
Abstract
Introduction
Innovation
The observation that nitric oxide (NO)-mediated activation of Ras in different subcellular compartments regulates different downstream signaling pathways adds an exciting new dimension to NO-mediated Ras signaling. These findings may explain the different signaling outcomes—cell proliferation, cell differentiation, and apoptosis—derived from NO or from other reactive nitrogen species-mediated interactions with Ras. Our findings may assist in the design of NO donors that target specific and compartmentalized isoforms of Ras.
In mammals, the genome encodes three ras genes that give rise to four proteins (H-Ras, K-Ras4A, K-Ras4B, and N-Ras) (28). Although highly homologous and indistinguishable in most in vitro assays, these proteins have been shown to produce isoform-specific biological outputs, attributed to differences in the plasma membrane and subcellular membrane localization (61). The C-terminal hypervariable region directs the post-translational modifications of the primary ras gene products that determine their subcellular localizations (27).
Ras proteins are anchored to the membranes by a series of post-translational modifications occurring in a CAAX motif (where C is cysteine, A is an aliphatic aminoacid, and X is any amino acid) present in the C-terminal domain of all Ras proteins (15). Three enzymes that work sequentially modify the CAAX sequence. Farnesyltransferase catalyzes the addition of a 15-carbon farnesyl lipid to the CAAX–Cys residue resulting in farnesylcysteine. Farnesylcysteine Ras is transported to the membranes of the endoplasmic reticulum, where the Ras-converting enzyme 1 (RCE1) processes it. RCE1 removes the AAX amino acids, and the C-terminus farnesylcysteine is modified by the enzyme isoprenylcysteine carboxylmethyltransferase, which catalyses the methyl esterification of the α-carboxyl group of the farnesylcysteine. The result of these modifications is the irreversible farnesylation of Ras and creation of a hydrophobic domain at the C-terminus that mediates Ras association with the membrane [reviewed in (2)].
On the other hand, palmitoylation is a reversible post-translational modification that occurs by formation of a thioester bond on one (N-Ras) or two cysteines residues (H-Ras) adjacent to the farnesylcysteine (60). The enzyme that catalyzes Ras palmitoylation resides on the Golgi apparatus. Palmitoylated H-Ras and N-Ras localize to the plasma membrane and the surface of the Golgi apparatus. In contrast to the other isoforms, K-Ras4B (K-Ras) is not palmitoylated and requires no further modification for full affinity (2).
Ras has long been thought to operate only at the plasma membrane. However, several publications have shown that the interaction of Ras with the plasma membrane is highly dynamic, and that Ras compartmentalization plays an important role in the process of growth factor-mediated signal transduction events (12, 13). Ras is present in endosomes, the endoplasmic reticulum, the Golgi apparatus, and mitochondria [reviewed in (20)]. These different subcellular locations can result in distinct activities through the activation of different signaling pathways (13). Ras activation at the Golgi does not require endocytosis or vesicular transport (13). This activation is mediated by Src-dependent signaling through phospholipase Cγ1 (PLCγ1) and the GEF RasGPR1, in contrast with the Grb2/SOS pathway that initiates signaling from the plasma membrane (7). Differential kinetic activation of Ras was found to be associated with compartmentalization of the GTPase. A rapid and transient activation of Ras associated with the plasma membrane is followed by a delayed and sustained activation of the Golgi-localized Ras (13, 61).
Accumulated experimental evidence over the last two decades supports the involvement of nitric oxide (NO) and other reactive nitrogen species (RNS) in signaling pathways associated with physiological and pathophysiological processes (30, 44, 45). NO, a signaling free radical produced physiologically by the ubiquitously expressed NO synthases (NOSs), can react directly with thiols to form a radical anion intermediate, which is oxidized to an S-nitrosothiol in the presence of an electron acceptor (26). Alternatively, NO may react with O2 to produce nitrosylating equivalents (53).
The reaction of NO with the most abundant cellular nonprotein thiol, glutathione (GSH), results in the formation of S-nitrosoglutathione (GSNO), the most physiologically relevant S-nitrosothiol (30). In addition, it is increasingly recognized that the S-nitrosylation of proteins and peptides plays an important role in a large number of signaling pathways (30). Over a thousand S-nitrosylated proteins of different classes have been identified. It has been demonstrated that this redox-based post-translational modification is essential for regulating the functions of these proteins. Thus, S-nitrosylation of proteins could provide the foundation for a regulatory mechanism based on physiological changes in the intracellular redox status (30, 45).
Although several studies have shown that NO can mediate cell death (47, 57), NO has also been proposed to be a physiological modulator of cell proliferation [reviewed in (58, 70)]. This paradoxical behavior may be explained by the action of distinct concentrations of NO attained under different experimental conditions within the cell systems used. In this context, low and high local concentrations of NO stimulate and inhibit, respectively, the proliferation of different cell types (67).
Early findings account for the NO stimulation of Ras activity through S-nitrosylation at the Cys118 residue of Ras (36). It was subsequently demonstrated that the S-nitrosylation and activation of Ras induces the Ras-MAP kinase–ERK1/2 signaling pathway in rabbit aortic endothelial cells, leading to cell cycle progression and proliferation (58, 59). The participation of Src kinase in NO-mediated proliferative and migration-associated signaling events has been recently demonstrated (16, 55). Ibiza et al. (31) recently provided evidence for a NOS-dependent mechanism that allows for differential activation of Ras at specific subcellular compartments in T cells. The authors proposed that Ras activation at the Golgi results in positive selection of T cells, whereas its activation at the plasma membrane results in negative selection of T cells.
Here, we investigated whether endothelial nitric oxide synthase (eNOS)-derived NO and low concentrations of the S-nitrosothiol GSNO regulate the activation of Ras and its compartmentalization during the stimulation of cell proliferation. Using genetically encoded fluorescent probes that report where and when Ras is active in living cells, we described the NO- and GSNO-mediated Ras signaling pathways at intracellular membranes. Although both signaling pathways are associated with S-nitrosylation of Ras, the activation and compartmentalization of Ras were dependent on the nature of the NO source and on the participation of Src kinase.
Results
GSNO promotes the S-nitrosylation and activation of H-Ras and Ras-mediated signaling events in HeLa cells
NO promotes guanine nucleotide exchange on the critical cellular signaling protein Ras by S-nitrosylation of a redox-active thiol group (Cys118) (36). To assess the ability of GSNO to activate Ras, we used the Ras-binding domain of Raf-1 kinase (RBD) that specifically binds the active, GTP-bound form of Ras (16a). Ras activity was determined in HeLa cells grown under low serum conditions for 24 h that were exposed to 100 μM GSNO for increasing time periods. Early and transient activation of Ras was observed after 5 min of stimulation with the nitrosothiol. Sustained activation of Ras was observed after 30 min of incubation with GSNO, which persisted for an additional 15 min of exposure to GSNO (Fig. 1A).

Mammalian genomes encode three ras genes that are translated into four protein isoforms: N-Ras, H-Ras, K-Ras4A, and K-Ras4B. These isoforms are expressed ubiquitously, although isoform ratios vary from tissue to tissue (28). To determine which isoform of Ras is predominant in HeLa cells, we assessed their mRNA levels by quantitative real-time polymerase chain reaction (PCR). Although all four Ras isoforms are expressed in HeLa cells, H-Ras is the predominant isoform (Fig. 1B). Because H-Ras is the predominant isoform of Ras in HeLa cells, we performed additional experiments to investigate the profile of H-Ras activation after stimulation with 100 μM GSNO. Similar to the activation profile determined for total Ras, a biphasic activation profile was observed for H-Ras after the stimulation of HeLa cells with 100 μM GSNO (Fig. 1C).
To evaluate the role of the S-nitrosylation of Ras on GSNO-mediated activation of the GTPase, lysates from HeLa cells exposed to 100 μM GSNO were subjected to the biotin-switch technique (BST) and western blotting with an anti-Ras antibody. We found that 100 μM GSNO induced a rapid and transient increase on S-nitrosylation levels of Ras, which fell after 45–60 min (Fig. 1D).
