
Abstract
Introduction
The association of ROS with cancer has been difficult to understand for numerous reasons. First, ROS play an important role in the initiation and progression of cancer (38, 45, 268, 332). Second, cancer cells exhibit greater ROS stress than normal cells do, owing in part to oncogenic stimulation, increased metabolic activity, and mitochondrial malfunction (27, 119, 299). Third, cell-cycle progression by growth factors and receptor tyrosine kinases require ROS (138). Fourth, chronic inflammation, one of the major mediators of cancer, is regulated by ROS (132, 259). Fifth, ROS controls the expression of various tumor suppressor genes, including p53 (47, 190, 275). Sixth, a high level of ROS can suppress tumor growth through the sustained activation of the cell-cycle inhibitor (256, 296). Seventh, most of the chemotherapeutic and radiotherapeutic agents kill cancer cells by augmenting ROS stress (258, 298). These contradictory statements imply that cancer cells die by the same mechanism which facilitates their survival. This paradox provides a great challenge for researchers whose aim is to exploit ROS stress for the development of cancer therapies.
Over the past several years, researchers have noticed that the role of ROS depends on their level. While a modest amount of ROS is required for tumor promotion, an excessive level serves to suppress tumors (113, 334). However, ROS effects cannot be regarded as a general phenomenon, as ROS constitute several molecular entities, each of which might have a differential effect, if examined separately. Both ROS-elevating and ROS-eliminating strategies have been developed; the former have been predominantly used (134, 135, 237, 272). ROS-elevating strategies are based on the fact that cancer cells with elevated ROS levels depend heavily on the antioxidant defense system. A further increase in the ROS stress level, either by ROS-generating agents or by agents that abrogate the inherent antioxidant system, should result in an overall increase in endogenous ROS, which when above a cellular tolerability threshold may induce cell death. This point is the so-called “threshold concept for cancer therapy” (168, 271). On the other hand, normal cells appear to have, under lower basal stress and reserve, a higher capacity to cope with additional ROS-generating insults than cancer cells do (271, 300). Therefore, it should be possible to preferentially accumulate ROS in cancer cells and kill them selectively. Kong and colleagues were the first to prove the idea of inducing death preferentially in cancer cells by an ROS-mediated mechanism (168, 169). ROS-depleting strategies are based on the use of antioxidants to scavenge ROS, thereby abrogating ROS signaling and suppressing tumor growth (63, 273). A number of pro-oxidant- and antioxidant-based anticancer agents have been developed, some of which have been approved by the U.S. Food and Drug Administration. For instance, procarbazine, motexafin gadolinium, elesclomol, 2-methoxyestradiol, and imexon are used to increase ROS content, and minodronate and histamine are used to eliminate ROS.
Although redox-based cancer therapy seems promising, it is likely that the biochemical and molecular changes caused by ROS stress may contribute to the emergence of drug-resistant machinery during disease progression. Under persistent intrinsic ROS stress, many cancer cells become highly adapted to such stress and become resistant to exogenous stress, partly due to the activation of redox-sensitive transcription factors such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), nuclear factor (erythroid-derived 2)-like factor 2, cellular Ju-nanna (c-Jun), and hypoxia-inducible factor-1α (HIF-1α) (243, 289, 297). The activation of these transcription factors, in turn, leads to enhanced activation of the antioxidant defense system and promotes the expression of cell survival proteins. For example, increased resistance of multi-drug resistant leukemia cells to the cytotoxic effects of H2O2 was found to be due mainly to elevated levels of catalase (182). Similarly, the resistance of bladder cancer cells to arsenic trioxide (As2O3) was associated with elevated superoxide dismutase (SOD) activity and reduced glutathione (GSH) content (125). Therefore, a combination approach based on the modulation of ROS stress and the breaking of signaling molecules associated with redox adaptation might be required to effectively eliminate cancer cells. In this context, nutraceuticals seem highly promising not only because they have the potential to generate ROS but also because of their ability to modulate signaling molecules associated with drug resistance.
In this article, we discuss how ROS modulate different stages of tumorigenesis, and the signaling molecules downstream of ROS and upstream of cancer, and contribute to chronic inflammation. Pro-oxidant- and antioxidant-based anticancer drugs are discussed. We argue that nutraceuticals derived from Mother Nature serve as excellent sources of anticancer agents. The outstanding questions raised by our current understanding are also discussed.
Sources of Biologically Relevant ROS
Broadly, there are two types of ROS: the free oxygen radical and the nonradical. While the free oxygen radical ROS contain one or more unpaired electron in their outer molecular orbital, the nonradical ROS lack unpaired electrons but are chemically reactive and can be converted to radical ROS (Table 1). Superoxide, H2O2, and hydroxyl radicals are the most well studied and common ROS in cancer.
Actually a reactive nitrogen species.
ROS, reactive oxygen species; RNS, reactive nitrogen species.
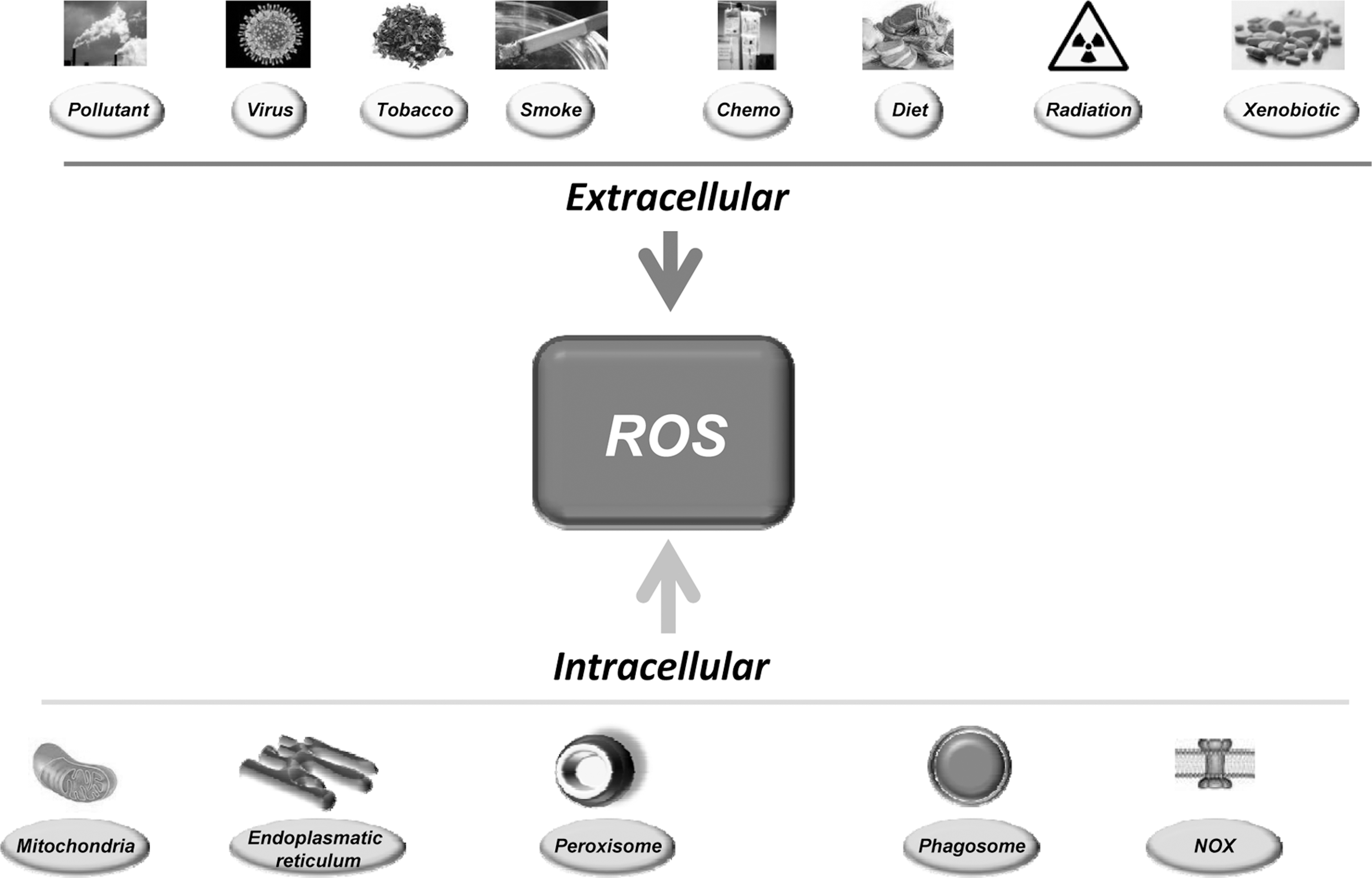
The sources of ROS are both extracellular and intracellular (Fig. 1). Extracellular ROS can be found as pollutants, tobacco, smoke, drugs, xenobiotics, or radiation. ROS are produced intracellularly through multiple mechanisms, the major sources being mitochondria, peroxisomes, endoplasmic reticulum, and the NADPH oxidase (NOX) complex in cell membranes (71, 137). Mitochondria house the electron transport chain, which transfers electrons from NADPH and succinate during respiratory ATP synthesis. The leakage of electrons from the electron transport chain during ATP synthesis results in the reduction of molecular oxygen to superoxide (100, 223). The mitochondrial permeability transition pore in the outer membrane of the mitochondria allows the leakage of superoxide into the cytoplasm (65, 287). Superoxide is dismutated to H2O2, either in the mitochondrial matrix (by Mn-SOD) or in the cytosol (by Cu-ZnSOD) (260). Peroxisomes are other major sites for superoxide and H2O2 production through the action of xanthine oxidase (37, 71, 284). H2O2, which is a highly diffusible oxygen species (219), can be converted to water by catalase, or in the presence of transition metals, it can be converted to highly reactive hydroxyl radicals. Superoxide can also react with the reactive nitric oxide (NO·) to form peroxynitrite (ONOO−) (295). Another major source of ROS, in the form of superoxide or H2O2, is NOX and its dual oxidase relatives, which are localized to various cellular membranes (26, 96, 174, 288, 291). NOX consists of NOX1, NOX2, NOX4, NOX5, p22phox, p47phox, and the small G protein Rac1. ROS are also generated in the endoplasmic reticulum during the process of protein folding and disulfide bond formation. The glycoprotein endoplasmic reticulum oxidoreductin 1, the protein disulfide isomerase, and NOX4 are the major sources of ROS in the endoplasmic reticulum (Fig. 2).
