
Abstract
Introduction
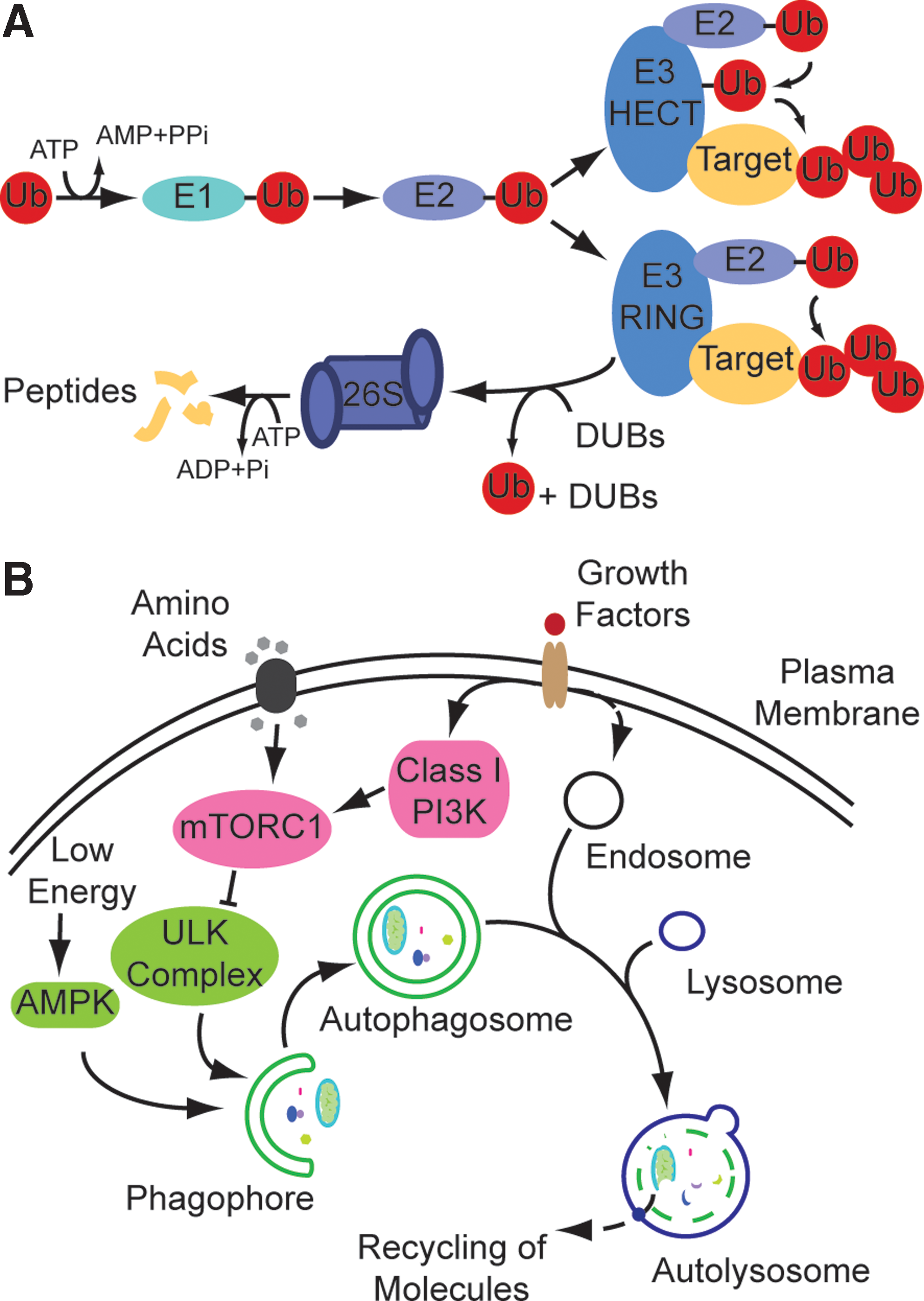
For decades, scientists were fascinated by protein synthesis trying to outline the central dogma of molecular biology on how a protein can be synthesized from DNA-encoded information. Protein degradation did not draw as much attention. It was believed to happen in lysosomes, acidic protease-containing compartments discovered and characterized by De Duve et al. (12), and from the 1950s to the 1970s lysosomes were regarded as the sole cellular degradative system. However, it was difficult to explain the observation of vastly different half-lives of intracellular proteins by the mechanism of action of lysosomes as proteins generally enter via vesicles and, thus, should display similar degradation rates. This changed with the discovery of the ubiquitin-proteasome system (UPS) by Aaron Ciechanover and Avram Hershko who could show that proteins are specifically marked for proteasomal degradation by the covalent attachment of ubiquitin (Fig. 1A). A lot of research focused on the understanding of the UPS (9) whereas autophagy, lysosomal degradation of intracellular proteins (Fig. 1B), received little attention as it was believed to be primarily unspecific and as molecular tools were missing to specifically study underlying mechanisms (31). This changed dramatically with the demonstration that autophagy was conserved from yeast to mammals (51). The genetically tractable organism yeast Saccharomyces cerevisiae allowed a rapid identification of parts of the molecular machinery paving the road for the boom years of autophagosomal-lysosomal research starting in the 2000s and still ongoing with a rise in respective scientific publications each year.
