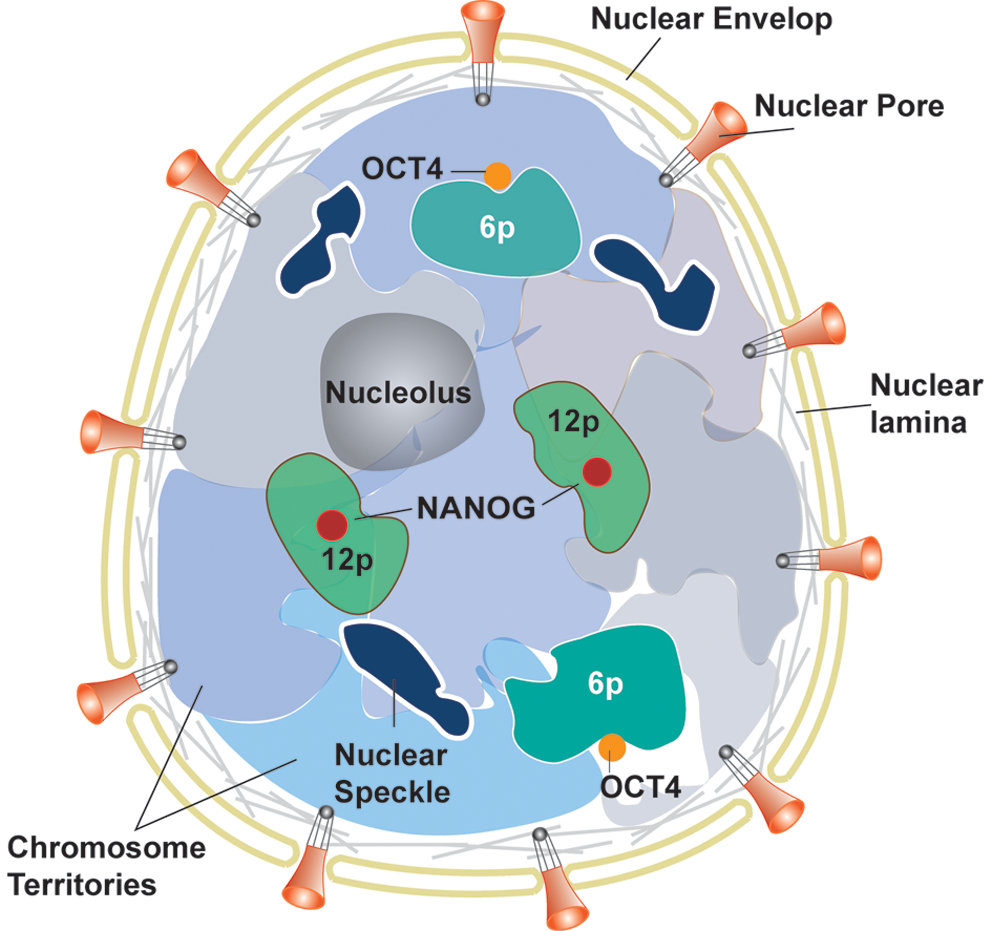
PS cells, including ES and iPS cells, have a dynamic and specialized higher order chromatin structure that is highly organized for the regulation of gene expression programs during development and differentiation, and is established by histone modification, DNA methylation, and the activity of ATP-dependent nucleosome remodeling complexes (107). PS cells have distinctive chromosomal regions that contain the genes that maintain pluripotency. A region of clustered pluripotency genes, including NANOG at chromosome 12p, has more central nuclear localization in ES cells than in differentiated cells (187). Although the functional significance of positioning in the nuclear center is unknown, the presence of the nuclear spots within the most gene-dense human chromosomes suggests that it may confer some transcriptional advantage (19). Chromosome 12p contains other pluripotency-related genes, STELLA and GDF3, which are also expressed in ES cells and downregulated upon differentiation (31). Another pluripotency gene, OCT4, at chromosome 6p forms a loop to relocalize to a position outside its chromosome territory (187). The data showing increased DNA methylation and histone deacetylation of the OCT4 enhancer/promoter in trophoblast cells compared with ES cells are consistent with a more long-range remodeling of chromatin architecture around OCT4, which might also contribute to its transcriptional regulation (58). Preferential association of inactive genes with the nuclear periphery has been reported in differentiated cells; however, neither NANOG nor OCT4 is associated with the nuclear periphery of clusters of inactive genes (198) (Fig. 2). In addition, genes and chromosome activities are directly related to their spatial positioning within the nucleus that is prone to alterations during early differentiation (78, 104). Furthermore, studies have indicated that nuclear structures such as nucleolus, heterochromatin (129), nuclear speckles (domains enriched in splicing factors) (32), and nuclear lamina experience morphological changes during the early stage of differentiation.
Histone modification in PS cells
N-terminal tail residues of histones can undergo posttranslational modifications such as acetylation, methylation, and phosphorylation (81). Given that ES and somatic cells contain almost identical genomic DNA, epigenetic regulation is one of the major influences on their pluripotency and differentiation potential. To maintain pluripotency in ES cells, differentiation-triggering genes should be inactive. Polycomb group proteins (PcGs) play important roles in silencing these developmental regulators. They form multiple polycomb repressive complexes (PRCs), the components of which are conserved from Drosophila to humans (148). The PcG proteins function in two distinct PRCs, PRC1 and PRC2. Genome-wide binding site analyses of PRC1 and PRC2 in mouse ES cells and PRC2 in human ES cells (18, 93) showed that the genes regulated by the PcG proteins are co-occupied by nucleosomes with trimethylated H3K27. These genes are transcriptionally repressed in ES cells and are preferentially activated when differentiation is induced. The pluripotency factors Oct4, Sox2, and Nanog co-occupy a significant fraction of the PcG protein-regulated genes when acting as transcription factors (18, 93). Most of the transcriptionally silent developmental regulators targeted by Oct4, Sox2, and Nanog are also occupied by the PcGs (11, 18, 93). PRC2 catalyzes H3K27 methylation, an enzymatic activity required for PRC2-mediated epigenetic gene silencing. H3K27 methylation is thought to provide a binding surface for PRC1, which facilitates oligomerization, condensation of chromatin structure, and inhibition of chromatin remodeling activity in order to maintain silencing. PRC1 also contains a histone ubiquitin ligase, Ring1b, whose activity appears to contribute to silencing of genes in ES cells (162). How the PcG is recruited to genes encoding developmental regulators in ES cells is unknown. Some of the most conserved vertebrate sequences are associated with genes encoding developmental regulators, and some of these may be sites for DNA-binding proteins that recruit PcG proteins (Fig. 3).
