
Abstract
Introduction
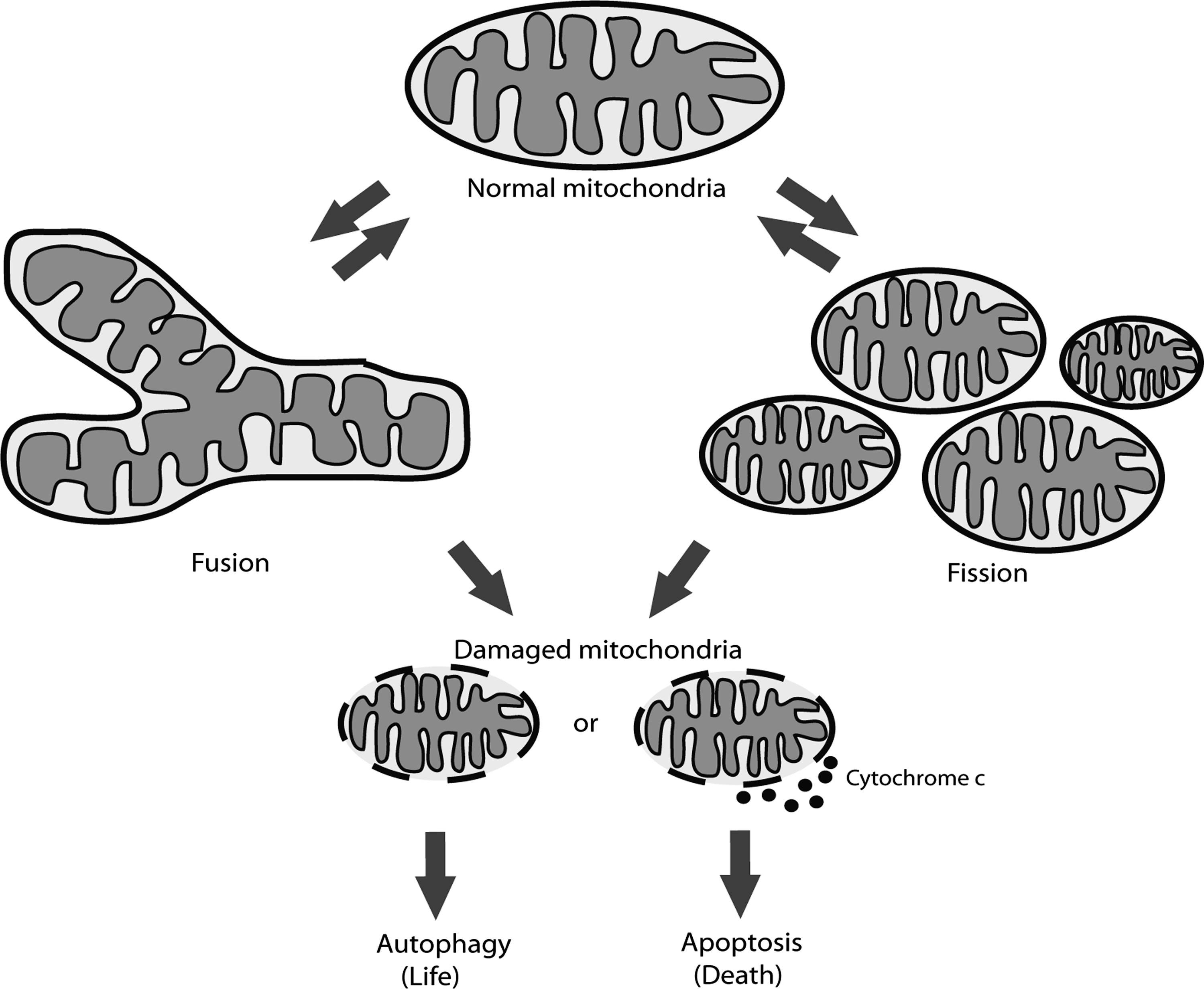
To maintain functional cellular population of mitochondria, selective degradation of mitochondria is required, in a manner coupled to fusion/fission events (Fig. 1). Through an evolutionarily conserved process of autophagy, which nonselectively or selectively catabolizes cellular components and organelles, mitochondria are degraded in response to alterations in metabolic demand or damage that cannot be otherwise eliminated (e.g., via fission and fusion events). Although the existence of autophagy has been known for many years, only recently has there been progress in the elucidation of the individual proteins involved. We currently can define two major types of autophagy depending on the cargo specificity. Starvation-induced autophagy nonselectively degrades cellular components to provide recycled biomolecules for the cell until normal feeding conditions are established. In contrast, selective autophagy specifically, under nonstarving conditions, degrades specific cargo, including ribosomes, peroxisomes, aggregated proteins, or even intracellular bacteria (for details see reviews by Yang and Klionsky [52] and Kirkin et al. [21]). The selective removal of mitochondria by autophagic machinery is termed mitophagy and, as in general autophagy, includes formation of double-membrane vesicles, autophagosomes, that engulf mitochondria marked for degradation and sequentially deliver the cargo to lysosomes. Recently, two research groups have described how starvation-induced autophagy influences mitochondrial morphology (14, 36). It was shown that during starvation mitochondria elongate due to phosphorylation of Drp1 by protein kinase A (PKA) in response to increased cAMP. Drp1 phosphorylation inhibits its activity as a mitochondrial fission protein, resulting in elongation of mitochondria that consequently escape autophagic degradation. Another key regulator of mitochondrial dynamics and degradation is transcription factor High-mobility group box 1 (HMGB1) that regulates heat-shock protein beta-1 (HSPB1) gene expression (43). Loss of either HMGB1 or HSPB1 results in fragmented mitochondria and deficient mitophagy. Therefore, the authors suggested that HMGB1 and HSPB1 regulate mitochondrial quality control and protect cells from abnormal mitochondria by activation of autophagy and mitophagy. Mitochondrial fission thus serves as a prerequisite to elimination under physiological and pathophysiological conditions and is integrated with the recognition process of mitochondrial capture.