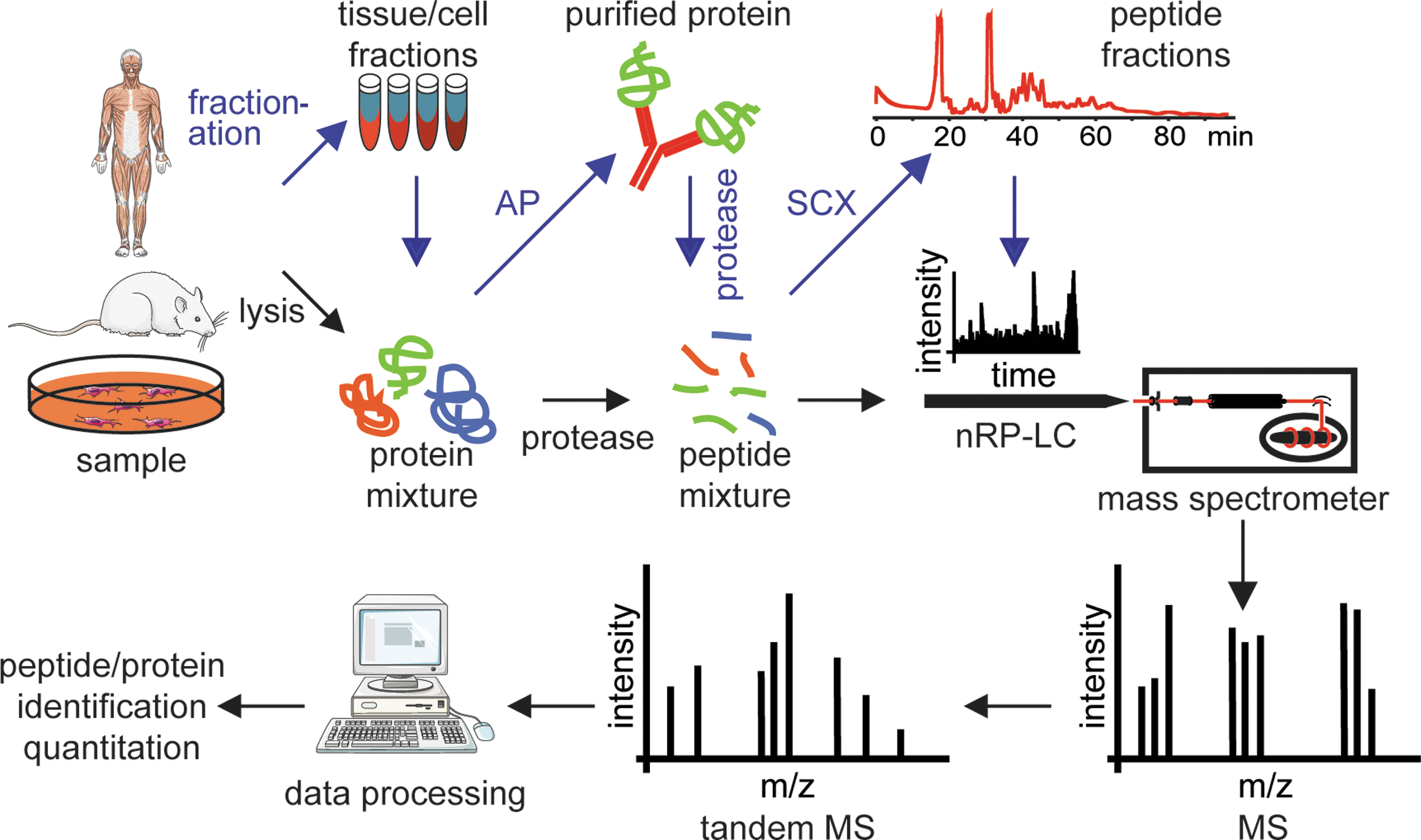
To study cellular protein dynamics on a global scale, mass spectrometry (MS)-based proteomics constitutes a versatile research platform (52). Whereas whole proteins can be analyzed by MS, so-called “top-down” proteomics, laboratories mostly analyze peptides derived from protein digests, “bottom-up” proteomics, due to its robustness, ease of handling, and potential for automation. Generally, trypsin, an endoprotease that cleaves after arginine and lysine residues if they are not followed by proline, is used for digestion. Generated peptides carry a basic C-terminal amino acid residue which is due to its positively charged side chain under acidic conditions beneficial for MS analysis. Standard proteomic workflows (Fig. 2) comprise the analyses of organelles (26), protein interactions (4), and protein modifications such as phosphorylation (57). Especially the development of sophisticated software for the analysis of raw data (10) but also for the statistical and graphical interpretation of processed data (42) allows for the design of complex, large-scale experiments. Hence, experimental setups similar to functional genomics approaches are now feasible.
