
Abstract
Introduction
Mitochondrial Aging—the Beginning of the End in AD?
Mitochondria play a pivotal role in cell survival and death by regulating both energy metabolism and apoptotic pathways; they contribute to many cellular functions, including intracellular calcium homeostasis, the alteration of the cellular reduction-oxidation (redox) potential, cell cycle regulation, and synaptic plasticity (47). They are the “powerhouses of cells,” providing energy from nutritional sources via ATP generation, which is accomplished through oxidative phosphorylation (OXPHOS) (65). However, when mitochondria fulfill their physiological function, it is as if Pandora's box has been opened, as this vital organelle contains potentially harmful proteins and biochemical reaction centers; mitochondria are the major producers of reactive oxygen species (ROS) at the same time being susceptible targets of ROS toxicity. Unstable ROS are capable of damaging many types of mitochondrial components; this includes oxidative deterioration of mitochondrial DNA (mtDNA), lipids of the mitochondrial membrane, and mitochondrial proteins, and it is thought that this damage that may accumulate over time from ROS generated from aerobic respiration may play a significant role in aging (Fig. 1). Moreover, it was previously demonstrated that nitrosative stress evoked by increased nitric oxide synthesis also leads to protein oxidation as well as mitochondrial and DNA damage, which are common mechanisms occurred in elderly (13, 34, 70).

Although most mitochondrial proteins are encoded by the nuclear genome, the mitochondrial genome encodes proteins required for 13 polypeptide complexes of the respiratory chain involved in ATP synthesis. Given that mtDNA exists in the inner matrix and this is in close proximity to the inner membrane where electrons can form unstable compounds, mtDNA, unlike nuclear DNA (nDNA), is not protected by histones (4) making it more vulnerable to oxidative stress and its mutation rate is about 10-fold higher than that of nDNA, especially in tissues with a high ATP demand like the brain (54). These mtDNA mutations occur in genes encoding electron transport chain (ETC) subunits including NADH dehydrogenase, cytochrome c oxidase (COX), and ATP synthase (83). Eventually, ROS-related mtDNA mutations can result in the synthesis of mutant ETC proteins that, in turn, can lead to the leakage of more electrons and increased ROS production. This so-called “vicious cycle” is hypothesized to play a critical role in the aging process according to the mitochondrial theory of aging. In addition to age-associated increase in mtDNA mutations, the amount of mtDNA also declines with age in various human and rodent tissues (2, 68). Furthermore, abundance of mtDNA correlates with the rate of mitochondrial ATP production (68), suggesting that age-related mitochondrial dysfunction in muscle is related to reduced mtDNA abundance. However, age-associated change in mtDNA abundance seems to be tissue specific, as several studies have reported no change in mtDNA abundance with age in other than muscular tissues in both man and mouse (20, 46).
How does the somatic mtDNA involved in aging phenotypes contribute to AD development? As only a small fraction of AD is caused by autosomal dominant mutations, this comes down to the question of what is causing the prevalent sporadic cases in the first place. Somatic mutations in mtDNA could cause energy deficiency, increased oxidative stress, and accumulation of Aβ, which act in a vicious cycle reinforcing mtDNA damage and oxidative stress (45). Indeed, defects in mtDNA associated with decreased cytochrome oxidase activity have been found in AD patients (9). Although a similarly impaired mitochondrial function and subsequent compensatory response have been observed in both nondemented aged and AD subjects, no clear causative mutations in the mtDNA have been correlated to AD; although some variations have functional consequences, including changes in enzymatic activity (40). Perhaps the main differences are that, in AD brains, defects are more profound due to Aβ and tau accumulation, because of decreased compensatory response machinery (Fig. 1).