To evaluate further the role of the S-nitrosylation on GSNO-mediated activation of Ras, we overexpressed H-RasWT and H-RasC118S (a non-nitrosatable mutant of H-Ras) in HeLa cells. Western blot analysis revealed that the transfection of HeLa cells with H-RasWT and H-RasC118S resulted in expression of H-Ras that was three- to fourfold higher than basal levels (Supplementary Fig. S1A; Supplementary Data are available online at
To rule out differences in NO generation in HeLa H-RasWT and HeLa H-RasC118S cells caused by the intracellular metabolism of GSNO, we examined the NO levels in both cell lines incubated with 100 μM GSNO. As determined by DAF2-derived fluorescence, the intracellular levels of NO increased to the same extent in all cell lines after 40 min of incubation with 100 μM GSNO compared with nontreated cells (Supplementary Fig. S1C). An estimate of the amount of NO released from GSNO-stimulated HeLa cells is shown on Supplementary Fig. S1D.
S-nitrosylation and activation of Ras promote the activation of the Raf/MEK/ERK pathway (36, 58). Therefore, we investigated the signaling events occurring downstream to Ras in HeLa cells exposed to 100 μM GSNO. ERK1/2 MAP kinase stimulation of phosphorylation was observed after 30 min of incubation with 100 μM GSNO, and maintained up to 60 min of incubation (Fig. 1E).
H-Ras is active at the plasma membrane and at the Golgi apparatus upon exposure of HeLa cells to low concentrations of GSNO
Using GFP fused to the Ras-binding domain of Raf-1 (GFP-RBD) as a spatiotemporal probe for active Ras in living cells, Chiu and coworkers demonstrated that Ras-GTP accumulates at the Golgi as well as at the plasma membrane in response to epidermal growth factor (EGF) signaling in fibroblasts (13). Using the same probe, we intended to establish the spatiotemporal profile of H-Ras activation in living HeLa cells exposed to GSNO. GFP-RBD was coexpressed with H-RasWT or H-RasC118S in serum-starved HeLa cells cultured in the absence or presence of 100 μM GSNO. When expressed alone, GFP-RBD localized primarily in the nuclear region without accumulating on any membrane structure (data not shown). This pattern was indistinguishable from that of GFP expressed alone (empty vector) or with untagged H-RasWT (Fig. 2A, upper panel).
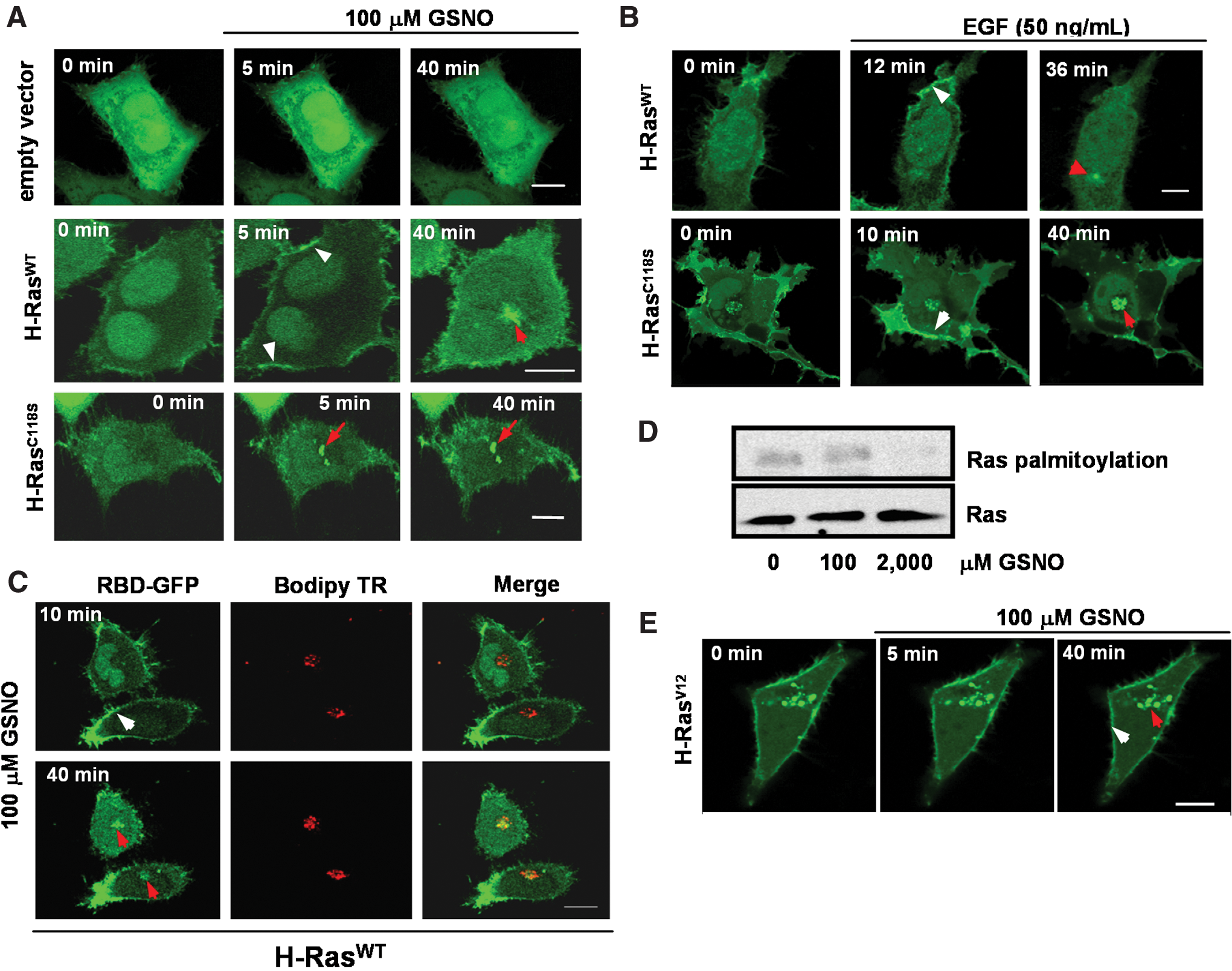
In H-RasWT-HeLa cells, we observed a rapid (5 min) and transient (reversed within 20–40 min) redistribution of GFP-RBD to the plasma membrane, followed by a delayed and sustained recruitment (up to 40 min) to the perinuclear region (Fig. 2A, middle panel). In cells overexpressing H-RasC118S, there was no activation of H-Ras at the plasma membrane, in contrast with the robust and sustained activation of the GTPase in the perinuclear region (Fig. 2A, lower panel). GFP-RBD was coexpressed with H-RasWT or H-RasC118S in serum-starved HeLa cells cultured in the presence of EGF (50 ng/ml). However, there were no differences observed between the activation pattern of Ras in HeLa H-RasC118S and the pattern observed for HeLa H-RasWT (Fig. 2B). These observations strongly support the S-nitrosylation of Cys118 in Ras as a key event in determining the GSNO-mediated activation pattern for the GTPase. The juxtanuclear accumulation of GFP-RBD was confirmed to be at the Golgi apparatus by showing colocalization with a Golgi-specific marker (BODIPY ceramide) in live cells coexpressed with untagged H-RasWT after GSNO stimulation (Fig. 2C).
Palmitoylated H-Ras is translocated by vesicular transport from the plasma membrane to the Golgi apparatus (14). Palmitoylation and depalmitoylation of H-Ras potentially can be regulated by S-nitrosothiols (6). We investigated whether low concentrations of GSNO used in this study would be able to interfere with H-Ras palmitoylation. We used the hydroxylamine-dependent biotin-switch assay to determine Ras palmitoylation levels in HeLa cells treated with 100 μM GSNO. We did not observe changes in Ras palmitoylation levels as compared to control levels in HeLa cells after the treatment (Fig. 2D). In contrast, Ras palmitoylation levels decreased after exposure of HeLa cells to 2.0 mM GSNO (Fig. 2D).
Additionally, we investigated whether low concentrations of GSNO would be able to stimulate protein trafficking. We coexpressed the constitutively active H-RasV12 mutant with GFP-RBD in HeLa cells. Cells were serum-starved overnight, cultured in the presence or absence of 100 μM GSNO, and subjected to live-cell imaging. We observed a dramatic redistribution of the fluorescent reporter to the plasma membrane and Golgi that was independent of stimulation with GSNO (Fig. 2E). These findings rule out any participation of GSNO in protein trafficking at the concentrations used in this study.