Innovation
The identification of specific lysosomal degradation pathways and underlying molecular players, as well as the recognition of the complexity of ubiquitin-mediated signaling has opened up a new “El Dorado” for cell biologists. Quantitative MS-based proteomics plays a vital role in addressing essential research questions and in shedding light onto responsible molecular networks related to these processes. Recent technical and software advances in the field will further contribute to making MS-based proteomics an indispensable research technique for studying protein turnover and for outlining degradation-dependent spatiotemporal protein dynamics. As the identification and quantification numbers of proteins are steadily increasing and through the development of new MS-based techniques specifically addressing protein turnover, synthesis, and degradation, classical research areas characterizing metabolic pathways are about to be rediscovered by today's researchers. An important and necessary technical advancement will be the development of mass spectrometers with a higher dynamic range as signal intensities in pulse labeling approaches may show large intensity differences currently leading to less-accurate quantifications compared with classical labeling experiments.
Finally, the generation of absolute quantitative data will allow construction of mathematical models describing cellular networks enabling scientists to perform cost- and time-effective in silico experiments to identify critical molecular players involved in protein degradation. These system biology approaches will greatly enhance our mechanistic understanding of cellular degradative protein dynamics.
In this review we will introduce current principles of quantitative MS-based proteomics and relate them to the study of cellular protein degradation. We will focus on the degradative inventory, the autophagosomal-lysosomal system and the UPS. In addition, global analyses of protein turnover and degradation will be summarized. The specific cleavage of substrates by proteases is regarded as a posttranslational modification (PTM) and has little to do with catabolic protein degradation. Nevertheless, as proteases are the main players in both processes, we will briefly outline current methods for the mass spectrometric analysis of protease cleavage sites, as well.
Quantitative MS-Based Proteomics
MS-based proteomics has become one of the most informative methods to identify proteins, determine protein composition of complex mixtures, and to study PTMs. However, due to technological limitations it was not trivial to deduce quantitative information from MS data or to compare different states of biological samples. In the past decade technical and methodological improvements have been made allowing absolute and relative quantification of proteins and PTMs in biological samples. The impact of quantitative proteomics on the range of applications in biological research is significant (52). Beyond simplistic profiling of differential proteomes, many novel quantitative MS-based proteomics strategies, for example, recording of signal transduction kinetics (14), have been reported.
