
Abstract
Introduction
Many obesity-related disorders have also been linked with the metabolic syndrome, including chronic kidney disease (CKD), fatty liver disease, polycystic ovary syndrome, obstructive sleep apnea, and hyperuricemia (6). Infact, patients with metabolic syndrome are at a high risk for renal manifestations, microalbuminuria, and increased glomerular filtration rate. Studies emphasize that metabolic syndrome enhances the probability of coronary heart disease (CHD) and diabetes (12). Metabolic syndrome has been found to be associated with a higher risk of heart failure (184). Metabolic syndromes such as CVD, CKD, and CHD have become global health concerns because of their high occurrence and significant effect on mortality and morbidity (130). There are multifactorial metabolic toxicities generating excess of reactive oxygen species (ROS) linked with metabolic syndrome such as T2D and accelerated atherosclerosis. Individuals with metabolic syndrome show significantly increased mortality from CAD, even though they lack diabetes (85).
With a drastic increase in the number of individuals with metabolic syndromes, nutritional genomics/nutrigenomics has evolved that deals with understanding the role of nutrients on metabolic pathways and homeostatic control of the organism (5). Nutrigenomics has uncovered how the genetic makeup of an individual alters the predisposition to metabolic syndromes, by understanding the link between epigenetic modifications with disease occurrence and progression. Some important epigenetic marks, such as methylation and acetylation, are etched on the promoters and alter gene expression. These modifications, both on the cytosine residues of DNA and on the histone tails, are reported to be altered by some of the components of the nutrients. Macronutrients such as carbohydrates, proteins, and fats along with micronutrients such as vitamins and minerals alter the repressor and/or activator complexes on the promoters, modulating the gene expression. The nutrient intake and dietary habits of an individual's grandparents influence the predisposition of that individual to different diseases and, thus, metabolic syndrome (200).
The present article mainly focuses on understanding the disease state of an individual at the level of nutrient intake, genetic profile, epigenetic alternations, and possible therapeutic interventions that can alter the regulatory effect of the nutrients on disease progression.
Metabolic Syndrome
In recent times, there is an alarming increase in the number of people having metabolic syndrome, and the prevalence varies according to the geographic location, race, gender, and urbanization. Occurrence of metabolic syndrome has been found to be age dependent (279). A follow-up study in middle-aged men and women suggests much higher percentage incidences of CVD and T2D (362). The current section will highlight the individual components and their contribution in disease manifestation (Fig. 1).

Insulin resistance
The most accepted and generalized feature that depicts the pathophysiology of the metabolic syndrome is IR. IR mainly describes the cluster of abnormalities such as glucose intolerance, endothelial dysfunction, enhanced inflammatory markers, and disturbed uric acid metabolism (358). IR is traditionally defined as a phenomenon wherein a normal or increased insulin level causes an attenuated biological response (54). Classically, it can be illustrated as a state of impaired sensitivity to insulin-mediated glucose utilization due to a defect in either insulin secretion by the pancreatic β cells or function that ensues in elevated levels of fasting glucose (1, 65, 178). Insulin plays a key role in maintaining normal blood glucose levels by facilitating cellular glucose uptake and regulating carbohydrate, lipid, and protein metabolism. The effect of insulin, insulin deficiency, and IR differ on the basis of the physiological function of the tissues and organs involved and their requirement for insulin in metabolic processes. Physiological interplay between insulin and many other hormones such as glucagon, glucocorticoids, nitric oxide (NO), and catecholamines influences the activity of insulin. Excessive secretion of such hormones leads to emergence of IR; albeit, in most of the cases, IR is supposed to be manifested via some postreceptor abnormalities in insulin signaling (358).
Insulin mediates its function via binding to insulin receptor, followed by tyrosine/serine phosphorylation of intracellular substrate proteins known as insulin responsive substrates (IRS) (358). Experiments in murine models have conclusively proved that the increased tyrosine or serine phosphorylation of the IRS1 due to hyperactivation of c-Jun N-terminal kinase (JNK) and endoplasmic reticulum X-box binding protein-1 causes IR (239). The prime contributor during the development of IR is overabundance of circulating free fatty acids that are usually generated from triglyceride stores of adipose tissue through the lipolysis which is mediated by hormone-sensitive lipoprotein lipase (LPL) (90). In the case of IR progression, increased accumulation of fat in adipocytes causes monocyte infiltration, releasing proinflammatory cytokines that further inhibit the antilipolytic activity of insulin. All these events create additional lipolysis and eventually lead to impaired insulin sensitivity. Proliferator-activated receptor gamma (PPARγ) is a nuclear hormone receptor that targets many genes involved in inflammatory processes and insulin sensitivity (160, 267, 281, 310). Increased PPARγ activity is linked with lipid-induced activation of genes, affecting fatty acid uptake, β-oxidation, and transport of fatty acid into peroxisomes.
Studies by Shuldiner and McLenithan (297) have reported that with age, many individuals show reduced expression of PPARγ coactivator-1α and coactivator-1β that contribute to the change in whole-body glucose and fatty acid oxidation with a subsequent occurrence of diabetes. Mice having reduced or negligible expression of the fatty acid-binding proteins such as aP2 and mal1 exhibit dramatic changes in phenotype with substantial protection from IR, diet-induced obesity, T2D, and fatty liver disease (203). Some of the transcription factors are also implicated in T2D etiology, such as hepatocyte nuclear factor 4 alpha plays an important role in controlling the expression of genes that regulate glucose transport and metabolism (308). Another critical cellular mechanism that modulates IR is oxidative stress. Oxidative stress induces upregulation of promoter activity of a purine pyrimidine base excision repair enzyme, endonuclease VIII like-1 (Neil1), that is helpful in the repair of oxidatively damaged bases in mammalian cells (78). Knockdown of Neil1 is associated with the increase in occurrence of hepatic steatosis and hyperinsulinemia (335). Thus, the interplay between various hormones, signaling cascades, signal transducers, and certain transcription factors influence IR underlying the significance to study at the molecular level.
Obesity and dyslipidemia
A majority of the world's population is suffering from obesity due to an imbalanced proportion of body fat. It is the sixth most important risk factor for contributing to the overall burden of disease worldwide (139, 172). Obesity is considered a prime contributing factor for the enhanced rate of IR, T2D, glucose intolerance, hypertension, CHD, dyslipidemia, and cancer (33, 304). Obesity occurs when the unused triglycerides are stored in the adipose tissues and released as free fatty acids, causing detrimental effects (237). Usually, an increase in the waist circumference has been routinely predicted as a marker of obesity, which results due to increased nonesterified fatty acid flux and fat deposits (224, 328). Exposure to excessive levels of glucocorticoids is the major cause of visceral obesity that is regulated by an enzyme 11β hydroxysteroid dehyrogenase type 1 (11β HSD-1). Transgenic mice overexpressing 11β HSD-1 show increased incidence of central obesity, hypertension, impaired glucose tolerance, hypertriglyceridemia, and elevated intra-adipose corticosterone levels (214). Mice homozygous for targeted deletion of 11β HSD-1 show anti-diabetic phenotype and are resistant to the development of metabolic syndrome (175). The major breakthrough in this field was to identify the function of adipocytes-derived humoral factor, such as leptin. Patients with obesity show increased serum levels of leptin (147). Studies by Yamada et al. (369) demonstrated that very limited expression of uncoupling protein 1 (UCP1) in epididymal fat markedly improved insulin and leptin resistance, thereby ameliorating glucose tolerance and decreasing food intake in mice with obesity and diabetes (369). The genetic basis of visceral obesity has been predicted to be the effect of a single gene mutation or defect in PPAR-1, lamin A/C, 1-acylglycerol-3-phosphate, O-acyltransferase, scipin, beta-2 adrenergic receptor, and adiponectin (74, 113, 143). Thus, obesity and metabolic syndrome are found to be associated with long-term parenteral nutrition administration due to the altered expression of PPAR and adipocytokines (318).
Along with obesity, dyslipidemia is another state of imbalance in lipid metabolism that is marked by either lipoprotein overproduction or deficiency. Dyslipidemia marks the increased levels of circulating proantherogenic low-density lipoprotein (LDL) cholesterol and diminished levels of antiantherogenic HDL. HDL is the key lipoprotein involved in reverse cholesterol transport between peripheral tissues and liver; therefore, alteration in HDL metabolism subsequently leads to a high risk for CHD. HDL displays high cholesterol efflux capacity and provides protection from LDL-mediated oxidative stress. Reduction in the levels of HDL-apoAI complexes due to decreased apoAI synthesis and/or substitution of apoAI by serum amyloid A (an acute-phase reactant and proinflammatory molecule) enhances the HDL catabolism (170). Such perturbation in apoAI synthesis manifests the increased clearance of HDL from circulation (40, 58). In addition to HDL, LDL also undergoes conformational changes via depletion of both esterified and nonesterified cholesterol (135). To this extent, small dense LDL is shown to be toxic to the endothelium, as it permeates the blood vessel walls, gets oxidized by binding to scavenger receptors on macrophages, thus further promoting atherosclerosis (240). Recent studies suggest that defective expression of LPL gene is considered the major contributing factor for the development of dyslipidemia, IR, and atherosclerosis (126, 152); whereas in diabetic patients, mutations in the LPL gene further increase the risk for CVD (222). Thus, obesity in conjunction with dyslipidemia predisposes the individual to elevated metabolic syndrome.
Hypertension
Hypertension or chronically elevated systolic and/or diastolic blood pressure is a common age-related disorder that inclines the individual for the coronary artery calcification (CAC) and CVDs such as stroke, ischemia, and heart failure (97, 141, 268). CAC has been linked with cardiac events for atherosclerosis and increases the likelihood for inducible myocardial ischemia (365). One of the significant markers for CVD is microalbuminuria, which is highly associated with increased blood pressure moving stroke incidence in diabetic patients (119). CVD represents diverse disorders that include myocardial infarction and hypertension-induced cardiac hypertrophy (197). Cardiac hypertrophy, in some cases, may occur due to normal physiological stimuli leading to congestive heart failure (37). Hypertension is predominant in individuals with obesity and diabetes (101). Insulin-resistant individuals lack the vasodilatory effect of insulin due to enhanced synthesis of NO by endothelial cells and increased fat deposition favors vasoconstriction, further worsening the disease manifestation (321). It is documented that the intrauterine environment of the mother during pregnancy has been found to influence blood pressure during the adult life of the offspring (18).
In this regard, the spontaneous hypertensive rat (SHR) is a commonly examined model to study and analyze hypertension, as it exhibits IR and other metabolic abnormalities that are common in humans. In some of the SHR strains, loss of function mutation in CD36 gene that encodes fatty acid translocase leads to altered cellular fatty acid utilization (8). CD36 deficiency also displays enhanced cardiac accumulation of glucose due to uncoupling of glucose oxidation from its cellular entry and is accompanied by enhanced O-glycosylation of proteins (O-glyNacylation) through the glucosamine-synthetic pathway. CD36-deficient patients mainly possess hyperlipidemia, increased remnant lipoproteins, hypertension, and impaired glucose metabolism that lead to a high risk for CHD development (370). Hypertension and many other metabolic syndrome factors are crucial in the ischemic stroke epidemiology depending on varied sex and race (156). The increased risk of stroke has been found more among women than in men, and it can be justified by the greater impact of metabolic disorders in postmenopausal women. Each factor of metabolic syndrome affects the stroke incidence to various degrees. In a population study of middle-aged Japanese men and women, the metabolic syndrome was identified to be the major determinant for ischemic CVD (155). This effect is also dependent on other confounding factors such as physical activity, smoking, and alcohol consumption (32). The presence of metabolic syndrome in individuals with pre-existing atherosclerosis renders the higher risk for ischemic stroke or transient ischemic attack (TIA) (173). TIA can be used as an indicator of stroke and provides a window of opportunity for clinical intervention (7). IFG and hypertension are the most critical components having the strongest relation with ischemic stroke or TIA incidence (176, 317, 337).
