
Abstract
The Autophagy Stress Response Pathway
Degradation by autophagy is driven by the formation of membrane vesicles. Nascent autophagosomal membranes form in the cytoplasm to envelop around components to be degraded (58). Complete engulfment produces a newly formed autophagosome vesicle which is transported and fused with lysosomal compartments to enable content degradation. Autophagosomes can form quickly (e.g., within 5–10 min of nutrient starvation) and a primary membrane assembly site has been proposed to be an ER-associated compartment termed the omegasome. Other membrane sources such as mitochondria and plasma membrane can also contribute to autophagosome formation. Depending on the physiological context, autophagosome cargo can be nonselective or encapsulated via specific mechanisms with adaptor proteins such as p62 and NBR1. Roles for membrane transport machinery such as Rab and SNARE proteins are also being clarified in the autophagy maturation process.
By promoting overall cell health, autophagy is postulated to be a key determinant of cellular and hence organismal aging. Further, autophagy plays roles in various tissues for a range of diseases including cancer, heart failure, and neurodegeneration (38). Development of autophagy-based therapeutics holds large potential fueling the need for better understanding in fundamental regulatory mechanisms. Here, we discuss recent insights into the regulation and function of uncoordinated-51 like kinase (ULK) signaling proteins.
Autophagy Signaling Overview for Mammalian Cells
Studies from yeast demonstrated three primary autophagy signaling systems. One complex is centered around the protein kinase Autophagy 1 (Atg1), which functions in parallel with a second complex containing Atg6. Atg1 and Atg6 coordinately control a downstream protein conjugation system that involves the ubiquitination E1-like enzyme Atg7. Autophagy regulation by the network of Atg proteins has been reviewed in detail elsewhere (40). Table 1 summarizes key autophagy regulatory proteins in yeast and mammals.
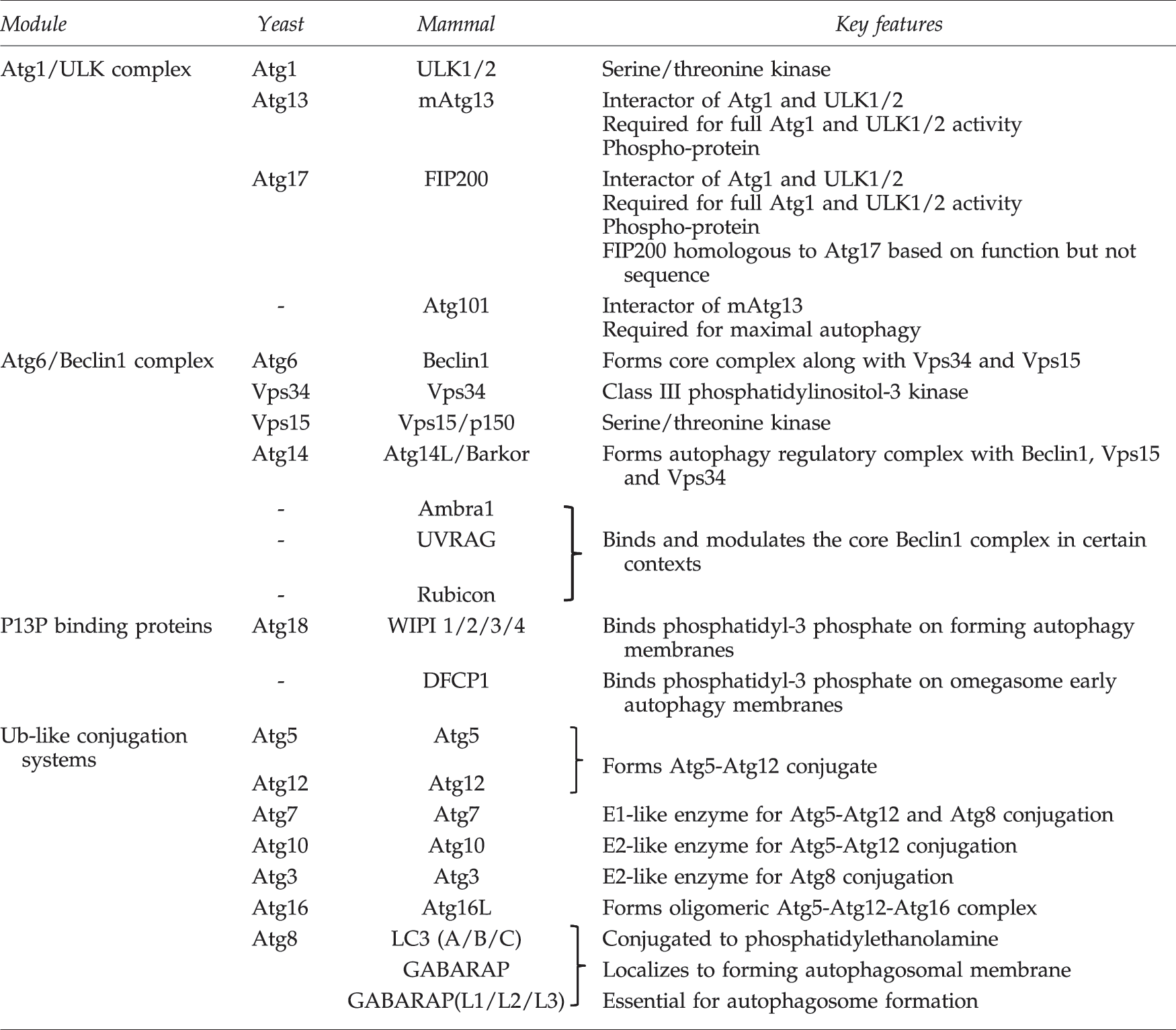
Atg, autophagy; ULK1, uncoordinated-51 like kinase 1; WIPI, WD-repeat protein interacting with phosphoinositides; DFCP1, double FYVE-containing protein 1; Ub, ubiquitin; LC3, microtubule-associated protein light chain 3; FIP200, focal-adhesion kinase family interacting protein of 200 kD.
