
Abstract
Introduction
Lipid peroxidation is one of the major sources of free radical–mediated injury that directly damages membranes and generates a number of secondary products. In particular, markers of lipid peroxidation have been found to be elevated in brain tissues and body fluids in several neurodegenerative diseases, and the role of lipid peroxidation has been widely investigated in the pathogenesis of Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), Huntington disease (HD), and Down syndrome (DS) (32, 111, 175, 176, 185).
This complex process involves the interaction of oxygen-derived free radicals with PUFAs resulting in a variety of highly reactive electrophilic aldehydes, including malondialdehyde (MDA), 4-hydroxy-2-nonenal (HNE), and acrolein (Fig. 1) (70, 118, 163). Following lipid peroxidation, HNE and MDA are the most abundant aldehydes produced, while acrolein is the most reactive. In addition, lipid hydroperoxyl radicals undergo endocyclization to produce fatty acids esters; two classes of these cyclized fatty acids are ispoprostanes and neuroprostanes (Fig. 1) (133, 134). Free radical damage to arachidonic acid leads to the formation F2-isoprostanes (F2-IsoPs), while F4-neuroprostanes (F4-NPs) are the stable product of free radical damage to docosahexanoic acid (DHA). Once formed, F2-NPs and F4-NPs can undergo hydrolysis to liberate free iso- and neuroprostanes that are detectable in body fluids (170).
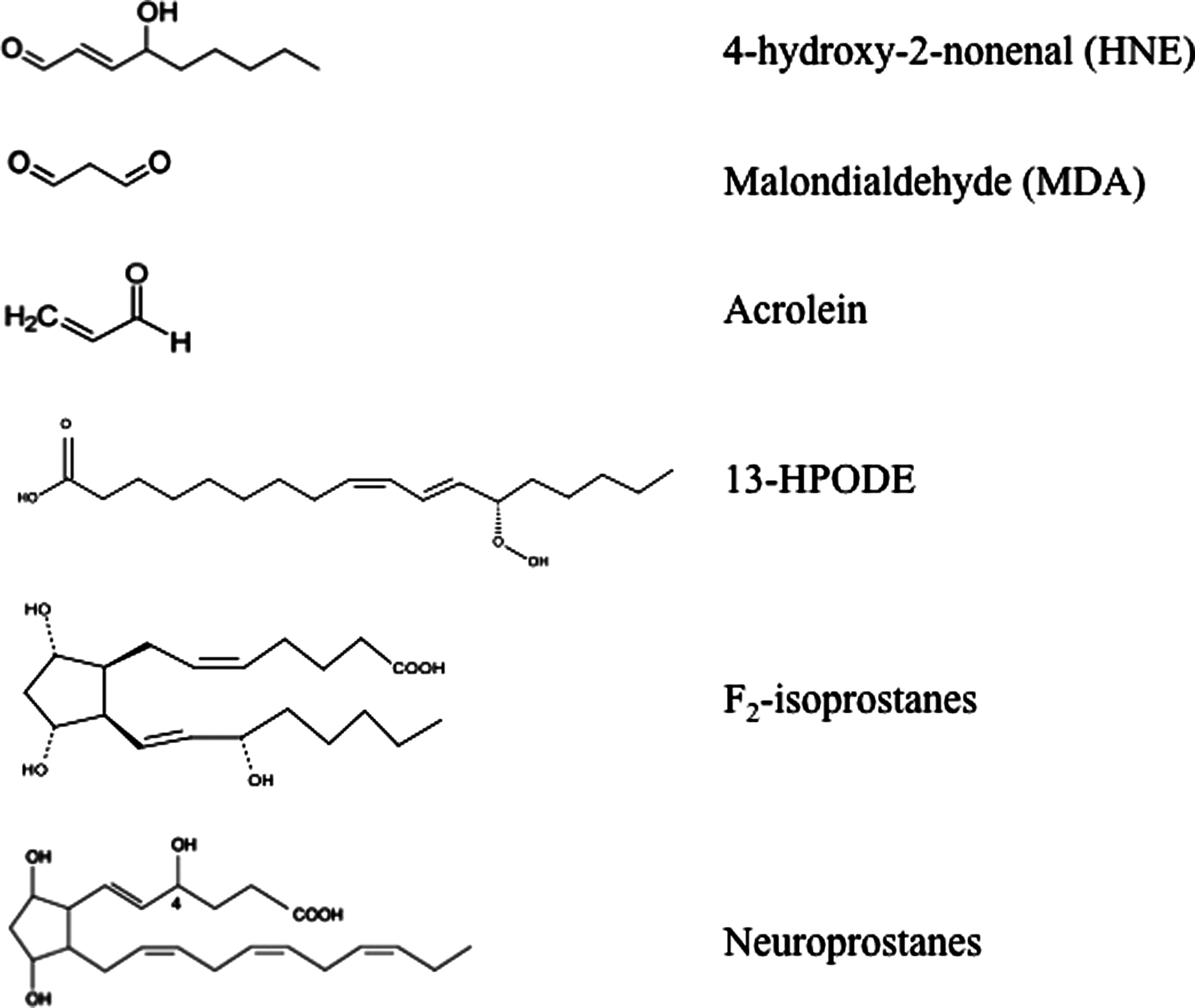
PUFAs and their metabolites display important physiological functions such as maintenance of membrane structure and fluidity, cell signaling, energy production, and regulation of gene expression. Among PUFAs, arachidonic acid (AA) and DHA are the major constituents of the brain phospholipid bilayer and are particularly vulnerable to free radical attack due to the presence of double bonds and associated allylic H atoms in their structure. In fact, the C–H bonds on the allylic carbon atom adjacent to the double bond become more labile and it results in a facile H atom abstraction from a methylene group (-CH2-) (86, 155). These reactions are self-propagating and proceed until substrate is consumed or termination occurs. In this way, lipid peroxidation is a self-sustaining process, which amplifies the effects of the original free radical and leads to the activation of a cascade of toxic reactions resulting in extensive tissue damage. Structural damage to membranes cause subsequent generation of oxidized products that, in turn, are chemically reactive and can covalently modify most biological macromolecules. Peroxidation of membrane lipids can have numerous effects: increased membrane rigidity, decreased activity of membrane-bound enzymes (e.g., sodium pump), altered activity of membrane receptors, and altered permeability (9, 213). In addition to effects on phospholipids, radicals can also directly attack membrane proteins and induce lipid–lipid, lipid–protein, and protein–protein cross-linking, all of which obviously have effects on membrane function (71). Several efforts have been devoted to understanding the role of toxic aldehydes in contributing to neuronal dysfunction in neurodegenerative diseases. Numerous studies have documented increased levels of reactive products of lipid peroxidation in diseased regions of brain in different brain disorders (32, 72, 194). Consistent with the importance of lipid peroxidation in neurodegenerative diseases, proteins modified by lipid peroxidation products were present in diseased regions of the brain but not in regions uninvolved in the disease (32).
Lipid Peroxidation: Chemistry and Products
Lipid peroxidation refers to the oxidative degradation of lipids which can be described in a five-step sequence (Fig. 2).
