
Abstract
Introduction
Recent findings indicate that cyanate amplifies vascular inflammation, linking it to uremia and smoking (10). Leukocyte-derived MPO may serve as an important catalytic source for protein carbamylation at sites of inflammation, based on the observation that protein carbamylation during peritonitis was markedly reduced in MPO-deficient mice (36). Moreover, carbamylated epitopes were found to colocalize with MPO in human atherosclerotic lesions (36).
We have recently reported that high-density lipoprotein (HDL) is selectively carbamylated in human atherosclerotic lesions (15). The carbamyllysine content of lesion-derived HDL correlated with the MPO-specific oxidation product 3-chlorotyrosine (3-CT), strongly supporting the concept that leukocyte-derived MPO generates significant amounts of cyanate at sites of inflammation. These findings are in line with the observations that (i) MPO associates with HDL in atherosclerotic lesions (6, 39); (ii) MPO/HDL interaction increases on the oxidation of HDL; and (iii) association of MPO with HDL does not alter MPO activity (22). Interestingly, the carbamyllysine content of lesion-derived HDL is several-fold higher compared with 3-CT levels (15), raising the possibility that MPO-generated chlorinating species are involved in cyanate formation.
Innovaion
The carbamylation of (lipo)proteins through cyanate generates novel risk factors for cardiovascular disease and is considered as playing a causal role in disease progression. We have recently observed that the phagocyte protein myeloperoxidase (MPO) preferentially induces high-density lipoprotein (HDL) carbamylation, rather than chlorination, in human atherosclerotic lesions, raising the possibility that MPO-derived chlorinating species are involved in cyanate formation.
Here, we report that MPO-derived reactive chlorinating species promote the carbamylation of proteins through the decomposition of urea and thiocyanate (SCN). Physiological concentrations of SCN prevented 3-chlorotyrosine (3-CT) formation in proteins, suggesting that MPO preferentially induces protein carbamylation—rather than chlorination—at sites of inflammation. Urea levels can reach 100 mM in patients with chronic renal failure; hence, an augmented decomposition of urea may generate high yields of cyanate. Significantly, elevated MPO activity was recently demonstrated in patients with inflammation undergoing hemodialysis, supporting the association between inflammation and cumulative oxidative stress. Moreover, here, we demonstrate that minimal carbamylation of HDL significantly impairs (i) HDL's ability to activate lecithin-cholesterol acyltransferase, an enzyme that is essential for cholesterol esterification and HDL maturation; (ii) the activity of the major HDL-associated anti-inflammatory enzyme paraoxonase; and (iii) the antioxidative capability of HDL. Thus, the carbamylation of HDL in the atherosclerotic intima might critically hamper the antioxidant and anti-inflammatory properties of HDL. These data raise the possibility that protein carbamylation links inflammation, uremia, and cardiovascular disease.
In the present study, we propose a role for MPO-derived chlorinating species in mediating the rapid decomposition of urea and SCN, as a novel mechanism inducing (lipo)protein carbamylation. Moreover, we describe that MPO-mediated cyanate formation might be an important mechanism in generating dysfunctional HDL.
Results
MPO predominantly generates carbamyllysine in the presence of SCN
HDL isolated from human atherosclerotic lesions is mainly carbamylated, rather than chlorinated (Fig. 1A), raising the possibility that in the presence of SCN, MPO-induced protein carbamylation is favored compared with chlorination. Carbamyllysine was detected on lesion-derived apolipoprotein A-I (apoA-I) by immunoblotting using an anti-HCit-antibody, but not on HDL isolated from peripheral blood (Supplementary Fig. S1; Supplementary Data are available online at
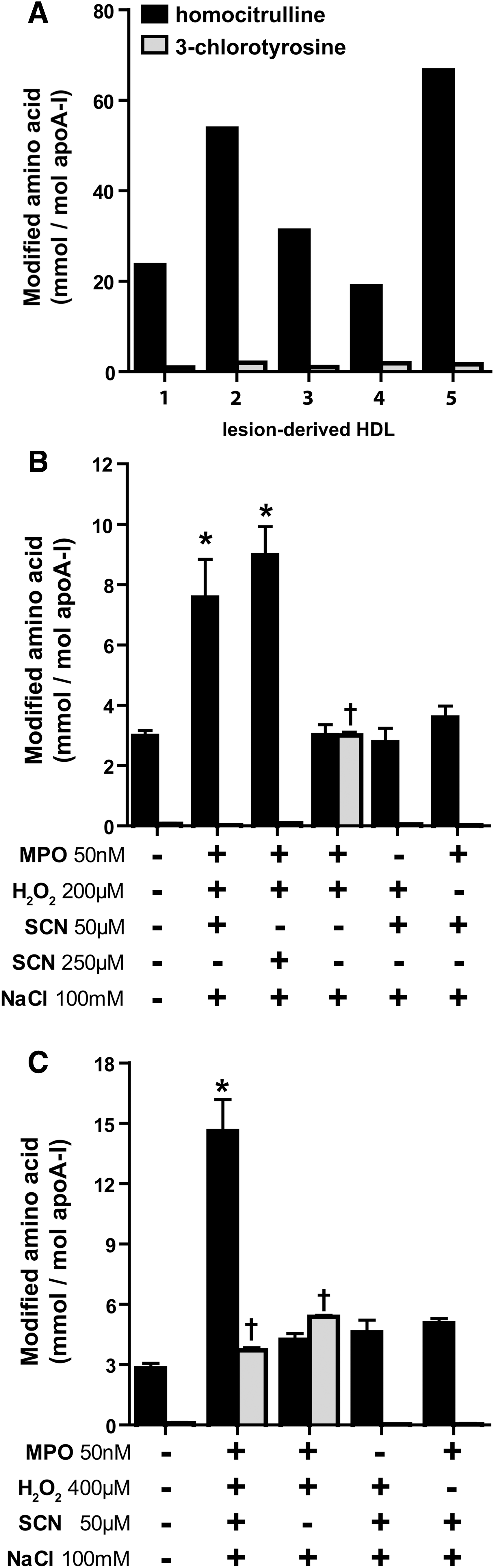
We first sought to investigate whether SCN could alter MPO-induced 3-CT formation in HDL. The incubation of MPO in the presence of physiological or pathological concentrations of SCN (50 or 250 μM, respectively), H2O2 (200 μM), and chloride (100 mM) significantly induced carbamyllysine formation, whereas 3-CT formation was not observed (Fig. 1B). When a higher excess of H2O2 (400 μM) than SCN (50 μM) was used, carbamyllysine formation increased further and, as expected, the formation of 3-CT was also observed (Fig. 1C). When SCN was removed from the reaction mixtures, 3-CT formation was prominent (Fig. 1B, C). In the absence of MPO or H2O2, neither carbamyllysine nor 3-CT formation was observed.
