
Abstract
Introduction
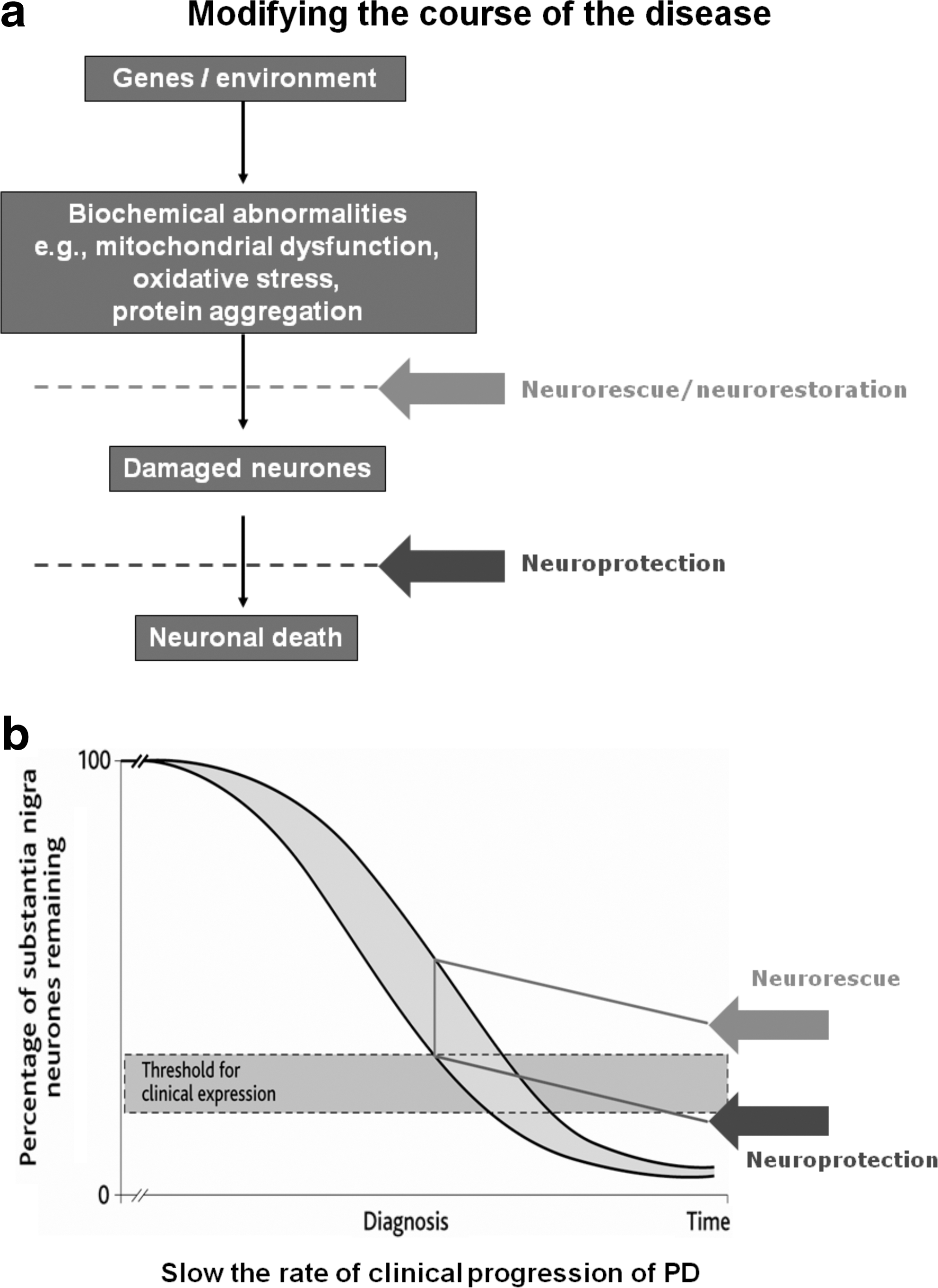
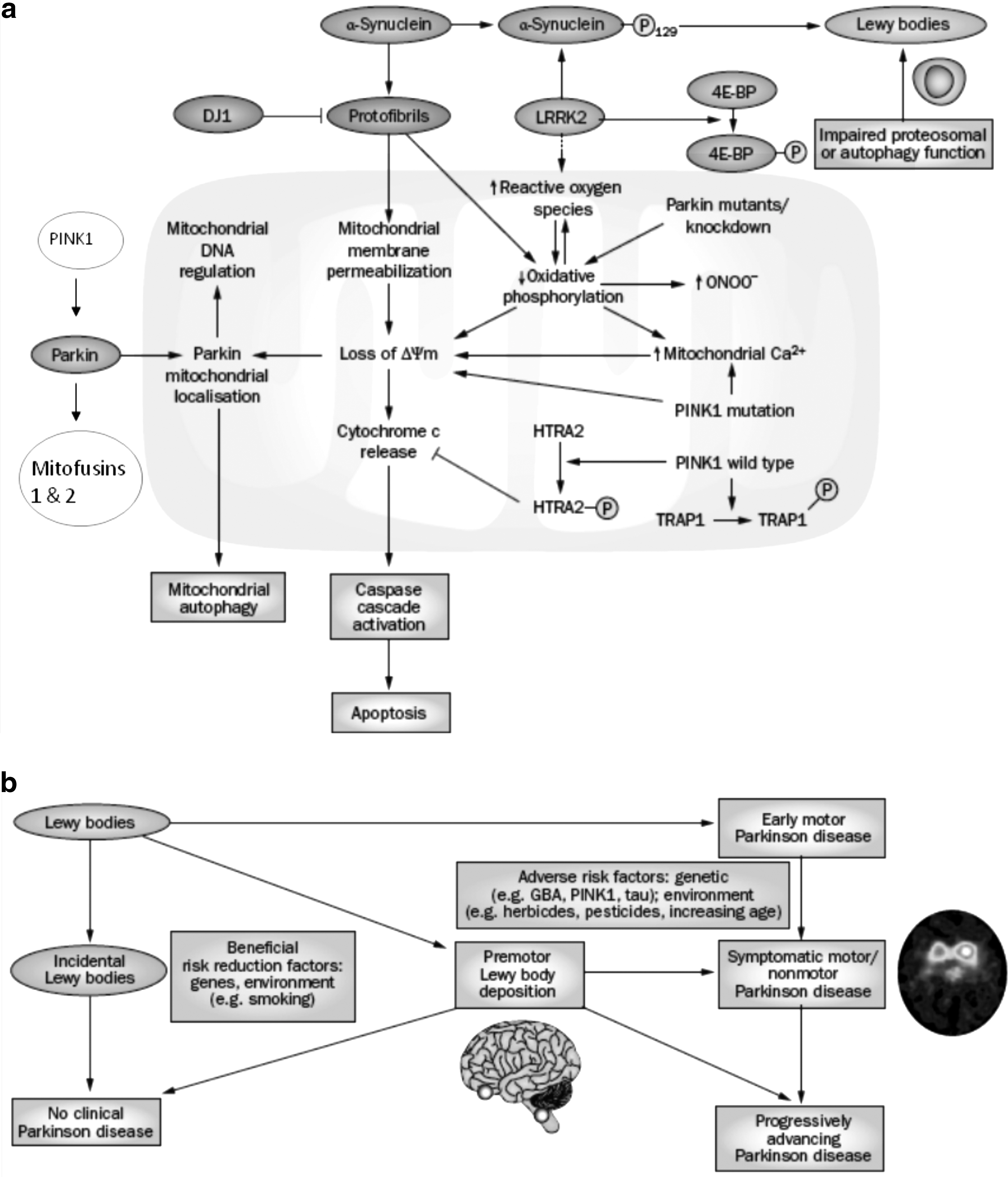
Adapted from Reference 62.
PD, Parkinson's disease.
This review will discuss attempts to manipulate mitochondrial function in PD that have already been undertaken, or in progress, and potential future targets for consideration.
Respiratory Chain Function and Oxidative Stress
The mitochondrion is a major source of reactive oxygen species by virtue of the reduction of oxygen by the respiratory chain and oxidative phosphorylation (OXPHOS) system. Defects of respiratory chain function, whether inherent or induced by toxins, cause an excess of free radical production. Senescence is associated with accumulating mitochondrial defects including declining mitochondrial OXPHOS function and increasing mitochondrial DNA mutations (30). This is of clear relevance to PD given that it is predominantly an age-related disease. Thus, any mitochondrial defect in PD will develop on the background of mitochondrial dysfunction due to age.
