
Abstract
Introduction
Despite this awareness, little detail of the mechanisms underlying CO toxicity is known. The original proposal that CO is toxic through hypoxemia via its binding to hemoglobin (14) is now considered unlikely as a sole explanation, since many features of CO toxicity are not observed following hypoxic/ischemic damage (42). Instead, mechanisms that also account for physiological (signaling) actions of CO are being considered as possible toxicity mechanisms. For example, CO can stimulate increased reactive oxygen species (ROS) primarily from mitochondria (3, 31, 50). This may be protective—a form of “oxidative preconditioning” (3, 47)—but could be deleterious by promoting oxidative stress. CO can also stimulate NO production, possibly by activating nitric oxide synthase (19, 21). Production of both ROS and NO by CO can also increase oxidative/nitrosative stress through formation of peroxynitrite [ONOO-; (15)]. Oxidative stress can cause neurodegeneration via disruption of Ca2+ homeostasis, a process implicated in aging and neurodegenerative diseases such as Alzheimer's disease (11, 23, 24). For example, apoptosis can arise from Ca2+-dependent activation of caspases, and the rise of [Ca2+]i can occur through oxidative triggering of Ca2+ influx and/or disruption of Ca2+ buffering / extrusion (2, 11, 20, 24). We have examined the effects of CO on neuronal Ca2+ signaling using the human neuroblastoma, SH-SY5Y, a line used extensively for studying Ca2+ homeostasis and neuronal signaling (18, 33 —35, 46). We reveal that CO stimulates formation of ONOO- and thereby disrupts Ca2+ signaling via plasma membrane Ca2+ ATPase downregulation. Such effects, confirmed in brain tissue of rats exposed to CO in vivo, may contribute to the neurological damage associated with CO poisoning.
Innovation
Although carbon monoxide (CO) is currently under intense investigation as a major intracellular signaling molecule, it is better established as a highly toxic agent. However, very little is known of the mechanisms underlying its neurotoxicity. The present study shows for the first time that acute CO exposure causes major disruption to neuronal Ca2+ homeostasis. This deleterious action arises from the ability of CO to stimulate increased production