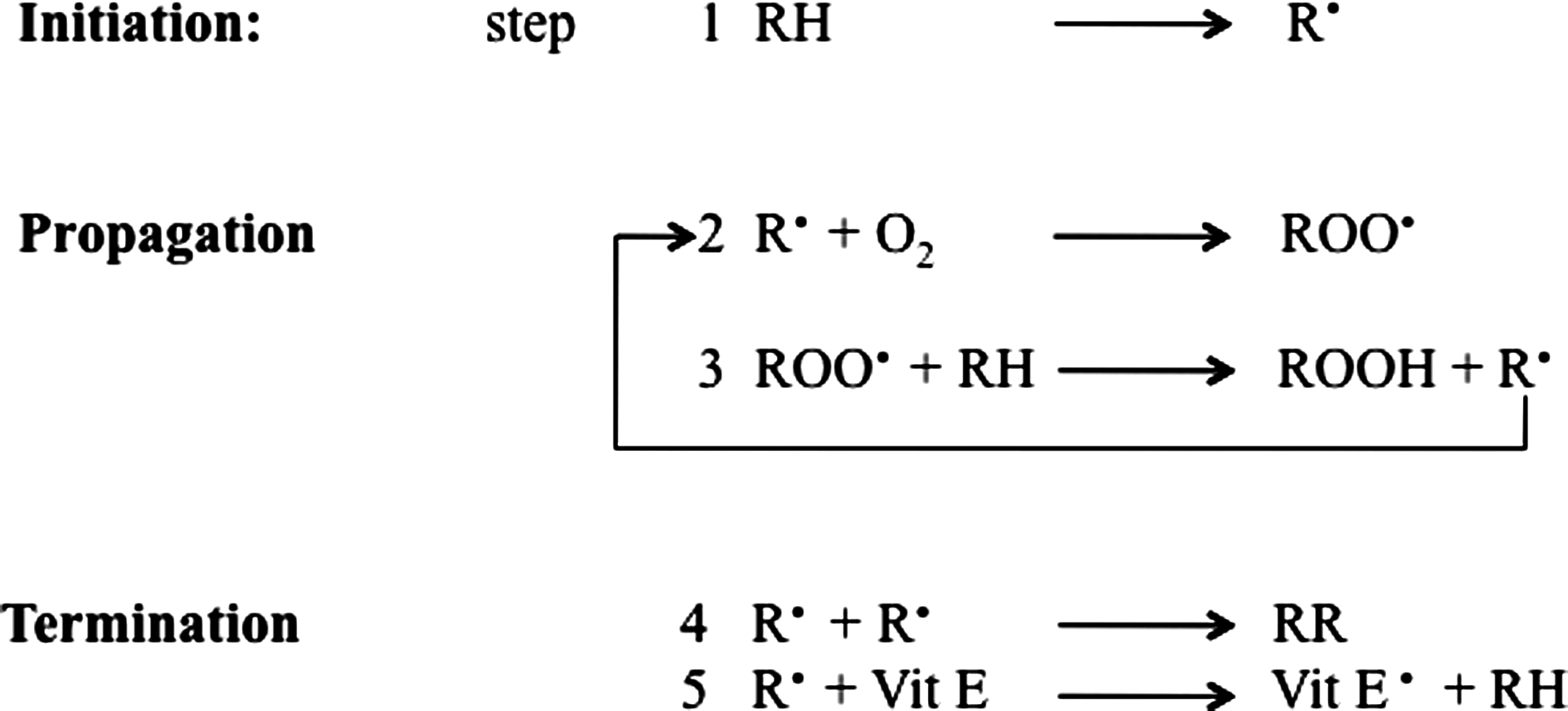
Step 1 involves initiation, in which the free radical (hydroxyl HO·, alkoxyl RO·, peroxyl ROO·, and possibly HO2 · but not H2O2 or O2 −·) attacks a methylene group in the PUFAs, leading to an allylic H atom abstraction with rearrangement of the double bonds to the conjugated diene form, and simultaneously producing a carbon-centered alkyl radical. In step 2, the alkyl radical reacts with paramagnetic molecular oxygen to form a peroxyl radical. Propagation occurs in step 3, in which the peroxyl radical abstracts another allylic H atom, thereby initiating a self-perpetuating chain reaction in which most of the membrane lipids are converted to a variety of hydroperoxides and cyclic peroxides. The hydroperoxides can be further degraded to hydrocarbons, alcohols, ether, epoxides, and aldehydes. Of these products, MDA, HNE, and acrolein can cause irreversible modification of phospholipids, proteins, and DNA resulting in impaired function (69). In step 4, termination occurs, in which direct interaction between different types of radicals block the chain reaction cascade. Finally, in step 5, there is termination by interaction between the radicals and antioxidants, giving rise to nonradical products or unreactive radicals.
Both exogenous and endogenous antioxidants such as vitamin E and vitamin C prevent the propagation of lipid peroxidation at the early stages of free radical attack (21, 56). Vitamin E (α-tocopherol) acts as a “chain-breaking” antioxidant and can terminate propagation steps of lipid peroxidation. When the hydrogen is abstracted in step 1, an α-tocopherol radical forms (step 5) that can be reverted back to vitamin E by the potent antioxidants vitamin C (ascorbic acid) and glutathione (GSH). Cadenas et al. (40) in 1983 proposed this mechanism by comparing the effects of toxic concentrations of HNE on rat hepatocytes with and without pretreatment with Vitamin E. The protective effects exerted by antioxidant towards HNE and other toxic aldehydes have been investigated by many groups to test the possibility of a therapeutic use of free radical scavengers and antioxidants against lipid peroxidation–mediated toxicity. In addition to small molecules, antioxidant enzymes such as heme oxygenase-1, catalase, superoxide dismutase, peroxiredoxin, and GSH reductase have been shown to lead to a significant decrease in lipid peroxidation products (189, 204). Lipid peroxidation also has been reported in phospholipids, specifically in the form of oxidized phosphatidylcholine, which is considered a marker for inflammation as observed in both stroke and multiple sclerosis (2, 164).
HNE
HNE is an α,β-unsaturated aldehyde that is formed by peroxidation of ω-6 PUFAs such as linoleic acid, linolenic acid, and mostly AA (Fig. 3). Although the precise steps of formation may be multifactorial, it is generally assumed that HNE is the product of a nonenzymatic process in which an initial hydroperoxide undergoes fragmentation to form HNE (161). However, evidence by Esterbauer's group showed that the formation of HNE from AA is greater in the presence of NADPH-dependent microsomal enzymes (68). In addition, iron ions also promote the formation of HNE by PUFAs.
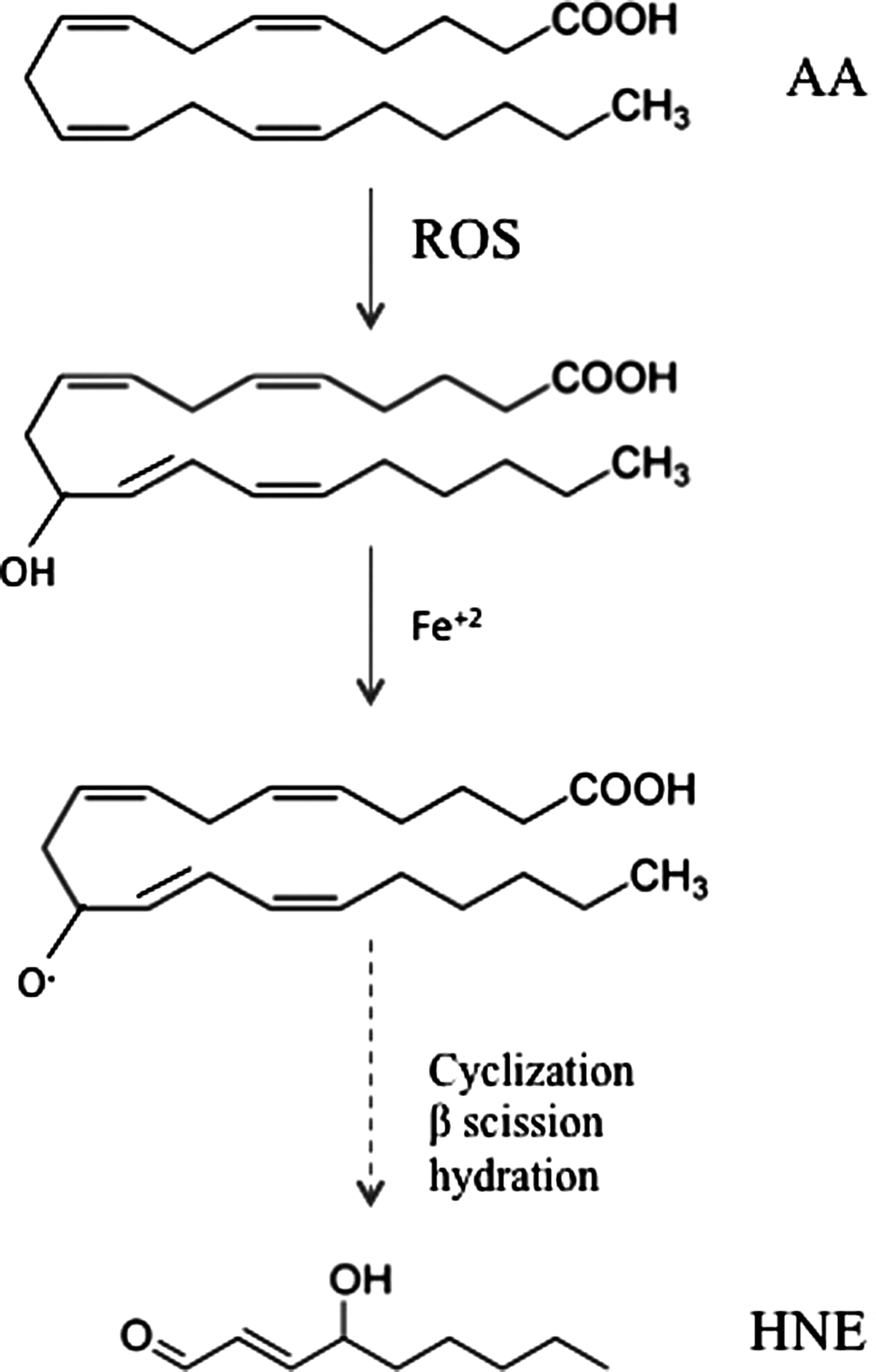
PUFAs undergo free radical–mediated mechanisms by which a lipid peroxyl radical is formed (182). Ultimately, the peroxyl radical is converted to an allylic carbocation via β-scission of the PUFA derivative. The peroxyl radical is further oxidized to a lipid peroxide. A hydration reaction breaks the C–O bond, resulting the a 9-carbon alkenal HNE. Once formed, HNE can covalently attach to proteins by Michael addition to Cys, His, and Lys residues, which can alter protein structure and cause a loss of protein function and activity (195, 202). HNE is not only a potent electrophile reacting with a variety of nucleophilic compounds, but also may act as a stress signaling molecule (70). HNE can accumulate at concentrations of 10 μM to 5 mM in response to oxidative insults and invokes a wide range of biological activities, including the selective suppression of basal and inducible nuclear factor-kappa B (NF-κB) activity (211). The transcription factor NF-κB can prevent neuronal death in experimental models of neurodegenerative disorders by inducing the expression of antiapoptotic proteins. HNE can disrupt ions homeostasis such as Ca2+, impair Na+/K+ATPase activity, disrupt the microtubule structure, and activate the caspase pathways leading to cell death (70). This reactive alkenal also causes an impairment of glucose transport in cultured rat hippocampal neurons and an alteration of the glutamate transport in rat neocortical synaptosomes (146). Therefore, uncontrolled and/or excessive production of HNE could interfere with normal cellular signaling and lead to development of pathological conditions as occur in several neurodegenerative diseases (129).