Quantification methods can be separated into two major approaches: label-free techniques and application of stable isotope labeling (33). In general, labeling strategies are considered to be more robust and accurate in quantifying protein abundances. The basic principle of quantification workflows with labels depends on direct comparison of differentially encoded experiment and reference analytes (Fig. 3). Since peptides marked by isotopically labeled tags or amino acids containing 13C and 15N behave physicochemically similar, samples can be simultaneously analyzed in one MS experiment. The corresponding isotope pairs can be discriminated by the introduced mass difference and quantified relatively. Common labeling techniques involve metabolic labeling of proteins by incorporation of stable isotopes (stable isotope labeling by amino acids in cell culture [SILAC]) (Fig. 3A) (35), chemical modification of peptides with isobaric tags for relative and absolute quantification (iTRAQ) (Fig. 3B) (47), and protein labeling with isotope-coded affinity tags (ICAT) (23).
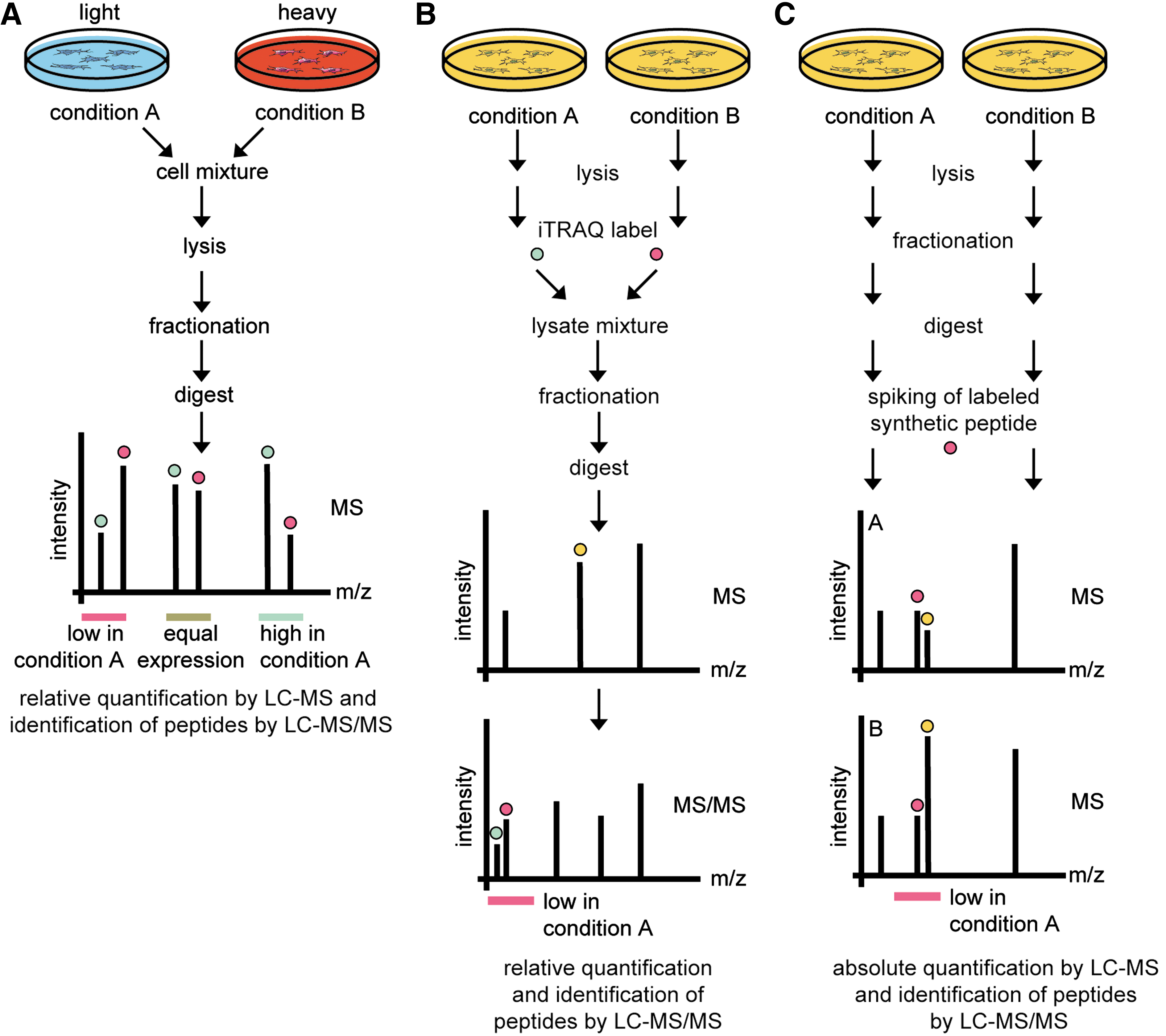
SILAC is an effective and simple strategy for incorporation of encoded amino acids in vivo. Distinct isotope variants (light, medium, and heavy) of both L-arginine and L-lysine can be incorporated metabolically into cells allowing their relative quantification after mixing (Fig. 3A). In contrast, covalent coupling of isotopically encoded tags in ICAT and iTRAQ strategies is performed on protein or peptide level (Fig. 3B). The ICAT method targets cysteine residues of proteins and allows differentially labeled samples to be analyzed during MS analysis. The isobaric tags used in iTRAQ are fragmented in the tandem MS mode to produce a reporter ion signature in the low signal region of the MS/MS spectrum to allow relative quantification. The iTRAQ system is now commercially available with eight isobaric tags allowing multiplexed analysis of eight samples in one experiment (37). Both, ICAT and iTRAQ methods are mainly used in clinical proteomics where metabolic labeling is not amenable to tissue and body fluid samples. Due to simple handling and its advantages in multistep purification and sample preparation workflows, we regard SILAC as the most convenient strategy. In addition, as SILAC quantification is performed on the MS level, compared with the MS/MS level in iTRAQ, more data points per peptide quantification are available leading to a more accurate protein quantification.
A widely distributed method for absolute quantification (AQUA) of peptides and their modifications employs synthetic isotopically labeled peptides as internal standards. Such AQUA peptides are spiked into protein samples and are used to quantify the amount of the endogenous counterpart peptides after protease digestion (Fig. 3C) (20). As reference peptides have to be synthesized, AQUA is commonly used in hypothesis-driven approaches.
Label-free methods are least accurate in quantitation but can be considered when metabolic and chemical isotope incorporation is infeasible or too costly [reviewed by Neilson et al. (33)]. Commonly, they rely on mass spectrometric peptide signal intensities (extracted ion current), peptide per protein, or total peptide fragment spectrum counting, or a combination of these.
Autophagosomal-Lysosomal System
Most organelles cannot be isolated in a pure form due to contaminating subcompartments that share similar physicochemical properties. Therefore, procedures have to be developed allowing to discriminate between true organellar and contaminating proteins. This is specifically true for the proteomic analysis of the autophagosomal-lysosomal system that is responsible for degradation of intracellular protein complexes, cytoplasmic proteins, and organelles (Fig. 1B). Virtually all cellular proteins can be part of the autolysosome at a specific time point making it more difficult to discriminate between organellar proteins, organellar content, and contaminating proteins (59).