of both reactive oxygen species and nitric oxide. These agents combine to form peroxynitrite which leads to damage/degradation of the plasmalemmal Ca2 + ATPase (PMCA), a key protein required for control of Ca2+ homeostasis. Loss of PMCA was also observed in rat brain samples following inhalation of CO at sublethal levels. These finds reveal for the first time a key homeostatic protein targeted for destruction by toxic levels of CO, and identify the underlying mechanism leading to PMCA degradation. Through identification of the underlying mechanisms, our study reveals potential pathways which may provide therapeutic targets to combat the neurological damage associated with CO toxicity.
Results
Effects of CO on Ca2+ signaling
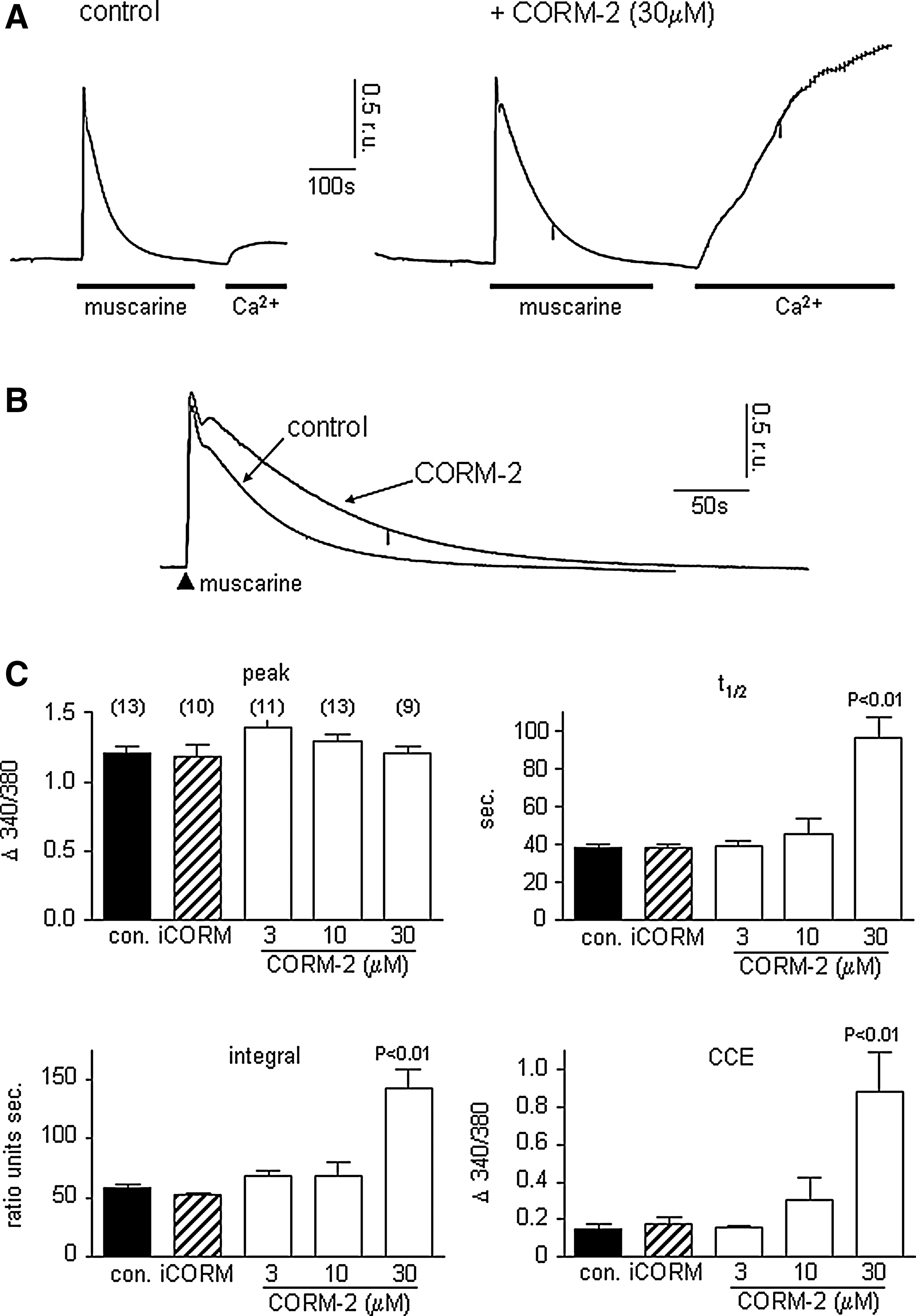
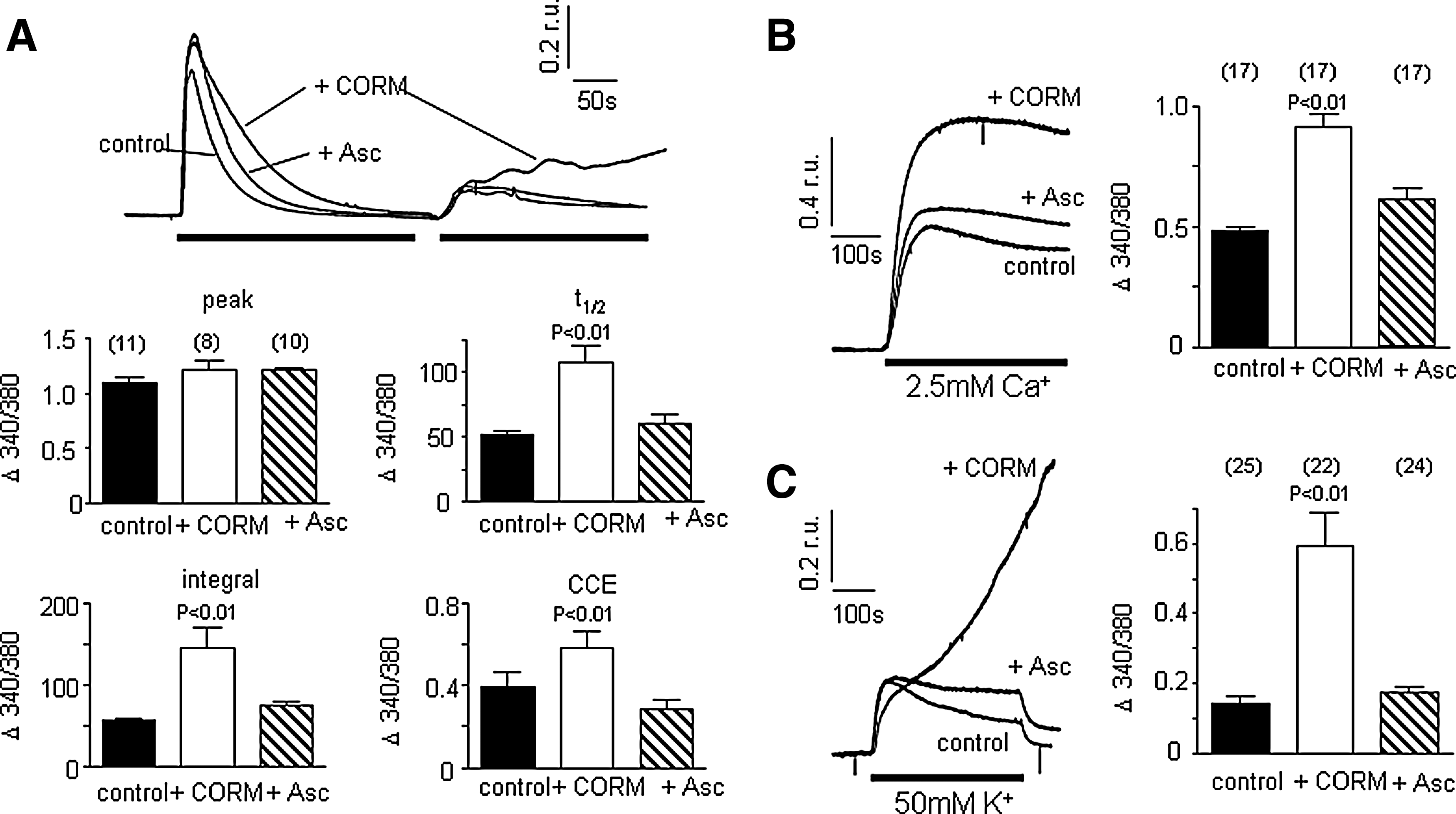
Activation of M3 muscarinic receptors in SH-SY5Y cells triggers release of Ca2+ from intracellular stores following generation of inositol trisphosphate (IP3) which in turn activates store-depletion mediated (capacitative) Ca2+ entry [CCE; (6, 30, 39, 40)]. These events can be resolved temporally by exposing cells to muscarine (100 μM) in the absence of extracellular Ca2+ (replaced with 1mM EGTA). This evoked a rise of [Ca2+]i due to mobilization from intracellular stores. Following removal of muscarine, restoring Ca2+ to the extracellular solution causes a second rise of [Ca2+]i resulting from CCE (Fig. 1A). The effects of the CO donor, CORM-2, on these responses are illustrated in Figures 1A and B. In the presence of CORM-2 (30 μM), two striking differences were noted: first, the transient rise of [Ca2+]i evoked by muscarine in the absence of extracellular Ca2+ decayed much more slowly, as highlighted in Figure 1B (quantified as transient integral and t1/2 in Fig. 1C). Second, CCE observed when Ca2+ was restored to the perfusate following washout of muscarine was dramatically enhanced (Figs. 1A and 1C). None of these parameters was significantly altered by iCORM (30 μM), the inactive form of CORM-2 which does not release CO (hatched bars, Fig. 1C). CORM-2 was applied in this and all subsequent figures for 3 min before additional experimental manoeuvres, and present throughout. At 30 mM, CORM-2 led to a CO concentration of 33 μM in solution.
