
Abstract
Although the etiology of Parkinson's disease (PD) remains unclear, ample empirical evidence suggests that oxidative stress is a major player in the development of PD and in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity. Nuclear factor E2-related factor 2 (Nrf2) is a redox-sensitive transcription factor that upregulates a battery of antioxidant response element (ARE)-driven antioxidative and cytoprotective genes that defend against oxidative stress.
Introduction
Innovation
Despite years of evidence for the involvement of oxidative stress in Parkinson's disease (PD), no effective antioxidant treatment has proven effective in the clinic. Using a novel reporter, we demonstrate that synthetic triterpenoids (TP) that were structurally modified to penetrate the blood–brain barrier are the most potent and direct activators of the redox-sensitive transcription factor nuclear factor E2-related factor 2 (Nrf2) in upregulating a battery of antioxidant response element (ARE)-driven antioxidative and cytoprotective genes both in vitro and in vivo. Oral administration of these TP blocked dopaminergic neurotoxicity in mice induced by the parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), by activating the Nrf2/ARE pathway in an Nrf2-dependent fashion, suggesting that targeting Nrf2-mediated gene transcription is a promising approach for therapeutic intervention in PD.
One means of increasing the antioxidant potential of a cell is through the nuclear Cap-n-Collar basic region leucine zipper transcription factor nuclear factor E2-related factor 2 (Nrf2) (21). Nrf2 functions as a critical mediator of an adaptive response to counteract oxidative stress. Under basal conditions, Nrf2 is tethered to its cytosolic inhibitor Kelch-like ECH-associated protein 1 (Keap1), which facilitates its ubiquitination and proteolytic degradation. In the presence of electrophiles or under oxidative conditions, Nrf2 escapes degradation and translocates to the nucleus, where it binds the antioxidant response elements (AREs), a consensus gene sequence present in the promoter region of a large number of genes encoding cytoprotective proteins (37).
There is increasing evidence that Nrf2/ARE signaling is impaired in PD. The capacity to induce an Nrf2-mediated cytoprotective response declines with age, which is the main risk factor for PD (37). In postmortem brains from PD patients, Nrf2 is localized in the nucleus, and the expression of ARE-driven genes such as NADPH quinone oxidoreductase 1 (Nqo1) and hemeoxygenase 1 (Ho-1) is increased (30, 33, 51), suggesting a neuroprotective response to block oxidative stress (43). Moreover, genetic studies have identified a functional haplotype in the Nrf2 gene promoter, which confers high transcriptional activity and a protective response, in two groups of European PD patients (46). Studies in animals demonstrate that Nrf2-deficient mice are hypersensitive to PD-causing toxins, whereas Nrf2 overexpression either by genetic means or through pharmacological activation renders a neuroprotective response (3, 4, 16, 42). These findings suggest that targeting the Nrf2 pathway could have important clinical benefits for the treatment of PD.
Synthetic triterpenoids (TP) such as 2-cyano-3-,12-dioxooleana-1,9 (11)-dien-28-oic acid (CDDO) and its derivatives are potent activators of the Nrf2/ARE pathway (8, 22, 23, 49). Many of these synthetic TP cross the blood–brain barrier (BBB) and are pharmacodynamically active in the brain (48, 49), but improved brain levels can be achieved with new structural analogs such as the ethylamide (TP-319) and trifluoroethylamide (TP-500) of CDDO (10, 36). Here, we compared the previously studied synthetic TP (TP-224) (48) with its two newer structural analogs (TP-319 and TP-500) modified to improve BBB permeability, and studied their abilities to activate the Nrf2/ARE signaling both in vitro and in vivo and attenuate experimental Parkinsonism in mice. Our current study is substantially innovative from the previous work (48), because (i) we used a novel Neh2-luc reporter assay for the first time to demonstrate that all three synthetic TP are equipotent and direct activators of the Nrf2 pathway (by disrupting the Nrf2-Keap1 binding) and more potent than the canonical Nrf2 activators (tert-butylhydroquinone [TBHQ] and sulforaphane); (ii) we performed docking experiments using a computer model for the intervening region (IVR) of Keap1 and showed the TP C1 potent alkylation site right against Cys226 and predicted that a cysteine residue that may be forced to form a disulfide bridge with Cys226 is Cys298 on Keap, and that all analogs of TP fit similarly into this site; and lastly, (iii) the addition of ethylamide (TP-319) and trifluoroethylamide (TP-500) groups to the parent TP molecule significantly increased BBB permeability, resulting in increased potency in activating the Nrf2 pathway and rendering neuroprotective effects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity in an Nrf2-dependent manner at nanomolar concentrations compared to TP-224. These results suggest that selective targeting of the Nrf2/ARE pathway by extremely potent, BBB-permeable synthetic TP serves as a credible strategy for therapeutic intervention in PD.