Hypochlorous acid induces decomposition of SCN and causes protein carbamylation
The lack of significant 3-CT formation indicates that SCN is a preferred substrate for MPO and is particularly effective in scavenging reactive chlorinating species, as demonstrated in previous studies (3, 24, 27, 37). Remarkably, when 100 μM hypochlorous acid (HOCl) was added to 30% plasma containing SCN (interstitial fluids contain about 30%–40% of plasma proteins) (30, 32), carbamyllysine formation was observed, whereas 3-CT formation was not increased (Fig. 2A). When HOCl was added at a higher concentration, the carbamyllysine formation further increased, depending on the SCN concentration. Of note, HOCl added at higher concentrations induced 3-CT formation, even in the presence of SCN (Fig. 2A).
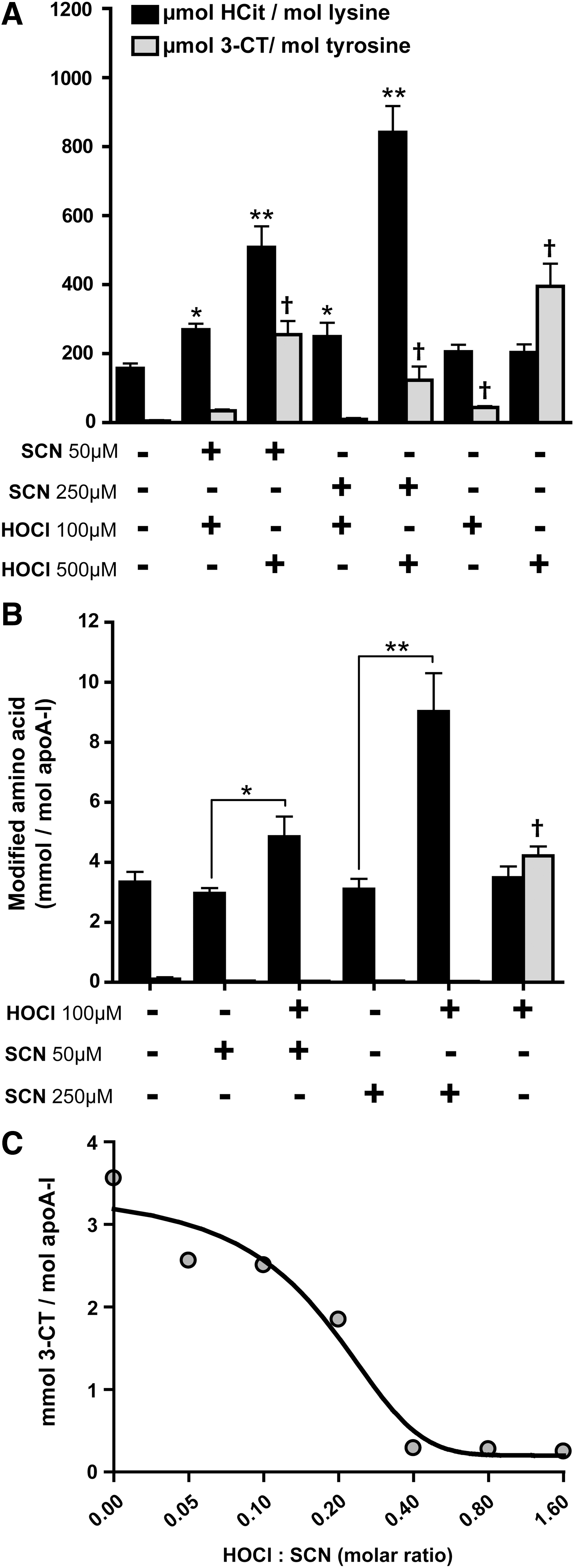
Similar results were obtained when HDL was incubated with SCN and HOCl in the absence of serum. Under these experimental conditions, 3-CT formation was completely prevented by SCN (Fig. 2B). The addition of HOCl in the absence of SCN induced 3-CT formation, as expected.
To estimate the HOCl-scavenging potential of SCN, a fixed amount of SCN was incubated with increasing concentrations of HOCl. We observed that SCN completely scavenged a 2.5-fold molar excess of HOCl (Fig. 2C). This indicates that over-oxidized products such as cyanosulfite or sulphate are formed on the oxidation of SCN with an excess of HOCl or chloroamines, as suggested in a recent study (37). These data indicate that only in the absence of adequate SCN, the overproduction of HOCl by MPO might result in relevant protein chlorination.