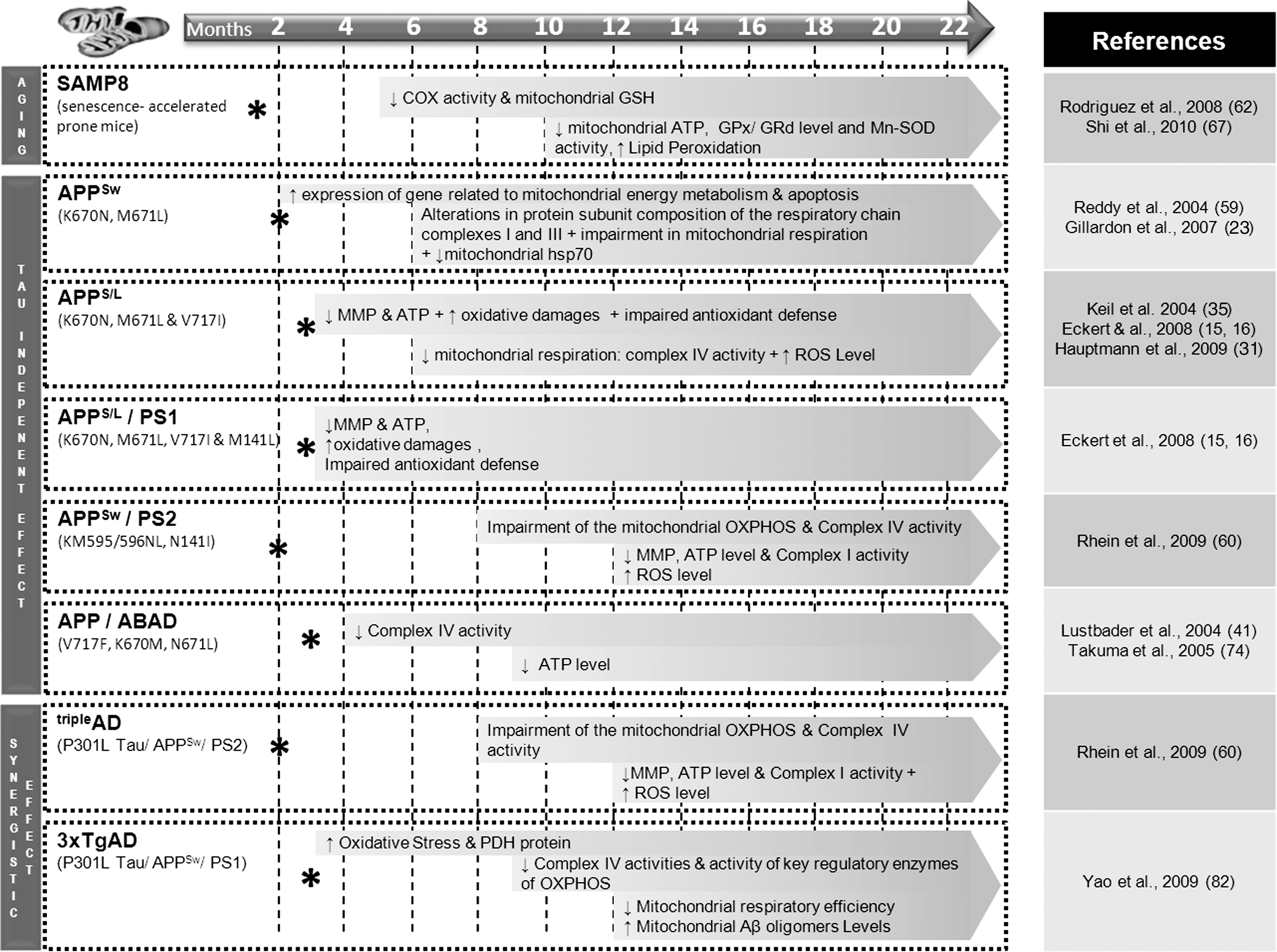
Many investigators have developed models for studying mitochondrial-related aging (36). Among them senescence-accelerated mice (SAM) strains are especially useful models to understand the mechanisms of the age-related mitochondrial decline. Behavioral studies showed that learning and memory deficits already started as early as 6 months and worsened with aging in SAMP8 mice (accelerated senescence-prone 8) (53, 77). Moreover, Omata and collaborators showed age-related changes in cerebral energy production in the 2-month-old SAMP8 followed by a decrease in mitochondrial function compared with SAMR1 mice (accelerated senescence-resistant 1) (51). Aging is not only connected with increased mitochondrial ROS production due to ETC impairment but also with a dysbalance of the protective antioxidant machinery inside mitochondria. For instance, age-related changes in levels of antioxidant enzymes, such as copper/zinc superoxide dismutase (Cu/Zn-SOD) and manganese SOD (Mn-SOD), have been found in liver and cortex of SAMP8 mice when compared with age-matched SAMR1 mice, supporting increased oxidative stress as a key mechanism involved in the aging process (37). More recently, Yew and collaborators have shown an impairment of mitochondrial functions including a decrease of COX activity, mitochondrial ATP content, and mitochondrial glutathione (GSH) level at a relatively early age in SAMP8 mice compared with SAMR1 mice (67, 78). Furthermore, the biochemical consequences of aging have been investigated using proteomic analysis in the brain of SAMP8 and SAMR1 mice at presymptomatic (5-month old) and symptomatic (15-month old) stages (84), revealing differentially expressed proteins with age in both mouse strains, such as Cu/Zn-SOD. Besides the progressive mitochondrial decline and increased oxidative stress, tau hyperphosphorylation was also observed at an early age in the brain of SAMP8 mice (1, 71). In addition, SAMP8 mice showed an age-related increase in mRNA and protein levels of amyloid-β precursor protein (APP). The cleavage product Aβ was significantly increased at 9 months in SAMP8 and amyloid plaques started to form at around 16 months of age (48, 73). Altogether, these data indicate that mitochondrial dysfunction is a highly relevant event in the aging process, which is also known as the primary risk factor for AD and other prevalent neurodegenerative disorders.
Age-Related Aβ and Tau Effects on Mitochondria in AD