Results
Synthetic TP are the most potent and direct activators of the Nrf2 pathway identified by the Neh2-luc reporter
Synthetic TP containing an activated cyanoenone Michael acceptor motif are extremely potent alkylating agents and are known to activate Nrf2-signaling pathway in a low nanomolar range (8). TP-224, TP-319, and TP-500 have similar structure, except for the substitutions at the C17 carboxygroup aimed at improving BBB permeability (Fig. 1A, Supplementary Fig. S1; Supplementary Data are available online at
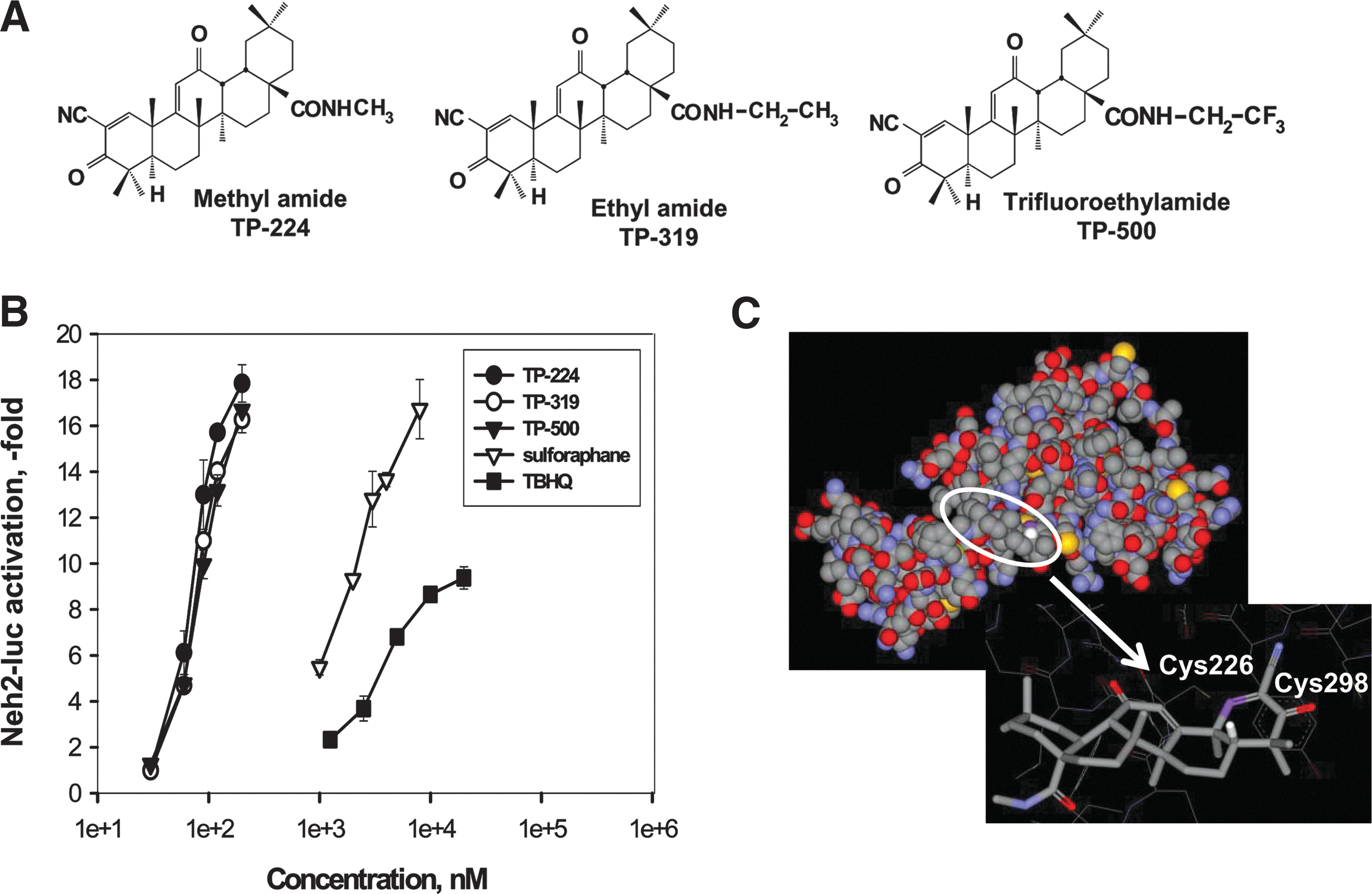
The advantage of the Neh2-luc assay is its immediate response and superior sensitivity, which permits direct monitoring of reporter activation kinetics within minutes of drug administration (35). The timecourse of reporter activation shows a distinct lag-period shortening with rising concentrations of TP-224 (and exactly the same for TP-319 and TP-500) that has been observed for other alkylating agents such as auranofin, but not andrographolide (Supplementary Figs. S1 and S2B–D). The subnanomolar potency of synthetic TP observed in in vitro experiments clearly points to the specific character of their alkylation of Keap1. The majority of Keap1 cysteines are localized in the IVR of Keap1, except for Cys151. No crystal structure of IVR is available; however, its model has been built and used for virtual screening purposes: 2 potent hits identified and confirmed (47). We reproduced their model using the same protein as a template for modeling, and performed docking of the triterpenoid TP-224. The best docking result is shown in Figure 1C, with triterpenoid C1 potential alkylation site right against Cys226. The latter has been identified among alkylation sites for TBHQ using ultrahigh-performance liquid chromatography–tandem mass spectrometry analysis (1), as well as for electrophilic nitrofatty acids (19), and in addition, has been proposed to form an intramolecular disulfide with Cys613 from the Kelch domain upon oxidation with hydrogen peroxide and nitrosative agents (12). In our model, a cysteine residue that may be forced to form a disulfide bridge with Cys226 is Cys298, which in its reduced form in the IVR model coordinates triterpenoid's C3 carbonyl oxygen. A bulky substitution at the C17 position in the triterpenoid will interfere with docking. The actual site of TP-224 interaction with Keap1 still has to be determined because of the speculative character of the IVR model; however, there is no doubt that there has to be a specific site for TP-224 binding to Keap1.
Synthetic TP activate the Nrf2 pathway in vitro
We previously showed that structural modification of oleanolic acid derivatives, specifically the CDDO class of synthetic TP, can activate Nrf2/ARE signaling both in vitro and in vivo (8, 10, 22, 36, 48, 49). In this study, we tested the relative potency of synthetic TP (TP-224, TP-319 and TP-500) in comparison to the classical Nrf2 activator TBHQ to induce the Nrf2 pathway in N27 rat dopaminergic cells. Immunoblot analyses show that treatment of TP (at doses 50, 100, and 200 nM) and TBHQ (at doses 5, 10, and 20 μM) dose-dependently upregulated protein levels for Nrf2 and its downstream target HO-1 in N27 cells at 4 h (Supplementary Fig. S3A, B). Significantly, TP (at nanomolar concentrations) were more potent in inducing the Nrf2 pathway than TBHQ (only effective at a micromolar range). Among the TP, TP-319 and TP-500 were more potent in activating the Nrf2 pathway than TP-224 in N27 cells (Supplementary Fig. S3) and in human neuroblastoma SHSY5Y cells (data not shown). We previously reported activation of Nrf2 and its downstream target genes by TP-224 in SHSY5Y cells (48); in the present study, we focused on the structural analogs TP-319 and TP-500 (Fig. 1A).