Even though the selection of mitochondria destined for degradation is still largely obscure, recent research evidences at least two mitophagy mechanisms that distinguish the removal of mitochondria damaged beyond repair from development-induced mitochondrial removal. In this review, focusing on mammalian cells, particularities of mitophagy mechanisms and important regulators and modulators are discussed (Table 1).
Mitophagy of Damaged Mitochondria
Recent work in the fields of neurodegenerative diseases and cancer are providing insight into the cellular consequences of accumulated, damaged mitochondria. Now, in addition to elucidating mechanisms of mitochondrial dysfunction (e.g., apoptosis), there is an increasing focus on mechanisms underlying recognition and removal of damaged mitochondria. Studies of Parkinson's disease (PD) revealed mutations in outer mitochondrial membrane PTEN-induced putative kinase 1 (PINK1) and E3-ubiquitin ligase Parkin (also known as PARK2), both of which are associated with defects in mitophagy when mutated (47). Further analysis of cells treated with mitochondrial uncoupling agent carbonyl cyanide m-chloro phenyl hydrazone (CCCP), which causes mitochondrial damage by decreasing the mitochondrial membrane potential, revealed robust translocation of Parkin from cytoplasm to mitochondria (29). Moreover, it was shown that Parkin exclusively translocates to defective mitochondria leaving the healthy mitochondria Parkin-free (reviewed by Narendra et al. [29]). Following Parkin recruitment, reduced mitochondrial mass occurred, i.e., mitophagy. Accordingly, it is proposed that Parkin influences mitophagy quality control pathway and governs selective damage removal. In vitro results in cultured cells further showed that PINK1 overexpression is sufficient to translocate Parkin even in the absence of uncoupler treatment (18, 19). How mitochondria-localized PINK1 recruits Parkin from cytoplasm has not yet been resolved, but possible recruitment mechanisms have been proposed. First, PINK1 directly interacts with Parkin (41, 46) in a manner that does not depend on either PINK1 kinase or Parkin E3-ligase activity (40). Second, the cytoplasm-facing PINK1 kinase domain is able to phosphorylate Parkin (19, 40), resulting in Parkin translocation to mitochondria as well as activation of Parkin to ubiquitinate its mitochondrial substrates. However, fly experiments also suggest additional regulators of Parkin translocation to mitochondria since Parkin overexpression in PINK1-depleted cells rescued PINK1 mutant phenotype (19). Thus, the upstream contributors that pre-translocate Parkin to mitochondrial vicinity for phosphorylation by PINK1 remain unclear.
