
Abstract
Introduction
This review focuses on macroautophagy (hereafter referred to as autophagy), which in the decade following the rapid identification of more than 30 genes belonging to the autophagic machinery in yeast screens (33), has emerged as a global regulator of cellular signaling. Autophagy is widely known to serve in the constitutive turnover of proteins and organelles. Moreover, during conditions of limited nutrient availability, nonessential cellular material is subjected to autophagic degradation in order to regain macromolecules for the synthesis of vital cellular components. The role has expanded to include the regulation of a wide range of other stress signals, including redox stress, calcium, damaged organelles, and intracellular pathogens (54). Furthermore, autophagy is implicated in development, tissue homeostasis, programmed cell death, innate immunity, pathogen defense, aging, and disease (54). Consequently, autophagy can no longer be considered as a general concept, but rather as a highly regulated process with distinct modes of degradation of specific substrates. Below, fundamental mechanisms of autophagic processes and regulatory events underling specificity are considered and discussed.
Molecular Mechanism of Autophagy
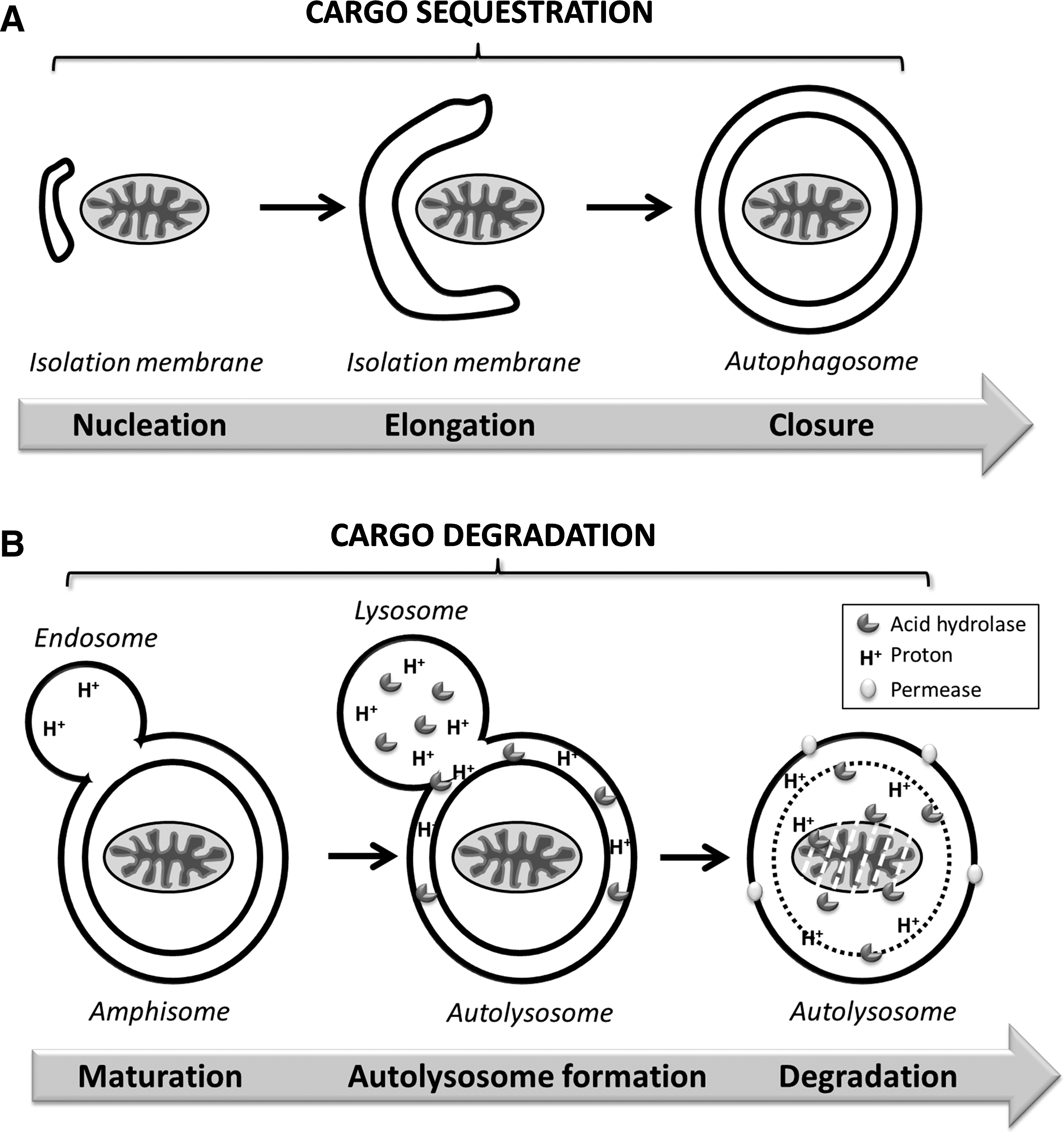
The autophagic process comprises a dynamic flow of formation and degradation of autophagosomes (autophagic flux) and can be divided into distinct stages (Fig. 1) (33). Following initiation of autophagy, the isolation membrane or phagophore is formed (nucleation). This pre-autophagosomal, crescent-shaped structure then engulfs the autophagic substrates through elongation and fusion to a double-membrane vesicle called autophagosome. Multiple fusion events with endosomes and finally lysosomes lead to maturation into the autolysosome, wherein the autophagic substrates together with the inner autophagosomal membrane are degraded by lysosomal hydrolases. Permeases in the lysosomal membrane allow for recycling of macromolecules. Each stage is highly regulated, and successful completion of the autophagic process can be interrupted at any stage.