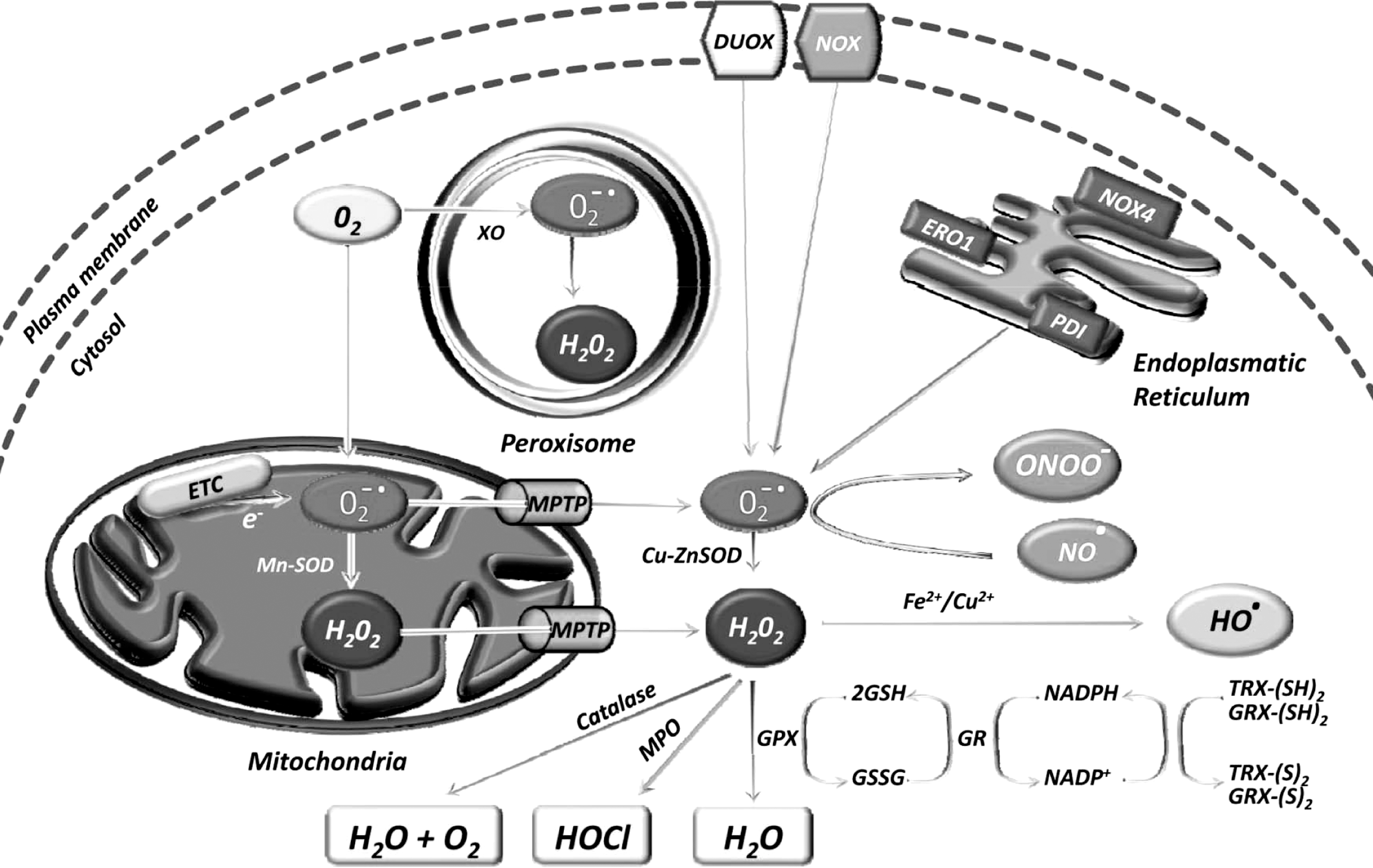
Under normoxic conditions, intracellular levels of ROS are maintained to protect cells from damage. Scavenging of ROS is facilitated by a dedicated set of antioxidants that may be both enzymatic and nonenzymatic in nature (Table 1).