AD is a progressive, neurodegenerative disorder, characterized by an age-dependent loss of memory and an impairment of multiple cognitive functions. From a genetic point of view, AD can be classified into two different forms: rare familial forms (FAD) where the disease onset is at an age below 60 years (<1% of the total number of AD case) and the vast majority of sporadic AD cases where onset occurs at an age over 60 years. Genetic studies in FAD patients have identified autosomal dominant mutations in three different genes, encoding the APP (over 20 pathogenic mutations identified) and the presenilins PS1 and PS2 (more than 130 mutations identified) (26). These mutations are directly linked to the increased production of Aβ from its precursor protein APP, suggesting a direct and pathological role for Aβ accumulation in the development of AD.
Mitochondrial dysfunction has been proposed as an underlying mechanism in the early stages of AD, since energy deficiency is a fundamental characteristic feature of AD brains (44) as well as of peripheral cells derived from AD patients (22). Understanding the molecular pathways by which the various pathological alterations including Aβ and tau compromise neuronal integrity, leading to clinical symptoms, has been a long-standing goal of AD research. The successful development of mouse models that mimic diverse aspects of the AD process has facilitated this effort and assisted in understanding of the age-dependent interplay of Aβ and tau on bioenergetics processes in vivo (Figs. 2 and 3).

Separate modes of Aβ and tau toxicity on mitochondria
Mitochondria were found to be a target for APP toxicity as both the full-length protein and Aβ accumulate in the mitochondrial import channels, and both lead to mitochondrial dysfunction (7, 42, 55, 56). Several evidences from cellular and animal AD models indicate that Aβ triggers mitochondrial dysfunction through a number of pathways such as impairment of OXPHOS, elevation of ROS production, interaction with mitochondrial proteins, and alteration of mitochondrial dynamics (52). Indeed, abnormal mitochondrial dynamics have been identified in sporadic and familial AD cases (43, 76) as well as in AD mouse model (6); a distortion probably mediated by altered expression of dynamin-like protein 1 (DLP1), a regulator of mitochondrial fission and distribution, due to elevated oxidative and/or Aβ-induced stress. This modification can disturb the balance between fission and fusion of mitochondria in favor of mitochondrial fission followed by mitochondrial depletion from axons and dendrites and, subsequently, synaptic loss.