Autophagy Induc tion
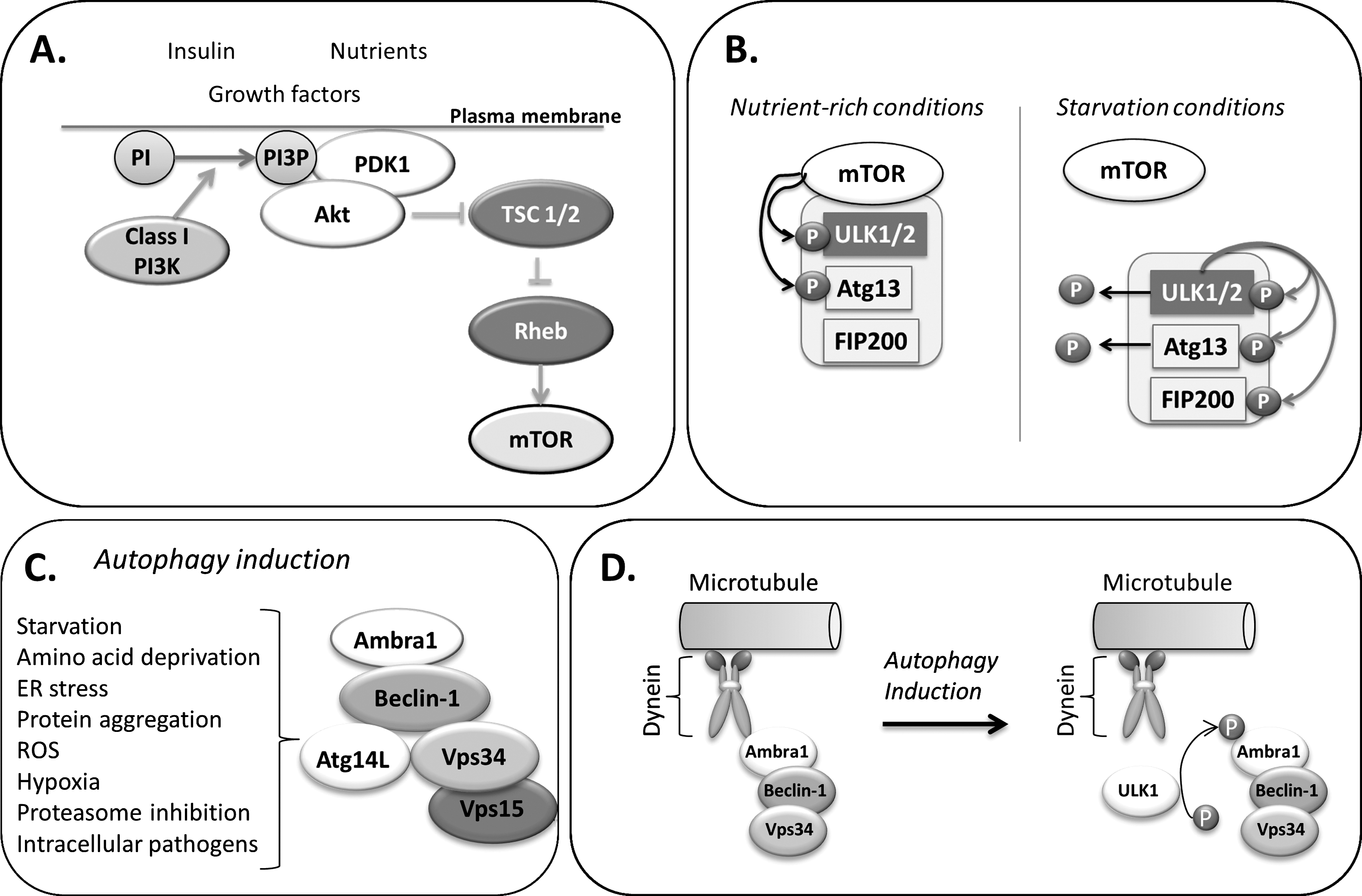
Autophagy induction is tightly regulated by the opposing actions of two distinct phosphatidylinositol 3-kinases (PI3K). The class III PI3K (PI3KC3/Vps34) positively regulates autophagy induction, while class I PI3K negatively regulates autophagy (see review (54)). Three main signaling complexes are controlling autophagy induction (Fig. 2).
Class I PI3K.Akt.mTORC1 complex
Cellular sensing of nutrient and growth factor availability, energy levels, and stress signals is mediated by the mammalian target of rapamycin (mTOR), a serine/threonine kinase and central downstream effector of the class I PI3-K/Akt pathway (Fig. 2A) (for a comprehensive review, see (54)). mTOR participates in two signaling complexes (C1 and C2), with the rapamycin-sensitive mTORC1 positively regulating anabolism (i.e., protein synthesis) and negatively regulating catabolism (i.e., autophagy). Under growth promoting conditions, generation of PI3P catalyzed by class I PI3K at the plasma membrane recruits both Akt and its activator phosphoinositide-dependent protein kinase 1 (PDK1). Active Akt phosphorylates to inhibit the tuberous sclerosis complex (TSC) 1/2, thereby relieving suppression of mTOR activator Rheb. Active mTORC1 promotes anabolism via activation of mRNA translation. S6 kinase 1 (S6K1), ones activated by mTORC1 phosphorylates factors involved in the translational machinery, such as the ribosomal protein S6 and eukaryotic elongation factor 2 kinase (eEF2K). In addition, mTORC1 phosphorylation deactivates the initiation factor eIF4E-binding protein 1 (4E-BP1) which then dissociates from the initiation factor eIF4E, thereby allowing for mRNA translation.