It has been proposed that mitochondrial fission precedes mitophagy, in order to transform normally elongated mitochondria into a form suitable for engulfment by the autophagic machinery (45). Blocking fusion is one possible action that would allow fission proteins to redesign the mitochondrial population. Therefore, numerous research groups investigating different cell systems have confirmed that Parkin ubiquitinates several mitochondria-residing proteins, including fusion-promoting mitofusins (Mfn1 and Mfn2). Through detailed analyses in fly and mammalian cells, it was shown that polyubiquitination of mitofusins by Parkin signals proteasome recruitment and mitofusin degradation (11, 34, 55) (Fig. 2). Removal of mitofusins is necessary for proper mitophagy induction and selective removal of damaged mitochondria because mitochondria that lack mitofusin are not able to fuse with healthy mitochondria as a repair mechanism (25). This programmed imbalance of mitochondrial morphology can thus function to isolate the pool of damaged mitochondria for subsequent selective autophagic sequestration and degradation.
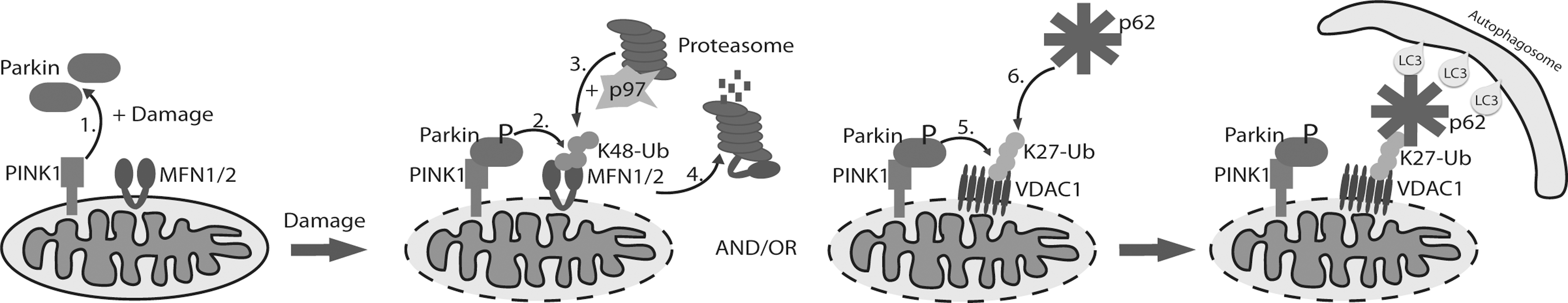
Intriguingly, Geisler et al. (12) demonstrated that the mitochondrial outer membrane–localized VDAC1 (voltage-dependent anion channel 1) is a Parkin target during mitophagy. In contrast to mitofusin ubiquitination, Parkin generates Lys 27 polyubiquitin chains on VDAC1 whose role is not to recruit proteasome and degrade VDAC1, but instead to engage the autophagy machinery by attracting the autophagy receptor p62. Recognition of polyubiquitinated substrates, in this case mitochondria, occurs via ubiquitin binding UBA domain of p62 on one side and LIR (LC3-interacting region) motif, which binds LC3/GABARAP proteins that are essential for autophagy initiation. This interaction thus generates the bridge between mitochondria and newly forming autophagosome (32). However, the role and requirement for p62 in mitophagy are controversial. Whereas p62 recruitment is shown to be necessary for PINK1/Parkin-induced mitophagy (31), the mitophagy can occur even in the absence of p62 at the mitochondria (28). Nevertheless, these studies do show that p62 is involved in mitochondrial clustering, an event that precedes mitochondrial engulfment (Fig. 2). Moreover, together with p62, the formation of ubiquitinated mitochondria clusters is aided by HDAC6, a ubiquitin-binding protein that signals through the actin-remodeling machinery to promote both aggregation of mitochondria (similar to clustering of polyubiquitinated proteins) and autophagosome–lysosome fusion (24). It is important to note that differences between proposed mechanisms regarding the recruitment of Parkin, p62, and other autophagy proteins might be due to different experimental systems. The ionophores CCCP and p-trifluoromethoxy carbonyl cyanide phenyl hydrazone (FCCP) are mitochondrial uncouplers often used to study damage-induced mitophagy. However, the signaling pathways activated by CCCP treatment are ill-defined and do not reflect the mitochondrial dysfunctional state during chronic neurodegenerative disease models. Detailed mechanisms of damage-induced mitophagy will need to incorporate physiological cellular systems and additional damage inducers in parallel. For example, AMPK and Ulk1 phosphorylation activities activate mitophagy both under starvation and mitochondrial stress (induced by CCCP) conditions, thereby demonstrating their roles as mitochondrial homeostasis sensors (8).
