
Abstract
Introduction
Redox homeostasis, the balance between cellular pro- and antioxidant status, is crucial for various physiological processes (7, 72). It controls the level of reactive oxygen species (ROS), derivatives of O2, that are constantly formed as side products of biological reactions that use electron transfers, such as oxidative phosphorylation, oxidases, and reductases activity. Superoxide radical (O2
Intracellular Sources of ROS
Mitochondrial generation of ROS
Under physiological conditions, mitochondria are a major intracellular source of ROS. Mitochondria are the site of oxidative phosphorylation that is carried out by four electron-transporting complexes and one H+-translocating ATP synthetic complex. During oxidative phosphorylation, electrons are transferred from electron donors to electron acceptors in redox reactions. During this process, some electrons may leak onto oxygen, resulting in the one-electron reduction of O2 to generate superoxide (O2
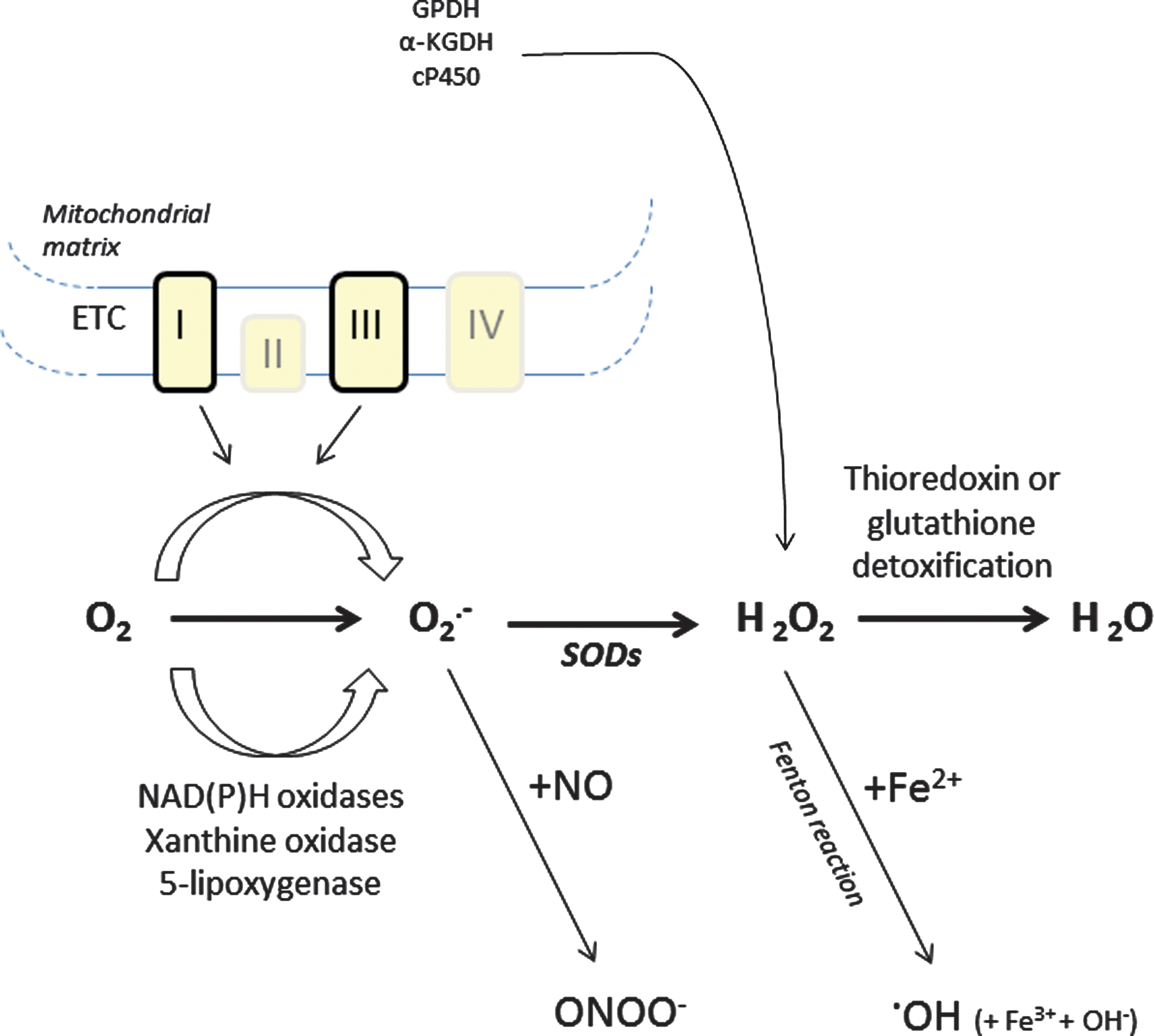
Nonmitochondrial production of ROS
The superoxide anion can also be formed enzymatically by enzymes such as NAD(P)H oxidases, 5-lipoxygenase, or xanthine oxidase (Fig. 1). NADPH oxidases (Nox) were first studied in phagocytic leukocytes such as