
Abstract
Introduction
The redox system plays an important role in the survival of the parasite in the host (150). All aerobic organisms are exposed to reactive oxygen species (ROS) such as superoxide anions (O2 •−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH) generated by their metabolism (150). Parasitic protozoa not only have to eliminate their endogenous toxic metabolites but they should also cope with the oxidative (or respiratory) burst of the host immune system. Redox imbalance occurs in the parasite when the endogenous antioxidants fail to cope with the excessive ROS (both endogenous and exogenous), and this leads to the development of oxidative stress (111). In general, antiparasitic drugs, which have the ability to inhibit vital redox reactions or promote oxidative stress in parasites, are considered redox-active antiparasitic drugs (135). In this article, we have focused on the redox-active antiparasitic drugs used against diseases caused by unicellular parasites such as malaria, trypanosomiasis, leishmaniasis, amoebiasis, and trichomoniasis, as well as diseases produced by multicellular parasites such as schistosomiasis. Here, for a better understanding, we first discuss the role of important redox-active enzymes and the redox reactions involved in different metabolic pathways in the parasites. Next, we concentrate on various redox-active antiparasitic drugs along with their mode of action. In this context, we believe that a review on highlighted inventions and innovative ideas in this field could be of importance to scientists, managers, and decision makers for developing novel antiparasitic drugs.
Redox Reactions and Redox Enzymes in Parasites
Redox systems have a variety of important functions in parasitic protozoa, and the key enzymes involved are vital for growth and development of the organisms. In this section, we discuss the reactions carried out by different redox-active enzymes in the parasite. We have summarized the important redox enzymes present in parasites that could be potential drug targets (Table 1).
GSH, glutathione.
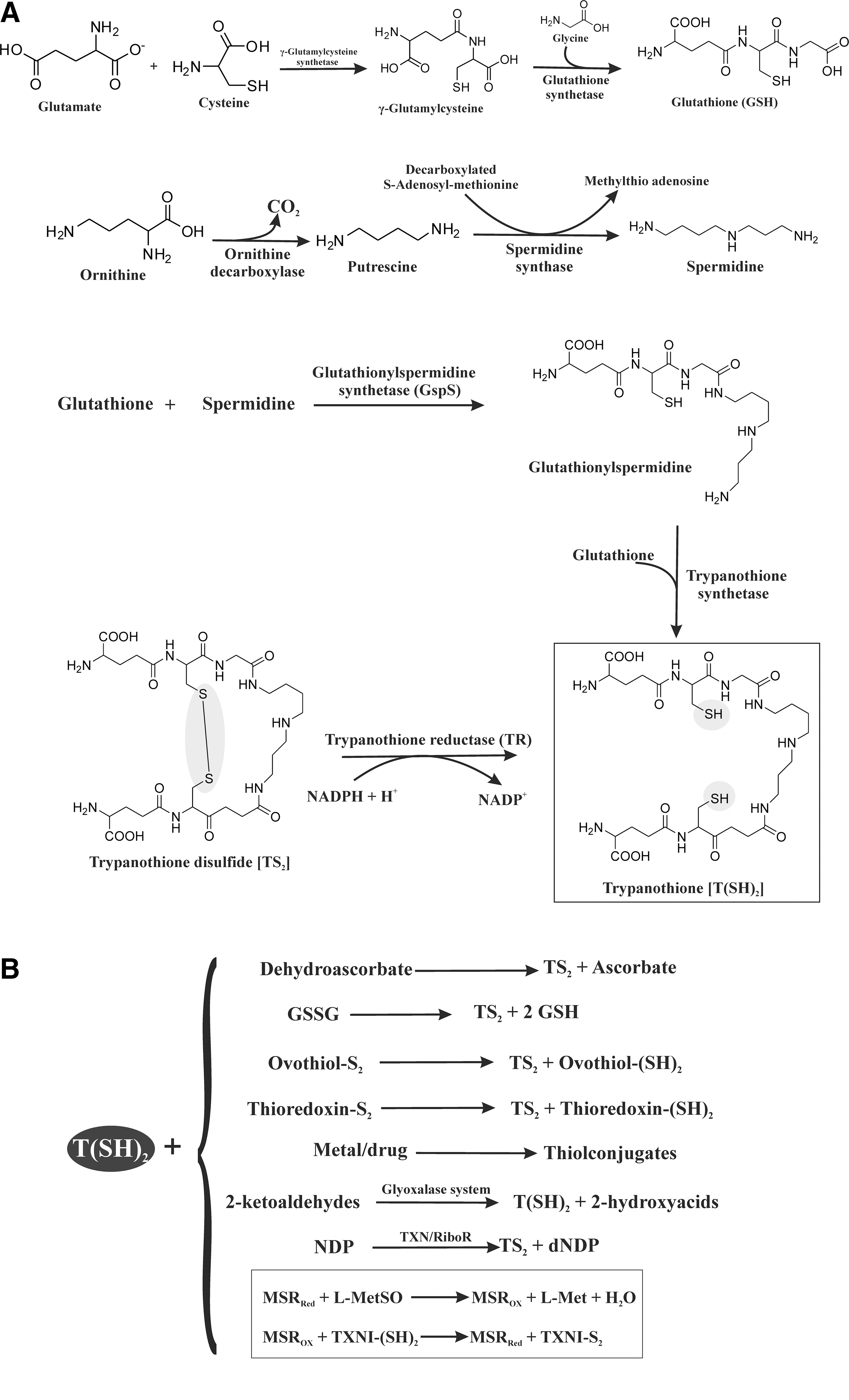
Trypanothione [T(SH)2] exclusively occurs in trypanosomatid protozoa (91) and carries out several redox reactions. T(SH)2 is synthesized by the conjugation of glutathione (GSH) with spermidine. This reaction is catalyzed by glutathionylspermidine synthetase (GspS) and T(SH)2 synthetase (75, 117). GSH is synthesized in two steps. First, the reaction of glutamate with cysteine is catalyzed by γ-glutamylcysteine synthetase, forming the intermediate γ-glutamylcysteine, which then reacts with glycine in the presence of glutathione synthetase yielding GSH. In African trypanosomes, spermidine is also synthesized in two steps. In the first step, decarboxylation of ornithine by ornithine decarboxylase gives putrescine, which in the presence of spermidine synthase yields spermidine (Fig. 1A). GspS catalyzes the reaction of GSH with spermidine to produce glutathionylspermidine, which reacts with another molecule of GSH catalyzed by T(SH)2 synthetase to produce T(SH)2. T(SH)2 is also synthesized by the reduction of trypanothione disulfide (TS2) (Fig. 1A). TS2 is reduced to T(SH)2 by the flavoenzyme TR at the expense of nicotinamide adeninine dinucleotide phosphate (NADPH) (Fig. 1A). T(SH)2 carries out numerous reactions in trypanosomatid protozoa (Fig. 1B) and is a spontaneous reductant of dehydroascorbate as well as of the disulfides of GSH, ovothiol, and the parasitic thioredoxin. T(SH)2 is involved in the detoxification of metals and drugs and in ketoaldehyde reduction. T(SH)2 also reduces nucleoside diphosphate (NDP) in the presence of tryparedoxin (TXN)/ribonucleotide reductase (RiboR) (Fig. 1B). Methionine sulfoxide reductase (MSR), a TXN-dependent enzyme, is a known antioxidant protein for maintaining the redox system in trypanosomes. T(SH)2 is the specific reductant of TXN, a multipurpose redox protein belonging to the TRX superfamily. MSR detoxifies methionine sulfoxide (MetSO) in trypanosomes by using the T(SH)2/TXN couple as a reducing system (Fig. 1B) (10).