Cyanate formation through HOCl-induced decomposition of urea
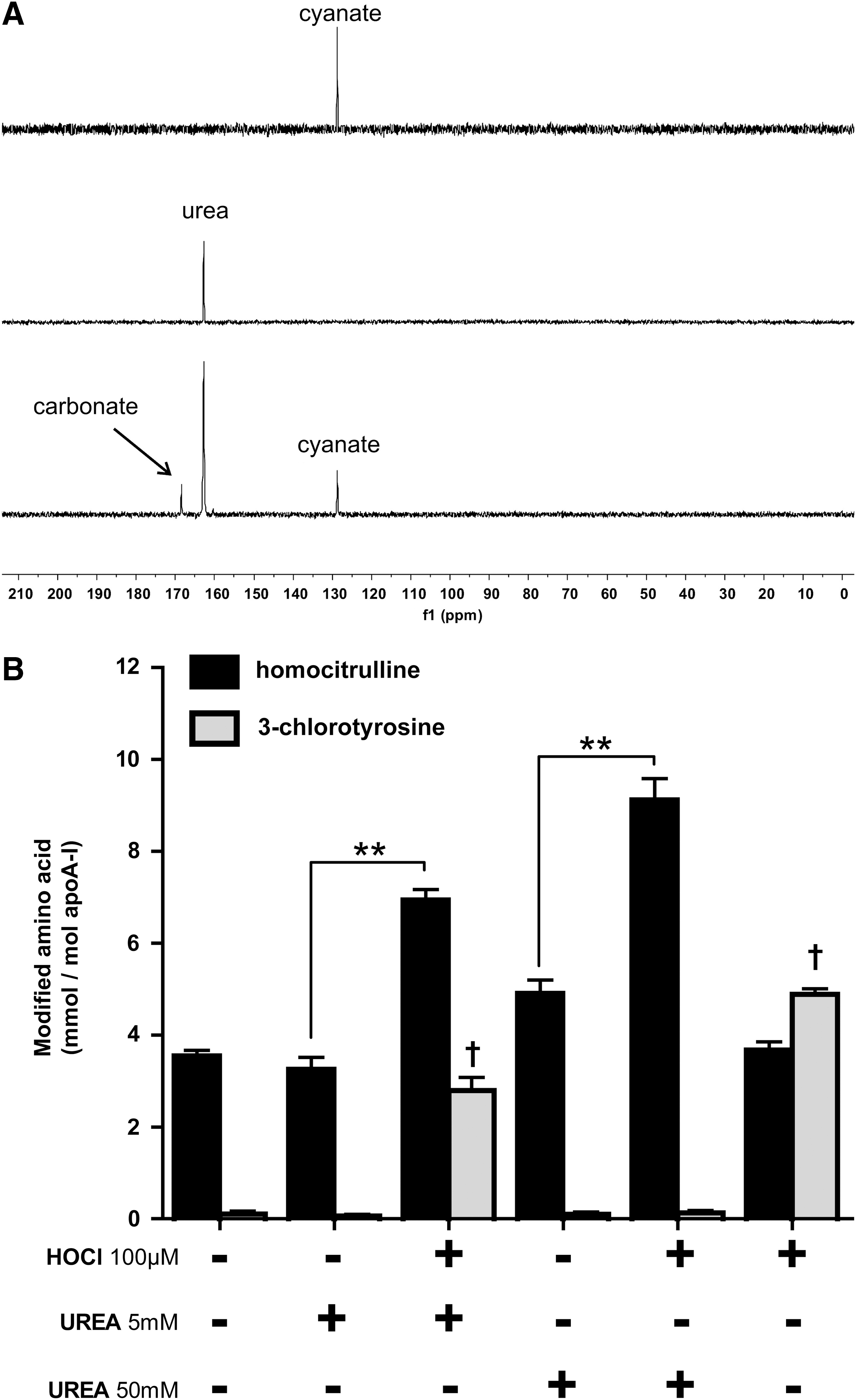
Besides the formation of cyanate from SCN oxidation, cyanate is known to be formed by the slow decomposition of urea. It is well known that excess HOCl decomposes urea, forming nitrogen and carbon dioxide. We tested whether cyanate is formed as a byproduct. For that purpose, we investigated the decomposition of urea by HOCl using solution-state nuclear magnetic resonance (NMR) spectroscopy, using a 1:0.5 molar ratio of 13C,15N-labeled urea: HOCl. The reaction was monitored by inverse gated decoupling 13C NMR spectra. On the addition of HOCl, the signal of urea (at 162.8 ppm) decreased within the dead time of the experiment (3 min), and peaks for carbonate and cyanate appeared at 168.5 and 128.7 ppm, respectively (Fig. 3A). Longer incubations did not further increase the signal intensity of cyanate, indicating the rapid formation of cyanate. About 25% of urea is converted to cyanate, demonstrating that high yields of cyanate are formed on the HOCl-induced decomposition of urea. The addition of HOCl (100 μM) to urea levels observed in control subjects (5 mM) induced significant protein carbamylation and 3-CT formation. Notably, in the presence of high urea concentrations (50 mM, observed in renal patients), the addition of HOCl significantly amplified protein carbamylation, but did not result in 3-CT formation, whereas the addition of HOCl alone induced only 3-CT formation (Fig. 3B). High urea concentrations on their own tended to induce protein carbamylation, but the extent of carbamylation significantly increased only in the presence of HOCl. Based on these results, it can be concluded that MPO-derived chlorinating species induce (lipo)protein carbamylation through the oxidation of SCN and/or the rapid decomposition of urea.