Role of ROS in Tumorigenesis
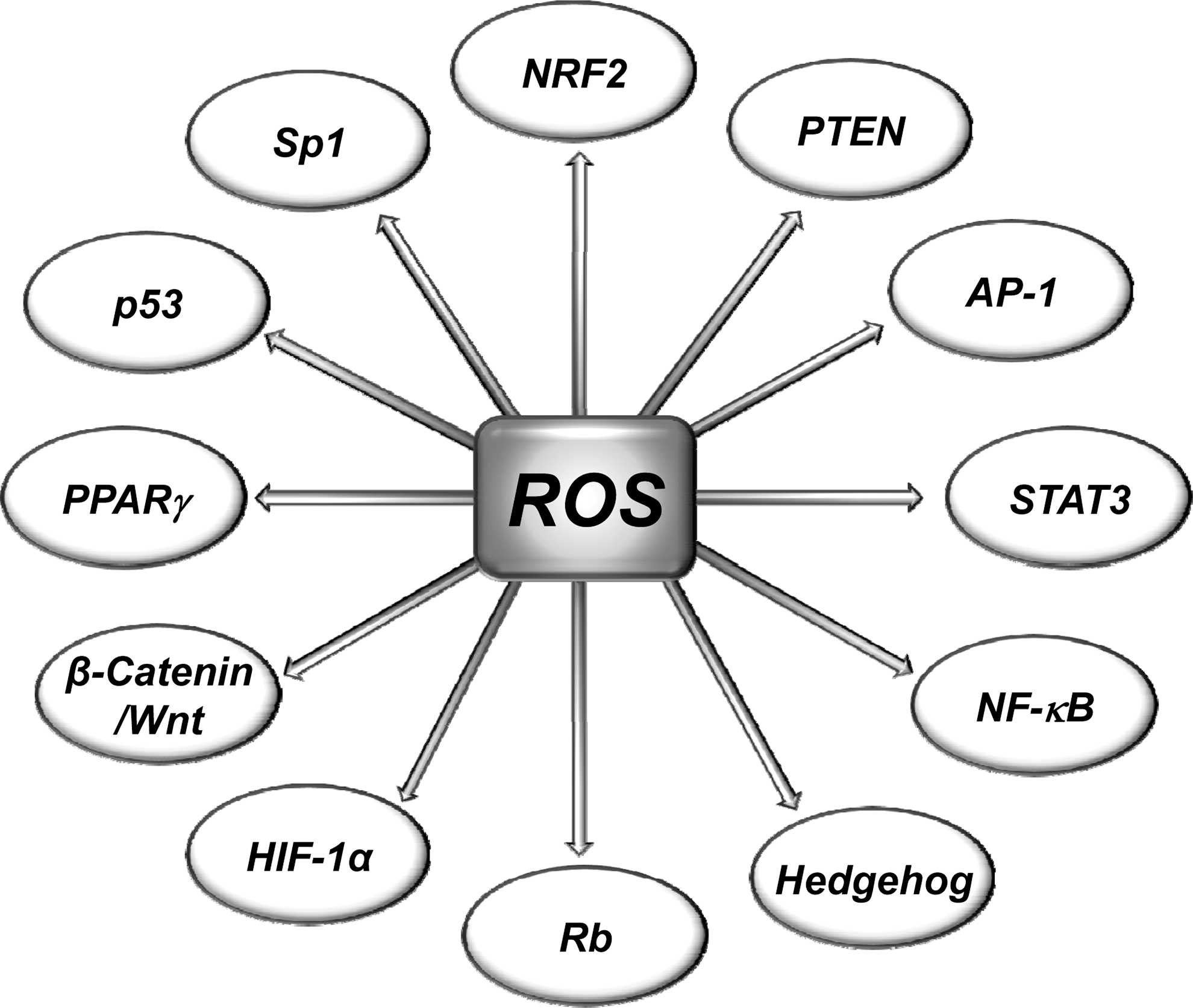
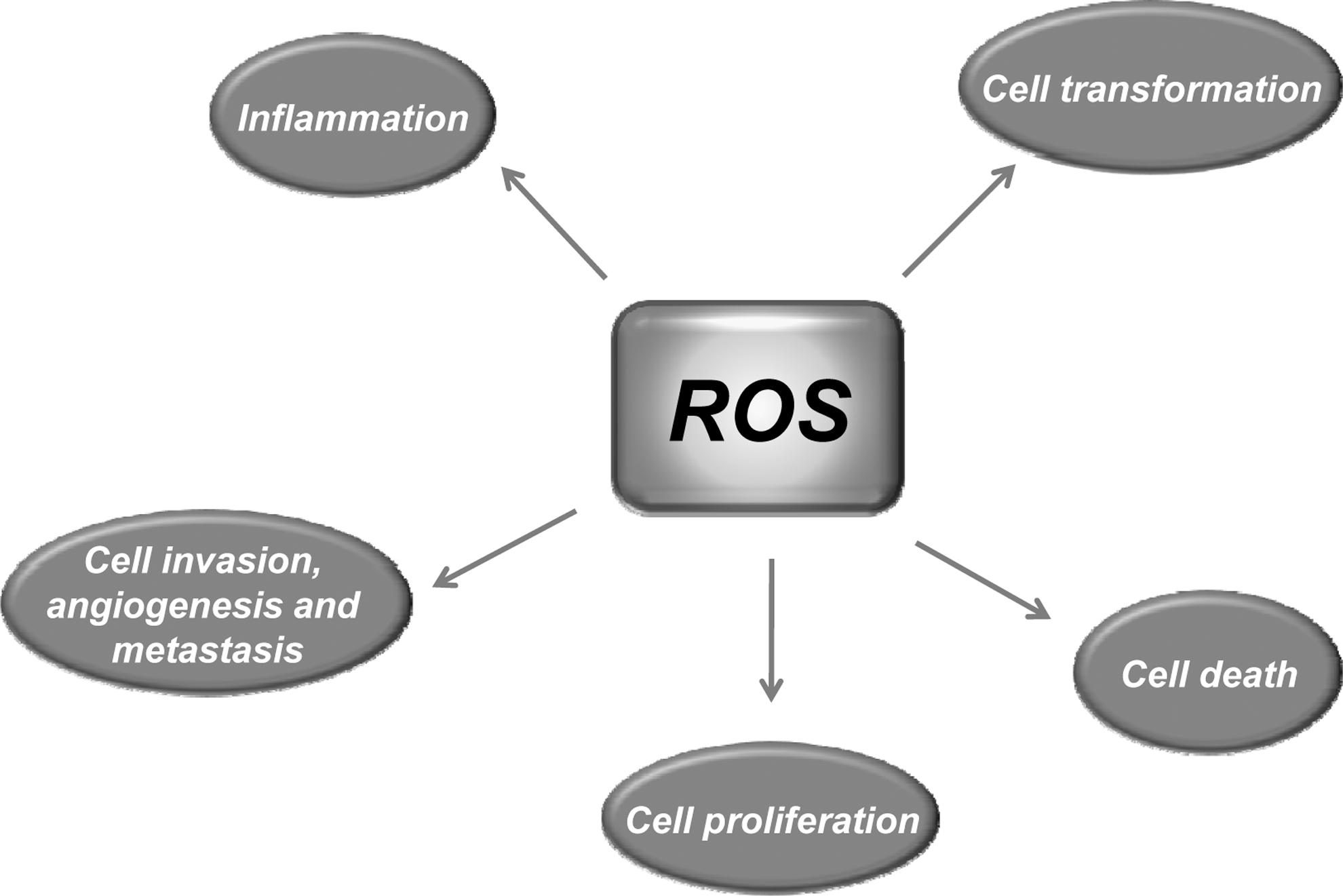
Most risk factors associated with cancer interact with cells through the generation of ROS. ROS, in turn, activate the transcription factors NF-κB, activator protein-1 (AP-1), HIF-1α, signal transducer and activator of transcription 3 (STAT3), and others (Fig. 3). These ROS-mediated transcription factors control the expression of genes involved in inflammation; cell transformation; and tumor cell death or survival, proliferation, invasion, angiogenesis, and metastasis (Fig. 4).
Role of ROS in cellular transformation
Cellular transformation in cancer biology is a process whereby normal cells acquire properties of malignant cells. The underlying causes of malignant transformation are the gain-of-function mutations in oncogenes and the loss-of-function mutations in tumor suppressor genes (319). The mutations lead to perturbations of a number of signaling molecules, including p53, Raf, retinoblastoma (Rb), protein phosphatase 2A, telomerase, Ral-GEFs, phosphatidylinositol 3-kinase (PI3K), Ras, Rac, cellular v-myc myelocytomatosis viral oncogene homolog (c-Myc), STAT3, NF-κB, and HIF-1α. Chemicals, viruses, radiation, hypoxia, and nutrient deprivation can also induce mutations in these genes, thereby giving rise to cancer cells (255).
Evidence accumulated over the past several years has indicated an association between ROS and malignant transformation (141, 311, 318). How elevated ROS levels lead to oncogene activation remains poorly understood, but DNA damage is known to play a role. For instance, the oncogenic transformation of ovarian epithelial cells with H-RasV12 or tyrosine kinase Bcr-Abl in hematopoietic cells was associated with an increase in ROS (301). In another study, transformation of fibroblasts with constitutively active isoforms of Rac and Ras was associated with production of superoxide; further study revealed that transformation could be suppressed by treatment with antioxidants (138). Mox1 is a phagocytic NOX, the over-expression of which has been shown to increase superoxide generation in mouse fibroblasts (288). The cells expressing Mox1 exhibited a transformed appearance and produced tumors in athymic mice (288).
In a recent study, cells genetically transformed to express the cancer phenotype were able to generate ROS in response to the small-molecule piperlongumine; normal cells, on the other hand, could rarely be induced to generate ROS (252). Wang et al. observed that chronic exposure of normal human lung epithelial cells to hexavalent chromium resulted in enhanced ROS production that correlated with an increase in NOX activity. Chromium exposure was also associated with malignant transformation that was suppressed by the over-expression of SOD1, SOD2, or CAT (321). In another study, sub-toxic doses of chromium transformed nontumorigenic lung epithelial cells into malignant cells (21). Exposure also led to an increase in NO production, which mediated S-nitrosylation and stabilization of the cell survival B-cell lymphoma-2 (Bcl-2) protein. Stabilization of the Bcl-2 was proposed to be a primary mechanism of malignant transformation (21).
The inflammatory cytokine tumor necrosis factor-α (TNF-α) has been shown to play a role in the transformation of mouse fibroblasts into malignant cells; this effect was partially suppressed by antioxidants (338). Apurinic/apyrimidinic endonuclease/redox effector factor-1 (APE/Ref-1) is a multifunctional protein involved in both DNA repair and redox regulation. Ref-1 was shown to induce malignant transformation in JB6 mouse epithelial cells through the mediation of ROS (342). Matrix metalloproteinase (MMP)-3, a stromal enzyme that is up-regulated in many breast tumors, has been shown to induce ROS, DNA damage, genomic instability, and the transformation of mouse mammary epithelial cells into malignant cells (251).
