
Abstract
Chronic inflammation is one of the foremost risk factors for different types of malignancies, including breast cancer. Additional risk factors of this pathology in postmenopausal women are weight gain, obesity, estrogen secretion, and an imbalance in the production of adipokines, such as leptin and adiponectin. Various signaling products of transcription factor, nuclear factor-kappaB, in particular inflammatory eicosanoids, reactive oxygen species (ROS), and cytokines, are thought to be involved in chronic inflammation-induced cancer. Together, these key components have an influence on inflammatory reactions in malignant tissue damage when their levels are deregulated endogenously. Prostaglandins (PGs) are well recognized in inflammation and cancer, and they are solely biosynthesized through cyclooxygenases (COXs) from arachidonic acid. Concurrently, ROS give rise to bioactive isoprostanes from arachidonic acid precursors that are also involved in acute and chronic inflammation, but their specific characteristics in breast cancer are less demonstrated. Higher aromatase activity, a cytochrome P-450 enzyme, is intimately connected to tumor growth in the breast through estrogen synthesis, and is interrelated to COXs that catalyze the formation of both inflammatory and anti-inflammatory PGs such as PGE2, PGF2α, PGD2, and PGJ2 synchronously under the influence of specific mediators and downstream enzymes. Some of the latter compounds upsurge the intracellular cyclic adenosine monophosphate concentration and appear to be associated with estrogen synthesis. This review discusses the role of COX- and ROS-catalyzed eicosanoids and adipokines in breast cancer, and therefore ranges from their molecular mechanisms to clinical aspects to understand the impact of inflammation. Antioxid. Redox Signal. 18, 323–360.
II. The Role of Obesity and Inflammation in Breast Cancer: Eicosanoids and Adipokines
VI. Adipokine Signaling in Breast Cancer and Their Relation to COX and ROS
VII. Clinical Studies on Breast Cancer Focusing on Obesity, Eicosanoids, and Adipokines
VIII. Unifying Theory of Eicosanoids and Adipokines in Breast Cancer
I. Introduction
Approximately 60% of all breast cancer patients are hormone-dependent (213), expressing estrogen receptors is therefore dependent on estrogen for tumor growth. Epidemiological evidence suggests that obesity, particularly in postmenopausal women, significantly increases the risk of hormone receptor-positive breast cancer (47). In postmenopausal women, adipose tissues biosynthesize aromatase, whose activity in breast cancer tumor tissues is greater than in normal breast tissue (43). Studies have shown that estrogen synthesis, proinflammatory mediators, in particular prostaglandins (PGs) and cytokines, and nutritional factors in combination with genetic susceptibility are probably the major causative elements in breast cancer. Some PGs may upsurge aromatase activity, and cyclooxygenase (COX) inhibitors decrease aromatase activity in breast cancer cells. This might subsequently regulate downstream inflammatory and anti-inflammatory PGs and estrogen synthesis. In addition, there is documented evidence that a diet rich in ω-6 polyunsaturated fatty acids (e.g., arachidonic acid) encourages the growth of metastatic human breast cancer cells in athymic nude mice by producing inflammatory PGs, whereas a fish oil diet containing ω-3 fatty acids (such as docosahexaenoic acid and eicosapentaenoic acid) inhibits such a formation (46, 199, 272, 296, 297). An epidemiological study in Mexican women provides further evidence that the risk of breast cancer increases with a higher intake of ω-6 fatty acids in premenopausal women, but not in postmenopausal women, and a decreased risk of breast cancer is significantly associated with greater ω-3 fatty acid intake in obese women, but not in normal- or overweight women (58). Thus, obesity seems to affect the association between ω-3 fatty acid intake and breast cancer probably through regulating PG formation and subsequent aromatase synthesis. The possible mechanisms involved are related to decreased inflammation and improved adipokine and aromatase/estrogen levels due to ω-3 fatty acids in adipose tissue in obese women. Such evidence may link fatty acids and their downstream derivatives with breast cancer, and is perhaps related to obesity and inflammation in postmenopausal women. Recent studies have suggested that combinations of several of these different underlying mechanisms relate obesity, adipocyte-secreted cytokines, aromatase, and inflammation to breast cancer (154, 213, 341).
The current leading hallmarks of breast cancer-related inflammatory factors appear to be eicosanoids and diverse enzymes/free radicals (COX, lipoxygenases, and reactive oxygen species [ROS]) that catalyze the formation of many of these compounds and adipokines, a cytokine produced by adipocytes. Eicosanoids are the common mutual expression for oxygenated derivatives of three different 20-carbon essential polyunsaturated fatty acids, namely dihomo-gamma-linolenic acid, a ω-6 fatty acid with three double bonds; arachidonic acid, a ω-6 fatty acid with four double bonds; and eicosapentaenoic acid, a ω-3 fatty acids with five double bonds. The classical eicosanoids are PGs, thromboxanes, prostacyclins, and leukotrienes. However, several other classes of eicosanoids, formed by reactive free radicals in vivo such as isoprostanes, have also been included among this group. Many of these compounds have been shown to have an important role in breast cancer owing to their potent inflammatory or anti-inflammatory properties.
In early studies, it was shown that COXs, and hence certain PGs, were involved in the development of various human cancers (32). Indeed, elevated levels of prostaglandin E2 (PGE2) were found in premalignant lesions and diverse forms of advanced cancers. PGE2 has carcinogenic effects in the human body by increasing tumor cell growth and invasion. There is much documented pharmacological and genetic evidence implicating COXs in various types of malignant neoplasia, and thus the general suggestions of involvement of PGs in cancer (154, 294, 344, 358, 379). However, earlier reports evidenced overexpression of cyclooxygenases-2 (COX-2) in breast and other types of cancer (40, 356). COX overexpression, particularly that of COX-2, appears to be related to 40% of incidents in all invasive breast carcinomas. This leads to higher levels of PGE2 biosynthesis, as reported in several studies, which may also enhance estrogen biosynthesis and release through the aromatase in the mammary gland. A similar process has been observed in epidemiological studies, in which various nonsteroidal anti-inflammatory drugs (NSAIDs) seemed to have a cytoprotective effect of varying degree in several types of cancer, including breast cancer (368). The underlying mechanism seems to be linked to the tumoral biosynthesis of specific eicosanoids that are essentially overexpressed in breast cancer tissues through COXs. Inhibiting the COXs and further regulating PGs as a potential target for chemoprevention have been performed in the last few decades to counteract some downstream pathways and their products that are responsible for cell proliferation and angiogenesis in miscellaneous cancer forms, but with a diverse outcome.
The contribution of deregulated oxygen free radicals to chronic inflammation and cancer is well documented. Plausible evidence of involvement of free radicals can be found in international disparities in breast cancer incidence rates and variations in existing migrant populations worldwide. It is widely recognized that diet and hence antioxidants protect against disease due to the putative role of various known and unknown antioxidants in the diet. Epidemiological studies addressing the levels of antioxidants have also provided indirect evidence of the role of free radicals and oxidative stress in breast cancer. However, there is no clear-cut evidence supporting a beneficial role of antioxidants in malignancy (29). The involvement of free radical-induced specific lipid peroxidation or DNA oxidation products such as F2-isoprostanes and 8-hydroxy-7,8-dihydro-2′-deoxyguanosine (8-OHdG), respectively, which are currently regarded as the most consistent parameters of oxidative stress, is less studied in breast cancer, except in a few studies, which will be discussed below.
According to the American Institute for Cancer Research, Washington D.C., advanced obesity is a potential cause of postmenopausal breast cancer (376). Current perceptions regarding the mechanisms of adipokines and obesity in breast cancer seem to be convincing, since a considerable interest has been shown in recent years in the function of leptin and adiponectin in mammary tumors. Both of these proteins/hormones, which are released by the adipocytes and the nonadipocyte fraction of adipocytes, are potential candidates in the process of carcinogenesis, specifically in obesity-related breast cancer (167). It is now recognized that the procarcinogenic effect of leptin and, inversely, the anticarcinogenic effect of adiponectin could be interrelated to inflammatory responses and tissue proliferation in the microenvironment.
In these contexts described above, the eicosanoids formed through COX and free-radical catalyzation, adipokines and estrogens are potential contestants in breast cancer. Collectively, the current review assesses the impact of eicosanoids and adipokines on breast cancer, and subsequently the impact of inflammation on breast cancer pathology.
II. The Role of Obesity and Inflammation in Breast Cancer: Eicosanoids and Adipokines
Obesity and inflammation are important causative factors in the development of several diseases, including breast cancer in which locally active eicosanoids and adipokines may have a crucial role in both the nonmalignant and malignant components of the tumor microenvironment. According to the American Institute for Cancer Research, Washington D.C., some convincing and probable causes of the increased risk of postmenopausal breast cancer are body fat, abdominal obesity, adult weight gain, and the intake of alcoholic beverages, which are all linked to inflammation. A crucial role in this context is performed by the adipose tissues, which comprise macrophages, undifferentiated fibroblasts, and mature adipocytes in homeostasis.
Breast tissue consists of fat, ducts, and glandular and connective tissues. It develops mainly in response to hormones such as progesterone, estrogen, prolactin, growth factors, and insulin during puberty, pregnancy, and lactation. These key hormones also have a strategic influence on breast cancer development. The importance of early-life events such as appropriate food intake together with the factors that affect both obesity and hormone status can also modify the risk of breast cancer. Cancer of the breast manifests typically a carcinoma of the epithelial cells lining the ducts (376). Because most of breast cancers are hormone-related, hormones are believed to play an important role in cancer development and progression, since they modulate the structure and growth of the epithelial tumor cells, in which obesity and inflammation may interplay. Several breast cancer types also initiate the production of hormones such as growth factors that act locally and can both stimulate or counteract tumor growth (205, 249). Some growth factors have been identified as a stimulator of COX-2 activation and secondary inflammatory response (65, 74).
Visceral fat is known to produce several adipokines and cytokines that have inflammatory properties and are linked to both obesity and breast cancer. Obesity causes subclinical inflammation in the adipose tissue microenvironment characterized by necrotic adipocytes surrounded by macrophages forming crown-like structures that increase the breast cancer risk (240). Inflammation accompanied by elevated aromatase expression generally persists in the tumor microenvironment and induces additional sequences of angiogenesis. Eicosanoids, adipokines, and other inflammatory compounds are probably biosynthesized and released from the damaged tissue site during this long-standing pathogenic process as a response to a complex network of various biochemical signaling processes and mediators. This tissue transformation from the healthy to the chronic inflammatory microenvironment further promotes tumor growth, angiogenesis, and metastasis (260), which may also play a part in the pathogenesis of breast cancer.
Inflammation is conceivably one of the key pathological mechanisms of tissue–cancer transition, and it could be brought about by excessive formation of several bioactive factors in the microenvironment. It is still unclear what influence such components have in triggering cancer and by what mechanisms they contribute to disease advancement. Numerous reports suggest that tumor tissues generally extradite chronic inflammatory mediators such as lipid autacoids derived from arachidonic acid metabolism, cytokines, and free radicals. One current hypothesis is that the underlying mechanisms of breast cancer can only be understood by studying several potent pathways of signaling cascades and the formation of different inflammatory mediators such as eicosanoids, adipokines, free radicals, and, more specifically, the inflammatory microenvironment of the affected tumor tissue (183, 341). Regrettably, systematic studies on these issues in human breast cancer are scarce. In the following sections, this review will discuss two major routes of inflammation that may have a direct mechanistic crosslink with adipokine regulation: eicosanoids, which are mainly formed through COXs and free radicals, since their contribution to the tumor microenvironment and systemic inflammation is indispensible (99).