Coenzyme Q10 is an electron carrier and antioxidant, and is an integral part of the respiratory chain. This compound has been used for many years in the treatment of primary mitochondrial disease, although no formal clinical trial of sufficient size has been performed to confirm efficacy (36). In early PD patients, a dose of 1200 mg/day, coenzyme Q10 appeared to slow progression in a pilot study over a period of 16 months (69). Other studies using similar bioavailable levels have either failed to show benefit (71, 79). Additional clinical trials of coenzyme Q10 in early PD are underway and due to report in 2012. Coenzyme Q10 has been used in other neurodegenerative disorders including Huntington's disease (HD) and Friedreich ataxia. In HD it showed a trend to slowing progression, but did not reach significance (78). In Friedreich's ataxia, a combination of coenzyme Q10 and vitamin E improved neurological progression compared with 4-year cross-sectional natural progression data (19) and low dose was as effective as high dose therapy in improving function in those with coexisting coenzyme Q deficiency (11).
Creatine has also been investigated in PD and at a dose of 10 g daily, was well tolerated, and satisfied the predetermined criterion for nonfutility based on time to requirement for symptomatic therapy for 66 early PD patients (45). Another blinded placebo controlled study of 2 g daily for 6 months then 4 g daily for 18 months in 31 PD patients compared with 17 placebo showed no significant differences compared with placebo (3).
For an agent that is to have an effect upon mitochondrial function, adequate penetration into both the central nervous system and into mitochondria is essential. There are attempts to produce modified agents with either a positive charge that can be concentrated into mitochondria or a mitochondrial targeting sequence. Mito-Q is an example of the former and in vitro studies have demonstrated good mitochondrial penetration and activity (37). This compound is under evaluation in clinical trial. An interesting paradigm for brain penetration and mitochondrial concentration is 1-methyl-4-phenyl 1,2,3,6 tetrahydropyridine (MPTP). This toxin is widely distributed after intravenous administration and has good brain penetration. It is converted to 1-methyl 4-phenylpyridinium (MPP+) by monoamine oxidase (MAO) in glia. MPP+ is actively concentrated into mitochondria where it acts as a complex I inhibitor and free radical generator (10, 41) and inducer of a parkinsonian phenotype (29).
Apoptosis
The mitochondrion is an important mediator of intracellular signals for apoptosis (23, 81, 83). In part this depends on a sequence of mitochondrial depolarization and loss of membrane potential with the release of cytochrome c and the activation of a series of proapoptotic molecules (caspase cascade). Impaired function of OXPHOS or increased oxidative stress could act to lower the membrane potential and thus lower the threshold for apoptosis. A mutation in the mitochondrial-associated apoptosis initiating factor has recently been associated with X-linked infantile onset mitochondrial encephalomyopathy, partially responsive to riboflavin (18).
There are many potential therapeutic targets in the mitochondrial apoptosis cascade. The principle would be to inhibit apoptosis and so prevent cell death. However, such a strategy would have to be specific for neurons and would raise concerns regarding tumorogenesis. Furthermore, simply preventing damaged cells from dying may not restore function to the pathways involved. Despite these concerns, several agents have demonstrated antiapoptotic activity and have been considered for use in slowing progression in PD. Among these are selegiline and rasagiline, two irreversible inhibitors of MAO, with specificity for MAO-B. MAO is responsible for the breakdown of dopamine and its inhibition therefore increases synaptic dopamine levels and reuptake into presynaptic vesicles.
Selegiline
A number of studies have demonstrated the neuroprotective properties of selegiline (L-deprenyl) and its metabolite, desmethylselegiline (DMS), in rodents and primates (17, 20, 26, 35, 74). Early studies by Heikkila and colleagues established that MAO-B inhibitors, including selegiline, could block MPTP-induced degeneration of the nigrostriatal pathway in mice by preventing the conversion of MPTP to its toxic metabolite, MPP+ (20). Tatton and Greenwood later showed that selegiline could increase survival of dopamine neurons in the substantia nigra in mice through a mechanism independent of blockade of MPTP conversion to MPP+ (74). Subsequent studies by Matsubara and colleagues showed that selegiline reduced lactate accumulation in the striatum following MPP+ and suggested that this reflected the improved function of OXPHOS (35).