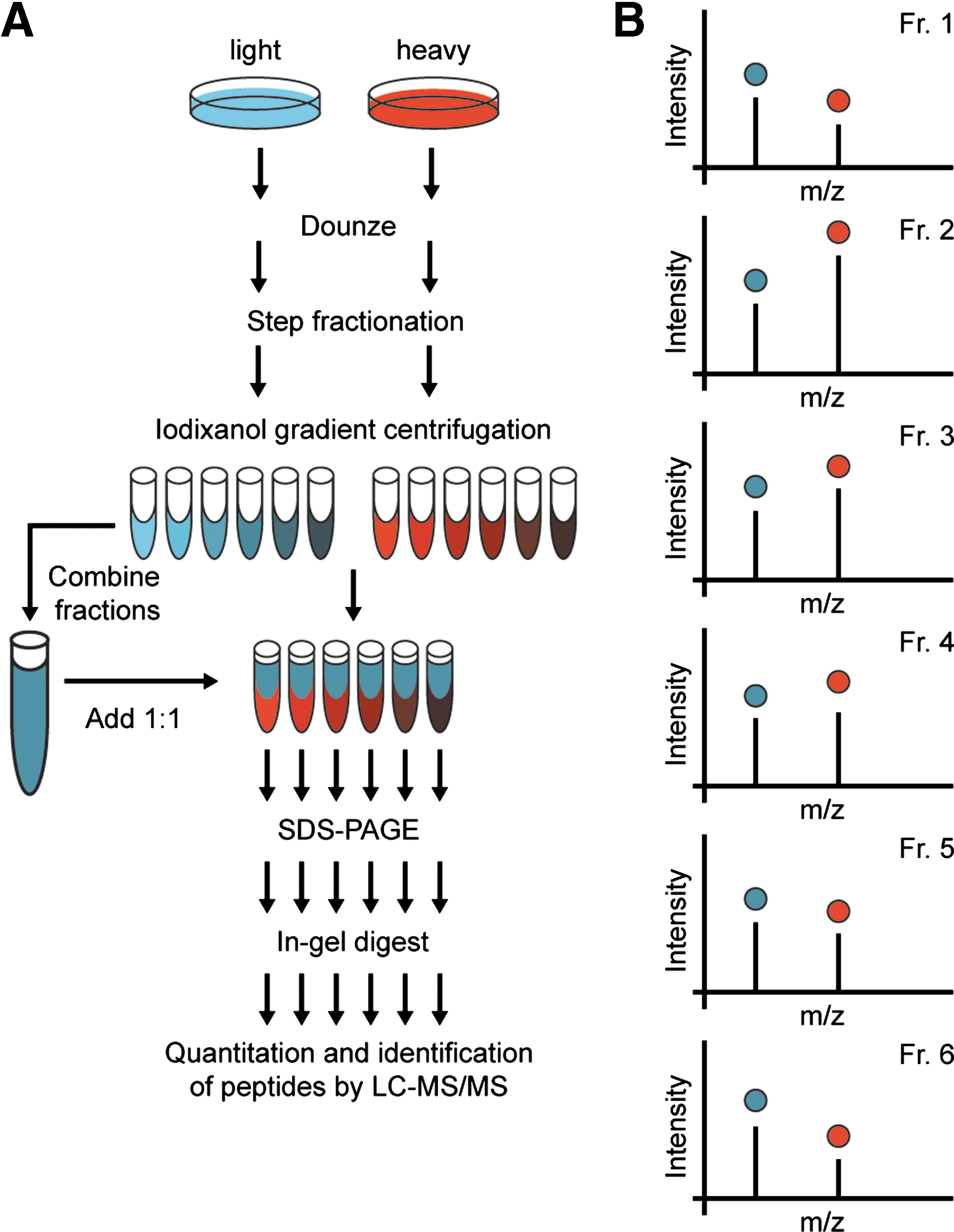
Two basic, quantitative MS-based proteomics principals exist for the analysis of organellar proteomes: first, purifications of organelles from differentially labeled cells, for example, by biochemical cell fractionation or by affinity purification (AP) followed by MS analysis; second, protein correlation profiling (PCP) where organelles are separated by density gradient centrifugation and all fractions are analyzed (2). In biochemical fractionation experiments purified organelles are generally compared with a differently labeled crude fractionation state allowing distinguishing between true constituents and other enriched proteins. In AP experiments enriched organellar proteins are identified after comparison to negative control experiments. In PCP each protein profile is characteristic for an organelle and allows assigning of proteins to certain organelles. Profiling strategies have been improved by implementation of SILAC labeling in PCP-SILAC (26) (Fig. 4) and ICAT in localization of organelle proteins by isotope tagging (16). Both, organellar purifications and density gradient profiling have been employed successfully for the proteomic analysis of autophagosomes and lysosomes as described below.
The endocytic uptake of iron-dextran particles followed by magnetic separation of iron-containing lysosomes was used to enrich lysosomes and in combination with SILAC it could be shown that an active form of the ErbB2 receptor leads to an enhanced recruitment of the motor protein kinesin heavy-chain protein KIF5B to lysosomes probably being involved in enhanced lysosomal trafficking in metastatic cancer (8). For this kind of analysis in vitro cell culture models have to be available as iron-dextran particles have to be endocytosed. An in vivo comparison of lysosomal membrane proteomes from different mouse models was quantitatively analyzed by the help of cleavable ICAT (58), and the combination of lysosomal purification by cell fractionation and iTRAQ was used for the detection of early changes in lysosomal protein expression/localization from cells undergoing apoptosis (38). The isolation of lysosomes by immunoprecipitation of the whole organelle using an antivacuolar-type H+-ATPase antibody in combination with SILAC was performed to study ErbB2-associated changes in the lysosomal proteome. This method is relatively robust not being influenced by changes in lysosomal density and stability and thus being suitable for the analysis of lysosomes from diverse disease conditions (34).