Reaction of HNE with proteins: biological consequences
HNE is an amphiphilic compound, with water-soluble, but much stronger lipophilic, properties that make HNE remain associated with the membranes where it is generated; however, HNE can also diffuse to different cellular compartments and interact with many different substrates. The high reactivity of HNE is due to the presence of three functional groups that frequently act synergistically (156). First a conjugated system of a C=C double bond and a CO carbonyl group provide a partial positive charge to carbon 3. This positive charge is further increased by the inductive effect of the hydroxy group at carbon 4 (Fig. 4). Therefore, nucleophilic attack, for example, by thiol or amino groups, occurs primarily at carbon 3 and second at the carbonyl carbon 1 (155). Besides biochemical modifications, biophysical changes of protein and lipid membrane properties also have to be considered. Alteration of membrane fluidity or loss of phospholipid asymmetry occurs in a dose-dependent manner (195). A physiologically relevant concentration of 1 mM HNE was found to cause significant protein conformational changes in synaptosomal membranes, while membrane fluidity increased only at a much higher concentration (50 mM) (195). In addition, HNE was found to modulate phospholipid asymmetry in synaptosomal membranes (46). This latter effect may have resulted from the formation of HNE adduct with the Cys residues of the enzyme flippase, which functions to maintain asymmetry of membrane lipid bilayer (46).

). At the same time, resonance between the double bond between C-3 and C-2 and the aldehyde dumps electron density away from C-3. The net effect is a partially positive C-3 that becomes a target for electrophilic reaction of lone pair of electrons on the S atom of cysteine.
Although proteins are the major targets of HNE reactivity, a great variety of biomolecules has been identified to form covalent adducts with HNE, including lipids, nucleic acids, and also cofactors and vitamins (39).
Proteins are particularly exposed to modification caused by HNE, and it is estimated that 1%–8% of the HNE formed in cells will modify proteins (101, 119). It is well established that HNE forms adducts with three different side chains in proteins, namely Cys, His, and Lys (Fig. 5). It can interact with thiol (SH) and amino (NH2) groups of these amino acids via Michael addition resulting in a covalent, bond between HNE and the amino acid. The differential reactivity of these residues compared with those of Arg and Glu for adduct formation showed that Cys residues displayed the highest reactivity, with Cys>His>Lys. This order does not imply that Cys residues are always the preferential targets of HNE in proteins. The tertiary structure of the protein determines the accessibility, and therefore reactivity, of an amino acid residue towards exogenous chemicals. No reactions of HNE were detected with Glu (60). Numerous proteins have been shown to be modified by HNE, including plasma membrane ion and nutrient transporters; receptors for growth factors and neurotransmitters; mitochondrial electron transport chain proteins; protein chaperones; proteasomal proteins; and cytoskeletal proteins (152, 155).

HNE-induced modification could lead to the alteration of both secondary and tertiary structure of proteins together with altered subunit association. Low levels of HNE modification appear sufficient to increase susceptibility of proteins to proteolysis and removal by the proteasomal system. But, while under normal conditions the proteasomal system is able to remove the majority of oxidatively damaged and modified proteins, under severe oxidative stress conditions accumulation of modified proteins occurs. This could take place either because of protein cross-linking or malfunction of the proteolytic machinery of the cell. Several other factors besides reactivity may determine the adduction site, such as polarity of the microenvironment and accessibility in the tertiary/quaternary structure (156).
Within this frame, reactivity of HNE towards amyloid beta-peptide (Aβ), the major component of senile plaques (SPs), has to be mentioned. Similar to proteins, Aβ possesses a nucleophilic side chain that can react with HNE. HNE covalently modifies Aβ(1 –40) via 1,4-conjugate addition and hastens Aβ aggregation and toxicity, likely by altering the oligomers formed, which in turn causes oxidative stress that leads to the formation of even more lipid peroxidation products, such as HNE and more toxic Aβ oligomers (187). Interestingly, in AD brain the major efflux protein for Aβ, LRP-1, is covalently modified by HNE, and the consequent dysfunction of this efflux protein conceivably may contribute to accumulation of this neurotoxic peptide (144).
Cells are endowed with several mechanisms to detoxify HNE and thereby prevent its potentially damaging actions. GSH is the most important defense against HNE, and it has the ability to efficiently bind HNE through its Cys residue. Also glutathione S-transferases (GSTs), participate and contribute to regulate the intracellular level of HNE together with the multidrug resistant protein (MRP-1). The latter catalyzes the export the GSH conjugate of HNE out of neurons. In addition to GSH, the dipeptide carnosine (β-alanyl-L-histidine) can quench HNE via intramolecular Michael addition (84). Proteins present in high amounts within or outside of cells may also play important roles in binding and thereby quenching HNE; examples include albumin (3) and apolipoprotein.
Lipid Peroxidation and Neurodegenerative Disorders: A Redox Proteomics Overview
The involvement of lipid peroxidation in neurodegenerative processes is widespread (32, 168). The number of people expected to suffer brain disorders—mainly AD and PD—is rapidly increasing as the worldwide population ages. Oxidative stress occurs in specific brain regions affected in the respective neurodegenerative disorders (130), and membrane lipid peroxidation and HNE are believed to contribute to neuronal dysfunction and death. However, the evidence that lipid peroxidation is a crucial factor is strongest for AD, while more detailed studies are lacking for PD, HD, and ALS. Proteomics analysis is a powerful tool for the identification of putative biomarkers of neurodegenerative diseases, including proteins on which HNE adduction has occurred (32, 168). These modifications yield a loss or reduction of enzyme activity and alterations in protein structure. By using proteomics to investigate these protein changes, several molecular mechanisms involved in degeneration of neuronal cells have been elucidated and likely will contribute to the establishment of prospective biomarkers to be used in clinical diagnosis.
The present review summarizes current knowledge on the role of HNE-induced protein modification and its effects on protein function. However, most of the studies have been performed for AD, while intriguing data are still lacking for many other neurodegenerative diseases. Further studies are needed to give a more detailed picture of the effects of lipid peroxidation in the pathogenesis of neuronal damage and to possibly establish a link between protein oxidation/dysfunction and neuronal death.
Redox proteomics
Proteomics involves the systematic study and identification of the entire set of proteins that are expressed within the cell. The term proteomics was first coined as an analogy to genomics in 1995 (209), but proteomics is much more complicated than genomics. While the genome is a relatively constant entity, the proteome continuously varies in response to both endogenous and exogenous stimuli. Protein expression is expected to be radically different in different cell types and in different stages of development. These differences become particularly intriguing when they are associated with a disease process. Thus, expression of specific proteins is often altered in disease conditions, and proteomics analysis is a powerful tool to help decipher biological processes and phenotypes of both normal and diseased cells.
Redox proteomics, arguably pioneered in the Butterfield laboratory (44, 50), is the branch of proteomics that allows the identification of oxidatively modified proteins, most often by coupling two-dimensional polyacrylamide gel electrophoresis, Western blot analyses, and mass spectrometry (MS) (50). The most common and abundant types of oxidative modifications to proteins are protein carbonylation, 3-nitrotyrosine formation, binding of HNE, and glutathionylation. Redox proteomics can be applied to study all the previously mentioned modifications, and several studies have been performed in our laboratory by following this approach. However, other non–gel-based approaches that utilize liquid or affinity chromatography in combination with MS have also been developed for these modifications by other groups (50).
In the following section, the most relevant results on HNE-modified proteins in neurodegenerative diseases are reported. By comparing results obtained in different forms of neurodegeneration, it is reasonable to hypothesize that HNE modification may be not a random event but affects specific proteins, which, in turn, display altered functions. The biological effects of such modifications are also discussed.