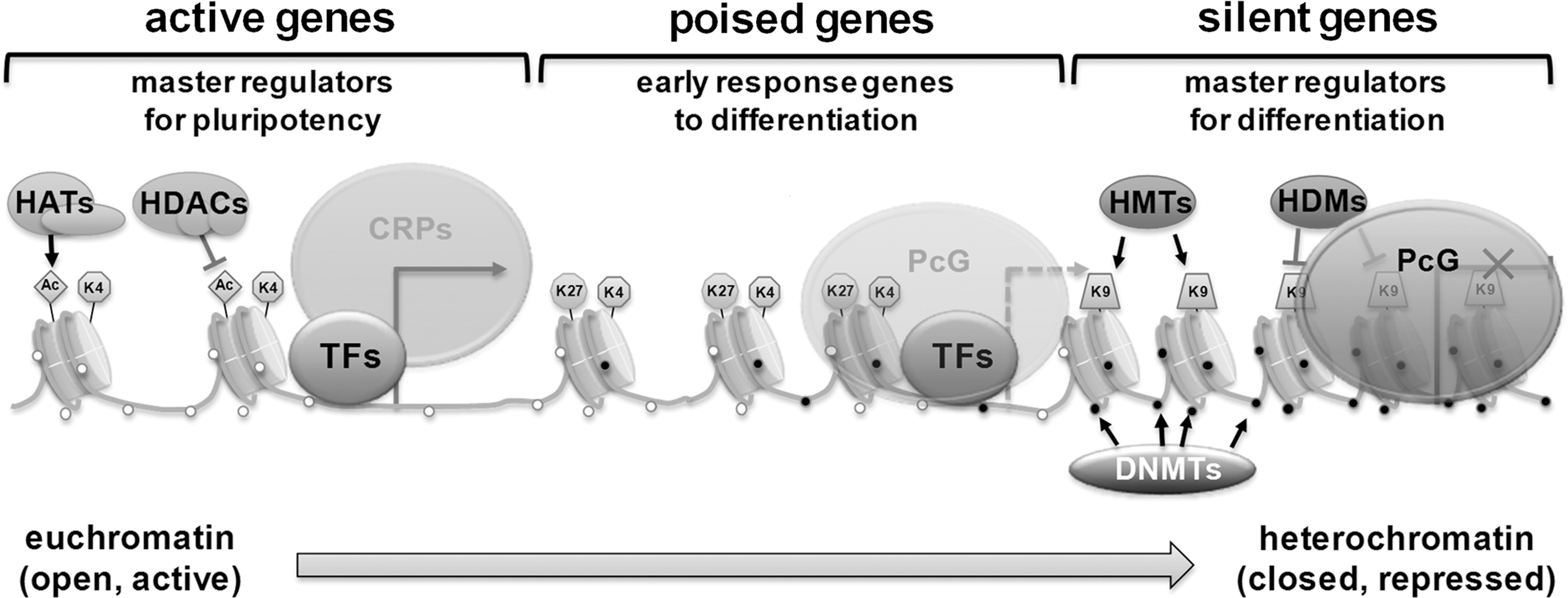
Recent studies demonstrated that the silent developmental genes that are occupied by Oct4, Sox2, Nanog, and PcG proteins experience an unusual form of transcriptional regulation (55). These genes undergo transcription initiation but not productive transcript elongation in ES cells. The transcription initiation apparatus is recruited to developmental gene promoters, but RNA polymerase is incapable of fully transcribing these genes, presumably because of repression mediated by the PcG. This explains why the silent genes encoding developmental regulators are generally organized in bivalent domains that are occupied by nucleosomes with histone H3K4me3 and H3K27me3 (4, 11, 55).
The presence of inactive RNA polymerase at the promoters of genes encoding developmental regulators may explain why these genes are especially poised for transcription activation during differentiation (18, 93). PcG complexes and associated proteins may serve to pause RNA polymerase machinery at key regulators of development in PS cells and lineage-committed cells where they are not expressed. When the cells are activated, PcGs and nucleosomes with H3K27 methylation are lost (18, 93, 116), allowing the transcription apparatus to fully function and transcribe these genes. The mechanisms that lead to selective activation of genes encoding specific developmental regulators are not yet understood (87). Beyond the specific regulation of development-related genes, ES cells maintain chromatin in a highly dynamic and transcriptionally permissive state. Fewer heterochromatin foci are detected in ES cell nuclei compared with differentiated cells. Fluorescence recovery after photobleaching and biochemical analyses reveal that ES cells, compared with differentiated cells, have an increased fraction of loosely bound or soluble architectural chromatin proteins, including core and linker histones and heterochromatin protein HP1. A hyperdynamic chromatin structure is functionally important for pluripotency (113). The status of histone modifications also indicates that the chromatin in ES cells is more transcriptionally permissive than in differentiated cells. Consistent with the global dynamics of chromatin, ES cell differentiation is associated with a decrease in global levels of active histone marks, such as acetylated histone H3 and H4, and an increase in repressive histone marks, histone H3 lysine 9 methylation (91, 113). Taken together, these unique epigenetic characteristics of ES cells facilitate rapid but regulated transcription, allowing differentiation down different cell fate pathways as needed by the organism.
Several studies of murine iPS cells have identified a small number of representative loci that have consistent chromatin and DNA methylation patterns (102, 170, 184). A ChIP-Chip study investigating H3K4me3 and H3K27me3 in the promoter regions of 16500 genes showed that iPS cells were highly similar to ES cells in epigenetic state (102). The H3K4me3 pattern was similar across all samples, indicating that reprogramming was largely associated with changes in H3K27me3 rather than H3K4me3 (102). Another ChIP-Seq study exploring genome-wide chromatin maps in several iPS lines derived by different methods (110, 184) demonstrated that global levels of repressive H3K27me3 and the characteristic bivalent chromatin structure are retained in the various iPS cell lines. The restoration of repressive chromatin marks appears crucial to stably silence lineage-specific genes that are active in somatic cells and inactive in pluripotent cells, suggesting that failure to establish the repressive marks results in incompletely reprogrammed cells. Activating H3K4me3 patterns are also crucial for complete reprogramming and have been observed to be restored genome wide, in particular around the promoters of pluripotency-associated genes such as Oct4 and Nanog, in the fully reprogrammed iPS lines (115).
Histone demethylation plays a critical role in ES cell epigenesis and pluripotency. The mechanism of histone demethylation was discovered only recently (81, 150, 151). Removal of a specific H3K27 tri-methylation can be achieved by two enzyme families: Jumonji (JMJD) and UTX (1, 35, 92). Jmjd1a and Jmjd2c genes, which are responsible for H3K9me2 and H3K9me3 demethylation, are up-regulated by Oct4. Jmjd2a induces Nanog up-regulation and Jmjd1a demethylates H3K9me2 at the Tc11, Zfp57, and Tcfcp211 promoters (100) (Fig. 1C).