macrophages and neutrophils (46, 63). All Nox family members are transmembrane carriers that use NADPH as an electron source and molecular oxygen as an acceptor. These chemical reactions are invariably associated with the formation of O2
ROS in cell fate signaling
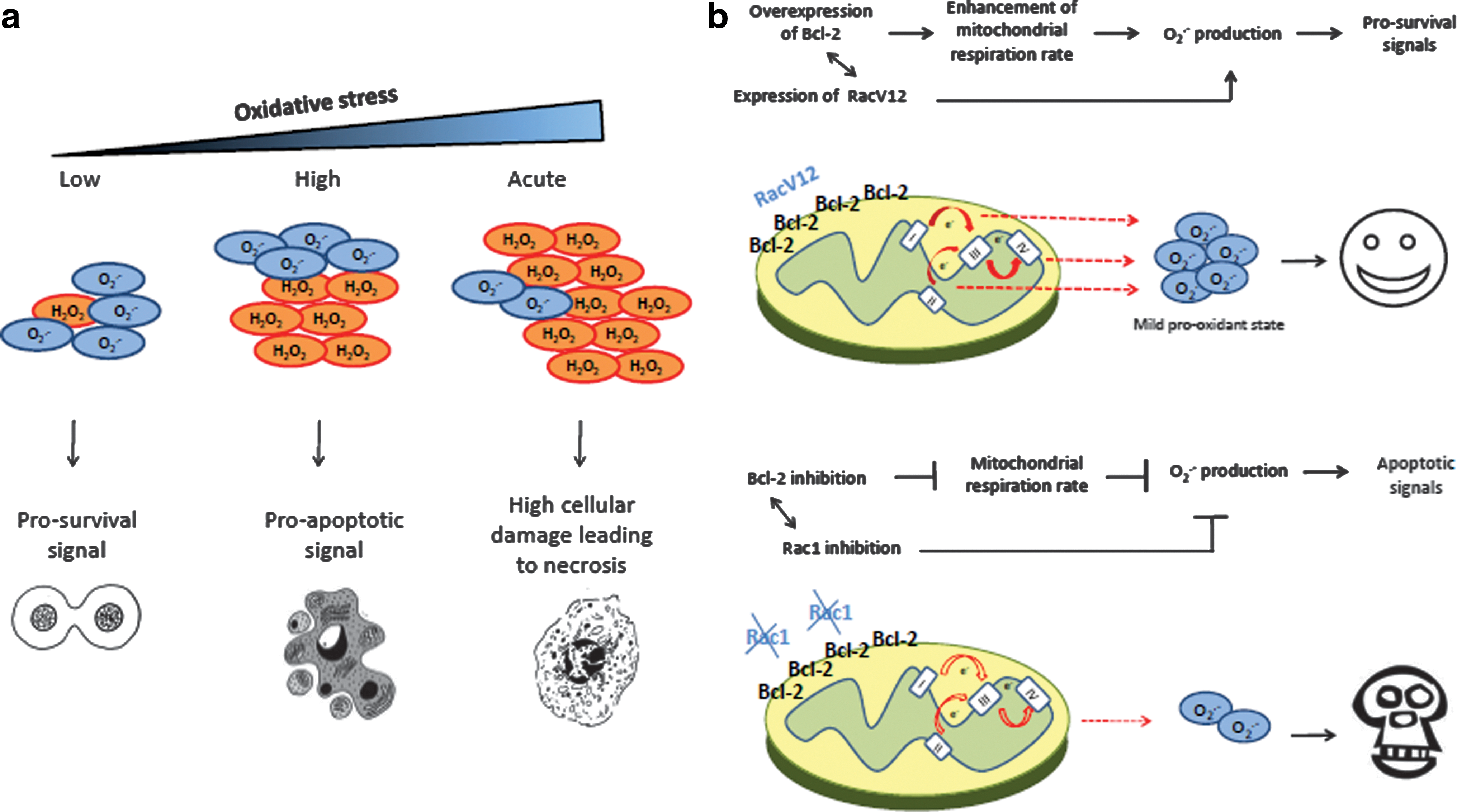
There is now overwhelming evidence that cellular redox status impacts cell fate decisions by regulating critical cellular processes such as growth and proliferation, gene expression, cell survival and death, and protein function. Traditionally, severe oxidative stress induced by ROS such as O2
Redox Regulation of p53
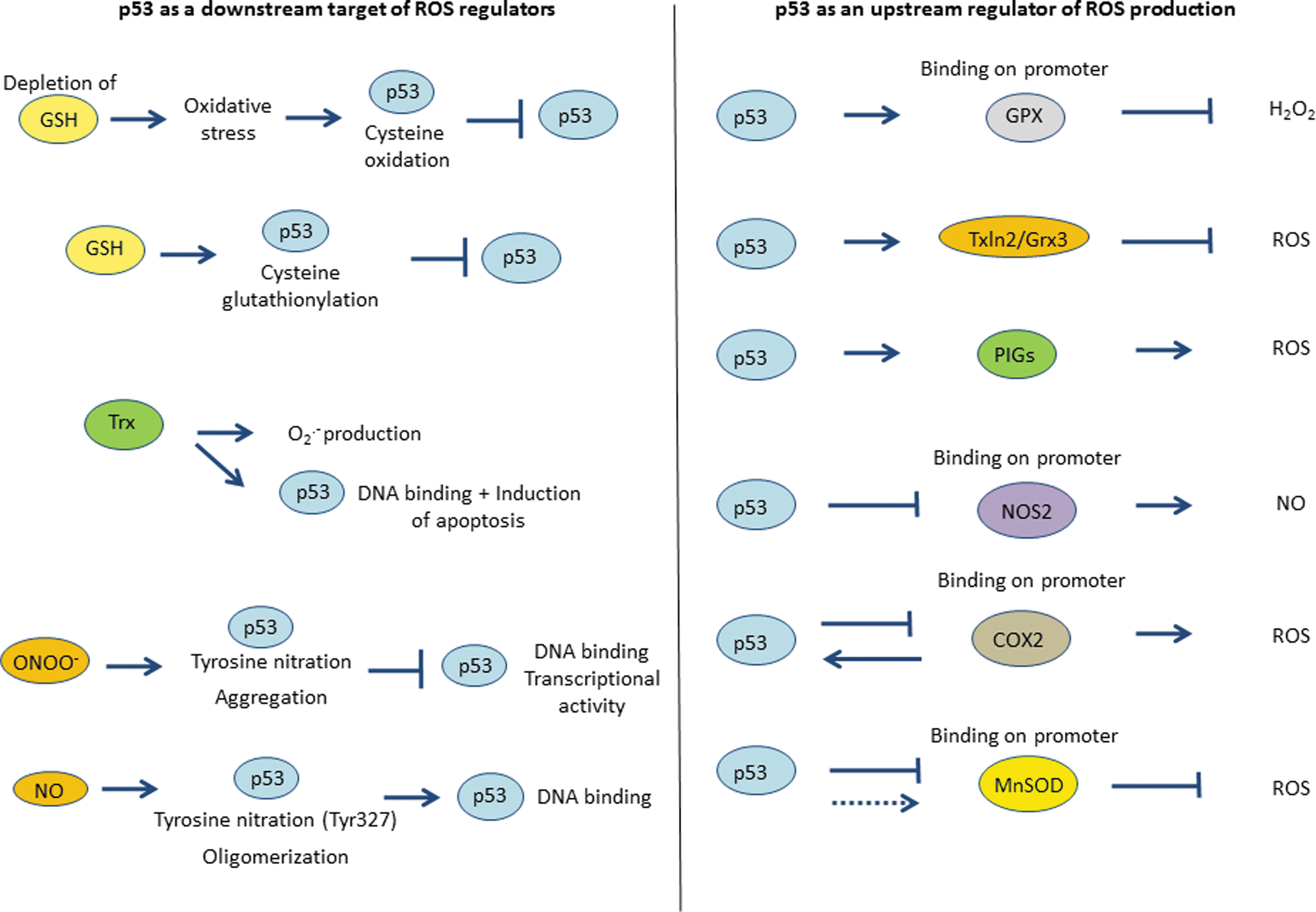
Modifications of amino acids by ROS are important to control the activity of many transcription factors, including HIF-1 alpha, Sp1, NF-κB, and p53 (23, 49, 89). Various endogenous and exogenous molecules have been shown to modify the redox status of p53. These include, but are not limited to, glutathione (GSH), thioredoxin (Trx)/thioredoxin reductase (TrxR), and nitrating compounds.
The GSH system
GSH is a major cellular antioxidant whose thiol group is a potent reducing agent. It inactivates electrophilic compounds and peroxides via catalysis by glutathione S-transferases and also serves as a substrate