Amyotrophic Lateral Sclerosis
ALS is a progressive neurodegenerative disorder that affects motor neurons of the cerebral cortex, brainstem, and the anterior horn of the spinal cord (49). Clinical manifestations of the disease are muscle weakness, wasting, spasticity, and weight loss. Death usually occurs within 2–5 years after the onset of symptoms. While the majority of cases are sporadic (sALS), about 5%–10% of patients have a positive family history (fALS) with genetic mutations of Cu/Zn superoxide dismutase (SOD1), an enzyme that is part of the cellular antioxidant defense systems. Among the proposed hypothesis of motor neuron degeneration in ALS, accumulating data suggest a role for oxidative stress–mediated cell injury and excitotoxicity. Considering that glutamate excitotoxicity is associated with increased oxidative stress and that failure of antioxidant defense systems can lead to excitotoxicity, a vicious cycle is initiated, thus suggesting common final pathways to motor neuron degeneration. Over 100 SOD1 mutations (7) have been identified in fALS patients, most of which result from substitution of one single amino acid, such as SOD1G85R, SOD1G37R, and SOD1G93A. Interestingly, it became clear that SOD1-mediated toxicity in ALS is due to a “gain” of toxic properties that are independent of the levels of SOD1 activity (73). mSOD1 proteins are or become misfolded and consequently oligomerize to form intracellular aggregates (49, 203), which also embody other essential proteins that once adducted cannot perform their proper function. In addition, toxicity is also caused by aberrant chemistry of the active Cu/Zn sites of the misfolded enzyme (20), which contributes to further exacerbate oxidative stress conditions by increasing the levels of reactive oxygen species (ROS) within the cell (20, 115). This latter mechanism participates in misfolding of SOD1 (165). Elevated levels of ROS and the formation of insoluble protein complexes of mSOD1 protein have been shown in spinal cords of the G93A transgenic mice and precede motor neuron degeneration (116, 117). Reasonably, protein aggregation and oxidative stress simultaneously occur, and this is consistent with the finding that SOD1 itself is oxidatively modified in the G93A-SOD1 transgenic mice (158).
Interestingly, recent studies showed that treatment with hydrogen peroxide or another oxidant induced wtSOD1 misfolding (24), which resulted in the toxic gain of functions similar to mSOD1. Following insult with an oxidizing agent wtSOD1, dimer conformation is altered, Cu/Zn ions are scattered, and finally dissociation into monomeric units occurs. Misfolded wtSOD1 displayed many properties that were thought to be characteristic of the mutant protein such as ubiquitinylation, association with chaperones, insolubility, and formation of protein aggregates. In addition, misfolded/oxidized wtSOD1 may be released outside the cell where it can induce other molecular pathways that lead to motor neurons degeneration, as it does mSOD1 (24). All these data support the view that oxidative damage, including mSOD itself, plays a central role in the pathogenesis and progression of ALS. Multiple pathological studies have reported evidence of increased oxidative stress in ALS post mortem tissue compared to control samples (17). Elevated protein carbonyl levels have been shown in the spinal cord (183) and motor cortex (72) from sALS cases, and increased 3-nitrotyrosine levels were found within spinal cord motor neurons in both SOD1–fALS and fALS patients (1). Markers for lipid peroxidation were detected in spinal cord from sALS patients but were absent in control spinal cords (184). Additionally, elevated levels of HNE (190) were observed in CSF samples from ALS patients. Transgenic mouse and cell culture models of ALS showed a similar pattern of oxidation, which was confirmed by the findings of increased levels of oxidative damage to protein, lipid, and DNA observed in the human disease (8, 72, 73, 117).
Redox proteomics studies from our laboratory identified proteins that were significantly modified by HNE in the spinal cord tissue of a model of fALS, G93A-SOD1 transgenic mice, including collapsin response mediator protein-2 (CRMP-2), heat shock protein (HSP)70, and possibly α-enolase (Table 1) (148). CRMP-2 is a member of the dihydropyrimidinase-related protein (DRP) gene family involved in axonal outgrowth and pathfinding through the transmission and modulation of extracellular signals (82). CRMP-2 colocalizes in the neurofibrillary tangles (NFTs) of AD human brain, suggesting that CRMP-2 plays a role in neuronal degeneration (214). CRMP-2 displays repair activity in the brain, and CRMP-2 oxidation may be responsible for loss of such function. The finding of increased level of HNE-modified CRMP-2 in G93A-SOD1 transgenic mice could provide a link between oxidation-mediated loss in protein function and neuritic regeneration and plasticity known to be altered in ALS (87).
HSP response is activated under stress condition (157), and SOD1 is aggregated with HSP70, HSP40, and α-crystallin in transfected cells (186). HSPs comprise a family of chaperone proteins that assist correct folding and transport of newly synthesized proteins (41). SOD1-immunoreactive inclusions in spinal cord of ALS patients and transgenic mice were found to be positive after staining with antibody against heat-shock cognate HSC70 (207). Moreover, overexpression of Hsp70 played a defensive role against protein aggregation and protected cell viability of G93A-SOD1 overexpressed motor neurons (29). Drug candidates with the ability to induce the heat shock response are currently investigated as potential anti-ALS therapy (105), supporting the role of HSP70 in the folding of SOD1 and prevention of aggregate formation. We suggested that diminished degradation of mSOD1 is possibly due to inactivation of HSP70, which is impaired by covalent binding by HNE. These data demonstrated that lipid peroxidation plays a crucial role in the pathophysiology of motor neuron disease, in particular in the context of mutations of SOD1.
Alzheimer Disease
AD, the most common form of dementia in the elderly, is characterized by the degeneration of neurons in brain regions involved in cognition (hippocampus, entorhinal and frontal cortex) and emotional behaviors (amygdala, prefrontal cortex, hypothalamus) (162). A definitive diagnosis of AD can only be made by post mortem brain evaluation and includes the detection of two major hallmarks: extracellular SPs and intracellular NFTs. SPs consist mostly of Aβ, a 40–42 amino acid peptide that derives from the proteolysis of the amyloid precursor protein (APP) catalyzed by beta- and gamma-secretases. NFTs are composed of hyperphosphorylated tau protein (64, 83). Synapse loss is also a characteristic feature of the disease with memory loss and cognitive decline.
AD has at least three stages: mild cognitive impairment (MCI), early-stage Alzheimer disease (EAD), and late-stage Alzheimer disease (LAD). Some patients show a presymptomatic phase before MCI, namely preclinical Alzheimer disease (PCAD). Due to the progressive nature of AD, patients are usually diagnosed on the basis of the severity of symptoms during the transition into each progressing stage. Braak staging (scoring) characterizes the severity of this disease based on the distribution of NFTs and neurophil threads; there are six levels of Braak staging (the higher the stage, the greater the severity of AD).
The identification of PCAD is a very recent finding and describes individuals who do not show memory deficits but have pronounced AD neuropathology (208). Braak scores are III or higher based on post mortem NFT levels (208). Due to current difficulties in collecting PCAD samples, experimental data on these subjects are still poor and will be available in the near future. However, our laboratory has described proteomics changes in PCAD brain (4) and in the transition from PCAD to amnestic MCI, including identification of proteins with elevated protein carbonylation that conceivably may be involved in memory loss of amnestic MCI compared with PCAD (5).
More detailed data are available for MCI, which is considered a prodromal phase of AD. Though MCI is considered the first clinical stage of AD, not all AD patients are diagnosed with MCI. Criteria for MCI include (a) no dementia, (b) slight cognitive deficit corroborated by a second person, and (c) undisturbed activities of daily living. MCI patients can be classified as having amnestic (memory-affecting) MCI or nonamnestic MCI (65, 153). Braak staging for MCI is typically III or IV, as NFTs are beginning to form in the hippocampus and neocortex. Pathologically, MCI brain shows mild degradation of the hippocampus, entorhinal cortex, sulci, and gyri as evaluated by using magnetic resonance imaging (MRI) technology (54), while early and late stage AD patients have considerably greater degradation in these areas (61). It is estimated that the rate of conversion of amnestic MCI to AD is roughly 10%–15% per year (173); however, in some cases individuals with MCI can revert to normal (153).
EAD, an intermediate stage between MCI and LAD, is characterized by increased frontal lobe atrophy, ventricular widening with progressive brain deterioration. Braak scores for this stage typically are between IV and V. As expected, EAD pathology shows decreased hippocampal volume that correlates with memory decline. There is a significant increase in the NFT load in EAD subjects compared with MCI subjects in both the frontal and the temporal lobes (126), which correlates with impairments in verbal abilities, visuo-spatial functions, and attention and executive functions. Similar to PCAD, EAD sample availability is very rare and therefore experimental evidence is still limited.
LAD is the final stage of the disease, and such patients show consistent symptom severity including memory loss, dementia, behavioral changes, and significant impairment of activities of daily living. Braak staging for these patients is approximately IV–VI, as NFTs have caused substantial neuronal death throughout the brain regions responsible for memory and learning, the hippocampus, and neocortex. Accordingly, synapse loss, Aβ accumulation, and SPs are profound (179). Markers of oxidative stress including DNA oxidation, protein oxidation, and lipid peroxidation are significantly higher in these patients. In contrast, levels of the antioxidant enzymes such as catalase, superoxide dismutase, GST, and GSH reductase are significantly decreased in LAD subjects.
The increased levels of lipid peroxidation throughout the progression of this dementing disorder highlights the great importance of this phenomenon in AD pathogenesis and progression.