Thus, while mechanisms underlying regulation and recognition are emerging, the challenge remains to decipher the role of all the players involved in removal of damaged mitochondria via autophagy. Further studies will be required to address this complexity by focusing on different cells or tissues, which may not only have different order of events, but utilize different components for recognition.
Mitophagy During Developmental Processes
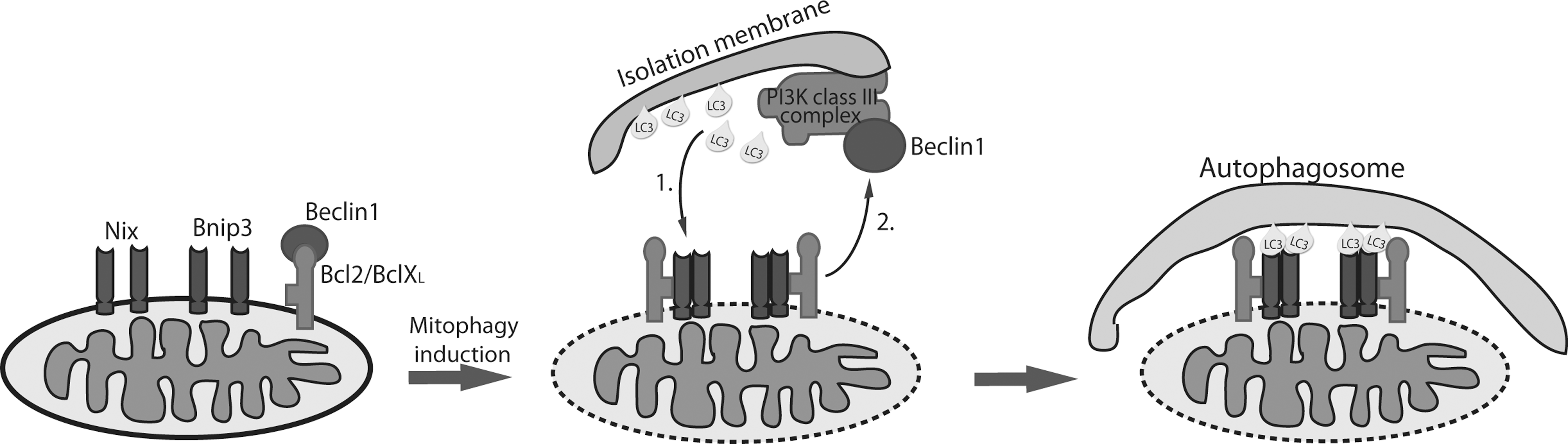
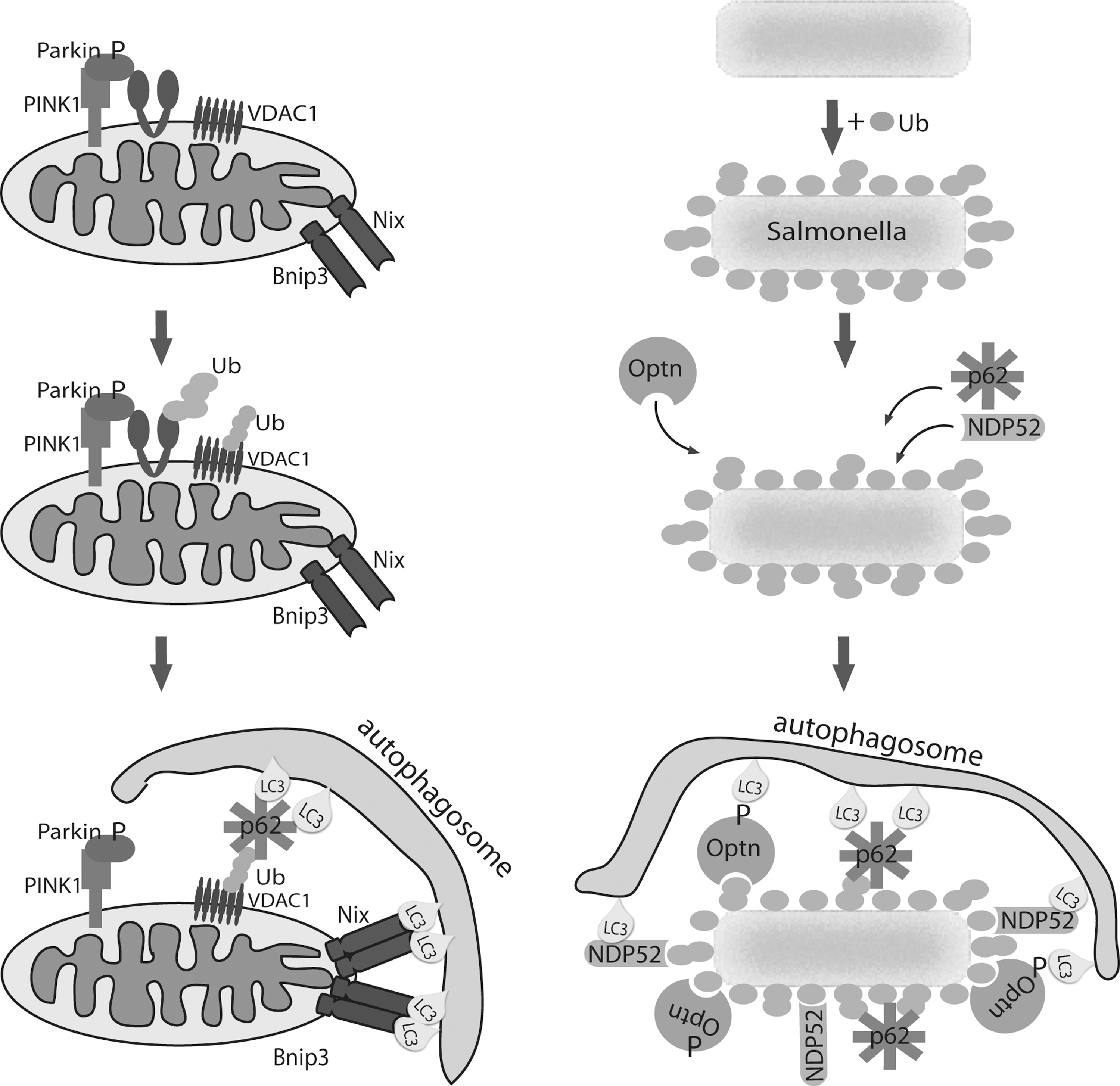
Removal of mitochondria occurs during development of some specialized cells and is essential for correct organ or tissue development. Unlike PINK1/Parkin-mediated mitophagy, mitophagy induced during differentiation is not a quality control mechanism for degradation of unhealthy mitochondria, but a programmed complete or almost complete mechanism for elimination of the mitochondrial population. The best-studied differentiation-induced mitophagy is the removal of mitochondria in red blood cells. Mature erythrocytes of mammals do not contain any mitochondria, allowing them to maximize their oxygen-carrying capacity and to live longer in the circulation because of lower risk of damage induced by reactive oxygen species (ROS) produced by mitochondria. Two groups independently identified the BH3-only protein Nix (known as Bnip3L), which localizes in the outer mitochondrial membrane, to be involved in programmed removal of mitochondria in immature red blood cells (reticulocytes) (37, 39). Reticulocytes of Nix knockout mice were unable to remove mitochondria, and the development into mature erythrocytes was inhibited even though the autophagic machinery remained intact. Interestingly, mitochondria in Nix−/− reticulocytes position themselves around autophagosomes but are not successfully engulfed and cleared. This work stimulated further investigation into the role of Nix in mitophagy, and it was shown that Nix directly interacts with Atg8 mammalian homologs LC3/GABARAP, which are essential for the formation of autophagosomes by incorporation into newly formed membranes (30) (Fig. 3). The interaction occurs through the LC3-interacting region (LIR) on the N-terminal part of Nix, the tetrapeptide WxxL motif found in other receptors involved in autophagic cargo recognition (21). This mechanism is analogous to how p62 and NBR1 specifically bind ubiquitinated protein aggregates