The mammalian Atg1 complex is based upon the kinase ULK1 (or closely related family member ULK2). Additional components include Atg13, focal-adhesion kinase family interacting protein of 200 kD (FIP200) and Atg101 which form a stable core complex with ULK1/2 (36). From a systematic “hierarchal” analysis of mouse embryonic fibroblasts (MEFs), ULK signaling was positioned to function upstream of the Beclin1 (mammalian Atg6) pathway (18). For example, Beclin1-dependent membrane assembly steps are inhibited in FIP200 knockout MEFs. Beclin1 forms a mammalian autophagy regulatory complex along with the class III phosphatidylinositol (PI)-3 kinase, Vps34, and associated subunits Vps15 and Atg14L/Barkor. Additional interactors such as Ambra1, UVRAG, and Rubicon can direct the core Beclin1-Vps34-Vps15 complex to function in autophagy and autophagy-independent membrane trafficking pathways. The primary role of the Beclin1 autophagy complex is to generate PI3P lipid products at ER-associated autophagosomal initiation sites that interact with PI3P-binding effector proteins such as WD-repeat protein interacting with phosphoinositides (WIPI) members and double FYVE-containing protein 1 (DFCP1). The role of omegasome membrane structures labeled by DFCP1 for autophagosome formation requires more clarification. However, Beclin1-dependent recruitment of WIPI proteins is essential for autophagy.
ULK- and Beclin1-directed assembly of autophagy initiation sites occurs upstream of two ubiquitin (Ub)-like conjugation systems that involve mammalian homologues of yeast Atg3, Atg5, Atg7, Atg8, Atg10, Atg12, and Atg16. One system conjugates a lysine residue of Atg5 to a C-terminal glycine residue in Atg12 through the action of Atg7 (E1-like enzyme) and Atg10 (E2-like). Atg5-Atg12 conjugates further interact with mammalian Atg16 to form oligomeric complexes that localize to autophagosomal membranes during early stages of formation. The second system conjugates glycine residues of mammalian Atg8 homologues to phosphatidylethanolamine through a lipidation reaction involving Atg7 and the E2-like enzyme, Atg3. Atg8 lipidation is dependent on the Atg5-Atg12-Atg16 complex. So far, the best characterized members of the mammalian Atg8 family belong to the microtubule-associated protein light chain 3 (LC3) family. Contributions of other mammalian Atg8 homologues such as GABARAP require further investigation. Generally, lipidation of Atg8 represents a relatively late step during the autophagy initiation sequence and higher levels of Atg8 lipidation correlate with autophagy activation in most mammalian cell systems. Lipid-modified Atg8 proteins localize to autophagosomal membranes during early formation stages where they regulate membrane fusion (44), although this role has raised some controversy (43). Localization of LC3 to autophagosome structures and LC3 lipidation are widely used experimental markers for autophagy. The strengths and weaknesses of LC3 and other autophagy assays have been summarized (39).
Mammalian Target of Rapamycin Complex 1 Suppresses Autophagy
Original evidence from yeast showed that Atg1 functioned downstream of the TOR (target-of-rapamycin) nutrient-sensitive signaling pathway. Rapamycin inhibits TOR leading to decreased phosphorylation of Atg13 and autophagy induction. In mammals, autophagy is controlled by mammalian target of rapamycin complex 1 (mTORC1), which is formed from the kinase mTOR along with regulatory cofactors RAPTOR, PRAS40, and mLST8. mTORC1 serves as a central control point that suppresses cellular catabolism via autophagy while stimulating protein synthesis and overall cell growth under conditions of high nutrient availability (Fig. 1). A distinct complex mTORC2 is formed with cofactors RICTOR, Sin1, and mLST8. mTORC2 is insensitive to nutrients with ascribed roles for regulating actin and Akt pathways.