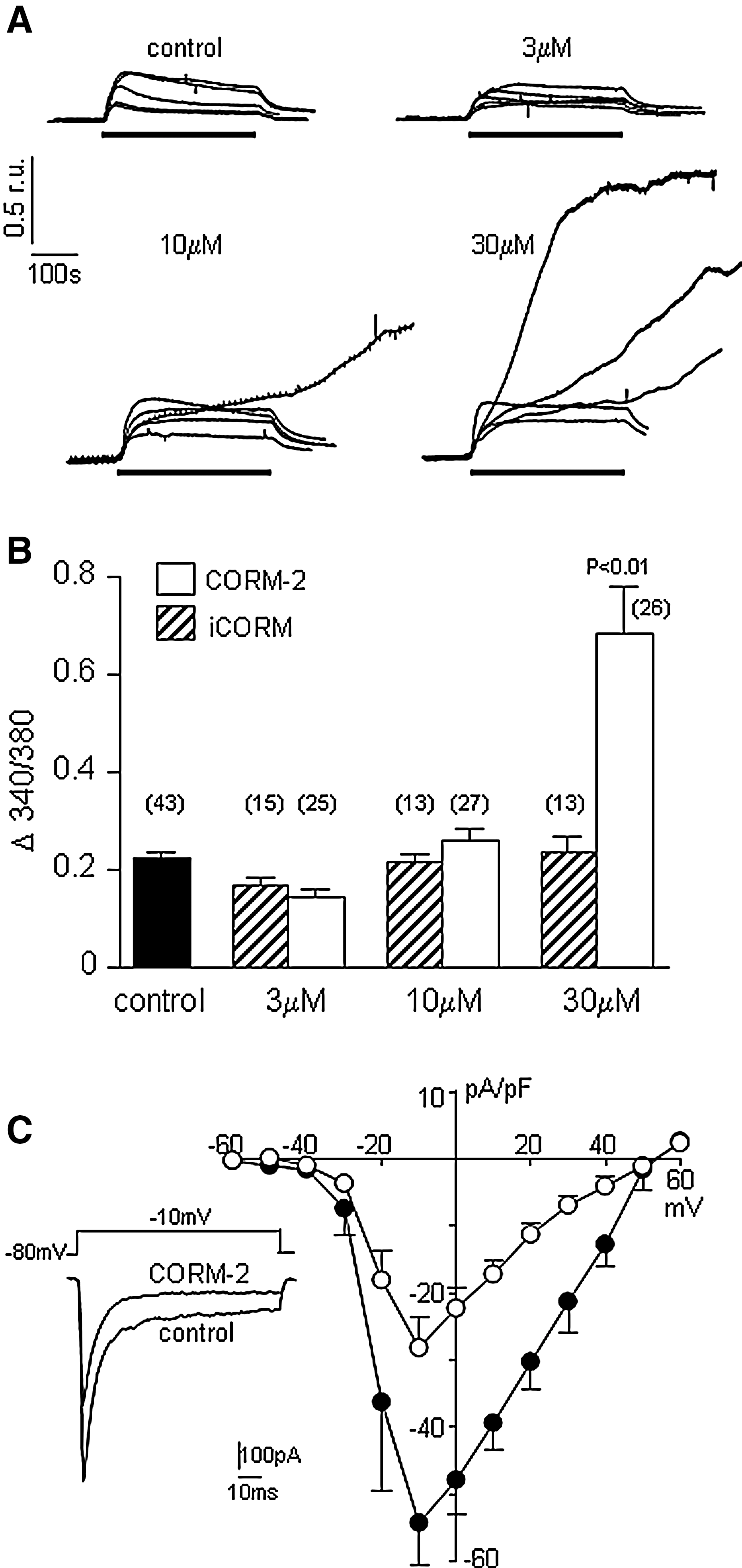
Depolarization of SH-SY5Y cells by exposure to 50 mM K+ causes a rapid rise of [Ca2+]i due to Ca2+ influx through L- and N-type Ca2+ channels (28, 35) as shown in Figures 2A and 2B. At 3 μM, CORM-2 did not significantly alter the amplitude of high K+-induced rises of [Ca2+]i. However, at 10 μM, CORM-2-evoked rises of [Ca2+]i were not significantly altered in amplitude in most recordings, but in 12 out of 27 recordings, [Ca2+]i rose dramatically during the exposure to high K+, and did not recover. In the presence of 30 μM CORM-2, the majority of recordings (24 out of 26) revealed this apparently uncontrolled rise of [Ca2+]i. Such effects of CORM-2 (but not iCORM) to evoke poorly controlled rises of [Ca2+]i were similar to its effects on CCE shown in Figure 1A. It was possible that these effects arose from increased voltage-gated Ca2+ entry on depolarization. To address this, we measured Ca2+ currents directly, as previously described (28, 35). Surprisingly, Ca2+ currents were significantly (p<0.001, n=6) inhibited by CORM-2 (30 μM), as shown in Figure 2C. Currents were significantly reduced by CORM-2 over the voltage range −20mV to +40mV (p<0.05—p<0.001, paired t-tests).
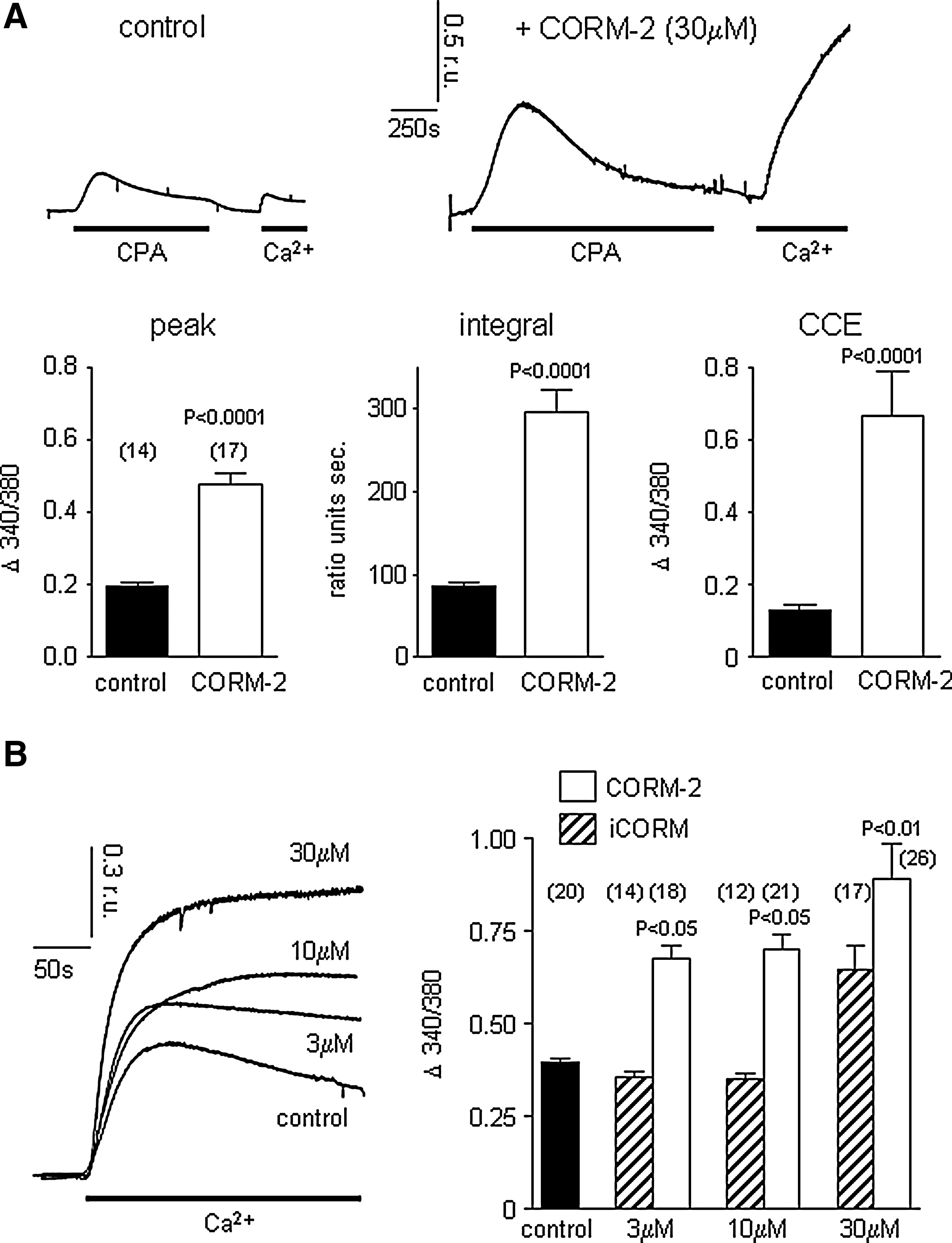
To investigate disruption of M3 receptor-mediated signaling further, we depleted stores directly with cyclopiazonic acid (CPA, which reversibly inhibits the endoplasmic reticulum (ER) Ca2+ ATPase). In the absence of extracellular Ca2+, CPA (10 μM) evoked transient rises of [Ca2+]i in control cells and, following its removal, restoration of Ca2+ to the perfusate evoked a rise of [Ca2+]i due to CCE (Fig. 3A). In the presence of CORM-2, both the CPA-evoked transient rise of [Ca2+]i and the resultant CCE were dramatically augmented (Fig. 3A), suggesting CO acts downstream of receptor activation and coupling to stores. Store depletion using thapsigargin (TG; applied for 20 min prior to monitoring [Ca2+]i) evoked larger CCE than was seen following CPA treatment (likely due to more complete store emptying), which was further enhanced by CORM-2, but not (at least at lower concentrations) by iCORM, again suggesting that CO augmented CCE in these cells (Fig. 3B).