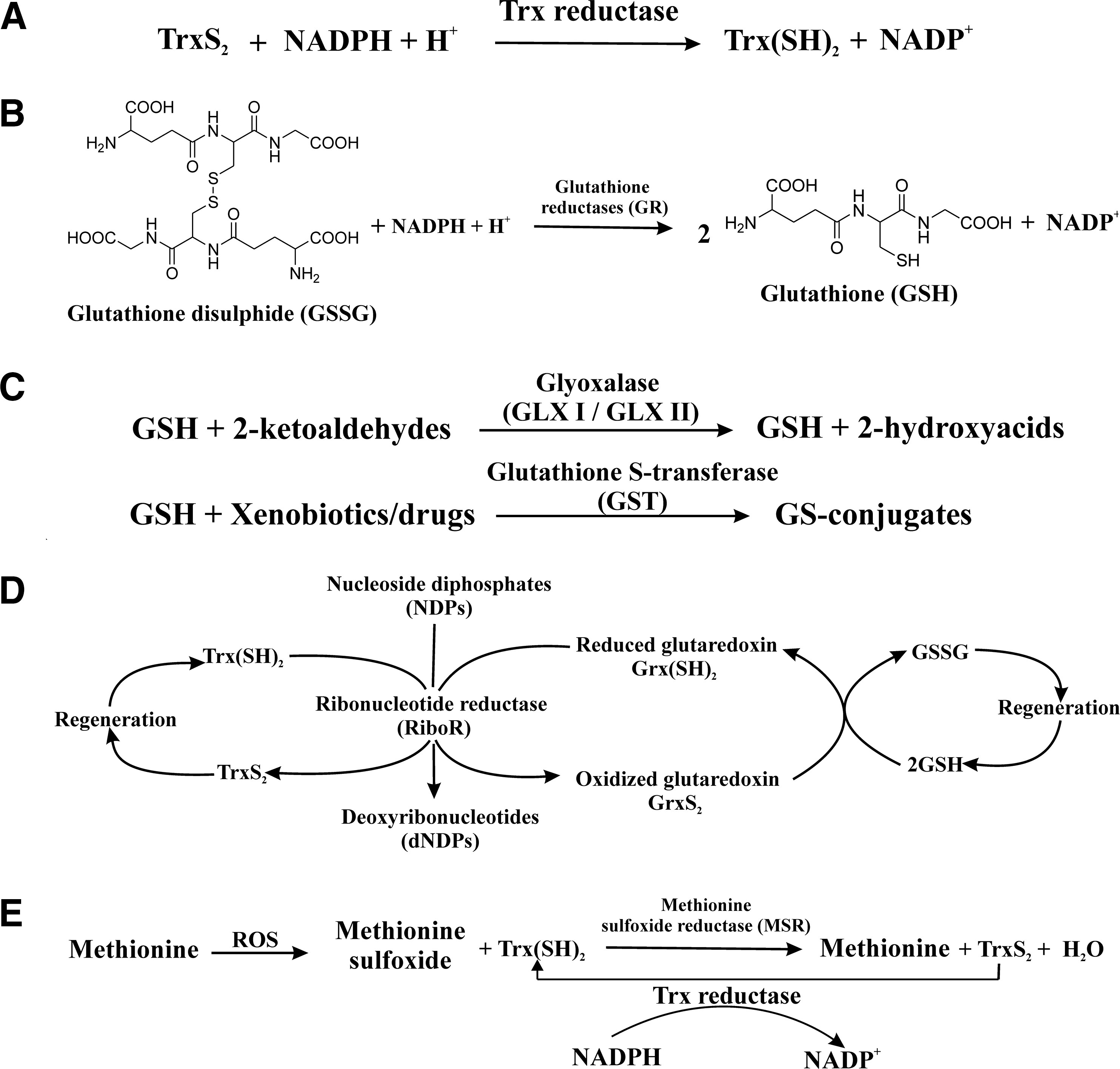
The biological reactions based on GSH and thioredoxin (Trx) in Plasmodium falciparum are shown in Fig. 2A–D. Two systems interact to protect malarial parasites against ROS. The GSH system comprises GSH, glutathione reductase (GR), glutathione S-transferase (GST), and different glutaredoxin-like proteins. The Trx system includes Trx, thioredoxin reductase (TrxR), and Trx-dependent peroxidases (73, 81 –83, 92–93, 123, 160). The newly discovered redox protein plasmoredoxin is one of the links between the two systems (19). TrxR reduces thiredoxin-S2 in the presence of NADPH (Fig. 2A). In order to maintain adequate levels of GSH, GR converts glutathione disulfide into GSH in the presence of NADPH (Fig. 2B) (155). GSH, which is known to safeguard P. falciparum from oxidative damage, also has an additional protective role through the promotion of heme catabolism (20, 39, 82, 90). GSH-specific reactions in the parasite involve the removal of 2-ketoaldehydes such as methylglyoxal by the glyoxalase (GLX) system (GLX I and II) (69, 102) as well as the detoxification of drugs/xenobiotics by GST (Fig. 2C) (147). A well-known function of both Trx and GSH is the reduction of NDPs to deoxynucleoside diphosphates catalyzed by RiboR. Reduced thioredoxin [Trx(SH)2] directly interacts with the enzyme, whereas GSH spontaneously reduces glutaredoxins, which subsequently reduce RiboR in P. falciparum (Fig. 2D) (122). In Schistosoma mansoni, methionine residue is oxidized by ROS to form the MetSO. The product is then converted back to its previous form by the action of the MSR system in the presence of Trx(SH)2 (Fig. 2E) (115).
Redox-Active Drugs Against Unicellular Parasites
The redox system is the backbone for the survival of parasites (150). Targeting this system would be a parasite-killing strategy (135). Compounds having a redox center and/or affecting redox biology and, hence, causing death of parasites are collectively called redox-active antiparasitic drugs. In this section, we have discussed the redox-active drugs against unicellular parasites based on our literature survey. P. falciparum, Trypanosoma cruzi, Trypanosoma brucei, Leishmania donovani, and Entamoeba histolytica are important parasites, and we have selected these protozoa, because they are the major infectious parasites of the world.
Redox-Active Antimalarial Drugs
The erythrocyte is the safest place for the malaria parasite to hide from its host's immune system, and the erythrocytic stages of Plasmodium spp. are responsible for clinical manifestation. The parasite is becoming increasingly resistant to conventional antimalarial drugs, and this has contributed to increasing morbidity and mortality (158). Malaria infection damages several major organs such as the liver, kidney, brain, spleen, heart, and lungs (42, 44, 49, 67, 113). In fact, severe malaria is characterized by multi-organ failure (64). Here, we have discussed the compounds that interrupt the redox system of the parasite and lead to cell death. In general, compounds that disturb the redox system of the parasite could be categorized into three different groups: (i) molecules which inhibit the activities of enzymes that are responsible for the maintenance of the redox balance of the parasite; (ii) molecules that prevent the inherent scavenging of pro-oxidant metabolic products (i.e., hemozoin [Hz] formation in malaria parasites) and lead to oxidative stress to induce parasite death; and (iii) molecules that produce ROS by themselves and, thus, lead to parasite death.
Drugs inhibiting enzymes in the redox system
GSH plays important roles in the maintenance of the redox and antioxidant status of protein-SH moieties in P. falciparum (84). An elevation of GSH content in parasites leads to an increased resistance toward chloroquine (CQ), while GSH depletion in resistant P. falciparum strains is expected to restore the sensitivity to CQ. GSH is involved in the reductive detoxification of free heme (11, 110) and also directly participates in the termination of radical-based chain reactions in which a single electron is transferred from thiyl radicals or disulfide radicals (53). GR is a key enzyme in the cell's defense mechanisms against oxidative stress, and based on this notion, several compounds were synthesized. They show antimalarial activity and inhibit P. falciparum GR (PfGR) (57, 65). A quinol-quinoline hybrid (
-, not evaluated; CQ-S, chloroquine-sensitive; CQ-R, chloroquine-resistant; MDR, multidrug-resistant; PfGR, Plasmodium falciparum glutathione reductase.