Carbamylated HDL is a less potent substrate for lecithin-cholesterol acyltransferase
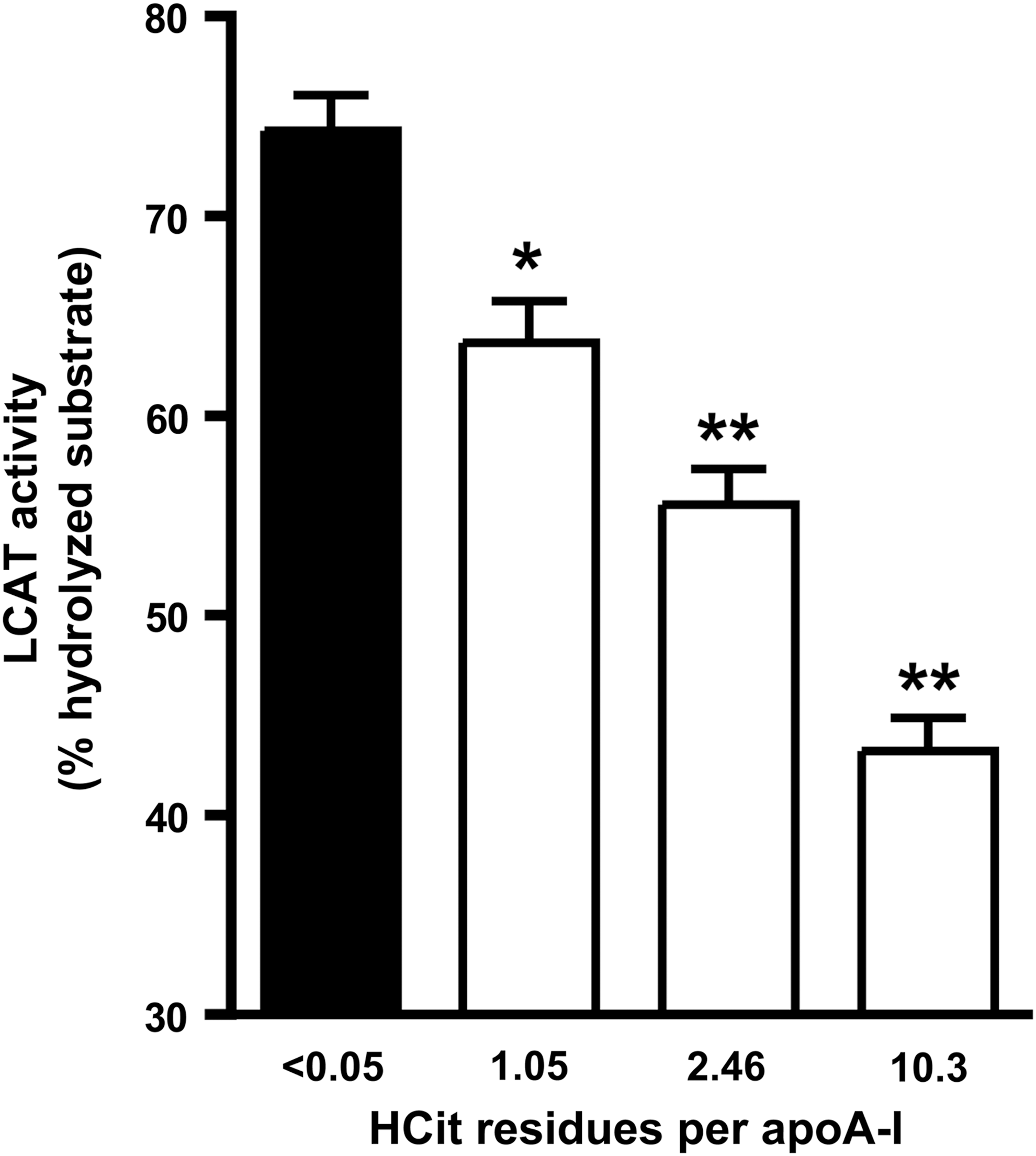
Lecithin-cholesterol acyltransferase (LCAT) drives the maturation of nascent HDL into spherical HDL, an essential step in reverse cholesterol transport. To assess whether carbamylation of HDL alters LCAT activity, a commercially available assay for serum/plasma LCAT activity was used to measure the efficiency of different HDL preparations to activate LCAT. For that purpose, HDL was labeled with a fluorescent LCAT substrate, which changes its fluorescence emission wavelength from 390 to 460 nm on hydrolysis by LCAT. The labeled substrate was rapidly incorporated into HDL (Supplementary Fig. S2). The labeled HDL preparations were incubated with a fixed amount of lipoprotein-deficient serum (LPDS) as a source for LCAT. As seen in Figure 4, carbamylation dose dependently decreased the ability of HDL to activate LCAT.
Carbamylation reduces the antioxidant properties of HDL and decreases the activity of HDL-associated paraoxonase
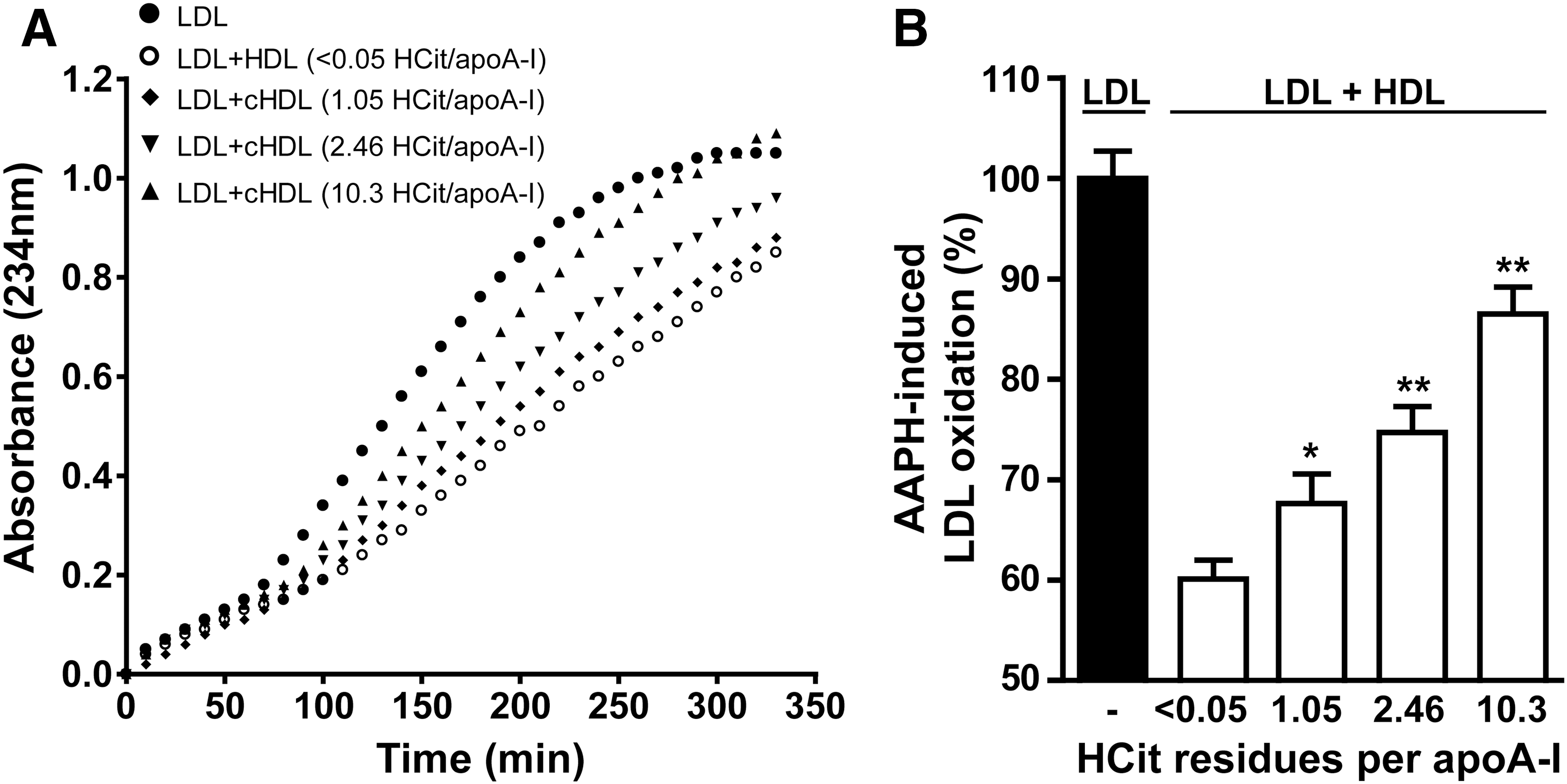