for glutathione peroxidase (GPX), which removes H2O2 accumulated in the cells (87). GSH is a tripeptide that can exist either in an oxidized (GSSG) or in a reduced (GSH) state (Fig. 3a). Maintaining optimal GSH:GSSG ratios in the cell is critical to survival; therefore, a tight regulation of this system is necessary. Russo et al. first investigated whether COS-2 and Hep3B cells treated by diethylmaleate (DEM), an agent that increases the concentration of free radicals by depleting cellular stores of GSH, showed modification of p53 DNA binding or transcriptional activity. Indeed, DEM-induced oxidative stress significantly reduced p53 DNA-binding and activation of its specific reporter genes (79). More recently, Velu et al. showed that p53 was a substrate for glutathionylation under oxidative stress and that cysteine 141 was the most reactive cysteine residue (91). Furthermore, a molecular modeling study showed that glutathionylation of this residue inhibits p53-DNA association and also interferes with protein dimerization (94). The glutathionylated p53 was detected in the nucleus, which was associated with the elimination of p53 activities and oncogenesis. These results show that S-glutathionylation of p53 cysteine residue was associated with a loss of function of the protein (Figs. 3b and 4). Along similar lines, Richie et al. examined the effect of GSH on spontaneous tumor development by using p53 knock-out transgenic mice. They treated the mice with a GSH-specific inhibitor of GSH synthesis (buthionine sulfoximine [BSO]) or a cysteine and GSH precursor (1,2-oxothiazolidine-4-carboxylic acid or OTCA) and demonstrated that GSH levels were decreased up to 88% in the BSO-treated group. After 10 weeks, mice in both groups showed a high frequency of lymphomas (80%) and other tumors (38%). Furthermore, the incidence of colon tumor was seen to be increased by fivefold in the BSO-treated group compared to the control group, showing that GSH and p53 have contributory roles in colon carcinogenesis (77).

The Trx system
Trx are a family of small redox proteins that undergo NADPH-dependent reduction by TrxR and reduce oxidized cysteine groups on proteins (Fig. 3a). Oxidation of cysteine residues on proteins results in disulfide bond formation, thereby inducing a conformational change in the protein. The Trx/TrxR system has been shown to reverse the inactivating effect of oxidative stress on target proteins (6, 28). Similar to GSH molecule, Trx might play a direct or indirect role in regulating the redox status of p53.