Histone acetylation activates transcription. The HAT p300 regulates the state of pluripotency by activating transcription. p300 is recruited to sites of multiple pluripotency factors such as Nanog, Oct4, and Sox2 (27). These multiple binding sites are involved in activation of the H3K4me3 mark. In ES cells, Tip60-p400 is the HAT and chromatin remodeling complex that positively or negatively regulates gene expression and DNA repair (146, 160). The complex is also a transcriptional activator that incorporates the histone H2AZ and catalyzes histone acetylation at target promoters (160). ES cells with Tip60-p400 deficiency show abnormal morphology and cannot self-renew or differentiate. There are significant similarities between genes affected by Tip60-p400 depletion and Nanog depletion. Furthermore, Tip60-p400 binding, Nanog binding, and the H3K4me3 mark have strong correlations with one another. Tip60-p400 localizes to both highly expressed genes and poised genes marked by bivalent modifications (43). Tip60-p400 binding is necessary in regulating histone H4 acetylation. In ES cells, Tip60-p400 can act as a repressor by catalyzing histone acetylation.
Histone deacetylation is mediated by nucleosome remodeling deacetylase (NuRD) complexes, which are responsible for HDAC activities as well as ATP-dependent chromatin remodeling. The NuRD complex consists of SNF2-like ATPase subunits, Mi2α or -β, deacetylase subunits HDAC1/2, and associated subunits including methyl-CpG-binding proteins MBD 1/2/3 and metastasis associated proteins MTA1/2/3 (37). Mi2β, in association with HDACs and MBD subunits, plays an important role as a transcriptional repressor but can also be an activator and is required for hematopoietic stem cell self-renewal and differentiation (179, 189, 196). Pluripotency requires MBD3, as ES cells without MBD3 cannot commit to specific developmental lineages (72). However, MBD3 is not required for expression of pluripotency factors but only for repression of gene transcription. Unrepressed promoters become hyperacetylated at H3K9 and H4, whereas there is no change in the acetylation status of other inactive promoters, suggesting that the NuRD complex is not globally required for ES cell gene silencing. Furthermore, MBD3-depleted ES cells can undergo initial stages of differentiation by forming embryoid bodies and trophectoderm but fail to differentiate further except when induced with retinoic acid. MBD3-depleted ES cells also fail to develop in utero as chimeric embryos when aggregated with wild-type morulae. Thus, MBD3 as part of the NuRD complex facilitates ES cell differentiation but is not absolutely required for ES cells to respond to all inductive signals.
DNA methylation in PS cells
DNA methylation suppresses gene expression; thus, in ES cell status, most of the pluripotency-related transcription factors are hypomethylated and lineage-committed genes are heavily methylated at their promoter regions (Fig. 3). H3K4me3 and DNA methylation are known to be linked. DNA methylation maps have been created for mouse and human pluripotent cells (89, 97, 109). High-CpG promoters of ES cells have H3K4me3 histone modifications and entirely lack DNA methylation (109,116). This may be related to the ability of Dnmt3 to bind to unmodified H3K4 (127). In ES cells, CpGs in low-CpG promoters, which are generally associated with tissue-specific genes (183), are mostly methylated, with the exception of a small subset (<10%) that are enriched for H3K4me3 or H3K4me2 (109). All pluripotency genes in ES cells have H3K4 methylation and DNA hypomethylation (65, 115, 116). However, the loci that are devoid of H3K4 and H3K27 methylation show extensive DNA methylation instead (46). Genome-wide bisulfite sequencing in human ES cells (89, 97) has confirmed the findings of these previous, more limited mouse (46, 109, 118) and human studies (183).
The Dnmt family maintains and directs catalysis of DNA methylation. The methyl-CpG-binding domain (MBD) family recognizes DNA sequences with high CpG content and mediates the effects of DNA methylation (60). During the division of the zygote, paternal and maternal methylation patterns disintegrate by down-regulating the expression of the Dnmt1 gene (136). During embryogenesis and formation of germ layers, there is a re-establishment of a new DNA methylation pattern. During this period, the epigenomic status of ES cells inside ICM is restored to redefine pluripotency. Studies of Dnmt1-deficient (Dnmt1 −/−) and Dnmt3a/Dnmt3b-deficient (Dnmt[3a −/−,3b −/−]) ES cells have demonstrated that restoring DNA methylation is essential for development (94, 126). These Dnmt mutant ES cells exhibited an early-lethal phenotype, defects in differentiation during embryoid body formation of Dnmt1 −/− (128), and teratoma formation of late-passage Dnmt[3a −/−,3b −/−] (29). Down-regulation of Oct4 and Nanog expression during the differentiation of NT2 cells, a neuronally committed human teratocarcinoma cell line, is related to methylation of 5′-flanking regions of both these genes (36).
While the role of non–CpG methylation is unclear in somatic cells, recent evidence suggests that the role of non–CpG methylation is important in maintenance of ES cell pluripotency. In adult somatic tissues, DNA methylation typically occurs at the site of the CpG dinucleotide. However, non–CpG methylation, including CpA and CpT methylation, is prevalent in mouse ES cells (108, 109) and human ES cells (89, 97). Non–CpG methylation level is mediated by Dnmt3a and Dnmt3b (39). Overexpression of Dnmt3a in Drosophila, which has no Dnmts or DNA methylation (53), induces CpG as well as non–CpG methylation. These data support the notion that non–CpG methylation results from de novo methylation activity of Dnmt3a, which is highly expressed in ES cells and poorly expressed in somatic tissues (135). In both mouse and human, non–CpG methylation is low in differentiated cells (97, 135) and reappears in iPS cell generation (97).
Recent studies have shown the importance of DNA hydroxymethylation in regulating gene expression. In fact, DNA hydroxymethylation is highly prevalent in ES cells compared with somatic cells. The ten–eleven translocation (TET) family direct catalysis of 5-methyl cytosine to 5-hydroxymethylcytosine (5hmC) (168) (Fig. 1D). 5hmC is detectable in undifferentiated mouse ES cells as well as in Purkinje neurons and granule cells (82). During the differentiation of ES cells, the amount of Tet1 and 5hmC decreases (168), and Tet1 knockdown by shRNA damages ES cells' ability to maintain and self-renew with diminishing Nanog expression, suggesting a crucial role of Tet1 in maintaining pluripotency (66). However, a more recent study showed that Tet1-deficient (Tet1 −/−) mouse ES cells still maintain pluripotency with expression of pluripotency markers Oct4, Sox2, and Nanog, and can drive normal embryo development in tetraploid complementation assays, although these mutant mice have a partial reduction of 5hmC and a slightly small body size at birth (34). Another study using shRNAs to knock down Tet1 and Tet2 also showed that depletion of these genes did not affect Nanog expression, self-renewal, or pluripotency (80).