Other than hypertension, abdominal obesity significantly affects the risk for ischemic stroke. It is observed that the loss of weight significantly reduces the prevalence of stroke (199). The impact of diabetes on the occurrence of stroke has been extensively studied which reveals that among all the components of metabolic syndrome, diabetes confer the highest risk for stroke (103, 168, 176). In diabetic patients, high blood pressure increases the risk for stroke, and risk is further elevated in synergy with IR (4, 38). Dietary habits have also been linked with the stroke incidence in adults (186). The frequency of stroke can be effectively controlled by altering the dietary regimes and, thus, modulating the metabolic syndrome.
Metabolic Syndrome and Oxidative Stress
Oxidative stress can be defined as a disequilibrium between free radical production and antioxidant defense mechanism of an individual that leads to the accumulation of oxidative products. ROS are highly reactive and short-lived derivatives of oxygen metabolism that are generated on incomplete reduction of oxygen. ROS mainly include superoxide anion (O2 ‾), hydroxyl radical (OH∙), hydrogen peroxide (H2O2), and some reactive nitrogen species such as NO and peroxynitrite radical (ONOO∙). ROS are integral players in the routine physiological mechanisms, and loss of reduction-oxidation (redox) homeostasis consequently leads to impairment in insulin metabolic signaling, reduced endothelial-mediated vasorelaxation, and associated cardiovascular functional abnormalities (356).
Antioxidative defense system has been evolved to help in the maintenance of oxidative homeostasis and on the disruption of this homeostasis, the oxidative stress ensues (329). Antioxidants provide protection from the vascular oxidative stress by neutralizing free radicals and, thus, exert beneficial effects on vascular function. The primary antioxidants enzymes are superoxide dismutases, catalase, and glutathione peroxidase. Other antioxidants include vitamin C, vitamin E, β carotene, and reduced glutathione. HDL possesses potent antioxidant capacity, and elevated levels of oxidative stress are intimately related with impaired antioxidative potential of HDL in insulin-resistant phenotype (138). Oxidative stress is an early event in the pathology of many chronic diseases rather than being a consequence as shown by the presence of elevated oxidative damage and reduced antioxidant protection in patients with metabolic syndrome (104). Oxidative stress causes a complex dysregulation of cell metabolism and is the final common pathway through which all the risk factors involved in chronic diseases exert their deleterious effects (253). In the case of metabolic syndrome, the oxidative stress gets amplified by a concomitant deficiency of antioxidants, leading to the pathophysiology of many metabolic disorders and is closely related with the pathogenesis of vascular alterations associated with metabolic syndrome (55). Patients with metabolic syndrome exhibit activation of biochemical pathways that causes increased generation of ROS and enhanced lipid peroxidation. Enhanced levels of oxidative stress markers such as 8-iso prostaglandin F2α, nitrotyrosine, oxidized LDL, myeloperoxidase, and erythrocyte glutathione peroxidase strongly predict an early risk for CVD and myocardial infarction (30, 39, 151, 245, 296, 355). The increased levels of 8-iso PGF2α in patients with hypertension and CAD are associated with more severity in these disorders, as the activity of superoxide dismutase is reduced in hypertension (278, 336). Increased oxidative stress in accumulated fat is an early instigator of metabolic syndrome and is accompanied by augmented expression of NADPH oxidase and decreased expression of antioxidative enzymes (114).
Total body weight and waist circumference shows positive association with oxidative stress-mediated endothelial dysfunction (250) and, thus, contributes to obesity mediated by the oxidative stress (144). Individuals with elevated oxidized LDL are at a higher risk of developing metabolic syndrome such as abdominal obesity, hyperglycemia, and hypertriglyceridemia (150). Even in diabetic as well as in dyslipidemic patients, oxidative stress is the underlying contributing factor for many pathogenic mechanisms (36, 352), impairing glucose uptake in muscle and adipocytes (204, 276), thus causing a decrease in insulin secretion from pancreatic β cells (215). Oxidative stress plays a crucial role in the pathogenesis of late diabetic complications and has also been discovered to be responsible for the IR development (154, 252). Oxidative stress shows close association with adiposity and IR (327). Hyperhomocysteinemia is also linked with oxidative stress in liver steatosis and impaired NO availability (3). Maintenance of the antioxidant potential of a cell primarily happens due to dietary intake and/or de novo synthesis. Diet constituents can efficiently modulate redox reactions and the extent of oxidative stress by governing the expression of many nuclear genes. Calorie restriction (CR) improves the cellular redox balance by controlling the generation of mitochondrial ROS (264). The evaluation of biomarkers for oxidative stress may further help in the identification and treatment of individuals with a future risk of diabetes and CVD.
Epigenetics in Metabolic Syndrome
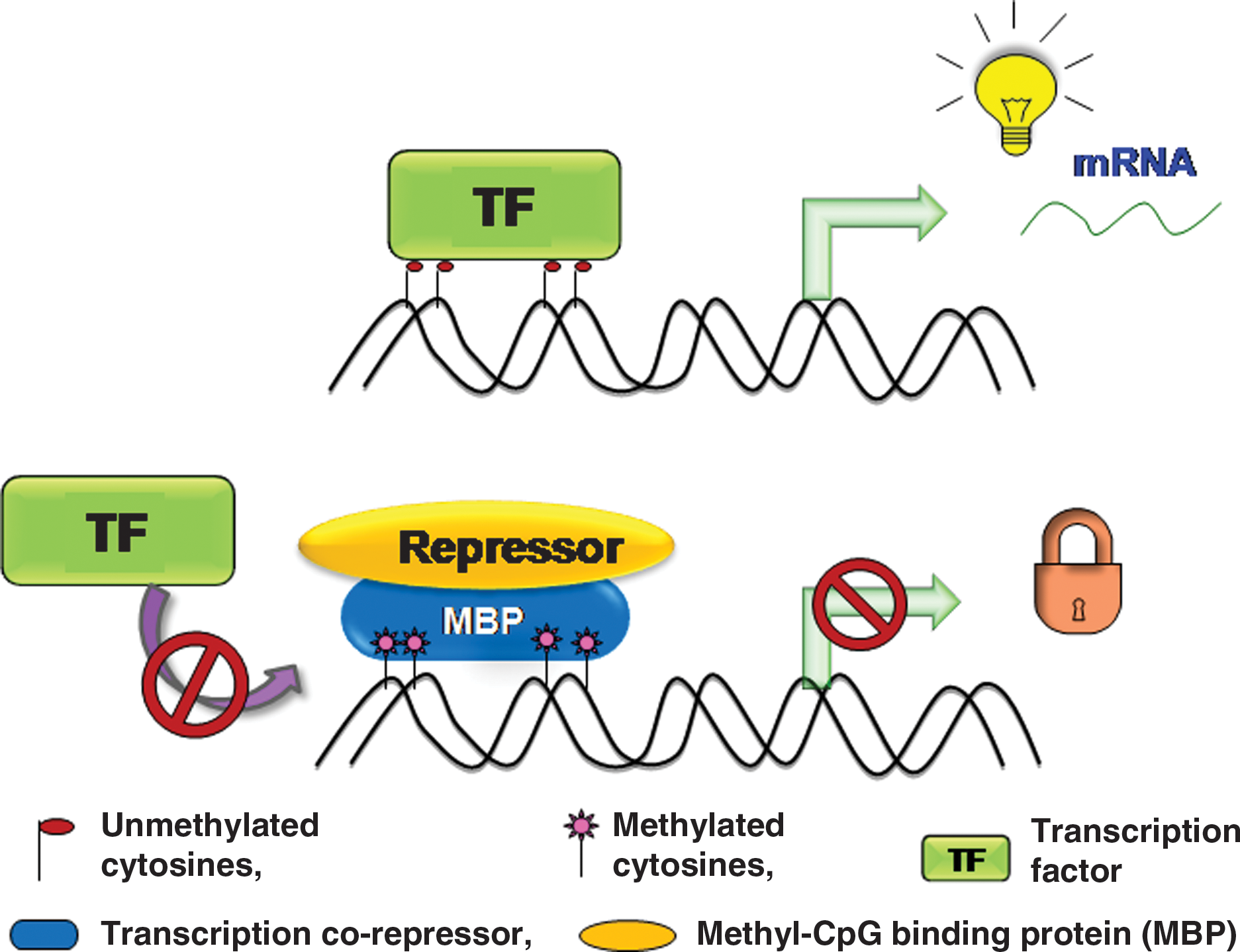
In 1942, Conrad Waddington for the first time coined the term “epigenetics,” which is a branch of biology that includes studying the causal interaction between genes and their products (341). Further refinement of this term was done by Nanney in 1958, describing epigenetics as a mechanism of cellular heredity that is independent of template-replicating mechanisms (230). Therefore, epigenetics is the collective cause of heritable changes in genome function that occur independent of any change in DNA sequence. Epigenetic regulation mainly controls the gene expression and also manipulates condensation and segregation of chromosomes during the cell cycle events. Epigenetic mechanisms are largely responsible for switching the genes “ON” and “OFF” and, thus, producing permanent changes in cell fate. Epigenetic modifications primarily include methylation of cytosine residues of the DNA as well as on the N-terminal tails of core histones that are substrates for many other posttranslational modifications such as acetylation, biotinylation, phosphorylation, ubiquitination, SUMOylation, and ADP ribosylation (142, 162, 181) (Fig. 2). Along with genetic influence, the epigenetic processes are also affected by environmental factors during specific periods of life in mature organisms, causing long-term changes in functional outcome (157). Epigenetic changes in the genome are highly dynamic and reversible depending on the inducible factors, while genetic changes are mostly irreversible. Such dynamic chromatin remodeling processes are required to facilitate the transcription of many genes that are packed into chromatin structures and also modulate the functional output of the genome by integrating environmental signals. DNA methylation and histone modifications represent two of the extensively studied and most crucial chromatin remodeling mechanisms. DNA methylation occurs via covalent modification of cytosine residues by addition of a methyl group to a 5′ carbon of the cytosine ring present within cytosine-phosphate-guanine (CpG) dinucleotides. Reports show that 90%–98% of CpG dinucleotide sites in the mammalian genome are methylated (149, 311). Methylation is one such repressive posttranslational modification that renders any gene silent in its expression, and demethylation reverses the effect of gene inactivation, making the gene functional again (Fig. 3). DNA methylation is one of the important epigenetic modifications that takes place in utero and during postnatal development, the phenomenon known as “fetal programming.”