Ravi et al. used antharacyclines, a class of ROS-producing antitumor drugs, to study the effect of Trx on p53. They showed that Trx increased p53-dependent apoptosis induced by daunomycin through elevated semiquinone production via redox cycling. Interestingly, using MCF-7 cells overexpressing Trx, they demonstrated significant induction of apoptosis upon daunomycin treatment, which was associated with intracellular generation of O2
It has been demonstrated that the covalent modification of TrxR by both endogenous and exogenous electrophiles causes the disruption of p53 protein conformation (62). Cassidy et al. reported that RKO colon cancer cells with damaged or deficient TrxR enzymatic activity by dint of electrophilic modification were less sensitive to electrophile-induced alteration in p53 conformation and apoptosis, compared to cells with normal TrxR activity. Transfection of TrxR-depleted cells with C-terminal mutants of TrxR lacking the catalytic selenocysteine led to the disruption of p53 conformation in a similar way to that induced by electrophiles in cells expressing wild-type TrxR (11). Furthermore, Stoner et al. demonstrated that p53 activity was unimpaired in yeast lacking Trx1 and Trx2 genes encoding cytosolic Trx. Subsequent analysis demonstrated that the inhibitory effect of TrxR was also deleted in yeast, suggesting that accumulation of oxidized Trx was necessary for p53 inhibition (81). These results provide evidence that formation of intramolecular disulfide bonds within p53 must be reduced by TrxR to facilitate the DNA binding activity of p53 (Figs. 3c and 4). To that end, Redox factor-1 (Ref-1) has been suggested as an intermediate in the inhibitory effect of oxidized Trx on p53 activity (42).
Reactive nitrogen species and p53
Reactive nitrogen species (NO or ONOO−) can also modulate the redox status of p53 via nitration of critical tyrosine residues present in its DNA binding-domain (Fig. 4). ONOO− is produced by excess of NO and O2
p53: A Transcriptional Regulator of Redox Effectors
p53 and ROS are both implicated in multiple physiological pathways. In order to better understand the relationship between p53 and ROS, it is fundamental to investigate how p53 expression and distribution influences intracellular ROS production. As a transcription factor, p53 can induce either transactivation or transrepression of its downstream targets. Among the redox-controlling genes specifically upregulated in a p53-dependent manner is GPX, PIGs, and glutaredoxin 3 (Grx3), while two other important redox modulators, cyclooxygenase 2 (COX2) and inducible nitric oxide synthase 2 (NOS2), are repressed upon p53 activation (Fig. 4).
GPX and Grx3
GPX is one of the primary cellular antioxidant enzymes that scavenge H2O2 and organic hydroperoxides using GSH as the hydrogen donor (Fig. 3a) (59). Tan et al. reported p53-binding site in the promoter of GPX. After determining a putative binding site by analysis of the sequence of the GPX gene, they showed that purified p53 protein or p53 obtained from nuclear extracts were binding to the promoter of GPX in U2-OS cells after exposure to the apoptosis-inducing agent, etoposide. Furthermore, p53 binding and transactivation of GPX promoter enhancement was blocked with a dominant negative form of p53 (84). These results, suggesting that GPX was a novel target of p53, were confirmed by a subsequent study conducted by Hussain et al. In their analysis, TK6 cells (p53 wild type) and WTK1 cells (p53 mutated) were exposed to doxorubucin and subjected to microarray analysis. The results showed an increase in the induction of GPX in TK6 cells compared with WTK1 cells, demonstrating that GPX was upregulated in a p53-dependent manner (41).