We tested the ability of TP-319 and TP-500 to induce nuclear translocation of Nrf2. Human neuroblastoma SHSY5Y cells were treated with 100 nM TP-319 or TP-500, and the control groups were treated with dimethylsulfoxide (DMSO) and processed for subcellular fractionation. As shown by immunoblot analysis, nuclear Nrf2 was significantly increased compared to DMSO-treated cytosolic and nuclear fractions after treatment with TP-319 or TP-500 for 1 and 2 h. The purity of cytosolic and nuclear fractions was confirmed by probing these preparations with anti-aldolase and anti-poly ADP ribose polymerase 1 (PARP1), respectively (Fig. 2A). We observed that both TP-319 and TP-500 administration resulted in abundant Nrf2 accumulation in nuclear fractions, and its migration was slightly higher than its actual molecular weight, a feature that we failed to observe with our prior studies with TP-224 (48). Next, we determined whether TP-319- and TP-500-induced nuclear translocation of Nrf2 resulted in significant increases in messenger RNA (mRNA) levels of downstream ARE genes. Assessment of mRNA and protein levels for several ARE genes in SHSY5Y cells treated with 100 nM of TP-319 or TP-500 showed significant upregulation of these ARE genes (data not shown). Similarly, N27 cells treated with 100 nM of TP-319 and TP-500 resulted in significant increases in mRNA levels of Nrf2-dependent ARE genes such as NQO1, glutathione cysteine ligase regulatory subunit (GCLC), glutathione cysteine ligase modulatory subunit (GCLM), and HO-1 as measured by quantitative reverse transcriptase–polymerase chain reaction (RT-PCR) at 2, 4, 6, 8, and 16 h (Fig. 2B). Consistent with upregulation of mRNA levels for ARE genes, Nrf2, Gclm, Gclc, Nqo1, Ho-1 and glutathione reductase (Gsr) protein levels were also significantly increased at indicated time points as shown by immunoblot (Fig. 2C) and densitometry analysis (Fig. 2D). Since both TP-319 and TP-500 significantly changed levels of genes that regulate glutathione metabolism, we measured cellular levels of reduced (GSH) and oxidized (GSSG) 24 h after treatment with the respective TP. Both TP-319 and TP-500 showed dose-dependent increases in levels of GSH (Fig. 2E) and corresponding increases in the ratio of GSH/GSSG that saturated at doses higher than 50 nanomoles. Together, our in vitro studies suggest that both TP-319 and TP-500 induce nuclear accumulation of Nrf2 to upregulate downstream ARE genes, leading to dose-dependent increases in cellular antioxidants such as GSH.

Synthetic TP activate Nrf2-mediated gene transcription in vivo
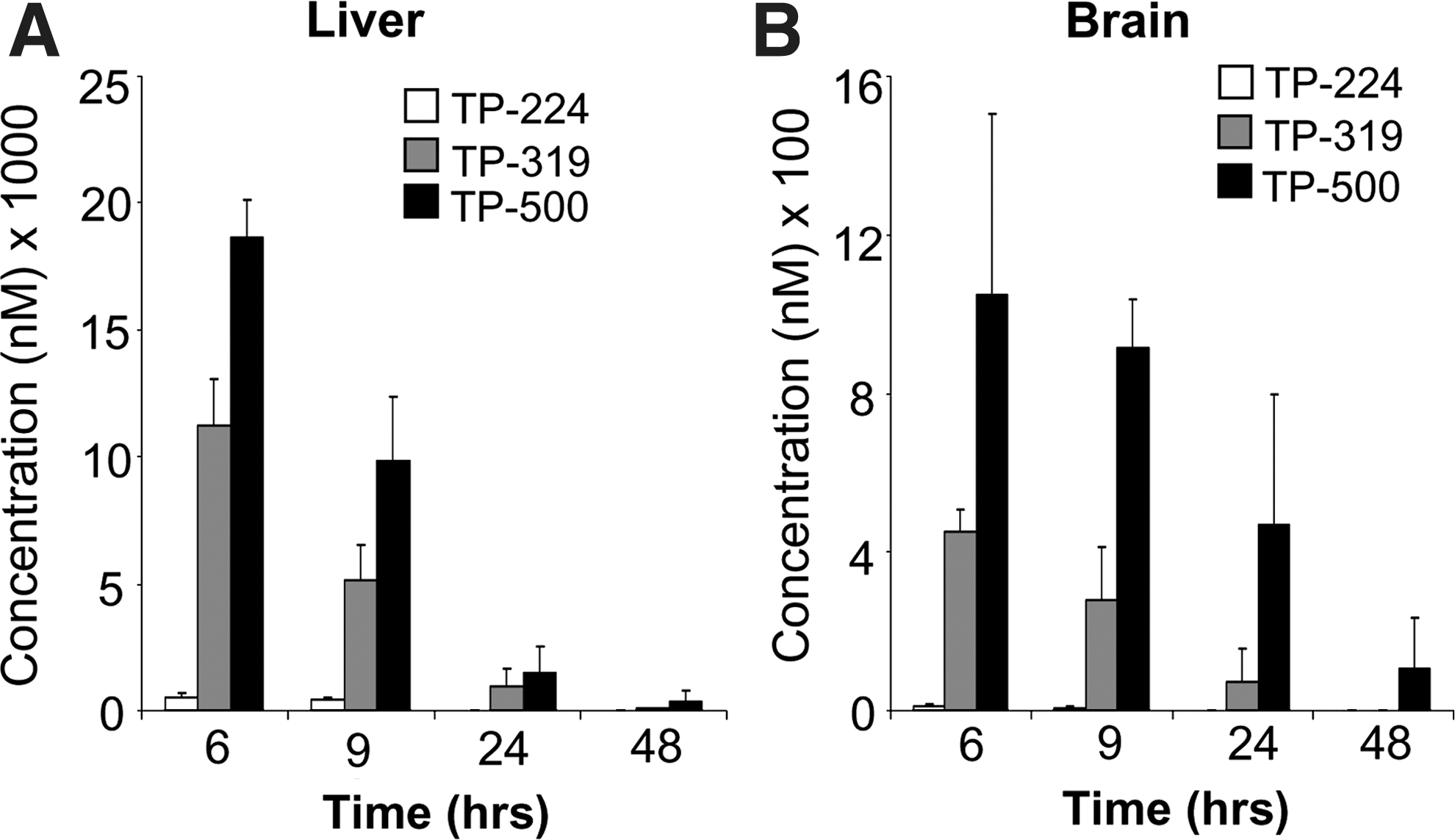
Next, we tested the ability of TP-319 and TP-500 to activate Nrf2/ARE signaling in vivo in mice. Ten-week-old C57Bl6 mice were given 4 μmol of TP-224, TP-319, and TP-500 twice, 12 h apart by oral gavage. Control animals received vehicle in the same frequency and volume as the triterpenoid drugs. Pharmacokinetic studies showed that in comparison to TP-224, TP-319 and TP-500 when administered at similar doses can achieve significantly higher levels in both the liver (Fig. 3A) and the brain (Fig. 3B) at concentrations that induced activation of the Nrf2 pathway in vitro (Figs. 1B and 2). Analysis of mRNA levels of downstream Nrf2-dependent ARE genes by quantitative RT-PCR analysis showed significant (5–25-fold) increases in mRNA levels in the liver for ARE genes such as GCLM, GCLC, HO-1, and Gsr at 6 and 9 h after the last dose of TP-319 and TP-500 (Fig. 4A). Striatal mRNA levels of Nqo1 and Ho-1 were also significantly upregulated, but genes involved in glutathione biosynthetic machinery such as Gclm and Gclc and Gsr were not upregulated (Fig. 4B). Similarly, in the ventral midbrain, a significant upregulation of mRNA levels of Nqo1 and Ho-1 was observed for both TP at 6 and 9 h in addition to significant increases in levels for Gclm when treated with TP-319 at 9 h (Fig. 4C). In the cortex, however, only the mRNA levels of Nqo1 were significantly upregulated at 6 and 9 h, whereas the mRNA levels of other ARE