III. COX and Free Radicals in Cell Function
A. COXs and PGs
Arachidonic acid, which is found in lipid bilayers, can be metabolized by several different pathways: enzymatic through COXs, lipoxygenases, and cytochrome P-450 monoxygenases; and nonenzymatic through free radicals (Fig. 1). The PGs are one of the most potent biologically active compounds formed from free arachidonic acid derived from membrane-bound phospholipids by phospholipase A2 (PLA2) (86, 110, 304 –306). PG synthase is an enzyme complex that predominantly bioconverts unesterified arachidonic acid to various PGs and thromboxanes (137 –139, 140, 305). COXs are the catalytic enzymes that are responsible for the formation of several primary PGs (333, 334, 360, 361). There are primarily two isoforms of COXs: COX-1 and COX-2. COX-3, which is an acetaminophen-sensitive splice, is recently discovered and is less well-known today (323 –327). The COXs (COX-1 and COX-2) differ very little in structure, mainly in the size of substrate-binding sites. PGs commonly possess numerous homeostatic functions and both inflammatory and anti-inflammatory properties, as evidenced in many studies with various species (14, 18, 290, 303, 304). COX-1 is usually believed to be constitutively formed and ubiquitous in most cells and tissues under normal physiological conditions, such as luteolysis, ovulation, sperm migration, regulation of estral cycle, cervix dilatation, pregnancy establishment, parturition, platelet regulation, renal function, and maintenance of gastric mucosa (18). COX-2 is typically absent or found in minute quantities in most cell types, but generally expressed at the site of inflammation after a proinflammatory or mitogenic stimulus by growth factors, interleukins, and cytokines, whose protein and mRNA have short half-lives (90). Initially, COXs (both COX-1 and COX-2) bioconvert arachidonic acid to extremely reactive, but unsteady, endoperoxide PGG2, by introducing two molecules of oxygen. Later, the 15-hydroperoxy group of PGG2 is bioconverted to the 15-hydroxyl group by peroxidase and forms unstable endoperoxide PGH2. Prostaglandin H2 is then further converted by PGE2 synthase or PG endoperoxide isomerases or serum albumin to PGE2 and prostaglandin D2 (PGD2). Prostaglandin F2α (PGF2α) is essentially formed by the reduction of PGH2 through PG endoperoxide synthase or reductase (Fig. 1). Nonetheless, PGF compounds can be formed, although not in significant portions, from other PGs via the enzymatic reduction of 9-keto group of PGE compounds by 9-keto reductases, which results in 9α-hydroxyl, yielding PGF2α compounds. PGF compounds may also be formed from PGD2 compounds by 11-keto reductases (Fig. 1). Thus, multiple precursors may present endogenously for the formation of PGF compounds. 15-Hydroxy prostaglandin dehydrogenase (15-PGDH) and Δ13-reductases are the major metabolizing enzymes of primary PG metabolism and are found in most tissues (24, 25, 136). All these oxidized and/or reduced metabolites of primary PGs and thromboxanes further metabolize through β- and ω-oxidation to shorter-chain metabolites than the parent compound in a species-specific manner, and efficiently excrete into the urine (22, 86, 126, 127, 136). Commonly, these more-degraded metabolites are less bioactive than their parent compounds.

The role of PGE2 as a lipid mediator is well documented in physiology and all types of cancer, including breast cancer (179, 293, 367). It is known to synthesize like other primary PGs from the readily available precursor arachidonic acid and acts locally through the autocrine and paracrine systems. The role of PGF2α in physiology and in acute and chronic inflammation is well established (18), but its involvement in cancer and specifically breast cancer is less documented. An in vitro study reported that PGF2α is involved in tumorigenesis (375). This study showed that PGF2α could increase carcinogen-induced transformation of fibroblast through induction of COX-2. Other studies showed that PGF2α stimulated motility and invasion of endometrial and colon tumor cell lines (281, 302). In addition, more recently discovered degradation products of PGD2, an antiangiogenic factor (246), anti-inflammatory PGJ2, and its dehydration product 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) have attracted much attention from scientists owing to their apoptotic role in various cancers, including mammary cancer (88). This is possibly because 15d-PGJ2 counteracts tumor development by inhibiting the nuclear receptor peroxisome proliferator-activated receptor-γ (PPAR-γ), as discussed below. Nonetheless, very little is known about the additional primary PGs and thromboxanes in breast cancer disease.
B. Free radicals and oxidized products
Oxidative stress is a state in which a severe disruption of the endogenous balance between the production of ROS or reactive nitrogen species and antioxidant defenses occurs in the body (133, 135, 322). Under these conditions, ROS usually appear at a higher intensity. Many of the ROS, such as superoxide radicals (O2·−), which are produced in cells by the mitochondrial electron transport chain, may react with lipids, proteins, and DNA, and exert effects on cancer in general (Fig. 2) (134, 278). Oxidative DNA damage is known to be caused by oxidizing the guanidine base and mutations of tumor suppressor genes that are believed to be crucial in mammalian carcinogenesis. It is thought to be one of the central underlying pathologies in various types of cancer, although its particular role in cancer seems to be more complex than earlier assumed, probably due to its subsequent influence on basal physiology (13, 16, 134, 178, 185, 193, 299). The idea that ROS may be involved in mutagenesis was originally suggested in the 1930s after being documented the instances of radiation injury and occurrences of cancer. It is presently well recognized that radiation-induced carcinogenesis involves initiation, promotion, and activation of proto-oncogenes and inactivation of tumor suppressor genes (134). Excessive formation of ROS is involved in mutagenesis by activating several signaling intermediates (134). A list of such signaling pathways is given elsewhere (289). Current evidence suggests that knockout of diverse antioxidant defense enzymes may lead to a greater oxidative damage and may promote age-related cancer development in experimental animal models.
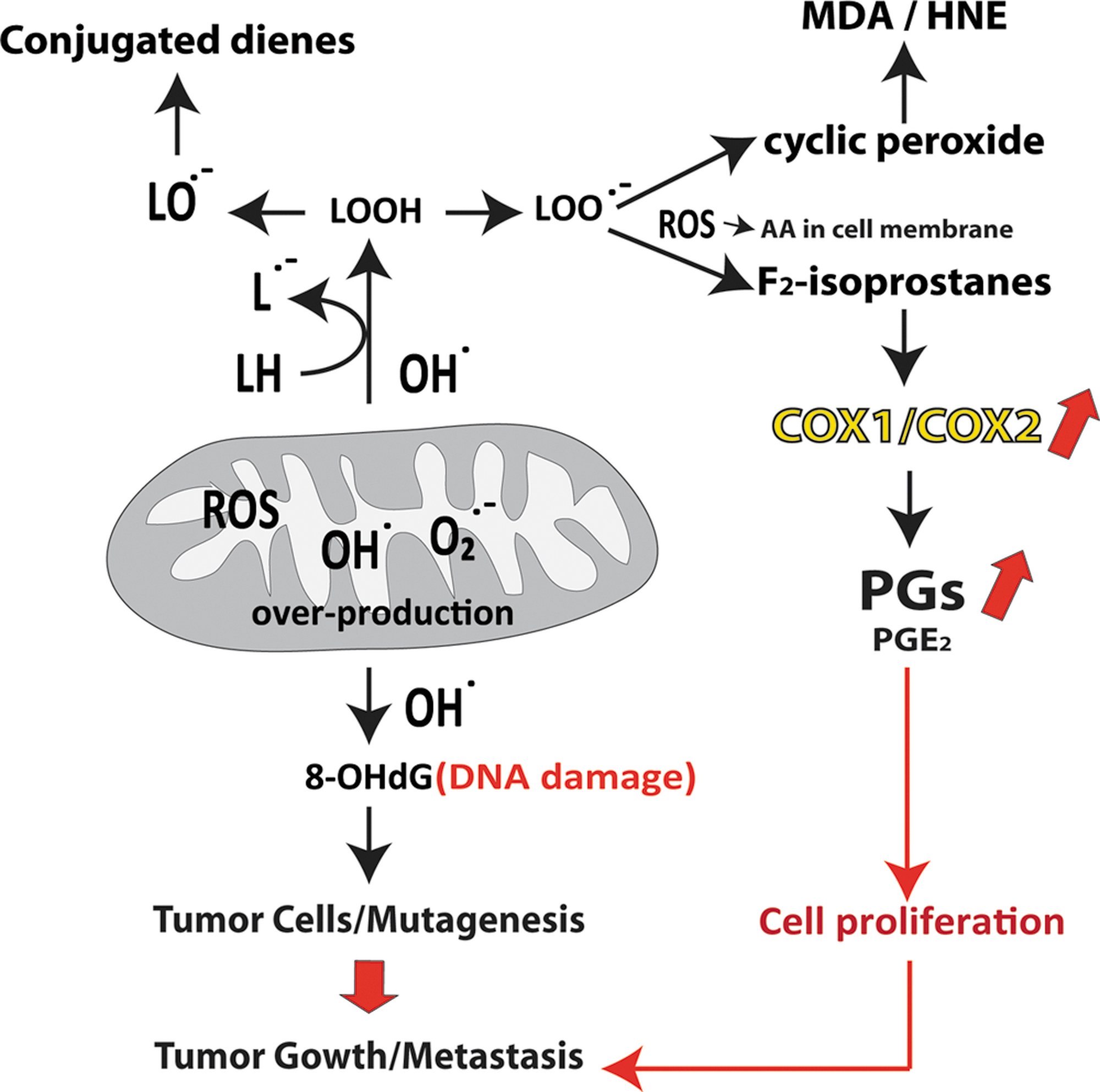
Lipids are also prone to oxidation by ROS, and excessive lipid peroxidation is involved in various types of cancer (Fig. 2). The identification of isoprostanes, a family of PG-like compounds generated in vivo by nonenzymatic free-radical catalyzed-peroxidation of arachidonic acid, heralded a new epoch of detection of nonenzymatic lipid peroxidation products and discovery of their bioactivities. A simplified biosynthetic diagram of isoprostane formation from the precursor esterified arachidonic acid is shown in Figure 1. The measurement of isoprostane is now considered to be the gold standard method for determining lipid peroxidation and oxidative stress in vivo (15, 17, 238, 244, 245). Isoprostanes can be determined reliably by various methods in various body fluids (15, 17, 19, 20, 238, 243) and also by immunohistochemical techniques that assess in situ oxidative organ damage (23, 52, 172) or by direct cellular quantification (335), which has opened up new possibilities in the study of the role of free radicals in cancer research. One of the major F2-isoprostanes, 8-iso-prostaglandin-F2α (8-iso-PGF2α), is a bioactive compound and is associated with several potential risk factors of atherosclerosis and some inflammatory diseases, but its involvement in breast cancer is less evaluated (15, 147, 148, 310).
Malondialdehyde (MDA) is another naturally occurring product of lipid peroxidation and is usually formed as a byproduct along with PGG2/thromboxane synthesis, which also possesses mutagenic and carcinogenic properties (223, 225). MDA is also exceptionally electrophilic and nucleophilic, and it reacts with DNA to form adducts to deoxyguanosine and deoxyadenosine. The main adduct to DNA is a pyrimidopurinone called M1G. A site-specific mutagenesis experiment indicated that M1G is mutagenic in bacteria and is repaired by the nucleotide excision repair pathway. M1G has been detected in breast tissues, the liver, the pancreas, and white blood cells of healthy human beings (174, 383).
DNA can also be damaged by ROS after exposure to ionizing radiation, and it plays a crucial role in carcinogenesis. Gene damage by radiation generally occurs as the result of direct absorption of energy by DNA and formation of highly reactive hydroxyl radicals (134). Following the formation of hydroxyl radicals and assault on DNA, they initiate formation of mutagenic purine, pyrimidine, and deoxyribose oxidation products. A very common biomarker of oxidative DNA damage is 8-OHdG. 8-OHdG is abundant and is generated by mutagenic lesions when ROS attack the guanine base in DNA. Oxidative DNA damage can also transmute the DNA structure, including the base and sugar lesions, DNA–protein crosslinks, and strand breakdowns. Chronic radiation exposure has shown to be carcinogenic in rats, leading to significant increases in 8-OHdG in the mammary gland DNA (132). In addition, urinary 8-OHdG levels were significantly higher among breast cancer patients than in age-matched controls (195). A significantly higher level of 8-OHdG was observed in cultured breast cancer cells than in normal breast epithelial cells in addition to significantly elevated levels of 8-OHdG seen in estrogen receptor-positive compared to estrogen-negative malignant tissue (250). Similarly, serum levels of 8-OHdG and superoxide dismutase were higher, and glutathione peroxidase levels were lower in breast cancer patients than in a control group (149). These findings strongly suggest that ROS are involved in DNA damage in breast cancer. However, in another study, no relationship was observed on the association of breast cancer risk with 8-OHdG (298). Neither does increased 8-OHdG lead to malignancy, although malignant tumors often show higher levels of DNA base oxidation (134). Collectively, oxidative damage is perhaps related to different stages or forms of breast cancer development, which is related to the different appearance and disappearance of 8-OHdG in various studies (229).
IV. Eicosanoids in Breast Cancer
Since the discovery, PGs are considered to be related to diverse cancer pathologies (128). There is ample evidence demonstrating the important role of eicosanoids in inflammation and cancer. Elevated levels of PGs are involved in controlling numerous physiological functions (18, 21, 160, 237). Similarly, higher levels of eicosanoids may also lead to dynamic pathologies such as oxidative stress and inflammation. They eventually result in distant organ damage when they are exposed for extended periods (14, 15, 241, 305, 311, 362). Owing to their dual properties (physiological and pathological), many of these compounds have been used regularly as reliable biomarkers of numerous physiological functions and to assess oxidative stress and inflammation in diseases (17, 18, 78, 102, 239, 242).