These effects of CO likely represent the cellular consequences of toxic CO exposure. To distinguish these effects from those arising from increased endogenous CO production, we investigated the effects of HO-1 induction by exposing cells to hemin (100 μM for 24 h). Such treatment caused a marked induction of HO-1, but was without significant effect on muscarine-evoked Ca2+ signals (Supplementary Fig. 1; supplementary data are available online at
NO-dependence of CO disruption of Ca2+ signaling
Multiple intracellular signaling pathways are modulated by CO, including those involving mitochondria and NO. To examine whether the effects of CO arose due to mitochondrial inhibition, we compared its effects with those of the mitochondrial inhibitor antimycin A. As shown in Supplementary Figure 2, antimycin A (3 μg/ml) increased the decay time and integral of the muscarine-evoked response (similar to the effects of CO). However, it also significantly raised basal Ca2+ levels and almost fully suppressed CCE. These latter effects clearly distinguish the effects of CO from those of direct mitochondrial inhibition.
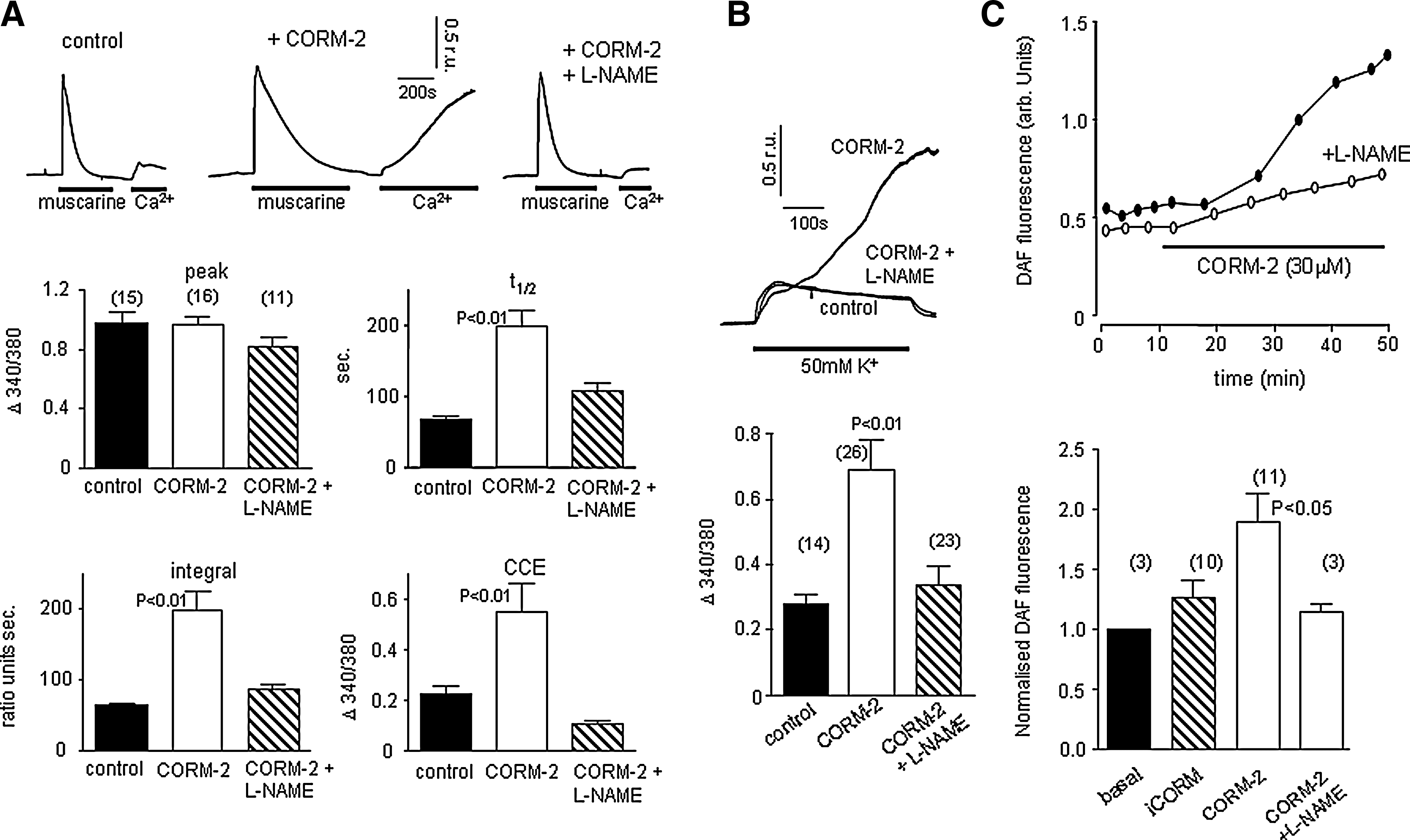
To investigate whether NO mediated the effects of CO described in Figures 1 –3, we examined whether the NOS inhibitor, L-NAME, could interfere with the actions of CO. Figure 4A shows that L-NAME (1 mM, applied for 1 h prior to, and during experiments) fully reversed the augmenting effects of CO on muscarine-evoked rises of [Ca2+]i and the subsequent CCE. Similarly, the dramatic rises of [Ca2+]i evoked by exposure of cells to 50 mM K+-containing solutions were reduced to levels seen in cells not exposed to CO (Fig. 4B). These findings (and also the observation that CO augmentation of TG-evoked CCE was prevented in the presence of L-NAME, see Fig. 5C), suggest that CO modulates Ca2+ homeostasis via production of NO. In confirmation of this idea, we found that CORM-2 increased fluorescence of the NO-sensitive fluoroprobe, DAF-2 (Fig. 4C). Importantly, this increased fluorescence, indicative of increased NO, was not observed when cells were exposed to iCORM, and was prevented in the presence of L-NAME (Fig. 4C). Thus, CO appears to modulate Ca2+ homeostasis via its ability to stimulate NO production. Such a conclusion would predict that exposure of cells to NO, or NO donor compounds, would mimic the effects of CO. However, S-nitroso N-acetylpenicillamine (SNAP; 10 or 100 μM) was unable to alter significantly the muscarinic-receptor mediated Ca2+ signaling (Fig. 5A), or rises of [Ca2+]i evoked by 50 mM K+ (Fig. 5B). Similarly, although L-NAME prevented the augmentation of CCE seen in the presence of CO (Fig. 4), SNAP did not alter CCE (Fig. 5C). A similar lack of effect was seen with three other NO donors (Supplementary Table 1). Thus, although effects of CO were NO-dependent, NO alone was unable to mimic the actions of CO.

