
Abstract
Introduction
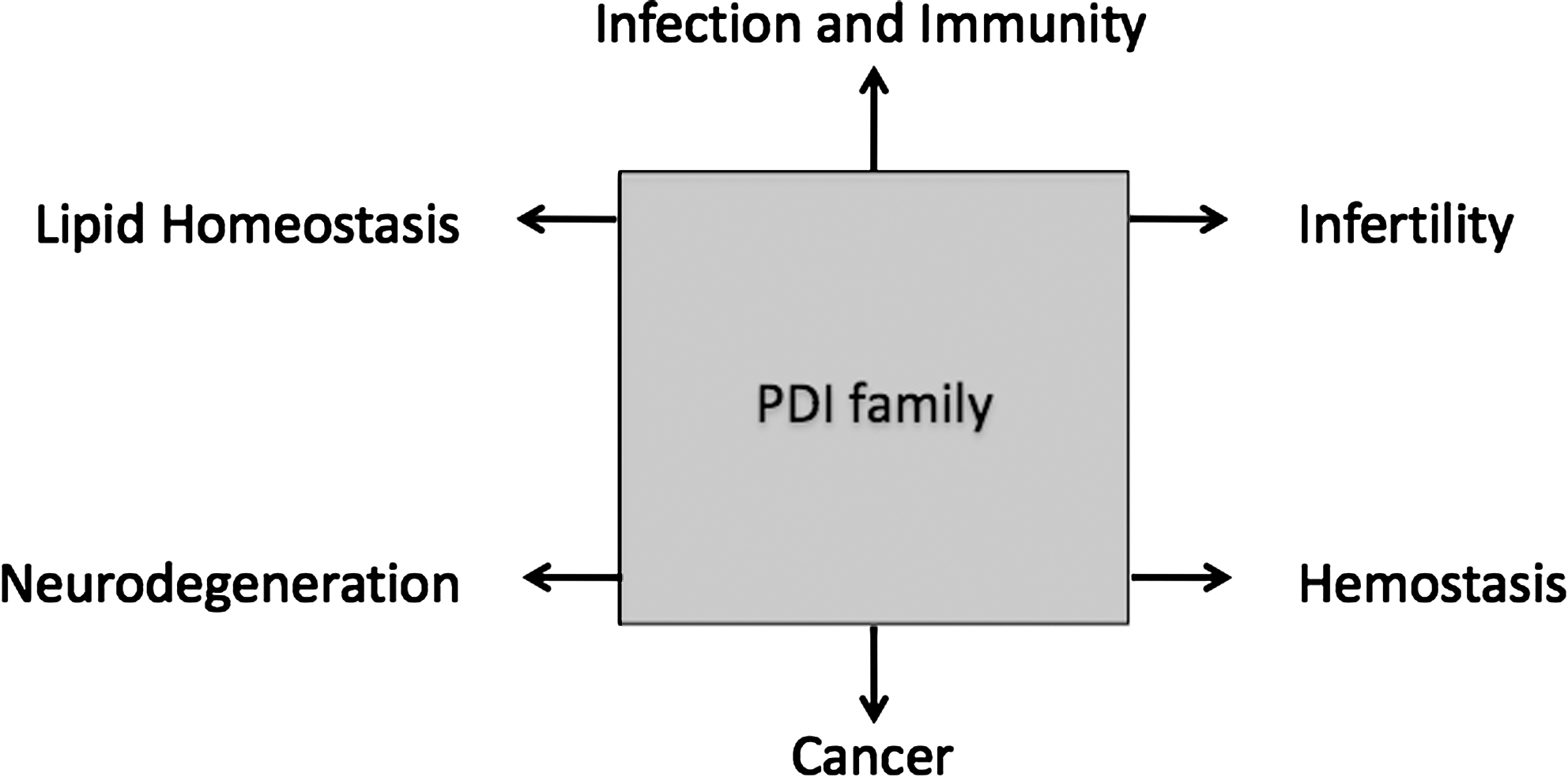
A Brief Introduction to PDI Function
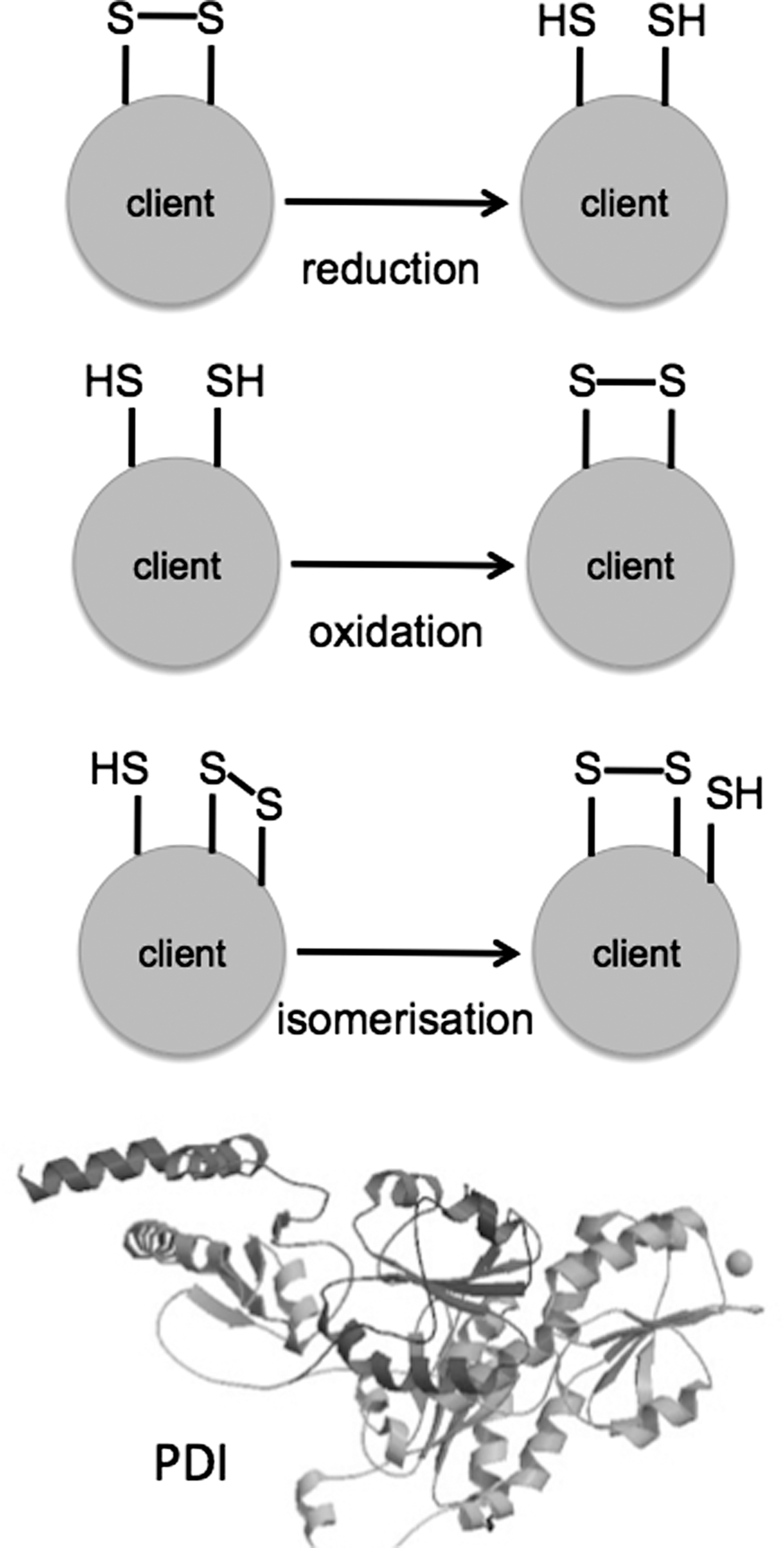
There have been many excellent recent reviews on the molecular function of PDI, and the reader is referred to these articles for more detail on this aspect of the PDI family for example (20), and references therein. PDI family members can function as molecular chaperones and as disulfide oxidoreductase/isomerases, which means that they can make, break, or rearrange disulfide bonds. These disulfide bonds (S-S) form between the -SH groups of cysteine residues in a variety of proteins that can be considered as PDI “clients” (Fig. 2). The S-S bonds equip the client protein with useful properties, including structural stability or an appropriately shaped active site. Intermolecular S-S bonds, between two protein chains, are also important to keep multimeric complexes together.
PDI usually resides in the endoplasmic reticulum (ER), although it can be released to function at the cell surface or extracellular matrix (examples of which will be described later). PDI is found in all multicellular organisms, including higher plants, and is expressed in yeast but not bacteria. Instead, bacteria have homologous Dsb proteins to facilitate oxidative protein folding and isomerisation in the periplasmic space (27). In Saccharomyces cerevisiae there are five PDI family members (Pdi1p, Mpd1p, Mpd2p, Eug1p, and Eps1p) (76). By the time we climb the evolutionary tree to Homo sapiens, this number has increased to at least 21 (Table 1) (20). Why should we need so many apparently similar enzymes? The answer is probably to do with the large number of different clients that require assistance in the secretory pathway of a multicellular organism. Different cells and tissues range widely in their secretory output, and extra PDI activity maybe required to support this. In addition to PDI's catalytic function as a thiol-disulfide isomerase, it also has molecular chaperone properties.