GST is one of the vital components of the GSH system. It uses GSH as a substrate and catalyzes the conjugation of GSH to a variety of electrophilic substrates (130). GST is a good target for designing antiparasitic drugs. Glycoconjugate (
PfTrxR, Plasmodium falciparum thioredoxin reductase; PfGST, P. falciparum glutathione S-transferase.
Drugs inhibiting Hz formation and inducing oxidative stress
Hemoglobin (Hb) is the major protein inside the erythrocyte, and the parasite has evolved a unique metabolic pathway to digest Hb. Hb degradation occurs inside the food vacuole (FV), which involves many enzymes (112). Heme is the degradation product of Hb, which is extremely toxic to the parasite. The released free heme (Fe+III) can offer a major toxic insult to the parasite through the generation of ROS (106, 112). A number of antimalarial drugs are known to act as inhibitors of Hz formation by binding to heme. Inhibition of Hz formation may develop oxidative stress in P. falciparum due to the accumulation of free heme and, thus, cause parasite death (106).
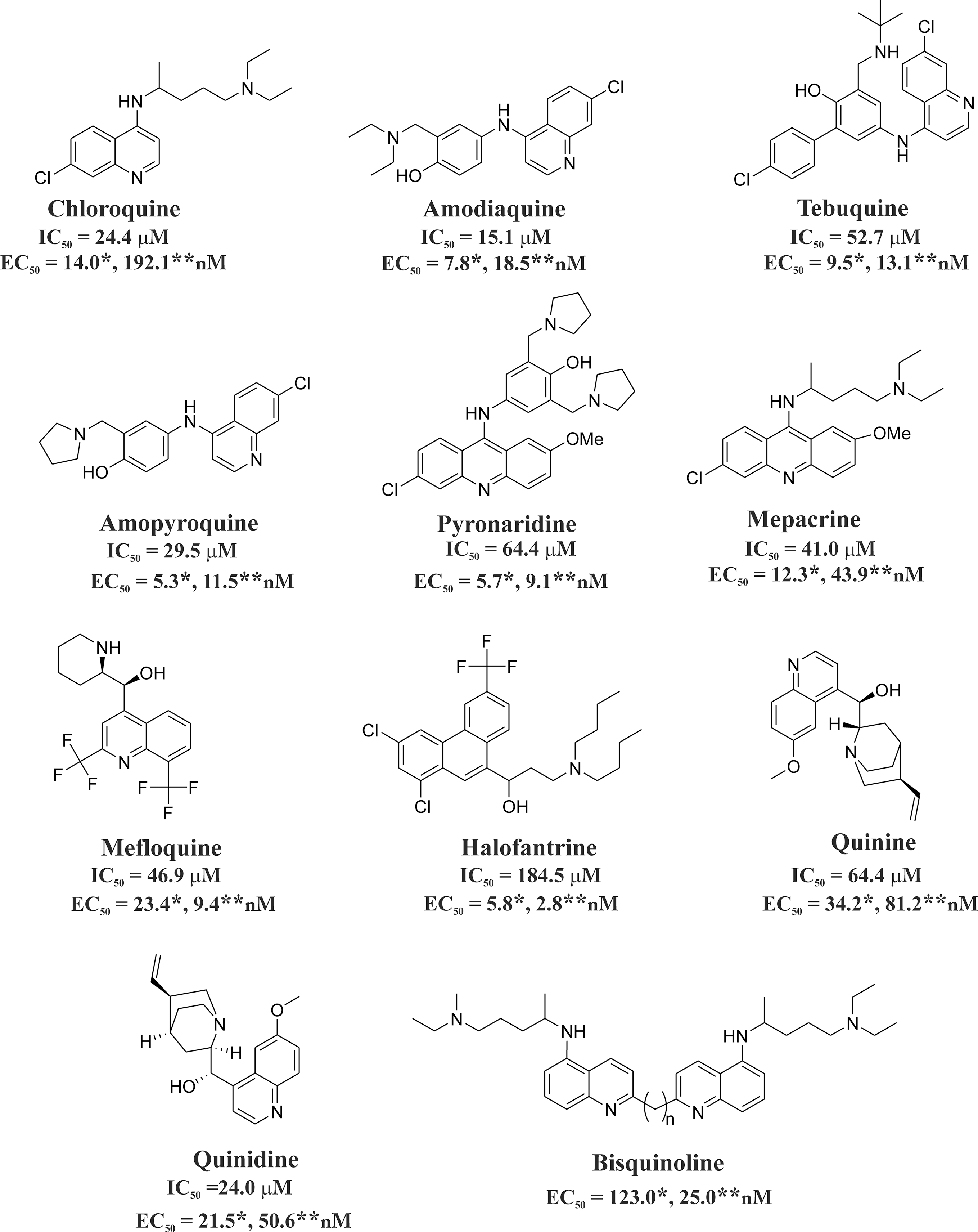
Quinolines, azoles, isonitriles, xanthones, and their derivatives adopt the aforementioned strategy to kill the parasites. Quinoline-containing derivatives such as CQ, amodiaquine, amopyroquine, tebuquine, mepacrine, pyronaridine, halofantrine, quinine, epiquinine, quinidine, and bisquinoline show antimalarial effects and inhibit Hz formation (Fig. 3) (48, 89, 114, 152). It is evident that amopyroquine (EC50=5.3 nM) and halofantrine (EC50=2.8 nM) are the most active antimalarial agents against CQ-S (3D7) and CQ-R (K1) strains, respectively (Fig. 3). Amodiaquine shows in vivo antimalarial activity against the Plasmodium yoelii NS strain in mice at a low dose (ED50=7.65 mg/kg) (114). Azole derivatives such as clotrimazole (CLT), ketoconazole, and miconazole (15, 125) inhibit Hz formation and show antimalarial effect against both CQ-S and CQ-R P. falciparum strains (62) (Fig. 4A). The azoles, including CLT (IC50=12.9 μM), ketoconazole (IC50=6.5 μM), and miconazole (IC50=21.4 μM), reversibly block the growth of ferriprotoporphyrin crystals and induce oxidative stress in malaria parasites (30). CLT shows low in vitro toxicity in human and murine cell lines (61–62).