Involvement of peroxynitrite formation on CO modulation of Ca2+ signaling
Since CO also increases ROS production, we investigated the possible role of ROS in the ability of CO to modulate Ca2+ signaling in SH-SY5Y cells. As illustrated in Figure 6A, the CO-induced increases in decay time and integral transient of the rise of [Ca2+]i evoked by muscarine mobilization from internal stores was completely reversed in the presence of the antioxidant ascorbic acid (200 μM). Similarly, the augmentation by CO of CCE observed following muscarine exposure and removal (Fig. 6A) and the CCE observed following TG treatment (Fig. 6B), were fully reversed by ascorbic acid. Finally, the effects of CO on 50 mM K+ evoked rises of [Ca2+]i were also fully prevented by ascorbic acid (Fig. 6C).
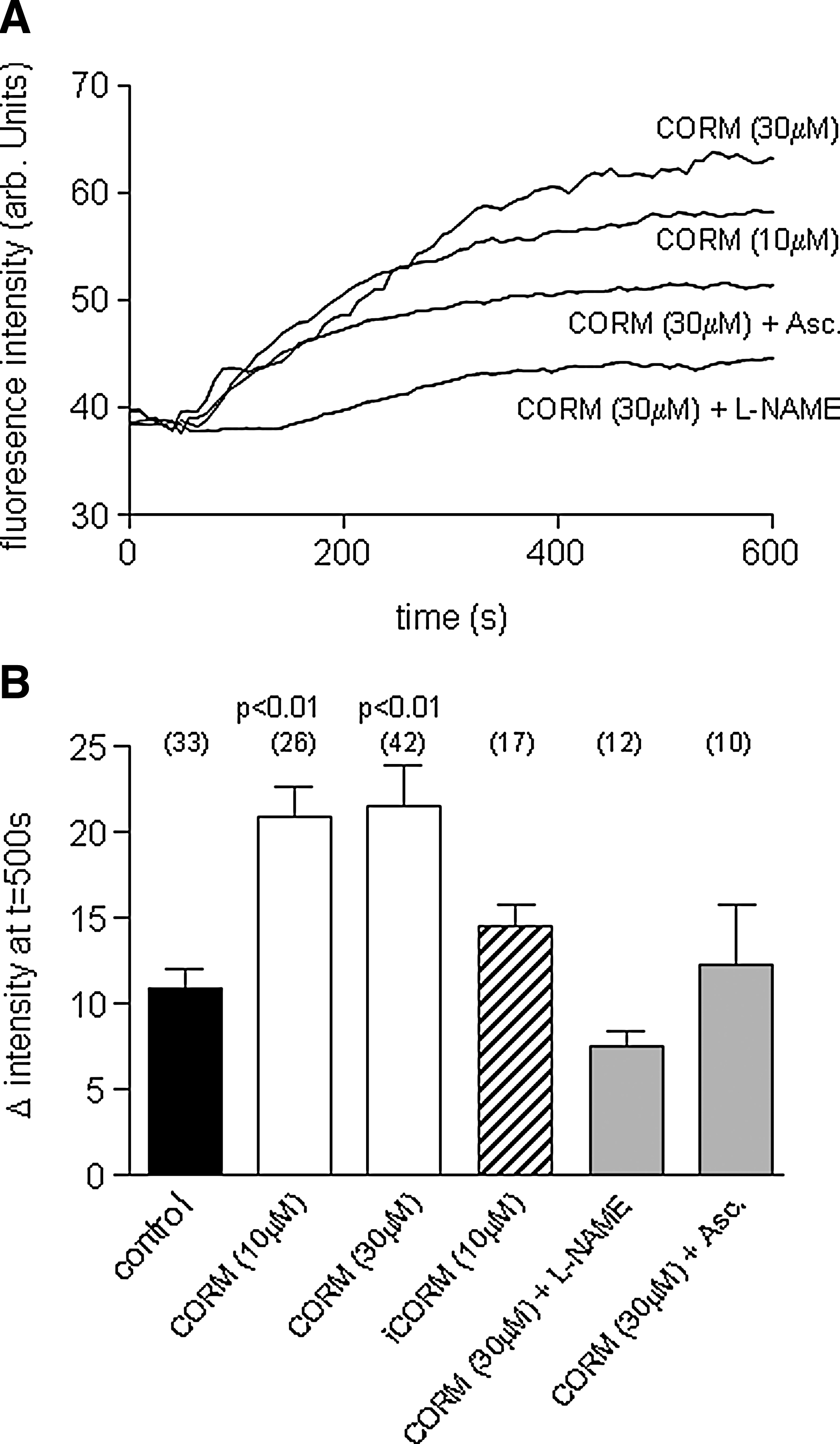
The observations that the effects of CO depended both on NO and on ROS production (Figs. 4 and 6) suggested to us the involvement of peroxynitrite (ONOO-) formation, since this is rapidly formed from NO and the ROS superoxide (O2 .-). To investigate this, we examined whether CO could evoke ONOO- production, using the ONOO- -sensitive fluoroprobe, APF (38). APF fluorescence increased in SH-SY5Y cells in response to CORM-2 (10 and 30 μM), but the increases seen in response to iCORM were not significantly different from the slight upward drift in fluorescence observed with time (Figs. 7A and 7B). Importantly, increased APF fluorescence was prevented in the presence of ascorbic acid or L-NAME, supporting the idea that APF is indeed reporting increased ONOO- levels in response to CO and, hence, that ONOO- may mediate the effects of CO on Ca2+ signaling. To test this further, we investigated the effects of the ONOO- decomposition catalyst, FeTPPs (5,10,15,20-tetrakis-[4-sulfonatophenyl]-porphyrinato-iron[III]; 50 μM), which rapidly converts ONOO- to nitrate (27). As illustrated and quantified in Figure 8, FeTPPs effectively reversed the effects of CO on muscarine-evoked rises of [Ca2+]i and subsequent CCE (Fig. 8A) and rises of [Ca2+]i evoked by 50 mM K+ (Fig. 8B).