Luminal lysosomal proteins are often tagged by mannose 6-phosphate (Man6-P) as a lysosomal targeting signal. Hence, Man6-P receptors have been used for affinity purifying Man6-P–containing proteins (28). This method was adopted to study the Man6-P proteome in 17 different rat tissues. Spectral counting was employed to compare protein abundances leading to the identification of 136 new potential lysosomal proteins (48). In a similar study human brain autopsy samples were used to identify the genetic basis of lysosomal storage diseases (49). Spectral counting was also used to identify lysosomal membrane proteins by comparing protein abundances between biochemically purified lysosomes and prepurified organelles leading to the identification of several new lysosomal proteins partially validated by image analysis (45).
In the described studies quantitative proteomics information leading to a set of potential new organellar proteins was generated by comparing two different experiments/biological samples. Generally, using gradient profiling of organelles yields more than two data points allowing the use of unsupervised clustering of protein profiles for pattern recognition that may generate information leading to the description of potential new organellar or suborganellar structures without any a priori knowledge. In a recent study, a combination of purification methods of native and density-shifted lysosomes was used to classify both membrane and luminal lysosomal proteins (13). Several density centrifugations were performed and protein distributions over the gradients were quantified by iTRAQ. To discriminate lysosomes from contaminating proteins the authors used Triton WR-1339 that specifically changes the biophysical properties of lysosomes and induces a density shift, which can be used to verify lysosomal candidates.
Autophagosomes have not been so extensively investigated as lysosomes; nevertheless, several interesting proteomic studies have already been conducted. For example, gradient profiling by PCP-SILAC was used to monitor the localization of the influenza A virus matrix protein 2 (M2) to autophagosomes in influenza A–infected cells (18). It could be further shown that the M2 protein blocks autophagosome-lysosome fusion. Also classical purification approaches were employed to enrich autophagosomal proteins. Membrane-associated proteins from rat liver autophagosomes were purified by density gradient centrifugation followed by organelle disruption. Autophagosomal and cytoplasmic membrane fractions were prepared and a comparative analysis identified 39 proteins as being significantly enriched in autophagosomal membranes (36). Cells expressing GFP-tagged LC3 were used for immunopurifying LC3-positive vesicles, which should also contain autophagosomes, by an anti-GFP antibody. One hundred one proteins could be identified including three autophagy-related proteins (LC3, ATG7, and ATG9) and four proteins that were also characterized as autophagosomal membrane proteins in the previous study, but with so far unknown autophagy-related function (19). The pure overlap maybe caused by both the different biological systems as well as the different purification procedures.
A global proteomics approach relying on protein abundance measurements during starvation-induced autophagy lead to the finding that organelle degradation via macroautophagy happens in an ordered fashion, contradicting the notion of macroautophagy being an entirely unspecific bulk degradation process (32). As whole cell lysates were analyzed organellar dynamics were identified in silico using the respective gene ontology. The biological experiments were performed hypothesis free with minimal sample preparation and highlight the strength of bioinformatics follow-up experiments.
Ubiquitin-Proteasome System
MS-based proteomics has contributed significantly to our understanding of the UPS [for review see Wang et al. (53)] (Fig. 1A). In the last decade, studies addressing the composition of 26S proteasome complexes highlighted their heterogeneity. By purifying tagged subunits the in vivo existence of hybrid proteasomes in yeast containing two different activator subunits could be confirmed (44). Heterogeneity is not only found in the composition of 26S proteasomes. Also the 20S core particles differ in their molecular makeup. The 20S particles isolated from mouse liver and heart differed in their molecular composition as well as in their PTMs, which may both contribute to their distinct cleavage specificities (15). PTMs commonly analyzed by MS-based proteomics are inter alia phosphorylations, acetylations, oxidations, glycosylations, and ubiquitinations, and all of these could be identified as modifying proteasomal subunits (53). For single sites the modifying enzymes have been elucidated and their function in regulating activity, assembly, and/or stability could be shown. However, detailed underlying mechanisms and especially the PTM crosstalk are still waiting to be discovered. Especially ubiquitin modifications are extensively investigated and are further discussed in the following paragraphs.
To our awareness the use of MS-based techniques contributed significantly of the complexity of ubiquitin-based signaling. It let to the finding that ubiquitin itself can be ubiquitinated at all seven lysine residues in vivo and that forked ubiquitin chains exist (40). In addition, MS-based quantification using synthetic labeled peptides reflecting all seven ubiquitin linkages revealed that K6, K11, K27, K29, and K33 linkages are abundant in yeast and that all contribute to protein degradation (55). A recent study using protein standard absolute quantification (PSAQ) MS used stable isotope–labeled free ubiquitin and ubiquitin conjugates to determine steady-state distributions of cellular ubiquitin pools (29). It could be shown that ubiquitin pools are unevenly distributed between free ubiquitin, mono-, and polyubiquitinated substrates and that the distribution is cell line specific and tissue specific.