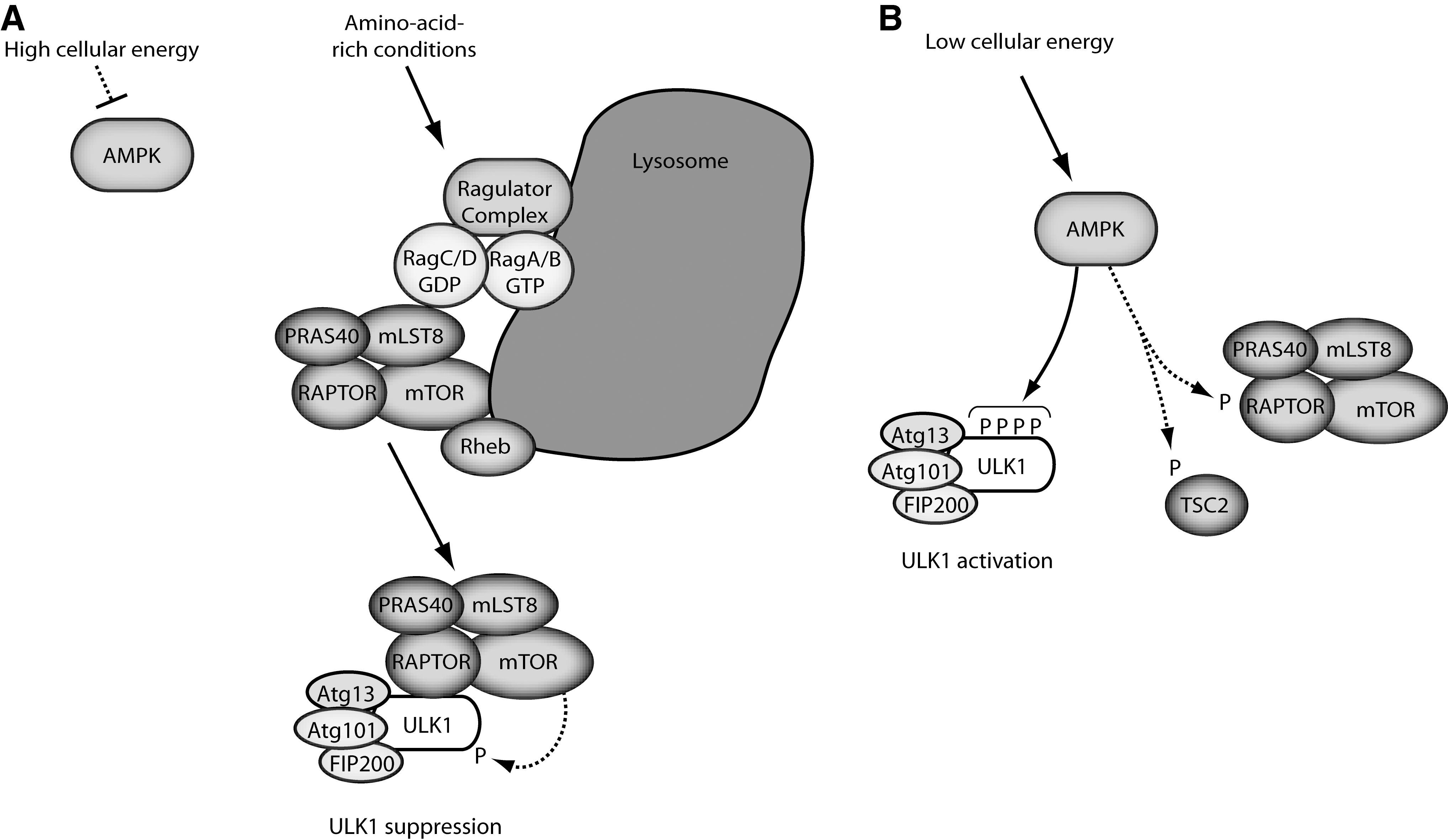
A primary characteristic of mTORC1 is sensitivity to intracellular amino acids and this regulation is critically dependent upon Rag family GTPases. The mechanism involves an amino-acid sensitive Ragulator complex (consisting of proteins MP1, p14, and p18) (49) that promotes translocation of mTORC1 to lysosomal compartments where heterodimeric Rag complexes (Rag A/B with Rag C/D) reside. Localization of mTORC1 onto lysosomes promotes further interaction with the GTP binding protein Rheb that activates mTORC1 catalytic activity (33). mTORC1 translocation onto cellular sub-compartments is a prominent regulatory mechanism that can function under other physiological contexts, for example during oncogene induced cell senescence (45).
Regulation of mTORC1 by Cellular Energy Levels and AMPK
Nutrient-dependent autophagy in mammalian cells is best characterized in terms of amino acid starvation. However, autophagy in response to energy deprivation (i.e., low glucose availability and cellular ATP) is becoming better understood (5, 25, 26). The working model has energy levels signaling to autophagy through an AMP-activated protein kinase (AMPK)-mTORC1 pathway. AMPK is activated under conditions of low cellular energy by the binding of AMP to allosteric activating sites. AMPK activation also requires phosphorylation by the upstream kinase LKB1. Binding of ADP to AMPK presents yet an additional mechanism that impedes AMPK dephosphorylation (61).
AMPK has been observed with cytoplasmic and nuclear localization (48) but roles and regulation of subcellular distribution are not well characterized. However, mechanisms of AMPK cross-talk to mTORC1 have been described. One signaling route involves AMPK-dependent phosphorylation and activation of TSC2 (tuberous sclerosis protein 2). The TSC2/TSC1 complex functions as a GTPase activating factor that negatively regulates Rheb (and hence mTORC1). Low energy and activated AMPK thereby stimulate TSC2/TSC1 to inactivate Rheb-mTORC1. AMPK has a second pathway to inhibit mTORC1 directly by phosphorylating RAPTOR. From these nutrient and energy inputs, mTORC1 and AMPK signal downstream to autophagy. Recent evidence of direct binding and phosphorylation between the mTORC1, AMPK, and ULK complexes has substantiated the underlying mechanism behind this control.
Regulation of the ULK Complex by Phosphorylation
In yeast, interaction with cofactors Atg13 and Atg17 is critical for Atg1 kinase activity. Further, yeast Atg1 complex formation is negatively regulated by TOR-mediated phosphorylation of Atg13 on at least eight serine residues (23). In mammals, FIP200 is the proposed functional homologue of Atg17 (14). As in yeast, maximal ULK1/2 kinase activity requires interaction with mAtg13 and FIP200 (12). The mammalian ULK complex also contains the essential protein Atg101, although the mechanistic role for this subunit needs more characterization (17, 35). In contrast to yeast, the core mammalian ULK1/2-FIP200-mAtg13-Atg101 is relatively stable under different nutrient conditions (14, 16). However, the ULK1 internal proline/serine-rich domain interacts with RAPTOR and this binding is promoted under amino acid-rich conditions (16). AMPK also binds the ULK proline/serine-rich domain in a nutrient sensitive manner (25, 28, 51).