Inhibition of Src kinase prevented GSNO-mediated activation of H-Ras at the Golgi
Recent studies using COS-1 cells stimulated by EGF showed that Ras can be activated at the Golgi apparatus by the recruitment of Src protein tyrosine kinase. Src phosphorylates PLCγ, which stimulates the production of diacylglycerol (DAG) and inositol triphosphate, resulting in the elevation of intracellular Ca2+ levels (7, 11). Thus, we investigated the participation of Src kinase, PLCγ, and Ca2+ in GSNO-mediated Ras activation in subcellular compartments.
Serum-starved (24 h) HeLa cells were cotransfected with GFP-RBD and H-RasWT and cultured for 30 min in the presence or absence of 2.0 μM PP2, a selective inhibitor of Src kinase (29), followed by treatment with 100 μM GSNO. GSNO-stimulated compartmentalized Ras signaling in live cells was analyzed by confocal microscopy. In the presence of PP2, we observed that GSNO promoted a rapid and persistent distribution of the probe (GFP-RBD) to the plasma membrane. We did not observe recruitment of the probe to the Golgi during analysis, demonstrating the involvement of Src kinase in this process. Activation of PLCγ and Ca2+ mobilization are placed downstream from Src kinase in the signaling cascade associated with compartmentalized Ras activation. The inhibition of PLCγ1 and the chelation of intracellular Ca2+ with BAPTA-AM prevented GSNO-mediated Ras activation at the Golgi apparatus (Fig. 3A).

We analyzed the S-nitrosylation and phosphorylation levels of Tyr416 of Src kinase in serum-starved HeLa cells under the same experimental conditions used to investigate Ras activation. A rapid and transient increase in the phosphorylation levels of Tyr416 (10 min) was observed after exposure of cells to 100 μM GSNO (Fig. 3B).
To evaluate the role of the S-nitrosylation of Src kinase on GSNO-mediated activation of the GTPase, lysates from HeLa cells exposed to 100 μM GSNO were subjected to BST and western blotting with an anti-Src antibody. We found that 100 μM GSNO induced a rapid and transient increase on S-nitrosylation levels of Src (maximum at 15 min), which fell after 45 min (Fig. 3C). Similar results were obtained when we used the anti-nitrosocysteine antibody to detect S-nitrosylation of Src kinase (Supplementary Fig. S2).
S-nitrosylation and phosphorylation at Tyr416 of Src kinase occurred almost simultaneously. Both post-translational modifications may cooperate resulting in Src kinase activation.
H-Ras is active at the plasma membrane upon stimulation of endothelial cells with bradykinin
We investigated the spatiotemporal characteristics of NO-mediated Ras signaling under conditions of endogenous NO production. Accumulating experimental evidence indicates that NOS enzymes are targeted to specific subcellular compartments. Cellular compartmentalization of NOS enzymes is essential for the specific nitrosylation of other signaling proteins. Within endothelial cells, the plasma membrane and the Golgi apparatus constitute the most prominent compartments for eNOS (49). With these facts in mind, we investigated the occurrence of eNOS-stimulated compartmentalized Ras signaling in endothelial cells. Human umbilical vein endothelial cells (HUVECs) were stimulated with 1.0 μM bradykinin (Bk), resulting in the phosphorylation and activation of eNOS. The activation of eNOS followed a biphasic pattern with maximal phosphorylation at 5 min (early phase) and 240 min (late phase). (Fig. 4A). A biphasic pattern of NO release paralleled the biphasic pattern of the phosphorylation and activation of eNOS (Fig. 4B). The NOS inhibitor L-NAME substantially inhibited Bk-induced NO synthesis in HUVECs (Fig. 4C). An estimate of the amount of NO released from Bk-stimulated HUVECs is shown on Supplementary Fig. S3.
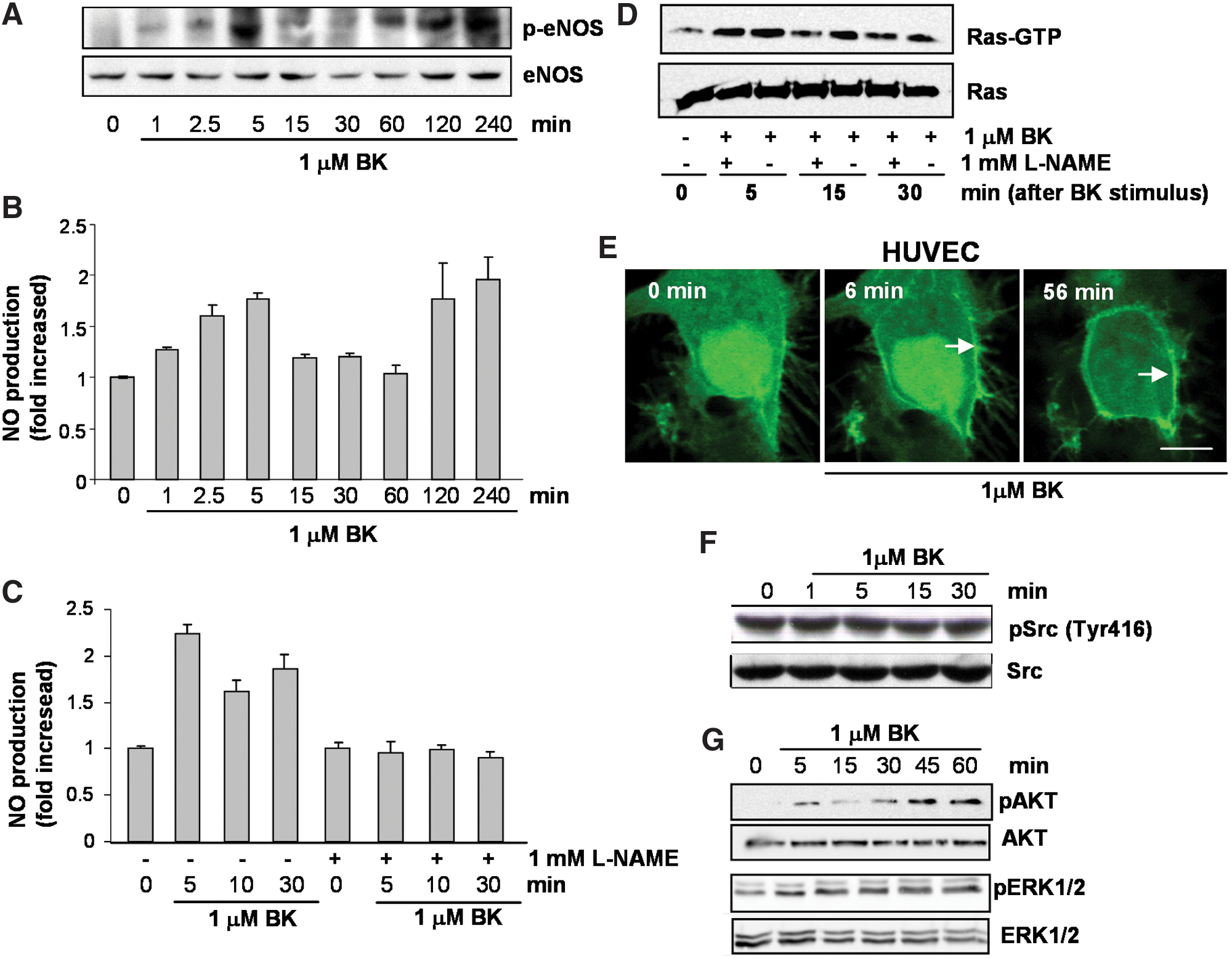
Ras activity was determined in HUVECs grown for 24 h in low-serum conditions that were stimulated with 1.0 μM Bk for increasing periods. Early and transient activation of Ras was observed after 5 min of stimulation with Bk. Sustained activation of Ras was observed after 15 min of stimulation with Bk that persisted for an additional 15 min. Pretreatment of HUVECs with L-NAME partially prevented Ras activation, confirming the dependence on eNOS (Fig. 4D).
HUVECs were cotransfected with GFP-RBD and H-RasWT, and Ras signaling in living cells was analyzed by confocal microscopy after stimulation with Bk. Endothelial NOS-mediated Ras signaling occurred exclusively at the plasma membrane level (Fig. 4E). We analyzed the phosphorylation levels of Tyr416 of Src kinase in serum-starved HUVECs under the same experimental conditions used to investigate Ras activation. In contrast with GSNO-stimulated Src-Ras signaling (Fig. 3B, C), the stimulation of HUVECs with Bk did not result in the activation of Src kinase (Fig. 4F). Downstream from Ras, we detected stimulation of phosphorylation levels on Akt and on the ERK1/2 MAP kinases. Increasing phosphorylation of Akt was observed from 5- to 60-min stimulation with Bk. A similar pattern of incremental levels of phosphorylation was observed for the ERK1/2 MAP kinases (Fig. 4G).