A. Prostaglandins
Deregulation of arachidonic acid-derived PGs is an important feature of several malignancies through their inflammatory and anti-inflammatory properties. These PGs are involved in various stages of tumor progression such as apoptosis, cell proliferation and angiogenesis, migration, and invasion. Proinflammatory PGs are the key mediators in the interaction between tumor epithelial cells and surrounding stromal cells in the development of the carcinogenic microenvironment by suppressing immune function and sequentially inducing chronic inflammation. This eventually unhinges the whole immune system in the body as the disease advances. PGE2 is one of the major vasoactive PGs formed at sites of inflammation and has been shown to be a potent indicator of COX-mediated inflammatory processes in vivo (Fig. 1). In breast cancer, PGE2 can upsurge aromatase synthesis in stromal fat cells, and its synthesis subsequently increases estrogen production that in turn can stimulate cell proliferation and other related biochemical processes in the tumor microenvironment (393). Since microsomal prostaglandin E synthase may be upregulated in breast cancer, it has been suggested that PGE2 is protumorigenic. Such evidence provides a pivotal role of COX-2 in breast cancer. Moreover, Subbaramaiah et al. demonstrated that human epidermal growth factor receptor-2 (HER-2)/neu overexpression induces the increase of COX2 expression and PGE2 synthesis in tumor cells, leading to enhanced aromatase activity that subsequently increases estrogen production and the enhancement of cell proliferation. HER-2/neu belongs to the HER family of transmembrane tyrosine kinases encoded by the neu proto-oncogene that is overexpressed in 20%–25% of human breast cancers. HER-2/neu overexpression correlates with an aggressive phenotype and a poor prognosis, and it is associated with increased levels of COX2 in human breast cancer (331, 342, 344).
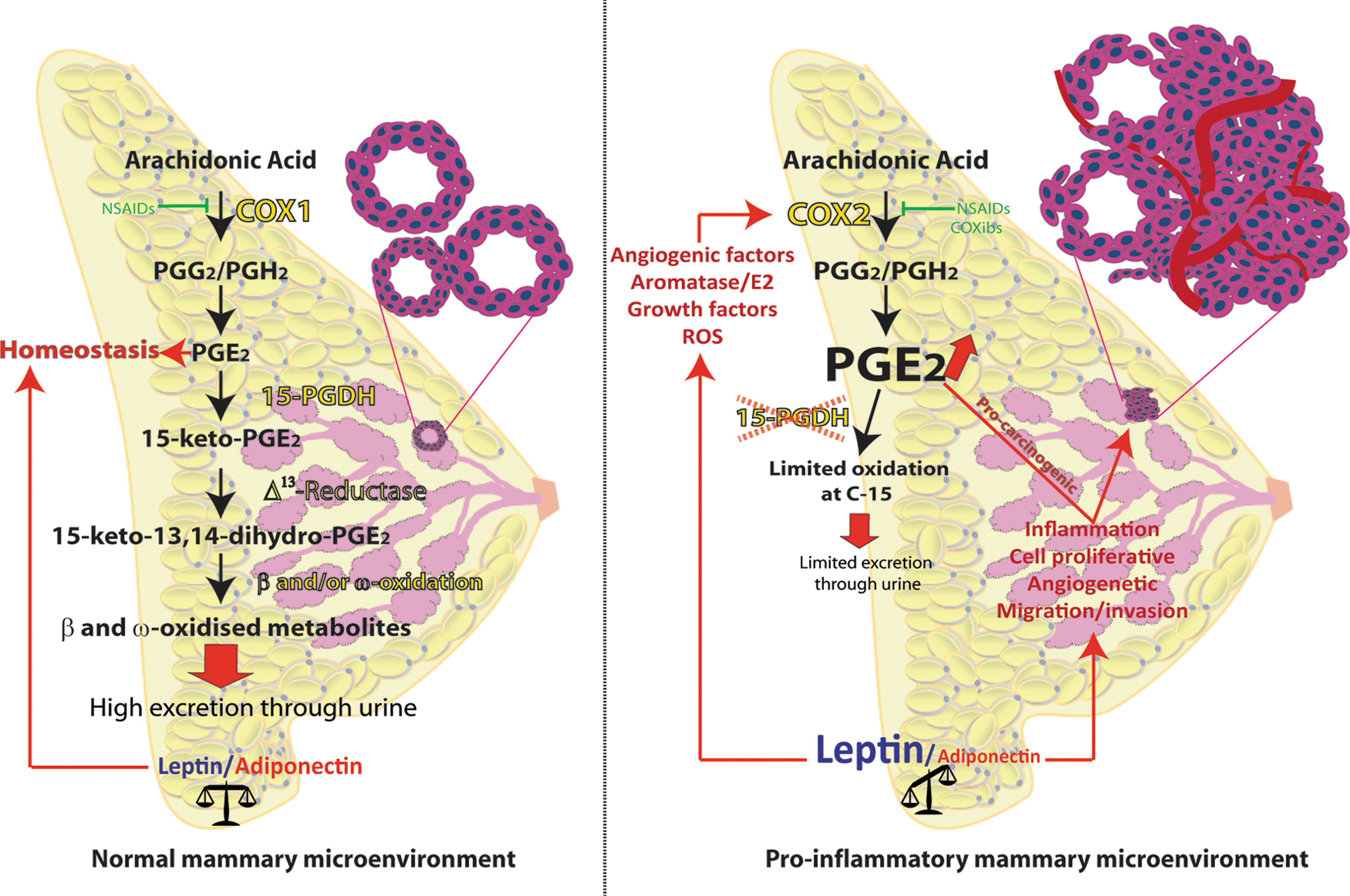
15-PGDH has recently been identified as an important regulating factor in diverse cancers due to its activity as the initial degrading enzyme of primary PGs, including PGE2, and possibly acts as a tumor suppressor (Fig. 3) (251, 252, 347). 15-PGDH is downregulated in colorectal cancer, signifying that PGDH plays a crucial role in modifying in vivo levels of PGE2, and PGDH overexpression could attenuate Ras-mediated tumorigenesis (276). Overexpression of 15-PGDH has been reported to protect against experimentally induced carcinogenesis. It also reduces the cancerous or transformed cells predisposed to apoptosis by inhibiting the pro-oncogenic effect of PGE2. Conversely, 15-PGDH is absent in some cancer cells, probably owing to epigenetic modification, such as DNA methylation and histone deacetylation, in the promoter region of 15-PGDH. A range of compounds, including the PPAR-γ agonists, histone deacetylase (HDAC) inhibitors, and NSAIDs, efficient in stimulating the expression of 15-PGDH have been identified. In normal homeostasis, PGE2 generally synthesizes at a relatively low amount (about 2 pg/ml in plasma) and is efficiently excreted through the urine after being rapidly metabolized by 15-PGDH initially, and subsequently by other downstream degrading systems. Hypothetically, in the normal mammary cellular environment, PGE2 levels are within basal levels (Fig. 3, left panel). In the proinflammatory mammary tumor microenvironment, 15-PGDH levels may fall. This in turn may maintain high PGE2 production in the mammary tumor tissues without PGE2 being metabolized efficiently. This possibility warrants further studies (Fig. 3, right panel). Low levels of 15-PGDH expression were seen in 40% of primary breast tumors, and a correlation was identified between 15-PGDH and ER expression (374). Transfection analysis revealed that transient upregulation of 15-PGDH levels in MDA-MB-231 cells resulted in decreased clonal growth, and stable upregulation significantly decreased the capacity of these cells to form tumors in athymic mice. In contrast, transient silencing of 15-PGDH on MCF-7 cells resulted in their enhanced proliferation. Further, 15-PGDH levels were downregulated by estrogen and upregulated by tumor suppressor gene CAAT/enhancer-binding protein alpha. In breast cancer cells, Calcitriol, an active form of vitamin D, reduced the levels of PGE2, a major stimulator of aromatase transcription, by inhibiting the expression of COX-2 and increasing 15-PGDH levels (192). It is also known that PPARγ agonists such as thiazolidinediones (TZDs) (pioglitazone and rosiglitazone) and HDAC, such as sodium butyrate, apicidin, and oxamflatin, induce 15-PGDH expression and suppress tumor-derived PGE2 in various cancer models (9, 146, 252, 355). Together, these findings suggest that 15-PGDH acts as a tumor suppressor of breast cancer and is able to modulate the ER pathway.
Evidence of the involvement of PGE2 and hence COXs in cancer comes mainly from epidemiological studies of regular intake of NSAIDs (222, 224). As a lipid mediator, PGE2 plays a substantial role in controlling basal physiology and also in different forms of cancer through its four distinctive categories of receptors, EP1, EP2, EP3, and EP4 (viz. PTGER1, PTGER2, PTGER3, and PTGER4), which act through G-protein-coupled receptors (GCPRs) (188, 255, 354). GCPRs are membrane-incorporated receptors that are commonly coupled to heterotrimeric guanine nucleotide-binding proteins (G proteins). EP1 is a GCPR that signals primarily through Gαq protein, through a transient rise in intracellular calcium. The three other PGE2 receptors (EP2, EP3, and EP4) signal through cyclic adenosine monophosphate (cAMP). EP1 can also secondarily inactivate PPAR-γ to a certain extent (370). However, the specific role of these distinctive PGE2 receptors in different organs and their involvement in diseases are not well demarcated and nor is their role in breast cancer. PGE2, which is predominantly formed by catalyzation through COX-1 and/or COX-2, is involved in controlling neuronal and kidney function, reproduction, gastric mucosal defense, and also in pain, fever, and inflammation. PGE2 is also involved in various types of tumorigenesis and angiogenesis and in the inhibition of apoptosis and the regulation immune function (27, 67, 314). Furthermore, the involvement of PGE2 in cancer is well acknowledged, since this particular PG is found in human colorectal cancer tissues (293). However, several studies have described a coherent involvement of PGE2 in breast cancer, in which it induces CYP19 transcription, resulting in increased aromatase activity (43, 61, 343, 344).
PGE2 synthase is induced essentially by cytokines, and it was later designated as microsomal prostaglandin E2 synthase-1 (mPGES-1), a member of the large membrane-associated proteins (MAPEG) (165, 231, 232). This enzyme is overexpressed in various types of cancer and induced overexpression of mPGES-1 in experimental animal models. It blocks apoptosis and further contributes to angiogenesis, tumor progression, and metastasis. In addition, mPGES-1 knockdown resulted in a diminished clonogenic capacity and slower growth of xenograft tumors (with a disintegrated tissue structure) in nude mice (142). Further, overexpression of mPGES-1 has been observed in nonsmall-cell lung cancer (NSCLC) in tumor tissues other than the adjacent normal tissues (384). This recent discovery of mPGES has shown its potential involvement in various cancers, including breast cancer, since mPGES-1 is overexpressed as assessed by immunochemistry methods (233). In addition, this overexpression has shown to be more intense than COX-2 activity. However, estrogen receptor expression, a biomarker of breast cancer, does not correlate with mPGES-1 expression. In NSCLC cells, interleukin-4 (IL-4), a cytokine, inhibits the production of PGE2 through COX-2, whereas expression of mPGES-1 is not affected. Although COX-2 inhibition was originally implicated as the initial target for chemoprevention, mPGES-1 is currently suggested as a potential entity for therapeutic intervention for regulating inflammation and cancer through the PGE2 route (164, 165).
Arachidonic acid can also be catalyzed through COXs to instable cyclopentenone PGs, PGA2, and PGD2. In a recent study, PGD2 was shown to be antiangiogenic in lung carcinoma (247). After biosynthesis, primary PGD2 may bioconvert to another highly reactive intermediate PG, PGJ2 (Fig. 1). 15d-PGJ2 is a major nonenzymatic dehydrated metabolite of PGJ2 and is recognized to be the endogenous ligand of PPAR-γ, which inhibits cell proliferation and induces apoptosis, as evidenced in several cell lines. 15d-PGJ2 may also be produced in the inflammatory cells and tissues as a consequence of upregulation of COX-2 (345). 15d-PGJ2 further possesses anti-inflammatory and cytoprotective effects by inhibiting nuclear factor-kappaB (NFκB) activation and PPAR-γ through binding to DP1 and DP2 receptors and also to receptor-independent mechanisms (175). Thus, it may additionally block the expression of the proinflammatory gene COX-2 (181) and stimulate the anti-inflammatory transcription factor Nrf2 (345). Furthermore, the carbonyl group of this compound has an electrophilic center that is susceptible to nucleophilic addition reactions (108). The cyclopentenone ring of 15d-PGJ2 covalently modifies cellular proteins, p50 and p65, subunits of NFκB (57, 340), and contributes to their biological effects (Fig. 1). The potent antiviral and antiproliferative properties of this PG are ascribed through this reactivity (109, 381). Current studies propose that inactivation of Akt (a serine/threonine-specific protein kinase) through mitochondrial ROS production, negative regulation of Akt transcript levels, NADPH oxidase activation, and activation of JNK leads to 15d-PGJ2-induced apoptosis in leukemia and colorectal cancer cell lines (319). Further, 15d-PGJ2 has shown to reduce the growth of mouse tumors (CT-26 cell growth) and HL-60 xenograph tumors. Whether this is the case in other cancer cell lines such as mammary cancer merits further exploration.