Success in developing mouse models that mimic diverse facets of the disease process has greatly facilitated the understanding of physiopathological mechanisms underlying AD. Thus, in 1995, Games and collaborators established the first APP mice model (called PDAPP) bearing the human “Indiana” mutation of the APP gene (V171F). They observed the accumulation of Aβ in the brain and subsequent amyloid plaque formation as well as astrocytosis and neuritic dystrophy (21). Interestingly, in this model cognitive deficits, such as spatial learning impairment, occur before the formation of Aβ plaques and increase with age (8). This phenomenon was also observed in Tg2576 transgenic mice bearing the human Swedish mutation of the APP gene (K670N, M671L). In fact, in most of the APP mouse models, the cognitive impairment begins concomitantly with Aβ oligomer formation in the brain (around 6 months of age), while neuritic amyloid deposits become visible only between 12 and 23 months and then the amount of deposits increases (23, 31, 35). Thus, memory deficits seem to directly correlate with the accumulation of intracellular Aβ oligomers and not with amyloid plaque formation. Crossing APP transgenic mice with those bearing a mutation in presenilin 1 gene enabled an earlier onset of amyloid plaques compared with APP mice. In one of the most aggressive models, double-transgenic APPS/L/PS1 (APPSwedish/London/PS1M141L) mice, Aβ accumulation begins very soon at 1–2 months of age while cognitive deficits and amyloid plaque formation are already observed at 3 months (3, 16). A stronger decrease of mitochondrial membrane potential as well as ATP level was also found in these mice.
Mitochondrial dysfunctions also appear to a very early stage in these transgenic mouse models. For example, in the APPSw transgenic strain Tg2576, an upregulation of genes related to mitochondrial energy metabolism and apoptosis was observed already at 2 months of age. Alterations in composition of the mitochondrial respiratory chain complexes I and III protein subunit as well as impairment of mitochondrial respiration were detected around 6 months, when soluble Aβ accumulated in the brain without plaque formation (10, 23, 59). To test the hypothesis that oxidative stress can underlie the deleterious effects of PS mutations, Schuessel and collaborators analyzed lipid peroxidation products (4-hydroxynonenal [HNE] and malondialdehyde) and antioxidant defense mechanisms in brain tissue and ROS levels in splenic lymphocytes from transgenic mice bearing the human PS1 M146L mutation (PS1M146L) compared with those from mice transgenic for wild-type human PS1 (PS1wt) and nontransgenic littermate control mice (66). In brain tissue, HNE levels were increased only in aged (19–22 months) PS1M146L transgenic animals compared with PS1wt mice and not in young (3–4 months) or middle-aged mice (13–15 months). Similarly, in splenic lymphocytes expressing the transgenic PS1 proteins, mitochondrial and cytosolic ROS levels were significantly elevated compared with controls only in cells from aged PS1M146L animals. Antioxidant defense mechanisms (activities of antioxidant enzymes including Cu/Zn-SOD, GSH peroxidase, and GSH reductase) as well as susceptibility to oxidative stress in vitro were unaltered. In summary, these results demonstrate that the PS1M146L mutation increases mitochondrial ROS formation and oxidative damage selectively in aged mice. Consistent with this observation, in Swedish amyloid precursor protein (APPSw)/PS2 double-transgenic mice, mitochondrial impairment was first detected at 8 months of age, before amyloid plaque deposition, but after soluble Aβ accumulation (60, 61). Taken together, these findings are consistent with the recently proposed hypothesis of the age-related Aβ toxicity cascade that suggests that the most toxic Aβ species that cause majority of molecular and biochemical abnormalities are in fact intracellular soluble oligomeric aggregates rather than the extracellular, insoluble plaques that may comprise the form of cellular defense against toxicity of oligomers (19). Interestingly, human amylin that aggregates in type 2 diabetic pancreas and shares with Aβ its amyloidogenic properties also causes an impaired complex IV activity, whereas nonamyloidogenic rat amylin did not (39).