GSNO-stimulated cell proliferation is dependent on the intracellular redox environment and on Ras activation
To confirm the biological significance of Ras activation with GSNO or with eNOS-derived NO, we studied cell proliferation as a readout of Ras function. The importance of steady-state concentrations of NO has been increasingly recognized as a major determinant of its biological functions (67). Studies using low concentrations of NO donors or stimulation of the constitutive isoforms of NOS have clearly demonstrated that NO, from NO donors or derived from active NOS, induces cell proliferation in different cell lines (19, 33, 48, 58). HeLa cells were cultured with different concentrations of GSNO (1.0–100 μM) for 5 h, as described in the Materials and Methods section. An 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was carried out 24 or 48 h after withdrawal of the stimulus. Cells preincubated for 5 h with low concentrations (1.0–100 μM) of GSNO were significantly stimulated to proliferate after 24 and 48 h. Maximum stimulation of HeLa cell proliferation was observed after incubation with 100 μM GSNO. However, exposure of HeLa cells to 1.0 mM GSNO inhibited cell proliferation (Fig. 5A). Extending our observations to other cell types, we showed that COS-1 cells also proliferate upon stimulation with 100 μM GSNO (Fig. 5B).
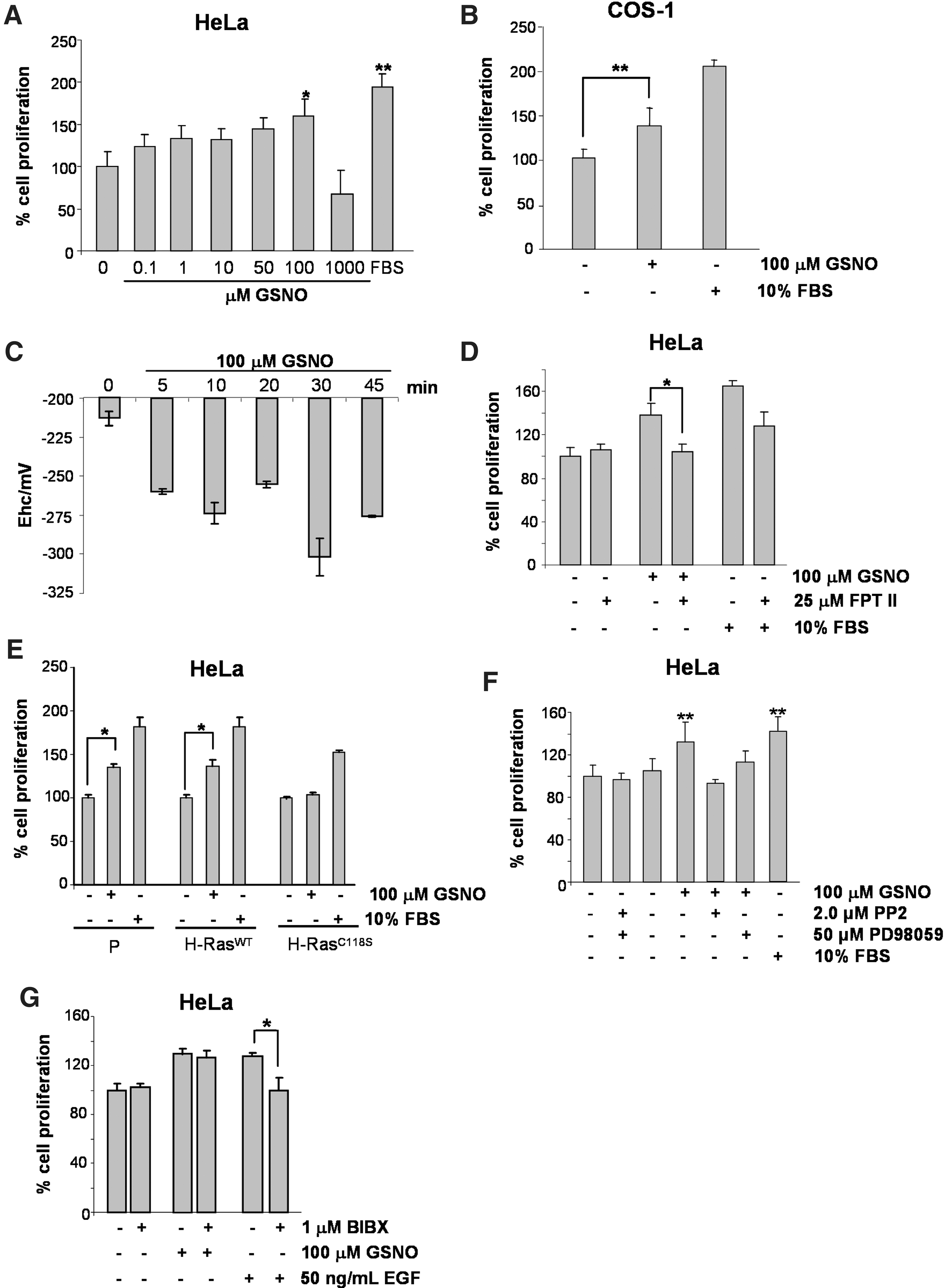
Changes in the half-cell reduction potential (Ehc) of the glutathione disulfide–glutathione (GSSG/2GSH) couple are thought to correlate with the biological status of the cell. In general, negative values for Ehc are indicative of cell proliferation, whereas positive values for Ehc are indicative of growth arrest and/or cell death (63). We evaluated the redox state of serum-starved HeLa cells stimulated with 100 μM GSNO for increasing periods. The intracellular concentrations of GSH and GSSG were determined, and the values of the redox state of the GSSG/2GSH couple were calculated and applied to the Nernst equation (63). The values obtained were more negative (−320 mV) after a 30-min exposure of cells to GSNO (Fig. 5C). The results indicated that a favorable redox state was achieved when HeLa cells proliferate after GSNO stimulus.
Ras and Ras GTPases must undergo farnesylation at their carboxyterminal domains before they can localize at the cytoplasmic side of the plasma membrane (66). FPT II, a potent and selective inhibitor of the enzyme farnesyl transferase, efficiently prevents farnesylation of Ras (56). To determine if GSNO-stimulated cell proliferation is dependent on Ras activation, HeLa cells were preincubated for 24 h with FPT II before GSNO exposure. Under these conditions, GSNO-stimulated HeLa cell proliferation was inhibited, indicating that Ras plays an important role in this process (Fig. 5D). The proliferation of parental HeLa, HeLa H-RasWT, and HeLa H-RasC118S cells was assayed in cells cultured in the presence and absence of 100 μM GSNO. HeLa and HeLa H-RasWT cells cultured with 100 μM GSNO proliferated significantly, with increases in proliferation rates of 43% and 55%, respectively. On the contrary, in HeLa H-RasC118S, we did not observe cell proliferation (Fig. 5E). The proliferation rate of the positive control cells (stimulation with 10% fetal bovine serum [FBS]) was ∼80% in both cases. These results suggest that the S-nitrosylation of H-Ras is an important signaling event in the NO-mediated proliferation of HeLa cells.
Recently, our group demonstrated that low concentrations of S-nitrosothiol S-nitroso-N-acetylpenicillamine (SNAP) stimulated the proliferation of mouse embryo fibroblasts. The SNAP-stimulated proliferation of mouse embryo fibroblasts was directly associated with the S-nitrosylation and activation of Src kinase (16).
PP2 is a pyrrolopyrimidine that is an effective inhibitor of Src-family kinases (29). Cell proliferation was determined in HeLa cells that were serum-starved for 24 h and cultured in the presence or absence of GSNO and PP2. PP2-mediated inhibition of Src kinase prevented the GSNO-induced proliferation of HeLa cells, implicating Src kinase in this process (Fig. 5F). Furthermore, GSNO-mediated proliferative signaling was dependent on the activation of the ERK1/2 MAP kinases. Preincubation of cells with the MEK inhibitor PD98059 prevented GSNO-stimulated proliferation of HeLa cells (Fig. 5F).