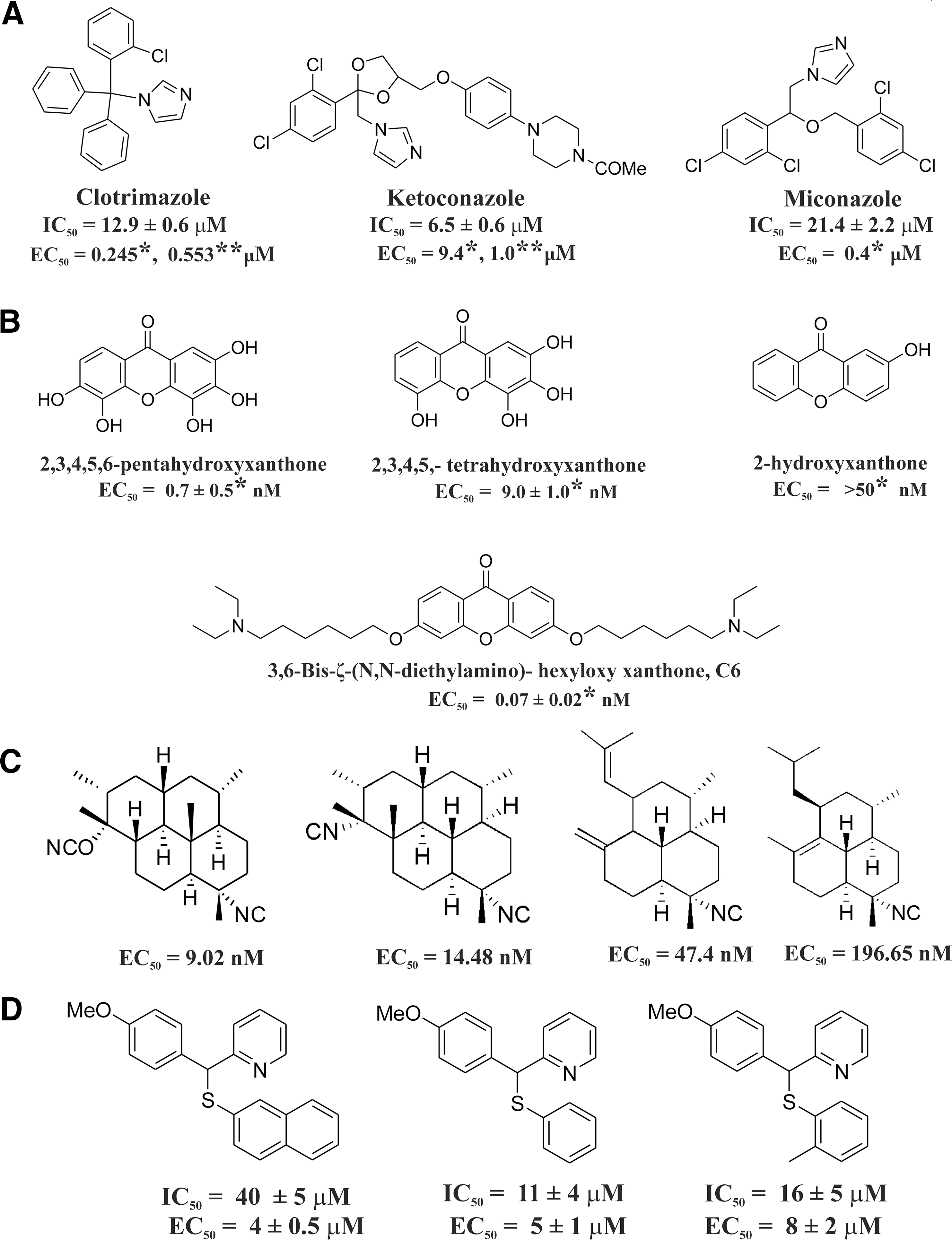
Xanthones have been identified as a novel class of antimalarial compounds (45). The antimalarial activity of xanthone and its derivatives (Fig. 4B) is based on the ability to interact with heme and, hence, prevent Hz formation (76, 162). Antimalarial activity is positively correlated with the number of hydroxyl substituents (51). 2,3,4,5,6-Pentahydroxyxanthone was found to be active against both the sensitive (3D7) and resistant strains (K1) of P. falciparum (76) (Fig. 4B) 1,3,6,8-Tetrahydroxyxanthone is very potent against P. berghei in vivo and was found to be better than the other oxygenated xanthones in this series (51). Xanthones bearing a hydroxyl group at any peri-position (1 or 8) show decreased antimalarial activity. These derivatives lose their affinity for heme due to intramolecular H-bond formation between the OH group and the carbonyls (77).
Several isonitrile derivatives (Fig. 4C) exhibit antimalarial activity and inhibit Hz formation (95, 161). Synthetic isonitriles were screened for their antimalarial activity against P. falciparum and MDR P. yoelii in the Swiss mice model (142). These isonitriles show antimalarial activity at a very low concentration (EC50=9.02 −196.65 nM) (Fig. 4C). A series of synthesized [(aryl)arylsufanylmethyl]pyridines (AASMP) derivatives (15, 94) (Fig. 4D) show antimalarial activity. These compounds inhibit Hz formation, form complexes (KD =12 to 20 μM) with free heme (ferriprotoporphyrin IX) at a pH close to the pH of the parasite FV, and exhibit antimalarial activity in vitro against P. falciparum (MDR strain). AASMP developed oxidative stress in the parasite by reducing the level of GSH and increasing the formation of lipid peroxide H2O2 and OH in P. falciparum. AASMP also exhibits profound antimalarial activity in vivo against CQ resistant P. yoelii. These AASMP derivatives suppressed the day 4 mean parasitemia by 30%, 50%, and 80% at doses of 25, 50, and 100 mg/kg, respectively, against MDR strain P. yoelii in BALB/c mice models (94). AASMP derivatives do not show any cytotoxicity against mammalian MCF-7 cells. Benzylmenadione (benzylNQ) derivatives have antimalarial activity against the CQ-R P. falciparum strain (Dd2) by inhibiting Hz formation and do not show any cytotoxicity against two human cell lines, the buccal carcinoma cell line cell (KB) and the human lung MRC-5 fibroblasts (Fig. 5A) (112). It is suggested that the benzylNQ are initially oxidized at the benzylic chain to benzoyl naphthoquinones in a heme-catalyzed reaction within the FV of the parasite. The major putative benzoyl metabolites are found to function as redox cyclers. The benzoyl metabolites (benzoylNQ, metabolite 1) are reduced by NADPH in GR-catalyzed reactions within the cytosol of infected red blood cells, and those benzoyl metabolites (reduced benzoylNQ, metabolite 2) can convert methemoglobin to oxyhemoglobin. This ultimately leads to the inhibition of Hz formation (Fig. 5A) (40). Since such inhibition is a validated antimalarial strategy (106), synthesis of a new inhibitor of Hz formation or identification of a new inhibitor against it from natural products will be helpful in developing novel antimalarials (106, 148). Liu et al. (103) suggested that 8-AQ causes heme toxicity by oxidizing Hb to methemoglobin. Both iron and 8-AQ donate an electron to the π* orbital of O2, which facilitates the formation of H2O2 in the parasite and leads to cell death (103). Since the pH in the FV is ∼5.2, in order to enter the compartment and exhibit antimalarial activity, compounds should be alkaline and stable in an acidic environment. Thus, synthesis of compounds (high pKa) specific for FV is necessary for antimalarial drug development, capitalizing Hz formation as a target.