genes were unaffected (Supplementary Fig. S4C). None of the brain regions, including striatum, ventral midbrain, and cortex, showed upregulation of mRNA levels for the ARE genes at 24 and 48 h after the administration of both the TP (Supplementary Fig. S4A, B, D). Consistent with mRNA upregulation in the liver, immunoblot (Fig. 5A) and densitometry analysis (Fig. 5B) showed significant increases in Nrf2 and downstream ARE target proteins such as NQO1 and HO-1 at 6 h, whereas Nrf2, NQO1, GCLM, GCLC, and HO-1 were markedly upregulated at 9 h. Immunoblot (Fig. 5C) and densitometry analysis (Fig. 5D) in the striatum confirmed significant increases in GCLM, GCLC, NQO1, HO-1, and Gsr protein levels at 9 h after TP-319 treatment, whereas these protein levels were unaffected at 6 h. In contrast to TP-319, administration of TP-500 resulted in increases in striatal levels of NQO1 and HO-1 by 6 h, whereas GCLC, NQO1, HO-1, and Gsr were markedly upregulated by 9 h without affecting Nrf2 expression. However, in the ventral midbrain, immunoblot (Supplementary Fig. S5A) and densitometry analysis (Supplementary Fig. S5B) showed significant increases only in NQO1 levels 9 h after TP-319 and at 6 and 9 h after TP-500 (Supplementary Fig. S5B). Collectively, these data provide in vivo evidence for the ability of both TP-319 and TP-500 to activate the Nrf2-mediated ARE gene transcription that is directly associated with their in vivo pharmacokinetic profiles.


Synthetic TP attenuate MPTP neurotoxicity in mice
Several lines of studies suggest that deficiency of Nrf2 in mice leads to increased vulnerability to MPTP neurotoxicity (3, 16, 20), whereas activation of Nrf2-mediated gene transcription either via a genetic or pharmacologic approach has shown profound neuroprotective effects against MPTP neurotoxicity (4, 15, 16). Hence, we sought to test if in vivo activation of Nrf2/ARE signaling by TP-319 and TP-500 can induce a neuroprotective response against MPTP neurotoxicity. We used an acute paradigm of MPTP administration (10 mg MPTP/kg×3, every 2 h), known to cause about a 50% loss of striatal DA and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) and a significant loss of tyrosine hydroxylase (TH) immunopositive neurons in the SNpc on the 7th day. Comparison of relative potency of TP in blocking MPTP neurotoxicity found that TP-224, TP-319, and TP-500 dose-dependently (at doses 4, 2, 1, and 0.5 μmol) protected against MPTP-induced loss of striatal DA, DOPAC, and HVA (Table 1). Significantly, TP-319 and TP-500 were more potent in blocking MPTP neurotoxicity than TP-224 at lower doses (0.5 μmol) (Table 1). Consistent with levels of striatal catecholamines, unbiased stereologic counts of total (i.e., Nissl-positive) and TH-positive neurons in SNpc showed a statistically significant loss of neurons in the MPTP group compared to controls. Analysis of total and TH-positive neuronal counts for both TP-319 and TP-500, when administered at either 4 or 0.5 μmol, showed a significant dose-dependent attenuation of MPTP-induced loss of total (i.e., Nissl-positive) and TH-immunopositive neurons as compared to MPTP-treated mice (data not shown). The neuroprotective effects of TP-319 and TP-500 against MPTP-neurotoxicity were not due to impaired metabolism of MPTP to its toxic metabolite 1-methyl-4-phenyl-pyridinium ion (MPP+). High-performance liquid chromatography (HPLC)–fluorimetric analysis of striatal MPP+ levels measured 90 min after a single intraperitoneal injection of 30 mg/kg free base MPTP showed no significant difference between the MPTP (12.12±2.09 ng/mg tissue) and TP (TP-319: 11.71±2.5; and TP-500: 12.58±3.79 ng/mg tissue).
Striatal levels of dopamine and its metabolites DOPAC and HVA were measured on the 7th day after MPTP. TP-319, TP-500, and TP-224 were administered at 4, 2, 1, and 0.5 μmol. Data represent mean±standard error of the mean, a p<0.001 compared to control, and b p<0.001 compared to the MPTP group (n=8 mice per group).
DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; TP, triterpenoids.