TMX5 has been noted in reviews but has not been described in a primary publication (35).
ERp90 Trx domains 3–5 are homologous to ERp57 abb′ domains, but ERp90 lacks typical CXXC motifs (55).
PDI, protein disulfide isomerase; ER, endoplasmic reticulum; AGR, anterior gradient; Trx, thioredoxin.
The main structural building block of PDI is the thioredoxin domain. There are two of these domains with active sites in PDI, denoted
The crystal structure of yeast Pdi1p has been solved (60) and, together with a host of nuclear magnetic resonance structures, this has paved the way for the elucidation of several mammalian PDI family crystal structures including ERp57 (
PDI Proteins and Disease
Hemostasis
Although the main function of PDI is in the ER, a number of studies have indicated that thiol-disulfide exchange at the cell surface is biologically important (Fig. 3), for example, in hemostasis (the control of bleeding) (31). Initial studies suggested that in platelets, blocking PDI family activity could inhibit platelet activation pathways, including aggregation, secretion, and binding to fibrinogen. Recent work in this area has shown that in a mouse model, thrombus formation and the production of fibrin after laser-induced arteriolar injury required PDI function (13). Fibrin production is under the control of tissue factor (TF). Most circulating TF is inactive, and it is proposed that contact with anionic phospholipids on the cell surface activates TF. Work by Reinhardt and colleagues suggested that PDI activates TF through its disulfide isomerase activity to drive fibrin production (53). These findings built on earlier work showing that secretion from platelets could be modified by the PDI inhibitor bacitracin (Fig. 4), or by using antibodies raised against PDI (16). However, these ideas have been called into question by investigators who looked at the intracellular distribution of PDI in platelets before and after activation, and could find no evidence for recruitment of PDI to the cell surface by immunoelectron microscopy (65).
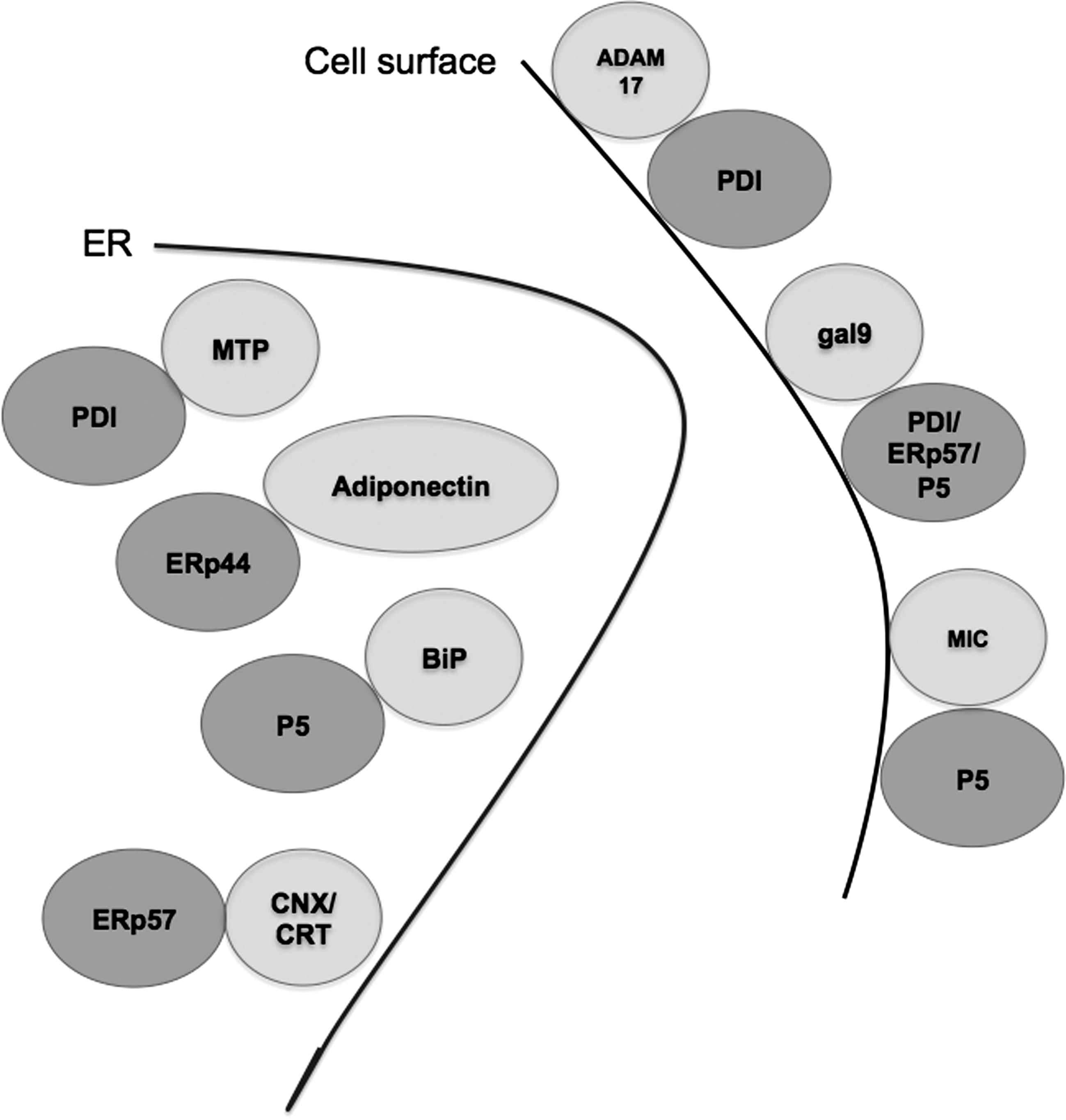