and mediate their removal by autophagy (20, 32). The Nix:LC3/GABARAP interaction proved to be highly important for maturation of reticulocytes and mitochondrial clearance since introduction of wild-type Nix into Nix−/− reticulocytes completely restored mitophagy. Intriguingly, rescue of Nix−/− reticulocytes was not complete when LC3/GABARAP-binding mutant Nix was used, suggesting the existence of either additional mitochondrial receptors or modifications, e.g., ubiquitination. Moreover, the presence of Nix-independent mitophagy pathway has been shown. The Nix-deficient phenotype was successfully restored by depolarization of mitochondria with CCCP (53), and the BH3 mimetic ABT737 induced loss of mitochondria in Nix−/− reticulocytes (37). Furthermore, Atg7 and Ulk1 proteins that are important in autophagosome formation and indispensable for basic autophagy, were also shown to be involved in programmed mitochondrial clearance in reticulocytes. Atg7 is an E1-like enzyme that activates two ubiquitin-like conjugations systems, Atg8 and Atg5-Atg12, involved in formation of autophagic vesicles. In the absence of Atg7, mitochondrial clearance in reticulocytes is impaired but not completely blocked, indicating the complexity of the developmental mitochondrial removal mechanism that might be distinct from basic autophagy machinery. Protein serine-threonine kinase Ulk1 was found indispensable for proper degradation of mitochondria in red blood cells, with Ulk1-deficient mice exhibiting an accumulation of defective mitochondria in terminally differentiated red blood cells that should be mitochondria depleted (23). In contrast to Atg7, which is crucial for general autophagy, Ulk1 is not essential for induction of autophagy, but rather it has a more specialized role in selective autophagy. At present, additional mitophagy receptors have not been described, though Bnip3, a protein homologous to Nix, has been implicated in hypoxia-induced autophagy. In addition to high sequence similarity to Nix, Bnip3 shares mitochondrial localization pattern as well as the LIR motif, suggesting it could be added to the list of mitophagy receptors (Fig. 3). Indeed, preliminary results showing similar interacting pattern of Bnip3 and LC3/GABARAP proteins to Nix are indeed encouraging and suggest that both Nix and Bnip3 might be equally important for mitochondrial selection and removal. Moreover, preliminary