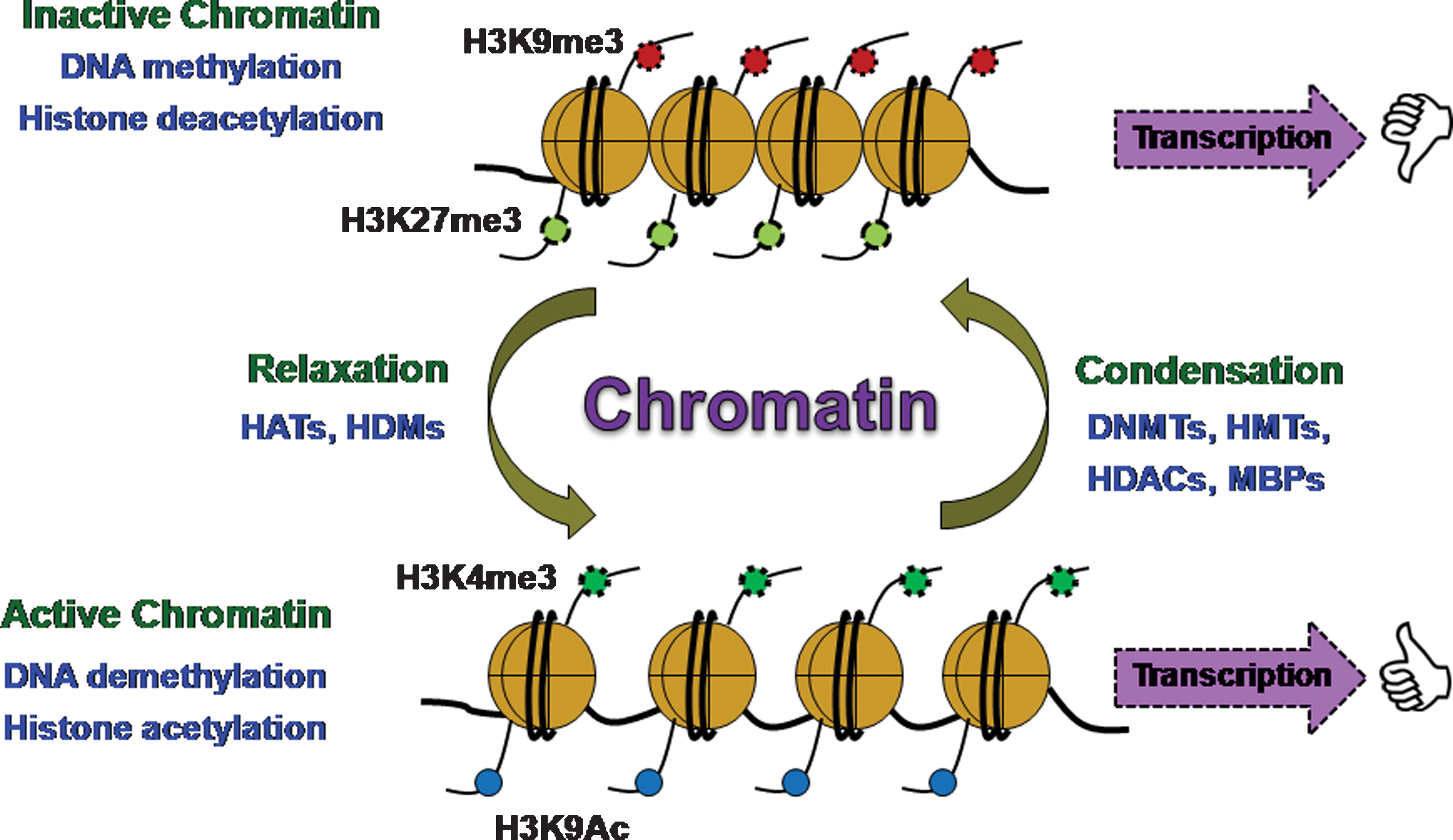
It has been reported that methylation patterns can alter with aging (105). Many genes have been found to show differential methylation patterns during the lifetime of an organism (Table 1). As an example, analysis of the state of methylation of apolipoprotein gene cluster apoA-I, apoC-III, apoA-IV in mice embryo shows differential methylation (223, 290). Extensive demethylation at the very early stage of the embryo, that is, morula stage, keeps this cluster actively expressed, but it is followed by de novo methylation immediately at the pregastrula stage. This phenomenon of differential methylation status clearly suggests that demethylation takes place before organogenesis. Subsequently, de novo methylation occurs at 5′ ends of gene promoters in a tissue- and age-specific manner. The 5′ end of the apoA-I gene undergoes de novo methylation in the kidney but not in the liver. The other two genes of this cluster, that is, apoC-III and apoA-IV, also have differential tissue specific methylation pattern, and the embryo-specific expression usually precedes the methylation in the adult which correlates with their activity (291). The apoA-I/C-III/AIV cluster mostly remains methylated in the tissues with less expression of lipoprotein genes, while some portion remains unmethylated in tissues where the expression is high as exemplified in liver. The tissue-specific methylation event in the later part of the life is the interplay between demethylation and de novo methylation events in the embryo. Differentiation of embryonic stem cells into various lineages is marked by the epigenetic processes that selectively activate a subset of tissue-specific genes. Such tissue-specific gene expression patterns allow the cells of identical genotypes to transmit a specific phenotype independent of either the presence or absence of stimulus (53). This phenomenon is termed as “cellular memory,” due to its function as a memory bank to maintain the genome function for cell identity after its differentiation.
FTO, fat mass and obesity associated; GR, glucocorticoid receptor; 11β HSD-2, 11β-hydroxysteroid dehydrogenase-2; PPARα, peroxisomal proliferator-activated receptor-alpha; PR, protein-restricted; UN, under nutrition.
There are a number of diseases that are reported to be associated with aberrant epigenetic modulation. The association between abnormal DNA methylation events in several cancers was first shown by Feinberg and Vogelstein (99). The specific and unique epigenetic modifications of some genes are involved in the onset of various disorders of metabolic syndrome. The role of epigenetics and their pathophysiological means during the course of metabolic syndrome are not fully characterized. Interplay between environment and genetic determinants that govern the metabolic syndrome should also be taken into consideration. Several studies emphasize that the occurrence of aberrant epigenetic programming in patients with metabolic syndrome occur either due to imbalanced nutrition at neonatal state or due to disturbed adult life metabolism (243). A recent study on a large cohort of population identified a link between metabolic syndrome incidence and geographical distribution of a population. This is attributed to the interplay between the expression of genes and the habitat of the individual, which is termed as “developmental plasticity.” Such plasticity is credited for fine tuning of the gene expression that ultimately leads to a phenotype which is most suitable for the environment (125). The Developmental Origin of Health and Disease (DOHaD) phenomenon is a subset of developmental plasticity and is underpinned by epigenetic mechanisms (124).
Waddington (342) for the first time established that the process of phenotypic induction through developmental plasticity induces permanent change in a range of organs via epigenetic mechanisms in Drosophila melanogaster; for instance, altered wing vein pattern on heat shock was observed. This phenotypic change was stably integrated and showed inheritance in the subsequent generations independent of further stimulus. Environmental factors modulate the early development, influence the health of the individual, and critically affect disease occurrence (367). These nongenomic factors tune the phenotype and have high adaptive importance. Any mismatch between an individual's phenotype and the surrounding environment triggers an abnormal state of high disease risk and, thus, the higher the mismatch, the greater is the risk. An impaired early environment produces a class of effects that alter cardiovascular and metabolic homeostasis, growth and body composition, reproductive function and longevity (123). Thus, both the genome and the epigenome interactively influence the mature phenotype of an individual, playing a critical role in determining the sensitivity to environmental factors in a later stage of life and, thus, the subsequent risk of disease.
Therefore, understanding of epigenetic mechanisms can be used as a tool to determine the pathophysiology of metabolic syndromes, providing potential strategies for timely diagnosis, prevention, and therapeutic intervention. The following sections will describe the alterations in both gene expression and epigenetic marks that play a crucial role in the occurrence and progression of disease (Table 2).
HMTs, histone methyl transferase; Dot1, disruptor of telomeric silencing; SRF, serum response factor; HDAC1, histone deacetylase.
Type 2 diabetes
T2D is a multistep disorder with strong genetic and environmental influences. The pancreatic β cells are the exclusive source for the synthesis and secretion of the insulin hormone that helps in controlling the blood glucose level. Several transcription factors have been identified that are responsible for controlling insulin production, mainly by governing the transcription of gene encoding insulin. Histone modifiers contribute to the activation or inactivation of transcriptional machinery by catalyzing the relaxation or compaction of chromatin. Covalent histone modifications play a crucial role in insulin gene expression as observed in the pancreatic β cells wherein the proximal insulin promoter is hyperacetylated at histone H3 by histone acetyltransferase (HAT) p300 (57). In addition to hyperacetylation, the proximal insulin promoter is hypermethylated at H3K4 in a β-cell specific manner. However, during unfavorable conditions, these epigenetic variations might rule out the transcription factor-mediated activation of the insulin gene.
Intrauterine growth supplementation critically controls the epigenetic mechanisms during the development of the fetus and can alter gene expression (300). Intrauterine growth retardation induces epigenetic modification of the genes involved in β cells development. These changes include hypomethylation of certain genes such as glucocorticoid receptor (GR) and PPARγ in the liver of offspring. Studies performed by Scarpulla (280) have established PPARγ coactivator-1α (PGC-1α encoded by PPARGC1A) as a master transcriptional coactivator of mitochondrial genes, and its expression is further regulated by environmental signals. Reduced expression of PGC-1α leads to impaired function of mitochondrial respiratory apparatus and subsequently lowering ATP synthesis. Reduced levels of ATP causes decreased insulin production in pancreatic β cells. Both genetic and epigenetic factors determine PPARGC1A mRNA expression in pancreatic islets of human patients suffering from T2D. Reduced PPARGC1A mRNA expression is due to increase in DNA methylation in diabetic islets compared with nondiabetic islets (193). Such crucial epigenetic modification of insulin promoter leads to the high occurrence of diabetes. Furthermore, in the PPARGC1A gene, a common polymorphism at Gly482 Ser has been found to be responsible for increased risk of T2D in an age-dependent manner (22, 92, 194). PGC1-α gene undergoes several polymorphisms, some of which are located in transcription factor binding sites and critically affect insulin response to glucose (235). A number of animal studies have revealed that during pregnancy, both maternal high fat nutrition and lactation induce DNA methylation on the PPARGC1A gene in the liver of offspring. Such epigenetic changes in the early postnatal life increase the susceptibility to metabolic disorders such as T2D and CVD in the later part of life (89).
Decreased expressions of many genes that regulate oxidative phosphorylation in skeletal muscles have been linked with IR and T2D. For example, expression of NDUFB6, a component of the respiratory gene, is decreased in diabetic patients, and the degree of DNA methylation is negatively correlated with muscle NDUFB6 expression (195). A single nucleotide polymorphism (SNP) in the promoter of NDUFB6 generates a CpG site that undergoes methylation in insulin-sensitive individuals. The DNA binding protein Pax2 regulates epigenetic modifications, wherein it maintains the chromatin in an active state by promoting the assembly of H3K4 methyltransferase and, thus, transactivation of glucagon promoter (244). Multiple lines of evidence prove the role of myeloid differentiation factor 88 in the progression of high-fat-diet (HFD) induced T2D and obesity as its deficiency increases the risk for diabetes in mice (153). Offspring of HFD-fed dams possess higher blood glucose levels, higher probabilities of glucose intolerance in adulthood due to alteration in the gluconeogenic capacity through epigenetic modification (309), lipid accumulation in the liver, and development of nonalcoholic fatty liver disease due to increased activation of certain kinases such as JNKs and inhibitor of nuclear factor kappa B kinase (17).