Glutaredoxins are a group of redox enzymes that are reduced by the oxidation of GSH. A recent study has described that Grx3, also named Txnl2, is a p53 target and is upregulated after NGF stimulation (10). Furthermore, Txln2/Grx3 has been shown to be involved in growth and metastasis regulation through redox mechanisms. Of note, knockdown of Txln2 in breast cancer cells resulted in an increase in ROS production, which correlated with inhibition of proliferation in vitro and inhibition of tumorigenesis in vivo (73). Moreover, Cheng et al. demonstrated that Grx3 is ubiquitously expressed in adult mice and embryos and its expression is induced under oxidative stress. Grx3−/− mouse embryonic fibroblasts had impaired growth and cell cycle progression (12).
p53-induced genes
PIGs were first discovered by Polyak et al. in a screen for genes induced by p53 during p53-induced apoptosis. In this study, Polyak et al. predicted that many of these genes encoded proteins that impacted cellular redox status, including two ROS-generating enzymes: PIG3, a homolog of the quinone oxidoreductase (NQO1), and PIG6, a homolog of the proline oxidase (POX) (71). In line with this, more recent reports strongly suggest that PIG3 and PIG6 are involved in the intracellular generation of ROS and ROS-mediated apoptosis (65, 78). However, PIG3 is not only restricted to the apoptotic response but also plays a role during p53-mediated growth arrest. It has recently been shown that depletion of PIG3 sensitized cells to DNA damage-inducing agents and impaired DNA repair (30, 51). Donald et al. showed that p53 was controlling PIG6/POX expression and consequent ROS production. POX induces ROS production when catalyzing the conversion of proline to pyrroline-5-carboxylate. When POX was conditionally expressed in p53-negative DLD-1 colon cancer cells, proline-dependent generation of ROS required the induction of POX (24). Furthermore, a recent study published by Abbas et al. has demonstrated a correlation between ROS production and PIGs overexpression in an in vivo model of p53 lacking apoptosis-inducing ability (hypomorphic allele p53R172P encoded by p53515C). In this study, they showed that Mdm2−/− p53515C/515C new-born mice had high ROS levels in whole bone marrow in comparison to Mdm2+/− p53515C/515C animals. These pups also presented higher PIG1, PIG8, and PIG12 levels compared with the control animals. These data give strong evidence that p53, via its role on PIGs, can be a key player in ROS production in vivo via its regulation by Mdm2 (1).
Nitric oxide synthase and cyclooxygenase
NOS catalyze the synthesis of NO from L-arginine, oxygen, and NADPH. High concentrations of the free radical NO can induce DNA damage and apoptosis in many cell types; therefore, regulation of NOS activity is fundamental to preserve cells integrity. Forrester et al. demonstrated that the expression of wt p53 in various human tumor cell lines or in murine fibroblasts resulted in the down-regulation of NOS2, the inducible form of NOS, through inhibition of its promoter (31). Later on, the same team showed that NOS2 was upregulated in p53 knock-out mice (5). Altogether, these results demonstrate that p53 is a transrepressor of NOS2 expression and attenuates NO production in a regulatory negative feedback loop.
Cyclooxygenase 2 (COX2), which catalyzes the conversion of arachidonic acid to prostaglandin, has been shown to generate ROS and has been linked to neuronal oxidative stress (44, 47). Similar to NOS, COX2 expression has been linked to p53 activity; COX2 protein and mRNA levels were markedly suppressed by wt p53 as well as the activity of the COX2 promoter was reduced by 85% in response to p53 (82). Thus, p53 might also play a role in modulating oxidative stress through its regulation of COX2. Interestingly, it should be pointed out that p53 and COX2 can reciprocally regulate each other (13, 52).