Protein-bound HNE: from MCI to late stage AD
Lipid peroxidation is an early event in MCI as indexed by increased levels of HNE and acrolein (38, 103, 210). Elevated levels of F2-IsoPs and F4-NPs have also been observed in the frontal, parietal, and occipital lobes, which could contribute to the slight memory deficits associated with this stage of the disease (159). An important role for HNE in AD is suggested by data from human patients and animal and cell culture models. Biochemical and immunohistochemical analysis of brain tissue samples from AD patients and age-matched neurologically normal control subjects have documented elevated levels of HNE in association with Aβ plaques and NFTs (125). Moreover, HNE levels are increased in the cerebrospinal fluid in AD demonstrating a robust ongoing process and also suggesting HNE as a potential biomarker of the disease (120). Both HNE and acrolein were found to be increased in a disease progression-dependent manner with statistically significant elevations in MCI and EAD (210).
Redox proteomics studies from our laboratory allowed the identification of HNE-modified proteins in MCI, EAD, and LAD, while redox proteomics data on identification of HNE-modified proteins are not yet available for PCAD. Two brain regions were investigated for MCI and late AD; namely, the inferior parietal lobule (IPL) and hippocampus (HP). Studies for EAD were performed only on IPL. The principal aims of our investigation were to establish a link between lipid peroxidation and both pathogenesis and progression of AD and to better understand the molecular mechanisms involved in degeneration of neuronal cells.
By following this approach, this section of the current review is designed to offer the reader the insight on the role of HNE modification in AD by the detailed analysis of the proteins identified in the three different stages of the disease. First, proteins oxidized in common in the three stages of the disease (MCI, EAD, and LAD) will be discussed; second, proteins at least in common for two stages (EAD and late AD); and ultimately proteins modified only in one of the three stages. Based on our findings, we suggest that MCI could represent the most “deregulated” stage; i.e., a condition in which the patient is at high risk to develop AD due to dysfunction of multiple proteins. However, other factors have to be involved in driving the progression to AD. Our data are also supported by studies from Moreira et al. (132), who suggested that oxidative damage is most pronounced early in AD.
As shown in Table 2 two proteins were found to be HNE-modified in the three stages of AD: ATP synthase and α-enolase. In addition, SOD2 and CRMP-2 were found to be HNE-modified in both EAD and late AD.
Proteins oxidized in all the three stages of AD.
Proteins found to be oxidized in both EAD and LAD (150, 167, 169). AD, Alzheimer disease; EAD, early Alzheimer disease; GS, glutamine synthase; HP, hippocampus; IPL, inferior parietal lobule; LAD, late-stage Alzheimer disease; MCI, mild cognitive impairment; MDH, malate dehydrogenase; NA, not available.
Energy dysfunction: ATP synthase and α-enolase
Although the brain is only 5% in weight compared with the rest of the body, it consumes 30% of total oxygen and depends almost exclusively on glucose metabolism for efficient ATP production. Glucose is the primary source of energy, and glucose metabolism is essential for proper brain function; a minimum interruption of glucose metabolism causes brain dysfunction and memory loss (91, 131). Positron emission tomography scanning shows a consistent pattern of reduced cerebral glucose utilization in AD brain (94, 166). Decreased ATP production could lead eventually to cellular impairment. Using redox proteomics we identified two crucial proteins essential for maintenance of ATP homeostasis: ATP synthase and α-enolase.
ATP synthase is a mitochondrial regulating subunit of complex V that plays a key role in energy production. ATP synthase goes through a sequence of coordinated conformational changes in its major subunits (α, β) to produce ATP. The oxidation of ATP synthase leads to the inactivation of this mitochondrial complex and is consistent with decreased enzymatic activity (197). Failure of ATP synthase could contribute to decreased activity of the entire electron transport chain (ETC) and impaired ATP production, resulting in possible electron leakage from their carrier molecules to generate ROS, suggesting an alternative rationalization for the well-documented existence of oxidative stress in AD (200). Altered expression of mitochondrial proteins, functional deficits, and lowered activity in various complexes of the ETC are seen in AD (12). These changes, coupled with the changes in complexes I, III, and IV, may cause electron leakage from the mitochondria to produce ROS.
α-Enolase catalyzes the conversion of 2-phosphoenolpyruvate to phosphoenolpyruvate in glycolysis, a 10-step process in which one molecule of glucose is converted to two molecules of ATP in the cytoplasm. α-Enolase demonstrates increased oxidation in AD and models of AD (36). HNE modification of this protein leads to lowered enzymatic activity, which has been reported in MCI (167), EAD (169), and AD (150). ATP is extremely important at nerve terminals for normal neural communication and decreased levels may be involved in loss of synapses and synaptic function. Both consequences can affect the propagation of action potentials, which may ultimately contribute to memory loss, a characteristic feature of AD pathology. Although the main function of enolase is its role in glycolysis, enolase can regulate by plasminogen (leading to plasmin that can degrade Aβ) and activate the MEK/ERK pro-survival pathway (36). Oxidative modification and dysfunction of α-enolase would disrupt neuronal energy metabolism and ion homeostasis, thereby impairing the function of membrane ion-motive ATPases and glucose and glutamate transporters and causing loss of membrane asymmetry and signal transduction, and could lead to diminution of both degradation of Aβ and activation of prosurvival pathways. Such oxidative and metabolic compromise may thereby render neurons vulnerable to excitotoxicity and apoptosis.
Interestingly, two other proteins were found to be significantly HNE-modified both in EAD and LAD: MnSOD (SOD2) and CRMP-2.
SOD catalyzes the conversion of superoxide anion and two protons to hydrogen peroxide and oxygen. Maintenance of this enzyme is critical to achieving oxidative balance; otherwise, the cell would be in a continual state of oxidative stress. There are four distinct forms of SOD, including Cu/ZnSOD (SOD1), MnSOD (SOD2), NiSOD, and FeSOD. SOD2 is located primarily in the mitochondria. Based on its location, oxidative impairment of mitochondrial resident SOD2 is likely a contributing factor to the mitochondrial dysfunction associated with AD. The activity of SOD2 is significantly reduced in EAD brain (169) and CSF compared with age-matched controls, which is consistent with the concept of mitochondrial dysfunction being a factor in the progression of AD. SOD2 was also found to be nitrated and subsequently inactivated in mice by peroxynitrite (6, 53, 75, 123). Overexpression of SOD2 increases Aβ degradation while partial deficiency promotes Aβ deposition, thereby likely contributing to cognitive decline observed in a transgenic mouse model of AD (63).
CRMP-2 has been reported to be HNE-modified in the spinal cord of ALS transgenic mice. As already discussed, CRMP-2 is a crucial adaptor protein that regulates cytoskeletal dynamics, microtubule organization, axonal transport, and other critical functions. CRMP-2 binds and stabilizes tubulin at the end of microtubules, thus promoting axon extension (82). Thus, overexpressing CRMP-2 in neuronal cells causes an increase of neurite length and can result in supernumerary axons (11, 97). CRMP-2 itself has no known enzymatic activity but interacts with several binding partners to affect microtubule dynamics, neurite outgrowth, neural differentiation, kinesin-dependent axonal transport, Ca2+ homeostasis, and neurotransmitter release, and other essential neurophysiology still being elucidated (93).
Although plaques and tangles are histological hallmarks of AD pathology, synapse loss is significantly correlated to clinical features of dementia (10). Synapse loss is evident even in MCI (181), in which loss of synapses generally correlates with local NFT density. Interestingly, CRMP-2 was found to be hyperphosphorylated in the AD brain by the same CDK5 and GSK3β kinases responsible for pathological tau phosphorylation (113). In fact, CRMP-2 phosphorylation in AD is so pronounced that it has been used as a marker of NFTs. Based on the role of CRMP-2 in maintaining neurite cytoskeletal integrity, it is likely that Aβ and tau pathways might converge to cause synapse loss through a CRMP-2–dependent mechanism (93). This hypothesis reasonably correlates with the three neuropathologic features of AD: Aβ, NFTs, and synapse loss. The processes responsible for co-aggregation of CRMP-2 with its binding partners within NFTs may be related to the redox status of the cell stress because oxidative stress is a well-known factor that can initiate/propagate protein aggregation phenomena through covalent cross-linkage and hydrophobic protein–protein interactions (93). Consistent with such a possibility, our studies showed that CRMP-2 is HNE-modified in both EAD and LAD, thus suggesting that its impairment has a crucial pathogenic role.
Although the following proteins have not been found to be specifically modified in each stage of the disease, it is possible to outline at least an altered “function” in common as a consequence of specific protein oxidation. In fact, we identified phosphoglycerate kinase (PGK)1, pyruvate kinase (PK), and lactate dehydrogenase (LDH) as HNE-modified in MCI, trioso phosphate isomerase (TPI) in EAD, and aldolase (ALDO1) in LAD. These proteins are all members of the glycolytic pathway, thus supporting a general trend of glycolysis impairment in the three stages of the disease.
PGK catalyzes the dephosphorylation of 1,3-bisphosphoglycerate to 3-phosphoglycerate. This reaction undergoes substrate level phosphorylation by phosphoryl transfer from 1,3-bisphosphoglycerate to ADP to produce one molecule of ATP. Impairment of this glycolytic enzyme results in decreased energy production and irreversible downstream effects, such as multidrug resistance (62). This result could conceivably be related to the identification of MRP-1 as a protein with elevated HNE binding in AD (198).