Another mode of epigenetic regulation of metabolic syndromes is at the posttranscriptional level that is mediated by microRNAs (miRNAs). miRNAs are short noncoding RNAs, ∼22 nucleotide long, which have been found to play a pivotal role in growth, differentiation, and organ functions. Thus, miRNAs serve as an additional layer of regulation of gene expression (14, 23, 376). A large fraction of all protein coding genes (∼30%) are targets of miRNAs and, therefore, it plays a critical role in normal and diseased states (169, 202, 319, 333, 373). Several reports indicate the crucial role of miRNAs in insulin synthesis as well as its secretion from pancreatic β cells (71, 159, 260). Impaired function of Dicer in pancreatic progenitor cells significantly affects β-cell development (202). Among these, miR-124a is highly expressed in pancreatic β cells and negatively controls insulin exocytosis (177, 198). miR-124a shares its function with miR-375 during the development of the pancreas (21, 169, 259, 260). miR-375 is one of the novel miRNAs that affects insulin synthesis and is highly expressed in pancreatic islets of humans and mice (258). miR-375 is essentially required for normal glucose homeostasis. miR-375 knockout mice are hyperglycaemic and exhibit increased gluconeogenesis.
Another miRNA, miR-192, has been found to play a key role in diabetic nephropathy that is a prime cause of kidney failure in patients with T2D. Recently, the role of miR-192 in diabetic complications was deciphered. miR-192 has been found to be elevated in glomeruli isolated from streptozotocin-injected diabetic mice as well as control diabetic mice (db/db) in comparison to nondiabetic mice (166). miR-192 is a regulator of TGFβ function and as well modulates the expression levels of extracellular matrix proteins collagen 1α-1 and -2 (Col 1a1 and -2). Glucose, being an important signal for β cells, also regulates the expression of many miRNAs such as miR-30d (316). Expression of miR-30d gets up-regulated on stimulation of high glucose, while its inhibition leads to downregulation of insulin gene transcription. miRNA miR-143 has been implicated in maturation of human adipocytes by controlling the expression of genes such as GLUT4, HSL, fatty acid-binding protein Ap2, and PPAR-γ2, which play a crucial role in adipocytes differentiation (96). These facts signify the importance of miRNAs, the dysregulation of which leads to various disorders that contribute to metabolic syndrome.
Obesity
Abdominal obesity is one of the metabolic disorders that is critically controlled by epigenetic events taking place at the target genes involved in fat metabolism. Epigenetic changes caused during the early life also increase the risk for obesity (146). Lack of methyl donors in the maternal diet or consumption of HFD during pregnancy may perturb the normal methylation pattern in the embryo, as is evident in the hypomethylation of melanocortin 4 receptor gene, POMC, and leptin genes (221, 254, 359). Reports suggest that in some cases impaired genomic repair processes leads to obesity and metabolic syndrome (228, 335). Previous research indicate the potential contribution of many genes such as fat mass and obesity associated (FTO) and nicotinamide phophoribosyltransferase in the development of obesity and related metabolic traits (107, 227). FTO encodes 2-oxoglutarate-dependent nucleic acid demethylase that links the nucleic acid methylation status to increased fat mass (118).
Loss of function in the FTO gene leads to a decrease in body weight in mice, and its overexpression causes an increased deposition of fat (68). This strongly suggests the relationship between obesity and epigenetics. FTO gene variants are associated with the predisposition of an individual to childhood/adult obesity, diabetes, and other metabolic syndrome components through their effect on body mass index (86, 284, 345, 346). Genetic and epigenetic analysis are used to identify haplotype-specific increased methylation of the FTO T2D and obesity susceptibility locus (27). Maternal food restriction during pregnancy has been found to have a strong effect on insulin-like growth factor-1 (IGF1) mRNA expression in intrauterine growth restricted offspring who are more prone to adult obesity and metabolic syndrome (323). It has been documented that hypomethylation in the dipeptidyl peptidase-4 (DPP4) gene promoter in severe obese patients having other metabolic disorders causes enhanced expression of DPP4 in the visceral adipose tissues compared with obese individuals without metabolic disorders (324). In this connection, genome imprinting plays a major role in obesity progression. Genomic imprinting determines the expression of the alleles on the basis of the origin, that is, maternal or paternal. Any failure in the imprinting or loss of imprinting (LOI) leads to obesity due to altered expression of growth factors. Such differential transmission of genes has been witnessed in the case of the Prader-Willi syndrome and Angelman syndrome. Both of these syndromes involve the deletion in chromosome 15, and deletion regions overlap within the patients (212, 288). However, these syndromes show a difference in the parental origin of deletion; for instance, the Prader-Willi syndrome shows paternal deletion, and the Angelman syndrome shows maternal deletion. This information emphasizes the importance of coordinated interplay between epigenetic events and nutrition for the obesity development.
Hypertension and CVD
Although genetic and environmental factors have been found to be responsible for hypertension (306), the etiology remains unclear. Fetal programming can be changed by epigenetic modifications that lead to altered expression of genes in adult life with an increased risk for hypertension. For example, a class of histone methyltransferase represented by disruptor of telomeric silencing affects gene silencing. It causes methylation of histone H3 lysine 79 (H3K79) and has been implicated in the development of renal fibrosis and hypertension (100, 233, 332). Recent studies have also shown the role of the enzyme 11 beta hydroxysteroid dehyrogenase type-2 (11β HSD-2) in essential hypertension (48, 102, 111, 210). Loss of 11β HSD-2 activity results in the activation of mineralocorticoid receptors by cortisol, leading to increased renal sodium retention, which causes increased blood pressure (174, 231, 307). It has also been depicted that renin-angiotensin system alters disease pathogenesis, as hypertension is associated with increased expression of the AT1b angiotensin receptor (AT1b) and decreased methylation of its promoter (34, 293, 294). Polymorphism in angiotensin II type 1 receptor gene is also associated with metabolic syndrome (10).
Hypertension is the major risk factor for CVD, and many epigenetic modifications have been found to be responsible for subclinical and clinical CVD. Among all, histone acetylation is the best check point for gene regulation in hypertrophic myocardium (374). Association of specific transcription factors with HATs and histone deacetylases (HDACs) govern the pattern of gene expression (158). To this extent, class II HDACs have been reported as signal-responsive repressors of cardiac hypertrophy through interaction with myocytic enhancer factor-2 and cause inactivation of prohypertrophic genes (218). Mice lacking the class II HDAC9 have been shown to be hypersensitive to hypertrophic stimuli and spontaneously develop cardiac hypertrophy (375). Recently, an unusual regulator of cardiac-specific gene transcription has been discovered that is termed as homeodomain-only protein (Hop) (62, 295). Hop is found to be associated with HDAC1, inhibits transcriptional activity of serum response factor, and, thus, enhances the expression of Hop, causing cardiac hypertrophy (136, 171). The role of transcription activators such as HAT in cardiac muscle development has indicated that deletion of either CBP or its coactivator p300 causes perturbation in the heart development, while increased expression of these activators leads to hypertrophy (132). Along with histone acetylation, DNA methylation is also demonstrated to be critical in CVDs. Mice deficient in DNA methyltransferases (DNMTs) or methylenetetrahydrofolate reductase show hypomethylation of their DNA and exhibit increased expression of inflammatory mediators and formation of aortic fatty streaks (64, 205). Expression of many other genes that are essential in maintaining homeostatic cardiac physiology are found to be altered by DNA methylation, such as the KVLQT1 gene that is involved in cardiac membrane transport (56, 303). Similarly, the cardiac troponin T gene that is associated with cardiac contractility function is mutated in hypertrophic cardiomyopathy due to cytosine methylation, and the cluster of mutations lie in CpG dinucleotides of exon 8 and 9 (81).
Diet As an Epigenetic Modulator
Humans and many other animals are sensitive to the environmental factors that make permanent changes in the functional outcome of many metabolic pathways, and this phenomenon is termed as “metabolic imprinting” (348). Diet is implicated in various pathways during the life time of an animal, and these pathways are further tightly controlled by epigenetic events; therefore, diet is a critical modulator of epigenetic programming in metabolic syndrome. Epigenetic events constitute the important mechanism by which the dietary factors can regulate the activation or inactivation of the gene (238). Epigenetic marks that mainly include the CpG methylation and covalent modifications on histone tails lead to heritable changes in genotypes and allow cell-specific gene expression.
By modifying the nutritional components in diet, the disease manifestation can be checked. Many non-nutrient dietary factors such as genistein and polyphenols can also modify epigenetic marks and induce persistent changes in gene expression. The altered expression of genes induced by diet is of very low intensity and hard to distinguish. Some nutrients act as ligands and bind to specific receptors, thereby directly regulating the expression of target genes (208). For example, unsaturated fatty acids bind to PPARs and regulate the genes that are involved in fatty acid metabolism and oxidation. “The Dutch famine,” a unique counterpart in animal models, describes the effect of poor prenatal nutrition during different stages of gestation on the prevalence of metabolic defects (82, 145). Exposure to famine during any stage of gestation is related with IFG that leads to dyslipidemia, altered blood clotting, increased risk for CVD, and obesity in women (241, 262, 263, 270, 271, 272). Prenatal nutritional constraint enhances the risk for many noncommunicable diseases and altered metabolic phenotype in the offspring. Early mismatched nutrition is a crucial factor for predisposing an individual to hypertension and atherosclerosis (35). Inadequate food intake pattern has been found to be associated with an increased risk for atherosclerosis and CHD (298). Interestingly, regulation of gene expression by different dietary components varies in different individuals because of the interindividual genetic variation that arises due to functional polymorphism in nutrition-related genes.