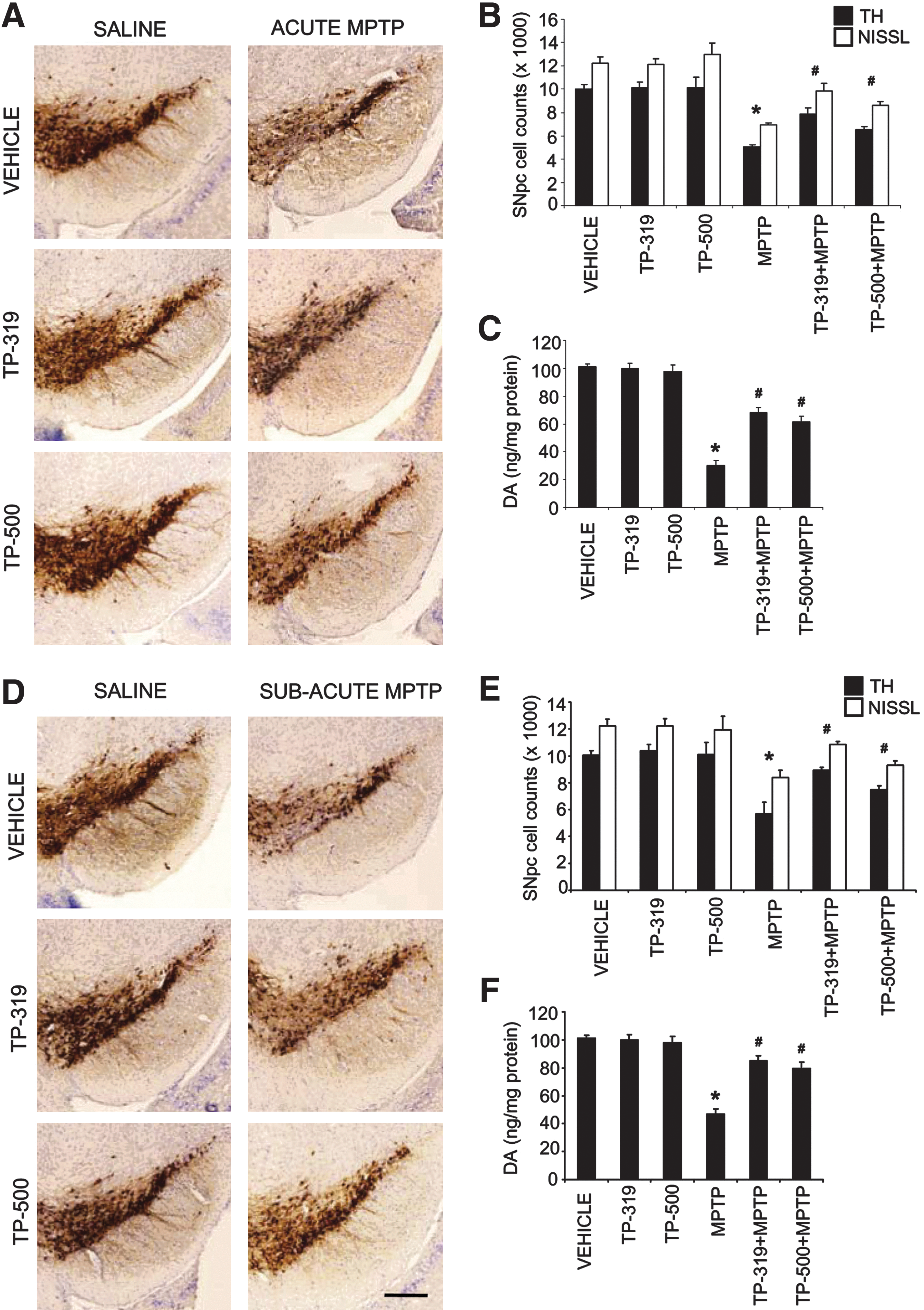
To fully characterize the neuroprotective effects of TP-319 and TP-500 in two well-established paradigms of MPTP neurotoxicity, we used an acute (15 mg MPTP/kg×4, every 2 h) and subacute paradigms of MPTP (30 mg/kg once daily for 5 days) (38). In both the neurotoxic paradigms of MPTP, a dose of 2 μmol of TP-319 and TP-500 was used (see the Methods section for dosing protocol). Neuropathologic evaluations were conducted 1 week after the acute MPTP paradigm and 3 weeks after subacute MPTP. In the acute (Fig. 6A–C) and the subacute (Fig. 6D–F) MPTP paradigms, immunohistochemical analysis for TH-positive neurons in the substantia nigra and striatal dopaminergic nerve terminals (data not shown) demonstrated a marked reduction when compared to saline controls. Both TP-319- and TP-500-treated mice showed significant reductions in the MPTP-induced loss of TH-positive neurons in SNpc (Fig. 6A, D) and striatal dopaminergic nerve terminals (data not shown) when compared to mice treated with MPTP alone. Consistent with TH-immunostaining in SNpc, stereological cell counts of total (i.e., Nissl-positive) and TH-positive neurons showed a statistically significant loss in the MPTP group compared to controls. Both TP-319 and TP-500 administration significantly attenuated the MPTP-induced loss of total and TH-positive neurons compared to MPTP-treated mice (Fig. 6B, E). Consistent with the loss of striatal dopaminergic terminals, HPLC–electrochemical analysis revealed a profound reduction in striatal DA (Fig. 6C, F) and its metabolites (DOPAC and HVA [data not shown]) after MPTP treatment, but both TP-319 and TP-500 significantly rescued against MPTP-induced loss of striatal DA (Fig. 6C, F) and its metabolites (data not shown). Together, these data conclusively prove that both TP-319 and TP-500 not only activate the Nrf2/ARE pathway in vivo but also possess great potential in protecting nigrostriatal dopaminergic pathway neurons in the acute and subacute paradigms of MPTP neurotoxicity.