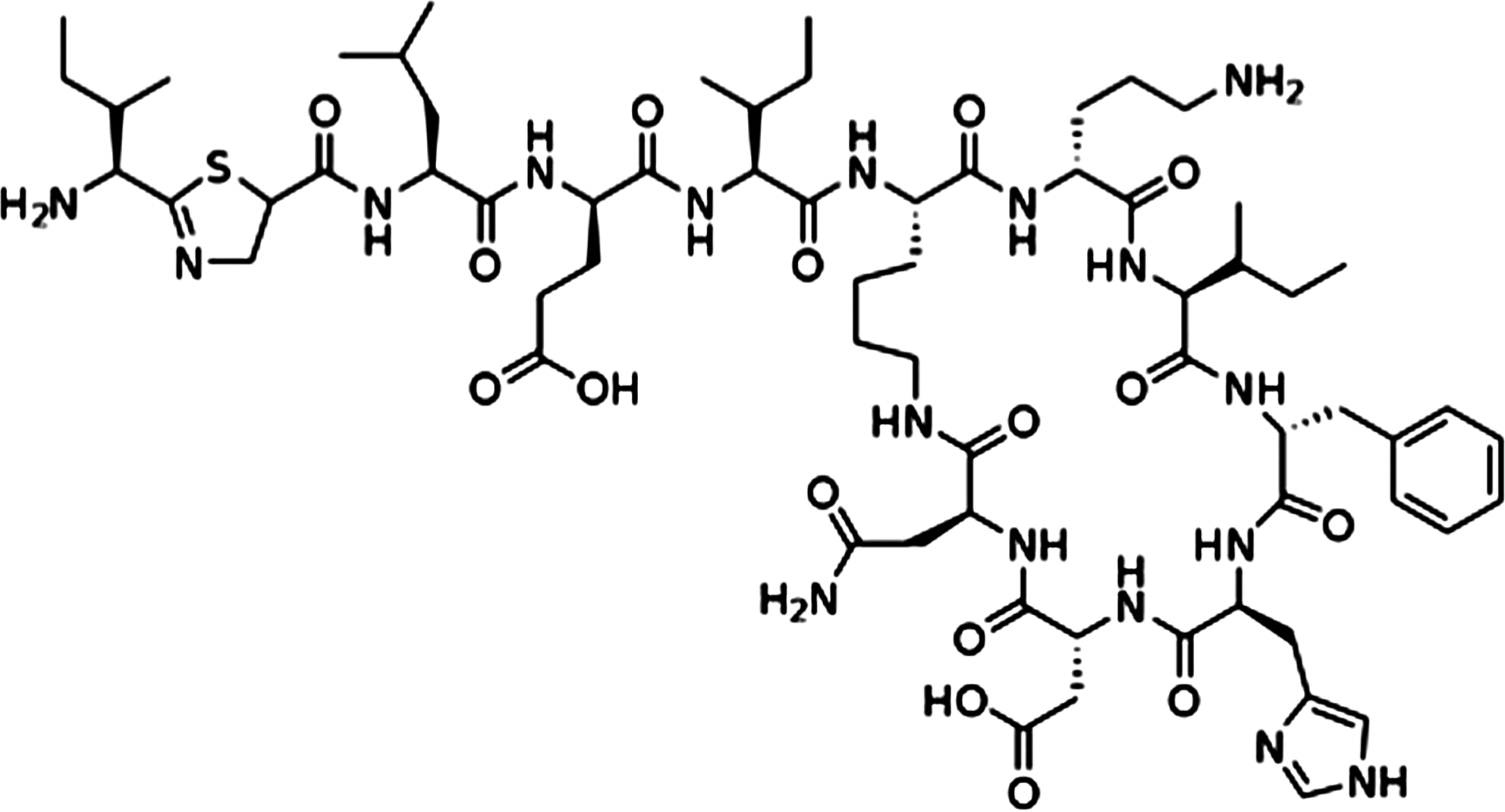
The debate over PDI function in platelets exemplifies a hurdle in the field. It is difficult to prove that PDI, an abundant and sticky protein, is genuinely recruited to the cell surface, and it is hard to be certain that PDI and not a relative (or a small-molecular-weight redox agent) is performing the oxidoreductase/isomerase function. Bacitracin is often used as a PDI inhibitor, but it is not specific (33). Similarly, some PDI antibodies used to block the activity or confirm the identity of PDI were developed before the full complement of PDI homologs was known. Thus to fully resolve the questions in the field, the specificity of the PDI antibodies used in these types of studies should be carefully considered, and the development of more specific PDI inhibitors is required.
It is also worth noting that vitamin K epoxide reductase (VKOR), which is localized to the ER and possesses a thioredoxin-like CXXC motif, maybe oxidized by PDI in the ER (67). VKOR is required for γ-carboxylation of vitamin K–dependent blood clotting proteins, and recycles vitamin K epoxide back to vitamin K (46). Mutations in VKOR genes can result in warfarin resistance and bleeding disorders (56). Taken together with the fibrin studies described previously, a picture is emerging of how redox regulation, both in the ER and outside the cell, is required for quality control in hemostasis.
Infectious disease
The notion that a function both within and beyond the ER is important for PDI's role in health and disease is further exemplified when considering the immune system. There is evidence that entry of some viruses into eukaryotic cells is governed by redox-regulated processes. One example is Newcastle disease, a bird virus. This single-stranded RNA paramyxovirus gains entry to its host cell through large conformational changes in its F fusion protein, which involves thiol/disulfide exchange (29). Overexpression of PDI and ERdJ5 (a PDI family reductase with an extra J domain) led to an increase in viral membrane fusion