Selegiline and DMS protected SH-SY5Y cells against apoptosis induced by peroxynitrate, a reactive oxygen species generated from N-morpholino sydnonimine (SIN-1), and suggested that protection was independent of MAO-B activity, since SH-SY5Y cells contain only MAO-A (17, 26, 33). It was suggested that (—)-deprenyl exerted antiapoptotic effects through the modulation of gene expression and induction of new protein synthesis rather than MAO inhibition (75, 76). Selegiline and DMS prevent neurotoxin and trophic factor withdrawal-induced cell death by virtue of their ability to induce antiapoptotic molecules, such as Bcl-xL, Bcl-2, glutathione, and superoxide dismutases 1 and 2 (73, 77). These molecules maintain closure of the mitochondrial permeability transition pore (PTP), whose opening is an important component of the signaling process leading to apoptosis (32, 67). Selegiline/DMS also exert antiapoptotic effects by downregulating proapoptotic molecules, including c-Jun (which activates the caspase cascade), nitrous oxide synthase, and Bax (which promotes apoptosis by inducing PTP opening), and by preventing nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase, an enzyme whose activation is involved in the early stages of apoptosis (32, 38, 67). Thus, it now appears that (—)-deprenyl-related propargylamines exert antiapoptotic effects via alteration of proteins involved in oxidative damage, mitochondrial membrane permeability, or apoptotic signaling pathways (77).
Several clinical studies have sought to evaluate the potential for selegiline to have a neuroprotective or disease modifying effect. The Deprenyl And Tocopherol Antioxidative Therapy Of Parkinsonism (DATATOP) study, a double-blind, multicenter, placebo-controlled clinical trial was undertaken in early PD (80). The clinical trial design combined selegiline with α-tocopherol. The DATATOP trial specifically evaluated whether chronic administration of deprenyl (10 mg/day) and/or tocopherol (2000 IU/day) to patients with early, untreated PD (duration of illness ≤5 years) would extend the time until sufficient disability developed to require L-dopa therapy (primary end point) (80). Analysis at 12±5 months of the study's start revealed the emergence of a major treatment effect: 176 of 401 subjects in the placebo group but only 97 of 399 subjects in the deprenyl groups had reached the primary end point (development of sufficient disability to require L-dopa therapy) (31). No beneficial effects of tocopherol were detected. The results suggested that chronic administration of selegiline (deprenyl) and/or tocopherol in otherwise untreated PD patients could extend the time until disability required therapy with L-dopa. The DATATOP results are now generally attributed to symptomatic benefits of selegiline (31, 59).
Rasagiline
Rasagiline is up to 15 times more potent than selegiline as an MAOB inhibitor in animal models in vivo and in vitro (2, 86). Rasagiline is metabolized to aminoindan via the hepatic cytochrome P450 (CYP) isozyme CYP1A2 (8). While some reports suggest that the aminoindan metabolite of rasagiline has neuroprotective activity, L-methamphetamine—the major metabolite of selegiline—has neurotoxic activity in vitro and blocks the neuroprotective action of selegiline and rasagiline (2).
In vitro studies support structure-activity analysis that suggests the antiapoptotic, neuroprotective activity of rasagiline resides in the propargyl moiety and is not related to MAO inhibition. TVP1022, the S-enantiomer of rasagiline, has 1000-fold weaker MAO inhibitory activity but exhibits similar neuroprotective effects in vitro (34, 84, 85). The neuroprotective activity of rasagiline has been demonstrated in vitro against a variety of neurotoxic insults, including the nitric oxide donor SIN-1, as well as glutamate, 6-hydroxydopamine (6-OHDA), MPTP, β-amyloid, 1,2,3,4-tetrahydroisoquinoline, and serum and growth factor deprivation (32).