Besides the important role of HDL in lipid metabolism, it also exhibits unique antioxidative activity. We investigated the ability of HDL to inhibit radical-induced low-density lipoprotein (LDL) oxidation. LDL was incubated with the radical inducer 2,2′-azobis-2-methyl-propanimidamide dihydrochloride (AAPH), and a characteristic oxidation curve with lag time and propagation phase was obtained (Fig. 5A). The oxidation of LDL could be markedly inhibited by the addition of native HDL. However, the ability of HDL to inhibit radical-induced oxidation was significantly decreased on carbamylation (Fig. 5B), which indicates that carbamylation impairs the antioxidative properties of HDL.
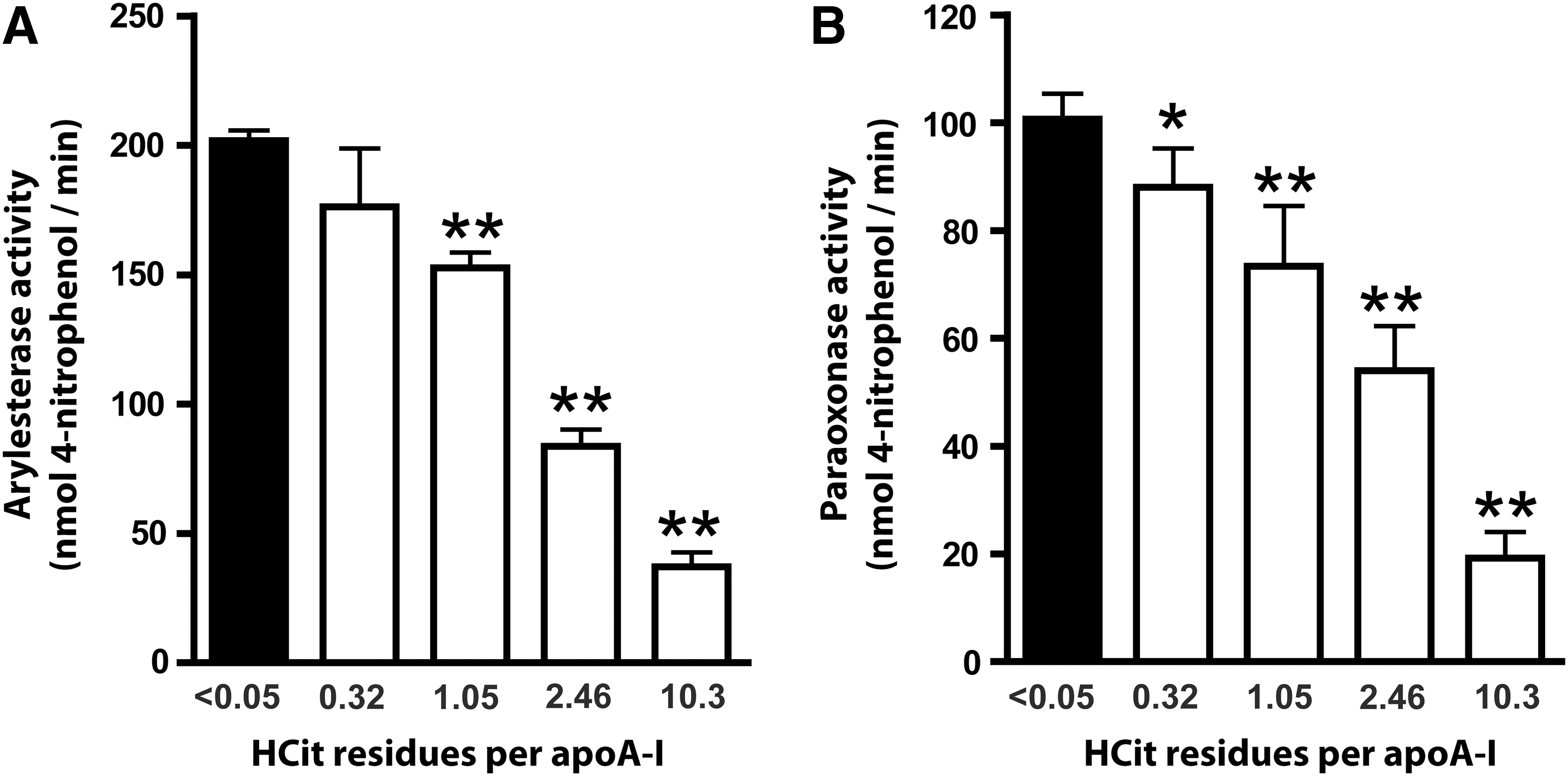
Recent data suggest that the HDL-associated enzymes paraoxonase (PON) and LCAT are involved in the antioxidant activity of HDL (4, 5, 13). We analyzed the impact of protein carbamylation on PON-associated PON and arylesterase activity. Strikingly, when HDL was exposed to cyanate-inducing minimal carbamylation, PON (Fig. 6A) and arylesterase activities (Fig. 6B) of PON were significantly reduced. PON activity further decreased when HDL was modified with higher cyanate concentrations.