and hinted at a route whereby viruses can take advantage of the PDI family to gain access to host cells (28). There are numerous other examples where bacteria and viruses can subvert PDI family members during infection. For example, the cholera holotoxin (secreted by Vibrio cholerae) is targeted by PDI in the ER (after its retrograde transport through the secretory pathway). PDI displaces the toxic A1 subunit and enables its transfer to the cytoplasm, where it can bind to ADP ribosylation factor 6 (see review by Tsai and colleagues in this Forum and references therein). Polyomavirus unfolding in the ER is mediated by the redox-inactive ERp29, which also has other functions, including the folding and secretion of thyroglobulin and a possible role in regulating the mesenchymal-epithelial transition in tumorigenesis (42, 78).
Lymphocytes (especially CD4+ T cells) increase the availability of cell surface thiols after immune activation (37). An intriguing study has recently shown that PDIs maybe involved in this process by interacting with galectin-9, a secreted lectin involved in the control of cell adhesion (9). Galectin-9 has a notable ability to negatively regulate CD4+ TH1 cells and trigger cell death, a process that is, in part, inhibited by α2,6-linked sialic acids (80). When galectin-9 was incubated with T cells that lacked Tim-3 (the primary galectin-9 receptor) the cell surface proteins that it bound to were not conventional membrane receptors, but rather PDI, ERp57, and P5 (9). The authors went on to show that PDI could enhance the functional activity of galectin-9 in two ways: by promoting T cell migration, and by enhancing infectivity when the T cells were exposed to the human immunodeficiency virus (HIV)-1. This study focused on the role of PDI, but it is notable that ERp57 and P5 were also identified, supporting a role for these proteins in disulfide-dependent events at the cell surface (Fig. 3). Further studies into how PDI and ERp57 interact with galectin will be important to determine the molecular specificity of these interactions.