Conditions of low nutrient and growth factor availability result in mTORC1 inhibition and consequently promote autophagy initiation. Indeed, mTOR inhibitor rapamycin induces autophagy, even in nutrient-rich conditions (33). However, although the control of mTORC1 over autophagy is primarily shown to be of suppressive nature, recent reports reveal that mTORC1 signaling can also be essential for autophagy, as both rapamycin and knockdown of S6K1 blunted the autophagic response to genotoxic stress in cancer cells (73). Moreover, mTOR-independent pathways have been reported in which upregulation of autophagy is uncoupled from mTOR activity. For instance, autophagic clearance of protein aggregates can be activated without changes in the activation status of mTOR (57) and involve calcium and cAMP pathways (71).
ULK1.Atg13.FIP200 complex
mTORC1 exerts negative control over autophagy via acting on the autophagy initiating multiprotein complex, which contains ULK1/2 (unc-51-like kinase 1/2), the mammalian homologues of the serine-threonine kinase Atg1, FIP200 (focal adhesion kinase family interacting protein of 200 kDa), and Atg13 (for a comprehensive review, see (54)). Within this complex, Atg13 mediates the interaction of ULK1 with FIP200 (31). Stability and basal phosphorylation of ULK1 and Atg13 is enhanced by Atg101, a further member of the complex (44). Under conditions of rich nutrient supply, mTOR is associated with the complex and phosphorylates ULK1 and Atg13, leading to their inactivation (22). During autophagy-inducing conditions, inhibition of mTOR results in its dissociation from the complex and dephosphorylation of ULK1 and Atg13 (Fig. 2B). Moreover, the cellular energy sensor AMPK can directly activate ULK1 via phosphorylation (32). Active ULK1 phosphorylates to activate Atg13, FIP200, and itself. The activated multiprotein complex then localizes to the nucleation site of the isolation membrane and is responsible for the recruitment of the autophagic machinery, whereby the exact mechanism is largely unknown. Interestingly, a recent study describes ULK1/2-independent autophagy induced by enhanced amino acid metabolism, further adding to the high variety of autophagic programs (5).
Class III PI3K/Vps34–Beclin-1.p150 complex
The core of the second multiprotein complex involved in autophagy induction consists of class III PI3K/Vps34 and Beclin-1 (Fig. 2C). Beclin-1 is the mammalian functional homologue of yeast Atg6/Vps30 and is essential to autophagy induction (for comprehensive reviews see (21, 54)). When complexed with Beclin-1, the generation of the sole product of class III PI3K/Vps34, phosphatidylinositol 3-phosphate (PI3P), mediates the induction of autophagy. Inhibition of PI3K activity with 3-methyladenine or wortmannin disables autophagy (33). Although the precise function of PI3P is still unresolved, it is likely that PI3P attracts effector proteins to the forming autophagosomal membranes. As such, WIPI-1 (mammalian Atg18 homolog) was found to bind to PI3P and be recruited to autophagosomes (52).
Autophagy induction by the Vps34–Beclin-1 complex additionally depends on the presence of Atg14L/Barkor, Vps15, and Ambra1 (for review see (21)). Beclin-1 interacts with Atg14L/Barkor via coiled-coil-domain interactions (65). Ambra-1 (activating molecule in Beclin1-regulated autophagy) is a necessary promoting factor for the interaction of Beclin-1 with Vps34 (14). Via binding to dynein, Ambra1 anchors the presumably inactive Beclin1-Vps34 complex to the microtubules (10). Upon phosphorylation by ULK1, Ambra1 dissociates from dynein and translocates to the ER together with the Vps34–Beclin1 complex to initiate autophagic nucleation (Fig. 2D).