Chromatin remodeling enzymes
ES cells are rich in euchromatin and diffused heterochromatin. Chromatin structural remodeling leads to highly compact heterochromatin during differentiation. The change from lower to a higher chromatin structure state is helped by chromatin remodeling protein complexes. Either the remodeling enzymes have the ability to alter the N-terminal characteristics of histones by adding or removing acetyl, methyl, ubiquitin, and sumo groups, or the remodeling proteins are composed of ATP-dependent enzymes, which use energy to transiently separate the association between DNA and histones. These modifications, in turn, may induce a conformational change in nucleosome and chromatin condensation state. This kind of ATP-mediated remodeling provides an accessible DNA, which is more prone to gene activation or repression by the transcriptional apparatus (154) (Fig. 3). An example is the SWI/SNF protein complex in mammals, which is composed of several different subunits such as Snf2. Different chromatin remodeling enzymes that are ATP-dependent are highly expressed in mouse ES cells and are down-regulated after leukemia inhibitory factor removal (84). Mice that lack remodeling proteins SNF2H (163), BRG1 (21), SNF5 (79), or SSRP (24) die before implantation. The SWI/SNF mechanism of catalysis and regulation of ES cell pluripotency is not very well understood and requires further investigation. It has been found that the SWI/SNF chromatin remodeling component, BAF250, is vital for ES cell pluripotency (50). BAF250b dysfunction lowers expression of pluripotency factors and increases that of lineage markers such as Gata2. The SWI/SNF complex also regulates nucleosome sliding and eviction (56, 95).
miRs in PS cells
The function of miRs in stem cells has been established in many systems, where disruption of Dicer or DGCR8, which are miR-processing enzymes, leads to defects in embryo development (12, 57, 73, 120, 123, 182, 188). Dicer-null mouse is embryonic lethal, indicating its critical role in early embryonic development (12). However, Dicer-null mouse ES cells are viable but show defective cell cycle progression. Dicer-null ES cells are defective in differentiation and fail to express differentiation markers such as HNF4a, BMP4, and GATA1 (73). Dicer-deficient cells further show decreased levels of DNA methylation and Dnmts (9, 158) and increases in telomerase recombination and elongation (9). This defect in DNA methylation leads to lack of differentiation by incomplete and reversible silencing of Oct4 (9, 158). DGCR8 knock-out mouse ES cells show phenotypes similar to Dicer-deficient mouse ES cells, showing reduced cell proliferation, abnormal cell cycle control, and defects in differentiation (182). DGCR8-null ES cells arrest in G1 phase and do not differentiate into teratoma, a feature characteristic of normal ES cells (16, 76, 112). Blelloch and colleagues have discovered specific functions for families of miRs controlling cell cycle and self-renewal in DGCR8-null mouse ES cells (111, 181) (Fig. 4).
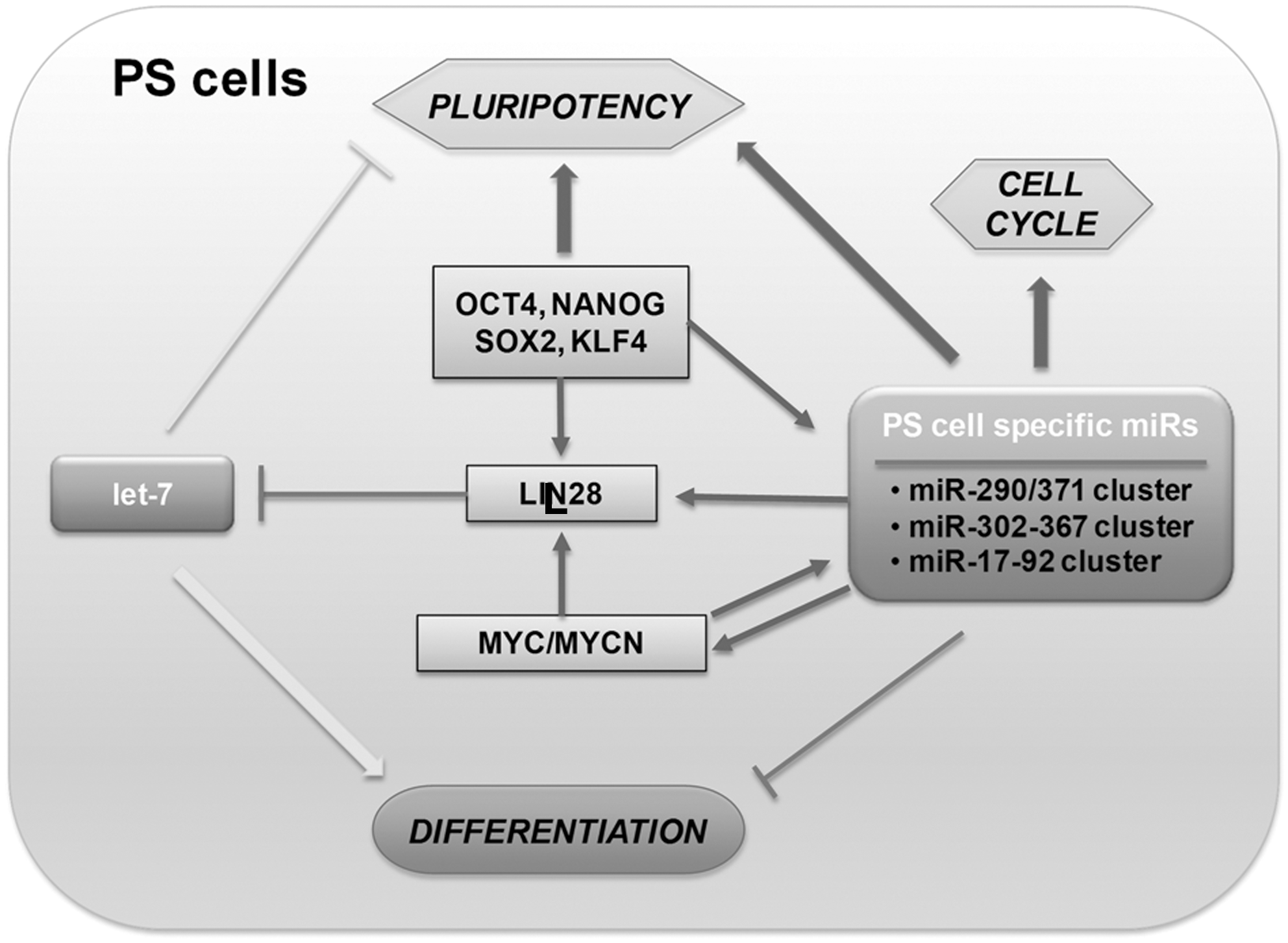
ES cells have been reported to express a small subset of unique miRs (23, 62, 86, 119, 166). The human miR-371 cluster is located on chromosome 19 and is analogous to the mouse miR-290 cluster, and the miR-302 cluster located on chromosome 4 is associated with both murine and human ES cells (62, 164, 166). Two additional clusters, miR-17 on chromosome 13 and the miR-106a cluster on chromosome X, have also been shown to be upregulated in ES cells (90).
miR-290 and miR-371 clusters
The miR-290 cluster is comprised of six miRs (miR-290 to miR-295), all of which are expressed at high levels in undifferentiated mouse ES cells but decreasing upon differentiation (62). Exogenous miR-290 family members can partially rescue self-renewal of Dicer-null cells (9, 158). The human homolog of the miR-290 cluster includes miR-371, -372, -373, and -373*. These miRs were identified as embryonic carcinoma and ES cell-specific miRs (69, 86, 166). The cluster is transcribed as a single polycistronic transcript and regulated by a common promoter (166). Mouse mutants with a homozygous deletion of the miR-290–295 cluster are embryonic lethal, demonstrating its importance during embryo development (2). The ES core transcriptional regulatory circuit, Oct4, Sox2, and Nanog, has recently been shown to be physically associated with promoters for the miR-290 cluster (105).