A number of in vitro studies have suggested several molecular mechanisms by which rasagiline prevents apoptosis (reviewed in 55, 58). Rasagiline significantly reduced cell death following serum deprivation, and prevented the appearance of cleaved forms of caspase-3 and the caspase substrate poly(ADP-ribose) polymerase (PARP). Results showed that GF109203×, a broad-spectrum protein kinase C (PKC) inhibitor, markedly reversed rasagiline's suppressive effect on the cleavage and activation of caspase-3 and PARP, suggesting that the PKC pathway mediates neuroprotection by rasagiline. Reverse transcriptase-polymerase chain reaction analysis revealed that treatment of PC12 cells with rasagiline for 24 h significantly increased expression of the PKC isoenzymes PKC-α and PKC-ɛ and antiapoptotic Bcl-2 family members Bcl-xL and Bcl-w, while decreasing the proapoptotic Bcl-2 family member, Bad. These authors suggested that the activation of PKC in association with the Bcl-2 protein family mediates the neuroprotective activity of rasagiline.
The potential for rasagiline to modify the progression of PD was assessed in the Attenuation of Disease progression with Azilect Given Once-daily (ADAGIO) study. This was an 18-month, double-blind, placebo-controlled, multicenter trial that used a delayed-start design (44). The delayed-start design can, with certain assumptions, define a difference between a pure symptomatic effect and one that maybe associated with modification of the progression of the disease (51, 53). Thus, in ADAGIO, 1176 patients with early PD were randomized to 1 or 2 mg of rasagiline or placebo and maintained blind for 9 months at which point patients were re-randomized to take 1 or 2 mg of rasagiline. In effect this involved those patients already taking a dose of active drug continuing on this, while those on placebo were transferred to one of the two doses of rasagiline.
The primary analysis comprised three hierarchical end points based on the change from baseline in the total Unified Parkinson's Disease Rating Scale (UPDRS) score. In the first, the rate of progression after week 12 had to be significantly slower during the first 9 months in the early start group. For the second, there had to be a significant difference at 18 months in UPDRS scores between the early and late starters. Finally, the rate of progression of UPDRS in months 9 to 18 should be at least parallel and not converging. The 1 mg dose of rasagiline met all three end points (Fig. 3); the 2 mg dose met the first, but failed the second and therefore failed the hierarchical analysis.

The mechanism by which 1 mg rasagiline caused its effect is not revealed by the ADAGIO trial. Indeed, there are several interpretations (54, 60, 61). One mechanism may be through the upregulation of protective and downregulation of harmful compensatory mechanisms that have developed over the long prodrome of PD and maintained normal motor function despite falling dopamine levels (56). Alternatively, rasagiline may be exerting a true protective effect, in line with the laboratory data presented previously. Nevertheless, a symptomatic action that accumulates over a period greater than 9 months, although unlikely, cannot be excluded (63).
Post hoc analysis of the ADAGIO study has shown that the only significant difference between the early and late starters was in the activities of daily living scores, with the 1 mg dose (49). This also showed that those patients with the lower UPDRS scores at baseline progressed slower than those with higher scores, and that the younger patients tended to progress faster (65).
Other inhibitors of apoptosis have also come to clinical trial, but have not demonstrated an ability to slow progression of PD (28, 42).
Autophagy
Mitochondria may divide or bud (fission) or fuse, and this fission–fusion process is regulated by several signaling proteins including mitofusins 1 and 2 that mediate fusion of mitochondrial outer membranes, and optic atrophy protein 1, a dynamin-like GTPase involved in fusion of the inner membranes. Dynamin-1–like protein 1 and fission 1 homologue regulate mitochondrial fission (9).
The fission–fusion process maintains the effectiveness, or quality, of cell mitochondria (6, 7, 12). Defective mitochondria, as identified by a fall in mitochondrial potential, may recover and return to the mitochondrial pool by fusion, or be destroyed by autophagy. The process of mitochondrial destruction by autophagy is referred to as “mitophagy.” Parkin and PINK1 proteins have recently been identified as playing an important role in the fission–fusion and mitophagy pathways (13, 47, 66). Parkin translocates from the cytosol to the mitochondrion in response to a fall in mitochondrial membrane potential (39). Recent data suggest that this is preceded by phosphorylation of parkin by PINK1 (13, 25, 39, 47), although other studies have failed to replicate this observation (40, 82). Parkin and PINK1 involvement in mitophagy includes the ubiquitination of MFN 1 and 2, and the mitochondrial voltage-gated anion channel by parkin (14, 16). HtrA2 is thought to participate in mitochondrial turnover and has also been identified as a phosphorylation target of PINK1 (46). The relevance of mitophagy to mitochondrial pathology is emphasized by evidence that defects of the mitophagy pathway contribute to PD pathogenesis (15).The phosphorylation of HtRA2 is dependent on PINK1, probably via a kinase cascade, rather than as a direct substrate (46). Mutations in the HtRA2 gene are a possible cause of familial PD (5). The mitochondrial chaperone TRAP1 has been shown to be a direct substrate of PINK1 (48). These data suggest that PINK1 might be involved in the degradation of mitochondrial proteins as well as mitochondria as a whole.