A role for the plasmalemmal Ca2+ ATPase in mediating the effects of CO
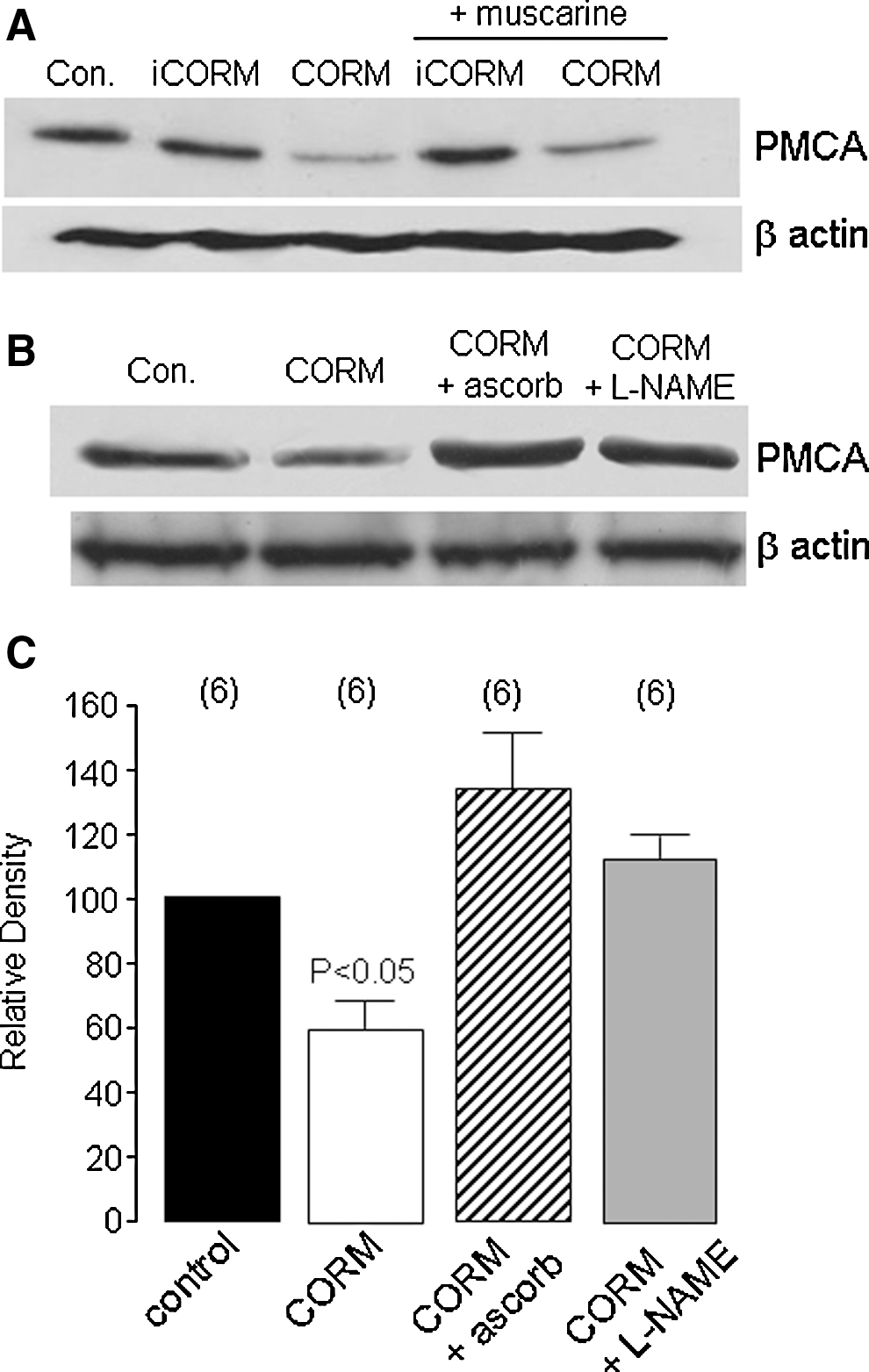
Our findings thus far indicated that CO disrupted all aspects of evoked Ca2+ signaling investigated, raising the possibility that it may act via disruption of the ability of cells to regulate or control rises of [Ca2+]i regardless of the source of Ca2+ rise. Given the known susceptibility of the plasma membrane Ca2+-ATPase (PMCA) to oxidative stress (13), we investigated the effects of CO on PMCA levels using a pan anti-PMCA antibody. Exposure of cells to CORM-2 (but not iCORM) for 20 min caused a significant reduction in protein levels, regardless of the presence of muscarine (Figs. 9A and 9C). This reduction in PMCA protein levels was recovered separately either by ascorbic acid (200 μM) or L-NAME (1 mM; Figs. 9B and 9C). These data strongly suggest that the disruption of Ca2+ signaling caused by CO may be attributed to the ONOO--dependent loss of PMCA.
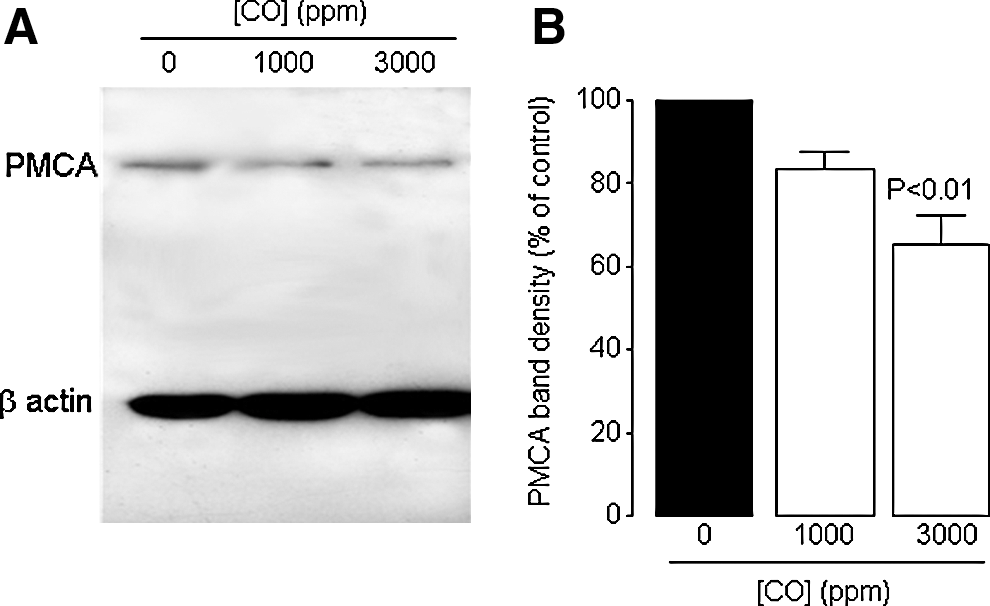
To investigate whether a similar degradation of PMCA occurred as a result of CO poisoning in vivo, we used the same antibody to probe whole-brain homogenates of rats exposed to either 1000 or 3000 ppm CO for 40 min. As shown in Figures 10A and 10B, a concentration-dependent loss of this important regulator of [Ca2+]i was observed, indicating that our findings in SH-SY5Y cells reflect the neurotoxic effects of CO in vivo.