In summary, ROS seem to play a role in the transformation of normal cells into cancer cells. The major conclusion to be drawn is that transformed cells appear to have greater ROS levels than normal cells do. However, how ROS transform normal cells is not precisely known. Further work in this direction is needed to fully elucidate the mechanism involved in ROS-mediated malignant transformation.
Role of ROS in tumor cell death
One of the chief characteristics of cancer cells is their inherent capacity to survive. Therefore, the major goal of cancer therapy is to selectively kill cancer cells without harming normal cells. There are three major ways by which a cancer cell can die: apoptosis, necrosis, and autophagy (283, 285, 329).
ROS and apoptosis
Apoptosis is a tightly controlled form of cell death and can be initiated by death receptors (extrinsic pathway) or through mitochondria (intrinsic pathway). Both extrinsic and intrinsic pathways of apoptosis depend on ROS (237). In the extrinsic pathway of apoptosis, ROS are generated by Fas ligand as an upstream event for Fas activation. In turn, ROS are required for Fas phosphorylation at the tyrosine residue, which is a signal for subsequent recruitment of Fas-associated protein with death domain and caspase 8 and for apoptosis induction (72, 216, 257, 305). In addition, ROS are required for the ubiquitination and subsequent degradation of the FLICE inhibitory protein to further enhance Fas activation (322). In contrast, the intrinsic pathway of apoptosis is characterized by the opening of the permeability transition pore complex on the mitochondrial membrane, which results in cytochrome c release, apoptosome formation, and caspase activation. Opposing effects of pro-apoptotic and anti-apoptotic Bcl-2 family proteins are required for opening of the permeability transition pore. ROS function to open the pore by both activating pore-destabilizing proteins (Bcl-2-associated X protein, Bcl-2 homologous antagonist/killer) and inhibiting pore-stabilizing proteins (Bcl-2 and Bcl-xL) (212).
Extensive research over the past several years from both cell culture and animal models has demonstrated the potential of ROS in inducing apoptosis in cancer cells. As of August 2011, a search on PubMed database (
ND, not determined; ↑a, activation; ↓b, inactivation; ↓, down-regulation; ↑, up-regulation.
ABITC, abietyl isothiocyanate; AGL, andrographolide; AKT, AKT8 virus oncogene cellular homolog; ATG5, autophagy protein 5; Bak, Bcl-2 homologous antagonist/killer; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma-2; Bcl-xL, B-cell lymphoma-extra large; Bim, Bcl-2-interacting mediator; BITC, benzyl isothiocyanate; CHL, chlorogenic acid; COX-2, cyclooxygenase-2; DMAPT, dimethylaminoparthenolide; DR5, death receptor 5; EGCG, epigallocatechin gallate; ERK1/2, extracellular signal-regulated kinase 1/2; ESB, erythrina suberosa stem bark; GA, 18 β-glycyrrhetinic acid; H2O2, hydrogen peroxide; IGF-I, insulin-like growth factor-1; JAK2, janus kinase 2; JNK, c-jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK1/2, MAPK/ERK kinase 1/2; MiMP, mitochondrial membrane potential; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; PDT, photodynamic therapy; PEITC, β-phenylethylisothiocyanate; PGE-2, prostaglandin E2; PI3K, phosphoinositide 3-kinase; PKC-δ, protein kinase C-delta; PL, piperlongumine; PTEN, phosphatase and tensin homolog deleted on chromosome 10; SAPK, stress-activated protein kinase; TRAF2, TNF receptor-associated factor 2; UDCA, ursodeoxycholic acid; XIAP, X-linked inhibitor of apoptosis protein.
Exogenous administration of H2O2 has been shown to induce apoptosis in lymphoma cells through activation of caspase-3 (114). H2O2 has also been shown to activate MAPK/ERK kinase1/2, extracellular signal-regulated kinase 1/2 (ERK1/2), and caspase, and to induce cell death in human bladder cancer cells (59). H2O2 generated by external sources has the potential to induce apoptosis in hepatoma cells (310), leukemia cells (28, 248), and osteosarcoma, breast, bladder, and lung cancer cells (252). NO generated by the small-molecule piperlongumine has also been shown to induce apoptosis in osteosarcoma cells and in breast, bladder, and lung cancer cells but not in normal cells (252). The authors of this study concluded that increased dependence of cancer cells on the ROS stress-response pathway could be a basis for the selectivity of piperlongumine-induced apoptosis in cancer cells (252). ROS has been shown to induce apoptosis in cancer cells in a caspase-independent manner as well. For instance, in human lung endothelial cells, ROS was shown to induce apoptosis in a caspase-independent manner but involved mitochondrial-to-nuclear translocation of apoptosis-inducing factor and endonuclease G (193).
Numerous agents have been shown to induce ROS and apoptosis in various cancer types. The most common signaling molecules modulated by ROS in these cell models are kinases, pro-inflammatory transcription factors such as NF-κB, caspases, cell survival proteins, pro-apoptotic proteins, and phosphatase, and tensin homolog deleted on chromosome 10 (PTEN). For example, the proteasome inhibitor bortezomib induces apoptosis in gastric cancer cells by inactivating NF-κB, activating c-jun N-terminal kinase (JNK), and inducing ROS generation (228). Some other common cancers for which ROS have demonstrated potential are listed in Table 2.
The potential of ROS in inducing apoptosis is evident from animal studies as well. Using mouse models of breast, bladder, and lung cancer, Raj et al. recently demonstrated that H2O2 generated by piperlongumine can selectively kill cancer cells (252). Chlorogenic acid has been shown to induce apoptosis in cells from chronic myeloid leukemia patients and also in nude mice bearing K562 xenografts in a ROS-dependent manner (254). In another study, andrographolide, a diterpenoid lactone, induced apoptosis in patient-derived lymphoma cells in an ROS-dependent manner; ROS induced apoptosis in these cells through activation of caspases-3, −8, and −9 that was inhibited by an ROS scavenger (341). Dimethylaminoparthenolide, a water-soluble parthenolide analog, has also been shown to induce apoptosis in prostate cancer cells in vivo by targeting NF-κB and generating ROS (276).
Some of the agents have been shown to generate ROS but they lack pro-apototic potential. Mi et al. found that the pro-apototic activities of benzyl isothiocyanate (BITC) and β-phenylethylisothiocyanate (PEITC) are not due to their ROS-inducing potential but to their ability to inhibit proteasomes and to bind covalently with target proteins (218).
Apart from their ability to kill cells, ROS are also required for cancer cell survival. In fact, the ability of cancer cells to distinguish between ROS as a survival or apoptotic signal is controlled by the dosage, duration, type, and site of ROS production. However, modest levels of ROS are required for cancer cells to survive, whereas excessive levels kill them (168, 268). Similarly, NOX-derived ROS in the cytoplasm in response to TNF-α play a protective role, whereas mitochondria-derived ROS promote apoptosis (73). Low levels of ROS have been shown to promote the survival of serum-deprived anaplastic large cell lymphoma cells (339). In prostate cancer cells, inhibition of ROS by antioxidants or NOX inhibitors was associated with an increase in apoptosis (41). ROS produced by NOX4 has also been shown to act as a mediator of cell survival (85, 220, 313). Similarly, in pancreatic cancer cells, ROS produced by NOX was shown to promote survival by inhibiting tyrosine phosphatase-mediated dephosphorylation of janus kinase 2 (178).
ROS and necrosis
Although an excess level of ROS is known to induce apoptosis, massive levels may lead to necrotic cell death. In some cases, ROS can induce both apoptosis and necrosis in cancer cells. For example, in Jurkat T-lymphocytes, H2O2 was found to have dual effects: At low H2O2 concentrations, the cells were found to undergo apoptosis by caspase activation, but at higher H2O2 concentrations, no detectable caspase activity was observed and the cells died of necrosis (114). In multiple myeloma cells, ROS generated in response to a peptide have been shown to induce necrosis (226). A switch from apoptotic to necrotic cell death has also been shown to be dependent on the ROS content in prostate cancer cells (93) and hepatoma cells (160). Similarly, 8-nitrocaffeine and its analog, which are candidate radiosensitizers for cancer therapy, were found to induce necrotic cell death in leukemia cells in an ROS-dependent manner (227).