The mammalian ULK complex is regulated by a series of phosphorylation events on ULK1/2, mAtg13, and FIP200. Understanding of ULK1/2 phosphorylation has advanced significantly in recent years. ULK1 and ULK2 family members share most homology along their entire length and are best characterized for autophagy. Both ULK1/2 have an N-terminal kinase domain flanked by an internal serine/proline-rich region, followed by a C-terminal domain that shows higher levels of conservation across family members (Fig. 2). Initial mass spectroscopy (MS) studies of immune-precipitated exogenous ULK1 could map 16 phosphorylation sites distributed all three functional domains (7).
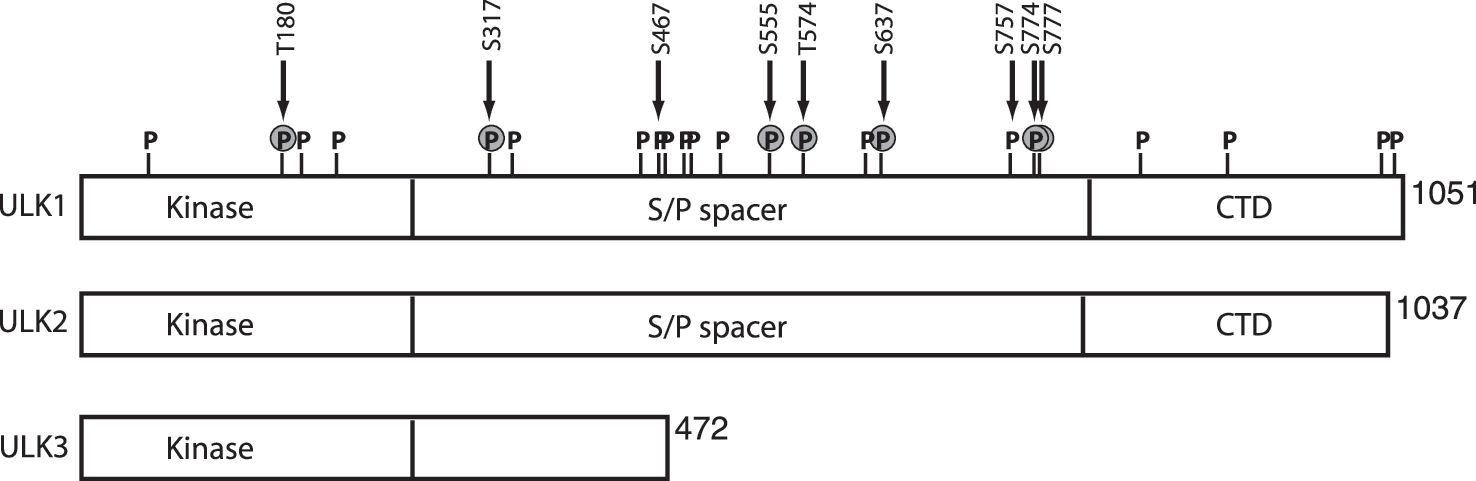
Functional roles of ULK phosphorylation were elucidated by multiple research groups that approached the issue from different angles. A search for new AMPK substrates identified ULK1 and further characterization in this system led to four sites (S467, S555, T574, and S637 of mouse ULK1) that resembled the AMPK phosphorylation recognition motif (Table 2) (9). Using phospho-specific antibodies and in vitro kinase assays, AMPK-mediated phosphorylation of ULK1 S467 and S555 were characterized in detail. These two serine residues showed increased phosphorylation in different cell types treated with AMPK-activating compounds. More definitively, serine to alanine substitution of the four AMPK sites yielded an ULK1 mutant that could no longer support basal or nutrient-dependent autophagy. Consistent with an essential role for the AMPK-ULK pathway, mouse hepatocytes lacking AMPK showed signs of impaired autophagy such as accumulation of the p62 cargo adaptor protein and damaged mitochondria. The requirement for AMPK in autophagy is further supported by other work using pharmacology, siRNA knockdowns, and genetic targeting of C elegans and Drosophila (9, 15, 31, 34). The S555 site may have a specific role in binding 14-3-3 proteins (1, 9) but the signaling function of this interaction is not clear.
Yeast T226/mouse, human T180.
Mouse S555/human S556.
Mouse T574/human T575.
Mouse S637/human S638.
Mouse S757/human S758.
In Atg13-deficient cellular background.
Mouse S777 not well conserved in human ULK1.
AMPK, AMP-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1.
Two other AMPK sites in ULK1 (S317 and S777, mouse protein) were identified when searching for modifications stimulated by glucose starvation of cells (25). Although the S317 and S777 residues were not close matches to the AMPK substrate motif, these two sites were phosphorylated in an AMPK-dependent manner after glucose starvation of MEFs. Using mutagenesis approaches, S317 and S777 were shown to be essential for AMPK to activate ULK1 and autophagy after AMPK stimulation (e.g., glucose starvation). Phosphorylation at S317 and S777 could be further confirmed with in vitro assays using purified AMPK. Thus, two independent approaches have identified a set of six AMPK-regulated sites in ULK1 with functional roles. The -MFSVG[pS]SSS- amino acid sequence surrounding mouse S777 is not conserved in human ULK1 and more clarification is needed for this phospho-site. More analysis is also needed to better define specific roles for each site and to explore possible roles for ULK2.