Discussion
Amid the widespread and developing interests in CO as a significant physiological signaling molecule and therapeutic agent, it is easy to lose sight of the fact that it is also a highly toxic gas. Exposure to toxic levels of CO can, when not lethal, cause delayed neurological/neuropsychiatric symptoms (26, 31, 32). At the cellular level, toxic levels of CO can cause apoptosis and necrosis, and toxicity has been attributed to oxidative damage. Although most cellular studies have employed non-neuronal tissue, such as endothelial cells (43, 45), one recent study has examined the toxic effects of CO inhalation on hippocampal neurons and, consistent with studies in endothelial cells, indicates that neuronal injury and death caused by CO arises from oxidative damage leading to apoptosis (12). Despite the known association of oxidative stress with disturbances of Ca2+ homeostasis that can lead to neuronal damage as observed in aging brain samples and those obtained from sufferers of neurodegenerative disorders (11, 24), the possibility that CO might perturb neuronal Ca2+ homeostasis has not been studied in depth.
The present study indicates that CO acts to modulate multiple aspects of Ca2+ signaling in human neuroblastoma SH-SY5Y cells. Thus, CO modified Ca2+ mobilization from intracellular stores (Figs. 1 and 3), store depletion-mediated (capacitative) Ca2+ entry (Figs. 1 and 3) and voltage-gated Ca2+ entry (Fig. 2). Our results further indicate that CO acts in this manner via ONOO- formation arising from increased levels of both NO and ROS (Figs. 4, 6 –8). The involvement of NO is indicated by the observations that L-NAME prevents the effects of CO to disrupt Ca2+ signaling, and that CO leads to increased levels of NO, as monitored fluorimetrically (Fig. 4). Our findings agree with previous studies suggesting that CO can stimulate NO formation in other systems (19, 21) and support a previous conclusion that CO neurotoxicity depends on activation of neuronal nitric oxide synthase (nNOS; (44)). Indeed, our findings provide a likely downstream effect that could account for CO neurotoxicity; the disruption of Ca2+ homeostasis. Importantly, however, the additional requirement of increased ROS in order to observe the effects of CO is clearly indicated by the fact that NO donors alone could not mimic effects of CO (Fig. 5, Supplementary table 1). Thus, NO formation alone is not sufficient to account for the effects of CO on Ca2+ homeostasis.
CO has previously been proposed to increase ONOO- formation in vascular endothelial cells, leading to the induction of apoptosis (43, 45). These studies indicated that caspase activation (indicating apoptosis initiation) could be prevented by an inhibitor of NOS, or by a ONOO- scavenger. Given these findings, and our observation that the effects of CO reported here were separately prevented by inhibition of either NO or ROS formation, we explored the potential involvement of ONOO-. We first monitored production of ONOO- using the fluoroprobe APF. This probe was originally produced to discriminate between ROS species, and was found to be useful in the detection of ONOO- and also hydroxyl radicals (OH•) and hypochlorite (-OCl) (38). Its ability to detect ONOO- with significant selectivity has subsequently been confirmed (e.g., Ref. 37). Our finding that APF fluorescence increased in response to CO in a manner that was dependent on both NO and ROS (Fig. 7) strongly suggested this fluoroprobe is reporting increased formation of ONOO-, and so we conclude that CO does indeed increase ONOO- in our cells. This conclusion was supported by the observation that the ONOO- decomposition catalyst, FeTPPs, prevented all of the effects of CO to disrupt Ca2+ homeostasis (Fig. 8).
The fact that CO modulated the rises of [Ca2+]i observed following activation of diverse Ca2+ entry/mobilization pathways suggested that it was unlikley to modify various proteins that contribute to each of these responses individually. Instead, we reasoned that CO was more likely to act via a part of the Ca2+ signaling pathway common to all manouevres used to raise [Ca2+]i in this study, namely the protein(s) responsible for restoring [Ca2+]i to baseline levels. Once cytosolic [Ca2+] has increased, it is removed either via re-uptake into organelles (primarily the ER, but also mitochondria) or via extrusion into the extracellular space. We discounted ER re-uptake as a likely site of action, since when ER uptake was inhibited by CPA (Fig. 3), the ensuing rise of [Ca2+]i was still augmented by CO. Instead, CO modulation of Ca2+ extrusion was investigated. Specifically, we investigated the possible involvement of the plasmalemmal Ca2+ ATPase, since a previous study suggested that ONOO- inhibits CaATPase activity in synaptosomal preparations (13). Furthermore, the Ca2+ ATPase is susceptible to oxidative damage/destruction (reviewed by Ref. 4). In agreement with such studies, and consistent with our functional studies, Western blots indicated that CO reduced Ca2+ ATPase protein levels (Fig. 9). Thus, we propose that CO disrupts Ca2+ signaling in SH-SY5Y cells via degradation of the plasmalemmal Ca2+ ATPase as a result of formation of ONOO-. We further propose that this mechanism contributes to the prolonged neurological damage associated with CO toxicity, since a similar degradation of the Ca2+ ATPase was observed in brain tissue taken from rats previously exposed to CO acutely (Fig. 10).
The present study has revealed a deleterious aspect of the influence of CO on intracellular signaling mechanisms, which contrasts with recognized protective effects of CO against apoptosis (e.g., in the endothelium (41)). Whether this is due to the fact that the present study employed neuronal tissue, whereas many previous studies have employed other tissues, is presently unknown. However, it is important to note that, whilst some of the beneficial effects of HO-1 can be mimicked by exogenous CO (for example, protection against experimental focal ischemia (49)), there are additional benefits observed following HO-1 induction (i.e., increased degradation of cytotoxic heme, production of biliverdin and hence bilirubin, a powerful antioxiant), which may be neuroprotective (1) and HO-1 location will likely influence the cellular actions of endogenous CO (see Ref. 10 for review). Thus, whilst the hopes for exogenous CO therapy remain justifiably high, potentially deleterious actions, such as those reported here, must be fully considered.
Materials and Methods
Tissue culture
SH-SY5Y cells were cultured as previously described (34, 35) in a 1:1 mixture of Ham's F12 medium and Eagle's minimal essential medium, supplemented with 10% (v/v) fetal calf serum, 1% nonessential amino acids, and 0.1% gentamicin (all from GIBCO Life Sciences, Paisley, Scotland, UK). Cells were incubated at 37°C in a humidified incubator gassed with 95% air and 5% CO2, passaged every 7 days and used for up to 14 passages.