ROS and autophagy
Autophagy is a self-catabolic process that involves sequestration of exhausted organelles and protein aggregates from the cytoplasm and their delivery into lysosomes for degradation. Autophagy is involved in both cell survival and cell death pathways, and the process is altered in cancer cells (120). Studies during the past 5 years have indicated a role for ROS as a signaling molecule in inducing autophagic cell death in cancer cells (20, 97, 139). For example, H2O2 production in human colon cancer cells has been associated with autophagic cell death (64). In a resistant pancreatic cancer model, gemcitabine and cannabinoid combinations triggered autophagic cell death through a ROS-mediated mechanism (77). Bufalin, which is isolated from a traditional Chinese medicine, was unable to induce apoptosis in colon cancer cells, contrary to its well-documented apoptosis-promoting activity in other cancer cells (336). Instead, bufalin activated an autophagy pathway, as characterized by the accumulation of LC3-II and the stimulation of autophagic flux. The induction of autophagy by bufalin was linked to ROS generation. ROS activated autophagy via JNK activation, which, in turn, increased the expression of autophagy protein 5 and Beclin-1. Further, bufalin-induced autophagy was attenuated by an ROS scavenger (336). Some other cancer types for which ROS have been shown to effectively induce autophagic cell death are breast cancer (281), nonsmall cell lung cancer (NSCLC) (184), glioma (239, 317), neuroblastoma (317), glioblastoma (53), and cervical cancer (53, 108).
In summary, ROS have dual roles: They can not only kill cancer cells but they can also promote tumor survival. The great challenge for cancer researchers is determining how to exploit this dual property of ROS for therapeutic development.
Role of ROS in tumor cell proliferation
Uncontrolled proliferation is one of the chief characteristics of tumor cells (115, 116). A precise set of cell cycle regulators such as cyclins and cyclin-dependent kinases (CDKs) control the progression of cell-cycle events. CDK activity is controlled by the opposing effects of cyclins and CDK inhibitors. CDK inhibitors such as p21 and p27 negatively regulate CDK activity, whereas cyclins are required for CDK activity and cell cycle progression. Another protein, c-Myc, is required for the G1-to-S-phase transition (118). The expression of c-Myc, in turn, is regulated by cdc25, a phosphatase that activates CDKs.
Intracellular ROS produced by exogenous stimuli as well as exogenous administration of ROS have been shown to enhance the proliferation of numerous cancer types (Table 3). For example, exogenous administration of H2O2 was shown to enhance the proliferation of hepatoma cells by increasing protein kinase B and extracellular signal-regulated kinase (ERK) activities (195). In another study, transformed bladder urothelial cells were found to be hyper-proliferative and produced elevated ROS levels in the presence of monomethylarsonous acid; the up-regulation in cyclooxygenase-2 (COX-2) expression observed in these cells was found to be ROS dependent (84). ROS produced by low concentrations of arsenite has been shown to enhance the proliferation of breast cancer cells by recruiting cells into the S phase of the cell cycle, enhancing the expression of c-Myc and heme oxygenase-1, and increasing NF-κB activity (262).
ATM, ataxia telangiectasia mutated; Cdc25c, cell division cycle 25 homolog c (S. pombe); CDK2, cyclin-dependent kinase 2; Chk, checkpoint kinase; c-Myc, cellular v-myc myelocytomatosis viral oncogene homolog (avian); DEN, diethylnitrosamine; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; GSH, glutathione; HO-1, heme oxygenase-1; LPA, lysophosphatidic acid; MKP-3, mitogen-activated protein kinase phosphatase-3; MMA, monomethylarsonous acid; NSCLC, nonsmall cell lung cancer; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; Rb, retinoblastoma; SOD, superoxide dismutase.
ROS produced by endogenous sources can also enhance cancer cell proliferation. For example, ROS produced by Romo1, a mitochondria-localized protein (61, 133), was shown to be indispensable to the proliferation of lung cancer cells (224). Such an induction in cell proliferation was found to be ERK dependent (224). Endogenous production of superoxide has also been shown to enhance tumor proliferation in hepatoma cells that was mediated through AKT8 virus oncogene cellular homolog (AKT) phosphorylation (78). Similarly, an increase in endogenous ROS due to reduction in the antioxidant defense system has been correlated with an increase in the proliferation of breast (70) and ovarian (127) cancer cells. In breast cancer cells, ROS-mediated tumor proliferation was found to be dependent on activation of PI3K pathway and reduction of PTEN activity (70).
The role of ROS in promoting tumor proliferation is further supported by observations that agents with the potential to inhibit ROS generation can also inhibit tumor cell proliferation. For instance, N-ethoxymethyl-3-amino-1,2,4-benzotriazine-1,4-dioxide, a novel N-ethoxymethyl-3-amino-1,2,4-benzotriazine-1,4-dioxide derivative, was found to inhibit the growth of NSCLC cells by inducing cell cycle arrest at the G2/M phase and suppressing ROS generation (196). Similarly, attenuation of ROS by a squamosamide derivative was associated with inhibition of the proliferation of liver cancer cells; decreased phosphorylation of AKT and Rb protein; down-regulated expression of cyclin D1, cyclin E, and CDK2; and enhanced expression of p27 (249). In another study, exogenous catalase inhibited the proliferation of numerous cancer types (245). Consistent with these observations, stable expression of human catalase in MCF-7 cells inhibited proliferation and reverted malignant features (245). Curcumin has been shown to inhibit the proliferation of lymphoma cells by increasing endogenous antioxidant enzyme activity and by inhibiting NF-κB activity (69). Inhibition of ROS generation by N-acetyl-
Some other common cancers for which ROS has been shown to enhance proliferation are listed in Table 3. The ability of ROS in promoting tumor cell proliferation is supported by animal studies as well (70, 127).
Although ROS promote tumor cell proliferation in general, an increase in the level of ROS has also been correlated with reduced tumor proliferation (Table 3). For example, silencing of the redox protein thioredoxin-like 2 (TXNL-2) in human breast cancer cells was associated with an increased ROS level, reduced NF-κB activity, and inhibited tumor proliferation (250). Recently, butein was shown to inhibit the growth of hepatoma cells, which was correlated with ROS content (221). The increase in ROS and the inhibition in growth were further correlated with the induction of G2/M cell cycle arrest; increased phosphorylation of ataxia telangiectasia mutated, checkpoint kinase (Chk) 1, and Chk2; and reduced cell division cycle 25 homolog c levels. Further, an antioxidant pretreatment abrogated butein's inhibitory effect on cell growth (221). ROS generated by gemcitabine and by thymoquinone have also been shown to inhibit the growth of pancreatic (76) and prostate (167) cancer cells, respectively.
Role of ROS in tumor cell invasion, angiogenesis, and metastasis
Tumor cell invasion, angiogenesis, and metastasis are inter-related processes that represent the final, most devastating stage of malignancy. The process involves cell growth, adhesion, and migration; proteolytic degradation of tissue barriers; and formation of new blood vessels (86). Several proteolytic enzymes, such as matrix metalloproteinases (MMPs) (146, 286) and the intercellular adhesion molecule, participate in the degradation of these barriers (9, 165). Other molecules involved in this process are serine proteases such as urokinase-type plasminogen activator and its receptor, vascular endothelial growth factor (VEGF) and its receptors, platelet-derived growth factor, fibroblast growth factors, epidermal growth factor (EGF), ephrins, angiopoietins, endothelins, integrins, cadherins, and transcription factors (e.g., AP-1, NF-κB) (3, 5, 103, 183, 229, 323) (Table 4).
AP-1, activator protein-1; BAEC, bovine aortic endothelial cell; Cav-1, caveolin-1; c-Met, hepatocyte growth factor receptor; CXCL14, CXC chemokine ligand 14; CXCR4, CXC chemokine receptor 4; EC, endothelial cell; EGF, epidermal growth factor; Ets-1 v-ets erythroblastosis virus E26 oncogene homolog 1; GT094, ethyl 2-((2,3-bis(nitrooxy)propyl)disulfanyl)benzoate; HIF-1, hypoxia-inducible factor-1; HNSCC, head and neck squamous cell carcinoma; IL, interleukin; iNOS, inducible nitric oxide synthase; ITGB3, integrin beta 3; LTB4, leukotriene B4; MMP, matrix metalloproteinase; PAK1, p21 activated kinase 1; PCBs, polychlorinated biphenyls; PMS, phenazine methosulfate; ROCK, rho-associated kinase; STMN1, stathmin 1; TGF-β1, transforming growth factor beta 1; TPA, 12-O-tetradecanoylphorbol-13-acetate; u-PA, urokinase-plasminogen activator; u-PAR, urokinase-plasminogen activator surface receptor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Accumulating evidence over the past several years from both in vitro and in vivo studies has indicated a role for ROS as a signaling mediator of angiogenesis and metastasis (306 –308). ROS has been shown to mediate these effects through induction of transcription factors and genes involved in angiogenesis and metastasis. However, the role of ROS in modulating tumor cell metastasis and angiogenesis has seemed paradoxical: High ROS levels suppress tumor angiogenesis and metastasis by destroying cancer cells, whereas sub-optimal concentrations assist cancer cells in metastasizing (232).