15d-PGJ2 also induced COX-2 expression in vitro through Akt-induced AP-1 activation in a human breast cell line, MCF-7 (180). It potentially upregulates the expression of COX-2 through activation of Akt, and subsequently of activator protein (AP-1). In addition, 15d-PGJ2 induced ROS, which led to increased phosphorylation of Akt, DNA binding AP-1, and further expression of COX-2. However, this effect was not evidenced by 9,10-dihydro-15d-PGJ2, a metabolite of this prostaglandin (15d-PGJ2), since an electrophilic carbon center located at the α, β-unsaturated carbonyl moiety of the cyclopentane ring is required, as with 15d-PGJ2, for COX-2 expression and upstream signal transduction. Thus, 15d-PGJ2 catalyzed by COX-2 overexpression may act as a positive regulator of COX-2 in breast cancer cell lines. A recent in vitro study investigated the effect of 15d-PGJ2 on the metastatic characteristics of breast cancer cells (88). 15d-PGJ2 decreased migration, stimulated focal-adhesion disassembly, and caused extensive filamentous actin. These effects are independent of PPAR-γ and modification of Keap1 or actin, which are recognized targets of 15d-PGJ2 at higher levels. Hence, 15d-PGJ2 appears to possess potential antimetastatic properties, at least when studied in vitro. In contrast, to our knowledge, no direct association of PGF2α or other PGs such as 6-keto-PGF1α and thromboxane B2 (metabolites of primary prostacyclin and thromboxane A2, respectively) has been established.
B. Isoprostanes
In the literature, there are very few studies on the role of F2-isoprostanes in breast cancer. The risk of breast cancer was doubled in those whose urinary F2-isoprostane levels were in the second, third, and fourth quartiles than those in the lowest quartile (298). Another study from Taiwan showed that individuals with levels in the second and third tertiles of F2-isoprostanes compared to those in the first tertile had significantly increased risk for hepatocellular carcinoma (378). Recently, a nested case–control study performed as part of the Shanghai Women's Health Study with F2-isoprostane and its major metabolites showed that urinary excretion of isoprostanes was not significantly different between breast cancer cases and controls (81). However, in overweight women, the levels of isoprostanes were positively associated with the breast cancer risk, but the mechanism of such a relation was not elucidated. These few studies reflect a variable outcome on isoprostanes and breast cancer development, perhaps due to differences in the study protocol and the criteria used for selecting the patient's tumor stage. Owing to the importance of isoprostanes as a gold standard method of determination of oxidative stress in vivo, and also because isoprostanes are bioactive compounds formed through free-radical pathways that are possibly related to breast cancer, further studies are warranted in this field together with adipokines to determine the role of oxidative stress in disease.
C. Eicosanoid inhibition
Tumor angiogenesis and the development of metastases are critical circumstances for tumor progression, which depends on the regeneration of new blood vessels. Numerous studies have established an association between COX-2 expression and the regulation of apoptosis and hence tumorigenesis. Early studies showed the potential benefit of NSAIDs in reducing the risk of breast cancer (144, 145, 309). Preclinical studies have consistently shown that NSAIDs prevent mammary carcinogenesis through their anti-inflammatory and antitumoral effects (202) probably through nonselective inhibition of COX-1 and COX-2, resulting in the inhibition of all the downstream PGs that in turn potentially reduce aromatase activity. The resulting decrease in estrogen biosynthesis could reduce the risk of breast cancer (Fig. 3, right panel). Since COX-2 is generally overexpressed in inflammatory sites and mammary tumors, it can be assumed that NSAIDs and selective specific COX-2 inhibitors (COXibs) could suppress the disease by inhibiting COXs and subsequent PG formation.
Extensive inhibition of COX-1, a key housekeeping enzyme that controls several physiological functions, could trigger various side effects. Nevertheless, the regular use of nonselective NSAIDs in various chronic inflammatory diseases is accompanied by undesirable gastrointestinal side effects that are believed to be due to the counteraction of COX-1. To circumvent these complications, much therapeutic research has been dedicated to developing selective COX-2 inhibitors, which could have beneficial effects without entailing these side effects to the body (189). Although the subsequent development of selective COX-2 inhibitors reduced some of these side effects, recent verdicts suggest that some COX-2 inhibitors increase the relative danger of cardiovascular morbidity (see below). COX-2 is overexpressed in breast cancer, and aromatase activity seems to play an active role in the development of hormone-dependent breast cancer. In addition, PGE2 is a potent inducer of aromatase activity and is intermittently expressed in human breast adipose stromal cells (Fig. 3, right panel). Accordingly, inhibition of PGE2 by COX-2 inhibitors may also inhibit aromatase activity. Owing to the major role of eicosanoids in cancer, in particular PGE2 in breast cancer, adequate blockage of this specific PG has attracted much interest as a therapeutic opportunity in recent decades. However, this appears to be inappropriate due to the aforementioned risk of cardiovascular morbidity or other related disorders. To our knowledge, no drug potent enough to counteract PGE2 has yet been successfully developed to treat breast cancer. Taken together, the studies on eicosanoids described above and the development of treatments that inhibit COX-PGE2-signaling pathway probably are not sufficient in themselves to have an impact on breast cancer prevention. This is due to the well-recognized fact that PGE2 is found in all tissues and is additionally related to several facets of our homeostasis, including their determining role in reproduction, renal and gastrointestinal function, fever, and thermoregulation (Fig. 3, left panel) (112, 141, 210, 350).
COX-2 has been identified in multiple segments of cancer such as angiogenesis, tumor promotion, apoptosis, and metastasis (82). COX-2 is preferentially inhibited by so-called anti-inflammatory COXibs (324). However, treatment of cancer through selective COX inhibition and regulation of PG signaling in chemoprevention has not yet been clinically successful, since the long-term use of selective COXibs leads to an escalation of undesirable cardiovascular side effects such as thrombosis. It is principally caused by inhibiting all the prostanoids that constitutively biosynthesize in the body through COXs rather than a specific PG that is related to the particular pathology (101, 105). Additionally, the cardiovascular side effects of COXibs are perhaps accompanied by the expression of vascular endothelial cells under basal status and inhibiting prostacyclin biosynthesis and release. Long-term use of anti-inflammatory drugs of both nonselective and selective COX-inhibitors in fertile women may hamper pregnancy, cervix dilatation at term, and parturition. It has also been evidenced in preclinical studies that COX-2-deficient mice have atherogenic properties as a result of the deposition of lipids in the circulation and liver.