How does tau interfere with mitochondrial function? In its hyperphosphorylated form, tau, which forms the NFTs, the second hallmark lesion in AD, has been shown to block mitochondrial transport, which results in energy deprivation and oxidative stress at the synapse and, hence, neurodegeneration (27, 33, 57). Till now, no mutations in microtubule-associated protein tau (MAPT) coding genes have been detected in relation to familial forms of AD. However, in familial frontotemporal dementia (FTD) with parkinsonism, mutations in the MAPT gene were identified on chromosome 17. This was the basis for creating a robust mouse model for tau pathology in 2001. These P301L tau–expressing pR5 mice (longest four-repeat 4R2N) show an accumulation of tau as soon as 3 months of age and develop NFTs around 6 months of age (24). A mass spectrometric analysis of the brain proteins from these mice revealed mainly a deregulation of mitochondrial respiratory chain complex components (including complex V), antioxidant enzymes, and synaptic protein space (11). The reduction in mitochondrial complex V levels in the P301L tau mice that was revealed using proteomics was also confirmed as decreased in human P301L FTDP-17 (FTD with parkinsonism linked to chromosome 17) brains. The functional analysis demonstrated age-related mitochondrial dysfunction, together with reduced NADH-ubiquinone oxidoreductase (complex I) activity as well as age-related impaired mitochondrial respiration and ATP synthesis in pR5 mice model. Mitochondrial dysfunction was also associated with higher levels of ROS in aged transgenic mice. Increased tau pathology resulted in modification of lipid peroxidation levels and the upregulation of antioxidant enzymes in response to oxidative stress (11). Thus, this evidence demonstrated for the first time that not only Aβ but also tau pathology leads to metabolic impairment and oxidative stress by distinct mechanisms from that caused by Aβ in AD.
Synergistic modes of Aβ and tau toxicity on mitochondria
Although Aβ and tau pathologies are both known hallmarks of AD, the mechanisms underlying the interplay between plaques and NFTs (or Aβ and tau, respectively) have remained unresolved. However, a close relationship between mitochondrial impairment and Aβ on the one hand and tau on the other hand has been already established. How do both AD features relate to each other? Is it possible that these two molecules synergistically affect mitochondrial integrity? Several studies suggest that Aβ aggregates and hyperphosphorylated tau may block the mitochondrial carriage to the synapse leading to energy deficiency and neurodegeneration (28). Moreover, the enhanced tau levels may inhibit the transport of APP into axons and dendrites, which suggests a direct link between tau and APP in axonal failure (14, 69). Remarkably, intracerebral Aβ injections amplify a preexisting tau pathology in several transgenic mouse models (5, 25, 29), whereas lack of tau abrogates Aβ toxicity (32, 33). Our findings indicate that in tau transgenic pR5 mice, mitochondria display an enhanced vulnerability toward an Aβ insult in vitro (12, 15, 16), suggesting a synergistic action of tau and Aβ pathology on this organelle (Figs. 2 and 3). The Aβ caused a significant reduction of mitochondrial membrane potential in cerebral cells from pR5 mice (11). Furthermore, incubation of isolated mitochondria from pR5 mice with either oligomeric or fibrillar Aβ species resulted in an impairment of the mitochondrial membrane potential and respiration. Interestingly, aging particularly increased the sensitivity of mitochondria to oligomeric Aβ insult compared with that of fibrillar Aβ (15). This suggests that while both oligomeric and fibrillar Aβ species are toxic, they exert different degrees of toxicity. Crossing P301L mutant tau transgenic JNPL3 mice (shortest four-repeat [4R0N] tau together with the P301L mutation) with APPSw transgenic Tg2576 mice revealed the presence of NFT pathology in spinal cord and pons already at 3 months of age (38). Aβ plaques were detected at the age of 6 months and had the same morphology and distribution than in the 1-year-old Tg2576 mice. Taken together, these studies illustrate the existence of a complex interplay between the two key proteins in AD.