miR-302-367 cluster
Another ES cell-specific miRs cluster is the miR-302–367 cluster, shown to be highly and specifically expressed in undifferentiated ES cell (69, 86, 119). This cluster, encoded by human chromosome 4, consists of nine different miRs: miR-302a, -302a*, -302b, -302b*, -302c, -302c*, -302d, -367, and -367* (88). The ES cell-specific transcription factors, Oct4, Nanog, Sox2, and Rex1, were shown to be upstream regulators of the miR-302–367 promoter (8, 25). In addition, miR-302–367 has been identified to posttranscriptionally regulate cyclin D1 and Cdk4, affecting cell cycle progression (25). In addition to their role in self-renewal and proliferation, miR-302–367 may indirectly induce the TGFβ/Nodal/Activin pathway via inhibition of intermediate negative regulators to maintain cells in the undifferentiated state (7).
miR-17–92 cluster
The miR-17–92 cluster is highly expressed in ES cells, and a single polycistronic transcript generates six miRs: miR-17, -18a, -19a, -20a, -19b-1, and -92a-1 (54, 62, 119). This cluster is a known OncomiR that promotes cell proliferation in several forms of cancer (59, 85, 124, 137, 176) and is activated by c-Myc, a repressor of other miRs such as miR-15 and let-7 (26). miR-20a is thought to play a role in cell proliferation and inhibition of apoptosis at the G1 to S transition via a feedback loop with E2F factors (124, 167, 174, 191). The role of c-Myc in combination with Oct4, Sox2, and Klf4 in inducing somatic cells to iPS cells suggests that these miRs play a key role in stem cell renewal and pluripotency. Indeed, Melton and colleagues demonstrated that a let-7 inhibitor was able to replace exogenous c-Myc in the production of iPS cell cultures (111).
let-7 family
ES cells are characterized by the presence of low levels of let-7, which is highly expressed in differentiating cell types (54, 86). Let-7g levels have been shown to be regulated by LIN28, a gene highly expressed in pluripotent cells (177). The LIN28 gene is in turn targeted by let-7 via target sites in its 3′-UTR (122, 130, 142, 177). Both LIN28 and let-7g genes are associated with Oct4/Sox2/Nanog/Tcf3 binding in ES cells, resulting in a regulatory loop where the core pluripotent transcription factors activate the LIN28 gene associated with maintenance of self-renewal and pluripotency while repressing let-7g to prevent differentiation (105). Exogenous addition of let-7 miRs can suppress self-renewal in DGCR8 null mouse ES cells but not in wild-type ES cells, and this suppression can be inhibited by introduction of ES cell–cell cycle-regulating miRs such as miR-294 (111).
Epigenetic Changes During Differentiation
The investigation of lineage specification and global epigenetic remodeling for specific cell types in vivo is a difficult challenge despite advances in experimental technologies. Therefore, PS cells have emerged as a powerful system to investigate epigenetic changes during differentiation. As human PS cells are the only available model system to study the mechanism of differentiation and cell fate decision in human developmental biology, they have been widely used for such purposes. Most of genome-wide DNA methylation and histone-modification studies were conducted comparing epigenetic changes during differentiation of PS cells into their differentiated lineages. During this cell fate change, PS cells lose their pluripotency by silencing pluripotency-related genes and gain differentiated cell phenotype by activating silenced or poised lineage-specific genes as the promoters and enhancers of these genes become accessible by epigenetic changes. The promoter-specific regulation of chromatin structure by specific enzymes leads to proper differentiation into specific cell types. However, the mechanism of recruiting enzymes that regulate chromatin remodeling and histone modification needs to be further elucidated.
Neural differentiation
During the differentiation of PS cells into the neural lineage, the promoters of many genes related to the neural lineage, including Nestin, Otx2, Nkx2-2, Astn, Olig1, and the miRNA mir-9-3, lose a repression mark, H3K27me3, and retain a monovalent activation mark, H3K4me3, resulting in triggering expression of these genes (116). This removal of H3K27me3 is mediated by upregulation of H3K27-specific HDM, Kdm6b (also known as Jmjd3), with downregulation of specific HMT, enhancer of zeste homolog 2 (Ezh2) at the promoters of neural genes (22). During the transition from human ES cells to NPCs, pluripotency-related genes such as NANOG, TERT, and LIN28, and non-neuronal lineage genes, including GATA4 and NODAL, gain another repressive mark, H3K9me2, concomitantly with the loss of H3K4me3, leading to their long-term repression (52). The transcriptional repressor RE1-silencing transcription factor (REST, also known as NRSF) targets a group of neuronal genes through its interaction with the 21-base pair RE1 element. It represses neuronal differentiation by recruiting histone modifiers and chromatin-binding proteins (5). A subset of REST target genes escape the repression and are selectively induced during the transition from ES cells to NPCs. Degradation of REST by β-transducin repeat-containing protein 1 (β-TRCP1) has been shown to be essential for ES cell commitment to a neural fate (185). Also, DNAs at the loci of neural-related distal regulatory elements is demethylated at CpG site upon commitment to the neural lineage, whereas non-neural gene loci gain CpG methylation in NPCs (118). The targeting mechanism of histone modifiers and DNMTs to specific loci is not well understood. However, it has been shown that PcG proteins preferentially target highly conserved promoters with long unmethylated CpG islands in ES cells (138, 147).
Endothelial differentiation
The role of histone acetylation in endothelial differentiation was identified by the studies using small molecule HDAC inhibitors such as trichostatin A (TSA). These inhibitors block the endothelial differentiation of adult progenitor cells by downregulation of HOXA9 expression (140). Other studies also showed similar negative effects of TSA or valproic acid (VPA), another HDAC inhibitor, on the expression of endothelial nitric oxide synthase (eNOS) at a high concentration (∼0.3 μg/ml) of inhibitors and on angiogenesis of human endothelial cells (114, 139). In contrast, HDAC inhibitors at a low concentration (0.1 μg/ml) in combination with DNMT inhibitor, 5′-aza-2′-deoxycystidine (5-aza-dC), induced differentiation into endothelial cells from bone-marrow-derived multipotent adult progenitor cells (103). This inhibitor of DNMT, 5-aza-dC, can also induce endothelial cell differentiation from mouse ES cells (6). Interestingly, nitric oxide (NO), which is generated by eNOS, results in the activation of class IIa HDACs and subsequently induces mesoderm differentiation of mouse ES cells (159). The elevated level of NO in endothelial cells by shear stress and direct exposure of mouse ES cells to NO upregulated vascular gene expression, including Acta2 (also known as smooth muscle actin), Taglin (smooth muscle protein 22-alpha), Pecam1 (CD31), Kdr (Flk-1), Mef2c, and Acta1 (alpha-sarcomeric actin) (64, 159).