Table 1 shows various effects of NSAIDs, COXibs, and aspirin in breast cancer as recorded in different studies. Although several NSAIDs have been seen to reduce the risk of breast cancer, results have not always been consistent. Perhaps, this inconsistency is related to the disparate etiology of breast tumors of diverse subtypes and also to the tumor grade and diversity of NSAIDs. Recent studies on present and lifetime users of aspirin reduced the risk of breast cancer, with no differences in the subtype (34, 36). However, a routine use of ibuprofen, a widely used NSAID, was significantly associated with an increased risk of ER+/PR+, HER2, and p53-breast cancers and luminal A and B breast cancers (34, 36). This evidence of the heterogeneous etiology of breast cancer subtypes and various types of NSAIDs (aspirin and ibuprofen) has varying effects in this disorder. Prospective studies of regular aspirin use have shown that it does not reduce the risk of breast cancer (93). In the Nurses' Health Study II, it has shown that neither aspirin nor NSAID is associated with a reduced risk of breast cancer (94). No conclusive correlation has been established between low-dose aspirin and tumor characteristics in breast cancer at diagnosis (388). Conversely, in a recent survey performed as part of the Nurses' Health Study, it was shown that the aspirin use was associated with a decreased risk of breast cancer among women living at least 1 year after breast cancer diagnosis (152). These variations in the observed effects of aspirin and NSAIDs are most likely due to differences between studies. Because of discrepancies in the reported effects of the nonselective and selective NSAID use on breast cancer, a study investigating the genetic polymorphisms of COX-2 was conducted to assess the role of these drugs in the different expressions of COX-2 (313). No major effects of the three COX-2 variant alleles on the breast cancer risk were seen. A total of 8 distinct haplotypes and 18 diplotypes were observed in a population of 1067 breast cancer cases and 1110 control individuals. No significant associations between COX-2 haplotypes/diplotypes and breast cancer risk were established. Among women with hormone receptor-positive breast cancer, the reduced risk for any NSAID use was only apparent among those who had at least one variant C allele of COX-2. These findings, collectively provide certain indications of C allele of COX-2, may interrelate with NSAIDs to reduce hormone receptor-positive breast cancer. However, none of these studies looked at the effect on inflammatory responses of these drugs adequately; an essential feature of future research, to have accurate assessments of the anti-inflammatory effects, would be the study of a range of putative biochemical pathways and relevant biomarkers.
COX, cyclooxygenases; COXibs, specific COX-2 inhibitors; NSAID, nonsteroidal anti-inflammatory drugs.
V. Adipokines in Cell Function
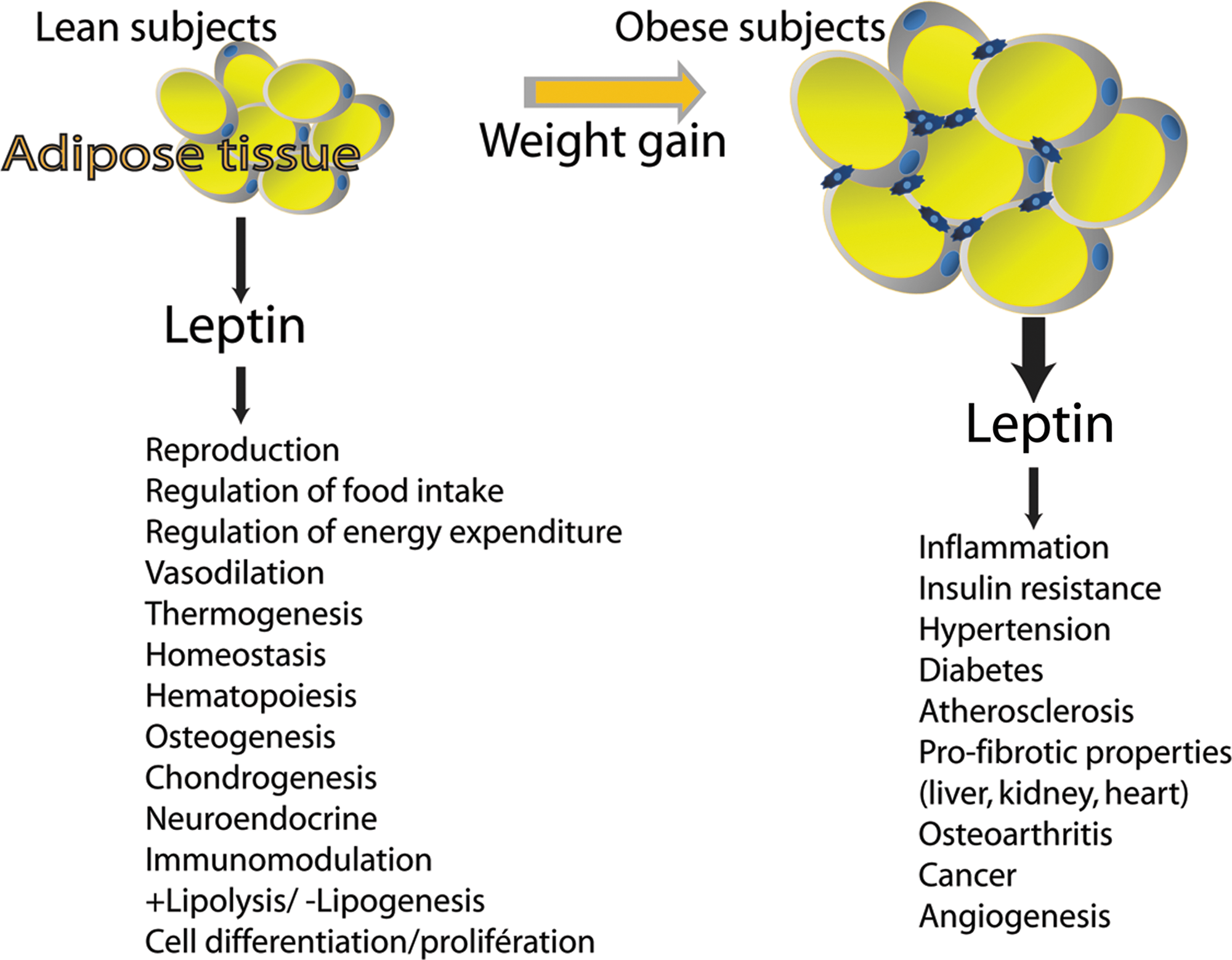
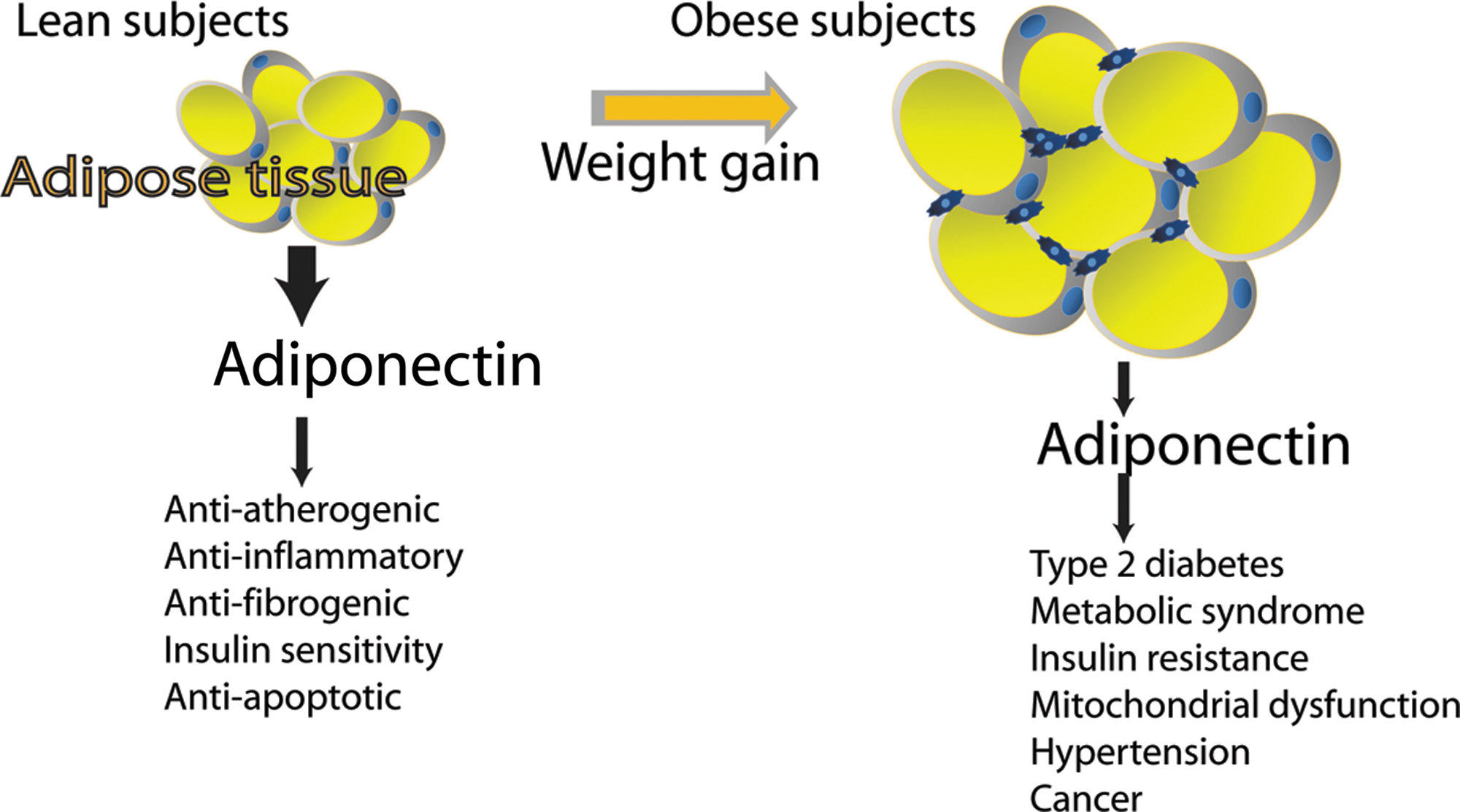
Adipose tissue acts as a central endocrine organ by releasing multiple bioactive substances, recognized as adipose tissue-derived secreted factors or adipokines with both potential proinflammatory or anti-inflammatory activity (264). Adipokines include hormones (e.g., leptin and adiponectin), inflammatory cytokines such as tumor necrosis factor-α (TNF-α) or IL-6, and other proteins (like plasminogen activator inhibitor-1 [PAI-1], adiponutrin, resistin, visfatin, and apelin). Among the many known adipokines, leptin and adiponectin and whose deregulation is involved in several syndromes are the most fully described. The identification and cloning of leptin were one of the initial breakthroughs of adipocyte-derived signaling. Until then, leptin had been proposed to have an important role in many aspects of physiology such as controlling food intake, restoring euglycemia, and regulating whole-body metabolism by stimulating energy expenditure (Fig. 4). Adiponectin acts in lipid metabolism to increase insulin sensitivity, fatty acid oxidation, and glucose uptake, and reduces the production of glucose by the liver (Fig. 5). Most adipokines with proinflammatory properties are overexpressed with growing adiposity, whereas the numbers of some other adipokines with insulin-sensitizing or anti-inflammatory properties, such as adiponectin, are generally inversely related to adiposity. Deregulated biosynthesis and release of these adipokines due to adipose tissue dysfunction may contribute to the pathogenesis of various obesity-linked complications such as cancer, diabetes, metabolic syndrome, and CVD. It is well documented that dietary components together with obesity may also contribute to central adiposity, adipokine production, and inflammation, all of which play a vital role in the aforementioned diseases (96, 186). Hence, dietary control of adipokine production from adipose tissues could be of importance in a variety of diseases.
A. Leptin
The Jackson Laboratory in the United States developed the so-called obese (ob/ob) mouse model in 1950 that opened up a new research area in obesity in the following decades (159). The ob mice usually weighed three times more than their wild-type counterparts. The identification of the gene mutated in this mouse model in 1994 led to the discovery of the adipokine called leptin (derived from leptos, which means “thin” in Greek) (389).
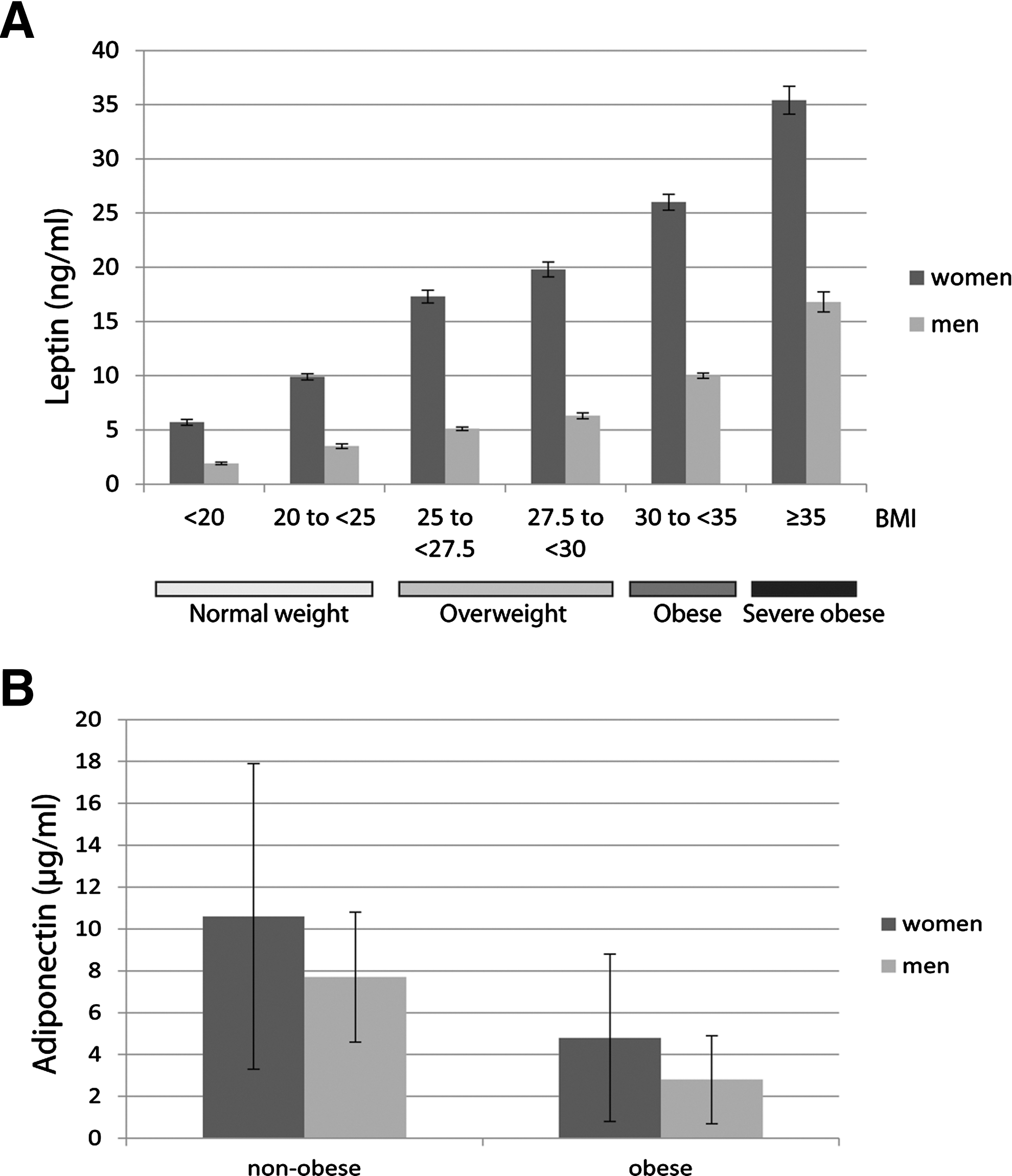
Leptin is a 16-kDa (167 amino acids) nonglycosylated circulating protein encoded by the ob gene located on 7q31.3 in humans (162), and its three-dimensional structure is comparable to that of a cytokine consisting of a four-α-helix bundle motif, which is mutual to the IL-6 family (386) (Fig. 6A). It is mainly expressed by white adipose tissue and more particularly by differentiated mature adipocytes. Leptin is also produced by several other tissues such as the stomach, skeletal muscle, bone marrow, pituitary cells, cardiac tissue, and the placenta (279). It has also been shown that circulating levels of leptin are correlated with body mass index (BMI), total body fat, and nutritional status (2). Leptin levels are ∼10 ng/ml in lean subjects, whereas obese subjects have an increased peripheral leptin concentration that can reach 35 ng/ml (Fig. 7A). Moreover, leptin levels are about 40% higher in women than men at any level of adiposity (Fig. 7A) (1, 5, 300, 301, 332). This gender difference has been attributed to higher leptin production in subcutaneous adipose tissue, stimulation of leptin by estrogen in females, and suppression of leptin by testosterone in males (51, 171). Leptin has been observed to possess pleiotropic properties, for example, in reproduction, immunomodulation, thermogenesis, and angiogenesis (Fig. 4) (59, 227, 321).

1. Leptin receptor
Leptin generally exerts its biological activity through binding to the long form of specific receptors (OB-R) that have similarities to the class I cytokine receptor superfamily (352). To date, at least six members of leptin receptors have been described, resulting from alternative mRNA splicing. They are usually referred to as OB-Ra, OB-Rc, OB-Rd, and OB-Rf for the short isoforms; OB-Rb for the long isoform; and OB-Re for the secreted leptin receptor lacking the transmembrane domain (TM) (Fig. 6B). They all essentially share an identical extracellular leptin-binding domain at the amino-terminus, but distinctly differ at the carboxy-terminus (351). Short leptin receptor isoforms (OB-Ra, OB-Rc, OB-Rd, and OB-Rf) that contain box-1 motif are able to bind Janus kinases (JAKs) and to activate signaling pathways. OB-Ra is abundantly present in cerebral microvessels where it mediates leptin transport across the blood–brain barrier (268). The soluble isoform (OB-Re) binds leptin and serves as an antagonist to OB-Rb-mediated signaling. OB-Re, the soluble receptor, binds and modulates leptin levels, and hence its bioavailability and activity in circulation serve as an antagonist to OB-Rb-mediated signaling (212). The short isoforms are widely expressed in multiple tissues, whereas the OB-Rb receptor is highly expressed in particular sites within the central nervous system (119). The biological effects of leptin are mainly exerted through the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway (Fig. 8) (107). Leptin stimulation activates the JAKs that are constitutively associated with the OB-Rb receptor, and subsequently with the phosphorylated tyrosine residues (Tyr985, Tyr1077, and Tyr1138) in the intracellular domain of the receptor.

The STAT proteins generally bind to phosphorylated Tyr1077 and Tyr1138, whereas phosphorylated Tyr985 recruits the SH2-containing tyrosine phosphatase (SHP2) and mediates activation of mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK). JAK2 can induce phosphorylation of the insulin receptor substrate 1/2 (IRS1/2) proteins and is responsible for the activation of phosphatidylinositol 3-kinase (PI3K) (44, 390). During prolonged stimulation, phosphorylated Tyr985 also binds to the suppressor of cytokine signaling-3 and mediates feedback inhibition of OB-Rb signaling (30). The antiapoptotic and proproliferative effects of leptin have been primarily attributed to PI3K/Akt or SHP2/MAPK/ERK signaling. Mutations of these three tyrosines or of only Tyr1138 alone induce leptin resistance and obesity in mutant mice, indicating that they contribute to the downstream pathways (170). OB-R receptor-deficient db/db mice exhibit a phenotype analogous to leptin-deficient ob/ob mice, indicating that the OB-Rb receptor is required for leptin signaling (63, 68).