Substrate Sequestration
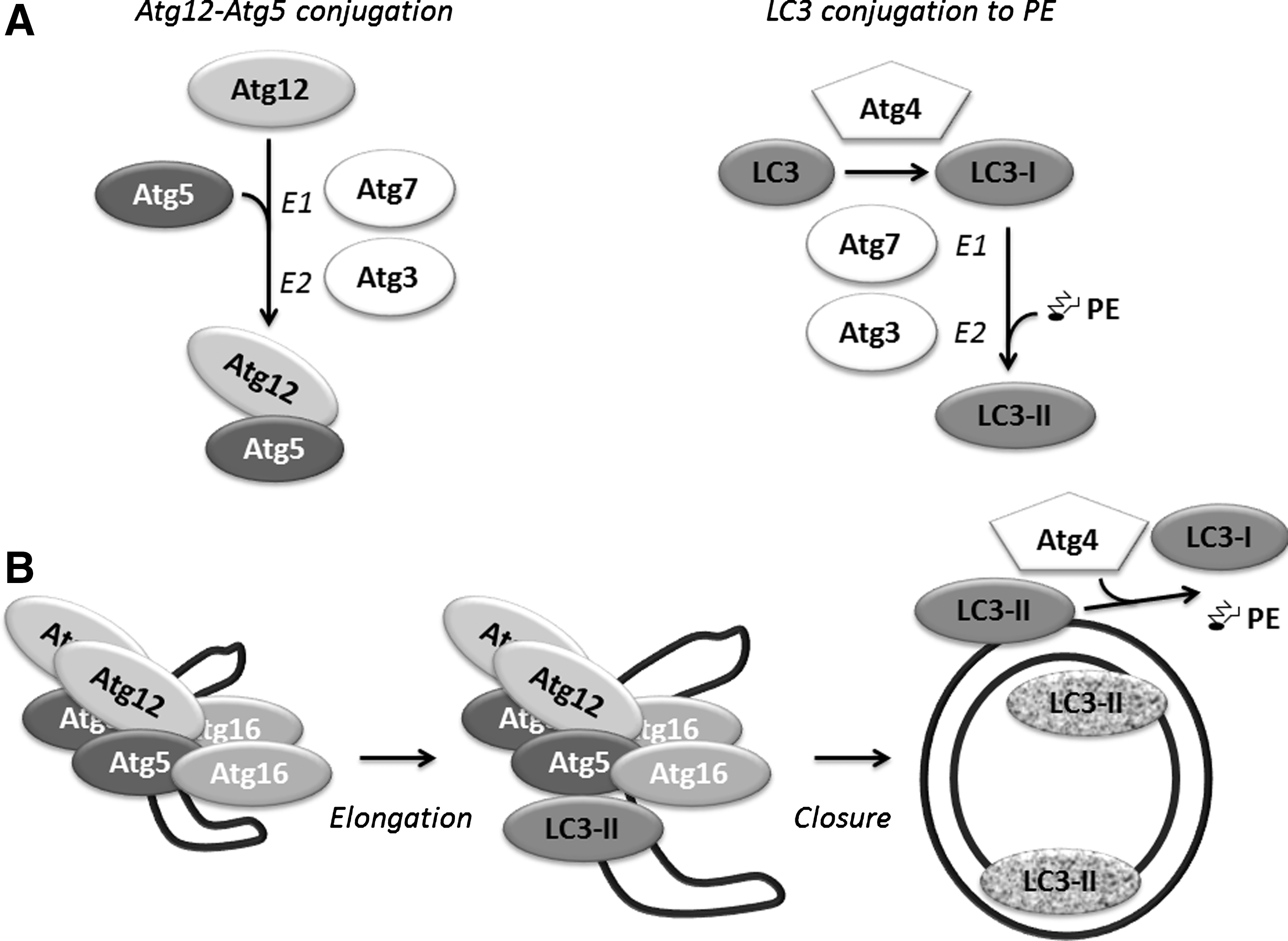
Following induction, cytosolic substrates are sequestered by the isolation membrane in a process consisting of membrane elongation and fusion events to generate the closed double-membrane autophagosome. Elongation of the isolation membrane depends on two ubiquitin-like conjugation systems (Fig. 3) (for detailed review see (54)).
Atg12 conjugation to Atg5
Atg7 activates the ubiquitin-like protein Atg12 in an ATP-dependent manner (similarly to ubiquitin-activating E1 enzymes), by binding to its C-terminal glycine. The C-terminal glycine of Atg12 is then transferred to Atg10, which in turn (similar to ubiquitin-conjugating E2 enzymes) mediates the conjugation of Atg12 C-terminal glycine to Atg5 lysine K130 forming an isopeptide bond. Two Atg12-conjugated Atg5 molecules then bind to Atg16L1 dimers and these complexes are incorporated into the isolation membrane. Atg12–Atg5.Atg16 complexes are essential for the elongation of the autophagosomal membrane, but dissociate from the completed autophagosome.
Atg8 lipidation
The second conjugation system involves the mammalian Atg8 protein LC3 (microtubule-associated protein 1 light chain 3) and its homologues (see review (54)). LC3 proteins are expressed as immature precursors (termed pro-LC3) and cleavage by Atg4 protease generates LC3-I. The thereby exposed C-terminal glycine residue is then activated by Atg7 (E1-like), transferred to Atg3 (E2-like), and finally conjugated with the lipid phosphatidylethanolamine (PE). The so-formed LC3-PE (or LC3-II) is inserted into the forming autophagosomal membrane, in a manner depending on Atg5–Atg12. LC3 lipidation is furthermore locally directed by Atg16L. LC3-II remains present throughout autophagosome maturation and its inner surface portion is finally subject to lysosomal degradation, while LC3-II located on the outer autophagosomal surface is converted back into LC3-I by Atg4.
Closure of autophagosomal membranes
Mammalian Atg8 homologues consist of the LC3 (LC3A, LC3B, LC3C) and GABARAP (GABARAP, GABARAP-L1, GABARAP-L2/GATE-16, GABARAP-L3) subfamilies, all of which are conjugated to PE (16). Both LC3 and GABARAP subfamilies are essential for autophagosome biogenesis. Interestingly, each subfamily has been suggested to exert distinct roles, whereby LC3 proteins mediate autophagic membrane elongation and GABARAP proteins may play a role more downstream in a step coupled to Atg5–Atg12.Atg16 dissociation and membrane closure (69). In an in vitro membrane fusion model both PE-conjugated LC3B and GATE-16 were shown to mediate membrane fusion via N-terminal residues (68). The role of LC3B and GATE-16 in phagophore to autophagosome maturation in vivo was supported by the accumulation of immature phagophores when expressing N-terminal LC3B and GATE-16 mutants in cells depleted of the respective wild-type proteins. However, a study in yeast demonstrated that Atg8 under physiological levels of PE was not able to mediate membrane fusion (45). Instead, autophagosomal membrane closure appeared to involve SNARE (Soluble NSF Attachment Protein (SNAP) Receptor) proteins, as in their model SNARE proteins participated in the recruitment of autophagosomal machinery to autophagosomal membranes and were required for organization of transmembrane Atg9 into tubulovesicular clusters.