Synthetic TP block MPTP-induced oxidative stress and inflammation
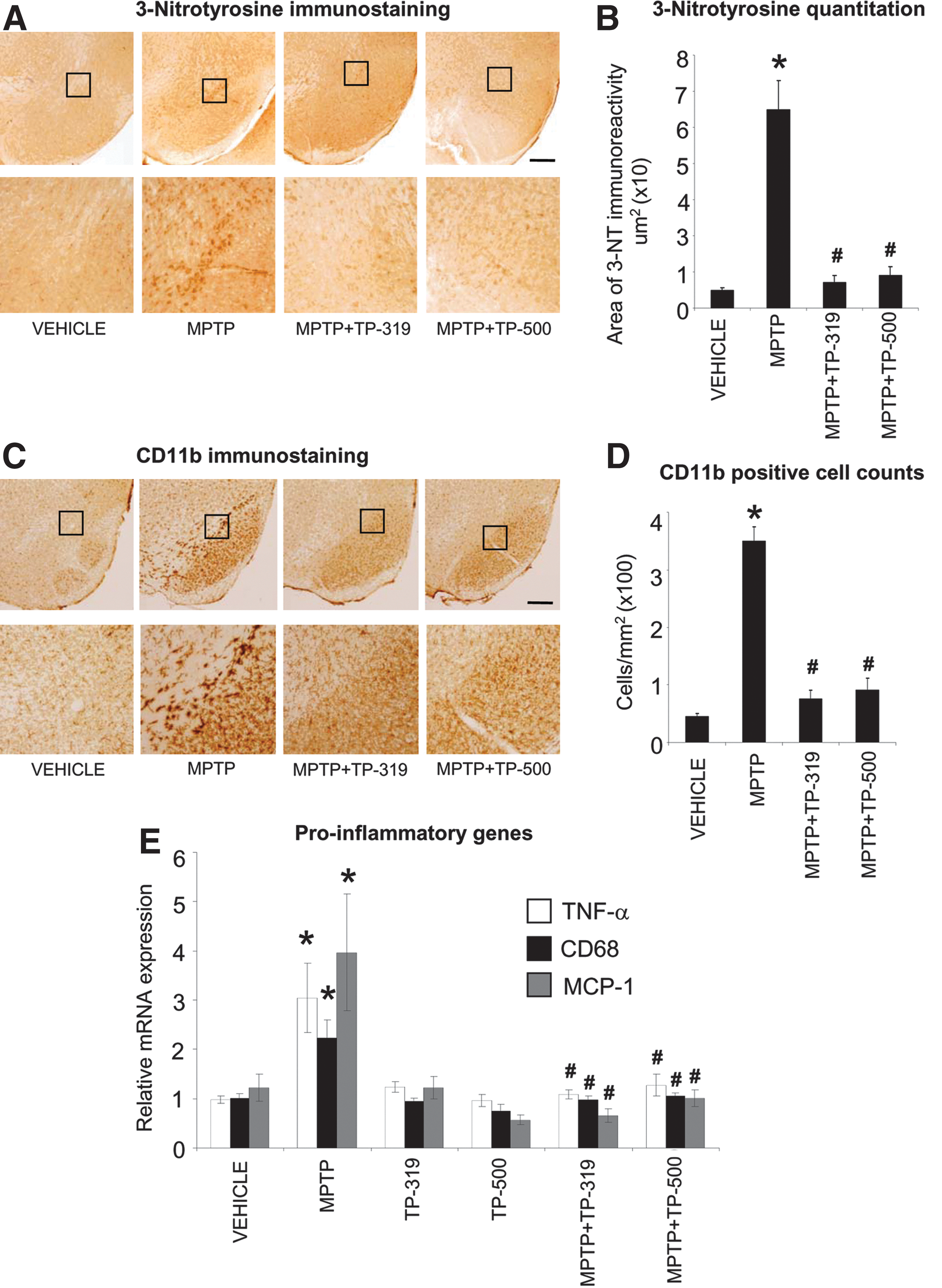
The MPTP-induced degeneration of nigrostriatal dopaminergic neurons is accompanied by increases in markers of oxidative stress and inflammation in the nigrostriatal pathway. Since Nrf2-mediated gene transcription controls both oxidative stress and inflammation (21, 31), we investigated if the neuroprotective effects of TP-319 and TP-500 are associated with the attenuation of markers of oxidative stress and inflammation. Accordingly, we examined the expression of 3-nitrotyrosine (a marker for protein oxidation produced by reactive nitrogen species) activated CD11b-immunopositive (microglial marker) cells and mRNA levels of the proinflammatory genes, respectively. As shown in Figure 7A, after 48 h of acute MPTP injection (15 mg MPTP/kg×4, every 2 h), there was a marked increase in accumulation of 3-nitrotyrosine immunoreactivity in the SNpc, which was completely abrogated in mice treated with TP-319 and TP-500. Quantitative analysis of 3-nitrotyrosine immunoreactivity in SNpc showed significantly higher levels after MPTP, which were markedly reduced when mice were treated with TP-319 or TP-500 (Fig. 7B). Administration of TP-319 and TP-500 alone did not affect 3-nitrotyrosine levels in the SNpc and were comparable to controls (data not shown). Similarly, MPTP administration markedly increased markers of inflammation such as reactive microglia at 36 h after acute MPTP administration (15 mg MPTP/kg×4, every 2 h), whereas in vehicle-treated animals, only a few faintly immunoreactive microglia (Fig. 7C) were observed in the SNpc. Both TP-319 and TP-500 administration resulted in a significant decrease in MPTP-induced microglial activation (Fig. 7C). Consistent with the qualitative changes in CD11b (microglial marker), morphometric analysis of reactive CD11b-positive microglia showed significantly higher numbers of reactive microglia (Fig. 7D) after MPTP, which were significantly reduced when mice were treated with TP-319 or TP-500 (Fig. 7D). Treatment with TP-319 and TP-500 alone showed similar numbers of activated microglia in the SNpc as in vehicle-treated controls (data not shown). Assessment of the mRNA levels of the proinflammatory cytokine tumor necrosis factor-alpha (Tnf), chemokine [C-C motif] ligand 2 (Ccl2, monocyte chemotactic protein 1, Mcp1), and microglial activation marker cluster of differentiation 68 (Cd68) showed significant increases in the ventral midbrain 24 h after acute MPTP administration (15 mg MPTP/kg×4, every 2 h) compared to vehicle (Fig. 7E). Both the TP significantly blocked MPTP-induced increases in proinflammatory genes (Fig. 7E). Taken together, these data suggest that the TP-319- and TP-500-induced neuroprotective response against MPTP-neurotoxicity is associated with a marked reduction in markers of oxidative stress and inflammation.