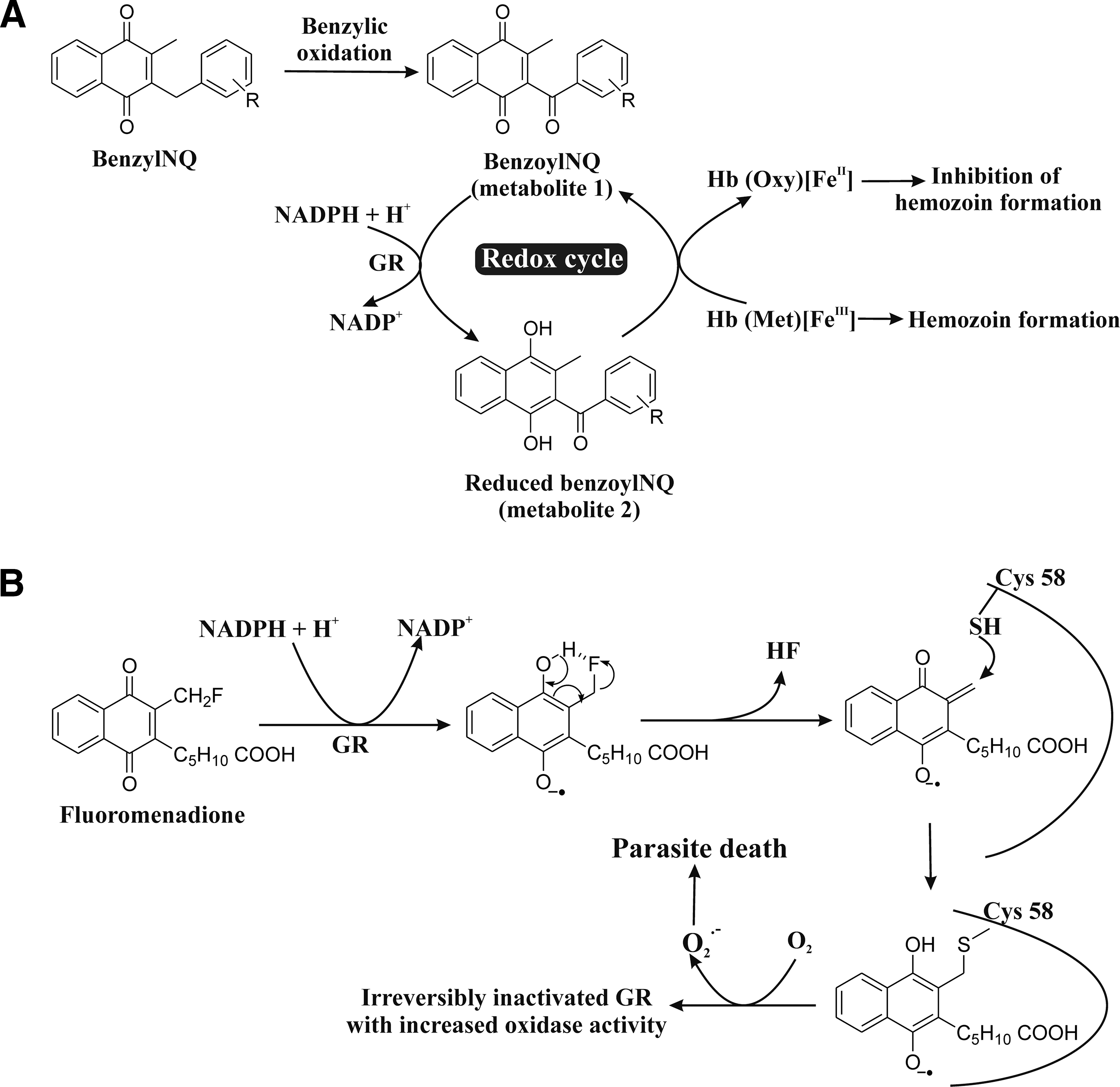
Drugs self-generating ROS and causing oxidative stress in the parasite
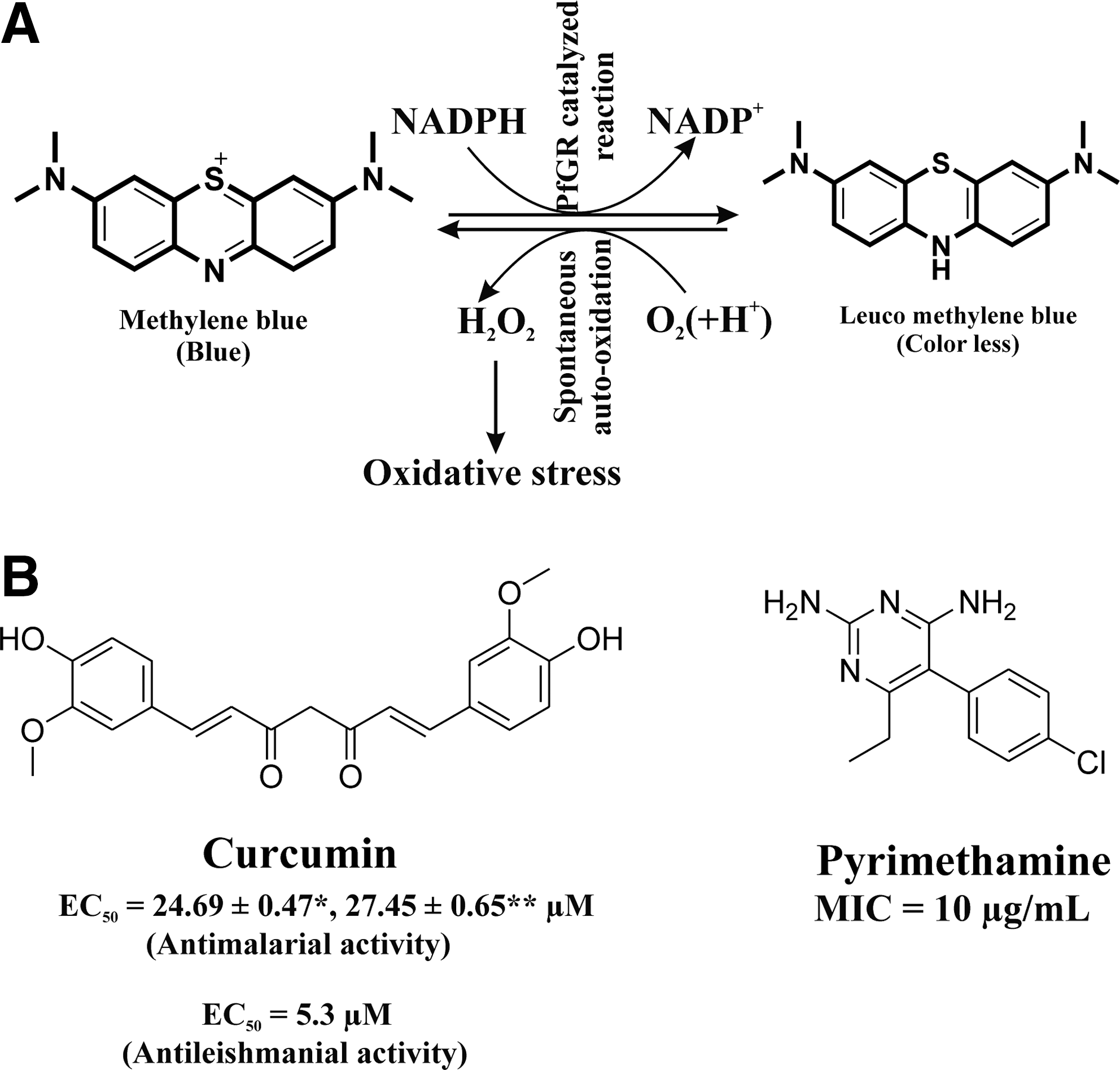
Many compounds kill Plasmodium spp. by self-generating ROS in the parasite. Fluoromenadione shows an antimalarial effect against the CQ-S strain 3D7 as well as the CQ-R strain K1 through the generation of ROS (O2 •−) but much lower cytotoxicity against human cells (18). Fluoromenadione is activated via GR-catalyzed one-electron reduction (two-electron transfer is also possible). Hydrogen fluoride is released from the activated fluoromenadione to form a quinone methide radical. Nucleophilic attack of the catalytic Cys58 of GR leads to covalent modification of the GR. The radical can react with oxygen, leading to the formation of O2 •−, which causes oxidative stress and parasite death (Fig. 5B) (40). MB, a well-known PfGR inhibitor, shows antimalarial activity by inducing oxidative stress in malaria parasite. MB not only inhibits the physiological reaction but also serves as a subversive substrate of PfGR. PfGR catalyzes the reduction of MB to leuco MB by NADPH. The product leuco MB is auto-oxidized back to MB with the concomitant production of H2O2 (ROS) resulting in oxidative stress in malaria parasite (Fig. 6A) (25, 40, 71). Curcumin (Fig. 6B) shows activity against both CQ-susceptible (3D7) and resistant (Dd2) P. falciparum strains by increasing ROS in the parasite (34). Curcumin's cytotoxic effect could be antagonized by co-incubation with antioxidants and ROS scavengers (34). Pyrimethamine (Fig. 6B) shows an antimalarial effect by inducing oxidative stress in parasites both in vivo and in P. yoelii 17XL-infected mice at a dose of 10 mg/kg (98). The minimum inhibiting concentration of pyrimethamine for activity against the P. falciparum NF-54 strain is 10 μg/ml (2).