Exogenous administration of H2O2 enhances metastasis by modulating multiple signaling molecules. For example, in colorectal cancer cells, H2O2 induced metastasis in a JNK- and mitogen-activated protein kinase (MAPK) mediated activation of AP-1 and MMP-7 up-regulation (121). H2O2 has also been shown to promote metastasis by up-regulating CXC chemokine receptor 4 (CXCR4) and pAKT and inactivating PTEN in prostate cancer cells (57); while in lung cancer cells, it stabilized caveolin-1 (263). Exogenous H2O2 can also induce angiogenesis of endothelial cells (253), bovine aortic endothelial cells (344), head and neck squamous cell carcinoma cells (206), and ovarian cancer cells (192).
In a few cases, endogenous H2O2 has been found to induce tumor angiogenesis. For example, Arbiser et al. demonstrated the potential of Nox-expressing prostate tumors to up-regulate VEGF, VEGF receptors 1 and 2, and MMP (18). These up-regulations were associated with vascularization and rapid expansion of the tumors. Further, induction of VEGF was eliminated by co-expression of catalase, indicating that H2O2 was required for induction of the angiogenic phenotype (18).
In one study, higher levels of ROS were observed in a colorectal cancer-derived metastatic cell line that correlated with an up-regulation in integrin beta 3 and stathmin 1 (180). Endogenous production of ROS has also been shown to induce angiogenesis and metastasis in ovarian (335), prostate (335), colon (154), and liver (186) cancer cells. ROS-generating agents can also induce angiogenesis and metastasis in cancer cells. Common agents under this category are 12-O-tetradecanoylphorbol-13-acetate in hepatocellular (177), lysophosphatidic acid in breast (80), leukotriene B4 in bladder (157), EGF in pancreatic (36), phenazine methosulfate in gastric (161), polychlorinated biphenyls in breast (194), transforming growth factor beta 1 in pancreatic (35), and protein kinase C-delta (PKC-δ) activator in prostate (159) cancer cells. The most common signaling molecules modulated by these agents through ROS production are MAPK, JNK, NF-κB, AP-1, MMPs, inducible NO synthase, cytokines, PI3K, p21 activated kinase 1, rho-associated kinase, VEGF, and the urokinase-type plasminogen activator receptor.
The potential of ROS in promoting tumor cell angiogenesis and metastasis has also been demonstrated in animal models of breast cancer (101), bladder cancer (157), lung cancer (140, 156, 162), melanoma (89), sarcoma (203), colon cancer (154), and prostate cancer (159) cells. In a transgenic mouse model that develops metastatic breast cancer (MMTV-PyMT), the invasive behavior of tumor cells was significantly reduced by catalase (101). In a mouse model of bladder cancer, ROS were shown to play a role in inducing metastasis through the stimulation of NF-κB (157). Ras-evoked lung metastasis was also recently shown to be induced through the generation of ROS and the up-regulation of NF-κB and MMP-9 in a mouse model (156). Of note is that the surgical procedure used to remove tumors has been shown to induce ROS generation and to enhance the growth of metastatic tumors in a mouse model of melanoma (135).
One study provided direct evidence for the causative relationship between ROS generation and tumor metastasis (140). After replacement of mitochondrial DNA derived from a highly metastatic mouse tumor cell line, a poorly metastatic cell line acquired the metastatic potential in mice. The transferred mitochondrial DNA contained mutations with deficiency in respiratory complex I activity and was associated with enhanced ROS production. Further, pretreatment of the highly metastatic tumor cells with ROS scavengers suppressed the metastatic potential in the mice (140).
In most cases, ROS have been demonstrated to induce angiogenesis and metastasis, but in a few studies, an increase in ROS levels has been shown to play a negative role. For instance, an increase in ROS after TXNL-2 silencing has been associated with reduction in NF-κB activity and metastasis of breast cancer (250). In another study, theaflavin, the bioactive flavonoid of black tea, suppressed breast cancer metastasis by activating the p53-ROS-p38MAPK pathway and inhibiting NF-κB activation and MMP-2 and MMP-9 expression (1). Similarly, the anti-metastatic potential of BITC and PEITC in human NSCLC cells has been associated with an increase in ROS generation and depletion in GSH content (333). Pathi et al. found that treatment of colon cancer cells with GT-094, a nonsteroidal anti-inflammatory drug, was associated with an increase in ROS and decreases in VEGF and VEGF receptors 1 and 2 (241). The combination of tyrosine kinase inhibitor dasatinib with oxaliplatin has also been shown to reduce angiogenesis in colon cancer cells in association with an increase in ROS generation (170). Fibulin-5 is a matricellular protein that has been shown to regulate angiogenesis (12, 13). In a recent study, the angiogenesis of pancreatic tumors was found to be suppressed in Fibulin-5–null (Fbln5−/−) mice compared with in wild-type littermates; this suppression was associated with an increase in ROS in these tumors (270).
Interestingly in one study, modulation of lung cancer metastasis was dependent on ROS type. The hydroxyl radical up-regulated caveolin-1 expression and promoted metastasis, whereas superoxide and H2O2 down-regulated caveolin-1 and inhibited metastasis (200).
ROS, Chronic Inflammation, and Cancer
Inflammation is a part of the body's defense system to counteract an insult incurred by internal or external stimuli. Acute inflammation is therapeutic, whereas chronic inflammation is a culprit for numerous chronic diseases, including cancer. It was Virchow in the nineteenth century who first noticed the presence of inflammatory cells within tumors and found that tumors arise at sites of chronic inflammation (24, 269). Experimental and epidemiologic research over the past several years has indicated close associations between ROS, chronic inflammation, and cancer (62, 105, 106, 132, 209, 210, 259, 269, 326). How ROS induce inflammation has also been investigated over the years. Induction of COX-2, inflammatory cytokines (TNF-α, interleukin [IL]-1, IL-6), chemokines (IL-8, CXCR4), and pro-inflammatory transcription factors (e.g., NF-κB)—all well-known mediators of inflammation and tumorigenesis—is regulated by ROS (131, 222). Further, mitochondrial ROS play a major role in inducing chronic inflammation and cancer (4, 149, 225, 324).
The involvement of chemokines and chemokine receptors in the invasion and metastasis of various tumors has been reported (236, 345). The metastatic potential of chemokines has been attributed to their ability to induce the expression of MMPs, which facilitate tumor invasion (197, 345). The silencing of endogenous chemokine receptors has been shown to inhibit the proliferation, adhesion, and invasion of salivary gland mucoepidermoid carcinoma cells (327). One study found a close association between the expression of IL-8 by human melanoma and ovarian cancer cells and their metastatic potential (129, 201, 337). In another study, serum IL-6 and IL-8 were associated with the incidence of lung cancer (244). Pro-inflammatory cytokines have been demonstrated to be predictors of prognosis for esophageal adenocarcinoma (230). Pro-inflammatory molecules have also been shown to be predictors of multiple myeloma (164), non-Hodgkin's lymphoma (316), colorectal cancer (176), bladder cancer (235), lung cancer (244), esophageal cancer (230), and renal cell carcinoma (17).
Although the examples just presented indicate a positive role of ROS-mediated inflammatory cytokines in cancer development, in a few cases, the suppression of inflammatory pathways has been found to be detrimental. For example, administration of TNF blockers to patients with rheumatoid arthritis was found to increase the risk for developing lymphomas (95). Similarly, suppression or deletion of NF-κB has been associated with progression to carcinogenesis (68, 92, 261, 274, 312). Thus, it can be concluded that the effects of ROS on inflammation may be either beneficial or detrimental depending on the cell type, species involved, and physiologic conditions.
Role of ROS in Cancer Prevention and Therapy: Lessons from Clinical Studies
Due to the dual role of ROS in cancer development, both pro-oxidant- and antioxidant-based agents have been developed for cancer prevention and therapy (15, 90, 271, 300, 318). Pro-oxidant-based anticancer agents can not only directly increase ROS production but also decrease the antioxidant defense system of cancer cells. The antioxidant-based agents can directly scavenge intracellular ROS, enhance ROS-scavenging enzyme activities, and inhibit NOX activity. In some cases, a combination of these approaches has been found to be very successful.