Additionally, in recent years triple-transgenic mouse models have been established that combine Aβ and tau pathologies (Figs. 2 and 3). In these models the contribution of both AD-related proteins on the mitochondrial respiratory machinery and energy homeostasis has been investigated in vivo. Indeed, our group demonstrated a mitochondrial dysfunction in a novel triple-transgenic mouse model (pR5/APPSw/PS2 N141I)—tripleAD mice—using proteomics followed by functional validation (60). Particularly, deregulation of activity of complex I was found to be tau dependent, whereas deregulation of complex IV was Aβ dependent, in 10-month-old tripleAD mice. The convergent effects of Aβ and tau led already at the age of 8 months to a depolarization of mitochondrial membrane potential in tripleAD mice. Additionally, we found that age-related oxidative stress also plays a significant part in the deleterious vicious cycle by exaggerating Aβ- and tau-induced disturbances in the respiratory system and ATP synthesis, finally leading to synaptic failure.
Our data complement those obtained in another triple-transgenic mouse model 3xTg-AD (P301Ltau/APPSw/PS1 M146L) (50). In these studies, mitochondrial dysfunction was evidenced by an age-related decrease in the activity of regulatory enzymes of OXPHOS such as COX, or of the Krebs cycle such as pyruvate dehydrogenase, analyzing 3xTg-AD mice aged from 3 to 12 months (82). Besides, these mice also exhibited increased oxidative stress and lipid peroxidation. Most of the effects on mitochondria were seen at the age of 9 months, whereas mitochondrial respiration was significantly decreased at 12 months of age. Importantly, mitochondrial bioenergetics deficits were found to precede the development of AD pathology in the 3xTg-AD mice. Figure 4 nicely shows that AD-specific changes including cognitive impairments, Aβ accumulation, Aβ plaques, and mitochondrial dysfunction seem to occur at an earlier onset from single, double up to triple AD transgenic mice models. Together, our studies highlight the key role of mitochondria in AD pathogenesis and consolidate the notion that a synergistic effect of tau and Aβ enhances the pathological weakening of mitochondria at an early stage of AD.

Aβ-Binding Alcohol Dehydrogenase: A New Lead to Decode the Mechanisms of Aβ-Induced Mitochondrial Dysfunction
A few years ago, Yan and collaborators showed that the Aβ peptide can directly bind a mitochondrial enzyme called Aβ-binding alcohol dehydrogenase (ABAD) that is overexpressed in the brains of Alzheimer's patients and AD mouse models (79). The interaction of Aβ with this enzyme exacerbates mitochondrial dysfunction induced by Aβ (decrease of mitochondrial complex IV activity, diminution of O2 consumption, and increase of ROS), as shown in double-transgenic mice overexpressing mutant APP and ABAD (81). Furthermore, these mice presented an earlier onset of cognitive impairment and histopathological changes when compared with APP mice, suggesting that the Aβ–ABAD interaction is an important mechanism underlying Aβ toxicity. The Aβ–ABAD complex could have a direct effect on the ETC because ABAD was found to be one of three proteins that comprise the fully functional mammalian mitochondrial RNAse P (63), a function that may not require dehydrogenase activity and that links ABAD directly to the production of mitochondrial ETC proteins and ROS generation.