2. Leptin regulation
Although many factors (such as nutritional or hormonal regulation) affect leptin production and secretion from the white adipose tissue, the details of the molecular mechanisms responsible for these regulations still remain to be clarified. Insulin, glucocorticoids, angiotensin II, and hypoxia increase leptin expression to some extent (80, 91, 256, 320, 363), whereas the PPAR-γ agonists, like the anti-diabetic TZDs, catecholamines (norepinephrine and epinephrine), and statins inhibit leptin gene expression (Fig. 9) (50, 84, 218, 292). In addition, it is known that the leptin gene is transcriptionally activated by CCAAT enhancer-binding protein-α (C/EBPα) and SP1 (226). Also, the leptin promoter is subject to epigenetic regulation in vitro and in vivo due to the presence of CpG sites in this region (234). Previous studies have shown that its methylation is affected by experimental diets such as high-fat diet in rats or low-protein diet in mice (173, 235).

B. Adiponectin
Adiponectin, one of the prominent adipokines specifically secreted by the adipose tissue (also referred to as GBP-28, apM1, AdipoQ, and Acrp30), was independently discovered by four independent laboratories (156, 215, 254, 308). Human adiponectin is a 30-kDa polypeptide containing 244 amino acids encoded by the ADIPOQ gene located on chromosome 3q27 (348). Adiponectin bears a significant homology to TNF-α, complement factor C1q, and collagen VIII and X. Its structure consists of an N-terminal signal peptide and a variable region, followed by a collagenous domain and a C-terminal globular domain (Fig. 10A) (312). Adiponectin is subject to consequential post-translational modifications (PTMs) such as glycosylation and hydroxylation in the collagenous domain. After secretion from the adipocytes, adiponectin circulates in serum in several isoforms due to the presence of a cysteine residue in the variable region (187). This cysteine residue mediates the formation of a disulfide bond responsible for the formation of adiponectin oligomers from trimers (low molecular weight), hexamers (medium molecular weight), and 12-mers or 18-mers (high molecular weight [HMW]) (Fig. 10B). Although, adiponectin is produced mainly by adipose tissue (308), many reports have shown that it is also synthesized by several other sites of the body, including cardiomyocytes, skeletal muscle, osteoblasts, the placenta, and the pituitary gland (48, 191, 277, 295). Peripheral plasma concentration of adiponectin, which is normally in the range of 5–30 μg/ml, is relatively high in healthy nonoverweight subjects. In contrast to leptin, plasma concentration of adiponectin is lower in obese individuals than in lean subjects (7) (Fig. 7B). Thus, the circulating levels are lower in overweight and obese subjects and are inversely related to visceral fat, waist circumference, and BMI. Nonetheless, contrary to leptin, adiponectin is considered to be an anti-inflammatory adipokine that has many pleiotropic protective properties. For example, adiponectin protects against the development of many obesity-related diseases, including diabetes, cardiovascular, and fibrotic disorders, through its antidiabetogenic, antiatherogenic, proapoptotic, and antifibrogenic effects. In addition, data from a meta-analysis demonstrated that higher adiponectin levels are associated with a lower risk of type 2 diabetes across diverse populations (204). Accordingly, mice lacking adiponectin exhibit severe diet-induced insulin resistance, whereas mice overexpressing adiponectin remain insulin sensitive even after consuming a high-fat diet (216). Moreover, low serum adiponectin levels have been associated with an increased risk of certain malignancies, including breast, endometrial, prostate, and colorectal cancers (236, 262).

1. Adiponectin receptor
Adiponectin exerts its pleiotropic actions through adiponectin receptors (AdipoR1 and AdipoR2), which are ubiquitously expressed (Fig. 10C). Both receptors contain seven TMs, but are structurally and functionally distinct from G-protein-coupled receptors (382). Additionally, T-cadherin has recently been identified as another receptor by its ability to bind HMW oligomeric complexes (158). Adiponectin signaling involves the activation of AMP-activated protein kinase (AMPK), p38 mitogen-activated protein kinase (p38 MAPK), and PPAR-α (Fig. 8). However, the detailed mechanisms of actions of adiponectin still remain largely unknown. The adaptor protein-containing PH domain, PTB domain, and leucine-zipper motif (APPL1), positively mediates adiponectin signaling by interacting directly with both the AdipoR1 and AdipoR2 receptors (221), whereas APPL2, an isoform of APPL1, acts as a negative regulator of adiponectin signaling in muscle cells (366). Two recently published investigations go a step toward elucidating the adiponectin-signaling pathway mechanisms (151, 163). The first study demonstrated that adiponectin signaling promotes the activation of PPAR-γ coactivator-1α (PGC-1α) and mitochondrial biogenesis in myocytes using AdipoR1-deficient mice in skeletal muscle (muscle-Adipo R1KO). According to the authors, adiponectin induces extracellular Ca2+ influx, which is necessary for the activation of Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ), AMPK, and SIRT1 deacetylase. The authors suggested that the decreased levels of adiponectin and AdipoR1 receptor in obesity might induce mitochondrial dysfunction and insulin resistance, both of which are observed in diabetes. The other study showed that adiponectin stimulates ceramidase activity through its two receptors, AdipoR1 and AdipoR2 (151). The enhanced ceramidase activity induces ceramide depletion and releases sphingosine. This eventually generates sphingosine-1-phosphate, known as a potent antiapoptotic and anti-inflammatory metabolite, in contrast to which ceramide mediates apoptosis, inflammation, and insulin resistance. Thus, it was suggested that the anti-inflammatory actions of adiponectin are directly related to ceramide depletion, and that modulation of sphingolipid metabolism is crucial for the actions of adiponectin (151).
2. Adiponectin regulation
Adiponectin gene expression is greatly (50–100-fold) increased during differentiation of 3T3-L1 adipocytes, and the half-life of adiponectin ranges 2.5–6 h in the plasma (150, 267). Furthermore, its expression is influenced by hormonal, pharmacologic, and dietary status. At the transcriptional level, several transcription factors, including PPAR−γ, C/EBPα, and forkhead transcription factor O-1 (Foxo-1), induce adiponectin gene transcription (280). Factors such as TNF-α, androgens, β-adrenergic agonists, glucocorticoids, and c-AMP analogs inhibit adiponectin gene expression and/or secretion (85, 97, 176, 258). In contrast, PPAR-γ agonists such as TZDs have been associated with increased adiponectin concentration (Fig. 11) (217).

Unlike leptin, hypoxia decreased adiponectin promoter activity, which suggests that it has a suppressive effect on adiponectin transcription (153). Additionally, adiponectin is subjected to numerous regulations at transcriptional and translational levels. Indeed, it is subjected to many PTMs known to be decisive for its oligomerization and secretion. Among these PTMs, the glucosylgalactosyl-modified lysines on the collagenous domain are involved in the conformation and the stability of the HMW adiponectin (273). However, adiponectin sialylation at the N-terminus is not required for multimer formation or secretion, but is essential for regulating its clearance (291). Additionally, the increase in the levels of plasma adiponectin by pioglitazone, a PPAR-γ agonist, results from post-translational regulation corresponding to the formation of HMW adiponectin species (283). Adiponectin assembly of trimers, hexamers, or HMW is mediated through oxidative oligomerization of multisubunit protein complexes in the endoplasmic reticulum (ERT). Thus, impairment of adiponectin multimerization affects its secretion, and consequently, its function has been associated with diabetes (364). Other studies have demonstrated that adiponectin downregulation is related with ER stress, which is observed in obesity, and tunicamycin, an ER stress inducer, suppresses adiponectin mRNA expression. Moreover, a recently identified adiponectin interactive protein, disulfide-bond-A oxidoreductase-like protein (DsbA-L), is involved in adiponectin stability and secretion by enhancing ER function (394). The cellular levels of DsbA-L are reduced in obesity and may contribute to decreasing HMW adiponectin levels (207). Lastly, resveratrol, a polyphenol with antioxidative, anti-inflammatory, and anti-insulin resistance properties, stimulates adiponectin cellular levels and multimerization through the activation of DsbA-L (75, 365).
VI. Adipokine Signaling in Breast Cancer and Their Relation to COX and ROS
We now have ample of evidence suggesting that adipokines have distinctive tasks in pathophysiology (167). In addition to their biological activities, it is also firmly established that adipose tissue interferes with breast cancer development, and obesity is closely associated with a higher risk of cancer development in different tissues, including the colon, prostate, and breast (particularly in postmenopausal women) (11, 124, 248). An essential part of the adipose tissue in this context is the breast glandular tissue, undifferentiated fibroblasts, matured adipocytes, and macrophages. Consequently, the adipose tissue microenvironment is an integral component in breast cancer development. Several laboratories have demonstrated that leptin and its receptor are not usually expressed in normal cells from healthy breasts. In contrast, they are clearly expressed in benign cell lesions such as flat epithelial atypia and atypical ductal hyperplasia. Leptin is detected in invasive ductal carcinoma (IDC) and ductal carcinoma in situ (DCIS) and in the healthy tissue surrounding the tumors, and is positively correlated with ERα expression (Fig. 12) (45, 161). A recent study indicated that positive rates of leptin and ObR receptor expression in the DCIS were significantly higher than those of the IDC, and that leptin expression was associated with high Ki-67 expression (169). Adiponectin has been identified in only 15% of breast tumors (Fig. 13) (166), and more recently, other workers demonstrated that adiponectin and AdipoR receptor expression was significantly higher in IDC than in to DCIS, and that high adiponectin expression was correlated with smaller tumor size and lower T-stage (169). All these physiological and pathological properties are clearly related to adipokine-signaling cascades, including diverse pathways of inflammation, as discussed below.


A. Leptin/adiponectin signaling
Leptin is considered to be one of the putative links between obesity and cancer development because of its high circulating levels in obese women. It has been observed to stimulate the proliferation of various benign and malignant cells, including breast, prostate, colon, gastric, and lung cell lines (89, 95, 104, 157, 167, 211, 265, 266, 336), via innumerable signaling pathways (168). A signaling pathway of leptin and adiponectin is given in Figure 8. Additionally, leptin shows proangiogenic actions, and stimulates the migration and invasion of different cancer cell lines, including the human breast cancer cell lines MDA-MB-231 (ERα-negative human breast cancer cell line) and MCF-7 (ERα-positive human breast cancer cell line) (117, 307). The involvement of leptin in breast carcinogenesis results from their antiapoptotic effect, which is undesirable in normal homeostasis in healthy circumstances. In the MCF-7 cell line, leptin reduces Bax expression and conversely increases the expression of the antiapoptotic genes Bcl2 and survivin in favor of cell survival (259, 274). The different leptin-signaling pathways involve several canonical and noncanonical signaling pathways promoting survival, cellular proliferation, mitogenesis, and angiogenesis (Fig. 8) (107, 168). A previous study demonstrated that leptin upregulates several genes associated with cell cycle and proliferation, DNA synthesis, and extracellular matrix proteins in the MCF-7 cell line (274). For example, it has been reported to increase the expression of cyclin D1 and c-Myc in human breast cancer cells (60). Conversely, the expression of genes associated with inhibition of proliferation and apoptosis, such as the tumor suppressors p53 and p21WAF1/CIP1, was downregulated in response to leptin (60).
A recent study showed that the proangiogenic effects of leptin in mouse mammary cancer cells mainly involved upregulation of vascular endothelial growth factor (VEGF) via HIF-1α and NFκB (395). Intriguingly, HIF-1α can also induce leptin expression itself in breast cancer cells and adipocytes (53, 129), indicating the existence of a feedback loop between leptin and HIF-1α. Recently, Guo and Gonzalez-Perez showed the Notch, IL-1, and leptin crosstalk outcome. They demonstrated that the leptin proangiogenic effects, via upregulation of VEGF/VEGFR-2, are mediated by leptin-induced Notch expression in breast cancer (130). Furthermore, interleukin-1α (IL-1α), one of the major proinflammatory cytokines, stimulates the expression of leptin when it is produced at high levels in breast cancer cells (194). Several studies have demonstrated the existence of functional crosstalk between leptin and HER2/neu. Specifically, leptin exerts its activity through activation of HER2 signaling. For example, in SK-BR-3 breast cancer cells, leptin treatment increased HER2 phosphorylation and transactivated HER2 via the epidermal growth factor receptor HER1 and JAK2 pathways (100, 336). Equally, the leptin/ObR system is coexpressed with HER2 in a large fraction of breast cancers and might contribute to enhanced HER2 activity and resistance to anti-HER2 treatments, as does trastuzumab. Further, cotreatment of leptin and insulin-like growth factor-1 (IGF-1) significantly increases the proliferation and invasion and migration of breast cancer cells. A bidirectional crosstalk between leptin and IGF-1 signaling that transactivates epidermal growth factor receptor (EGFR) was observed in breast cancer when inhibition of EGFR activation potently inhibited leptin- and IGF-1-induced invasion and migration of breast cancer cells. Several other mechanisms driving crosstalk between leptin signaling and multiple oncogenic pathways in breast cancer have been described and are recently reviewed elsewhere (131).