PK catalyzes the final step in glycolysis, the conversion of phosphoenolpyruvate to pyruvate, with the concomitant transfer of the high-energy phosphate group from phosphoenolpyruvate to ADP, thereby generating ATP. Under aerobic conditions, pyruvate can be transported to the mitochondria, where it enters the tricarboxylic acid (TCA) cycle and is further metabolized to produce considerably more ATP through oxidative phosphorylation. Anaerobically, pyruvate can be reduced to lactate in mitochondria-deficient cells or under hypoxic conditions, such as those found in active muscle tissues or brain ischemia. Additionally, enzymatic activity is reduced, thus suggesting that oxidative modification leads to impairment of protein function (167).
LDH anaerobically reduces pyruvate to lactate through lactic acid fermentation using NADH as a cofactor. The NAD+ generated in this process is used in glycolysis to oxidize glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate. Lactate is a substrate for gluconeogenesis and given that glucose is the major supplier of energy to the brain, proper lactate production is crucial (104). LDH enzymatic activity is significantly reduced in MCI hippocampus (167), which provides supplemental evidence for the correlation between protein oxidative modification by HNE and enzyme activity impairment. Dysfunction of this enzyme could yield excess pyruvate and a reduction in the production of glucose.
ALDO1 cleaves fructose 1,6-bisphosphate and produces the two glycolytic intermediates, glyceraldehyde-3-phosphate and dihydroxyacetone phosphate. Fructose 1,6-bisphosphate is neuroprotective and preserves GSH in cortical neurons during oxidative stress conditions (206). ALDO1 catalyzes a critical step as it generates two substrates that are used to eventually produce two molecules of ATP and more in the TCA cycle and ETC. Consequently, HNE modification results in decreased energy metabolism. Levels of ALDO1 are significantly decreased in AD hippocampus (22) and PD (81). Enzyme activity is reduced (22) and impairment can cause increased levels of fructose 1,6-bisphosphate, inhibition of complete glycolysis, and ATP depletion.
Similar comments could also be extended to TCA cycle proteins because we identified malate dehydrogenase (MDH) in EAD and aconitase in LAD to be HNE-modified.
MDH catalyzes the reversible oxidation of malate to oxaloacetate by NAD+ in the TCA cycle. MDH links glycolysis to the electron transport chain by transferring NADH to NADH dehydrogenase (Complex I) through the malate-aspartate shuttle resulting in the production of ATP. In contrast to elevated HNE binding to MDH, MDH levels were increased in AD patients, but the level of protein oxidation of MDH was not significant, which possibly reflects a compensatory mechanism in response to oxidative stress (107). Activity of MDH increases during aging (30, 141), which further supports the hypothesis of mitochondrial dysfunction in AD.
Aconitase catalyzes the isomerization of citrate to isocitrate in the TCA cycle. As an iron-sulfur protein, its Fe-S cluster participates in the hydration–dehydration reaction that occurs. The three cysteine residues in the Fe-S core can undergo Michael addition and form acrolein-, HNE-, and MDA-conjugated adducts, thereby increasing lipid peroxidation markers (114, 188). Enzymatic activity of this enzyme is significantly reduced in AD, consonant with protein dysfunction (150). The TCA cycle takes place in the mitochondria; therefore, aconitase impairment results in mitochondrial dysfunction, a common theme of neurodegenerative diseases (178, 212). As noted above, decreased ATP production can lead to voltage-gated channel and ion-motive pump disruption as well as synapse loss, an early event in Alzheimer's disease pathology (180).
The proteins we identified to be HNE-modified exclusively in MCI included actin, carbonyl reductase (CR1), elongation factor Tu (EF-Tu), initiation factor 1 alpha (eIF-α), neuropolypeptide h3 (Nh3), and HSP70.
Actin is a key protein that plays a central role in maintaining structural integrity, cell morphology, and structure of the plasma membrane (18). In the central nervous system, actin is distributed widely in neurons, astrocytes, and blood vessels. It is particularly concentrated in growth cones, dendritic spines, and presynaptic terminals. HNE conjugation of actin can lead to loss of membrane cytoskeletal structure, decreased membrane fluidity, and trafficking of synaptic proteins and mitochondria. Moreover, actin is involved in the elongation of the growth cone, and loss of function of actin could play a role in the loss of synapses and neuronal communication documented in AD (128, 180).
CR1 is an enzyme that reduces carbonyl-containing compounds to their resultant alcohols, thereby reducing the level of protein carbonyls. Subsequent malfunction or down-regulation of this enzyme would be consistent with increased protein carbonyls in AD (92) and amnestic MCI (167), which, because of the polarity of the carbonyl moiety, could expose normally buried hydrophobic amino acids to the protein surface resulting in a disruption of conformation. CR has been shown to reduce the levels of HNE (59, 142). CR expression is altered in DS and AD subjects (15). This enzyme was found to be modified in persons with corticobasal degeneration, a neurological disorder whose symptoms closely mirror that of PD (48). The gene for CR is located in close proximity to the gene for the antioxidant enzyme, SOD1 (112); these genes, in addition to APP, are located on chromosome 21, which is trisomic in DS patients (77, 106). A potential link between DS and AD by irregular meiotic recombination in chromosome 21 (154) has been postulated. Current research supports a possible relationship among CR, DS, and Aβ in neurodegeneration.
EF-Tu and eIF-α are intimately involved in protein synthesis machinery. Human mitochondrial EF-Tu is a nuclear-encoded protein that functions in the translational apparatus of mitochondria. Mammalian EF-Tu acts as a GTPase by hydrolyzing one molecule of GTP for each A site amino-acylated tRNA of the ribosome. As discussed before, mitochondria play pivotal roles in eukaryotic cells in producing cellular energy and essential metabolites as well as in controlling apoptosis by integrating various death signals (143). Mitochondrial protein synthesis inhibition, either by deleting mtDNA or by blocking translation in the organelle, is associated with the impairment of differentiation in different cell types, including neurons (205). Loss of neuronal differentiation can lead to an incomplete development of the neuron, which would result in reduced neurotransmission.
eIF-α, which binds aminoacyl-tRNA to acceptor sites of ribosomes in a GTP-dependent manner (151), is involved in cytoskeletal organization by bundling and binding actin filaments and microtubules. The expression level of eIF-α is regulated in aging, transformation, and growth arrest. Because of eIF-α regulation in differing states of cell life and its key position in protein synthesis and cytoskeletal organization, this protein is an important determinant of cell proliferation and senescence (201). Inhibition of eIF-α promotes apoptosis (27, 151), indicating that eIF-α activity is critical to normal cell function.
Taken together, increased levels of HNE-bound eIF-α and EF-Tu suggest an impairment of protein synthesis machinery, either in cytosol or mitochondria, associated with an impairment of the rate and specificity of ribosome functions. Numerous studies have provided indirect evidence that suggests alterations in protein synthesis may occur in AD (58, 74, 176). The dysfunction of the protein synthesis apparatus, mediated in part by redox proteomics identified oxidatively dysfunctional EF-Tu and eIF-α, secondary to oxidative stress–induced lipid peroxidation and formation of HNE, could compromise the ability of brain cells to generate the countless factors needed to regulate cell homeostasis, thus contributing to impaired neuronal function and to the development of neuropathology in AD.
Nh3 is critical for modulation of choline acetyltransferase, an enzyme essential in the synthesis of acetylcholine. The loss of choline acetyltransferase leads to reduced levels of acetylcholine causing poor neurotransmission (51, 140). N-methyl D-aspartic acid (NMDA) receptors activate the production of this enzyme, and modulation of the NMDA receptor mediates cholinergic deficits (99). AD patients have considerable cholinergic deficits, consistent with dysregulation of acetylcholine levels and loss of cholinergic neurons (51, 52, 172). The oxidative modification of this protein further supports the involvement of cholinergic neurons in AD, an early hypothesis of this disorder (78). Nh3 undergoes HNE modification in MCI hippocampus and nitration in LAD (33). Nh3 has several other names, including phosphatidylethanolamine binding protein (PEBP), hippocampal cholinergic neurostimulating peptide, and Raf kinase inhibitor protein. As a phosphatidylethanolamine binding protein, PEBP may be important in phospholipid asymmetry. Apoptosis is initiated when phosphatidylserine resides on the outer leaflet of the membrane. Loss of function and changes in conformation of PEBP conceivably could lead to loss of phospholipid asymmetry, a signal for neuronal apoptosis, which further supports the role of PEBP as a parapoptosis inhibitor (191). HNE modification of PEBP may impact lipid asymmetry as loss of activity is observed in AD and MCI and mouse models of familial AD (13, 14, 80) and can potentially disrupt cellular homeostasis. PEBP levels are decreased in AD, which promotes amyloid beta accumulation in the Tg2576 transgenic mouse model of AD (80). RAF kinases are serine/threonine protein kinases involved in cell signaling in the mitogen activated protein cascade and NF-κB. As demonstrated by the various functions through its numerous monikers, Nh3 is a highly important protein and oxidative modification likely is detrimental to neurons.