Role of nutraceuticals and antioxidants in cancer prevention
According to one report, 90%–95% of cancers are caused by life style factors and only 5%–10% are caused by genetic defects (16). These proportions indicate that cancer is a disease which can be prevented largely by life style changes. Since ROS are involved in the transformation of nonmalignant cells to malignant cells, a potential approach to cancer prevention might be to control ROS production at the transformative stage.
Due to their effect on multiple targets as well as their cost-effectiveness, efficacy, safety, and immediate availability, plant-derived nutraceuticals and antioxidants have attracted the attention of clinicians and researchers during the past two decades. Nutraceuticals can act as either a pro-oxidant or an antioxidant on the basis of the concentration and cancer type. Although they have been proved beneficial for both cancer prevention and treatment, in this section, we discuss the role of nutraceuticals for prevention.
Curcumin is one of the most widely studied nutraceuticals that has potential against numerous cancers. Depending on the concentration and cancer type, curcumin can exhibit both antioxidant and pro-oxidant activities (8, 19, 33, 91, 280). For instance, in one study, curcumin at 12.5 μM reduced ROS formation in human myeloid leukemia cells but at higher concentrations, curcumin elevated ROS levels (50). A number of clinical trials have evaluated the potential of curcumin for cancer prevention. For example, curcumin was found to benefit patients with ulcerative proctitis and Crohn's disease (122). In one study, the regimen of curcumin (8 g/day) given for 3 months to patients with high-risk or premalignant lesions was found to be safe and effective (56). Curcumin also seems a promising and safe medication for maintaining remission in patients with quiescent ulcerative colitis (117). One study investigated the effect of oral curcumin in combination with piperine on pain and on the markers of oxidative stress in patients with tropical pancreatitis (83). Twenty patients were randomly allocated to receive 500 mg of curcumin with 5 mg of piperine thrice a day, or placebo for 6 weeks. The effects on the pattern of pain and on the malondialdehyde and GSH content in red blood cells were assessed. Curcumin in combination with piperine was correlated with a significant reduction in the erythrocyte malondialdehyde content and a significant increase in GSH levels in patients with tropical pancreatitis (83). Curcumin in combination with quercetin has been found to reduce the number and size of ileal and rectal adenomas in patients with familial adenomatous polyposis, an autosomal-dominant disorder characterized by the development of colorectal adenomas and eventually colorectal cancer, without appreciable toxicity (66). In addition, curcumin in combination with isoflavones has been found to suppress the production of prostate-specific antigen, which is a biomarker of prostate cancer (136).
Vitamins, selenium, carotenoids, pomegranate, green tea, and soy have also been effective in human clinical trials for cancer prevention (175, 302). Lycopene is one of the main carotenoids in the regional Mediterranean diet and can account for 50% of the carotenoids in human serum. Lycopene is present in fruits, including watermelon, apricots, pink guava, grapefruit, rose hip, and tomatoes. Scavenging of ROS is one of the mechanisms for the anticancer effects of lycopene. In one study, consumption of tomato sauce before prostatectomy decreased the serum prostate-specific antigen level and oxidative DNA damage and increased the lycopene concentration in prostate tissue (51). In another study, tomato sauce suppressed the progression of disease in patients diagnosed with prostate carcinoma (158). In a study conducted in Japan, 244 subjects with atrophic gastritis were randomly allocated to receive vitamin C (50 or 500 mg) for 5 years. Vitamin C was found to reduce oxidative stress among subjects with atrophic gastritis (266). An inverse correlation between vitamin C and cancer risk has been demonstrated by other studies as well (44, 153).
Green tea is popular for its epigallocatechin gallate, a polyphenolic compound that contributes to the potential health benefits associated with green tea consumption (152). In a study conducted in China, the risk of prostate cancer declined with increasing frequency, duration, and quantity of green tea consumption (145). Pomegranate has been used for centuries for medicinal purposes. The fruit is known for its isoflavonoid contents, such as quercetin, kaempferol, and luteolin (98). A phase II clinical trial evaluated the effects of pomegranate juice consumption in men with a rising prostate-specific antigen level after surgery or radiotherapy for prostate cancer. The mean prostate-specific antigen doubling time significantly increased after treatment with pomegranate juice from a mean of 15 months at baseline to 54 months post-treatment. Further, a decrease in cell proliferation and an increase in apoptosis were observed in pomegranate-consuming patients (238). Selenium supplementation has also been found to reduce the incidence of prostate, colorectal, and lung cancers (82).
In addition to the reports just cited supporting the clinical efficacy of antioxidants, numerous preclinical studies have demonstrated the efficacy of antioxidants against cancer (39, 272). For instance, over-expression of Mn-SOD retarded the growth of prostate cancer cells both in vitro and in vivo (315). Over-expression of glutathione peroxidase has been associated with a decrease in pancreatic cancer growth in mice (191). Delivery of PEG-conjugated antioxidant enzymes has also shown promise in preventing tumor growth in a mouse model of melanoma (134, 233).
In summary, the use of nutraceuticals and antioxidants seems promising for reducing the risk of cancer. In addition, antioxidants have been shown to enhance the effectiveness of cancer chemotherapy by minimizing the associated side effects (10, 63). However, antioxidant-based cancer therapy has two caveats. First, the use of antioxidants can disturb the ROS-dependent normal cell function and promote tumor growth, especially when ROS are required for apoptotic cell death of precancerous and transformed cells. Second, the use of antioxidants would interfere with radiotherapy and chemotherapy, which are largely dependent on ROS that induce cytotoxicity in tumors (63, 273).
Role of ROS in cancer therapy
Chemotherapy
Sydney Farber and colleagues were the first that introduced the concept of chemotherapy for cancer treatment in 1948 (87, 88). The group found that an injection of a synthetic folic acid antagonist might be of value in the treatment of acute leukemia. Since then, a number of chemotherapeutic agents have been developed. Most of these agents work through ROS generation (330). Some have already been approved by the U.S. Food and Drug Administration (Tables 5 and 6), while others are still in clinical trials (Table 7).
ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; GTD, gestational trophoblastic disease; HL, hodgkin's lymphoma; MPE, malignant pleural effusion; NHL, non-Hodgkin's lymphoma.
BCR-ABL, breakpoint cluster region gene on chromosome22 and Abelson murine leukemia viral oncogene homologue; CTLA 4, cytotoxic T-lymphocyte-associated antigen 4; ER, estrogen receptor; GnRH, gonadotrophin releasing hormone; HDAC, histone deacetylase; HER2, human epidermal receptor 2; MM, multiple myeloma; PDGFR, platelet derived growth factor receptor; RANKL, receptor activated NF-κB ligand; SERD, selective estrogen receptor down regulator; SERM, selective estrogen receptor modulator; Src, sarcoma; TLR, Toll like receptor.
↓, down-regulation; ↑, up-regulation.
2-ME, 2-methoxyestradiol; ATN-224, choline tetrathiomolybdate; BSO, buthionine sulfoximine; GSH, glutathione reduced; NOX, NADPH oxidase; ROS, reactive oxygen species; SOD, superoxide dismutase; TrxR, thioredoxin reductase.
The cancer drugs approved by the U.S. Food and Drug Administration may be basically classified into two categories: nontargeted and targeted (265). The nontargeted drugs may be cell-cycle specific or cell-cycle nonspecific. The cell-cycle specific drugs act at specific phases during the cell-cycle progression, whereas the nonspecific drugs may act at any point (Table 5). Targeted cancer drugs block the growth and spread of cancer by interfering with signaling molecules, growth factors, and receptors associated with tumor growth and progression. Some of these targeted drugs are monoclonal antibodies such as rituximab, ibritumomab tiuxetan, ofatumumab, and alemtuzumab (Table 6). Procarbazine was one of the first drugs developed based on its ROS-generating properties (258). Procarbazine undergoes oxidation in aqueous solution and results in H2O2 production that is believed to be essential for the cytotoxic effects of the drug (30, 31). The drug is now approved for the treatment of Hodgkin's lymphoma, non-Hodgkin's lymphoma, and primary brain tumors (40, 112, 213, 214). As2O3 is another approved anti-cancer agent that has shown potential against acute promyelocytic leukemia. As2O3 has the ability to induce superoxide production in cancer cells (147, 234, 242, 277). As2O3 has also been shown to irreversibly inhibit mammalian thioredoxin reductase (TrxR) and impair mitochondrial functions (199).