The PDI family is indirectly involved in protecting cells from infectious disease through the ER quality control of proteins involved in immune defense. Two very well-studied examples are the major histocompatibility complex (MHC) class I molecule—the main player in our defense against viruses and tumors—and the immunoglobulins (Ig), secreted from B cells to protect us from extracellular foreign antigens. Both ERp57 (working with tapasin) and PDI are involved at multiple stages of the MHC class I quality control process, from monitoring the folded state of the peptide binding domain through to the disposal of incorrectly folded MHC class I heavy chains via its chaperone binding activity (34, 38).
Antibodies also call upon the PDI family for their quality control, and an important player here is ERp44, an
Lipid homeostasis
ERp44 has another important role to play, together with Ero1α, in the quality control of adiponectin (72). Adiponectin is a major secreted hormone of adipocytes, and low circulating levels of the high-molecular-weight version of this adipokine are involved in cardiovascular disease and type 2 diabetes. ERp44 prevents the secretion of adiponectin oligomers by keeping the partially folded chains in the ER (Fig. 3) (73). In contrast, Ero1α can facilitate the oxidative release of adiponectin from the ER (50). The transcription factor peroxisome proliferator–activated receptor-γ induces Ero1α and represses ERp44, enabling transcriptional regulation to control levels of adiponectin through ER quality control (40, 50). Although ERp44 has a critical role in thiol-mediated retention, it can also regulate the inositol phosphate 3 receptor type I, a channel that governs calcium release from the ER (21). Thus further work is required to understand the full scope of ERp44 functions and its client interactions.
It has been known for some time that PDI is required for the assembly of procollagen, where it moonlights as the β subunit of the enzyme prolyl 4-hydroxylase [reviewed in (43)]. Another, sometimes overlooked, example of the multifunctional nature of PDI is its heterodimerization with microsomal triglyceride transfer protein (MTP) in the ER, for example (74). MTP is made by the liver and intestine, and helps to transport fats by guiding the assembly and secretion of apolipoprotein B–containing lipoproteins. Too much circulating apolipoprotein B is often bad news, as it can predispose people to atherosclerosis, obesity, and diabetes. In the ER, PDI facilitates the transfer of apolipoprotein to MTP (11) and mutations in MTP that prevent it from interacting with PDI directly result in clinical abetalipoproteinemia (low lipid levels) (49, 52, 54). Mimicking or controlling PDIs contribution to the function of MTP might help provide new avenues for therapy in these diseases, and perhaps should be revisited in light of our improved understanding of oxidative protein folding pathways in the ER.
Cancer
PDI and its homologs are emerging as important players in other processes, such as cancer, that require changes in cell adhesion or migration. It was shown that P5 facilitates the detachment of tumor-associated proteins by reducing disulfide bonds on the tumor cell surface (32). The MHC I–related chain (MIC) protein that P5 targeted turns out to be a ligand for NKG2D, an NK cell receptor (Fig. 3). Thus the absence of MIC/NKG2D interactions partly liberates the cancerous cell from immune surveillance. In an unrelated study that provides clues to a functional link between PDI and P5, the metalloprotease ADAM17 (also known as TACE) was shown to direct PMA-induced release of MIC (10). It turns out that ADAM17 activity (judged by the shedding of an epidermal growth factor reporter protein) can be controlled at the cell surface by PDI (8, 75). Willems and colleagues demonstrated that PDI could maintain ADAM17 in an inactive conformation, and that changes in the local redox environment, such as mobilization of reactive oxygen species, could facilitate the activation of ADAM17 (75). The open conformation of ADAM17 is proposed to be zinc accessible, and is “closed” by interaction with PDI, most likely via shuffling ADAM17 disulfides (Fig. 3). It will be very interesting to learn whether other PDI family members can regulate ADAM activities in a similar way, and how targeting specific S-S bonds in ADAM17 is achieved at the molecular level.
A number of techniques are in use to probe PDI family client specificity. Using substrate trapping methods, P5 was revealed to bind strongly to BiP and maybe involved in the quality control of a subset of BiP clients in the ER, such as Ig domain proteins (30, 41). However, no effect of P5 knockdown was seen on the oxidative folding of a range of client proteins in HepG2 cells (57). Thus an alternative suggestion is that P5 mainly operates as a reductase in vivo, even though it has an oxidant activity in vitro (36). P5 has an atypical domain arrangement, meaning that it is likely to select its clients rather differently to PDI. BiP is upregulated in a number of cancers (12), so it will be interesting to see whether a BiP/P5 quality control pathway is specifically required for the regulation of integrins that are involved in metastasis.