Origin of Autophagosomal Membranes
The membrane origin of the autophagosome remains unsettled, and is presently reported to derive from multiple sources. The apparent absence of organelle markers had initially suggested that autophagosomes are not directly formed from other organelles but rather de novo as a distinct organelle called phagophore (64). Recent work indicates multiple subcellular origins of autophagosomal membranes. The endoplasmic reticulum can act as a source of membrane for autophagy, and the plasma membrane provides membrane components through a mechanism involving interaction of Atg16L1 with the vesicle coat-protein clathrin (53). Furthermore, during starvation mitochondria contribute membrane to the forming autophagosome (17). It is thus tempting to speculate that different sources may contribute differentially to autophagosomes of distinct composition, and form a component of specific autophagy.
Substrate Recognition
The original view of autophagy, as a mechanism for random sequestration of cytosolic components for lysosomal degradation, is rapidly evolving. Autophagy can target specific proteins, protein aggregates, intracellular pathogens, and organelles (54). Moreover, quantitative mass spectrometry revealed that protein degradation during amino acid starvation is temporally ordered, with cytosolic annotated proteins being degraded prior to organellar annotated proteins (36). This suggests triage mechanisms for nonessential cellular material, to preserve vital cellular components.
Recently, the discovery of autophagy receptors has spurred advances in the understanding of the specific targeting of certain cargos (25, 48, 50). These autophagy receptors contain an LC3 interacting-region (LIR), defined by a hydrophobic W/YxxL/I core motif, which interacts with a conserved surface of Atg8 homologues (see review (30)).
p62/SQSTM1 (sequestosome 1) is such a LIR-containing autophagy receptor that couples autophagy to survival and death pathways. In addition to its LIR motif, p62 contains a C-terminal ubiquitin-associated (UBA) domain that binds to mono- and poly-ubiquitinated proteins. Using both its UBA and LIR domains, p62 delivers ubiquitin-tagged cargo to autophagosome-mediated lysosomal degradation (for detailed review see (30)). Phosphorylation of p62 in its UBA domain enhances autophagic degradation of poly-ubiquitinated proteins (42). Targeting of p62 to autophagosome formation site does not require its binding to LC3, but requires self-oligomerization (27). Moreover, it was recently found that p62 is a component of the mTORC1 complex via amino acid-dependent interaction with raptor (11). Given the wide-reaching roles for p62 in the cell fate decision, pharmacological regulation of autophagy may offer strategies to target integrated pathways.
Intriguingly, BH3-only Bcl-2 family proteins Bnip3 and Bnip3L/Nix, which trigger the mitochondrial apoptotic pathway and mitochondrial autophagy (19, 48, 59), both contain LIR motifs [own unpublished results; (48)]. As such, Bnip3 and Nix directly couple apoptosis and autophagy initiation events. Importantly, secondary modifications of LIR motifs and LC3 itself may contribute to specificity and strength of substrate binding. Serine/threonine phosphorylation of autophagy receptor optineurin by TANK binding kinase 1 (TBK1) enhances autophagic degradation of cytosolic, ubiquitin-coated Salmonella (70). Conversely, phosphorylation of LC3 by protein kinase A (PKA) inhibits its participation in autophagy (6).
However, Atg8-mediated substrate recognition is not always dependent on the presence of Atg8–LIR interaction, suggesting additional targeting mechanisms. A recent proteomics study demonstrated that mutation of the LIR interacting surface of LC3B or GABARAP affected binding of only 38% of tested LC3B-interacting proteins, and 60% of tested GABARAP-interacting proteins, respectively (2).
Fusion with Endo-Lysosomal Compartment and Multivesicular Bodies
Formation of the degradative autolysosome is a highly coordinated membrane trafficking event. Following completion of autophagic sequestration, autophagosomes are transported along the microtubules in a dynein-dependent manner in order to fuse with the endo-lysosomal compartment (47). LC3 may hereby function as an adaptor protein between microtubules and autophagosomes as its C-terminal end interacts with autophagosomal membranes, while its amino-terminal end mediates binding to tubulin and microtubules (35). The cytoplasmic deacetylase HDAC6 can be involved in this retrograde transport of autophagosomes along the microtubules (28). Moreover, plus end-directed transport of autophagosomes is achieved via interaction of FYCO-1 (FYVE and coiled-coil domain-containing 1) with LC3, PI3P, and Rab7 (49).
Prior to lysosomal degradation, autophagosomes mature to amphisomes via fusion with components of the endocytic pathway, including early endosomes, late endosomes, and multivesicular bodies (MVBs) (reviewed in (47)). These multiple fusion events are prerequisite for successful autophagic degradation, as inhibition of endosome maturation or dysfunctional MVBs cause the accumulation of nondegraded autophagosomes (13, 55).
Fusion events between autophagosomes and endo-lysosomes are thought to involve the canonical vacuole-to-vacuole fusion machinery, consisting of Rab GTPases and SNAREs (for detailed review see (47)). Rab7 is a key component in the regulation of autophagosomal maturation, aided by lysosomal membrane proteins Lamp1 and Lamp2. Membrane trafficking activity of Rab7 involves its cycling between GTP-bound (active) and GDP-bound (inactive) states, with the class C-VPS/HOPS (homotypic fusion and vacuole protein sorting) complex acting as its guanine nucleotide exchange factor (GEF).