Diet in early life epigenetic reprogramming
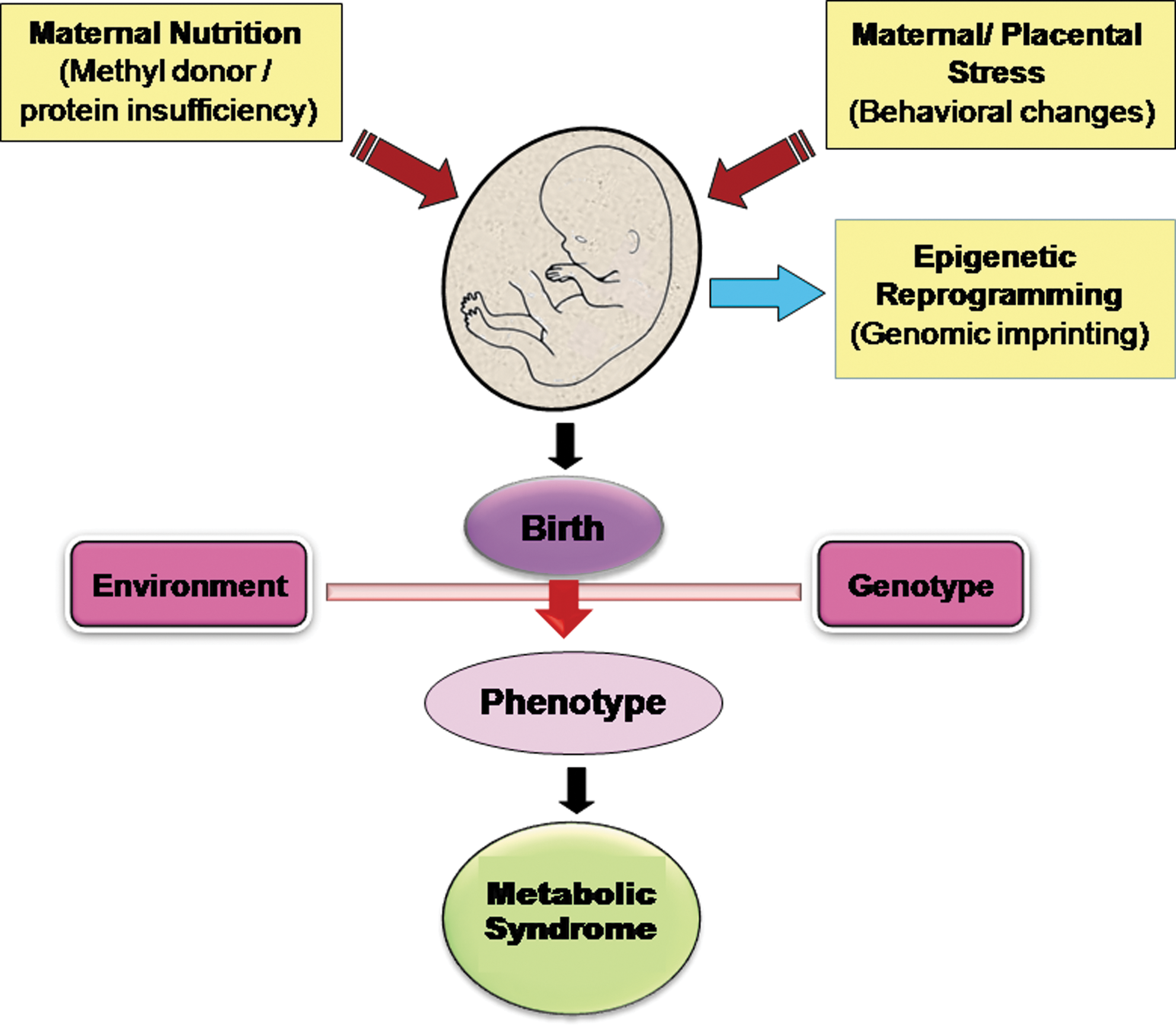
The epigenetic changes caused by dietary components are highly critical during the early life development, as it is reported that permanent physiological adaptations are programmed in the fetus in response to a poor environment in utero and contributes to disease risk in adulthood (20). Poor nutrition during the first trimester contributes to restricted fetal growth, which results in increased susceptibility to metabolic disorders in the later part of life, although the effect depends on the timing of nutritional deprivation during gestation (269). There is a strong evidence of increased mortality from stroke in adults if there has been any change in food availability during the intrauterine life (46). “The thrifty phenotype hypothesis” or “fetal origin of disease” was put forward by Hales and Barker. This theory explains the association between the poor fetal and infant growth with an increased risk of impaired glucose tolerance and the metabolic syndromes in the adult life (19, 134). Thrifty phenotype hypothesis also imposes the role of certain factors such as obesity, ageing, and physical inactivity in the emergence of pathological changes. Imbalanced nutrition during prenatal and postnatal phases of life is responsible for proper metabolic and structural adaptations that lead to persistent modifications in the phenotype of the offspring (24). For example, poor nutrition in early life due to maternal malnourishment can cause permanent alterations in insulin secretion by pancreatic β cells and perturbed glucose metabolism. Although pathogenesis of T2D depends on both the genetic and environmental factors, the thrifty hypothesis proposes that the environmental factors are a crucial determinant for T2D. Fetal malnutrition is one of the key factors in IR incidence.
Several population studies and research in ethnic groups have reiterated the earlier concept of thrifty hypothesis (302), and the “fetal origin” term was replaced with the DOHaD. This hypothesis supports the concept of developmental plasticity and underpins the significance of early life exposure to environmental factors that decides the health outcomes (122) (Fig. 4). Such exposures mark the individual at the molecular level and are associated with altered gene expression and phenotypic changes without any change in the DNA sequence. In humans, failure of the leptin uptake during the childhood via breast milk has long-lasting consequences on health (137). The adipocyte-specific gene leptin acts similar to a multifunctional hormone that plays a pivotal role in the regulation of nutrient uptake and energy expenditure by controlling the electrophysiological modulation of orexigenic neurons. Surprisingly, in human preadipocytes, leptin gene promoters follow a specific chromatin remodeling pattern that allows expression of leptin only in mature adipocytes but not in premature adipocytes (220). About thirty two CpG repeats in the proximal promoter of leptin gene are extensively methylated rendering the gene silent, while demethylation in terminally differentiated adipocytes results in the expression of leptin gene. Metabolic health of the offspring is determined by the dietary components of the mother. Low glycemic index diet of the mother is associated with the better metabolic profile of the offspring and reduced risk for metabolic disorders (325). Protein is essentially required in the diet for efficient homeostasis and proper growth. Dietary restriction during gestation strongly effects the disease development in the offspring during adulthood. Pregnant female rats fed with low-protein and HFD gave birth to offspring that are at a higher risk for IR, hypertension, obesity, and diabetes (117).
Any change in the dietary protein during the intrauterine life drastically perturbs the pancreatic development and delays the insulin secretion. In rats, a drastic change in the phenotypic expression was found due to feeding pregnant dams with protein-restricted (PR) diet. This was attributed to the hypomethylation on the promoter of specific genes that subsequently resulted in increased expression (182). Experiments done in the rats have shown that maternal protein restriction during pregnancy causes enhanced expression of GR, reduced expression of 11β HSD, altered expression of PPAR, acetyl-CoA carboxylase, and fatty acid synthase, impaired lipid homeostasis, and disturbed cardiovascular function (206). Another novel study in rats highlights the importance of maternal nutrition on the health of offspring wherein it was found that maternal protein restriction elevates the long-term cholesterol dysregulation in the offspring due to epigenetic modification (305). This augmentation of cholesterol happens due to reduced expression of hepatic cholesterol 7α-hydroxylase (Cyp7a1) because of posttranslational modifications at the Cyp7a1 promoter like diminished acetylation and enhanced methylation of histone H3 lysine 9 (H3K9). Animals born to the mothers fed with low-protein diet exhibit lower levels of leptin mRNA and protein due to altered epigenetic changes at the leptin gene promoter (161). Consumption of high-fiber and high-protein diet during pregnancy also causes an increased expression of UCP1 and PPAR-γ coactivator as well as enhanced mRNA levels of IL-6 and resistin. All these early life changes might have long-term consequences for obesity risk in the later part of life (216). GR activity is critical for regulating the blood pressure, while PPAR is central in lipid and carbohydrate homeostasis (28, 29, 45, 87, 371). During pregnancy, dietary protein restriction affects epigenetic modification of hepatic GR receptor (188), but such modulations can be reversed in the later part of life. Folate supplementation in diet prevents the hypertension in low-protein fed dams during their adulthood (322). All these studies establish the significance of early life nutrition for the proper and balanced epigenetic events leading to the healthy metabolic status of an individual.
Transition of methylation marks by dietary factors
During the early development, extensive chromatin reprogramming occurs in the germ cells in such a way that histone 3 lysine 9 (H3K9) mono and di methylation decreases, which are the two major repressive modifications (285). Some components of maternal diet can influence the epigenetic programming during the pregnancy by inducing alterations in the histone methylation patterns, further contributing to altered gene expression. Thus, maternal diet can have prolonged consequences on health and disease risk (187). In utero nutritional condition is known to regulate the posttranslational modification of histones, particularly the DNA methylation. Some of the food components are involved in one-carbon metabolism, which, in turn, influence the supply of methyl groups.
S-adenosylmethionine (SAM) is a crucial methyl donor for many of the methyltransferases that modify DNA, RNA, histones, and other proteins (67, 196). Folate holds the central position in one-carbon metabolism, during which the carbon unit from serine or glycine is transferred to tetrahydrofolate to form 5,10-tetramethylenetetrahydrofolate, which subsequently gets converted to SAM in an ATP-dependent manner using vitamin B6 an B12 as cofactors (273, 283). Many nutrients such as vitamin B6, vitamin B12, folate, methionine, betaine, and choline are involved in one-carbon metabolism and SAM-substrate methylation (213). Deficiency of these nutrients in the diet or SNPs in the genes encoding certain critical enzymes such as methionine synthase contribute to impaired synthesis of SAM and, thus, DNA hypomethylation (79, 286, 289, 353). Studies done in some animals such as sheep have shown that restricting the supply of folate, vitamin B12, and methionine during the periconceptional period leads to widespread epigenetic alterations and, thus, results in obesity, altered immune response, IR, and elevated blood pressure in offspring (301). Any change in the methionine metabolism causes an elevation in the production of homocysteine, which, in turn, causes the increased endogenous methylation of DNA (265).
Epigenetic reprogramming in one-carbon metabolism pathways and elevation in methyl group donor supply cause a persistent change in the global methylation pattern, thus silencing the expression of many growth-promoting genes. Acute methyl deficiency has been linked with histone H3 lysine 9 (H3K9) and histone H4 lysine 20 trimethylation (H4K20me3) by affecting the Suv4 and Suv39 methyltransferases (256, 282). Histone H3 lysine 9 trimethylation (H3K9me3) is a key repressive and a relatively stable epigenetic mark that is involved in the metabolic memory. Decreased H3K9me3 has been proposed to underlie the sustained proinflammatory phenotype of vascular smooth muscles in diabetic animals even after achieving glycemic control (339). Increased inflammatory gene expression due to decreased H3K9me3 promotes vascular smooth muscle cell migration and proliferation in diabetes that is a key process of atherosclerosis (121). Detrimental effects of low folate consumption by mother before and during pregnancy increases the risk of neural tube defects in the offspring (109), while high folate consumption during the pregnancy causes increased IR in children (368).