Some clues about P5 function have also come from the zebrafish. In this model organism, morpholino knockdown of P5 expression resulted in a marked embryonic developmental phenotype: the heart, pancreas, liver, and gastro-intestinal system all lost their normal, asymmetric patterning (23). In other organs, such as the brain, all gene products that would normally be expressed asymmetrically lost their polarity. The study did not identify the P5 target(s) responsible for the zebrafish phenotype. In the light of recent data, it would be interesting to ascertain whether a failure in the quality control one or more integrins occurs in the absence of P5 during zebrafish development.
Cancer, colitis, and the anterior gradient proteins
Anterior gradient (AGR) 2 and AGR3, also known as hAG-2 and -3 in humans, were assigned as members of the PDI family by phylogenetic analysis in 2005 (48). These proteins drew the attention of cancer researchers because they were induced in estrogen-positive breast tumors (59). AGR2, unlike AGR3, was also expressed in malignant prostate epithelial cells (17). The functional significance of hAG expression in these cancers is still not clear, but a homolog in Xenopus laevis (XAG2) is expressed in the cement gland, which is required for anteroposterior fate determination during development in the African clawed toad (1, 58). A yeast two-hybrid analysis suggested that the GPI-anchored C4.4a protein and α-dystroglycan might be partners for AGRs, consistent with a role in quality control of adhesion proteins and the extracellular matrix (17). Subsequently, AGR2 −/− mice have been analyzed. These animals have a defect in intestinal mucus production, and are susceptible to experimentally induced colitis (47), so it would be exciting to see whether AGR2 mutations are linked to intestinal disease in humans.
Although AGR2 has a cysteine residue, which may form mixed disulfides with the mucin MUC2 (47), the AGRs lack redox-active CXXC motifs, and thus cannot directly transfer oxidizing equivalents to client proteins on their own. It is therefore likely that AGR2 works together with other PDI family members (or other oxidizing agents) to ensure the quality control of mucin, and perhaps other clients. The AGRs have ER retention/retrieval motifs (51), but further work needs to be done to establish how they function in the secretory pathway and whether they are active extracellularly. Since AGR2 is strongly expressed in normal lung tissue (59), the protein may also be required for maintenance of the respiratory mucosa.
Neurodegeneration
Nitrosative stress can modify the reactive cysteine residues of PDI by S-nitrosylation (Fig. 5), leading to misfolding and neurological disease (39, 44). In an exciting study, Uehara et al. examined tissue from the brains of patients with sporadic Parkinson's and Alzheimer's diseases. They found that PDI was S-nitrosylated, and that cysteine thiols in both redox-active domains of PDI were potentially subjected to modification (62). S-nitrosylation decreased both the chaperone activity and the isomerase activity of PDI, and overexpression of wild-type PDI protected cells from death in various cellular models of neurodegeneration. In addition to providing a potential new therapeutic route for Parkinson's and Alzheimer's diseases, these studies also raise questions about whether/how S-nitrosylation is controlled in the ER (or at the cell surface) and whether S-nitrosylation has a functional or regulatory role to play in other cell types. Interestingly, PDIp, the pancreas-specific PDI, has also been shown to be induced in experimental models of Parkinson's disease (14), but whether PDIp or other members of the PDI family are also subject to S-nitrosylation is not known.