Further studies of AMPK-mTORC1 cross-talk revealed an additional phosphorylation mechanism. The fundamental observation was that AMPK-ULK1 binding was not regulated by glucose availability, but by mTORC1 (25). An additional site (mouse ULK1 S757) was mapped that could be phosphorylated by mTORC1, which resulted in destabilization of AMPK-ULK1 binding. The emerging model showed a balance of mTORC1 and AMPK function under different conditions (Fig. 1). With high amino acid availability, interactions between activated mTORC1 and ULK1 are promoted (16) leading to mTORC1-dependent phosphorylation at S757 of ULK1. S757 phosphorylation limits AMPK-ULK1 interaction thereby preventing AMPK from phosphorylating ULK1 and activating autophagy. Glucose starvation controls from the opposite angle. Activated AMPK suppresses mTORC1 via TSC2 and RAPTOR phosphorylation, thereby enabling AMPK to bind and activate ULK1.
Although the AMPK-ULK interaction model explains many data, it does not readily explain how amino acid starvation activates the ULK1 complex. mTORC1 would be inhibited, but AMPK signaling appears also repressed after amino acid deprivation (51) and so it is unclear what kinase is activating ULK1 in this context. A further independent study provided additional complexities. Comparative phospho-proteinomics were performed to search for modifications after amino acid starvation (autophagy activation), identifying 13 sites on human ULK1 (51). Of these, S638 and S758 showed dramatic de-phosphorylation under amino acid starvation. Due to mouse/human ULK1 differences, S638 and S758 correspond respectively to S637 and S757 discussed above (Table 2). Using time course analyses with phospho-antibodies, de-phosphorylation of S638 was observed to occur faster than S758 de-phosphorylation after amino acid starvation, revealing step-wise aspects of the mechanism. Knockdown approaches confirmed roles for mTORC1 and AMPK. S758 was concluded to be an mTORC1 site (fitting with model) while S638 was regulated by both mTORC1 and AMPK. Thus, de-phosphorylation of the S638 candidate AMPK site correlated with amino acid starvation and autophagy activation. In this system, AMPK-ULK1 interaction was dissociated after amino acid starvation which does not fit the simplistic model. Additional work on S758 revealed further surprises. A ULK1 S758A mutant showed normal function when introduced into otherwise wildtype cells. When introduced into Atg13-deficient cells, ULK1 S758A enabled a faster autophagic response to amino acid starvation. These findings suggest that ULK1 phosphorylation might regulate autophagy kinetics and also the existence of a core complex (ULK1/2-FIP200-Atg101) without mAtg13, both intriguing concepts. Overall, mechanisms linking nutrients, mTORC1 and AMPK to ULK1-dependent autophagy have been resolved to greater detail. Other recent work has demonstrated an Akt-dependent phosphorylation site at S774 of ULK1 (mouse) which may function in linking the insulin-Akt pathway to autophagy repression (1). Further investigation will define more integrated signaling models.
Homology between yeast and mammalian Atg1 proteins is limited, but lessons can be learnt across systems. Yeast Atg1 also shows complex patterns of phosphorylation. One MS analysis identified 29 Atg1 phosphorylation sites, most of which were in noncatalytic regulatory regions (24). However, functional data could be shown for phospho-sites T226 and S230 which are located in the kinase domain activation loop. Mutation at these sites disables Atg1 kinase function and autophagy. It had been previously shown that T226 showed increased autophosphorylation in cells with activated autophagy (65), suggesting that Atg1 was regulated by modification of the kinase activation loop. Further MS analyses identified nine additional novel phospho-sites in Atg1 (63), including S34 within the glycine-rich loop of the kinase ATP-binding pocket. Phospho-mimetic mutation of S34 to aspartate or glutamate impaired Atg1 function suggesting that phosphorylation at S34 may serve to repress Atg1-mediated autophagy. These functional data highlight regulatory roles for kinase domain phospho-sites. Mutation of T180 in the ULK1 activation loop (homologous to yeast T226) also decreased autophosphorylation (1), indicating conservation of some kinase regulatory mechanisms. Autophosphoryaltion of yeast Atg1 is also in part controlled by Atg13-mediated oligomerization of Atg1 (64). As ULK1 exists in large complexes (up to 3 MDa) (16), trans-autophosphorylation between ULK1/2 proteins is conceivable.
Together, the multiple MS studies reveal that Atg1/ULK proteins are phosphorylated on a large number of sites (7, 9, 24, 51, 63), leading to the arduous task of assigning functional roles. Phospho-sites are so far best characterized in the ULK1 internal regulatory region while analysis of yeast Atg1 has provided further insight of sites within the kinase domain. The C-terminal domain of ULK1/2 interacts with mAtg13, FIP200, and membranes (3, 16, 21). As such, phospho-sites in the C-terminal domain might potentially modulate ULK1 binding events. Work in C elegans has shown phosphatase PP2A to bind and regulate the function of Atg1 (47). PP2A is also known to negatively regulate yeast autophagy (66). A role of PP2A or any other phosphatase for balancing the ULK complex has not yet been described.