Redox-Active Drugs Against Trypanosomiasis
Trypanosomes are parasitic protozoa within the order Kinetoplastida that comprise the causative agents of African sleeping sickness (Trypanosoma brucei gambiense and T. b. rhodesiense), South American Chagas disease (T. cruzi), and Nagana cattle disease (Trypanosoma congolense) (60, 88). Human African trypanosomiasis (HAT) or sleeping sickness is caused by two subspecies of T. brucei. In West and Central Africa, T. b. gambiense causes the chronic form of sleeping sickness, while in East Africa, T. b. rhodesiense causes the more fulminant form (79). T. cruzi is the causative agent of Chagas disease, the most important parasitic infection in Latin America. Approximately 8 million people are thought to be infected (124). T. cruzi infects many mammalian species (163) and is transmitted to humans primarily by the infected feces of hematophagous triatomine bugs coming in contact with mucosal membranes or damaged skin. Transmission also occurs via blood transfusion, congenitally, or, rarely, by ingestion of food contaminated by infected triatomine feces (5, 164). Chemotherapy against all forms of trypanosomiasis is very limited and unsatisfactory (85). Sleeping sickness is transmitted by the tsetse fly in sub-Saharan Africa with an estimated incidence of 70,000–80,000 cases and ∼30,000 deaths per annum (145). Once the infection has spread to the central nervous system, the disease is invariably fatal without treatment (145). Trypanothione reductase (TR) is an NADPH-dependent flavoprotein disulfide oxidoreductase unique to and essential for growth of trypanosomes, whose function is to convert TS2 into the physiologically relevant reduced form T[SH]2 (72, 146). In trypanosomes, T[SH]2 serves as a substitute for many of the metabolic and antioxidant functions ascribed to GSH in mammalian cells (32). Mammalian GR is the nearest homolog to TR. However, both enzymes of the host and parasite have significant differences in their active site architecture, resulting in a pronounced ability to discriminate between their respective disulfide substrates. These features make TR an attractive target for selective drug design (144). T. brucei TR and T. cruzi TR are the key enzymes for controlling the redox system of their respective parasites (Table 1) (36). Thus, inhibitors of T. brucei TR and T. cruzi TR are suitable candidates for drug development against HAT and American trypanosomiasis, respectively (28, 90).
Drugs inhibiting enzyme activity in the redox system
In this section, we discuss the drugs that inhibit the activity of T. brucei TR. Quinolines (
Drugs self-inducing oxidative stress in the parasite
Chinifur [

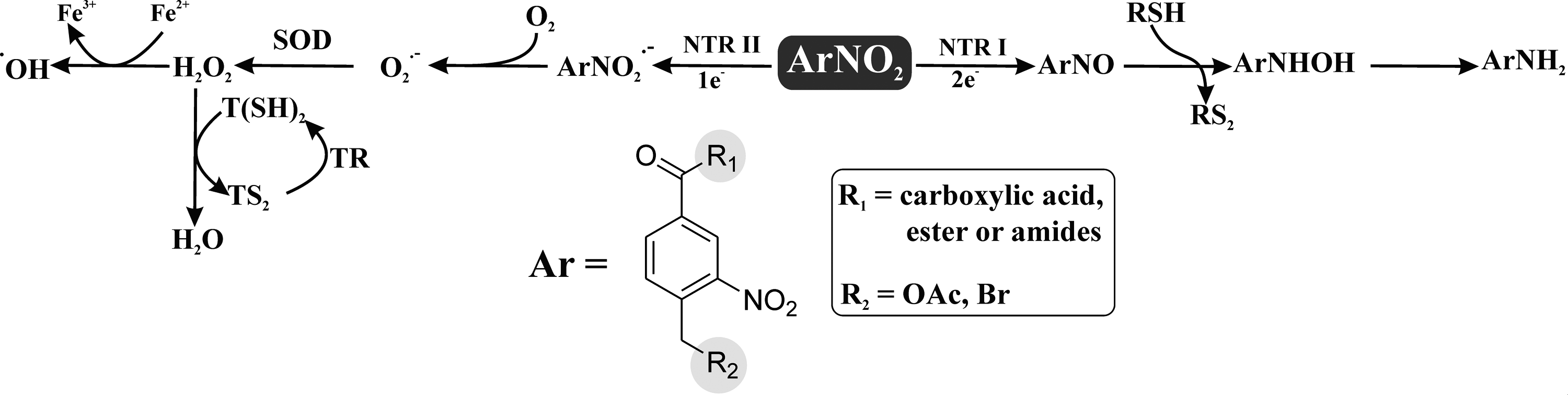
Molecules having nitro group as well as metal complexes have the potential to become drugs against trypanosomiasis. N-oxide-containing heterocycles such as benzofuroxans [
Redox-Active Antileishmanial Drugs
The leishmaniases are a group of diseases caused by infection with the protozoan parasite Leishmania, transmitted by the sand fly. The most fatal form of the disease is visceral leishmaniasis, which results from infection with L. donovani and L. infantum. The amastigote form replicates in macrophages of the liver, spleen, and bone marrow, causing persistent fever, hepatosplenomegaly, weight loss, and pancytopenia. If untreated, it eventually becomes fatal and is thought to account for 41,000 deaths per year (66, 119). According to current WHO statistics, about 12 million people living in 88 countries, mainly of 5 continents, that is, Asia, Europe, Africa, South America, and North America are suffering from leishmaniasis with 1.5–2 million new cases annually (139). This disease is endemic in the low-income population of Central and South American countries. Thus, there is an urgent need for new and less toxic treatments for leishmaniasis (139). Targeting the redox system of the parasite via small molecules would be a good strategy for drug development against Leishmania. In this section, we refer to a series of such small molecules that inhibit the enzyme activity, which maintains the redox system of L. donovani.
L. donovani TR is a validated drug target against leishmaniasis (Table 1). Doxorubicin (
ROS, reactive oxygen species.
Antimonial drugs (
Camptothecin (CPT) (Fig. 9A), an inhibitor of DNA topoisomerase I, shows an antileishmanial effect against the L. donovani AG83 strain by inducing ROS both in the amastigotes and promastigotes of L. donovani (137). CPT inhibits growth of L. donovani AG83 to 65% at a concentration of 5 μM. CPT-induced cellular dysfunction in L. donovani promastigotes is characterized by several cytoplasmic and nuclear features of apoptosis. It has been proposed that CPT-induced apoptosis-like death is due to mitochondrial dysfunction and ROS generation. (137). Baicalein (BLN) (Fig. 9A) shows its antileishmanial potency against L. donovani AG83 strain through ROS generation in parasites without involving caspases (24). BLN inhibits 73% growth of L. donovani AG83 promastigotes at a concentration of 10 μM (24). Menadione and withaferin A, potent inhibitors of protein kinase C (PKC), show an antileishmanial effect by inducing ROS in Leishmania chagasi and L. donovani, respectively, (Fig. 9A) (107, 136). In L. donovani AG83 promastigotes, the inhibition of PKC by withaferin A causes depolarization of ΔΨ and generates ROS inside cells. Loss of ΔΨ leads to the release of cytochrome c into the cytosol and subsequently activates caspase-like proteases and oligonucleosomal DNA cleavage and causes parasite death (136). At 6 h, withaferin A inhibits 85% growth of L. donovani AG83 promastigotes at a concentration of 15 μM (136). Mitochondria are the principal site for the generation of cellular ATP by oxidative phosphorylation (127). 3,3′-diindolylmethane, a DNA topoisomerase I inhibitor, inhibits mitochondrial F0F1-ATP synthase of L. donovani. This, in turn, causes depletion of mitochondrial ATP levels and significant stimulation of mitochondrial ROS production, leading to oxidation and fragmentation of DNA and hence, death of the parasite (Fig. 9B) (127). Quinone compounds act as subversive substrates and are reduced by L. donovani TR to give reduced semihydroquinones (139). These semihydroquinones are further oxidized by oxygen to quinone and O2 •−, which kills the parasite (Fig. 9C) (80).