Cardiac differentiation
Inhibition of DNA methylation by small molecule chemical, 5-aza-dC, also promotes cardiac differentiation of ES cells in a time-dependent manner (193). In addition, inhibition of HDAC by TSA promotes cardiac differentiation by increased expression of acetylated Gata4, Nkx2-5, and Mef2c (74). In Nxk2.5-GFP mouse ES cells, TSA exposure between days 7 and 8 of differentiation doubled the percentage of GFP+ cells, and boosted expression of Nkx2-5, Myh7 (β-MHC), and Naap (atrial natriuretic factor [ANF]). TSA-treated mouse EBs, which showed enhanced acetylation of histone 3 and 4, have increased GATA4 acetylation and DNA binding to the ANF promoter and enhancedcardiomyocyte differentiation (74). Another cardiac differentiation-related histone methylatransferase is Dot1L, which catalyzes H3K79 methylation despite lacking a SET domain (44). Dot1L knockout mice are embryonically lethal due to dilated heart and angiogenesis defects in yolk sac. ES cells from Dot1L knockout blastocysts show global loss of H3K79 methylation as well as other repressive histone marks, H3K9me2 and H4K20me3, and display aneuploidy, telomere elongation, and proliferation defects (68). In addition, overexpression of cardiac-enriched subunit of the SWI/SNF-like chromatin remodeling BAF complex, Baf60c (also known as Smarcd3), when combined with ectopic expression of Gata4 and Tbx5, was able to induce cardiac differentiation of mouse embryos (171).
Reprogramming of Cell Fate and the Role of the Epigenome
In 2006, Shinya Yamanaka discovered that adult somatic cells can be reprogrammed to ES-like cells called “iPS cells” using viral transduction of pluripotency-related transcription factor genes (169, 170). The mechanism by which ectopic transcription factors override the existing epigenetic state and change it into a specific alternative state without passing through normal development or complete resetting of all marks is still largely unknown; however this triumph suggests the flexibility of the epigenome and its influence on cell fate determination. The key factors involved in the epigenetic remodeling have not been identified, and many questions regarding the dynamics of this process still remain.
Epigenetic changes during reprogramming
The term “epigenetics” was first coined by Waddington in 1942 to describe the role of genetic interactions in phenotype. Later, he presented the epigenetic landscape model in 1957 by using the metaphor of a ball traveling down a canal that starts from the stage of the fertilized totipotent embryo and ends with various lineage-specific cells. In this concept, cells move downward through different one-way branched valleys inside a canal and select their ultimate irreversible cellular fates (178). The four transcription factors essential in reprogramming reverse the cells' path in this canal by unlocking specific epigenetic gateways, which normally induces stabilization into a differentiated form (194) (Fig. 5).
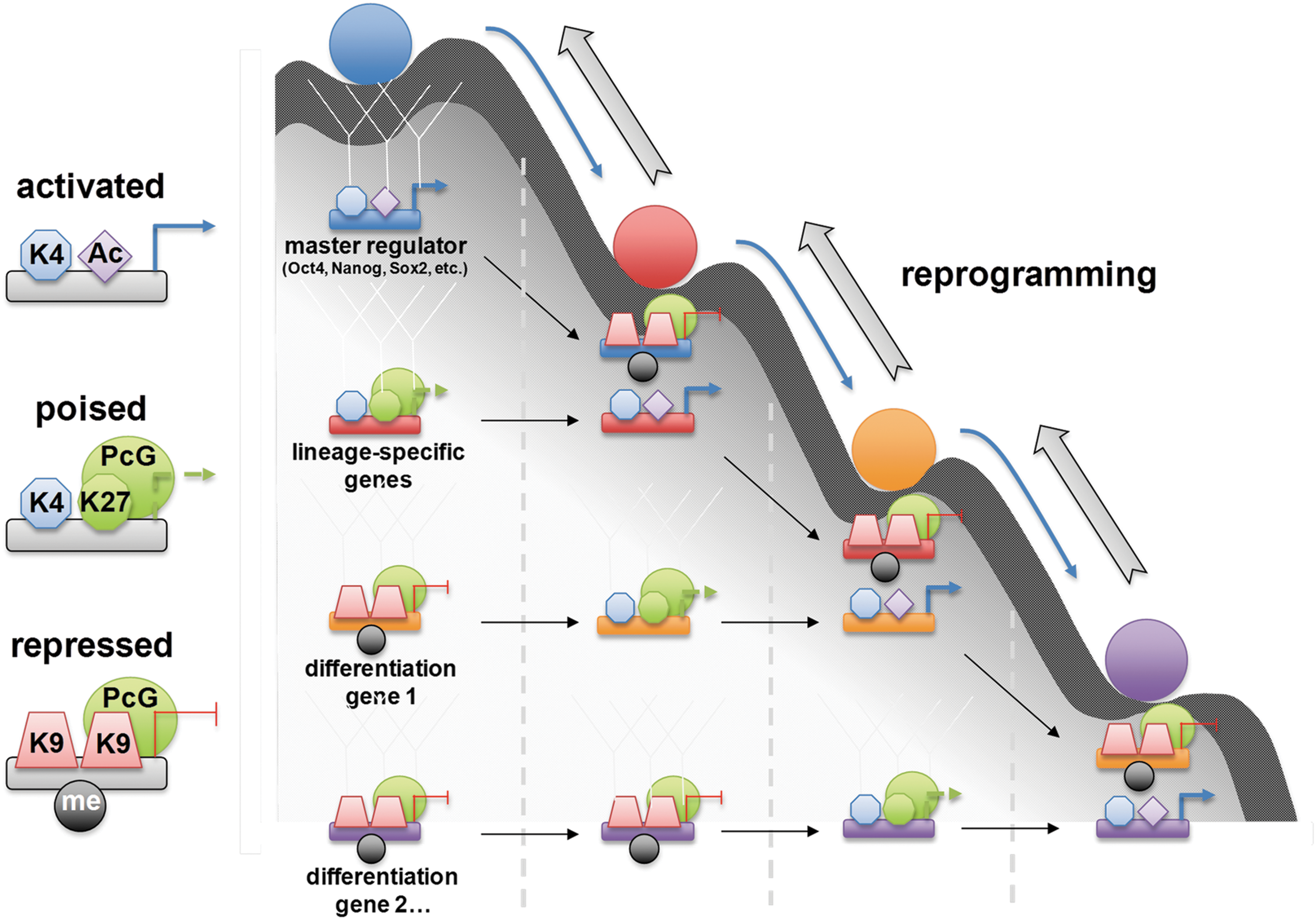
Yamanaka proposed a stochastic model for iPS cell generation. A group of cells are prevented from moving back up the slope toward the state of pluripotency by some epigenetic barrier; once they overcome such barriers, they will achieve the ability to self-renew. The second group goes through partial reprogramming. This will cause them to lose their pluripotency and roll toward a specific lineage. The third group with improper expression of ectopic factors trans-differentiates. The fourth group undergoes apoptosis or death instead of traveling on the slope. Due to large differences in epigenetic status between ES cells and their differentiated progeny, Oct4 and Sox2 cannot find their targets in somatic cells. It has been proposed that c-Myc and Klf4 alter the structure of chromatin, enabling these two core factors access to their targets, thereby increasing the expression of downstream genes (170). c-Myc induces up-regulation of Gcn5 (histone acetyl transferase gene), which is a key player in histone structure, and therefore might improve the accessibility of target genes to Oct4. Klf4 is also acetylated by p300 (acetyl transferase protein) and has the ability to control gene transcription through regulation of histone acetylation (42).