We and others have demonstrated that NO can signal through a cascade of reactions initiated by the association of the growth factor EGF to its cognate receptor (52, 70). We investigated the participation of EGF/EGFR in the GSNO-stimulated proliferation of HeLa cells. Preincubation of HeLa cells with BIBX1382 (1.0 μM), a specific inhibitor of EGFR (65), did not prevent the GSNO-stimulated proliferation of HeLa cells, ruling out the participation of EGFR in this process (Fig. 5G).
eNOS-derived NO-stimulated proliferation of endothelial cells is also dependent on Ras activation
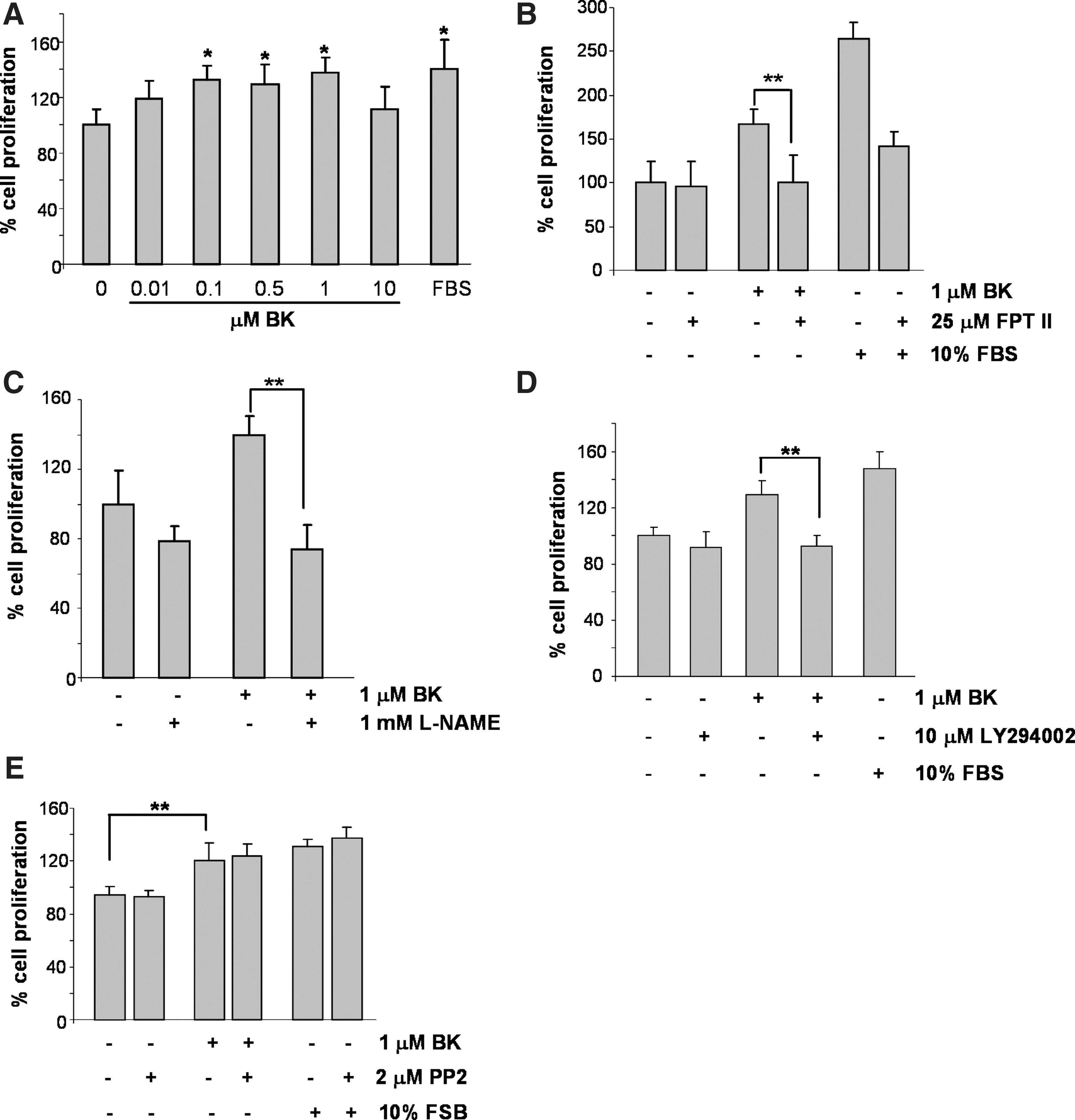
It was early shown that increasing concentrations of Bk promoted DNA synthesis and proliferation of human venular endothelial cells (45). We reproduced these observations, showing a positive effect on proliferation of HUVECs by low concentrations of Bk (0.01–1.0 μM). On the other hand, incubation of cells with 10 μM Bk resulted in growth arrest (Fig. 6A). In addition, the farnesyl transferase inhibitor of Ras, FPT II, the PI3K inhibitor, LY294002, and the general NOS inhibitor L-NAME effectively block eNOS-derived NO-promoted proliferation of HUVECs stimulated with 1.0 μM Bk (Fig. 6B–D). However, in contrast with GSNO-mediated proliferative signaling, the inhibition of Src kinase showed no effects on the Bk-stimulated proliferation of HUVECs (Fig. 6E).
Discussion
Post-translational modifications of proteins are crucial events in cell signaling. They include phosphorylation on Ser/Thr or Tyr residues and redox-based modifications such as the S-nitrosylation of Cys residues (30, 51, 64). Increasing experimental evidence indicates that cysteine S-nitrosylation and tyrosine phosphorylation may cooperate with each other by influencing the same signal transduction pathways (45). Early findings on the S-nitrosylation of a single residue of Ras, Cys118, have provided the foundation for understanding the structural basis of the NO-Ras interaction (36).
In addition to S-nitrosylation, S-glutathionylation of Ras at Cys118 was associated with GTPase activation in angiotensin-stimulated vascular smooth muscle cells (1). The authors suggested that Ras S-nitrosylation might be an intermediate during GSH modification of Ras. Taking under consideration the fact that we found that S-nitrosylation was a transient modification of Ras (Fig. 3C and Supplementary Fig. S2), our findings may concur with these observations.
S-glutathionylation and S-nitrosylation may occur simultaneously in the same protein in the presence of appropriate interactions between reactive oxygen and nitrogen species. The chemical nature of the nitrosylating agent, and features such as histidine, and basic and acidic residues flanking the reactive cysteine, will determine the occurrence of S-nitrosylation, S-glutathionylation, or both (25).
Mammalian cells express four protein isoforms of Ras, H-Ras, K-Ras4A, K-Ras4B, and N-Ras, all of which have very similar structures. Using the specific anti-SNOCys antibody, we showed that low concentrations of GSNO can transnitrosylate H-Ras, the predominant Ras isoform in HeLa cells. These findings suggested that transnitrosylation from the nitrosothiol to the GTPase would generate an S-nitrosylated form of H-Ras that is activated. On the contrary, although we also showed that the activation of Ras in HUVECs upon Bk stimulation was dependent on NO, we were unable to show that this activation was directly associated with the S-nitrosylation of Ras. These differences could be explained by the fact that in many circumstances, the actions of endogenously generated NO from NOS cannot be reproduced by exogenous NO donors.
A step forward was taken in the present study, in which we showed spatiotemporal changes in the Ras activation pattern after stimulation of HeLa cells with GSNO and after stimulation of HUVECs with Bk. Using a fluorescent reporter of Ras activation in living cells, we demonstrated marked differences in the compartmentalization of Ras after the activation of this protein by GSNO and eNOS-derived NO.
The pattern of GSNO-mediated compartmentalized H-Ras activation is very similar to that described by Bivona and coworkers (7) in response to an EGF stimulus. GSNO stimulated GDP-GTP exchange of Ras at the plasma membrane and at the Golgi. A transient activation of Ras at the plasma membrane was followed by a sustained activation of Ras at the Golgi. The mechanism proposed by Bivona et al. involves the activation of the EGFR on the cell surface, which in turn recruits the protein tyrosine kinase Src. Simultaneously, another enzyme, PLC-γ1, is recruited and phosphorylated by Src. Phosphorylated PLC-γ1 induces the formation of DAG and promotes increased levels of intracellular Ca2+.
Our results showed that H-RasWT is activated both at the plasma membrane and at the Golgi apparatus by low concentrations (100 μM) of GSNO (perinuclear labeling). Low levels of GSNO led to S-nitrosylation of Ras at the plasma membrane on Cys118 (Fig. 1D), whereas Ras activation observed at the Golgi apparatus was independent of the direct action of the nitrosothiol (Fig. 2B). This is corroborated by our findings in HeLa cells that overexpress H-RasC118S, the non-nitrosatable form of Ras. Incubation of H-RasC118S HeLa cells with GSNO leads to a sustained activation of Ras only at the Golgi apparatus.