ULK Transcript Regulation
Under different physiological contexts, ULK expression is transcriptionally modulated, demonstrating another level of regulation. ULK1 and ULK2 transcripts show widespread patterns in adult mammalian tissues with notable enrichment in brain, heart, and skeletal muscle (56, 62). ULK1, but not ULK2, is upregulated during erthryocyte maturation and this may underlie its specific role for autophagic mitochondrial clearance (26). ULK1 expression increased in several cancer cell lines after DNA damage (13). DNA-damage upregulation of ULK1 was p53-dependent and p53-binding sequences could be identified in 5-prime transcriptional regulatory regions of ULK1 and ULK2. Upregulation of ULK1/2 in this context promotes autophagy-associated cell death. Evidence from esophageal cancer cells and tissues further support the potential of using ULK1 expression as a prognostic biomarker (20). Other findings from myoblasts have demonstrated a role for transcription factor FoxO3 in upregulating ULK2 (69). By comparison, oncogene-induced cell senescence primarily upregulates the shorter ULK3 isoform (68) (Fig. 2). Multiple transcriptional networks might converge to regulate differential expression patterns within the ULK family. Further characterization of expression patterns could provide more insight into specific physiological roles of ULK isoforms.
ULK Regulation of Autophagy and Vesicle Trafficking
It is only beginning to be understood how ULK function fits into the mechanism of autophagosome formation. With autophagy activation, the ULK complex translocates to ER-associated autophagosome formation sites as supported by evidence from multiple subunits (12, 14, 17). Detailed analysis has characterized the partial ULK1 co-localization with PIP3-dependent membrane assembly sites (e.g., marked by Atg14L, DFCP1, or WIPI1) (18). Further, ULK1 has been shown to associate with ER-related membranes containing the p62 adaptor protein (19). These p62-associated membranes are putative early autophagosome precursor compartments as they form independently of ULK1/2 (FIP200−/− contexts). Together, the data suggest that ULK signaling may regulate membrane dynamics during early formation steps as compartments enriched in p62 and VMP1 (vacuole membrane protein 1) become abnormally trapped in FIP200−/− cells (Fig. 3 summarizes downstream pathways).
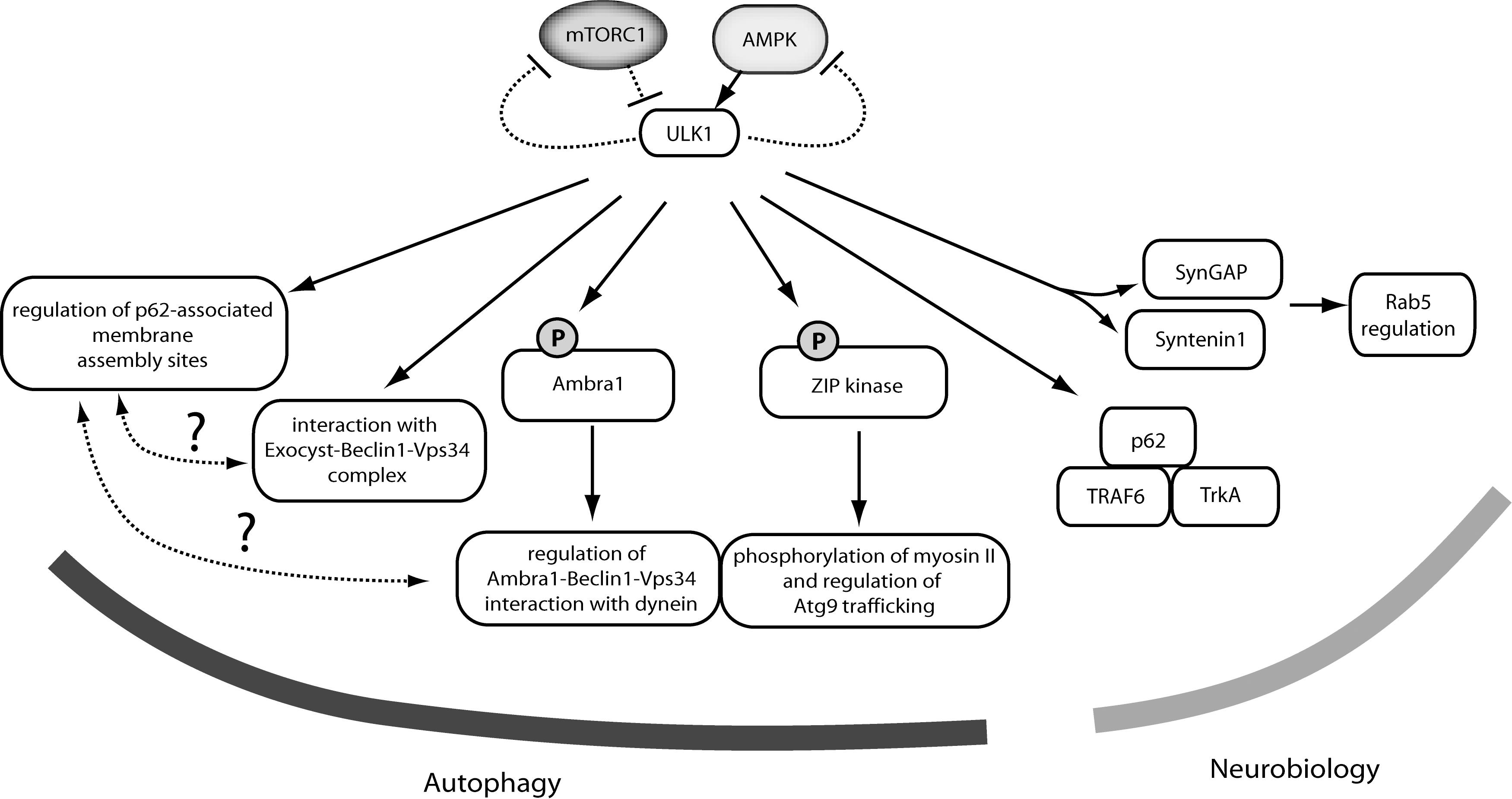
Consistent with roles in coordinating membrane flow, ULK1 regulates translocation of the autophagy transmembrane protein Atg9 (67). In mammalian cells, Atg9 transits from Golgi-associated compartments toward a distinct cytosolic pool after autophagy activation, acting as an additional membrane donor for forming autophagosomes. Atg1/ULK-dependent membrane trafficking for autophagosome assembly is a process conserved from yeast. Recent work combining Drosophila and mammalian systems could demonstrate Atg1/ULK1-dependent phosphorylation of a myosin light chain kinase protein (ZIP kinase in mammals, spaghetti-squash activator in Drosophila) (52). Once activated by ULK1, ZIP kinase phosphorylates myosin II regulatory light chain which controls starvation-induced trafficking through direct interaction with Atg9.