As mentioned previously, leptin exerts its action through interaction with the long-form Ob-Rb receptor (Figs. 6B and 8). The Ob-Rb is believed to be primarily responsible for active signal transduction to various biochemical cascades. Moreover, leptin and its receptors are overexpressed in 70%–80% of breast cancer incidents, but are absent in the normal epithelial breast tissues of healthy subjects (Fig. 12) (45, 116, 161). In addition, high levels of leptin have been associated with increased incidence of breast cancer metastasis and poor prognosis of the disease (73). However, tumor surgical excision did not influence circulating leptin levels in patients with breast cancer, suggesting that the tumor leptin production is minor (62). In postmenopausal breast cancer patients, serum leptin levels and BMI were significantly increased in patients with higher pathological tumor classification and stage and in the presence of distant metastases (214).
The clinical study of Ishikawa et al., confirmed for the first time a direct link between leptin signaling and human breast cancer (161). Since then, countless in vitro and in vivo studies in humans or experimental animal models have corroborated this direct association. Indeed, many experimental animal models of breast cancer have been developed to study the mechanistic link between distinctive key molecules that are involved in the process. Tables 2 and 3 describe several mouse models of breast cancer in which of leptin or adiponectin levels either are manipulated by dietary, genetic, or pharmacological means. These models suggest that leptin and adiponectin could play a role in breast cancer development. Mouse mammary tumor virus (MMTV), which was discovered as a milk-transmitted, infectious cancer-inducing agent in the 1930s, has been frequently used to create mouse models of breast cancer. Mice that overexpress human transforming growth factor (TGF)-α under the control of the MMTV promoter (MMTV-TGF-α mice) were bred onto the leptin-deficient and LepRb-deficient genetic background (71, 72). Although serum leptin levels of MMTV-TGF-α/LeprdbLeprdb mice were 12–20-fold higher than levels of lean MMTV-TGF-α/Lepr+Lepr+ mice, the leptin-deficient mice did not develop mammary tumors because they lacked ductal mammary epithelium. From these observations, it was concluded that LepobLepob and LeprdbLeprdb were not suitable for the study of mammary tumorigenesis. However, genetically modified db/dbNse/Nse mice, where central leptin signaling is reconstituted only in the brain, do not develop obesity and diabetes (69). The db/dbNse/Nse mice were crossed with a mammary tumor model, such as the MMTV-PyMT. The PyMT/db/dbNse/Nse mice developed smaller tumors with reduced PI3K, ERK1/2, and Jak2/STAT3, demonstrating that leptin signaling through its receptor promotes tumor growth (269). In addition, several models of diet-induced obesity in mice show that the mammary tumor weight and/or volume is higher in animals given a high-fat diet than in mice on a normal or restricted diet, and which coincides with the rise in plasma leptin.
CR, caloric-restricted diet; ERK, extracellular signal-regulated kinases; HFD, high-fat diet; LFD, low-fat diet; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; NRD, normal diet; ND, not determined; STAT, signal transducer and activator of transcription; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; WT, wild-type.
MMTV, mouse mammary tumor virus; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homolog deleted on chromosome 10.
The lower adiponectin levels observed in obese women are believed to be associated with increased risks of breast cancer, but the exact molecular mechanisms underlying this association are not fully elucidated (190, 220, 357, 372). Likewise, tumors in women with low serum adiponectin levels are more likely to show a biologically aggressive phenotype. Different breast cancer cell lines express one or both of the adiponectin receptors, and adiponectin could essentially block the proliferation of several different types of human breast cancer cells, including MDA-MB-231, T47D, and MCF-7 cells (6, 190). However, another study demonstrated that the effects of adiponectin on the proliferation and apoptosis of human breast cancer cells are dependent on 17−β estradiol levels (275). The antiproliferative activity of adiponectin has also been demonstrated in other cell lines, including endometrial cancer, gastric cancer, and prostate cancer cells. Nevertheless, adiponectin is able to promote survival and enhance proliferation in other cell types such as cardiac cells that protect against ischemia–reperfusion injury and cardiac fibrosis (316). In addition, adiponectin inhibits proliferation in breast cancer cells through the activation of AMPK and inhibition of Akt and could negatively regulate the Wnt/beta-catenin pathways in MDA-MB-231 human breast carcinoma cells (Fig. 8) (371). Further, adiponectin treatment enhanced the mRNA and protein expression of a Wnt antagonist, WIF1, which might at least partially mediate the inhibitory effects of adiponectin on Wnt/beta-catenin signaling. It has been suggested that epigenetic regulations are involved in the stimulatory effects of adiponectin on WIF1 expression, but the molecular mechanisms involved in this regulation remain unknown (206). The impact of reduced adiponectin expression on mammary tumor development has been evaluated in different mammary cancer mouse models (Table 3). Adiponectin-haplodeficient MMTV-PyVT mice show earlier tumor onset and accelerated tumor growth compared to those with MMTV-PyVT/adiponectin(+/+) (200). However, MMTV-PyVT/adiponectin knockout mice exhibited tumor growth retardation, due to vascular starvation of the tumors (201). Restricted diet slightly increased plasma adiponectin levels and decreased tumor progression in a 4T1 murine mammary carcinoma model implanted in syngeneic Balb/c mice (83).
B. Adipokines in relation to COX and ROS
The two adipokines, the leptin, which is also a mitogen, and adiponectin, an antimitogenic protein, appear to play a vital role in breast cancer, in which the impact of inflammation and oxidative stress might be key pathological networking elements that act on the initiation of angiogenic processes. Figure 14 shows a hypothetical interaction model of adipokines, aromatase/estrogens, and eicosanoids in breast cancer. However, the orchestrated mechanisms of action of adipokines, aromatase/estrogens, and eicosanoids in breast cancer have not yet been clearly elucidated, and their coherent existence in human breast cancer microenvironment has not been established. Accordingly, the two primary pathologies, inflammation and oxidative stress, are closely connected (14), and they are perhaps the crucial cornerstones of cancer in general. While these two pathologies and the biological role of adipokines seem to be decisive, they have only rarely been investigated together in breast cancer studies. A local function of the adipocyte microenvironment and chronic oxidative stress and inflammation in the mammary gland may also be important in breast cancer. The mammary epithelial tissue, either healthy or tumoral, is directly in communication with fat cells, and the intimate interactions between these cellular types involve not only adipokines but also local proinflammatory mechanisms and processes of oxidative stress that could stimulate angiogenesis and cell proliferation. Leptin can transform the microenvironment mainly through its ability to potentiate both the migration of endothelial cells and angiogenesis and to sustain the recruitment of macrophages and monocytes, which subsequently release VEGF and proinflammatory cytokines (4). The proinflammatory cytokines are already known to be related to both COX and ROS production through activation of NFκB. As a consequence, an obscure role of inflammatory eicosanoids in epithelial-derived tumors and their microenvironment is well recognized (368, 369).
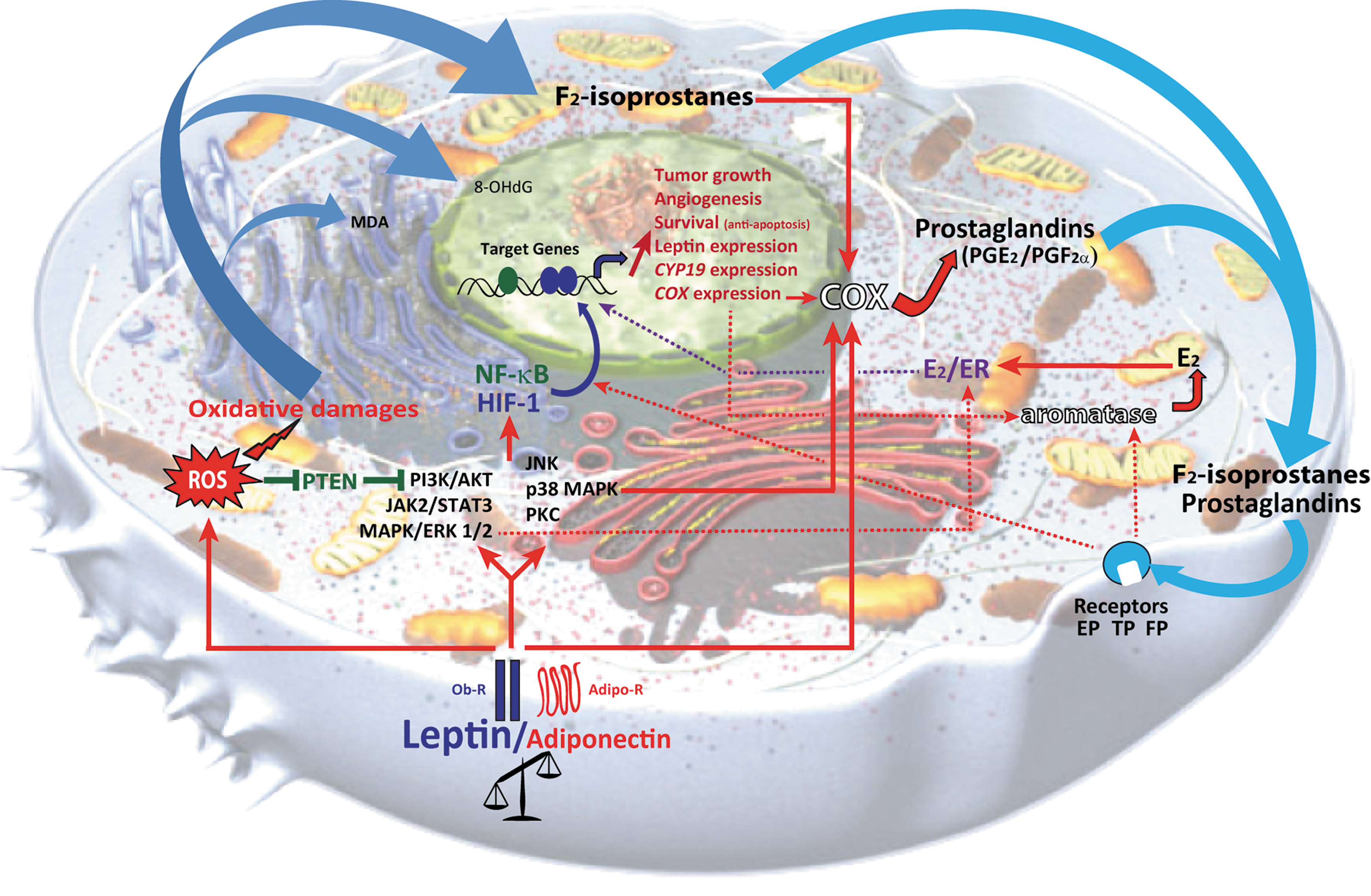
ROS, and hence oxidative stress, seem to be an important characteristic feature in cancer pathogenesis according to numerous investigations during the last 30 years. However, only a few studies have shown the relationship between adipokines and ROS. Augmented oxidative stress may instigate deregulated production of leptin and adiponectin in cultured adipocytes, and leptin induces oxidative stress in human endothelial cells (33, 111). Exogenous leptin increases lipid peroxidation/oxidative stress and inhibits the endogenous antioxidant system in the mouse brain (197). In addition, higher levels of systemic oxidative stress (as measured by F2-isoprostanes) and inflammatory biomarkers (CRP, IL-6, and TNF-α) have been shown to correlate with lower levels of adiponectin in obese adolescents (having high insulin resistance and high BMI), indicating that obesity is closely linked to adipokines and chronic oxidative stress and inflammation (Table 4) (329). When the relationship between systemic oxidative stress and body fat distribution, adipokines, and inflammatory markers was estimated in middle-aged healthy women, BMI and waist circumference were positively correlated with a major F2-isoprostane, leptin, and high-specific C-reactive proteins (hsCRP), and PAI-1 was also positively correlated to 8-epi-prostaglandin F2α (377). Circulating leptin was positively correlated with urinary F2-isoprostane in nondiabetic hypercholesterolemic patients, but no relationship with adiponectin was observed (317). Significant relationships were established among obesity-related indices, oxidative stress, inflammation, and adipokines in other studies (253). Hyperleptimia induced by exogenous leptin administration in rats increased urinary isoprostanes and decreased paraoxonase-1 and the platelet-activating factor acetylhydrolase (26). In adolescents, leptin concentration was positively related to low-density lipid oxidation (318). Similarly, elevated oxidative stress as evaluated by hydroperoxides, conjugated diene, and carbonyl proteins together with high leptin levels were found among breast cancer patients compared to controls (10). Likewise, exposure of endothelial cells to leptin led to ROS formation (64, 330, 380), indicating that leptin may induce F2-isoprostane production through ROS from esterified arachidonic acid in the membrane. More specific studies are required to elucidate these aspects in breast cancer.
T, thin; H, heavy; IS, insulin-sensitive; IR, insulin-resistance (n=numbers) [adapted From Ref. (329)].
8-iso-PGF2α, 8-Iso-prostaglandin F2α; IL-6, interleukin-6; PGF2α, prostaglandin F2α; TNF-α, tumor necrosis factor-α.