Histone modification during reprogramming
Studies using cDNA microarray and ChIP approaches revealed that Oct4 regulates the expression of over 350 genes in ES cells, including several epigenetic modifiers (106). Two HDMs, Jmjd1a and Jmjd2c, have been identified to be part of the groups of the genes regulated by Oct4 (100), and Jmdj2c is recruited to the Nanog promoter. In this way, ES cell transcription factors regulate the expression of chromatin remodeling genes and, in turn, help unveil chromatin conformation in promoter regions of target genes allowing self-regulation of the epigenetic network. During the reprogramming of fibroblasts into pluripotent cells, genes in an open chromatin state, which is marked by H3K4me3 or H3K4me3 and H3K27me3 (bivalent), are efficiently reactivated, whereas genes that are silenced by H3K27me3 alone remain mostly repressed. In both near promoters and intergenic regions, most of the high CpG promoters are enriched with monovalent H3K4me3 (activation mark) or bivalent H3K4me3 and H3K27me3 (repression mark) in ES cells (Fig. 6). However, MEFs show only H3K27 (115). One group of genes shows the reactivation and associated gain of H3K4me3 at key pluripotency factors. These can be further subdivided into two classes: the first includes genes such as Lin28, Sox2, and Fgf4, which are repressed by H3K27 and lack detectable H3K4me3; the second includes the genes Oct4, Nanog, and Dppa, which are repressed by DNA methylation and also lack detectable H3K4me3. The other major group describes genes, including MyoD, which are repressed by H3K27me3 and highly enriched for developmental transcription factors. To create a truly pluripotent cell line, these loci must remain repressed but need to acquire H3K4me3 to reestablish their bivalency and thus their developmental competence for all germ layers and cell types (115).
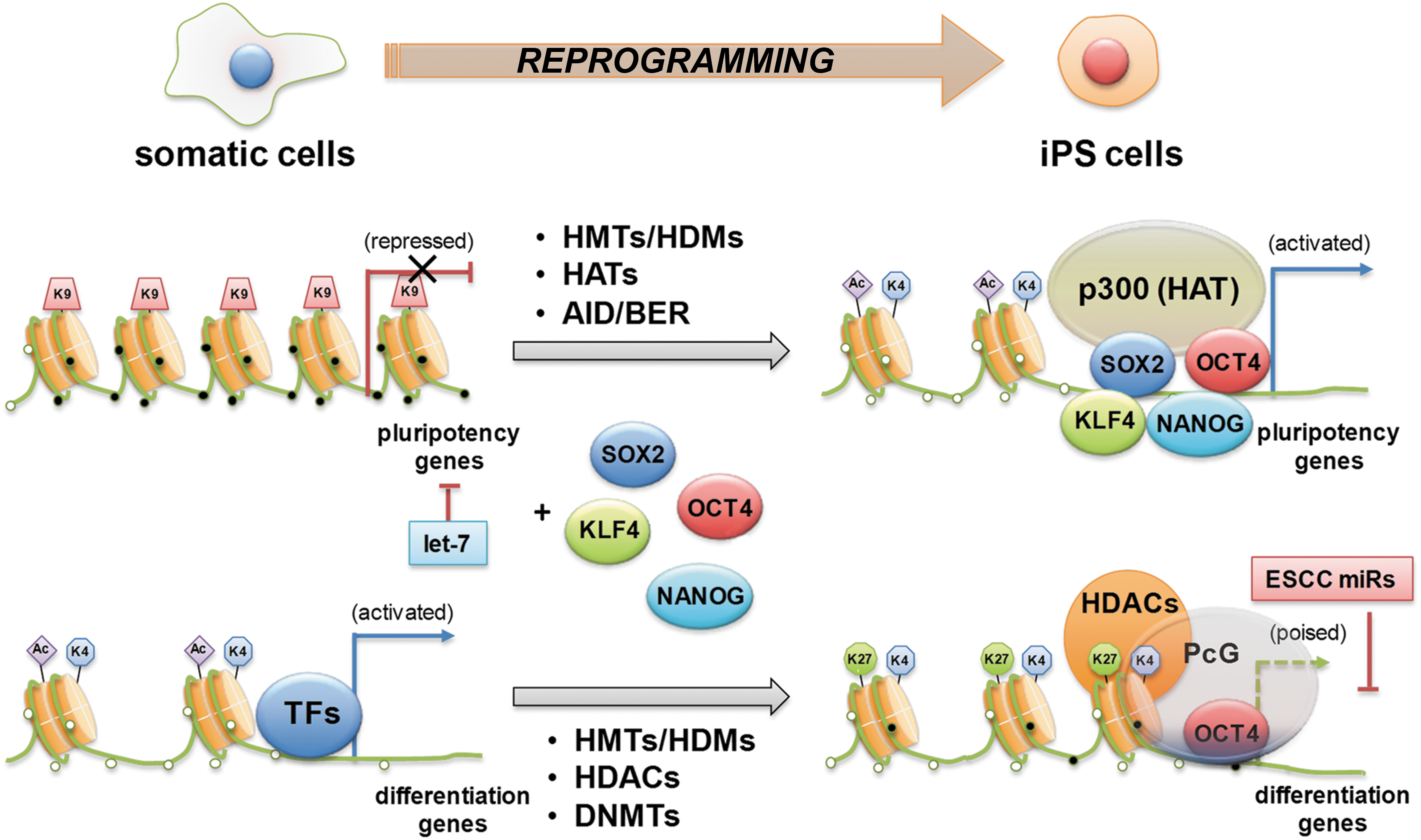
DNA methylation during reprogramming
The efficiency of iPS cell reprogramming is extremely low (0.01–0.1%) (61). Treating differentiated cells with DNA methylation inhibitor 5-Aza-CR or siRNA or shRNA against Dnmt1 to revert repressive epigenetic marks of pluripotency genes can significantly increase the efficiency of iPS generation (115). These data suggest that epigenetic modifications are acquired to overcome the epigenetic repression of one or more key loci during the artificial process of reprogramming. DNA methylation is a critical epigenetic barrier to cellular reprogramming, and the removal of DNA methylation (i.e., demethylation) at the promoter of pluripotency genes is the key mechanism of reprogramming (155) (Fig. 6). The molecular mechanisms of DNA demethylation have been proposed as passive or active processes. Passive DNA demethylation occurs by DNA replication in the absence of methylation of newly synthesized DNA strands when maintenance methylation is impaired (197). Active DNA demethylation depends on demethylating enzymes, which is independent of replication (197). Activation-induced cytidine deaminase (AID, also known as AICDA) is the only enzyme that has been proposed as a DNA demethylase in mammalian reprogramming (13, 131). It is suggested that AID-mediated DNA demethylation occurs due to deamination of methylated cytidine residues in single-stranded DNA, followed by DNA repair (134). A recent study using a cell fusion reprogramming strategy showed that a rapid increase in Oct4 and Nanog expression was accompanied by DNA demethylation at their promoters (13). Moreover, AID knockdown by siRNA decreases the expression of pluripotency genes such as Oct4 and Nanog by reducing DNA demethylation (13). These data support the importance of active DNA demethylation by AID in reprogramming.