Synthetic TP attenuate MPTP neurotoxicity in an Nrf2-dependent manner
To determine if the neuroprotective effects exerted by TP-319 and TP-500 are indeed via the Nrf2-dependent pathway, we evaluated their ability to block MPTP neurotoxicity in wild-type (WT) and Nrf2 knockout (KO) mice. Because Nrf2 KO mice show increased sensitivity to MPTP neurotoxicity as compared to WT mice (3, 4), we delivered a milder dose of MPTP (10 mg MPTP/kg×4, every 2 h) using an acute intoxication regimen in Nrf2 KO and age-matched WT littermate mice. Both TP-319 and TP-500 were administered at a dose of 2 μmol (see the Methods section for dosing protocol). One week after MPTP administration, immunohistochemical analysis in SNpc demonstrated a marked reduction in TH-immunopositive neurons in WT mice (Fig. 8A) when compared to controls. MPTP administration in Nrf2 KO mice also showed increased vulnerability to the loss of TH-positive neurons as compared to MPTP-treated WT mice. Both TP-319 and TP-500 markedly reduced the MPTP-induced loss of TH-positive neurons in the SNpc in WT mice, but failed to protect against MPTP neurotoxicity in Nrf2 KO mice (Fig. 8A). Unbiased stereologic counts of total (i.e., Nissl-positive) (Supplementary Fig. S6A) and TH-immunopositive neurons in SNpc (Fig. 8B) showed a statistically significant loss of neurons in the MPTP group compared to the saline control. Both TP-319 and TP-500 significantly attenuated the MPTP-induced loss of total (Supplementary Fig. S6A) and TH-immunopositive neurons (Fig. 8B) in WT mice, but not in Nrf2 KO mice. HPLC–electrochemical analysis also revealed a profound reduction in striatal DA (Fig. 8C) and its metabolites [DOPAC and HVA (Supplementary Fig. S6B, C)] after MPTP treatment in WT mice compared to saline-injected WT controls. MPTP treatment in Nrf2 KO mice produced a significantly higher depletion of striatal DA (Fig. 8C) and its metabolites [DOPAC and HVA (Supplementary Fig. S6B, C)] as compared to MPTP-treated WT mice. However, both TP-319 and TP-500 treatment rescued the MPTP-induced loss of striatal DA (Fig. 8C) and its metabolites only in the WT mice, but not in Nrf2 KO mice. The lack of neuroprotection in Nrf2 KO mice after synthetic TP against MPTP neurotoxicity was not due to differences in the conversion of MPTP to its toxic metabolite MPP+, in the Nrf2 KO mice compared to WT mice (Supplementary Table S1). These results demonstrate that the ability of TP-319 and TP-500 to attenuate MPTP neurotoxicity is dependent on the Nrf2 pathway.
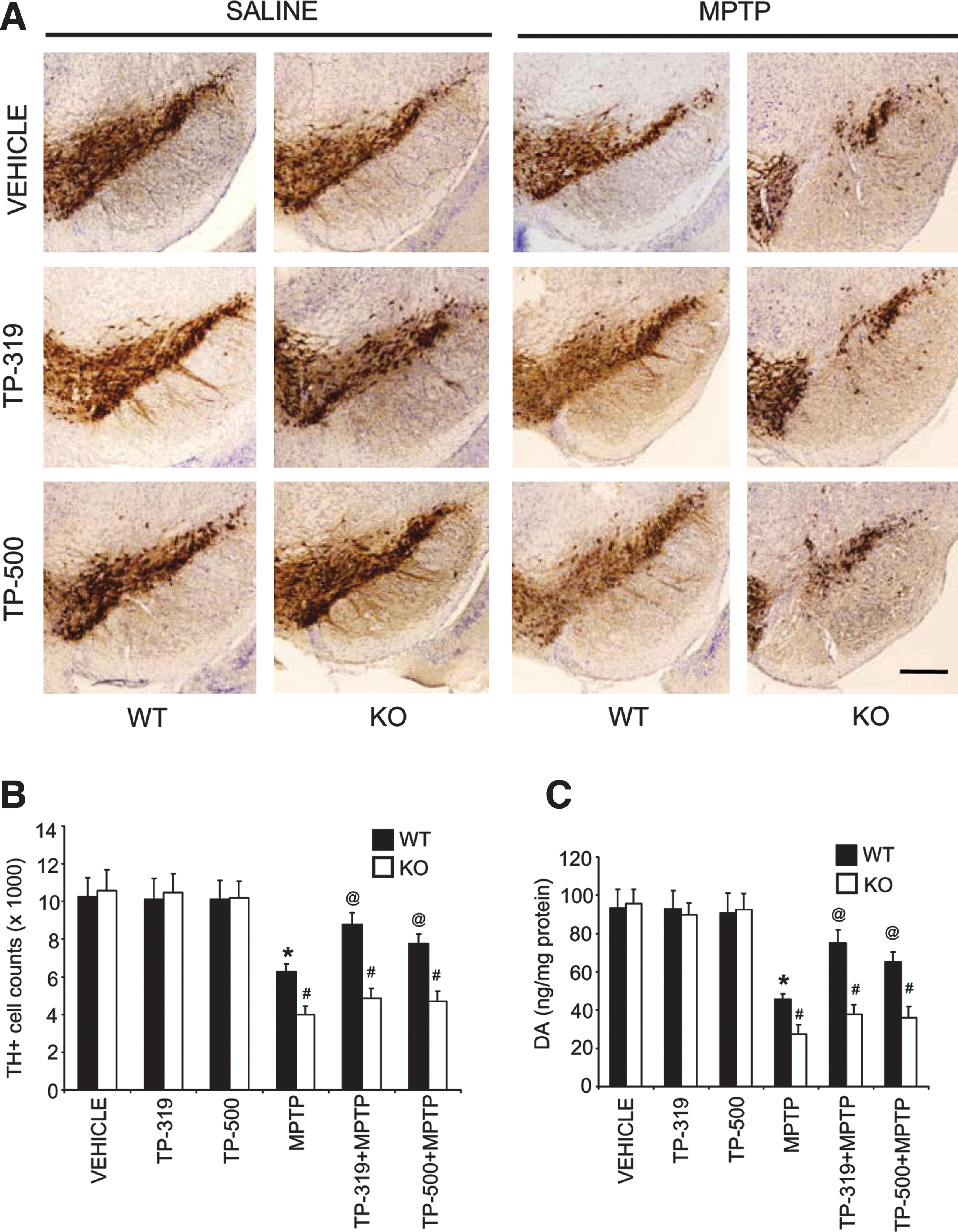
Selective activation of Nrf2 mediated gene transcription by synthetic TP in vivo
We evaluated Nrf2-mediated gene transcription in WT and Nrf2 KO mice to determine if the triterpenoid-mediated neuroprotective effects are due to their selective activation of the Nrf2/ARE pathway. Age-matched Nrf2 KO mice and WT littermates, were administered 4 μmol of either TP-319 or TP-500 by oral gavage twice 12 h apart. Control animals of both genotypes received vehicle in the same frequency and volume as the two triterpenoid drugs. As shown by quantitative RT-PCR analysis, mRNA levels of ARE genes such as Gclm, Gclc, Ho-1, and Gsr were significantly increased (2–12-fold) in the liver of WT mice at 9 h after the last dose of TP-319 and TP-500 (Fig. 9A). However, this induction of mRNA for target ARE genes was absent in Nrf2 KO mice (Fig. 9A). Immunoblot (Fig. 9B) and corresponding densitometry analysis (Fig. 9C) also showed significant upregulation of protein levels for GCLM, GCLC, and NQO1 in the liver of WT mice after TP-319 and TP-500 treatment. However, basal levels of GCLM, GCLC, and NQO1 protein were significantly lower in vehicle-treated Nrf2 KO mice compared to WT, and administration of either TP-319 or TP-500 failed to upregulate protein levels for any of these genes in the KO mice, suggesting that induction of the ARE pathway is dependent on the presence of a functional Nrf2. In the nigrostriatal system, TP-319 and TP-500 significantly upregulated the mRNA levels of Nqo1 and Ho-1 in the striatum of WT mice, whereas Nrf2 KO mice failed to show any induction of mRNA levels for these ARE genes (Fig. 9D). Similarly, TP-319 significantly upregulated Nqo1 and Ho-1, whereas TP-500 significantly increased Nqo1 in the ventral midbrains of WT mice compared to controls. Both TP-319 and TP-500, however, failed to upregulate ARE genes in the ventral midbrains of Nrf2 KO mice (Fig. 9D). Similar to the liver, the nigrostriatal system also showed reduced basal gene transcripts for ARE genes Gclm, Gclc, Ho-1, and Gsr in striatum and Gclc and Gsr in the ventral midbrains of Nrf2 KO mice (Fig. 9D). Importantly, the differences in induction of Nrf2-mediated downstream ARE activation between WT and Nrf2 KO mice were not due to differences in pharmacokinetic profiles of the triterpenoid drugs in these target organs between the two genotypes (Supplementary Fig. S7). Hence, the observed neuroprotective activity of TP-319 and TP-500 against MPTP neurotoxicity is directly associated with selective activation of Nrf2-mediated gene transcription.