Due to technical advances on the mass spectrometric instrumentation side as well as quantitative proteomics strategies and software developments, an increasing number of global studies addressing ubiquitin signaling pathways are being conducted. Global protein–protein interaction studies shed light onto the network topology of deubiquitinating enzymes (50) and ubiquitin ligases (5) highlighting their complex interactions. In addition, the global mapping of ubiquitination sites could be increased from 110 sites in yeast 8 years ago (40) to 753 sites in human U2OS osteosarcoma cells this year (11). For the efficient identification of ubiquitination sites ubiquitinated proteins have to be enriched (1). Common strategies include immunoaffinity purifications using antiubiquitin antibodies or tagged ubiquitin versions, as for example, MYC-, HA-, FLAG-, or His-tagged ubiquitin, and pull downs by ubiquitin-binding domains (1). In the mentioned yeast study all endogenous ubiquitin genes were exchanged with His-tagged ubiquitin (40). Manipulation of mammalian cells is however more challenging and epitope-tagged ubiquitin is usually expressed ectopically as a surplus to its endogenous counterpart. After tryptic digestion of purified proteins ubiquitination sites are marked by a di-glycine remnant that can be identified by MS. However, other ubiquitin-like modifiers like NEDD8 and ISG15 also leave the same mark and, hence, cannot be discriminated from ubiquitin modifications at the tryptic peptide level. Recently, a purification strategy using a monoclonal antibody directed against the K-G-G mark was used successfully to profile ubiquitination sites (54). For further details we would like to point out a recent review specifically discussing MS strategies for analyzing ubiquitinated proteins (39).
Protease Cleavage Sites
Similar to lysosomal and proteasomal protein degradation, proteases in the extracellular milieu, such as matrix metalloproteinases, were believed to degrade all components of the extracellular matrix being important for basement membrane degradation and, thus, cell migration (7). However, recent developments for the unbiased analysis of protease cleavage sites by MS-based proteomics shed new light on underlying processes highlighting the tightly controlled and specific action of extracellular proteases (7). The characterization of proteases, their substrates, inhibitors, and interactions by proteomics and genomics approaches is termed “degradomics” and is a steadily growing research field (3, 7). As in the other fields of proteomics, degradomics has greatly benefited from the introduction of relative quantification strategies allowing the direct comparison of two or more cellular states, for example, protease-treated samples versus untreated control samples. In the following paragraph we briefly outline important technological proteomic advances for the analysis of protease cleavage sites.
Depending on their mechanism of action proteases can be divided into two groups: exoproteases that process protein termini and endoproteases cutting inside proteins. Regardless which protease group is studied, protease cleavage leads to the generation of new termini, so called neo-N- and neo-C-termini that can be identified by MS-based proteomics approaches characterizing the respective protease cleavage site. Similar to other PTMs, processed peptides are present in substoichiometric amounts and have to be enriched for an efficient identification. Each peptide carrying a neo-N-terminus has a counterpart carrying the respective neo-C-terminus and the pair can be used for experimental validation of the cleavage site. However, the enrichment of new C-terminal peptides has been proven to be experimentally more challenging (43). For the analysis of new termini two principal approaches can be differentiated: positive selection of newly formed termini followed by MS analysis, or negative selection, meaning the removal of all internal peptides, followed by MS analysis of the remaining newly formed termini containing peptides. During negative selection procedures neo-termini may not be lost due to inefficient enzymatic or chemical reactions as it might be the case for positive selection approaches. This might be one reason why negative selection procedures like N-terminal–combined fractional diagonal chromatography (21) and N- and C-terminal amine-based isotope labeling of substrates (30, 43) are widely used and have been applied successfully for unbiased, proteome-wide studies characterizing protease cleavage products.
Regardless which experimental approach has been used to enrich neo-terminal peptides, peptides carrying neo-termini are commonly only semitryptic, which has to be considered for the identification of termini by database searches. Special software tools have been developed to specifically address the needs of degradomics experiments. For detailed information we would like to refer to recent reviews (3, 25).
Protein Turnover
Protein turnover analysis was classically performed by administration of radiolabeled tracers, which are incorporated into the proteins of interest during synthesis [reviewed Hinkson and Elias (24)]. Bulk protein half-life dynamics were measured by the loss of scintillation counts. Accordingly, the half-life of a single protein was measured after protein purification followed by autoradiography. Recently, time-lapse microscopy in combination with the bleach-chase approach was used to study the influence of chemotherapeutic drugs on protein degradation (17). Global protein stability profiling studies have been performed by western blotting of proteins from TAP-tagged yeast strains (22), or by tracking of substrate-GFP fusion protein stabilities at single-cell resolution using flow cytometry (56).