The Vps34–Beclin-1 core complex, in addition to mediating autophagy induction, plays a role in fusion of autophagosomes with endo-lysosomes, based on distinct co-regulatory factors binding to Beclin-1 (Fig. 4) (reviewed in detail in (21)). UVRAG (UV radiation resistance-associated gene) competes with Atg14L for binding to Beclin-1 (26). When bound to Beclin-1, UVRAG promotes interaction with the class C-Vps complex, thereby enhancing Rab7 activity and autophagosome fusion with endo-lysosomes (38). In addition to promoting autophagosome maturation, Vps34–Beclin-1-UVRAG complex may also contribute to autophagy induction via Bif-1/Endophilin B1-mediated activation of Vps34 (66).
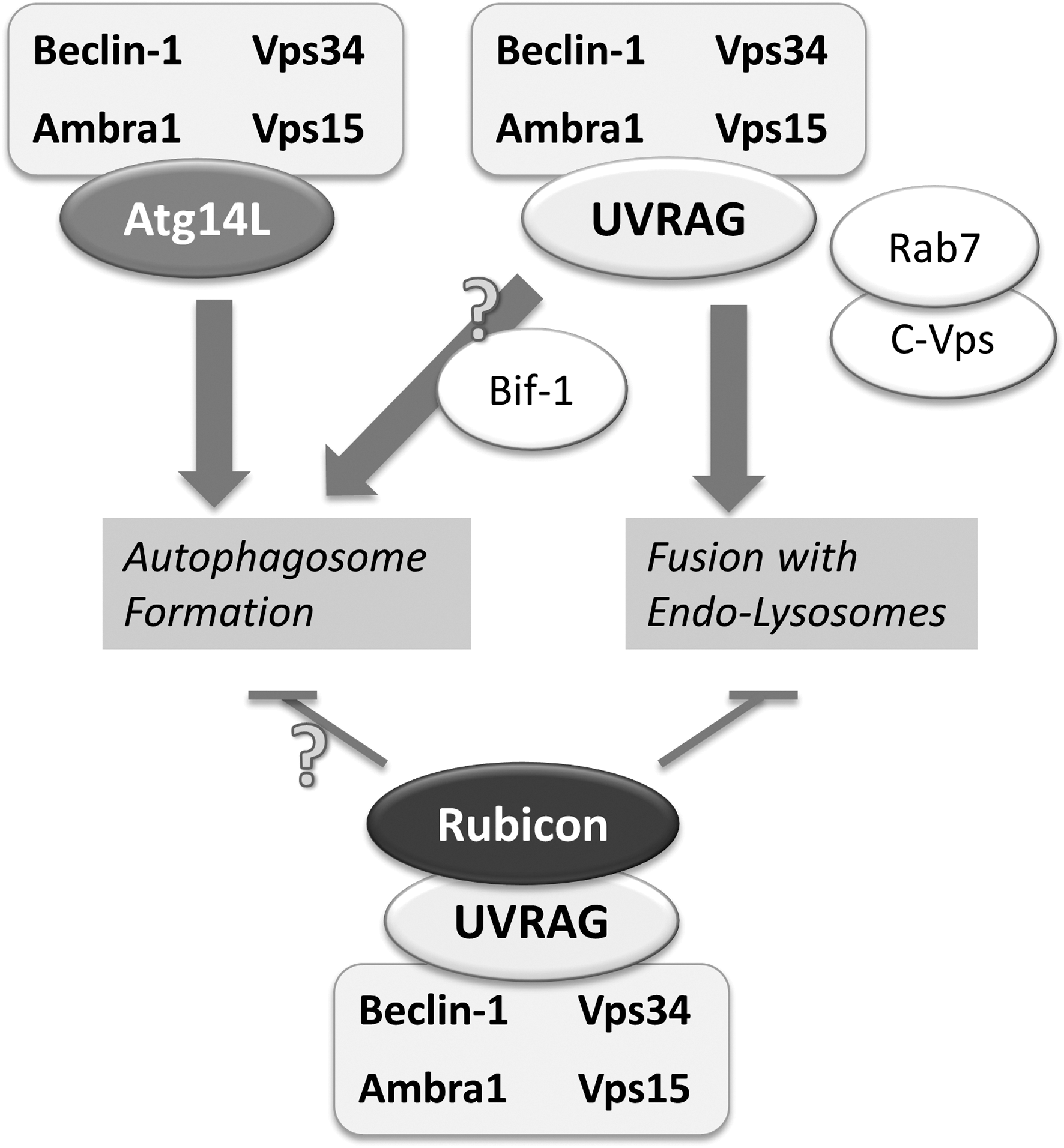
Rubicon (Run domain protein as Beclin-1 interacting and cystein-rich containing), another co-factor that binds to the Vps34–Beclin-1 complex when associated with UVRAG, serves as a negative regulator of autophagosome formation and maturation (43). As a negative regulatory mechanism opposing Atg14L, Rubicon binding to Vps34 lowers its activity and limits autophagy induction.
Degradation
Following formation of the autolysosome, lysosomal acid hydrolases degrade both cargo and inner autophagosomal membrane. Intracellular trafficking of lysosomal proteases through the secretory pathway is essential for their maturation and subcellular localization (56). Recently, it has been uncovered that the syntaxin-5 SNARE complex is required for the efficient degradation of autophagic substrates in that it mediates the anterograde transport of lysosomal proteases from ER to Golgi (56).