Importantly, it now appears that mutations of parkin or PINK1 that cause PD interfere with mitophagy efficiency and result in accumulation of defective mitochondria (14). This can be reversed by upregulation of parkin or PINK1 and the removal of defective mitochondria. The recent demonstration of abnormal expression of autophagy proteins in PD brain has further highlighted the importance of degradation pathways to the pathogenesis of this disease (1). These observations offer intriguing opportunities for the development of future therapies for disorders that involve impaired mitochondrial function. In this context it is of note that upregulation of the translation inhibitor 4E-BP ameliorates the effects of PINK1/parkin mutants in Drosophila, and rapamycin, a drug that activates 4E-BP and autophagy, is also protective in these mutants (72).
Mitochondrial Biogenesis
An interesting strategy for reversing the effects of mitochondrial dysfunction might be through the regulation of mitochondrial protein transcription and biogenesis. Peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1α (PGC-1α) is an important regulator of mitochondrial activity (see box) and functions together with SIRT1 to influence genetic programs for mitochondrial biogenesis. SIRT1 is one of the family of situins, known to catalyze NAD+-dependent protein deacetylation, yielding nicotinamide and O-acetyl-ADP-ribose and to regulate longevity, apoptosis, and DNA repair (4). SIRT1 interacts directly with and deacetylates PGC-1α and increases PGC-1a activity leading to the induction of gene transcription. PGC-1α and SIRT1 promote adaptation to caloric restriction by regulating the genetic programs for gluconeogenesis and glycolysis (50). Activation of PPARγ transactivates target genes with the support of PGC-1α. Drugs that activate PPARγ include rosiglitazone, pioglitazone, and troglitazone—currently available for treatment of diabetes mellitus. Resveratrol (RSV) is a natural polyphenolic compound found in the skin of grapes and has been shown significantly to increase SIRT1 activity (21). RSV enhances PGC-1α activity, increasing mitochondrial biogenesis and rendered animals resistant to diet-induced obesity and insulin resistance (27). RSV or bezafibrate, another PGC-1α agonist, has demonstrated protective properties in animal models of PD (22, 24).
There is evidence that PGC-1α upregulation enhances antioxidant activity and inhibition of this effect enhances cell death (70). A recently identified parkin interacting protein, termed PARIS, downregulates PGC-1α but this action is itself inhibited by parkin. Thus this represents a direct axis between a cause of familial PD and PGC-1α (68).
Conclusion
Mitochondria are considered an important component of PD pathogenesis, and as such represent a reasonable target for therapeutic intervention to modify the progression of the disease. Several attempts have been made to enhance mitochondrial OXPHOS, reduce oxidative stress, or decrease mitochondrial-mediated apoptosis. None have proven unequivocally effective. This probably reflects in part the rather nonspecific nature of these targets. Perhaps more promising pathways include those of intervening in mitochondrial biogenesis and mitophagy, that is, in the turnover of mitochondria to ensure a healthy population of organelles. However, the proportion of PD patients in whom mitochondrial dysfunction plays a prominent or primary role remains unknown. Thus the clinical trials quoted previously very likely contained a heterogeneous PD population in terms of etiopathogenesis. A single therapeutic agent designed only to improve mitochondrial function, including apoptosis, may thus not be beneficial in those in whom these play no part. This also reflects the complexity of clinical trial design for neuroprotection: the population studies, the end points selected, and so on (43, 65, 66). Although much work needs to be done in this area, developments in clinical trial design for disease modification must also progress in parallel.
Footnotes
Author Disclosure Statement
The author has received honoraria for educational symposia and advisory boards for Teva-Lundbeck.