Some of the anti-cancer agents that work through ROS generation and in development process are listed in Table 7. Motexafin gadolinium is an anticancer drug that selectively localizes in tumors. The molecular mechanism for ROS production by this drug appears to be inhibition of TrxR (207, 208). The drug exhibited modest anti-tumor activity in patients with chronic lymphocytic leukemia in a phase II trial (189). The efficacy of motexafin was demonstrated in another phase III trial with NSCLC patients who had brain metastases (217). Some compounds have exhibited anticancer activity in clinical trials through ROS generation, but their mechanism of ROS production is unknown. Elesclomol (STA-4783) is one such compound that has shown therapeutic activity against malignant melanoma; it was shown to prolong the progression-free survival of patients in a phase II clinical trial (163, 304). The progression-free survival induced by elesclomol in melanoma patients was further increased when given in combination with paclitaxel (304).
Anticancer agents have also been shown to enhance ROS stress in cancer cells by inhibiting the antioxidant defense system. SOD has emerged as one of the important targets under this category. For example, in a phase II clinical trial, a low dose of a SOD inhibitor (ATN-224) exhibited activity in patients with biochemically recurrent prostate cancer (187). 2-Methoxyestradiol is another known inhibitor of SOD that has the potential to increase superoxide radical levels (128). In a phase I clinical trial of patients with metastatic breast cancer, 2-methoxyestradiol, alone and in combination with docetaxel, was well tolerated (143). Another phase II randomized clinical trial evaluated the safety and efficacy of this drug for patients with prostate cancer; it was well tolerated and exhibited some anticancer activity (294).
Some of the anticancer agents target the GSH system. Examples include PEITC (301) and buthionine sulfoximine (BSO) (258). Although to our knowledge no clinical data on the efficacy and safety of PEITC with cancer patients are available, this drug has been shown to deplete cellular levels of GSH and to inhibit the activity of glutathione peroxidase (301). BSO is a specific inhibitor of glutamylsynthetase and, thus, can inhibit GSH synthesis. In a phase I study, administration of BSO and melphalan was found to be safe and significantly reduced the GSH content in cancer patients (23). Maeda et al. recently reported that the combination of BSO and As2O3 resulted in the effective treatment of advanced solid tumors (204). Imexon also has GSH-depleting, ROS-accumulating, and apoptosis-inducing potential, as revealed from a phase I study of patients with non-Hodgkin's lymphoma (79) and melanoma (325).
Two anticancer agents that are based on the modulation of NOX activity are minodronate and histamine (Table 7). Minodronate was found to be safe in a 60-year-old man with multiple myeloma in a phase I trial (279). In a phase III clinical trial, histamine was used as an adjunct to IL-2 therapy in melanoma patients. This agent was safe, well tolerated, and associated with a statistically significant prolongation of survival compared with IL-2 alone (2).
Radiotherapy
Similar to chemotherapy, radiotherapy employs ROS to eradicate cancer cells (22, 246). Radiotherapy uses X-rays, γ-rays, and, to a lesser extent, heavy particle radiation, such as with protons and neutrons. Radiation kills cancer cells by inducing apoptosis and mitotic failure and by inhibiting their proliferation (25, 175).
The role of ROS in mediating radiation-induced cancer cell killing is evident from a number of preclinical and clinical studies. For example, in a recent study, HIF-2α inhibition was found to enhance the response of lung cancer cells to radiation treatment that was associated with an accumulation of ROS and increased p53 activity (32). In another study, radiation induced death in prostate and breast cancer cells (74). Some other cancer types for which ROS have been shown to play a role in radiation-induced cancer cell death are lung adenocarcinoma (171), nonsmall-cell-lung cancer (290), prostate cancer (166), and breast cancer (7, 173). The role of ROS in mediating the anti-tumorigenic response of radiotherapy is evident from animal studies as well (278).
Clinical studies also support the role of ROS in mediating radiation-induced cancer cell death. For example, an elevated level of cellular damage induced by radiation was associated with increased ROS stress in patients with head and neck squamous cell carcinoma (109). ROS have been shown to play a role in the radiation-induced death of cells from cervical cancer patients as well (34). Other clinical studies for which ROS have been shown to play a role in radiation-induced therapy include patients with prostate cancer (148), NSCLC (104, 107, 144, 290), rectal cancer (81), and breast cancer (309).
Role of ROS in Eliminating Chemoresistance and Radioresistance
One of the major hurdles in treating cancer is that tumor cells, although initially sensitive, gradually develop resistance to chemotherapy and radiotherapy, in part owing to the induction of multidrug resistance proteins. Extensive research over the past several years has indicated that ROS-generating anticancer agents can reduce the chemoresistance and radioresistance of cancer cells. In this regard, nutraceuticals have shown promise in sensitizing tumor cells to chemotherapeutic and radiotherapeutic agents.
Curcumin has been shown to eliminate chemoresistant cells by sensitizing them to chemotherapy, in part by inhibiting pathways that lead to treatment resistance (94, 99). For example, curcumin treatment in conjunction with 5-fluorouracil (5-FU) or with both 5-FU and oxaliplatin resulted in significantly greater growth inhibition and more apoptosis in HCT116 and HT29 colon cancer cells than that caused by curcumin alone or 5-FU alone (240). In another study, curcumin given with tamoxifen resulted in synergistically induced apoptosis and autophagy in chemoresistant melanoma cells that correlated with an increase in ROS generation (48). An interesting finding from that study was that noncancerous cells were unaffected by the combination treatment (48). A number of other in vitro and in vivo studies have provided evidence for curcumin's use singly or as an adjunct to current chemotherapeutic drugs (99).
Other neutraceuticals have demonstrated usefulness in reducing tumor cell resistance to chemotherapy or radiotherapy. Emodin, an active component of Chinese medicinal herbs, was shown to enhance the sensitivity of gallbladder cancer cells to cisplatin in an ROS-dependent manner (320). Resveratrol is another nutraceutical that has shown potential in overcoming the chemoresistance of tumor cells (110). Our laboratory has identified a number of nutraceuticals over the past 5 years that can sensitize cancer cells to TNF-related apoptosis inducing ligand through an ROS-dependent mechanism. Some of these agents are nimbolide (111), ursolic acid (247), gossypol (293), γ-tocotrienol (151), and celastrol (292).
In addition to its role as a potent chemosensitizer, curcumin shows promise as a radiosensitizer in a wide variety of cancer cells (99). A sesquiterpene lactone was shown to sensitize prostate cancer cells to radiation by increasing ROS stress (166). 2-Methoxyestradiol has been shown to sensitize radioresistant breast cancer cells to γ radiation by generating ROS (264). The rare sugar D-allose was recently shown to enhance the efficacy of radiation against human head and neck cancer cells through ROS generation (124), and the natural compound Withaferin A sensitized renal cancer cells to radiation, also through ROS generation (340). As2O3 enhances the radiation response of cervical cancer cells (60, 150). Some other common cancers for which ROS has been shown to play a role in eliminating radioresistance are colon cancer (346), nasopharyngeal cancer (11), lung cancer (179), hepatoma (179), and leukemia (179).
Summary, Conclusion, and Future Perspectives
ROS are integral components of cell signaling pathways and have been shown to regulate cell transformation, survival, proliferation, invasion, angiogenesis, and metastasis. Paradoxically, ROS can also suppress tumor progression, and most chemotherapeutic and radiotherapeutic agents work by augmenting ROS stress in cancer cells. Due to the dual role of ROS, both pro-oxidant- and antioxidant-based anticancer agents have been developed. However, modulation of ROS signaling alone seems not to be an ideal approach, because some cancers are highly adapted to ROS stress, the redundant pathways supporting cancer growth are complex, and some ROS-generating anticancer drugs are associated with toxic side effects. Combinations of ROS-generating agents with agents that can break the redox adaptation could be a better strategy for enhancing cancer cell cytotoxicity. Due to their ROS-generating and multi-targeting properties, nutraceuticals might offer an advantage in selectively killing cancer cells. However, only a limited number of nutraceuticals have shown clinical efficacy, and none has been approved for human use. Future attempts in this direction will hopefully lead to the development of novel drugs.
Footnotes
Acknowledgments
The authors thank Elizabeth L. Hess of The University of Texas MD Anderson Cancer Center's Department of Scientific Publications for carefully editing the article and providing valuable comments. Dr. Aggarwal is the Ransom Horne, Jr., Professor of Cancer Research. This work was supported by a core grant from the National Institutes of Health (CA16672), a program project grant from the National Institutes of Health (NIH CA124787-01A2), and a grant from the Center for Targeted Therapy at the MD Anderson Cancer Center.