Discussion
Carbamylated (lipo)proteins are risk factors for cardiovascular disease and seem to play a causal role in disease progression. It was long thought that cyanate formation occurs to a significant extent only during renal dysfunction through the slow decomposition of urea. Approximately, 0.8% of urea decomposes to cyanate within one week in aqueous solution (8). However, recent studies have unambiguously demonstrated that humans are exposed to additional significant amounts of cyanate formed by the pyrolysis/combustion of coal, biomass, or tobacco (28), and that cyanate formation is catalyzed by the leukocyte heme-peroxidase MPO (2, 36). Here, we provide novel evidence of an alternative mechanism for cyanate formation. MPO-derived chlorinating species rapidly decompose SCN and/or urea, forming cyanate, therby promoting (lipo)protein carbamylation (Fig. 7). Physiologic concentrations of SCN prevented 3-CT formation in HDL, suggesting that MPO preferentially induces protein carbamylation—rather than chlorination—at sites of inflammation. This is in good agreement with the finding that the carbamyllysine content of lesion-derived HDL was more than 20-fold higher in comparison to 3-CT levels, a specific oxidation product of MPO (15). We quantified that apoA-I recovered from atherosclerotic tissue contains approximately 70 mmol HCit/mol apoA-I (Fig. 1A), indicating that approximately 7% of lesion-derived apoA-I possess at least one HCit residue. Our in vitro experiments revealed that one HCit-residue/apoA-I renders HDL dysfunctional; therefore, particularly those HDL in macrophage-rich compartments with high MPO activity—but not all HDL in the lesion—are likely to be rendered dysfunctional through carbamylation. It should be noted that more then 10-fold higher 3-CT levels of apoA-I—as observed in our study—were reported when human atherosclerotic tissue was stripped of adventitia before isolating HDL (39), raising the possibility that HDL in subendothelial compartments with high MPO activity may be even more extensively carbamylated.
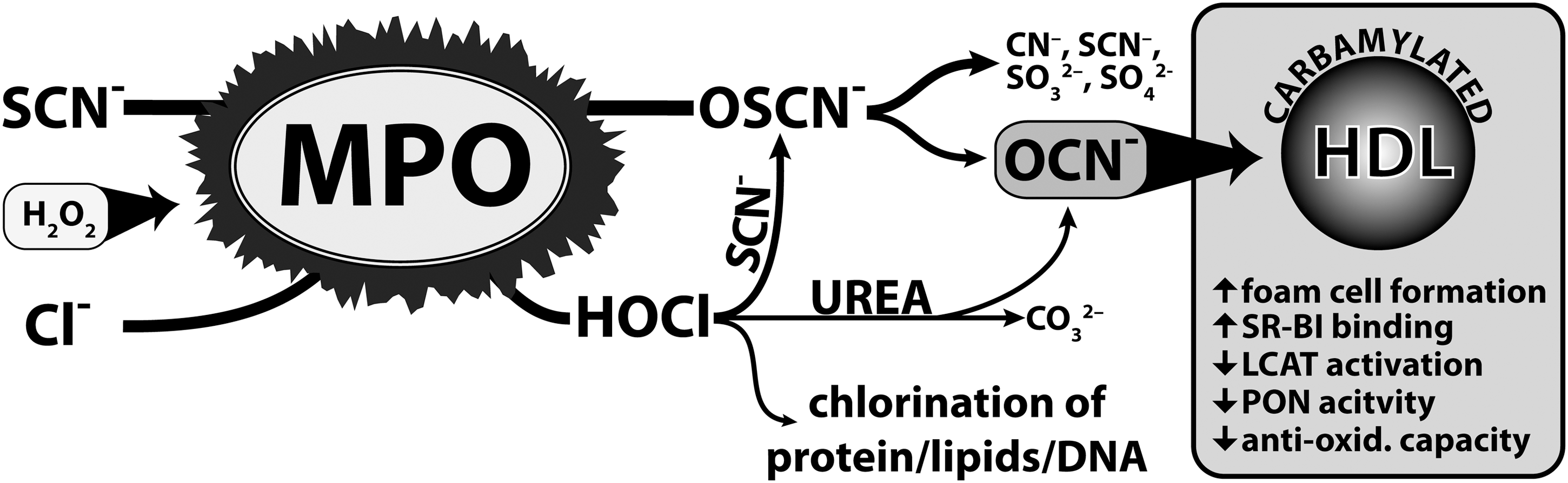
In line with our observations, it was recently demonstrated that SCN effectively scavenges HOCl and protein-associated chloramines, forming cyanate and sulphate (37). This is in agreement with a study showing that the rate constant for the reaction of SCN with chloramines is comparable to the rate constants for the best proteinaceous nucleophiles cysteine and methionine (26). Interestingly, hypothiocyanous acid (HOSCN) itself was reported to react even faster with chloramines than SCN (37), likely explaining that in the present study, low concentrations of SCN completely blocked 3-CT formation, as lysine-chloramine formation is reported to be involved in 3-CT formation (9).