biochemical results suggest that Nix and Bnip3 bind individual LC3/GABARAP proteins with different affinity. For example, LC3B was shown to be the weakest Nix interactor, while Bnip3 binds LC3B with high affinity (own unpublished results and N. Brady, personal communication). This suggests nonredundancy of two mitophagy receptors and their possible recruitment and additional regulatory mechanism mediating interactions with LC3/GABARAP.
To fully understand the role of mitophagy in the development of red blood cells, further investigations must identify the upstream signals that lead the cells to start programmed mitochondrial removal at a very precise time point, in the absence of cell death or developmental delays. The major questions under investigation now are how mitochondria become a target, and what are the sensors that activate mitophagy. One of the obvious scenarios is depolarization of the mitochondrial membrane and exposure of inner mitochondrial proteins to cytoplasmic environment for recognition by autophagy initiators. Potentially, depolarization of the mitochondrial membrane could be achieved through dimerization of Nix and Bnip3 receptors to form channels in the outer membrane. Dimerization of Nix and Bnip3 has been confirmed (4, 15), but it remains to be determined if such interactions can promote the change of membrane potential, which could initiate and regulate mitophagy. Moreover, future investigations should address if depolarization is a cause or consequence. Zhang et al. (53) demonstrate that depolarization is a consequence rather than a cause because analysis in Atg7−/− cultured reticulocytes showed that before elimination, mitochondria are polarized compared with depolarized ones in wild-type cells. Thus, the role of decreased mitochondrial membrane potential in mitophagy remains a fundamental question for continued investigation.
Two additional examples of development-induced mitochondrial clearance present in mammals are mitochondrial loss in differentiated eye lens and almost complete elimination of mitochondria in mature sperm (1, 42). Due to the complexity of analyzing developmental mitochondrial clearance in these systems, there is no evidence that mitophagy is the pathway responsible for performing these specialized differentiation steps. Further analysis of these systems might give additional clues to better understand the mechanism of mitophagy.