The ZIP kinase pathway provides an attractive mechanism that links ULK1 kinase function with downstream autophagy. As with Atg1, ULK1 activity increases with autophagy induction (starvation or rapamycin treament) (14, 21). Several other ULK1 substrates suggest additional alternative downstream pathways. Atg13 and FIP200 can serve as ULK1/2 substrates (3, 16, 21) but functional roles are not understood. Paxillin is another Atg1/ULK candidate to be further explored (4).
Ambra1 is an interesting ULK1 substrate that mechanistically bridges with Beclin1-Vps34 signaling (6). ULK1-mediated phosphorylation was required for proper dissociation of a dynein-light-chain-Ambra1 complex from a larger dynein microtubule-dependent motor. Thus, ULK1 might regulate autophagy by releasing Ambra1-Beclin1-Vps34 activity that had been sequestered on microtubule-associated dynein motors. Intriguingly, other evidence supports a direct connection between ULK and Beclin1-Vps34 pathways. Components of the exocyst complex (better understood for secretory membrane transport) were shown to directly interact with ULK1 and Beclin1-Vps34 (2). The model from this work has ULK1 directing formation and localization of different sub-complexes of Beclin1-Vps34 with either the Exo84 or Sec5 exocyst subunits. These dynamic interactions were starvation dependent and orchestrated by the small GTP binding protein RalB. Other evidence supports a link between ULK1 S758 phosphorylation and the modulation of Vps34 activity (51). Thus, ULK1 has several substrates and binding partners that can mechanistically link to autophagosome formation. The coordination of ULK, Beclin1, and Atg9 pathways can be further defined with future studies.
In addition to activating autophagy, ULK1 signaling has other consequences. ULK1 can phosphorylate all three subunits of AMPK and these modifications repress AMPK activation toward further downstream signaling (32). The roles of ULK1 signaling to AMPK under different physiologic contexts requires more analysis. Another proposed feedback loop links Atg1 to the negative regulation of TOR as first demonstrated in Drosophila (50). Mammalian cells have a homologous feedback link between ULK to mTORC1 (29) and a putative mechanism includes ULK1-dependent phosphorylation of RAPTOR leading to decreased mTORC1 activity toward downstream (8). As mTORC1 primarily serves to repress ULK function, feedback from ULK1/2 to inhibit mTORC1 might form a positive reinforcement circuit. The ULK1 feedback loop can also suppress the ability of mTORC1 to promote cell proliferation and this role is fully separate from autophagy (22).
A comprehensive model of ULK function would include pathways for neuronal vesicular transport that function in parallel with autophagy. C elegans with mutation of their Atg1 homolog (unc-51) were originally characterized for uncoordinated behavior with an underlying axonal defect. Roles for Atg1/ULK in neuronal development are supported by findings from flies and mammals. A number of possible mechanisms have been proposed. The ULK1 C-terminal domain binds syntenin and SynGAP, proteins implicated in Rab5-dependent vesicular trafficking (41, 57). Drosophila Atg1 directly phosphorylates factors involved in vesicular transport such as unc-14, unc-76, and VAB-8L (55). Drosophila Atg1-dependent ERK signaling plays a role in synaptic formation (59). Work from Zebrafish showed a neurological role for ULK2 involving a potassium channel pathway (53). Further work is needed to determine which ULK neuronal model predominates under varying contexts.
Other data support an additional mammalian neuronal signaling complex containing ULK1/2, TrkA NGF receptor, TRAF6, and p62 (70). NGF-stimulation causes TRAF6-mediated ubiquitination of ULK1/2 and resulting interaction with the Ub-binding domain of p62. However, there is no evidence so far linking the neuronal ULK1/2-p62 pathway to autophagy and it remains unclear how ULK roles in autophagy and vesicle trafficking are coordinated. The role of other ULK complex components (mAtg13, Atg101, FIP200) in neuronal trafficking is also uncertain. C elegans mutants of the Atg13 homolog epg-1 are not described to have a neuronal phenotype (54). FIP200−/− mice show embryonic lethality but with abnormal heart and liver development (11). Mice with neuronal-specific FIP200 deletion do show pathology, but the phenotype suggests defective autophagy rather than neuronal trafficking (30). In this respect, ULK1/2 might regulate neuronal pathways without its core autophagy interacting partners.