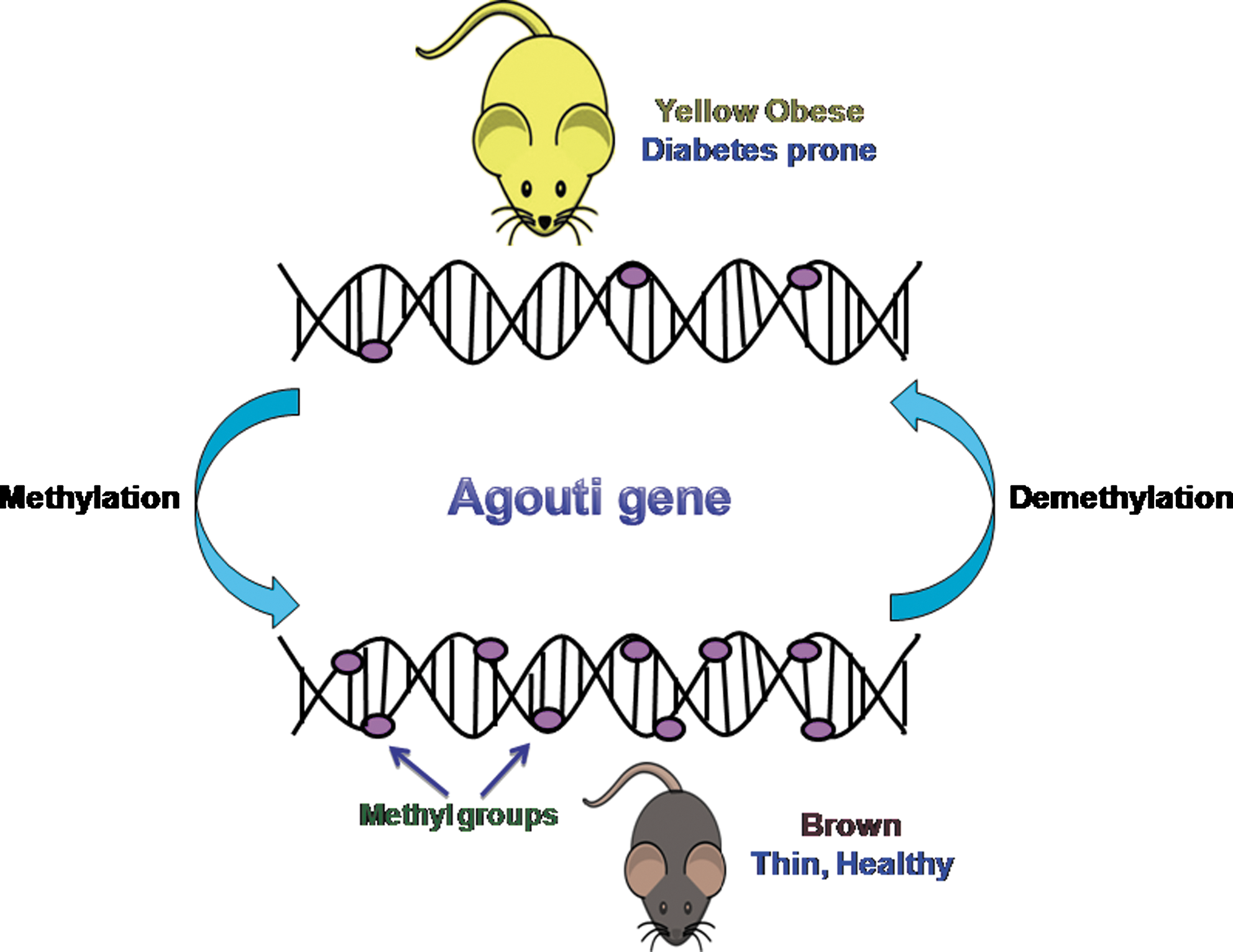
Supplementation of the diet with folic acid prevents altered metabolic phenotype, suggesting that the cause of defect lies in one-carbon metabolism. Thus, reduced levels of folic acid induce certain modifications that are associated with hypertension and dyslipidemia. Experiments in the mice agouti model further corroborate that dietary methylation in utero affects the phenotype of offspring by favoring epigenetic modifications and, thus, altering the levels of DNA methylation (350, 364). Hypomethylation of the Agouti gene promoter facilitates its enhanced expression, leading to the yellow phenotype in mice. These “viable yellow” (Avy/a) mice are larger in size and are more prone to obesity and hyperinsulinemia, eventually resulting in a shorter life span than their non-yellow siblings (Fig. 5). The differentiation of these Avy/a embryos toward the pseudoagouti brown phenotype is favored on dietary supplementation with folic acid, betaine, zinc, and vitamin B12 in maternal diet during pregnancy, thus reducing chances of obesity, cancer, and diabetes. Therefore, maternal nutrition positively affects the health and longevity of the offspring through epigenetic alteration. Prenatal under-nutrition involving maternal Magnesium (Mg) deficiency drastically affects glucocorticoid metabolism due to increased methylation in the CpG dinucleotides in the 11β HSD-2 promoters in the liver (312).
The role of vitamin biotin has also been indicated to be involved in chromatin modifications and gene silencing by causing biotinylation of histone H4, thus offering an exciting mechanism of alteration in chromatin structure and gene function mediated by certain micronutrients (140). Alcohol and zinc are also reported to be essential in the diet, as they influence the availability of SAM (79, 256, 273). Alcohol consumption affects DNMT expression, leading to an altered methylation pattern, and perturbed genomic imprinting and excessive alcohol intake predispose an individual to several cancers (73, 95, 108, 331). Deficiency of many bioactive food components such as zinc, selenium, arsenic, genistein, and vitamin A induce either global hypomethylation or hypermethylation (26, 78, 80, 274, 343). Interestingly, some bioactive dietary components affect DNA methylation by interfering with DNMT activity, as found in many animal models, wherein the diminished carcinogenesis by green tea was demonstrated (98). Cadmium and zinc are also been reported to inhibit the DNMT activity (257, 314). Altered epigenetic marks on hepatic GR and PPAR genes are attributed to reduced DNMT1 expression (189, 190). Persistent alteration in the dietary components during the embryonic stage is correlated with stable changes in the gene expression of the offspring (366). In vascular endothelial cells, transient hyperglycemia has been implicated in the induction of long-lasting epigenetic changes on nuclear factor kappa B (NFκB) promoter by modifying histone H2 methylation, thus making it an independent risk factor for diabetic complications (93). Till date, the role of dietary components on RNA-mediated transcriptional or posttranscriptional gene silencing has not been fully elucidated. A few reports suggest that some hepatic miRNAs show dysregulation if the mice are fed with a choline-deficient and amino-acid defined diet. Under these conditions, miRNAs similar to miR-155, miR-221/222, and miR-21 showed upregulation, while miR-122 expressed downregulation (344).
Nutritional components in diet and dietary habits of an individual strongly influence genome imprinting. Genome imprinting plays a major role in the incidence of a disease, as it was discovered that many multifactorial syndromes show different incidence in males and females with frequent occurrence in one gender than in the other (60, 129, 201). Neural tube defects show varied frequency, with more occurrences in females than in males (209). Such influence of epigenome supports the hypothesis “parent of origin effect.” Sometimes, the disease occurrence in the later part of life is strongly favored by epigenetic modifications than the environmental factors, namely the nutritional components in the diet, stress, xenobiotics, and nursing behavior. Effect of diet on genomic imprinting was studied by understanding the expression of imprinted gene insulin-like growth factor 2 (IGF2) (351). The maternally inherited allele of IGF2 is epigenetically silenced, rendering the expression exclusive from the paternal allele. LOI on the IGF2 locus causes biallelic expression of this mitogenic growth factor, as evident in Beckwith-Weiderman syndrome (354). Dietary deficiency of methyl donors and cofactors during postweaning development causes hypomethylation on the Igf2 differentially methylated regions and LOI that is persistent even after returning to a normal diet. Thus, nutritional stimuli during preadulthood persistently alter epigenetic marks on imprinted genes. Therefore, susceptibility to epigenetic-based metabolic disorders can be effectively regulated by modifying the dietary nutrition during infancy and childhood.
Dietary components affect acetylation
Histone acetylation is another crucial epigenetic modulation that is affected by many bioactive food components. HDACs impinge on gene expression by primarily affecting the lysine residues of core histones. Although expression of various HDACs are affected by the nutritional components of the diet, implication of dietary HDAC inhibitors to control gene expression is still in infancy. Class I, III, and IV HDACs are classified as metal-dependent enzymes, while HDAC I, II, and HDAC 11 are zinc-dependent enzymes; therefore, zinc is essential for the activity of these HDACs (115). The presence of HDAC inhibitors in the diet and metals such as zinc affect the HDAC activity. Many short-chain fatty acids, butyrate, nickel, diallyl sulfide, and sulforaphane have been found to affect gene expression by modulating histone acetylation status (41, 77, 127, 183). Butyrate is the smallest known HDAC inhibitor that directly enters into the active site of the HDAC and reactivates the epigenetically silenced genes. Sulforaphane that is present in the broccoli and broccoli sprouts significantly inhibit the HDAC activity (77).
Feeding of mice with HFD has been found to induce hepatic mitochondrial protein acetylation by downregulating mitochondrial protein deacetylase, SIRT3 (148). These mice are prone to obesity, IR, and dyslipidemia. CR studies carried out in many organisms emphasize that dietary restriction improves age-related health disorders by favoring metabolic changes such as decreased blood glucose, insulin, glycogen, fat, and reduced body weight (69). Such beneficial effect can be explained by the role of diet and dietary components in modulating the acetylation status of different genes. Certain dietary agents may enhance the HDAC activity and, thus, repress the gene expression. CR causes the overexpression of a class III HDAC SIRT1. SIRT1 deacetylates not only histones but also some transcription factors such as p53, FOXO, and NFκB. Thus, CR provides protection from apoptosis by changing cell redox potential (191, 192). Furthermore, in adipose tissues, CR amends adipokine dysfunction by activating PPARγ via SIRT1 (313). CR also downregulates mammalian targets of rapamycin signaling that has been linked with T2D (219), and it induces a dramatic change in the expression of many genes related to carbohydrate metabolism and transport in the visceral white adipose tissue (357). After fertilization but before formation of a single celled nucleus, the male pronucleus exhibits greater transcriptional activity than female pronucleus. This is reasoned to hyperacetylation of H4 in male pronucleus and global deacetylation of histones in the oocytes during meiosis (2, 9). During the progression of meiosis, the recruitment of heterochromatin proteins is facilitated by global histone deacetylation, and a diet having deacetylase inhibitors drastically hampers the development of fetus (83).
Transgenerational response
Differential response of individuals to nutrients and other environmental factors is highly influenced by interindividual genetic differences and varied developmental exposures or sometimes both. Nutritional value of the diet dictates epigenetic modifications of some genes during the slow growth period (SGP) before prepubertal peak in the growth velocity of the childhood (164). The intergenerational feed-forward control loop that describes the effect of grandparental nutrition on the grandchild's health is termed as “transgenerational response” (TGR). During TGR, the nutritional state directly modifies the gametic imprint on one or many genes that gets inherited to the future generations (164). TGR is described as “cellular memory,” as it depicts the effect of environmental and nutritional exposure of the parents and grandparents on the offspring, thus serving as a mechanism for transmitting this information to the next generations (Fig. 6). TGR is an indicator of very early programming events in the SGP of the child that causes stable epigenetic inheritance. TGR in probands (individuals under study) are characterized by the access to food during the SGP of either the parents' or grandparents' life (47).