p53 and Regulation of Mitochondrial ROS Production
In addition to its role in response to cellular stress and DNA damage, p53 has been described as a modulator of mitochondrial respiration (60) and mitochondrial ROS production (45). In p53 null mouse and p53 knock-down fibroblasts, both the mitochondrial DNA and the mitochondrial mass are decreased. This is correlated with a disruption of cellular ROS homeostasis, characterized by a reduction in O2
In addition, Achanta et al. demonstrated that in response to oxidative mtDNA damage, p53 translocated to the mitochondria, where it interacted with mtDNA polymerase γ and enhanced the DNA replication function of the enzyme. As some components of the ETC are encoded by mtDNA and that a well-controlled functioning of the ETC is necessary to prevent spurious generation of O2
Another way by which p53 could regulate cellular ROS production is via its ability to modulate antioxidant genes expression, such as the mitochondrial MnSOD/SOD2. A few recent studies have investigated the link between p53 activity and MnSOD expression. Pani et al. demonstrated that E1A-Ras-transformed fibroblasts lacking p53 showed increased MnSOD expression in comparison to wild-type controls. This is associated with a higher resistance to apoptosis induced by serum deprivation or by treatment with ROS-generating agents, such as paraquat and adriamycin. Furthermore, transfection of HeLa cells with wt p53 led to a drop in MnSOD mRNA level and activity (66). Taken together, these results describe MnSOD as a downstream target of p53, which can be specifically downregulated by the tumor suppressor. In addition, Drane et al. showed that p53 repression of MnSOD was mediated via an effect on the gene promoter. Indeed, using a plasmid pluc-SOD2 in which the luciferase reporter gene was placed under the control of SOD2 promoter, they showed that luciferase activity was three times higher in MCF-7 cells expressing wt p53 than those expressing mutant p53 (26). Further studies have supported these findings and demonstrated that p53, NF-κB, and Sp1 were acting as co-factors for MnSOD activation (21).
In contrast to the repressive effect of p53 on MnSOD expression, a positive effect of p53 on MnSOD has also been reported. To that end, arsenite-induced apoptosis of human leukemia cells resulted in a slight upregulation of MnSOD, which was dependent on intracellular ROS-mediated activation of the MAP kinase, extracellular signal-regulated kinase (Erk2). After its nuclear translocation, Erk2 was responsible for the phosphorylation of p53 dowsntream MnSOD gene induction (55). It has been postulated that this bi-modal effect of p53 on MnSOD could be a function of the level of activation of p53. For example, in PC3 human prostate cancer cell line low level of p53 increases MnSOD gene transcription in a p65-dependent and NF-κB-dependent manner, but, on the other hand, robust activation of p53 abrogated p65-driven transcription of MnSOD resulting in its downregulation (22). Taken together, these results indicate that the effect of p53 on MnSOD expression remains ambiguous and further investigations are needed to elucidate the nature of the crosstalk between p53 and MnSOD. Analysis of the domains involved in p53-MnSOD interaction could clarify the complex role of p53 in MnSOD regulation and, thus, its involvement in mitochondrial ROS production.
The ROS regulatory activity of p53 at the mitochondria could be compromised by pro-apoptotic proteins of the Bcl-2 family such as Bax and PUMA that induce mitochondria leading to ROS production by a less efficient ETC (80). Interestingly, both these pro-oxidant proteins (Bax and PUMA) are transcriptional targets of p53 (57, 61). Furthermore, p66Shc, another downstream target of p53, under pro-apoptotic stress generates H2O2 production utilizing reducing equivalents of the mitochondrial ETC through the oxidation of cytochrome c. Finally, this leads to opening of the mitochondrial permeability transition pore and to apoptosis (32).
Conclusion
In this review, we have summarized the results of recent studies focused on exploring the interactions between p53 and ROS pathways. Indeed, as p53 and ROS participate in many cellular processes, it has become evident that there is a close crosstalk between these two signaling networks. p53 has been shown to transactivate genes implicated in the regulation of cellular redox status. That p53 can directly or indirectly impact antioxidant genes such as GPx, SOD2, sestrins, and aldehyde dehydrogenase-4, as well as prooxidant genes such as PIGs, is rather intriguing and clearly underscores the wide spectrum of cellular proteins and signaling circuits impacted by this remarkable transcription factor. It has been proposed that these genes can be regulated at different stages after p53 induction; antioxidant genes, such as GPX, activated at an early time point (83) would be part of an initial protective response against oxidative stress, whereas prooxidant genes, such as PIGs, would be activated as a late response and leading to ROS generation (30). Thus, p53 has a definite role in redox homeostasis but is also a target of ROS and susceptible to redox modification at its cysteine or tyrosine residues. These modifications can directly affect the stability, DNA-binding, and transactivation potential of the protein. However, p53-mediated ROS generation is a function of its level of activity as well as subcellular localization, and this phenomenon is regulated by a feedback loop because ROS alter p53 subcellular localization and transcriptional activity.