Mitophagy and ROS
Constant energy production in mitochondria generates ROS as byproducts of oxidative phosphorylation. If not removed, ROS cause DNA and protein damage. Extensive amounts of mutations and lesions in mtDNA and misfolding or aggregation of mitochondrial proteins lead to mitochondrial dysfunction, and therefore robust control for ROS production and removal of ROS-damaged mitochondria are required (22). An accumulating body of work demonstrates the involvement of ROS in autophagy, with the involvement of mitochondria as the main source of ROS. Mitophagy may thereby be important in removing the ROS-damaged mitochondria to extend cellular lifespan. Intriguingly, mitochondria, autophagy, and mitophagy are highly coupled, and ROS may act as a signal that induces autophagy in order to prevent cell death (38). It has been demonstrated that mitochondrial response to ROS damage leads to mitochondrial depolarization and recruitment of autophagic machinery (9). Studies investigating oxidative stress conditions, like hypoxia, noted that increased expression of Bnip3 and Nix is driven by common hypoxia transcription factor HIF1 (2). However, regulation of Bnip3 and Nix might be different in some specialized tissues like heart where distinct pathways are responsible for their induction (10). Bnip3 and Nix, both BH3-only proteins, through binding to Bcl-2 and/or Bcl-xL disrupt the interaction between Bcl-2 or Bcl-XL and Beclin1 (33) thus freeing Beclin1 to induce autophagy (2). Beclin1 activates the Atg6/PIK3-class III complex important for autophagosome membrane construction and is therefore essential for autophagy initiation (16). Further, involvement of Nix and Bnip3 in hypoxia-induced autophagy and their prosurvival role were confirmed by knock-down experiments, in which lack of both proteins resulted in increased cell death similar to effects seen in Beclin1 or Atg5 cells (2). To address whether Nix is involved in the recruitment of autophagic machinery and is ROS dependent, Ding et al. (7) induced mitochondrial damage with CCCP and investigated ROS production and recruitment of Parkin and p62. The authors showed increased ROS (as a consequence of depolarization) and autophagy induction only in the presence of Nix. Curiously, CCCP-induced autophagy was completely suppressed by antioxidant N-acetylcysteine, which creates a situation resembling Nix-deficiency, suggesting the importance of ROS upstream of Nix. In addition, Parkin, ubiquitin, and p62 localization to mitochondria were mediated by Nix-dependent Parkin translocation to mitochondria. Taken together, the authors speculated that Nix is upstream of both mitochondrial “priming” by Parkin/p62 and autophagy machinery recruitment, thus linking PINK1/Parkin-mediated mitophagy with hypoxia/Nix/Bnip3-mediated mitophagy (7). These findings are intriguing considering involvement of Parkin and PINK1 in PD development in which mitochondrial dysfunction–induced oxidative stress may contribute to disease progression. Interestingly, clinical characteristics of both Parkin and PINK1 mutations cannot be distinguished. However, recent analysis of PINK1-deficient neurons demonstrated the presence of fragmented and depolarized mitochondria accompanied by increased ROS. This phenotype was rescued only by wild-type PINK1 but not PD-related PINK1 mutants, indicating that PINK1 is required for maintaining normal membrane potential and thus, normal ROS levels (49). PD PINK1 mutation patient fibroblasts treated with valinomycin and hydrogen peroxide, to induce mitochondrial stress, could not recruit Parkin to mitochondria compared to the cells from healthy individuals. This suggested that PINK1 is essential for Parkin recruitment and that recruitment is not dependent on membrane potential (35). Furthermore, inability of PD PINK1 mutations to recruit Parkin resulted in impaired mitophagy (13), demonstrating the importance of PD-related PINK1 mutations in disease pathogenesis. Thus, lessons from PINK1-deficient cells suggest a vital role of PINK1 in maintaining mitochondrial homeostasis by operating as a sensor of mitochondrial damage and initiator of mitophagy. It would be of great interest to identify additional proteins, besides Parkin, that are modified and activated by PINK1 and necessary for mitophagy.