As discussed previously, HSPs serve as molecular chaperones and help guide damaged proteins to the proteasome. HSPs vary in size and are named according to their molecular weight. HSP70 has been reported as being oxidized in AD by using redox proteomics (45). Several other HSPs have been found to be oxidatively modified in AD (45) and HD (67), including HSP90 and HSP60, whereas other heat shock proteins, HSP27 and HSP32, are induced in amnestic MCI (57). Impairment of this protein may exacerbate protein misfolding and aggregation and eventual proteasomal overload and dysfunction known to occur in AD (102). Aβ-treated synaptosomes show that HPSs are oxidatively modified (25), further illustrating the importance of Aβ in inducing lipid peroxidation in AD brain (110) and the importance of proper functioning of HSPs in the cell.
Redox proteomics-identified proteins that are HNE-modified exclusively in LAD among the various stages of AD include α-tubulin, glutamine synthase (GS), and peroxiredoxin VI (Prx VI) (150).
α-Tubulin is an isoform of tubulin that alternates with β-tubulin to form a prominent cytoskeletal structure, the microtubule. Microtubules are used to transport cargo (i.e., vesicles and organelles) from the cell body to the periphery and vice versa. Upon HNE modification, α- tubulin is structurally altered, and microtubules depolymerize (79). Therefore, cargo cannot reach its destination and the cytoskeleton is altered (136, 137). This could contribute to the notion that synaptic domains are the first to be damaged in AD neurons (109).
Peroxiredoxins are a family of antioxidant enzymes that are pivotal in antioxidant defense as already discussed. Prx VI is a 1-Cys peroxiredoxin that plays a role as a second messenger for growth factors and cytokines. Prx VI, a GSH peroxidase that exhibits Ca2+-independent phospholipase A2 activity (177), is cytosolic and is expressed in astrocytes and in neurons at low levels (47). In addition, the decrease in the activity of this enzyme may also lead to decreased phospholipase A2 activity. Phospholipase A2 is a target for regulation by Pin1, which has been reported to be oxidatively modified and down-regulated and to have decreased activity in both AD and MCI brain (37, 196, 197). Prx VI has been found to be protective against mitochondrial dysfunction, a feature that pinpoints its effectiveness as an antioxidant (66). Prx VI also plays important roles in cell differentiation and apoptosis. Consequently, HNE modification may lead to tau hyperphosphorylation and NFT formation, in addition to development of oxidative stress.
GS is an important enzyme in maintaining the glutamate–glutamine cycle. GS converts the acidic amino acid glutamate to the basic amino acid glutamine. Glutamate is taken up from the extracellular fluid of neuronal tissues via glutamate transporters so that levels are upheld. Once GS is conjugated with HNE, it becomes structurally altered and can no longer preserve glutamate levels, resulting in possible excitotoxicity and neurodegeneration (35). In addition, GS levels are significantly decreased in AD brain (92) and contribute to neuronal excitotoxic death.
Down Syndrome
DS is the most common genetic cause of mental retardation and results from trisomy of chromosome 21. DS should be considered a multifactorial disease in which an abnormal expression of trisomic genes arises not only from genetic, but also environmental factors (89). Thus, trisomy also affects disomic genes, which ultimately results in a deregulation of a plethora of functions with a high variability within the DS population (171). Neuropathological features of DS include decreased brain weight and neuronal number, abnormal neuronal differentiation, and structural changes in synapses (16). Most DS patients (aged above 40 years) show progressive cognitive decline, dementia, and neuropathological hallmarks of older AD patients (124). NFTs are detected both in DS and AD brains as is Aβ deposition. There is accumulating evidence that neuronal cell death may be induced by oxidative stress in DS (95). Although oxidative stress has been demonstrated to contribute to the DS phenotype (43, 100, 145, 215), a direct link between the accumulation of oxidative damage and clinical features of DS has not yet been elucidated. Chronic oxidative injury in the brain could represent one of the major risk factors for subsequent neurodegeneration in aged DS patients (95, 138, 193). Overexpression of some of the genes located on chromosome 21 results in increased conditions of oxidative stress. SOD1 is at the top of the list: increased levels of SOD1 leads to overproduction of H2O2, which is in turn a source of other ROS. In addition, overexpression of SOD1 in DS subjects has been found to alter the levels of other antioxidant enzymes, such as catalase and GSH peroxidase, which may induce oxidative damage if dysfunctional. Accordingly, Busciglio and Yankner (31) demonstrated increased production of ROS and elevated levels of lipid peroxidation in neurons of DS fetuses. A proteomics study from Gulesserian et al. (85) showed that oxidative stress in fetal DS did not result from overexpression of SOD1 protein but appeared to be the consequence of low levels of reducing agents and enzymes involved in removal of hydrogen peroxide, such as GSTs and thioredoxin peroxidases.
Included in the list of potential chromosome 21 gene candidates leading to DS, another important player is APP. Its overexpression leads to overproduction of Aβ(1 –42), which is proved to induce oxidative stress (34, 42, 98, 160), and post mortem studies on DS brain evidenced accumulation of Aβ(1 –42) that correlated with age (90).
Elevated levels of thiobarbituric acid reactive substances were demonstrated in the cortex from DS fetal brain compared with controls (139). Further, increased levels of isoprostane (8,12-iso-iPF2α) have been measured in urine samples from adults with DS (160).
Ishihara et al. (98) reported increased level of ROS and mitochondrial dysfunction in primary cultured astrocytes and neurons from Ts1Cje transgenic mice. The authors also identified by a redox proteomics approach the putative target proteins that were modified by lipid peroxidation-derived products (98). ATP synthase mitochondrial F1 complex b subunit, α-enolase and TPI were identified as proteins modified by 3-hydroperoxy-9Z,11E-octadecadienoic acid (13-HPODE). Neurofilament light polypeptide, internexin neuronal intermediate filament α, neuron specific enolase, Prx VI, PGK1, and TPI were shown to be HNE-modified proteins (Table 3). Thus, dysfunction of these proteins as a consequence of oxidative damage may affect ATP production, the neuronal cytoskeleton system and antioxidant network function. Interestingly, redox proteomics studies from our laboratory identified α-enolase, Prx VI, PGK1, and TPI to be HNE-modified in AD and MCI brain (32, 150, 167), suggesting that these proteins might contribute to cognitive dysfunction and neurodegenerative processes occurring in DS. These findings are consistent with the notion that DS and AD may share common pathways of neurodegeneration, a hypothesis that needs to be further elucidated.
13-HPODE, 3-hydroperoxy-9Z,11E-octadecadienoic acid.
A recent study from our laboratory, investigated the levels of oxidative stress in the amniotic fluid from DS-affected pregnancies compared with healthy controls. Amniotic fluid, similar to CSF for the brain, is in direct contact with the fetus and can be considered a reliable index of the physiological condition of the fetus. Therefore, amniotic fluid could be used for the identification of disease biomarkers to be coupled with current genetic screening. We evaluated a set of oxidative stress biomarkers in DS amniotic and found increased levels of oxidative stress, as indexed by increased protein oxidation, lipid peroxidation, reduction of GSH and Trx levels and induction of the HSP response. By a redox proteomics approach, we identified selective proteins that showed increased oxidation in DS fetuses compared with healthy controls. Our results demonstrated that oxidative stress is an early event in the pathogenesis of DS (147).
Parkinson Disease
PD, the second most common form of dementia in the elderly, is pathologically characterized by a decline in motor function in the form of resting tremors, muscle rigidity, akinesia, and bradykinesia. PD, similar to several other age-related neurodegenerative diseases, is considered a “protein misfolding disease.” In the case of PD, protein aggregates of α-synuclein, a protein that is important for proper mitochondrial function and synaptic vesicle formation (23), are found. These aggregates are the major component of Lewy bodies located primarily in the putamen and substantia nigra. These brain regions are largely involved in learning and motor control. Evidence of the involvement of α-synuclein in neurodegeneration came from studies showing that three independent mutations in this synaptic protein lead to the development of familial PD (175). Additional studies demonstrated that duplications and triplications of the α-synuclein gene also cause an early onset of PD (96). α-Synuclein is a small, unfolded cytoplasmic protein that is highly expressed in the CNS, which has the ability to bind lipids, including lipid membranes (108, 192) and fatty acids (122). This binding activity is thought to be responsible for its neuroprotective role, specifically in response to oxidative stress, and it was also demonstrated to prevent oxidation of membrane PUFAs only in the monomeric, but not fibrillar, form of the protein (217). The first evidence to show the involvement of lipid peroxidation in PD as a cause of nigral cell death was from Dexter and co-workers (55). It is well documented that HNE and MDA adducts accumulate in Lewy bodies in neocortical and brain stem neurons (216). HNE was also found to alter dopamine transport (76). These findings may contribute to the loss of dopamine, which is paramount to the PD pathogenesis. Indeed, under conditions of elevated lipid peroxidation, dopamine, in addition to other catecholamines, is oxidatively converted to the corresponding o-quinone, which may initiate a cascade of spontaneous reactions, including intramolecular cyclization and interaction with molecular targets ultimately resulting in cytotoxic responses and altered cellular functions. Further, dopamine may interact with products of lipid peroxidation, notably glyoxal and other aldehydes, leading for example to tetrahydroisoquinolines via Pictet-Spengler chemistry (135). Dopamine loss coupled with protein aggregation and HNE elevation causes a profound effect on the learning and physical capabilities of persons with PD.