In addition, GSNO-mediated compartmentalized Ras activation was dependent on Src kinase. Accordingly, blocking Src activity inhibited Ras activation at the Golgi apparatus, but not at the plasma membrane. Early observations accounted for changes in the phosphotyrosine content of Src promoted by exogenous NO sources (3, 46). GSNO-mediated activation of Src kinase was initiated by cysteine S-nitrosylation, followed by tyrosine phosphorylation at Tyr416. Recently, our group demonstrated that SNAP promotes the S-nitrosylation and activation of Src kinase in mouse embryo fibroblasts. The S-nitrosylation of Src kinase leads to structural changes in the protein, allowing the phosphorylation of Tyr416, a marker of Src kinase activation (16). Almost concomitantly with the description of our findings, the Cys498 residue of Src kinase was identified as the site of S-nitrosylation that is associated with kinase activation in the human breast cancer cell line MCF-7 (55).
Although Src kinase clearly mediated GSNO-stimulated compartmentalized Ras activation, this process was independent of EGFR activation. GSNO-mediated Src activation is an early signaling event coinciding with the period for Ras activation at the plasma membrane. The cytoplasmic guanine nucleotide-releasing proteins, Ras-GRPs, were strongly implicated in GSNO-stimulated Ras activation via the inhibition of this process by an inhibitor of Src kinase, by a specific inhibitor of the downstream enzyme PLC-γ and by a Ca2+ chelator. Ras-GRPs are activated by elevated levels of the second messengers DAG and Ca2+ (18). PLC-γ is a known substrate of Src (38) and upon activation, generates the second messengers needed for the activation of Ras-GRPs. Phorbol 12-myristate 13-acetate promotes oxidative stress and induces the translocation of RasGRP3 from the cytosol to the endoplasmic reticulum and the Golgi in HEK-293 cells (41). Based on these data, Ras-GRPs are strong candidates as upstream elements in the GSNO-mediated activation of Ras at the Golgi.
It has been demonstrated that Ras/ERK1/2 MAP kinase signaling can proceed from the plasma membrane and from the Golgi apparatus (20). Although our data do not allow us to discriminate between both cell compartments from where the signals are coming from, GSNO-mediated sustained activation of the ERK1/2 MAP kinases suggests that signals from both compartments are converging.
As opposed to GSNO-stimulated Ras signaling in HeLa cells, eNOS-derived NO-stimulated Ras signaling in HUVECs occurred only in one compartment, the plasma membrane. Although both sources of NO may trigger the S-nitrosylation and activation of Ras, the downstream elements of these signaling cascades are divergent. In contrast to GSNO-stimulated Ras signaling, in which Src kinase plays a major role, Ras signaling stimulated by eNOS-derived NO is independent of Src kinase activation.
An extensive number of reports in the literature present compelling experimental evidence that the signaling events mediated by endogenously generated NO can be recapitulated by NO generated from exogenous sources. However, in many circumstances, this equivalence of NO sources is not observed. Our findings on GSNO-derived versus eNOS-derived NO-stimulated Ras signaling provide an additional argument, suggesting that not all sources of NO are bioequivalent. The differences between NO pools generated from different sources start with the spatial distribution of NO within the cells. Iwakiri et al. (32) showed that NO generated by the S-nitrosothiols GSNO and SNAP is diffusely distributed inside COS-7 cells loaded with DAF-2. Conversely, activation of eNOS was concomitant with the generation of a localized and concentrated NO pool (24). Our findings on the activation of Ras by eNOS-derived NO are consistent with this localized production of NO pools. Furthermore, Ras activation was dependent on NO production, but independent of EGFR mediation (not shown). This is a strong argument in favor of a compartmentalized NO production associated with a compartmentalized activation of Ras. In addition to the compartmentalization of NO generation by eNOS, this enzyme's localization to both the plasma membrane and the Golgi apparatus in endothelial cells determines specific functions for eNOS in each compartment. Although the mechanisms underlying this distribution are not completely understood, the nature of eNOS agonists is important to the compartmentalized activation of the enzyme. Agonists that utilize Akt activation as their primary triggering event, such as insulin and estrogen (17), would preferentially activate Golgi eNOS. On the other hand, calcium-dependent agonists such as thapsigargin and Bk (used in this study) would preferentially activate eNOS from the plasma membrane (23).
We showed that NO signaling through Ras localized at the plasma membrane leads to the activation of Akt and of the ERK1/2 MAP kinases. Our findings are in agreement with the stimulation of B2R receptors by Bk that leads to downstream activation of the PI3K/Akt pathway in human endothelial cells. Activated Akt phosphorylates eNOS at Ser1177, a major activation site for the enzyme (16). Furthermore, B2R agonists such as Bk also promote the activation of ERK1/2 MAP kinases (9).
The general scheme describing the differences in the compartmentalization of Ras signaling mediated by GSNO-derived NO versus eNOS-derived NO is shown in Figure 7.

Growth factors such as EGF associate with their specific receptors to trigger signaling cascades associated with proliferation. However, reactive species such as NO and nitrosothiols do not have specific receptors to signal. Other elements must be taken into consideration to attribute a specific signaling role to these species. One of these elements is related to changes in the redox environment. Early studies demonstrated that the redox state of the GSSG/2GSH couple can serve as an important indicator of the redox environment. Changes in the redox potential (Ehc) of the GSSG/2GSH couple associated with a more-reducing environment (Ehc ∼−240 mV) suggest a positive correlation with cell proliferation (63). Accordingly, in HeLa cells exposed to low concentrations of GSNO, we determined changes in the redox potential of the GSSG/2GSH couple that were compatible with cell proliferation. In addition to the promotion of changes in the redox environment, it has been extensively demonstrated that NO and other reactive species can activate and assemble a cascade of the same signaling proteins used by growth factors to promote cell growth [reviewed in (45)].
The proliferation of mammalian cells almost invariably involves the small GTPase Ras (66). Local and temporal changes in the activation state of Ras follow the stimulation of different cell types with growth factors (7, 8, 31). Recently, our group described the SNAP-mediated S-nitrosylation of Ras and activation of the Ras-ERK1/2 signaling pathway in rabbit aortic endothelial cells. Activation of this signaling pathway leads to cell cycle progression and proliferation (58, 59).
The inducible isoform of NOS is the isoform commonly associated with tumorigenesis (37). Inducible NOS (iNOS) upregulation and sustained NO generation are induced during the inflammatory response and correlate positively with lung tumorigenesis. It was recently shown that the expression of oncogenic K-Ras is directly associated with the expression of iNOS in a mouse lung cancer model (50). Inflammatory cytokines and lipopolysaccharide stimulated iNOS and promote the formation of intracellular S-nitrosothiols, in particular GSNO, in RAW 264.7 murine macrophages (71). Therefore, activation of iNOS in tumor cells could promote the generation of GSNO and activation of Ras, a situation that resembles our experimental model.
Using GFP-RBD probes that precisely report the spatiotemporal signaling of Ras in living cells, it was shown that Ras-GTP accumulates at the plasma membrane as well as at the Golgi in response to proliferative signaling stimulated by growth factors such as EGF (7). Although similar to EGF-stimulated Ras signaling, we showed that GSNO-stimulated Ras signaling did not involve the participation of EGF and EGFR.
S-nitrosothiols may interfere with palmitoylation of H-Ras. Studies by Baker et al. (6) and Mallis et al. (42, 43) demonstrated in NIH/3T3 cells that GSNO-mediated alterations on H-Ras palmitoylation were observed at concentrations of 4.0–5.0 mM. Alterations were not observed at lower concentrations. Accordingly, we did not observe changes on palmitoylation when cells were exposed to 100 μM GSNO as compared to control nontreated cells (Fig. 2D). On the other hand, confirming Baker's and Mallis' observations, exposure of HeLa cells to 2.0 mM GSNO resulted in inhibion of palmitoylation of H-Ras (Fig. 2D). Furthermore, in HeLa cells cotransfected with H-RasV12, an oncogenic isoform of Ras, the accumulation of Ras-GTP at the plasma membrane and at the Golgi was unaffected by exposure to GSNO. This result argues against GSNO-stimulated Ras-GTP trafficking (6) from the plasma membrane to the Golgi. Our findings strongly suggest that low concentrations (up to 100 μM GSNO) do not alter H-Ras traffic or turnover. In fact, we are observing the redox modulation of Ras-mediated signaling from two cell compartments, the plasma membrane and the Golgi apparatus.