Redox-Active Drugs Against Amoebiasis and Trichomoniasis
E. histolytica, which is responsible for amoebiasis infects,>10% of the world's population, primarily in the tropics and regions with poor sanitation. Approximately 10% of those infected will become clinically symptomatic, resulting in an annual toll of 50 million to 100 million cases of invasive colitis and liver abscess and up to 100,000 deaths (99, 104, 151). Targeting the redox system of E. histolytica is a strategy for the development of drugs against amoebiasis. Different nitroimidazoles such as metronidazole, tinidazole, ornidazole, secnidazole, sulnidazole, carnidazole, misonidazole, ipronidazole, ronidazole, nimorazole, and panidazole show activity against amoebiasis (13, 47, 56, 121) (Fig. 10A). EC50 value of metronidazole and tinidazole for their antiamoebic activity against E. histolytica HM1: IMSS strain are 1.84 and 10.2 μM, respectively (17, 70).
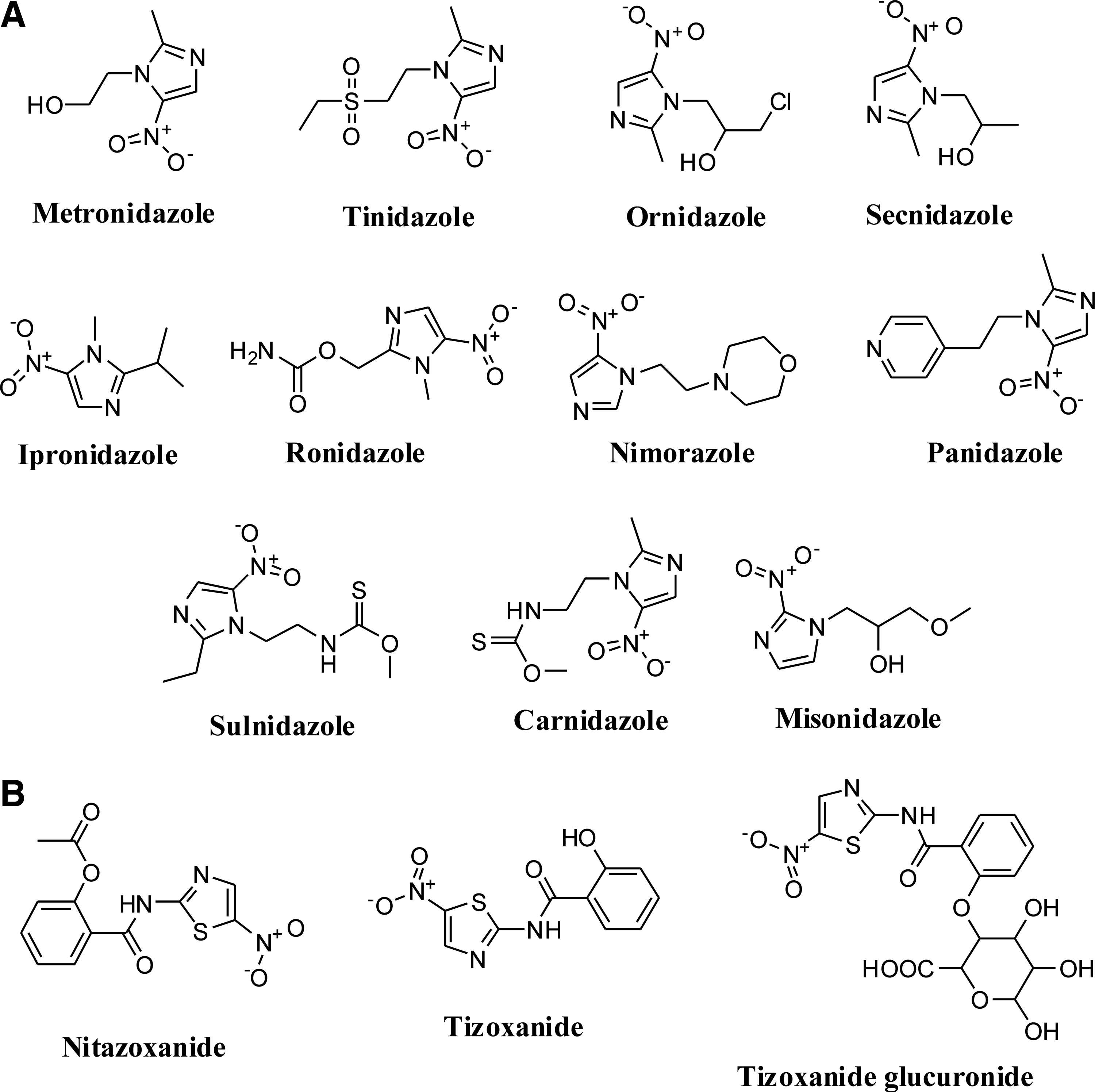
Nitroimidazoles, through the formation of highly reactive nitro radical anions, damage susceptible pathogens by radical-mediated mechanisms (59). Unlike aerobic organisms, these pathogens possess the electron transport protein ferredoxin (small Fe-S protein). Once the drug has entered the parasitic trophozoite and is within the cell, ferredoxin donates electrons to the nitro group of the drug. The drug becomes activated by the reduction of the nitro group. The active drug binds covalently to DNA, resulting in DNA damage and, subsequently, to the death of the trophozoite. The reductive activation of metronidazole, an important nitroimidazole drug, may also lead to toxic radicals reacting with essential cellular components. In addition to these effects, the drug also inhibits trophozoite respiration. O2 is a competitor of 5-nitroimidazoles. O2 is able to generate both a decrease in the reductive activation of a 5-nitroimidazole drug and an increase in the catalytic recycling of the activated drug (1, 121).
Trichomoniasis is a common sexually transmitted disease caused by Trichomonas vaginalis, a protozoan parasite. T. vaginalis infection has been associated with problems in pregnancy, premature birth, and low birth weight (157). T. vaginalis pyruvate: ferredoxin/flavodoxin oxidoreductases (PFORs) are key enzymes that maintain the redox system of T. vaginalis (Table 1). T. vaginalis derives energy from the oxidative fermentation of pyruvate. PFOR, an important enzyme of the intermediary metabolisms of these organisms, catalyzes pyruvate decarboxylation. This process releases electrons that reduce ferredoxin, and the latter, in turn, catalytically donates its electrons to biological electron acceptors (121). Nitazoxanide shows activity against trichomoniasis and inhibits T. vaginalis PFORs in a noncompetitive manner (74). Tizoxanilide and tizoxanilide glucuronide are also redox-active antiparasitic drugs against trichomoniasis (Fig. 10B) (1). Recent studies in anaerobic protozoa (T. vaginalis) have shown that nitazoxanide inhibits T. vaginalis PFOR. Unlike nitroimidazoles, nitazoxanide is independent of reduced ferredoxin, that is, it appears to interact directly with PFOR. The different mechanisms of action and resistance may explain the therapeutic efficacy of nitazoxanide against organisms showing resistance to 5-nitroimidazoles (e.g., T. vaginalis), especially metronidazole. In helminths, the mechanism of nitazoxanide activity is not yet fully understood, but the enzymes involved in anaerobic electron transport appear to be potential targets. The products of nitazoxanide activation do not induce mutations in DNA (63).