Recently, it has been shown that inhibition of Aβ–ABAD interaction by a decoy peptide can restore mitochondrial deficits and improve neuronal and cognitive function (81). Our findings, using SH-SY5Y neuroblastoma cells treated with Aβ1-42, a cellular model of AD, seem to confirm these observations (Lim et al., unpublished observations). We employed a novel small ABAD-specific inhibitor to investigate the role of this enzyme in Aβ toxicity. The inhibitor significantly improved metabolic functions impaired by Aβ, and specifically reduced Aβ-induced oxidative stress and cell death. Furthermore, we have shown previously that the production of estradiol, a well-known neuroprotective neurosteroid and ABAD substrate, is increased after 24 h in the presence of a “nontoxic” concentration of Aβ and is decreased when using a toxic concentration of this peptide (64), suggesting that Aβ is able to modulate (directly or indirectly) neurosteroid levels. Accordingly, new findings from our group demonstrate that the levels of estradiol in the cytosol and in mitochondria can differently be influenced by Aβ peptide (500 nM, 5 days of treatment) (Fig. 5A, B). We observed that cytosolic estradiol is reduced in the presence of Aβ, but at the same time mitochondrial estradiol load was significantly increased. We suggest that this increase is due to an Aβ-induced decrease of ABAD activity, thus limiting the conversion of estradiol in estrone within mitochondria (Fig. 5C). Inhibition of ABAD activity by Aβ peptide was already demonstrated by Yan and collaborators (80) using 17β-estradiol as substrate of the enzyme. One mechanism that could explain this inhibition is the fact that Aβ–ABAD interaction changes the conformation of the enzyme, avoiding the binding of the cofactor NAD+, and this reduces the metabolic activity of ABAD (41). However, the total amount of estradiol is about 500-fold higher than in the mitochondrial fraction. Even if Aβ induced an increase in estradiol within mitochondria, the reduction of total estradiol level by other enzymes of the complex steroidogenic pathway may therefore be more relevant for cellular dysfunction. Besides, it was also speculated that estradiol exhibits a “prooxidant effect” in the presence of ongoing oxidative stress (49). Thereby, estradiol is hydroxylated to catecholestrogens that can enter a redox cycle generating superoxide radicals, leading to a continuous formation of ROS that amplifies oxidative stress.