Another important observation in the present study is that the major MPO product HOCl decomposes urea, generating high yields of cyanate. Our findings may be of particular relevance, as urea levels can reach 100 mM in patients with chronic renal failure, and elevated MPO activity was demonstrated in patients undergoing hemodialysis with high-grade persistent inflammation (29). Therefore, the increased decomposition of urea may generate high yields of cyanate at sites of inflammation. In line with our data, it was previously reported that MPO-containing neutrophils and monocytes of patients with chronic renal disease are highly enriched with carbamylated proteins compared with control subjects (20). The localization of phagocytes in the immediate vicinity of endothelial cells at sites of inflammation may contribute to the cyanate-induced expression of endothelial intercellular cell adhesion molecule-1, as recently observed (10). This raises the possibility that cyanate increases endothelial-leukocyte interaction, thus forming a vicious circle. In this regard, it was shown that serum MPO levels correlate with levels of inflammatory markers and with mortality in patients undergoing hemodialysis (17).
In line with our previous results (15), we observed that carbamyllysine levels are markedly elevated in HDL isolated from human atherosclerotic tissue. These results are in line with previous findings that MPO binds to HDL within human atherosclerotic lesions and might produce reactive species in closest proximity to HDL (6, 15, 23, 39). Interestingly, HDL derived from atherosclerotic lesions was mainly carbamylated—rather than chlorinated—suggesting that MPO-generated chlorinating species are involved in cyanate formation.
In the present study, we report that the carbamylation of HDL significantly impaires its ability to activate LCAT, an enzyme that is essential for cholesterol esterification and maturation of HDL. apoA-I is recognized as a major activator of LCAT in HDL; however, the mechanism by which apoA-I facilitates cholesterol esterification and particle maturation remains unclear. Previous studies have shown that the mutation of charged residues in apoA-I results in marked reductions of LCAT activity, indicating that the elimination of positive charge through the carbamylation of apoA-I lysine residues may cause reduced LCAT activity (1). Significantly, cyanate targets not only lysine residues, but also phosphatidyethanolamine (15) and interacts with cysteine groups with an even higher reactivity (34). Therefore, the decrease in the LCAT-activating capability of cyanate-treated HDL might be the result of the modification of several reactive groups in HDL-associated apoproteins, enzymes, and phospholipids.
In a previous study, we observed that carbamylation increases the binding affinity of HDL to its physiological receptor, scavenger receptor B-I (SR-BI), thereby causing intracellular cholesterol accumulation and lipid-droplet formation in macrophages (15). According to our present study, these data raise the possibility that the loss of functional HDL/SR-BI interaction and LCAT-activating ability—as induced by carbamylation—results in impaired reverse-cholesterol transfer capacity.
The antiatherogenic effect of HDL has been attributed to several mechanisms, including the prevention of LDL oxidation. HDL-mediated inactivation of LDL-associated phospholipid-hydroperoxides involves initial transfer from LDL to HDL, which is governed by the rigidity of the surface monolayer of HDL, and the subsequent reduction of hydroperoxides by redox-active methionine residues of apolipoproteins (12, 19, 38). HDL-associated enzymes may, in turn, contribute to the hydrolytic inactivation of short-chain oxidized phospholipids. PON is a HDL-associated enzyme hydrolyzing various lactones and organophosphates and was shown to metabolize lipoprotein-associated oxidized lipids (5, 21). Significantly, PON was shown to selectively decrease lipid peroxides in human coronary and carotid atherosclerotic lesions (4) and to protect against atherosclerosis and coronary artery diseases (31). Therefore, the decrease in antioxidant activity of carbamylated HDL may be the result of decreased PON activity. However, other cyanate-induced modifications of the protein and phospholipid moiety of HDL may contribute to the impaired antioxidative capacity.
In summary, the present study has identified novel mechanisms of protein carbamylation, highlighting the link between inflammation and cardiovascular disease with uremia, smoking, and air pollution. The carbamylation of HDL in the atherosclerotic intima might critically impair antioxidant and anti-inflammatory properties of HDL, thereby promoting the development of atherosclerosis.
Materials and Methods
13C,15N-labeled urea was purchased from Cambridge Isotope Laboratories (Andover, MA). HCit (carbamyllysine) was purchased from Bachem (Weil am Rhein, Germany), and 13C6-HCit was purchased from Ascent Scientific (Bristol, United Kingdom). The LCAT activity kit was purchased from Merck (Darmstadt, Germany). MPO was supplied by Enzo Life Science (Vienna, Austria). All other reagents were obtained from Sigma (Vienna, Austria), unless otherwise specified.
Blood collection
Blood was sampled from healthy volunteers after an informed consent, according to a protocol approved by the Institutional Review Board of the Medical University of Graz (14).
Isolation of lipoproteins
For lipoprotein isolation, serum density from normolipidemic blood donors was adjusted with KBr to 1.24 g/ml. To optimize lipoprotein separation, long centrifuge tubes (16×76 mm; Beckman Instruments, Krefeld, Germany) were used. A two-layer density gradient was generated by layering the density-adjusted serum (1.24 g/ml) underneath phosphate-buffered saline (PBS; 1.006 g/ml). The tubes were sealed and centrifuged at 90,000 rpm for 4 h in a 90TI fixed-angle rotor (Beckman Instruments). After centrifugation, the two clearly separated HDL- and LDL-containing bands were collected, desalted via a PD10 column (GE Healthcare, Vienna, Austria), and directly used for experiments. HDL-like particles from human atherosclerotic tissue were isolated as previously described (15).