Role of miRs in reprogramming
The importance of miRs for ES cells is further indicated by the discovery that specific miRs mimics and inhibitors induce the reprogramming of somatic cells into iPS cells (3, 70, 117). Several miRs, including ES cell-specific cell cycle-regulating (ESCC) miRs (miR-291a-3p, miR-291b-3p, miR-294, miR-295, and miR-302), Myc-induced miRs (miR-200, miR-141, miR-429, and miR-17-92 clusters), miR-92b, and the miR-520 cluster, have been shown to positively regulate the self-renewal and pluripotency of ES cells. Among these, ESCC miRs (miR-291-3p, miR-294, and miR-295) increase the efficiency of reprogramming of fibroblasts into iPS cells induced by Oct4, Sox2, and Klf4 (70). Additionally, the inhibition of tissue-specific miRs, let-7, has been shown to promote reprogramming (111). miR-125, which inhibits the expression of Lin28, is also expected to positively influence reprogramming (190, 192) (Fig. 4). More importantly, ectopic expression of only miRs (miR-302/367 and miR-200c/302/369) without transcription factor over-expression was shown to have sufficient capability for rapid and efficient reprogramming of mouse and human somatic cells into iPS cells (3, 117).
The equivalency of ES cells and iPS cells is controversial, and this discrepancy is possibly mediated by the difference in the epigenetic status between these two types of PS cells (30, 125). During the reprogramming, miRs also have roles in epigenetic modifications that favor reprogramming. For example, the Dlk1–Dio3 gene cluster, which includes ∼50 conserved miRs, is often silenced in iPS cells (98, 161), and the treatment by HDAC inhibitors of iPS cells led to activation of the Dlk1–Dio3 gene miR cluster to enhance the developmental potential to be equivalent to that of ES cells (161). These findings suggest that the appropriate epigenetic status of cells needs to be achieved during the reprogramming, and further reinforce the critical roles of miRs for establishing and maintaining pluripotency. Additionally, inhibition or knock-down of Dnmt enhanced iPS cell generation by inducing the demethylation of promoters of pluripotency-related genes (115). Dnmt inhibitors were also used to generate iPS cells with only two factors (Oct4 and Klf4) or three factors (Sox2, Oct4, and Klf4) (63, 152). The targets of miR-29b, which induces global DNA hypomethylation and re-expression of Ink4b tumor suppressor gene, are Dnmt1, 3a, and 3b (51). Therefore, miR-29b expression and Dnmt inhibitors is expected to enhance reprogramming efficiency by promoting demethylation of promoter regions of pluripotency-related genes. These results indicate a critical role for miRs in reprogramming somatic cells into iPS cells.
Epigenome reprogramming by small molecules
Strong evidence that epigenetic modifications are closely related to the reprogramming of somatic cells is augmentation of reprogramming efficiency by the use of small molecules in generating iPS cells. In somatic cells, reprogramming factors have highly methylated endogenous loci. However, in ES cells and iPS cells, these factors are hypomethylated (65). Their promoters need to be demethylated in order to be reactivated and thereby derive iPS cells. Direct reprogramming steps require downstream epigenetic modifiers because the direct reprogramming factors lack demethylation activity. Differences in epigenetic modifiers such as CpG methylation between ES cells and lineage-specific ES cells emphasize importance of epigenetic landscape in iPS cell generation. Doi et al. (40) found that characterization of CpG methylation can help in distinguishing the identity of cell types such as fibroblasts, ES cells, and iPS cells. Because chromatin remodeling is important for reprogramming, small molecules are being used to overcome these epigenetic blocks and enhance iPS cell generation. Consequently, DNA and HMTs, chemical inhibitors of HDACs, and genetic factors were used in different combinations (63, 153). Ding and colleagues (83) found that a small-molecule inhibitor of G9a HMT, BIX-01294, could enhance the induction of reprogramming in neural stem cells while replacing Oct4. Since G9a is a down-regulator of Oct4 during early development, they suggested that BIX-01294 enhances iPS cell formation by inhibiting G9a and subsequently releasing Oct4 from negative regulation. iPS cells were generated from MEFs with only two factors, Oct4 and Klf4, in the presence of BIX-01294.
In a four-factor system, VPA increases the efficiency and kinetics of reprogramming by a hundred fold. VPA can chemically induce iPS cell generation with only Oct4 and Sox2 from human fibroblasts (63) and it could replace either Klf4 or c-Myc in reprogramming steps. These results indicate that DNA methylation, histone methylation, and histone deacetylation contribute to epigenetic hurdles which limit effective iPS cell generation.
Conclusion
Recent advances in unbiased next-generation sequencing methods for whole epigenome sequencing allowed mapping of epigenetic marks at single-nucleotide resolution and led to novel insights into the role of epigenetic changes in transcriptional regulation and cell fate determination. In particular, epigenome sequencing of ES and iPS cells shed light on the mechanisms governing maintenance and induction of pluripotency. In addition, other advances have been made in regard to the identification of PS cell-related miRs, their role in reprogramming and maintenance of PS cells, and their interaction with other epigenetic modulators.
However, much more work needs to be done to fully understand the role of epigenetic modifications in PS cells. Particularly, the dynamic changes of chromatin state in responses to the ectopic expression of transcription factors during the generation of iPS cells should be comprehensively investigated to develop more efficient methods of reprogramming. New approaches for defining the epigenetic landscape are needed for a better understanding of the chromatin structure and its role in establishment of cell identity. A comprehensive epigenomic map will also be helpful to determine the quality and utility of directly reprogrammed or in vitro differentiated cells. Epigenome reference maps and data by the enormous increase in sequencing capabilities will significantly facilitate our understanding of the epigenetic landscape of PS cells and their dynamic changes during reprogramming and differentiation.
Footnotes
Acknowledgments
This work was supported in part by NIH grants DP3DK094346, RC1GM092035, and NIH contract, HHSN268201000043C (Program of Excellence in Nanotechnology Award); NSF-EBICS (Emergent Behaviors of Integrated Cellular Systems) grant; and Stem Cell Research Center of the 21st Century Frontier Research Program grant SC4300, funded by the Ministry of Science and Technology, Republic of Korea. We would like to thank Andrea Wecker and Julie J. Kim for editing of the article and Juhyun Kim for assisting in preparation of figures.