Succeeding degradation of autophagic cargo, macromolecules such as amino acids are transported back into the cytosol. Thus far, mechanistic insight into the recycling of macromolecules is limited to few known transport proteins in the lysosomal membrane. Importantly, local release of amino acids can recruit mTOR to (auto)lysosomes, in a Rag GTPase- and p62-dependent manner, to become activated by Rheb (11, 46). This renewed mTOR activity results in the attenuation of autophagy and signals lysosomal reformation from autolysosomes (72). Intriguingly, this process is coordinated spatially, as intracellular lysosomal localization controls mTOR signaling capacity (34).
Transcriptional Regulation of Autophagy
Recent work implicates several transcriptional factors in the regulation of autophagy (Fig. 5). The transcription factor EB (TFEB) regulates lysosome biogenesis, and is necessary for starvation-induced autophagy initiation (62). TFEB-induced expression of vATPase can be positively regulated by mTOR, which may in part mediate lysosomal reformation following autophagy termination (51). SREBP-2 (Sterol regulatory element-binding protein-2) is a transcription factor which regulates lipid metabolism, and in response to sterol depletion, activates autophagy to increase intracellular sterol levels via autophagic degradation of lipid droplet (61). Transcriptional regulation of major cell fate decision pathways integrates autophagy. The forkhead box O (FoxO) family of transcription factors which mediate both survival and death signaling and are negatively regulated by Akt, regulate autophagy gene expression (40). FoxO3 activates autophagy in muscle cells independently of mTORC1 activity (40). Under starvation conditions, FoxO1 and FoxO3 induce transcription of GABARAP-L1 and Atg12 via binding to the promoter regions of the respective genes (60).
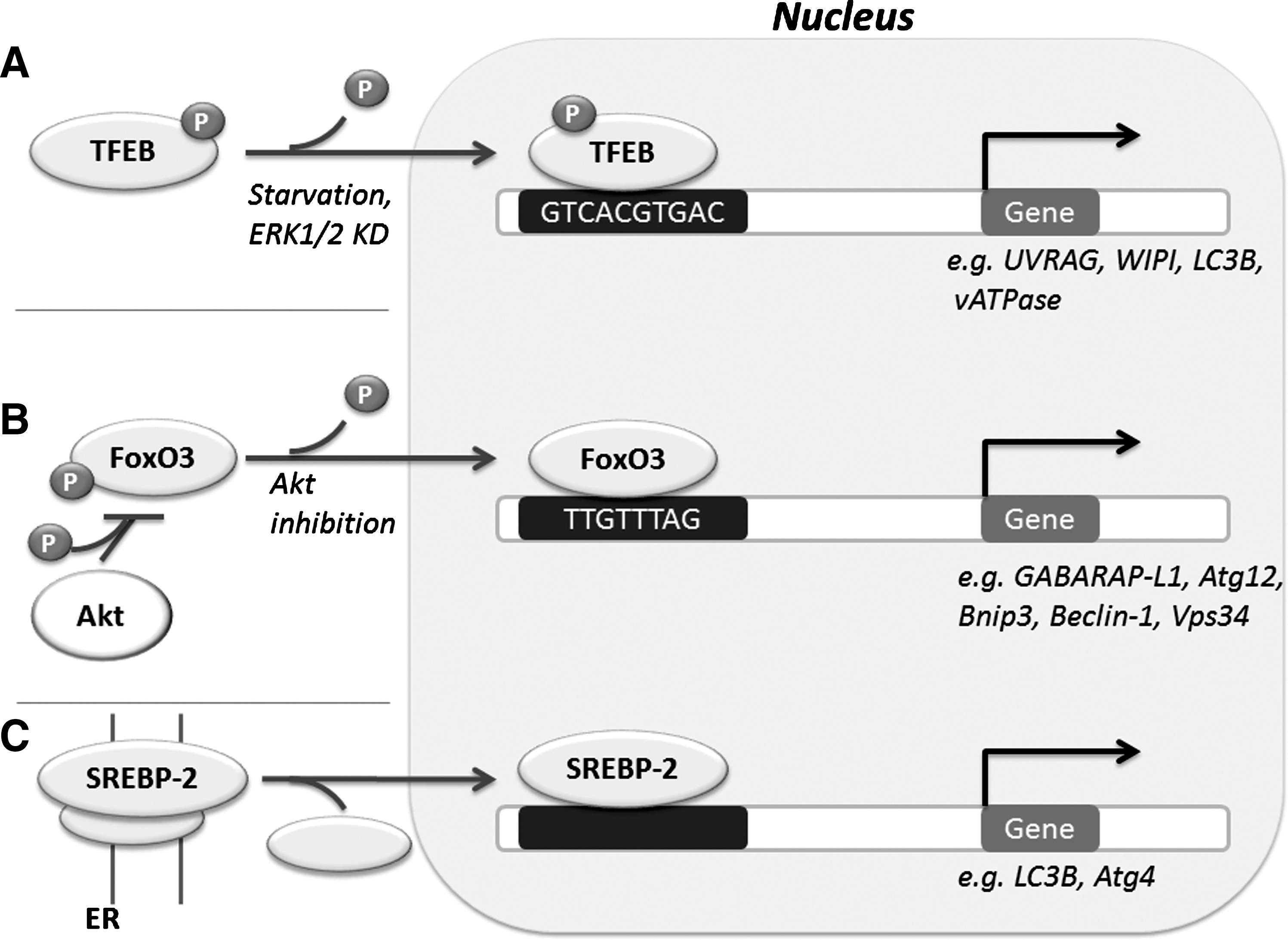
Moreover, transcriptional regulation of autophagy is directly implicated in death and survival signaling. For instance, pro-survival NFκB can promote Beclin-1 expression (7). Conversely, reduced autophagic degradation of p62 promotes oncogenic NFκB activity (41). The Kelch-like ECH-associated protein 1 (Keap1), which negatively regulates NF-E2-related factor 2 (Nrf2), is an autophagy substrate and thus integrates autophagy with reactive oxygen species (ROS) signaling (30). However, the relationship between autophagy and ROS is paradoxical: autophagy is promoted by ROS (58), yet reduced autophagy results in oncogenic ROS levels and overactivation of NFκB signaling (41). Conversely, autophagy reduces ROS through activation of the Nrf2 antioxidant response (29).