Mitophagy Fine-Tuning—Lessons From Xenophagy
Although, focused mitophagy research is increasingly contributing to our understanding of this perplexing process, fine-tuning and detailed molecular mechanisms are still not resolved. Well-supported and accepted endosymbiotic theory explains how mitochondria in eukaryotic cells evolved from bacteria. Hence, recent research on autophagy of intracellular bacteria, referred to as xenophagy, might provide novel insights to mitophagy regulation and induction (6). Xenophagy defends the cell from bacterial invasion by enabling selective recognition of the target. Again, the specific target is recognized by autophagy receptors that serve as a bridge between cargo and autophagic machinery. To date, two bacteria-specific receptors have been described, NDP52 (44) and Optineurin (Optn) (51), targeting Salmonella. Interestingly, recruitment of both receptors is ubiquitin-dependent since coating of bacteria with ubiquitin precedes NDP52 and Optn bacterial localization. Another common feature of receptors is possession and necessity of ubiquitin-binding domains and LIR motifs that allow simultaneous interaction with ubiquitin-coated bacteria on one side and ATG8/LC3/GABARAP proteins on the other to successfully mediate bacterial degradation. As expected, due to the presence of ubiquitin, p62 receptor is also found localized to Salmonella (54). Strikingly, it was shown that NDP52 and Optn co-localize, but neither exists in the same area as p62, but instead form non-overlapping domains on bacteria that suggest independent recruitment of receptors (3, 51). Studies have also shown how recruitment of individual receptors is nonredundant by knocking them down individually. Briefly, individual depletion of NDP52, Optn, and p62 or LIR motif mutations in Salmonella-infected cells resulted in increased cytosolic bacterial proliferation and impaired xenophagy. Recently, study on Shigella and Listeria (also intracellular bacterial species), in which NDP52 and p62 are recruited to ubiquinated bacteria, confirmed the same recognition mechanism (27). If Optn recognizes these additional species, pathogen cargo selection by autophagy may be a fundamental event.
Wild et al. (51) further determined the mechanism that recruits Optn to Salmonella. They found that mobilization of Optn is phosphorylation-driven by identifying a serine residue adjacent to LIR motif as a target of protein kinase TBK1. Phosphorylation of Optn enhances its LC3-binding affinity and therefore, autophagic clearance of bacteria. This finding suggests a novel mechanism of selective cargo regulation through phosphorylation of LIR-containing autophagy receptors.
However, it is not surprising that phosphorylation would be the key regulator of selective autophagy fine-tuning since phosphorylation events have already been described to govern the autophagy activation and regulation (for detail see the review by McEwan and Dikic [26]). To this end, highly conserved phosphorylation sites on LC3 proteins (but not GABARAP), neighboring the domain interacting with LIR motif of autophagy receptors, are shown to be phosphorylated by PKA in order to reduce LC3 activity (5). This and other examples (26) indicate how phosphorylation can be both an activator and inhibitor depending on target protein.
Therefore, a current pressing question concerns if other autophagy receptors would be regulated, in respect to interaction with LC3/GABARAP, through phosphorylation of serines adjacent of LIR. For example, Nix and Bnip3, similarly to Optn, have serine residues juxtaposed proximal to their LIR motifs. This suggests a very important issue if mitophagy is also fine-tuned via the phosphorylation status of mitophagy receptors (Fig. 4). If this can be proven, further studies will have to focus on the identification of kinases and phosphatases specific for individual mechanisms and how they contributes to delicate regulation of autophagic machinery.
Concluding Remarks
It is now well established that mitophagy has a central role in the regulation of mitochondrial dynamics to ensure proper mitochondrial and, therefore, cellular functions. Major advances have been made recently that identified key proteins responsible for recognition of damaged mitochondria and their elimination by autophagic machinery. Damage-induced mitophagy is largely governed by PINK1 and it is speculated that PINK1 senses mitochondrial membrane potential change or increased ROS production, therefore protecting mitochondrial integrity. However, clearance of mitochondria during development could be induced by a different, PINK1-independent mechanism, which includes mitophagy receptors Nix and Bnip3, but it is becoming evident that the mechanisms are overlapping. How these events are elicited and regulated and what determines if all or only some mitochondria will be eliminated remain obscure. Lessons from autophagy of intracellular bacteria suggest fine-tuning of the mitophagy process through phosphorylation of adaptors that mediate the mitophagy process cooperatively. Future studies analyzing individual components of mitophagy may decipher the complex network that will help to understand physiological and more importantly pathological conditions, such as Parkinson's disease, and allow as to manipulate mitophagy to fight diseases.
Footnotes
Acknowledgments
I apologize to all scientists whose important contributions were not referenced in this review as a result of limitations in number of references. Many thanks to Nathan Brady, Ivan Dikic, Ana Kilic, and Jelena Korac for comments and discussions.