Redox-Active Drugs Against Multicellular Parasites
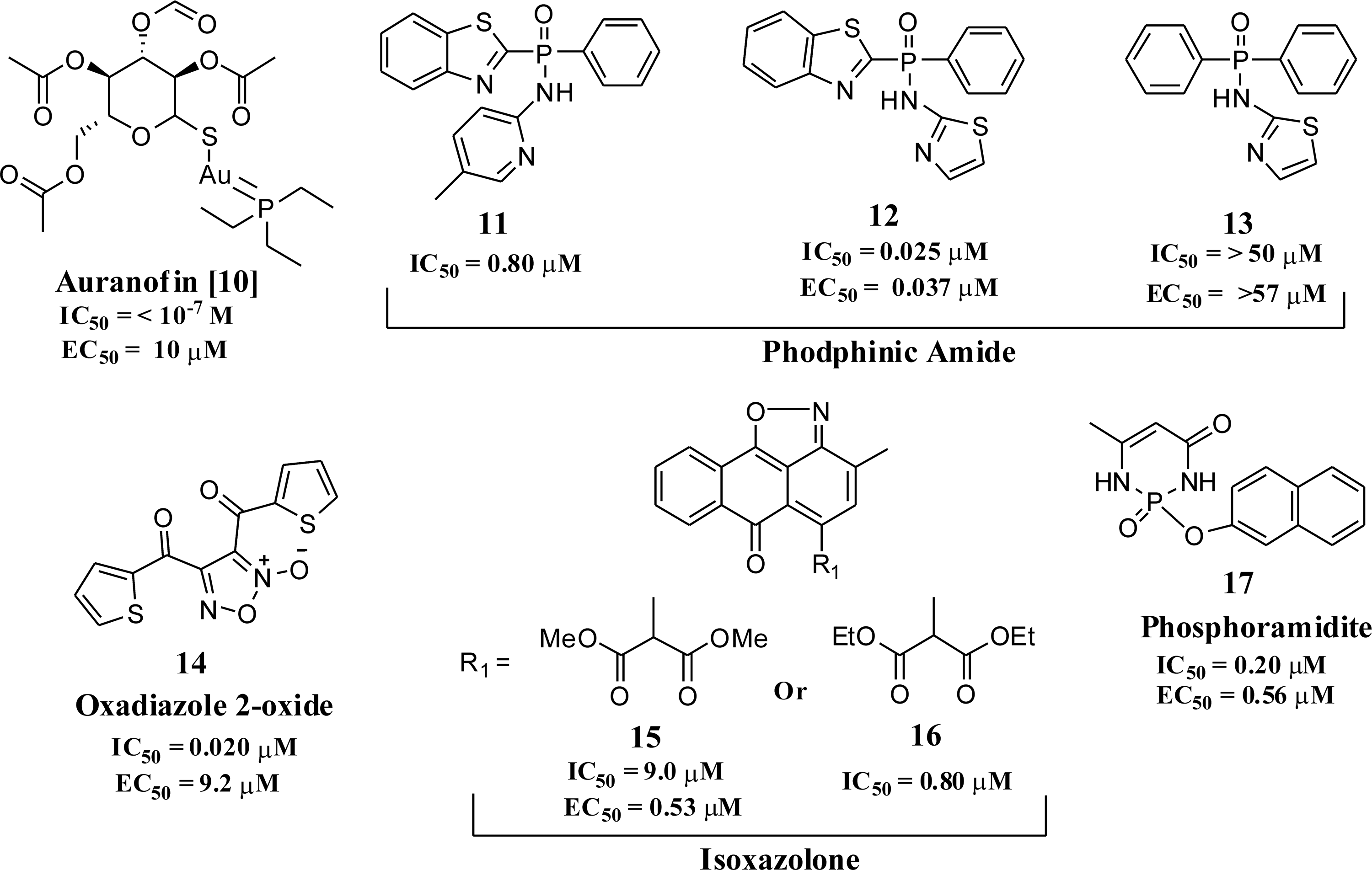
The muticellular parasite S. mansoni is responsible for schistosomiasis, affecting more than 200 million people in >70 countries (115–116). The parasites can survive for up to decades in the human host, as it has a unique set of antioxidant enzymes that continuously degrade the ROS produced by the host's innate immune response. Since schistosomes do not have catalase (108), other mechanisms should exist within the parasite to degrade H2O2. The principal component of this defense system, thioredoxin-GR (TGR), has been recently identified and validated as a target for antischistosomiasis drug development (Table 1) (96). TGR is a multifunctional selenocysteine-containing enzyme that catalyzes the inter conversion between reduced and oxidized forms of both GSH and Trx, which are major contributors to the maintenance of redox balance (6). Auranofin [
Conclusions and Future Perspectives
The major problem of current chemotherapy for parasitic diseases is the emergence of drug resistance against available drugs. This situation warrants the identification of functionally validated targets in order to discover novel, chemotherapeutically viable molecules that treat resistant parasites. The available parasite genome data provide a scope to search for new targets. Different approaches, such as evolutionary patterning, gene networks, system biology, and synthetic biology, will be extremely helpful for the identification of suitable targets. There are so many reported molecules having antiparasitic activity; however, not all of these are therapeutically viable drug-like molecules due to various limitations such as toxicity, low bioavailability, rapid inactivation under in vivo conditions, and development of resistance. Thus, synthesis of new molecules by cutting down the toxophore part of active antiparasitic molecules and simultaneously adding valuable moieties/scaffold to them with a view to overcome the above-mentioned limitations might be fruitful for developing novel antiparasitic drugs for a future generation. Furthermore, studies on drug synergism should receive special attention, which can open new avenues to improve the efficacy of antiparasitic drugs in combination with others. Since parasites such as P. falciparum, L. chagasi, T. brucei, T. cruzi, and S. mansoni are very susceptible to oxidative stress (33, 55, 107, 115, 133, 150), the identification of new scaffolds that affect the redox systems of these parasites and induce oxidative stress will be a valid rationale to develop new drugs. However, extreme care should be taken so that the designed scaffolds/molecules cannot affect the redox system of the host and the products formed from these molecules after metabolic turn-over by the parasitic machinery should not be toxic. Ethnopharmacology is one of the important areas for new antiparasitic drug development. Ethnopharmacology is the interdisciplinary scientific exploration of biologically active agents traditionally employed or observed by man. Medicinal plants are important source of indigenous medical systems in many parts of the world, and these resources will be useful for the development of antiparasitic drugs. We have tried to incorporate the maximum number of known redox-active antiparasitic molecules, but the number of reported lead molecules may be much higher than indicated in this article. It can be anticipated that effective research in the near future capitalizing redox-active enzymes and molecules in parasites will open new avenues for the development of novel antiparasitic drugs that combat resistant parasites.
Footnotes
Acknowledgments
The authors thank Council of Scientific and Industrial Research (CSIR), New Delhi, for providing funds to carry out this work. They thank Sumanta Dey, Samik Bindu, Souvik Sarkar, Rahul Kumar, Manish Goyal, Mohd. Shameel Iqbal, and Athar Alam for critically reading and editing the article.