Besides these findings, redox proteomics data on HNE-modified proteins have not been currently undertaken. Further studies will be valuable towards this direction.
Huntington Disease
HD is a progressive neurodegenerative disorder in which mutation in the huntingtin protein (Htt) occurs via multiple CAG (Gln) repeats. The CAG repeat region shows a range of 11–35 repeats in normal individuals, while a repeat number greater than 35 indicates a high probability of developing HD (174). Although the function of the Htt protein is not clearly understood, it has been shown to interact in cell signaling and vesicle transport (88). HD was first described by George Huntington (1872) as chorea, to indicate a movement disorder characterized by involuntary jerky limb movements. Although progressive, this neurological disease takes approximately 10 years to fully manifest into rigidity and bradykinesia. HD is inherited in an autosomal dominant manner, which indicates that each offspring of a parent with HD has a 50% chance of inheriting this devastating disease. As observed in some other neurodegenerative diseases, movement decline is also associated with cognitive decline and psychiatric problems. Neuronal degeneration is evidenced throughout the basal ganglia, thalamus, brain stem, and mostly in striatum and cortex, which demonstrate consistent protein aggregation and cell death. Accordingly, the polyQ expansion can cause a conformational change in the Htt mutant protein leading to intranuclear and intracytoplasmic aggregates, which are a pathological hallmark in the brains of both HD patients and mouse models thereof (19). The mechanisms by which mutant Htt causes neuronal dysfunction and degeneration are not fully understood. It is not unreasonable to propose that the HD mutation may increase ROS, resulting in a primary defect associated with oxidative stress (28). As such, a significant body of evidence from studies in both HD patients and experimental models of HD support a role for oxidative stress and mitochondrial dysfunction in mediating the neuronal degeneration observed in HD brain (19). Increased levels of oxidative damage indices, including protein nitration, lipid peroxidation, DNA oxidation, and exacerbated lipofuscin accumulation, occur in HD. Strong evidence exists for early oxidative stress in HD, coupled with mitochondrial dysfunction, each exacerbating the other and leading to an energy deficit (26). A significant increase in HNE adducts is observed in the striatum and found to be colocalized in Htt inclusions and also MDA has also been found to be elevated in HD brain (28, 111). It is worthy to note that HNE immunoreactivity was colocalized with mtHtt inclusions in the striatal neurons of R6/2 HD mice. Administration of the antioxidant compound nordihydroguaiaretic acid (NDGA) markedly reduced HNE adduct formation in the nuclear inclusions of R6/2 striatal neurons. NDGA also protected cultured neurons against oxidative stress-induced cell death by improving ATP generation and mitochondrial morphology and function (111). As indicated for PD, redox proteomics studies in HD models have thus far focused on protein carbonylation (149). Specific redox proteomics studies for the identification of HNE-modified proteins have not yet been performed, and adding new data for this important issue is desirable.
Concluding Remarks
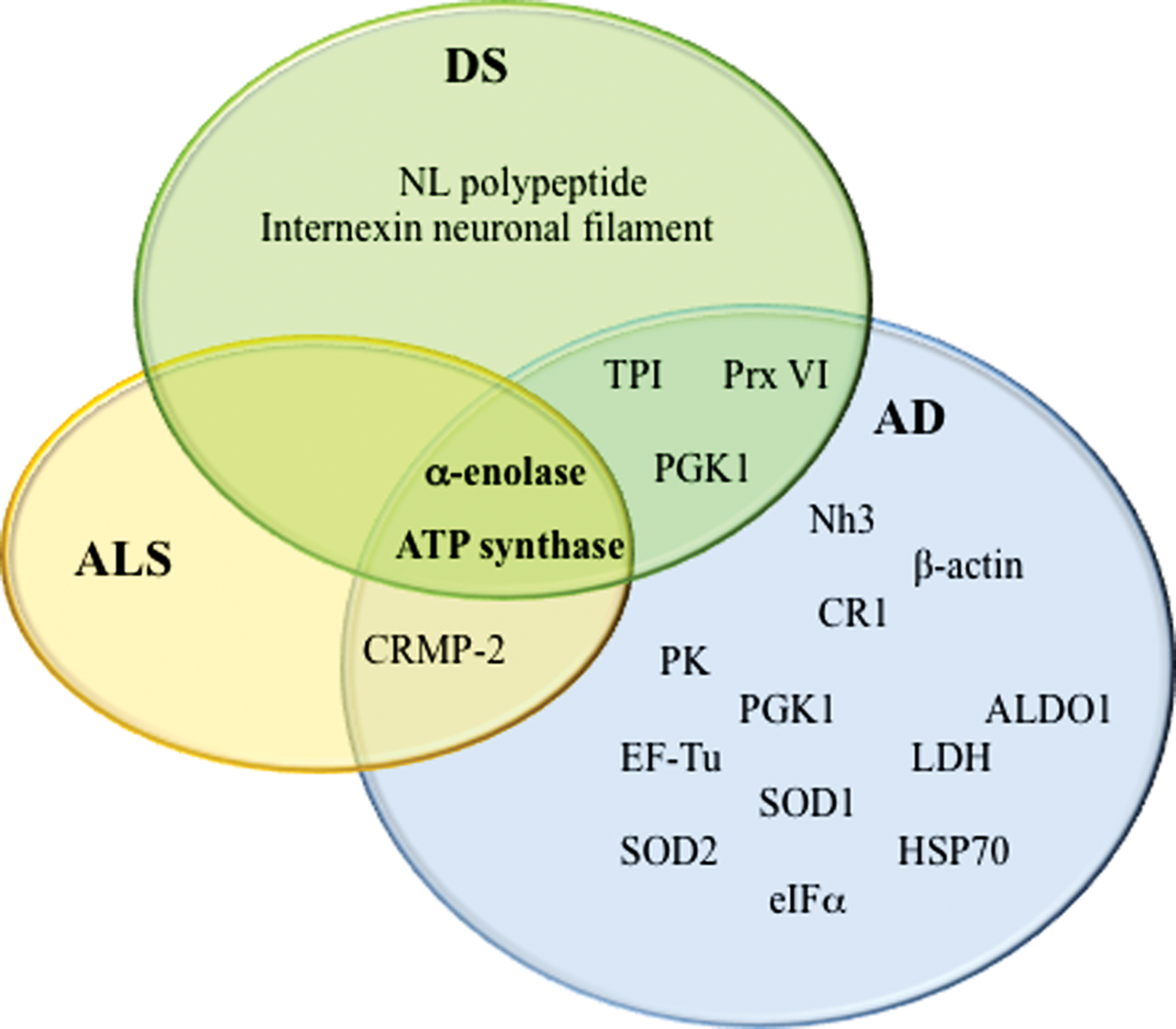
Many studies support a fundamental role of lipid peroxidation in neurodegenerative diseases. HNE is the most abundant by-product of lipid peroxidation, and its toxic properties have been extensively demonstrated for AD, PD, HD, ALS, and DS. The precise mechanisms of HNE neurotoxicity remains to be established. These mechanisms involve a cascade of chain reactions initiated by the covalent interaction with a nucleophilic compound. Among several substrates, proteins are major targets of HNE reactivity resulting in impairment of protein function. In the last decade, redox proteomics studies from our laboratory contributed to better understanding of the biological effects of these complex processes. Identification of specific HNE-modified proteins in the brain of diseased subjects has led to the determination of which selective cellular functions are altered, how they possibly translate into clinical symptoms, and how they relate to pathology of these disorders. By comparing results obtained in different neurodegenerative diseases, it potentially could be possible to identify both similarities and specific differences in addition to better characterize selective neurodegenerative phenomena that involve protein dysfunction. So far, results obtained by following this approach in our laboratory and others suggest that lipid peroxidation is an early event in the pathogenesis of neurodegenerative diseases and that therapeutic strategies towards prevention of lipid peroxidation reactions could be neuroprotective. As show in Figure 6, α-enolase and ATP synthase are the only HNE-modified proteins that overlap AD, DS, and ALS. However, most of the proteins identified in the diseases discussed in this review, though not the same, often share at least the same functions, consistent with the hypothesis that altered energy metabolism, reduced antioxidant defense, and mitochondrial dysfunction are characteristic hallmarks of neurodegenerative disorders and provide support for the physical impairments and progressive memory loss associated with the diseases. Proteomics and redox proteomics in particular have contributed significantly to broaden the knowledge in regard to potential biomarkers for disease diagnosis and may also provide insight into damaged metabolic networks and potential targets for modulation of disease progression.
Footnotes
Acknowledgments
This work was supported in part by NIH grants to D.A.B. [AG-05119; AG-029839]. We thank former Ph.D. students and postdoctoral scholars in the Butterfield laboratory for their excellent research cited in this review.
Disclosure Statement
No competing financial interests exist.