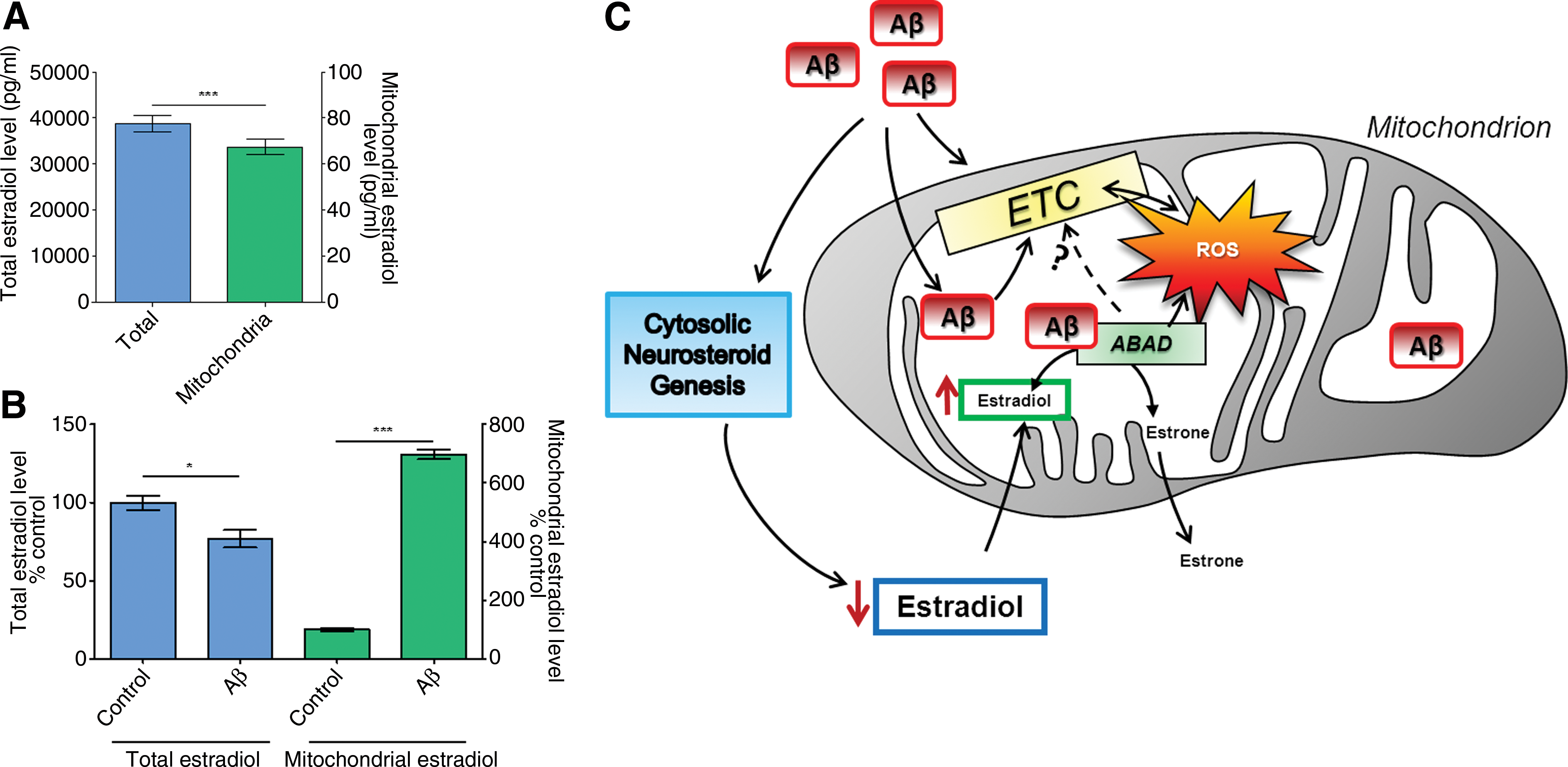
Thus, inhibition of the Aβ–ABAD interaction seems to be an interesting therapeutic target to block or prevent Aβ-induced mitochondrial toxicity because it could normalize the imbalance between ROS and estradiol levels in mitochondria and thereby help in improving mitochondrial and neuronal function.
Conclusion
We discuss here the recent findings regarding the possible shared mechanisms involving mitochondrial decline driven by brain aging and the close interrelationship of this organelle with the two main pathological features in the pathogenic process underlying AD.
According to the mitochondrial aging theory, ROS-induced damage and mtDNA mutations accumulate over time inducing ETC impairment and weaken mitochondria function in a rather unspecific way; thus, laying the ground for the two common hallmarks of AD, plaques and NFTs, or Aβ and tau, respectively, which destruct independently as well as synergistically this vital organelle via specific mode of actions on complexes I and IV.
Given the complexities of AD, the key role of mitochondrial dysfunction in the early pathogenic pathways by which Aβ leads to neuronal dysfunction in AD is particularly challenging with respect to establishing therapeutic treatments. Besides the modulation and/or removal of both Aβ and tau pathology, strategies involving efforts to protect cells at the mitochondrial level by stabilizing or restoring mitochondrial function or by interfering with energy metabolism appear to be promising. Transgenic AD mice, and particularly triple-transgenic models that combine both pathologies in an age-dependent manner (Fig. 4), are valuable tools in monitoring therapeutic interventions at the mitochondrial level. Eventually, this may lead to therapies that prevent the progression of the age-related mitochondrial decline thereby reducing the vulnerability to Aβ and/or tau at an early stage of the disease.
Footnotes
Acknowledgments
This work has been supported by grants from the Swiss National Science Foundation (#31000_122572), Sciex_NMSCH and Synapsis Foundation.