Proliferative concentrations of GSNO induced H-Ras activation at the plasma membrane and Golgi in HeLa H-RasWT, whereas in HeLa H-RasC118S, activation of H-Ras was observed only at the Golgi apparatus. Because GSNO could not induce the proliferation of HeLa cells expressing H-RasC118S, our findings indicate that the activation of Ras at the plasma membrane and subsequent activation at the Golgi are essential to S-nitrosylation-mediated Ras activation, which is in turn linked to cell proliferation.
GSNO promoted the S-nitrosylation of H-Ras and Src kinase localized at the plasma membrane, resulting in the activation of both proteins. The S-nitrosylation and activation of Src kinase proved to be important for the pattern of compartmentalized Ras activation stimulated by GSNO. The initial accumulation of Ras-GTP at the plasma membrane followed by the accumulation of active Ras at the Golgi is an important event for cell proliferation triggered by GSNO. These observations are in agreement with previous reports that describe the capacity of NO and other RNS to signal through the EGFR-mediated signaling cascade even in the absence of EGF (46, 70).
In contrast with GSNO-mediated Ras signaling, eNOS-derived NO-mediated Ras signaling occurs only at the plasma membrane. The Bk-mediated signaling pathway directly influences Ras signaling that is stimulated by eNOS-derived NO and associated with endothelial cell proliferation. Bk associates with the B2R receptor in endothelial cells, releases downstream mediators such as Ca2+, and promotes phosphorylation and activation of eNOS. Recruitment of Src kinase is not observed upon activation of B2R by Bk in endothelial cells (35). In addition, Bk-mediated eNOS activation promotes the transactivation of EGFR, which seems to play an important role in endothelial cell proliferation (M.S. Moraes, T. Paschoalin and H.P. Monteiro, unpublished data).
Our group showed recently that low concentrations of SNAP induced endothelial cell proliferation with the participation of Ras (58). Low concentrations of NO and other RNS were shown to promote proliferation in human ovarian carcinomas (19). Clinical and experimental studies support a positive relationship between tumor malignancy and NOS activity in certain tumors (22, 39). In contrast, high concentrations of SNAP and GSNO stimulate the Ras signaling pathway and promote apoptosis in HeLa and THP-1 tumor cells, respectively (4, 68). This interesting paradox could be explained by the pattern of compartmentalization operating in NO-mediated Ras activation.
In summary, we have demonstrated differences in the compartmentalization of Ras-signaling events mediated by exogenously added GSNO versus NO derived from eNOS activation. Such differences may explain how compartmentalization of the NO-Ras signaling axis could perform an oncogenic versus an antioncogenic role in tumor biology.
Materials and Methods
Reagents, plasmids, and antibodies
GSNO, PP2 (Src inhibitor), BIBX1382 (EGFR inhibitor), and 4-amino-5-methylamino-2,7-difluorofluorescein diacetate (DAF2-DA) were purchased from Calbiochem. Bradykinin, EGF (from mouse submaxillary glands), L-NAME (Nω-nitro-
Dr. Harry M. Lander (Cornell University, USA) generously provided the expression vectors pcDNA H-RasC118S and pcDNA H-Raswt. Dr. Mark R. Philips (New York University School of Medicine, USA) generously provided the expression vector pEYFP-RBD. The region RBD was amplified by PCR from the RBD-YFP vector and used for cloning in the pEGFP plasmid that was used in this work. Dr. Hugo A. Armelin (Universidade de São Paulo, Brazil) generously provided the expression vector pSVE H-RasV12.
Cell culture and transfection
HeLa, COS-1, or HUVEC cells were cultured in a minimum essential medium (MEM), in the Dulbecco's modified minimal essential medium (DMEM) or the RPMI Medium 1640, respectively, supplemented with 1 mM sodium pyruvate, 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C, 5% CO2. Cells were transfected with expression vectors (pcDNA H-RasC118S, pcDNA H-Raswt, pEGFP-RBD, and pSVE H-RasV12) using Lipofectamine® (Invitrogen). DNA/Lipofectamine® complexes were added to cell cultures for 24 h at 37°C in 5% CO2 with a serum-starved medium. Cells were then suspended in a complete medium for 24 h and selected for permanent transfection using a medium supplemented with Geneticin® (750 μg/ml) or were used in confocal microscopy experiments.
Assay of Ras activation
Ras activation was determined using the GST-RBD fusion protein, which binds very tightly to the GTP-associated form of Ras (69). Cells were grown to near confluence (90%), serum-starved overnight, and treated with different concentrations of GSNO. At the end of the incubation period, cells were lysed in an RIPA buffer (20 mM Tris, pH 8.0, 137 mM NaCl, 1% NP40, 10% glycerol, 1% sodium dodecyl sulfate (SDS), 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM PMSF, and 200 (M Na3VO4). Cell lysates were centrifuged at 14,000 g for 15 min, and the supernatant was used for the pulldown assay. The protein content of the cell extract was determined with the Bradford reagent (Bio-Rad). A 500 (g sample of protein was incubated with glutathione–Sepharose® beads associated with Raf-RBD-GST for 1 h with gentle rocking. The samples were spun at 7200 g for 10–20 s, and the resins were washed three times with a lysis/binding/wash buffer (7200 g for 30 s). The final pulldown was assayed by western blot probed with mouse monoclonal anti-pan-Ras or anti-H-Ras antibodies. The remaining lysates were probed with the same antibody to determine the levels of total and endogenous Ras (69).
Affinity capture of palmitoylated proteins
To detect palmitoylated Ras, we used the method described by Burgoyne et al. (10) with some modifications. HeLa cells were lysed into 100 mM Tris (pH 7.4) buffer containing 1% SDS and 100 mM maleimide. Samples were incubated overnight at room temperature to block free thiols. Maleimide was removed from the soluble fraction after precipitation with cold acetone (three volumes). Proteins were precipitated for 20 min at −20°C and collected by centrifugation at 2000 g for 5 min. The clear supernatants were removed, and the protein pellets were gently washed (four time) with 70% acetone. Samples were resuspended in buffer containing 100 mM Tris (pH 7.4), 0.5% SDS, 400 mM hydroxylamine, and 0.25 mM biotin-HPDP (Thermo Scientific) and incubated overnight. Reactions were terminated by a second precipitation step with ice-cold acetone. Biotinylated proteins were purified by incubating samples for 3 h with streptavidin–agarose beads. Beads were rinsed 5 times with buffer (100 mM Tris, pH 7.4, and 1% Triton X-100) after being placed into empty spin columns. Proteins modified by biotin-HPDP were eluted from the streptavidin–agarose beads upon addition of 100 mM 2-mercaptoethanol. Purified proteins were resolved on SDS-PAGE gels, and immunoblotted with an anti-pan-Ras antibody (Calbiochem).
Quantitative determination of GSH and GSSG
HeLa cells at a density of 1×106 per well were lysed in a lysis buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 1% Triton X-100. After centrifugation at 14,000 g for 10 min at 4°C, cell supernatants were immediately transferred to Eppendorf tubes and stored at −80°C. GSH and GSSG concentrations were determined essentially as described by Rahman et al. (54). After obtaining the concentrations of GSH and GSSG, these concentrations were used to calculate the Nernst equation for the reduction potential of the GSSG/2GSH redox pair:
The values obtained are a good estimate of the cellular redox environment (63).