Mouse Loss-of-Function Studies for Autophagy Genes
Targeting Atg1 homologs in yeast, C elegans and Drosophila lead to strong autophagy deficiency. In mammalian cells, roles have been widely elucidated using knockdowns of ULK1, ULK2, or both in a variety of cell contexts. Complete genetic targeting of ULK-dependent autophagy in mammals has been complicated by the presence of up to five putative family members (ULK1-ULK4 and STK36) in vertebrates. All evidence indicates that the ULK1/ULK2 isoforms control nutrient-dependent autophagy. ULK3 is a relatively smaller protein that regulates a specific form of autophagy associated with cell senescence (68) (Fig. 2).
Loss of key autophagy proteins such as Atg5, Beclin1, and Atg7 clearly ablates autophagy with embryonic or neonatal lethality (37). In contrast, ULK1−/− mice have relatively normal viability and autophagy (26). This lack of strong phenotype indicates that ULK1-dependent processes are not strictly essential for development and overall survival. However, cells derived from ULK1−/− mice such as erythrocytes, fibroblasts, and hepatocytes show signs of defective autophagy and pathological phenotypes such as impaired mitochondrial clearance (9, 21, 26). We and others have detected differences in ULK1/ULK2 regulation or function under certain experimental contexts (3, 13, 27). The precise contributions of ULK1/2 isoforms for cellular and organismal physiology need further clarification. ULK1 might control particular cell-specific subclasses of autophagy, although essential autophagy functions may be maintained by joint function of ULK1 and ULK2. The idea of ULK1/2 redundancy is further supported by the lack of strong phenotype after ULK2 knockout (5, 27).
Mice with combined ULK1 and ULK2 disruption show postnatal lethality consistent with autophagy impairment comparable to Atg5, Atg7, and Atg16L loss (5). Embryonic fibroblasts from ULK1/2 double knockout mice show clearly targeted autophagy after amino acid starvation, consistent with a full block of mTORC1-ULK signaling. Surprisingly, autophagy in fibroblasts with double ULK1/2 knockout was fully functional after glucose starvation. Prolonged glucose starvation (e.g., overnight) triggered a compensatory pathway in which amino acids are metabolized to generate carbon backbone intermediates, producing intracellular ammonia as by-product (10). Ammonia in this context acts as a soluble factor that is sufficient to trigger robust autophagy. Ammonia-induced autophagy is independent of mTORC1 and the ULK1/2 complex but further details on mechanisms are still unclear.
Other components of the ULK core complex have been targeted to provide complementary evidence. FIP200−/− MEFs are well characterized to show defective basal and amino-acid starvation dependent autophagy (14). FIP200−/− MEFs represented loss of ULK function for the hierarchal classification of autophagy signaling (18). Without FIP200, high molecular weight ULK1 complexes are not formed and ULK1 fails to display maximal catalytic activity (14, 16). Interestingly, FIP200 is implicated in several other signaling pathways. FIP200 regulates focal adhesion kinase signaling and mTORC1 dependent cell growth via direct interactions with TSC. FIP200 (also known as RB1CC1 or RB1-inducible coiled-coil1) can induce RB1 expression and regulate TNFalpha signaling via a ASK1-TRAF2 dependent mechanism (11). It is unclear how the different roles of FIP200 might coordinate with ULK function and autophagy. Interaction data suggests that FIP200 could mechanistically link a cytosolic pool of p53 to ULK1 for autophagy regulation (42). Involvement in multiple physiological roles could explain why FIP200−/− knockout mice show embryonic phenotypes unlike other autophagy knockout mouse models (11, 37). However, conditional FIP200 deletion can produce phenotypes of autophagy disruption, for example, in cells within the mammary tissue and hematopoietic stem cells (60).
Knockout of autophagy factors has provided a wealth of insight into different regulatory pathways. Atg5−/− MEFs have a full block of LC3 lipidation with strongly defective autophagy and this cell system has allowed clear assignment of autophagy functions in a variety of contexts. Intriguingly, alternative forms of autophagy have been proposed based on Atg5-deficient cells treated with DNA damaging agents (46). “Alternative autophagy” was independent of Atg5 and LC3 lipidation, but still dependent on ULK/FIP200 and Beclin1/vps34 pathways (46). These data originally suggested ULK and Beclin1 functions might be primary and absolutely essential for autophagy. However, LC3 conversion is not strictly dependent on the ULK complex. Low amounts of lipid-conjugated LC3 are still observed in FIP200−/− and ULK1/2 double knockout cells (5, 14). Further, mTORC1-independent autophagy (e.g., triggered by ammonia) is fully functional without ULK1/2. Thus, ULK signaling may primarily serve to activate LC3 lipidation and autophagosome formation pathways in response to mTORC1-dependent cues such as amino acid availability. Overall, while a basic scheme for autophagy signaling is available, one model likely cannot account for all forms of mammalian autophagy.
Conclusions
Our current view of Atg1/unc-51/ULK function is based on almost 20 years of data representing yeast to mammals. A primary role for autophagy is established but Atg1 family members also regulate vesicle transport in neurons of higher metazoans. Recent studies have provided important insights into the multiple binding and phosphorylation mechanisms linking mTOR and AMPK to the ULK autophagy complex. Better understanding of the signaling network between mTOR, AMPK, and ULK will improve our ability to target these pathways to modulate autophagy. The expanding list of Atg1/ULK1 substrates features several candidates with potential mechanisms that link to autophagosome formation and vesicular transport, leading to much scope for future work.
Footnotes
Acknowledgments
We apologize to those authors whose works we could not cite due to space limitations. E.Y.C is supported by the Royal Society of London for Improving Natural Knowledge.