Monitoring [Ca2+]i
Cells were plated onto two 10 mm glass coverslips (thickness 0) after dilution to the desired concentration with culture media. Prior to experiments, cells were incubated in HEPES-buffered saline containing 4 μM Fura-2AM (Invitrogen) at room temperature (22–24°C) and left in the dark for 40 min. HEPES-buffered saline was composed of (in mM): NaCl 135, KCl 5, MgSO4 1.2, CaCl2 2.5, HEPES 5, and glucose 10 (pH 7.4, osmolarity adjusted to 300 mosmol with sucrose, 21–24°C). After 40 min, the fura-2 containing solution was replaced by fresh HEPES-buffered saline and left for 15 min for de-esterification before commencing experiments. Fragments of coverslips were then placed in a perfusion chamber and [Ca2+]i was measured using a Cairn Research ME-SE Photometry system (Cairn Research, Cambridge). Cells were superperfused (3–5 ml/min) under gravity and [Ca2+]i was indicated by the ratio of fluorescence emitted at 510 nm due to alternating excitation at 340 nm and 380 nm using a monochromator. Drugs used to investigate Ca2+ homeostasis were applied to cells via switch of inflow chamber to one containing the HEPES-buffered saline and required drug. Ca2+-free perfusate contained 1 mM EGTA and no added CaCl2. The high K+ solution used in experiments was made by isotonic replacement of Na+ in the perfusate (final [K+] 50 mM).
Electrophysiology
Fragments of coverslip with attached cells were transferred to a perfused (3–5 ml/min) recording chamber mounted on an Olympus CK40 inverted microscope. Whole cell patch clamp recordings were made using patch pipettes of 4–7 MΩ resistance. Series resistance was monitored after breaking into the whole cell configuration throughout the duration of experiments. If a significant increase occurred (>20%), the experiment was terminated. The perfusate (pH 7.4, NaOH; 22–24°C) was composed of (mM): NaCl (95); MgCl2 (0.6); CsCl (5); HEPES (5); BaCl2 (20); D-glucose (10); TEACl (20). The intracellular solution (pH 7.2, CsOH) consisted of (mM): CsCl (120); TEACl (20); EGTA (10); MgCl2 (2); HEPES (10); MgATP (3); NaATP (2). CORM-2 was bath applied at the stated concentrations.
Signals were acquired using a Axopatch 200B controlled by Clampex 9.0 software via a Digidata 1322A interface (Axon Instruments, Inc., Foster City, CA). Data were filtered at 1 kHz and digitized at 2 kHz. To evoke ionic currents in SHSY5Y cells, a series of 100 ms depolarizing steps from −80 mV (holding potential) to +60 mV, in 10 mV increments, were employed. Offline analysis was carried out using the data analysis package Clampfit 9 (Axon Instruments) and data are expressed as mean±SEM.
Monitoring NO production
DAF-FM was used to detect NO production fluorimetrically. Cells plated on coverslips were incubated with 5 mM DAF-FM (Invitrogen) in HEPES-buffered saline supplemented with 100 μM L-arginine and 0.002% pluronic acid for 40 min in the dark at room temperature. Thereafter the DAF-FM-containing solution was replaced by HEPES-buffered saline solution with 100 μM L-arginine in a perfusion chamber and NO was measured using a Cairn Research ME-SE Photometry system (Cairn Research, Cambridge). Cells were continuously superperfused under gravity and NO was indicated by the fluorescence emitted at 515 nm following excitation at 495 nm. In these experiments, the light path shutter was opened for 10 sec at 5 min intervals in order to avoid photobleaching of the dye.
Monitoring peroxynitrite production
Cells were plated on to coverslips in 24-well plates as before and incubated with 2- [6-(4'-amino)phenoxy-3H-xanthen-3-on-9-yl]benzoic acid (APF; 10 μM) dissolved in HEPES buffered saline in the dark for 1 h at 37°C. The coverslip was then cut into fragments and one fragment was placed on a glass slide containing 200 μl of HEPES buffered saline with 10 μM APF. APF was chosen both for its sensitivity to ONOO- and for relative insensitivity to O2 - and also because it is resistance to light-induced auto-oxidation. Changes in fluorescence intensity were measured over 10 min using a ZEISS (Oberkochen, Germany) laser scanning confocal microscope (LSM 510). The fluorophore was excited at 488 nm and emission monitored at 510 nm. Zeiss AIM software was used to obtain the images. Identical settings were used for each test condition. For APF experiments with L-NAME, the cells were incubated with both 10 μM APF and 1 mM L-NAME for 1h at 37°C prior to starting the recordings.
Western blot analysis
Cells were grown to near confluence in 25 cm2 flasks, treated with test substance as indicated, washed with PBS, and then solubilized in situ in 300 μl mammalian protein extraction reagent (Pierce Perbio, UK) containing a Complete mini-protease inhibitor tablet (Roche Bioscience, UK). Cell proteins (20 mg protein per lane) were separated on 7.5%, 0.75 mm polyacrylamide SDS gels and electrophoretically transferred to PVDF membranes (BioRad). Blots were incubated in 5% non-fat milk protein and then probed with a monoclonal antibody raised against plasma membrane Ca2+ ATPases (1:100; PMCA (H8); Santa Cruz Biotechnology Inc, Santa Cruz, CA). Bands were visualized using an enhanced chemiluminescence detection system (ECL) and hyperfilm ECL (GE Healthcare, UK). Approximate equal loading of proteins was confirmed by blotting with monoclonal β-actin antibody (1/2000, Sigma) in 5% non-fat milk protein in TBS. Band densities were measured using ImageJ and expressed as a percentage of the control (untreated) band density.
In vivo CO exposure
Male Sprague-Dawley rats (260–280 g) were individually put in a plastic chamber (26.5 cm in diameter, 28.5 cm in height) and exposed to 1000 or 3000 ppm CO for 40 min, as previously reported (16). After reoxygenation for 2 h, the brain was removed under pentobarbital anesthesia (50 mg/kg, i.p.) and immediately frozen on dry ice. The control rats were exposed to room air alone for 40 min. Brain cortices were homogenized in ice-cold RIPA buffer containing a complete mini-protease inhibitor tablet (Roche Bioscience, UK) at 3 ml per gram of tissue, disrupted by sonication, and incubated on ice for 30 min. Crude homogenates were then cleared by centrifugation (10,000 g 10 min). The supernatant was removed and centrifuged (15,000 g 15 min) again, and the total cell lysate removed and immediately frozen in liquid N2. The cleared brain homogenates were probed by Western blots as described above.
Drugs and dyes employed
The following compounds were employed in this study, as described in the Results section. Muscarine, CORM-2, L-NAME, thapsigargin, ascorbic acid, antimycin A and APF were all obtained from Sigma Aldrich. Cyclopiazonic acid (CPA) and S-nitroso N-acetylpenicillamine (SNAP) were from Ascent Scientific. DEA NONOate and DPTA NONOate were from Cayman Chemicals. The NO donor, s-nitrosocysteine, was synthesized in-house immediately before use in experiments, as described by (5). Fura-2AM and DAF-FM were from Invitrogen, Fe-TPPS from Calbiochem.
Statistical analysis
Data are presented as individual examples and analyzed results presented as mean±SEM. Statistical analysis was carried out using one way ANOVA followed by a post hoc test unless indicated otherwise. P values of less than 0.05 were considered significant.
Footnotes
Acknowledgment
This work was supported by the Alzheimer's Society, UK.
Disclosure Statement
None of the authors have any conflicts to disclose regarding this submission.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.