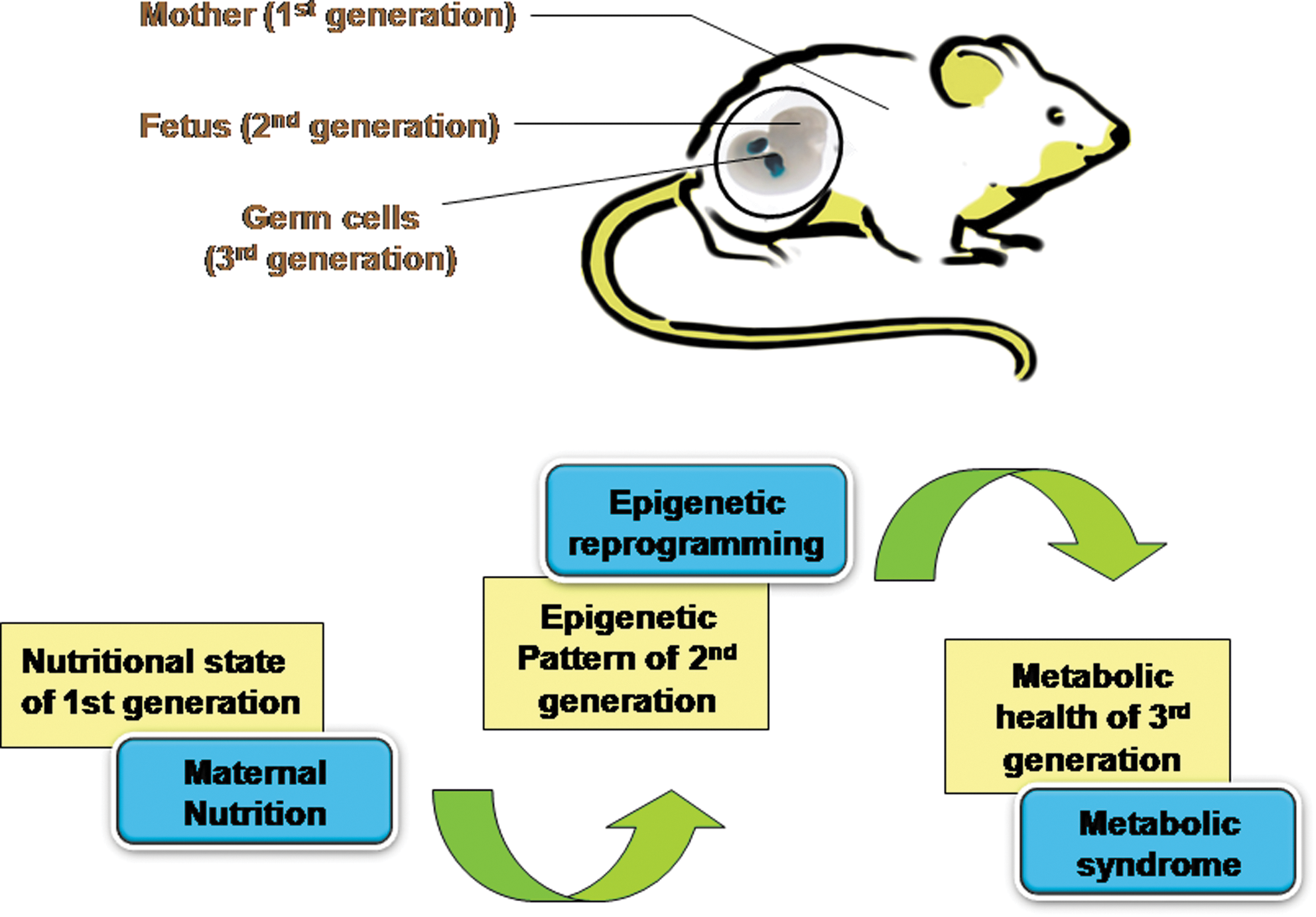
TGR from a mother's nutrition during her childhood can be a key factor in influencing the probability of CVD, diabetes mellitus, and hypertension in the adult stage of her progeny. Moreover, such an effect of the mother's nutrition encompasses a wider range of disease determinants (211, 349). Excessive food uptake in children and adolescents may program the lipid metabolism and other metabolic pathways for a life time (217). Overeating during a child's SGP, before attaining the prepubertal peak in growth velocity, markedly affects the risk of death from CVD and diabetes in the descendants' life. There will be less CVD mortality in the probands, if the food were not readily available during the father's SGP, and there will be increased diabetic mortality if the paternal grandfather were exposed to surfeit of food (163). The paternal grandfather's food intake specifically controls the mortality risk ratios of grandsons, while that of granddaughters is influenced by the paternal grandmother's food intake. Therefore, the nutritional state of the grandparents before their puberty critically affects the health of grandchildren and shows sex-specific transmission (163).
Studies indicated that the nutrition of the grandmother during her pregnancy influences the grandchild's body weight during birth, emphasizing the direct evidence of nutritional influence in the family lineage (200).
The effect of paternal grandparental nutrition on the occurrence of CVD, diabetes, and mortality in the grandson strongly implicates male line TGRs in humans. Paternal childhood smoking hampers the early growth in sons, and paternal betel nut chewing links to early onset of metabolic syndrome in the offspring (63, 246). X and Y chromosomes can be attributed for such sex-specific transgenerational effects. Patterns of smoking, diet, and healthy life style are considered the prime regulators of the disease predisposition across more than one generation (42).
A novel mechanism of disease etiology involving the epigenetic transmission through the germ line has been discovered by Anway et al. (16). In their study, it was reported that exposure of the male rat to endocrine disruptor, vinclozolin, affected the male fertility, and this effect was further transmitted to future generations without any subsequent exposure to the chemical. Their observation supported that transient embryonic exposure to endocrine disruptor during the stage of gonadal sex determination results in transgenerational disease states in adults. Similar results were observed by Nilsson et al. (234), where vinclozolin exposure caused the transgenerational increase in pregnancy abnormalities and the onset of disease in female adults.
Nutrigenomics and Nutrigenetics
Nutrigenomics is the study of interaction between nutrition and its components with genomics by applying high-throughput technology in nutrition research (5). Nutrigenomics is an important technological tool with the help of which it is possible to study the effect of nutrients on the metabolic pathways at the molecular level and how the homeostatic control is disturbed in the initial stages of diet-related diseases. Most importantly, it emphasizes how individual's sensitizing genotype contributes to the extent of metabolic disorders (334) (Fig. 7). The effect of the nutrients on health and metabolic syndrome incidence cannot be studied without the complete knowledge of the mechanism by which nutrients act at the molecular level (229). Micronutrients (vitamins, minerals, etc.) and macronutrients (carbohydrate, fatty acids, amino acids, etc.) act as potent dietary signals that play an important role in the homeostasis by regulating the metabolic programming of the cells (70, 106).
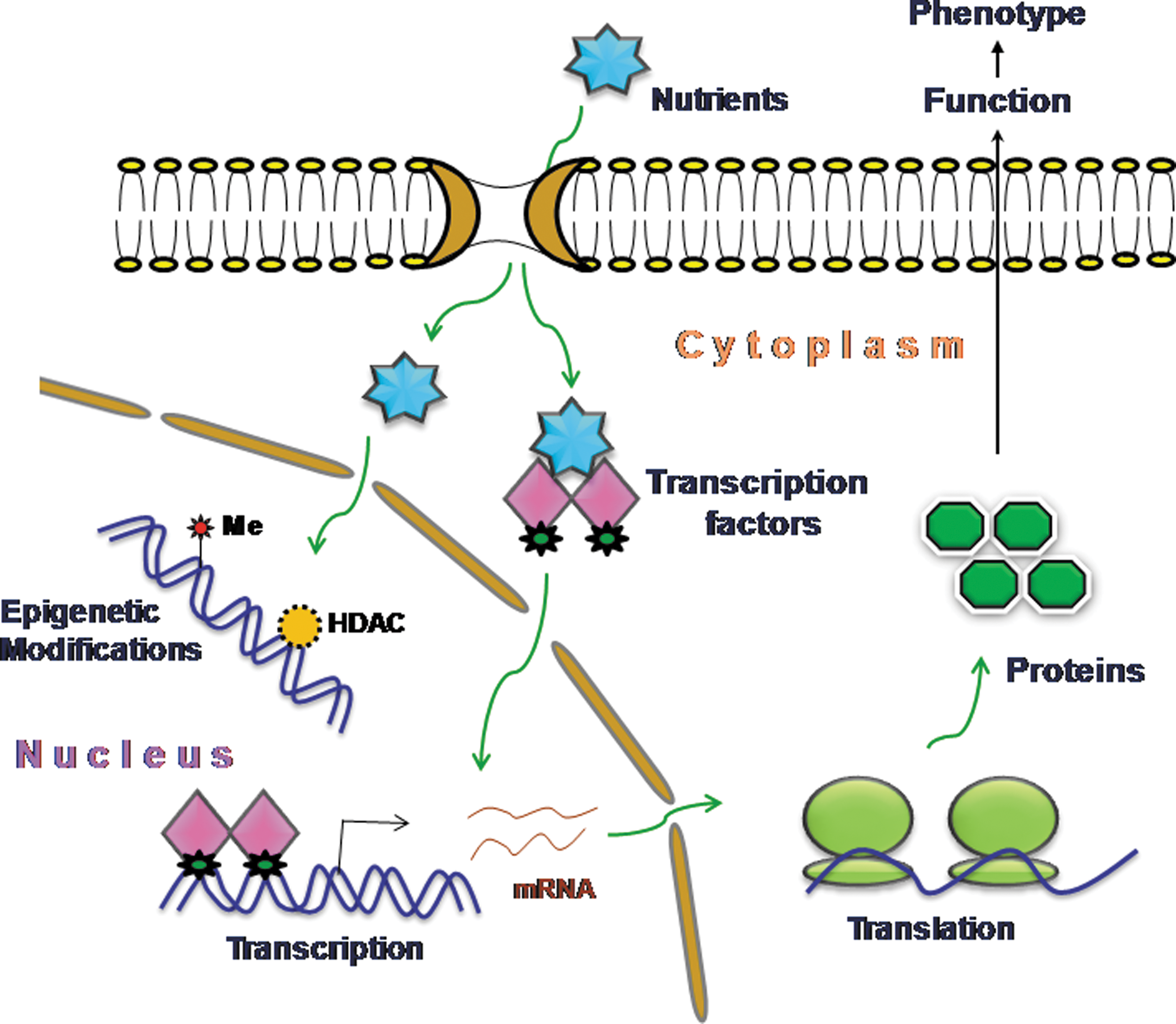
The molecular structure of the micronutrients and amino acids decides the specificity of the signaling pathway for activation, and any minute change in the molecular configuration leads to a drastic change in the response of cellular sensing machinery. Therefore, many nutrients exert different effects on the cellular function as in the case of fatty acids that show a varied nutritional effect (43, 120). Nutrigenomics assists in the identification of downstream effects of cell specific nutrients. In certain cases of T2D, a defect in the amino-acid-dependent signaling is the underlying mechanism for disease progression rather than defective insulin-dependent signaling (326). Nutrients eventually act as stimulators for cellular sensor system and, thus, influence the expression of many genes involved in metabolite production. Therefore, nutrigenomics aims at discovering the genes playing a significant role in diet-related diseases and as well deciphering the mechanism of such genetic predisposition. Genetic predisposition is recognized as a critical contributor for the occurrence of CVD, hypertension, and T2D, because inherited genotypic difference dictates the variation in the phenotype and, thus, modulates disease risk (360). Transcription factors are the critical regulators by which nutrients modulate the expression of target genes. The nuclear hormone receptor superfamily of transcription factors consists of 48 members in humans and is considered the most important nutrient sensor (207). Many members of this superfamily such as retinoic acid receptor, retinoid X receptor, fatty acid receptor (PPAR), liver X receptor (LXR), vitamin D receptor, oxysterols (LXR), bile salts (farnesoid X receptor-FXR), constitutively active receptor, estrogen receptor, pregnane X receptor, and carbohydrate-responsive element binding protein bind to their respective nutrients and their metabolites (61). Binding of the ligand to specific receptors leads to dynamic conformational changes in the receptor, favors the assembly of co-activators and dissociation of the co-repressors that eventually modulates the transcriptional activation (5). PPARs get activated after binding to its ligand and control the expression of genes involved in lipid metabolism, lipoprotein metabolism, and reverse cholesterol transport in a tissue-specific manner (31). PPARα has been found to be a fatty acid sensor and acts predominantly during the stage of food deprivation. Increased occurrences of certain diseases are linked with fatty acids and fatty acid metabolism, underlying the impact of fatty acids on the human health (11).