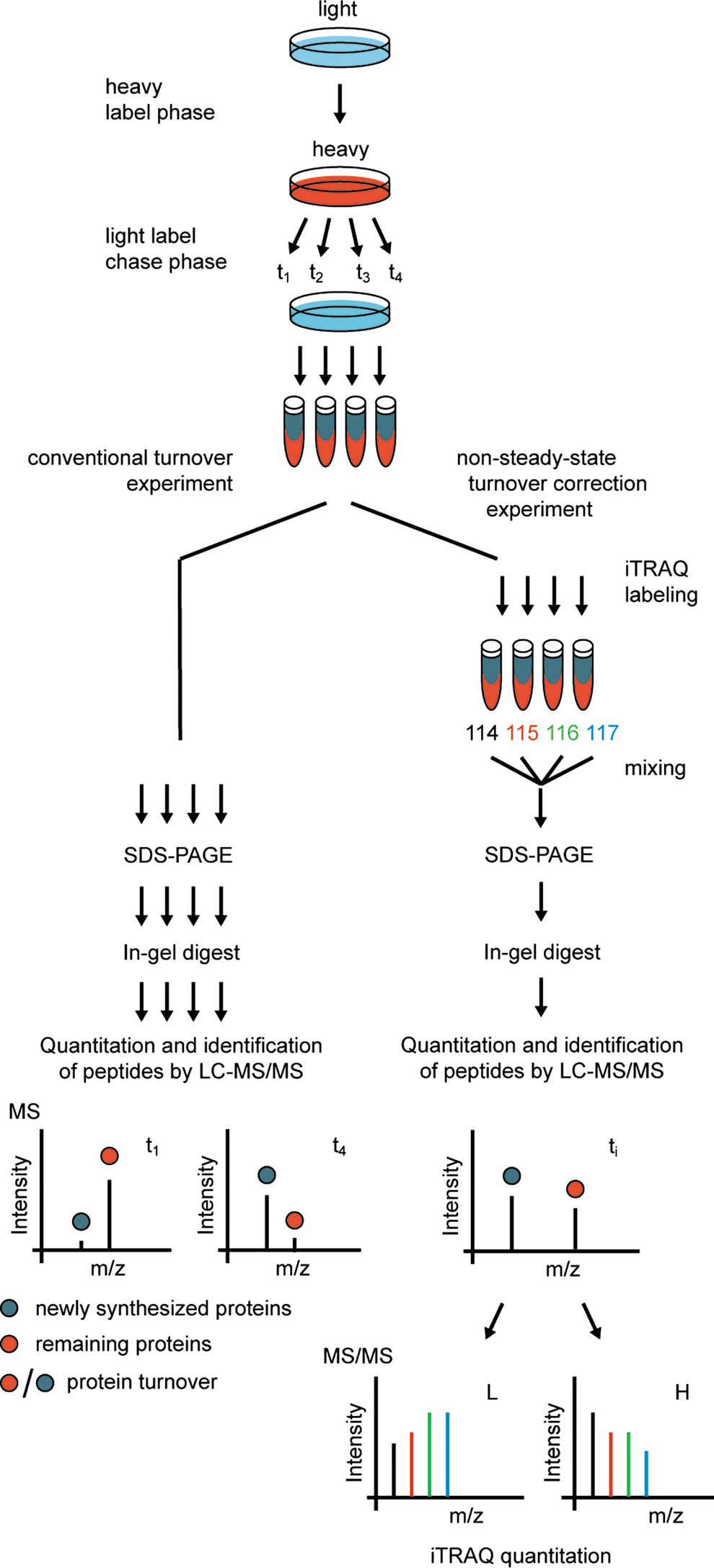
Lately, classic labeling principles have been transferred to quantitative proteomics by metabolic pulse labeling (41) where the decrease of stable isotope-labeled peptide signals is used to determine protein turnover rates under steady-state conditions. Exponentially fitted isotope decay curves obtained in turnover experiments are then used to estimate protein degradation constants. Such methods are able to record the half-lives of thousands of proteins in vivo at arbitrary temporal resolution and minimal cell perturbation. However, turnover studies are generally performed on growing or experimentally perturbed cells and therefore describe the overall flux of protein synthesis and degradation under non-steady-state conditions. To overcome this problem, a novel SILAC-based pulse experiment has been performed. Recording of time-resolved protein turnover under non-steady-state conditions in combination with measurements of absolute protein quantities provides a possibility to deduce both protein synthesis and degradation (46). Using an elegant triple SILAC approach, protein turnover, synthesis, and degradation were analyzed in different subcellular compartments identifying proteins that show different regulation depending on their subcellular localization (6). Another sophisticated approach with the aim to overcome the problem of non-steady-state conditions in growing cells has also been published recently (27). The authors developed a multitagging proteomic approach combining both SILAC and iTRAQ labeling in Streptomyces coelicolor providing abundance-corrected protein turnover rates (Fig. 5). Comparing MS-based approaches with classical radiolabeling, a big advantage is the measurement of specific protein turnover rates for thousands of proteins. However, as relative quantification errors are generally in the range of 15%–25% in MS-based proteomics experiments, the methods are still less accurate than classical radiolabeling-based approaches with errors typically being smaller than 10%.
Footnotes
Acknowledgments
The research leading to these results has received funding from the Excellence Initiative of the German Federal and State Governments through FRIAS and BIOSS, from the Deutsche Forschungsgemeinschaft (GZ DE1757/2-1), and from the Federal Ministry of Education and Research through GerontoSys II–NephAge (031 5896 A). We thank all Protein Dynamics group members, as well as M. Boerries, H. Busch, A. Schlosser, and colleagues from the ZBSA, for helpful discussions and support. We apologize to colleagues whose work was not covered in this review due to space limitations.