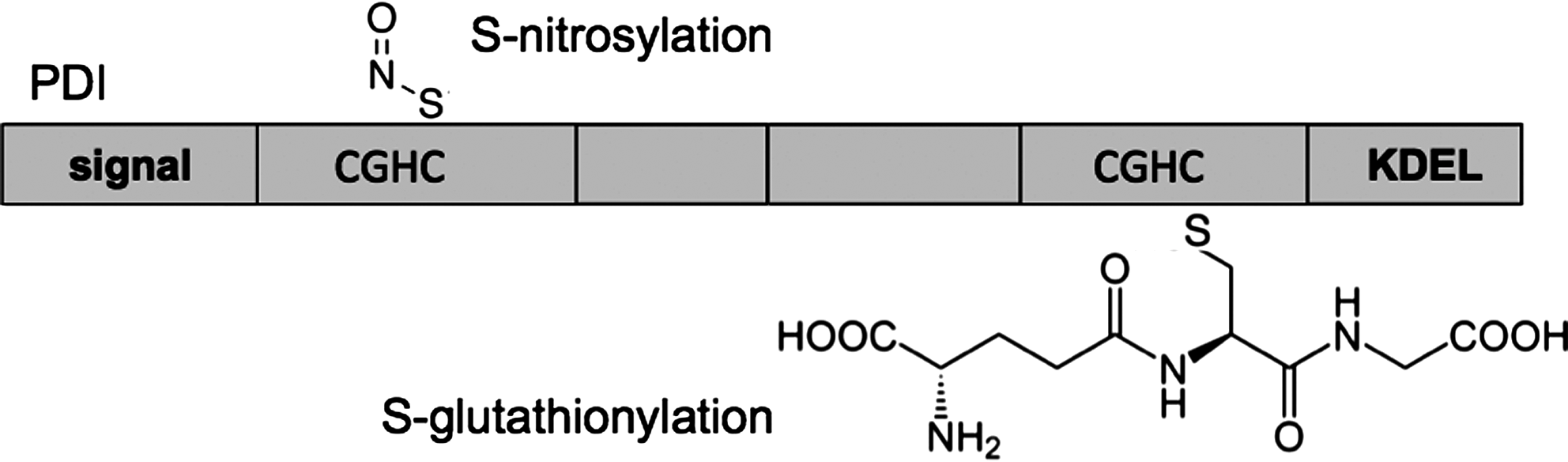
PDI has also been implicated in the pathology of Huntington's disease, where it localizes to mitochondrial-associated ER membranes and has a proapototic function (22), and in familial amyotrophic lateral sclerosis (ALS), a motor neuron disease linked to mutations in Cu/Zn superoxide dismutase (SOD1) (6). Proteomic analysis of the transgenic SOD1 (G93A) rat model of ALS revealed that various PDI family members were upregulated in the spinal cord, and that PDI colocalized with misfolded SOD intracellularly. PDI inactivation by S-nitrosylation (68) and concomitant activation of the unfolded protein response (7) has also been detected in the motor neurons of human ALS patients. The data suggest that PDI can be protective in ALS unless it is “switched off” by S-nitrosylation, and this is supported by the observation that the small molecule PDI mimic BMC protects against SOD1 inclusion body formation in a cellular model (68). Thus the PDI pathway could be a therapeutic target for ALS, although careful monitoring for detrimental effects on the folding of other clients needs to be considered.
S-glutathionylation of PDI (Fig. 5) might also occur after cells have been subjected to nitrosative stress (61). Treating immortalized cells with PABA/NO, a drug that raises intracellular nitric oxide levels, led to S-glutathionylation (but not S-nitrosylation) at cysteine residues within the PDI active sites. These modifications also inhibited the reductase activity of PDI, and gave rise to structural defects, as judged by changes in circular dichroism spectra (61). However, whether nitrosative stress leads to specific modification and/or regulation of PDI by S-glutathionylation in vivo still requires further study.
Infertility
PDI family members have attracted interest in fertility research, where disulfide bond rearrangements are necessary for sperm adhesion proteins to bind to their counterparts on the egg cell. The Primakoff group showed that PDI inhibitors such as bacitracin could inhibit sperm–egg fusion in vitro (15). They found that the PDI homolog ERp57 was expressed at the sperm surface, and that antibodies against ERp57 also blocked sperm–egg fusion. As a PDI homolog with poise toward reductase activity, ERp57 is an attractive candidate for the remodeling of sperm surface integrins. Although Ellerman et al. (15) and others (2, 79) have visualized ERp57 on sperm, alternate PDIs such as ERp29 (77) might also be involved in preparing sperm for fusion. Further work needs to be done on identifying the targets, using more specific reagents, to fully understand the redox control of sperm head proteins. Nevertheless, ERp57 holds promise as a marker for male infertility, as its expression was found to be downregulated in a cohort of male IVF patients with low-fertilization rates (79).
Whereas most PDIs have widespread expression in many cells and tissues, one PDI homolog, named PDILT, is only expressed in the testis. PDILT was identified as an unusual PDI relative with a hydrophobic C terminal tail extension that was conserved in vertebrates (63). Although PDILT shares the same
Summary
The number of PDI family members has expanded rapidly in the last few years, and with it the importance of these proteins in health and disease has grown. Here, I have discussed some recent examples from the fields of hemostasis, immunity, lipid biology, cancer, neurodegeneration, and infertility. The development of more specific tools and reagents, the advent of high-resolution crystal structures, and the development of inducible and tissue-specific knockout mice for some members of the PDI family should help us to define their specific functions in health and disease.
Footnotes
Acknowledgments
The author would like to acknowledge Arthritis Research, United Kingdom, the BBSRC, the Leverhulme Foundation, the MRC, and the Wellcome Trust for funding projects in the laboratory. The author apologizes to many scientists whose work could not be cited due to space considerations.