In metabolically active organs such as liver and adipose tissues, transcription factors sense even a minute change in the nutritional components in diet and regulate the expression of target genes at the transcription level. FXR acts as a nutrient sensor for the increased levels of bile salts and elevates the expression of many genes such as PPAR, phospholipid transfer protein, short heterodimeric partner, apolipoprotein E, and APOC11 (15, 66, 251, 255).
Nutrigenetics has emerged as a novel tool that examines the role of genetic variations on the interaction between diet and disease risk. It emphasizes that inter-individual genetic variation is the key determinant of difference between nutrient requirements (128). Nutrient availability dictates the change in the chromatin architecture as implicated in starvation wherein the condensation of macronuclear chromatin and decreased global gene expression is observed (242, 292). A change in the chromatin structure due to nutritional status is modulated by the histones. Castro et al. (52) have shown the difference in the rat liver chromatin and nuclear proteins on nutrient variation, such as a high-carbohydrate, fat-free diet (diet 1) or a low-carbohydrate, protein-free diet (diet 2). On nutrient deprivation, there is an increase in the RNA polymerase activity and increased incorporation of orotic acid into nuclear RNA (275). Starvation and malnutrition modifies the linker histone H1 and other histones of the core nucleosome, subsequently affecting the enzymatic activity of the topoisomerases and helix-destabilizing proteins (50, 51, 226). Protein depletion in the diet resulted in a decrease of total nuclear proteins in the mice liver and on replenishing the protein in the diet, there is a slow recovery of histone proteins compared with other lost proteins (49, 261).
Metabolic Health Programming by Nutrients
The correction of epigenetic defects by nutrients in the food has come as a very interesting and attractive therapy that ameliorates the effects of deleterious genes involved in metabolic disorders. Epigenetic marks can be effectively reversed by nutrients and can be applied for diagnosing the common metabolic diseases such as diabetes, CVD, hypertension, and obesity. DNA methylation by DNMTs and histone deacetylation by HDACs are epigenetic modifications that mainly silence the expression of many genes and, therefore, several drugs which inhibit the activity of these enzymes restore the normal transcription of regulatory genes (13). Since folate is the key source for the methyl group, it is an attractive nutrient for modifying the epigenetic programming. Studies done by Van Der Put (330) revealed that 65% occurrence of neural tube defects and 50% reoccurrence can be checked by periconceptional folic acid supplementation. Importance of methyl group supplementation was established by research in Avy mouse, which provided an important clue for understanding the role of altered DNA methylation in modifying gene expression with subsequent phenotypic changes (88). Genistein, a phytoestrogen, causes permanent alteration of maternal epigenome in utero. This was determined by assessing the coat color, DNA methylation, and body weight in Avy mice. Maternal dietary genistein supplementation during the pregnancy results in the change of coat color phenotype from heterozygous viable yellow agouti (Avy/a) offspring to brown pseudoagouti. This effect on phenotype is attributed to increased methylation of a retrotransposon that is upstream to the Agouti gene promoter, causing decreased Agouti expression and protection from obesity. In rats, reduced maternal protein uptake leads to decreased methylation on the promoters of GR and PPARα, resulting in enhanced expression of these genes in the liver of offspring, but such effects can be efficiently reversed by dietary folic acid supplementation (188).
Folate supplementation during the juvenile-pubertal phase of life alters the phenotypic expression as well as those epigenotypes that were usually induced by a maternal PR diet (44). Such folate supplementation during the juvenile-pubertal period influences the methylation of certain genes such as PPARα in the liver and a concomitant hypomethylation of insulin receptor gene in adipose tissues. Leafy vegetables, peas, beans, sunflower seeds, fortified breads, cereals, and the like are considered to be enriched with folic acid, and their consumption in a sufficient amount favors balanced DNA methylation pattern. In the past few decades, the substantial increase in the intake of methyl-consuming compounds has been reported, primarily due to pollution, high meat consumption, and food additives, which might be one more causative agent for metabolic syndrome (377).
Balance in the metabolic pathways of hepatic folate, methyl group, and homocysteine determine the optimum health, and any disruption in these pathways leads to numerous pathologies (361). Metabolic dysregulation in diabetic state leads to imbalanced hepatic methyl group metabolism, which marks an increased glycine-N-methylase expression and activity, leading to hypomethylation. These disorders can be effectively prevented by consuming a methyl group donor-enriched diet and, thus, would be an effective epigenetic therapy. Several epigenetic drugs are already used for the treatment of cancer, and many new drugs are under study for their use in other diseases. Many promising results on these epigenetic drugs for their application in metabolic disorders provide an alternative option in therapeutic armamentarium for people suffering from metabolic abnormalities (340). In spite of many drugs being used as key modulators of epigenome, the biological source of key nutrients can be the potential tool to attenuate the progression of metabolic syndrome (167). For improving IR, the alternative strategies such as supplementation of many plant-derived products in diet have become quite popular and accepted. Plants produce many compounds that have medicinal value and can be used to cure diabetes (347).
Studies by Devalaraja et al. (84) have revealed the role of exotic fruits and their constituents in ameliorating metabolic derangements such as diabetes and obesity. According to the food and drug administration, there is availability of ∼29,000 nutritional supplements in the market (232). Metformin is a highly prescribed and recommended glucose-lowering medicine that is used in the treatment of polycystic ovary syndrome, T2D, and obesity (299, 315, 363). Many medicinal plants from the genus Artemisia have been routinely used in the traditional treatment of diabetes (266). Atremisia dracunculus L or Russian terragon has a long history of medicinal use, as its ethanolic extract has been found to significantly decrease the blood glucose levels and, thus, helps in the treatment of diabetes by improving the insulin action. The molecular mechanism behind this effect of ethanolic extract can be explained by improved carbohydrate metabolism through enhancement of the insulin receptor signaling and modulating the levels of PTP1B, a specific protein tyrosine phosphatase (347). Similarly, the fruit extracts of Ligustrum lucidum have been discovered to be of great significance in curing diabetes, as they possess antidiabetic, anticancer, antioxidant, and neuroprotective activities (185). This fruit extract contains bioactive component NZ-01, which causes upregulation of glycine-N-methyltransferase. Ginger rhizome has been remarkably proved as an anti-obesity agent due to the presence of a major chemical component [6]-gingerol analog that forms a stable analog Aza-[6]-gingerol after digestion by gastric fluid (236).
Ginger consumption is considered to be beneficial in reducing body weight and fat deposition along the visceral and subcutaneous regions. Grapes are a rich source of phenolic compounds such as flavanoids (catechin and epicatechin) and polyphenols (3,5,4′-trihyroxistilbene—popularly known as resveratrol) (75). Resveratrol and sirtuins have pharmacological significance for the treatment of metabolic syndrome (25, 372). Resveratrol activates longevity proteins such as SIRT1 and, thus, possesses antiapoptotic activities (248). Wine is a rich source of resveratrol, and individuals who consume wine in a moderate amount are at a lower risk of mortality from the risk for CAD (110, 112, 277). Resveratrol exhibits antioxidant, anti-inflammatory, and antiatherogenic effects. Cardioprotection with grapes and grape seeds proanthocyanidins was reported to be beneficial in ischemia reperfusion injuries (72). Resveratrol induces the expression of thioredoxin-1 (Trx-1), heme oxygenase-1 (HO-1), and vascular endothelial growth factor (VEGF), thus improving neovascularization in the infarcted rat myocardium (165). A recent study on high-fat-fed rats has proved the significance of resveratrol in improving cardiac function by decreasing the infarct size as well as reducing cardiomyocyte apoptosis, by modulating the lipid levels (249). Combination studies of resveratrol with statin improved the cardiac function due to the synergistic effect. Resveratrol plays an important role in cardioprotection and alleviation of cardiac dysfunctions by modulating NO, Trx and HO (320). Enhanced production of NO by resveratrol helps in anti-ischemic function in resveratrol therapy and is useful in amelioration of endothelial dysfunction during hypercholesterolemia. Resveratrol regulates some biomarkers such as cav-1, HO-1, eNOS, and VEGF (247, 338). Vinifera grape skin (ACH09) extract helps in the prevention of metabolic syndrome in normally fed offspring from HFD dams (94).
In a very interesting study, the regular intake of functional yogurt NY-YP901 has been found to be helpful in the improvement of metabolic syndrome (59). Blueberry consumption has been found to be highly effective in attenuating CVD development and reduces IR in obese rats (287). Blueberries are a rich source of anthocyanins that modulate the activity of PPAR, thus altering the energy-substrate metabolism. Replenishment of nutrition during the postnatal state causes the normalcy of growth; still, certain diseases have been reported to reappear in the later part of life, such as obesity (225). Many more studies in this filed have clearly suggested that there is a phase of epigenetic plasticity in the life of an individual before attaining adulthood, where nutritional interventions can be used to reverse the effect of prenatal nutrition on offspring health.
Conclusion
Environment and genetic interactions predispose a majority of the world's population to various metabolic disorders, and these syndromes exhibit perturbed epigenetic regulations that are inherited to the subsequent generations. Epigenetic marks such as methylation occur on the cytosine residue of CpG dinucleotides in DNA and on the histone tails, wherein acetylations are also reported to occur. Dynamics of methylation and acetylation on some specific genes make an individual prone to metabolic diseases. miRNA-mediated post-transcriptional regulation of some genes also prompt an individual to metabolic disorders. Since nutrients considerably influence the epigenetic marks that modulate the gene expression, improper nutrition during prenatal and postnatal stages predisposes an individual to metabolic disorders, emphasizing the temporal regulation of the nutrient-dependent epigenetic marking. A diet that is deprived of methyl group donors such as folate and choline leads to aberrant expression of many genes and, thus, contributes to altered phenotypes. Similarly, PR diet and HFD makes an individual prone to IR, hypertension, and obesity. A majority of these altered epigenetic marks are inherited by the offspring. Gametic imprinting causes TGR, signifying the effect of nutrients on disease occurrence. Nutrigenomics has evolved as a major branch that understands the impact of nutrients on gene expression either through epigenetic modifications or through the activation of distinct transcription factors. Recent studies have revealed that a majority of metabolic disorders such as IR, dyslipidemia, and obesity are caused by improper nutrients and can be reverted to normalcy by supplementing the individual with an enriched diet. Thus, many medicinal plant-derived products and some fruit extracts are interesting targets of nutrient therapy to ameliorate dysregulated epigenetic events in metabolic disorders. Even though extensive studies on nutritional epigenetics and metabolic disorders have rendered the field with deluged information, the therapeutic potential of this information is yet to be amply explored.