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a phosphoinositide-3-phosphatase, is a tumor suppressor that is not expressed or inactivated in many advanced cancers, including breast cancer (203, 353). PTEN negatively controls the PI3K/Akt-signaling pathway by catalyzing the dephosphorylation of phosphatidylinositol 3,4,5-trisphosphate (PIP3) at the D3 position, resulting in phosphatidylinositol 4,5-bisphosphate (PIP2). Lower PTEN expression and/or activity results in elevated activation of the PI3K/Akt-signaling pathway, which induces high cell survival and proliferation (49). ROS/H2O2 generally mediates PTEN inactivation by forming a disulfide bond within the active site, rendering it catalytically inactive. For instance, exposure of MCF-7 cells to H2O2 oxidizes and inactivates PTEN, which triggers hyperactivity of the PI3K/Akt-signaling pathways (76, 77). A previous study showed that oxidation of cellular PTEN involves enzymatic peroxidation of arachidonic acid by COXs in pancreatic cancer cell lines (77). In addition, leptin can affect PI3K activity and intracellular cAMP concentration by inhibiting PTEN (Fig. 8) (257). In view of these observations, inactivation of PTEN may be achieved via leptin, ROS production, and enhanced COX-2 expression, and therefore the role of PTEN in breast cancer pathology is an important factor.
The involvement of leptin in inflammation is provided by studies performing regulation of macrophages, monocytes, and T-cell signaling (209, 385). Leptin promotes COX-2 induction and PGE2 production in adenocarcinoma cells and macrophages (261, 282) and produces proangiogenic effects in human endothelial cells through p38MAPK and PI3K/Akt signaling (117). One in vitro study showed that in MCF-7 cells, leptin treatment induced COX-2 upregulation (284). However, in other cell lines, as in PC-3, a human prostate cancer cell line, PGE2 induced stabilization and nuclear localization of HIF-1α (208). It is suppressed by NS398, a NSAID and a selective COX-2 inhibitor, demonstrating that the effect of NS398 on HIF-1α seems to be COX-2/PGE2 dependent (392). Thus, we can assume the existence of a link between leptin and COX-2/PGE2 through HIF-1α in breast cancer cells. Leptin is known to promote COX-2 induction in macrophages, adenocarcinoma cell lines, and in human vascular endothelial cells (117, 122, 261, 282), and it preferentially stimulates PGE2 and PGF2α production in rat hypothalamic cells. This latter mechanism of action of leptin as a proangiogenic factor is possibly responsible for a molecular link between angiogenic response and leptin stimulation. Further, leptin stimulated cell proliferation and induced functional activation of COX-2 through JAK/STAT3-, MAPK/ERK-, and PI3K/AKT-dependent pathways in human endometrial cancer cells (115). Thus, leptin may promote cell proliferation in breast cancer cells through a similar mechanism, and hence it may serve as a critical factor in breast carcinogenesis in obesity. To date, there have been few in vivo studies associating the proinflammatory effects of leptin and COXs and related PGs in breast cancer.
C. Leptin and estrogens: link to inflammation
Breast cancer is a multifactorial disease among women, and obesity is one of the main primary risk factors, especially in menopausal women. Estrogens and family history with genetical inheritance appear to be the two additional major risk factors. Estrogens perform a substantial role in breast cancer development, specifically in estrogen receptor-positive breast cancer in postmenopausal women. This association is partially due to the increase in estrogen production by fat cells. The ovaries are generally the main source of production of estrogens through the conversion of androstanedione to estrogens, which are released into the peripheral circulation, and hence regulate various fundamental human reproductive functions throughout the premenopausal period in women (103, 339, 346).
Following menopause, the ovaries profoundly shrink estrogen synthesis. This process then mainly occurs in the adipose tissue, whose mass increases with obesity and in which leptin-influenced aromatase activity plays a strategic role in breast cancer development. Aromatase is an integral membrane enzyme that catalyzes the removal of the 19-methyl group and aromatization of androgens to estrogens. Aromatase activity is generally higher in human breast cancer cells (estrogen receptor-positive) than in normal breast tissues (66, 328). Consequently, a concurrent correlation may exist in the cellular microenvironment between breast tumor alignment and the aromatase-mediated estrogens in estrogen receptor-positive breast cancer in postmenopausal women. Further, estrogen levels in the breast cancer tissues of these women have been shown to be higher than peripheral levels (37, 315). This possibly has a significant impact on the tumor progression, and hence aromatase inhibition, which subsequently controls estrogen production, and would become a therapeutic advantage in the treatment of breast cancer in postmenopausal women after initial treatment (39, 120).
Estrogens are thought to act through nuclear estrogen receptors (ER), of which there are two known subtypes, namely ERα and ERβ. In previous studies, our laboratory and others demonstrated that Ob-R expression is positively correlated with ERα expression in primary breast carcinoma (113, 166). Leptin has been shown to activate ERα through the MAPK pathway in breast cancer cells (55). Leptin stimulates aromatase gene expression through Stat3 activation in MCF-7 cells and enhances the sensitivity of this cell line to estrogens through the upregulation of ERα (54, 89, 113). Additionally, a significant association between CYP19 gene expression and COX-2 gene has been reported in breast cancer (38, 40).
Several studies have suggested that a mechanistic link might exist between leptin signaling, inflammation, and estrogen synthesis. Initially, leptin through Stat3 activation stimulates aromatase gene expression in MCF-7 cells (54, 89). Subsequently, elevated levels of Stat3 phosphorylation are associated with an increase in COX-2 in immortalized breast epithelial cells and in invasive breast cancer tissues (155, 337, 338). In addition, COX-derived PGE2 or exogenous PGE2 increases aromatase expression and activity in breast tissue, possibly through macrophage-derived proinflammatory mediators (342). Further, saturated fatty acids, which have been associated to obesity-related inflammation, stimulated NFκB activity in macrophages, leading to enhanced levels of TNFα, IL-1β, and COX-2, and they all contributed to the induction of aromatase in preadipocytes (341). Hence, a sequence of reactions may ultimately occur in the breast tissues when malignant: Leptin→Stat3 activation→increase COX-2/PGE2→increase aromatase→increase estrogens→ERα expression→increase leptin Stat3 activity. Consequently, all these dynamic parameters relate to a codependent alliance in which NFκB, leptin, aromatase, estrogens, and COX may have a central role in breast cancer development and progression. Unlike leptin, adiponectin has been identified as a protective adipokine against breast cancer due to its antiproliferative, anti-inflammatory, antiangiogenic, and proapoptotic effects (168). The signaling antagonism pathways of leptin and adiponectin in breast cancer are summarized in Figure 8.
It was earlier shown that aromatase expression and activity in breast cancer patients are higher in the adjacent tumor tissue than in the normal breast tissue (42). A strong association between aromatase and COX gene expression has also been described (41). In addition, NSAIDs and selective COX inhibitors can efficiently suppress CYP19 gene expression and reduce aromatase activity. A recent study in Lancet Oncology included a consensus statement on breast cancer prevention (79). The authors indicated that results-of-trials aromatase inhibitors, which are pending, might show them to be efficacious. Additional agents that are presented to this statement such as aspirin, NSAIDs, COX-2 inhibitors have limited therapeutic effects or are in the early phase of study evaluation.
Taken together, an unbalanced eicosanoid axis oxidative stress and inflammation as a result of NFκB activation, adipokines, and aromatase/estrogens could form an important mechanistic link to breast cancer pathology. The study of adipose tissue and its vital secretory products together with inflammation and oxidative stress interactions are now emerging as a critical step in a better understanding of the mechanisms by which obesity could influence the growth and proliferation of tumor stroma and malignant cells.
VII. Clinical Studies on Breast Cancer Focusing on Obesity, Eicosanoids, and Adipokines
As mentioned earlier, obesity and overweight have a substantial impact on the risk of breast cancer primarily involving hyperinsulinemia, deregulation of adipokine levels, and various known and unknown inflammatory promoters. Although obesity related to nutrition is a well-acknowledged risk factor for postmenopausal breast cancer, the mechanisms underlying the association are still unclear. Obese and overweight patients generally have a worse prognosis of survival compared to normal-weight breast cancer patients. A recent study assessed whether low levels of adiponectin and a greater level of insulin resistance are associated with breast cancer mortality and all-cause mortality (92). Elevating homeostasis model assessment-estimated insulin resistance (HOMA) scores were correlated with reduced breast cancer survival and with reduced all-cause survival after adjustment for potential confounders. Higher levels of adiponectin were related to longer breast cancer survival after adjustment for different covariates. However, a continuous determination of adiponectin was not associated with either breast cancer-specific or all-cause mortality.
In a nested case–control study of postmenopausal breast cancer patients and controls in a cohort with prospectively collected serum samples, lower levels of adiponectin and higher levels of PAI-1 were correlated with increasing levels of estradiol, decreasing levels of sex hormone-binding globulin, and increasing BMI (118). Breast cancer risk was not related to circulating levels of adiponectin. The association was not confounded by BMI, parity, age at first full-term birth and age at menopause, current postmenopausal hormone use, and circulating sex steroid hormones. Additionally, adipokine associations were not modified by BMI. The lack of association with risk assessment may be due to a measurement error of the laboratory assays, as suggested by the authors. Although dissimilar, these studies collectively demonstrate that there is a link between low levels of adiponectin and increased breast cancer mortality in breast cancer survivors.
Increased serum levels of leptin, overexpression of leptin, and its two main receptor isoforms have been documented in breast cancer patients. In a recent study, the relationship between tissue leptin levels and breast cancer was evaluated (177). Mean tissue leptin levels in breast cancer tissue samples were significantly higher than those found in normal breast tissue. Nonetheless, no correlation was found between tissue leptin levels and menopausal status, hormone receptor and HER2/neu status, lymph node involvement, and histopathologic characteristics.
VIII. Unifying Theory of Eicosanoids and Adipokines in Breast Cancer
Endogenously constituted controlled inflammation and free-radical formation in homeostasis are the absolute fundamental requirements of our existence. Once unbalanced, they may potentially act to gradually induce chronic inflammation and initiate inflammatory tissue modifications in the local microenvironment in a lingering fashion unless alleviated therapeutically. This elementary reaction persists in the breast tumor milieu. Some of the primary eicosanoids originated through COX and ROS catalyzation of arachidonic acid such as PGE2, PGF2α, F2-isoprostane, and 8-iso-PGF2α are basically proinflammatory, whereas others, PGA2, PGD2, PGJ2, and 15d-PGJ2, appear to have anti-inflammatory and antiangiogenic properties. Their hypothetical crosslink with adipokines and aromatase/estrogens, although less well documented, is vital to understand their role in breast cancer and requires further systematic investigation. COX-2 is overexpressed in breast cancer (40, 270, 271), and is associated with aromatase gene expression in human breast cancer and provides additional evidence on COX-related estrogen formation locally (41) and its networking to adipokines and ROS. In addition, neither NSAIDs nor COXibs have shown a clear beneficial effect, for many possible reasons such as inhibition of all PGs instead of those specific ones related to the disease, distinctive subtypes of breast cancer, duration of disease, therapeutic dose, and specific types of these drugs, and more importantly, at which phase of disease and grade of tumor these types of drugs should be administered to achieve the utmost beneficial effect. Nonetheless, the role of aromatase inhibitors and hormone therapy is at present indisputable in the breast cancer pathology. Mutually, it compels across-the-board preclinical, experimental, and clinical studies to determine the impact of eicosanoids of the COX and ROS origin and of adipokines/cytokines in conjunction with aromatase-induced estrogen synthesis, and thus the mechanistic role of inflammation and oxidative stress in human breast cancer. Collectively, more research would plausibly facilitate in identifying the key molecules of disease-origin, including the underlying disease mechanism and subtype variance, and that could open up the avenue for future drug development in hormone-dependent breast cancer.
IX. Conclusion and Future Prospects
Eicosanoids derived through COXs and ROS from arachidonic acid together with adipokines/cytokines and aromatase/estrogens conceivably have specific roles in mammalian carcinogenesis and hormone-dependent breast cancer development. Although most human breast cancers are hormone-dependent, therapeutic approaches should include the development of specific antagonists to regulate estrogen synthesis. However, much will inevitably be overlooked in the future prevention and treatment of breast cancer, because inflammation originates via multifactorial events. This review considers that these factors should be simultaneously investigated for a fuller understanding of the disease. Elucidating their role in molecular mechanisms and providing new data could form the theoretical basis of therapeutic opportunities for breast cancer chemoprevention. This review describes a novel model of interactions that exist between the significant pathways of chronic inflammation via eicosanoids and their relation to adipokines in breast cancer. These potential parameters remain to be critically evaluated from molecular endpoints to clinical applications and could have a therapeutic impact on future cancer prevention and treatment.
A comprehensive study of the role of adipokines in relation to inflammatory and anti-inflammatory eicosanoids and to estrogens at the microenvironmental, experimental, and clinical levels are needed to understand the pathology of breast cancer and to assess future treatment.
Footnotes
Acknowledgments
The authors are indebted to the Conseil Regional d'Auvergne and the University d'Auvergne, Clermont-Ferrand 1, France, for a Chaire d'Excellence Research Grant to support the work. Authors also apologize for not mentioning many excellent publications in the field due to the limited space.