Cell stimulation and imaging
Cells to be examined by live fluorescence microscopy were plated at 2–4×105 per plate containing a 25-mm glass coverslip. The cells were transfected with expression vectors (pcDNA H-RasC118S, pcDNA H-RasWT, pSVE H-RasV12, and pEGFP-RBD) using Lipofectamine® reagent (Invitrogen). To ensure cotransfection of untagged Ras proteins (pcDNA H-RasC118S, pcDNA H-RasWT, or pSVE H-RasV12) and the fluorescent RBD reporter construct (pEGFP-RBD), a DNA ratio of 5:1 untagged:fluorescent was used (5). DNA/Lipofectamine® complexes were added to cell cultures for 24 h at 37°C in 5% CO2 with a serum-starved medium. The efficiency of cotransfection was determined by using indirect immunofluorescence microscopy. We estimated efficiency of cotransfection in 30% of the population of the observed cells. (Supplementary Fig. S4). Cells were stimulated under continuous observation by adding EGF (50 ng/ml), GSNO (100 μM), or Bk (1.0 (M) maintained at 37°C using a PDMI-2 microincubator (Harvard Apparatus). In some experiments, cells were treated with PP2 (2.0 (M), U73122 (10 (M), or BAPTA-AM (10 (M). The treatment of cells with different inhibitors was initiated 30 min before stimulation. Where indicated, 5.0 (M BODIPY TR marker (Molecular Probes) was added 30 min (at 4°C) before stimulation, according to the manufacturer's instructions. Live cells were imaged with a Zeiss 510 inverted laser-scanning confocal microscope LSM-510 NLO (Carl Zeiss). Cells were imaged as previously described (7, 8). Multiple images (at least 5–6 slices of Z=0.45 (m) were captured before and then every 3 min after the induction period for a total of 45–90 min. One or more sections showing the activation of Ras (Ras-GTP) in both the plasma membrane and Golgi were chosen.
Cell proliferation assay
HeLa, HeLa H-RasWT, H-HeLa RasC118S, HUVECs, or COS-1 cells (8×103) were seeded in a 96-well culture plate and subjected to a 24-h period of starvation with MEM 0.5% FBS at 37°C, 5% CO2. Cells were exposed to different concentrations of GSNO, 50 ng/ml EGF, 1 μM BK, or 10% FBS for 5 h. In the assay that was used, PP2 (a Src kinase inhibitor) or BIBX1382 (an EGFR inhibitor) or LY294002 (PI3K inhibitor) or PD98059 (MEK inhibitor) was added 30 min before stimulation with GSNO, EGF, Bk, or FBS. Cells were washed and subsequently incubated with MTT (Sigma) (0.5 mg/ml) reagent at 37°C, 5% CO2, for 2 h to measure the daily proliferation rate (16). The medium was discarded, and 100 μl of cold isopropanol was added to monitor the absorbance spectrophotometrically at 570 nm. The experiment was repeated three times. Cell proliferation was evaluated by a cell count assay. Cells were cultured in six-well plates to attain 40%–50% confluence and stimulated during 5 h with GSNO or FBS in a culture medium without supplements. After 24–48 h stimulation, cells were trypsinized and counted using an inverted microscope.
Western blotting
Cells were lysed in an RIPA buffer containing protease and phosphatase inhibitors except for detection of S-nitrosylated proteins using the anti-nitrosocysteine antibody. In this particular case, cells were lysed in 50 mM Tris, pH 8.0, 500 mM NaCl, 0.1 mM EDTA, 1% NP-40, 50 μg/ml leupeptin, 25 μg/ml aprotinin, 1 mM PMSF, and 10 mM N-ethylmaleimide.
For western blotting, proteins (50 (g) were separated on 10% or 12% SDS–polyacrylamide gels and transferred to nitrocellulose membranes. After blocking, the membranes were incubated overnight at 4°C with primary antibodies against the following: H-Ras, pan-Ras, Src, Src pTyr416, nitrosocysteine, Akt, Akt pSer473, ERK1/2 MAP kinases, phosphor-ERK1/2 MAP kinases, and β−actin. The appropriate secondary antibody (anti-mouse or anti-rabbit) conjugated with horseradish peroxidase was used in the second step of the procedure (room temperature incubation for 1 h). Blots were developed using the Super Signal® (Pierce) system.
Determination of NO levels
NO generation was evaluated by the use of the cell-permeable specific fluorescent NO indicator 4-amino-5-methylamino-2,7-difluorofluorescein diacetate (DAF2-DA). Cells were preincubated with 5 μM DAF2-DA dissolved in a serum-free medium for 30 min at 37°C in the dark. Once deacetylated, DAF2-DA remains free at the cytoplasm to react with NO and generate the fluorescent benzotriazole DAF2. Cells were incubated with 100 μM GSNO and analyzed in a FACSCalibur flow cytometer (Becton Dickinson Co.); excitation wavelength was set at 495 nm, and emission wavelength was set at 515 nm. Results are expressed as DAF-2-derived fluorescence (mean fluorescence intensity). Unlabeled samples were used as blanks. Experiments were performed in triplicate.
NO generation was also evaluated by a chemiluminescence assay as described in (5). Confluent HUVEC monolayers were incubated in a starvation medium for 24–48 h, and were stimulated with Bk for increasing periods, in the presence or absence of inhibitors. Culture supernatants were collected for NO measurements. To detect NO in this system, we performed a chemical reduction of NOx—a mixture of NO2 − and NO3 −—to NO, by using 0.1 mM VOSO4 and 2.0 M HCl. The NO measurements were based on a gas-phase chemiluminescence reaction between NO and ozone. The medium was used as blank. Samples were analyzed on a Sievers Nitric Oxide Analyzer Model 280 (Sievers Instruments), a highly sensitive detector for quantifying NO. The sensitivity for measurement of NO and its reaction products in liquid samples is ∼1.0 pmol. All measured NO was expressed as means (fold)±standard deviation (SD). Experiments were performed in triplicate.
Quantitative real-time-PCR
Total RNA was extracted from cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions. The RNA was reverse transcribed to cDNA using the SuperScript First-Strand Synthesis System for real time (RT)-PCR (Invitrogen). RT-PCRs were performed using SYBR Green PCR Master Mix (Applied Biosystems) in a GeneAmp 5700 Sequence Detection System (Applied Biosystems). For quantification, we added 2.0 μM of each specific primer. Sequences optimized for RT-PCR are shown in Table 1. Reactions were run in triplicate in parallel with an endogenous control of a ribosomal gene (RPL13A) used for calibration. RT-PCRs were performed in triplicate. The parameters for the PCR were 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. The relative expression ratio (experimental/control) was determined based on the 2-[Delta][Delta]Ct method (40). The data are represented as means±SD. All primers used in real-time PCR experiments were validated and calibrated.
Detection of S-nitrosylated proteins
The BST to detect S-nitrosylation of c-Src and Ras was performed as described in (21). To detect S-nitrosylation of c-Src or Ras in HeLa cells after GSNO treatment, cells were serum-starved for 24 h and treated with GSNO for increasing periods. Cells were lysed in a lysis buffer containing 25 mM Hepes, pH 7.4, 50 mM NaCl, 0.1 mM EDTA, 1% NP-40, and 0.5 mM PMSF supplemented with a cocktail of protease inhibitors (Roche Diagnostic). Cell debris were removed by centrifugation, and samples (600 μg protein extract) were diluted to 1.8 ml with HEN buffer (100 mM Hepes, 1 mM EDTA, 0.1 mM neocuproine, pH 8.0) and SDS and methylmethane thiosulfonate (MMTS) were added to the final concentrations of 2.5% and 0.1%, respectively. After frequent vortexing and incubation at 50°C in the dark for 20 min, lysates were precipitated with 3 volumes of acetone at −20°C for 1 h. Proteins were centrifuged at 2000 g for 15 min, and the protein pellet was gently washed with 70% acetone (four times). Pellets were suspended in 240 μl of HENS buffer (HEN buffer with 1% SDS). Samples were further incubated with 30 μl biotin-HPDP (2.5 mg/ml) in the presence or absence of 20 mM ascorbate at room temperature in the dark for 1 h. After acetone precipitation, proteins were resuspended in 250 μl of HENS buffer, followed by addition of 750 μl of neutralization buffer (25 mM Hepes, 100 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, pH 7.5). About 50 μl of streptavidin–agarose beads (prewashed) were added to each sample and incubated overnight at 4°C. Beads were washed with a washing buffer (neutralization buffer with 600 mM NaCl) four times. To detect S-nitrosylated proteins, 50 μl of 2×SDS sample buffer was added to the beads and immunoblotted with anti-pan-Ras or anti-Src antibody.
Statistical analysis
Data are expressed as means±SD. The statistical analysis of significance was assessed by one-way analysis of variance with the Student's t-test used for comparison. p<0.05 was considered statistically significant.
Footnotes
Acknowledgments
The authors acknowledge the financial support from FAPESP (Fundação de Amparo a Pesquisa do Estado de São Paulo/Brazil) Proc. 07/59617-6 and 09/52730-7 to HPM, and Proc. 11/14392-2 to WLB. The financial support from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico/Brazil) to HPM is also acknowledged.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.