Modification of HDL
HDL was carbamylated with potassium cyanate as previously described (15). MPO-mediated modification of HDL was performed in 50 mM phosphate buffer (pH 7.0). Reaction samples contained 2.5 mg/ml HDL, 50 nM MPO, 100 mM NaCl, 200 or 400 μM H2O2, and 50 or 250 μM SCN. The reactions were allowed to take place for 48 h at 37°C. Afterward, the samples were placed on ice to stop the reaction, and unreacted compounds were removed by gel filtration. Carbamylation was assessed by liquid chromatography tandem mass spectrometry (LC-MS/MS).
HOCl-induced decomposition of SCN/urea
SCN (50–250 μM) or urea (5–50 mM) in PBS were mixed with HOCl (100 μM) and immediately added to a 2.5 mg/ml HDL solution. The reaction mixture was incubated for 16 h at 37°C. For experiments with human plasma, total plasma was diluted with PBS to 30%. The diluted plasma was incubated in the absence or presence of 50 or 250 μM SCN and 100 or 500 μM HOCl for 16 h at 37°C. Afterward, the unreacted compounds were removed by gel filtration, and protein carbamylation was assessed by LC-MS/MS.
NMR spectroscopy
All spectra were recorded on a Bruker 700 MHz Avance III NMR spectrometer, equipped with a 5 mm TCI cryo probe. Inverse gated proton-decoupled 13C-spectra were acquired with 128 scans and an interscan delay of 2 s. The pulse width corresponded to a flip angle of ∼30° to reduce any saturation in the spectra. A solution of 50 mM 13C, 15N-labeled urea was mixed with a 750 mM stock solution of HOCl, yielding a final concentration of 25 mM HOCl. The experiments were carried out in a PBS buffer (pH 7.4) at 298 K.
Quantification of carbamyllysine and 3-CT
Proteins were hydrolyzed with a fast, low-volume hydrolysis method as described (7). To quantify carbamyllysine and chlorotyrosine formation in HDL, electrospray ionization LC-MS/MS was used as previously described (15).
PON/arylesterase activity assay
Ca2+-dependent PON activity was determined as the rate of hydrolysis of paraoxon into 4-nitrophenol, which can be monitored by an increase in absorbance at 405 nm (11). Native or carbamylated HDL (10 μg protein) was placed into a 96-well plate, and the reaction was initiated by the addition of 200 μL buffer containing 100 mM Tris, 2 mM CaCl2, and 1 mM paraoxon (pH 8.0). The assay was performed at 37°C, and readings were taken every 5 min at 405 nm on a plate reader to generate a kinetic plot. The slope from the kinetic chart was used to determine ΔAbs405nm/min. Enzymatic activity was calculated with the Beer–Lambert Law from the molar extinction coefficient of 17,100 L·mol−1·cm−1. PON activities were expressed as nM 4-nitrophenol formed per minute.
Ca2+-dependent arylesterase activity was determined with a photometric assay using phenylacetate as the substrate (11). Native or carbamylated HDL (2 μg protein) was added to 200 μL buffer containing 100 mM Tris, 2 mM CaCl2 (pH 8.0), and phenylacetate (1 mM). The rate of hydrolysis of phenylacetate was monitored by the increase of absorbance at 270 nm, and readings were taken every 30 s at room temperature to generate a kinetic plot. The slope from the kinetic chart was used to determine ΔAbs270nm/min. Enzymatic activity was calculated with the Beer–Lambert Law from the molar extinction coefficient of 1310 L·mol−1·cm−1 for phenylacetate.
Carbamylation of HDL and assessment of LCAT activity
LCAT activity was measured using a commercially available kit (Merck), which was adapted to measure the substrate efficiency of HDL preparations. For that purpose, native or carbamylated HDL was preincubated with LCAT substrate for 90 min at 37°C, and the unbound substrate was removed using gel filtration. The binding efficiency was ∼95% of the added substrate, determined by fluorescence measurement of the substrate at 470 nm after gel filtration. The labeled HDL preparations were used to determine the substrate efficiency in response to LCAT enzymatic activity. LPDS was used as the source for LCAT.
The experimental setting was as follows: 100 μg labeled HDL and 10 μL LPDS were completed with LCAT buffer to 200 μL. LCAT buffer contained 150 mM NaCl, 10 mM TRIS, 4 mM mercaptoethanol, and 1 mM EDTA.
Endpoint readouts were obtained after incubation for 5 h at 37°C. Results are expressed as the percent of substrate hydrolyzed.
Determination of the antioxidative capacity of HDL
The antioxidative activity of HDL was determined as previously described (69, 120). LDL (100 μg/ml protein) was oxidized in vitro with the free radical initiator AAPH (1 mM in PBS) in the presence or absence of HDL (100 μg/ml protein) at 37°C. The oxidation was monitored at 234 nm. A time point in the propagation phase (at 200 min) was selected to assess the antioxidant activity of HDL. Oxidation in the presence of HDL was calculated as relative to the oxidation of LDL alone, which was set to 100%.
Statistical analyses
Data are shown as mean±SEM for n observations unless otherwise stated. Comparison between 2 groups was performed with Student's t-test and between multiple groups using one-way ANOVA with Bonferroni post hoc test. Significance was accepted at probability levels of p<0.05 and p<0.01. Statistical analyses were performed with GraphPad Prism version 4.03.
Sources of Funding
Michael Holzer and Sanja Curcic were funded by the Ph.D. Program Molecular Medicine of the Medical University of Graz. This work was supported by the Austrian Science Fund FWF FWF (Grants P21004-B02, P22521-B18, P22976-B18, P20020, and P22630).
Author Disclosure Statement
No competing financial interests exist.
Footnotes
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.