Thus, transcription factor regulation adds further complexity to autophagy regulation, coupling metabolic, pro-death, and survival signaling pathways, and may be fundamental determinants for the role of autophagy in disease.
Calcium Second Messenger Regulation of Initiation and Maturation
In addition to the ROS second messenger system, calcium plays fundamental roles in autophagy. Calcium is a diffusible second messenger which participates in many aspects of cell signaling through binding of target proteins, and is stored within the endoplasmic reticulum (ER) [reviewed in (20)]. Activation of G protein-coupled receptors is a major pathway for calcium release, whereby IP3 activates release to the cytosol by IP3R calcium channel. The sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pumps calcium into the ER. It binds calcium on the cytosolic side and releases it into the ER in an ATP dependent manner.
Calcium regulates both autophagosome formation and maturation steps (20). Calcium-activated calmodulin activates CamKKII (calcium/calmodulin-dependent kinase kinase II), leading to AMPK-mediated inactivation of mTOR (23). However, the IP3R negatively regulates autophagy (8). At least in part, this control is localized at ER/mitochondrial junctions, where the release of calcium stimulates ATP production, thereby inhibiting AMPK activation of autophagy (4). Calcium is involved in autophagosome to lysosome fusion, with disrupted calcium homeostasis interfering with Rab7-mediated autolysosome formation (15), and inhibiting autophagy through calpain activation (71). These conflicting findings likely signify a complexity which has not yet been addressed, and which may depend on local fluxes. The integrated actions of calcium release and uptake result in different calcium transients and oscillatory behavior, which depend on cell types (cancer cell versus cardiac cell). Calcium control of autophagy represents an attractive target for pharmacological modification.
Bcl-2 Family Members and Autophagy: Crosstalk of Apoptosis and Autophagy
Key pro-death and pro-survival apoptosis members interact with components of the autophagy initiation machinery, to form pathway crosstalk. Beclin-1 contains a Bcl-2 homology domain 3 (BH3) that can bind to anti-apoptotic Bcl-2 members (21). BH3-only Bcl-2 proteins activate autophagy through the disruption of the Bcl-2–Beclin-1 complex. Notably, the BH3-only protein Nix contains a LIR domain, which directly binds to Atg8 members (48) adding a critical regulatory component for engagement of the specific mode of mitophagy.
Additional regulatory mechanisms underlie the apoptosis/autophagy cell-fate decision process. Bcl-2 binding may be inhibited by phosphorylation (67) and additional factors, including Ambra1, may be required to couple mitochondrial Bcl-2 control (63). Importantly, high Bcl-2 activity does not always suppress autophagy. Bcl-xL overexpression occurs during Nix-mediated mitophagy, autophagy (59), a Beclin-1 Bcl-2 binding mutant reduced autophagic flux in heart cells (18), and endogenous Beclin-1 does not necessarily immunoprecipitate physiological levels of Bcl-2 (74). Many other BH3-only proteins impact autophagy, with varying consequences. For example, de-regulated H-Ras induces caspase-independent cell death mediated by BH3-only protein Noxa and Beclin-1 (12), whereas the BH3-only protein response is suppressed by autophagy during DNA damage (1). Furthermore, a component of Bcl-2 regulation of autophagy is due to Bcl-2 and Bcl-xL impact on ER-to-cytosol calcium release (20). Bcl-2 inhibits autophagy by attenuating calcium exit from the ER (3).
It should also be noted that caspases 3 and 8 cleave to inactivate Beclin-1 (37, 75), indicating that during apoptosis initiation and execution, the autophagy capacity of a cell is severely limited. Indeed, Bcl-xL overexpression permits autophagy during apoptosis, reducing Beclin-1 cleavage (39).
Concluding Remarks
Autophagy is fast moving from being regarded as a general phenomenon linked to cause and progression of disease, to being well-described in terms of regulatory molecular machinery and cellular function. As discussed above, its regulation and roles emerge as network biology problems, where no regulatory process is isolated, but rather needs to be considered in the context of its integration within transcriptional, signaling, and metabolic pathways.
These pathways share core protein components, second messenger systems, transcriptional and post-transcriptions regulation, and are highly organized in terms of subcellular compartmentalization. A systems understanding of autophagy will be required in order to exploit mechanistic knowledge for translational applications. This current challenge facing the field will require considerable development in terms of quantitative and multi-dimensional platforms (24), as well as mathematical tools for knowledge integration and hypothesis testing.
Footnotes
Acknowledgments
The author sincerely regrets in many instances not being able to directly cite original work due to reference limitations. This work was supported through SBCancer within the Helmholtz Alliance on Systems Biology funded by the Initiative and Networking Fund of the Helmholtz Association.
Author Disclosure Statement
No competing financial interests exist.