Discussion
The Nrf2 signaling cascade has emerged as a promising pathway for neurotherapeutics by transcriptional modulation of both oxidative stress and inflammation. Current therapies for PD are largely symptomatic and may partially improve the quality of life for patients, but none has been convincingly shown to stop or slow the insidious degenerative process. The pathogenesis of neuronal degeneration in PD is yet to be fully understood; however, oxidative stress appears to be a prominent pathogenic component in PD (17). Based on this, one would postulate that increasing the capacity of dopaminergic neurons to cope with oxidants could serve as an important strategy to prevent the onset and/or to delay the progression of PD. In the past, antioxidants such as vitamin E, vitamin C, N-acetylcysteine, and glutathione have been tested in animal models and in clinical trials for several neurodegenerative disorders, including PD (18, 34). However, such approaches that rely predominantly on stoichiometric scavenging of oxidants unfortunately had little or no benefits. An alternative promising therapeutic strategy is to restore redox homeostasis in neurodegenerative disorders by activation of the transcription factor Nrf2, a member of the Cap-n-Collar family of basic leucine-zipper transcription factors that regulates a coordinated adaptive genetic program (26). The ability to selectively induce a battery of cytoprotective and antioxidative enzymes that are directly under the genetic control of a single transcription factor such as Nrf2 may have significant advantages over conventional strategies. One can potentially induce multiple antioxidant pathways and associated neuroprotective responses, which would not be dependent on the antioxidant effects of single or combinations of molecules that are frequently consumed while scavenging toxic-free radical species in a diseased or aging brain. This study identified that synthetic TP such as TP-224, TP-319, and TP-500 are thus far the most potent and direct Nrf2 activators. Among these TP, TP-319 and TP-500 are orally active and extremely potent, with profound neuroprotective properties at nanomolar concentrations in an experimental model of PD by virtue of their ability to activate the Nrf2-signaling cascade.
TP are synthesized by the cyclization of squalene, a triterpene hydrocarbon found ubiquitously in nature (6). Structure–activity analysis shows that α, β-unsaturated carbonyl groups in CDDO at key positions on rings A and C are essential for the striking anti-inflammatory potency, and enable Michael addition with a nucleophilic target (6, 23) similar to Keap 1 (a protein with multiple nucleophilic–SH residues and a cytoplasmic repressor of Nrf2). This reaction activates the phase-2 response, an intrinsic mechanism used by cells to deactivate electrophiles or oxidative stress. We used a novel Neh2-luc reporter and identified that synthetic TP are the most potent and direct activators of the Nrf2 pathway reported to date. The Neh2-luc reporter is extremely sensitive, allowing reporter activation kinetics within minutes of drug administration (35). With increasing concentrations of TP, the reporter activation showed a distinct lag-period shortening similar to alkylating agents such as auranofin, but not andrographolide (Supplementary Figs. S1 and S2B–D). Auranofin is a bulky molecule, known to alkylate active protein thiols (or selenols such as in thioredoxin reductase) by a ligand-exchange mechanism, the so-called sulfhydryl shuttle (29), while andrographolide, a labdane diterpenoid recently patented for the treatment of Amyotrophic Lateral Sclerosis and other neurodegenerative disorders (UK Patent application GB2464813), has an α,β-unsaturated lactone moiety alkylating protein or peptide-active -SH or -NH2 groups by Michael addition followed by dehydration (53). The observed lag-periods for TP may be interpreted as a switch in the rate-limiting step, meaning that there is a step preceding their direct alkylation of Keap1 reactive cysteines. The TP studied are chemically complex molecules, where alkylation should occur at C1 of the ring A [see Fig. 1 in (8) by analogy with the alkylation site in acetylenic tricyclic bis(cyanoenone) TBE-1 (9)].
Our computer-modeling experiments using the Keap IVR model found the best docking result with TP C1 as a potent alkylation site against Cys226. The latter has been identified among alkylation sites for TBHQ using mass spectrometry analysis (1), as well as for electrophilic nitrofatty acids (19). The access to this position is much more restricted than to the freely rotating α,β-unsaturated lactone moiety of andrographolide, and thus we may suppose either formation of a specific protein–triterpenoid complex before alkylation actually takes place or a ligand exchange mechanism as in the case of auranofin. The idea of specific interaction between Keap-Nrf2 and TP is supported by the high potency of TP toward the Nrf2-Keap1 system, which is orders of magnitude higher than their potency for targeting the NF-Kb pathway by alkylating critical Cys179 in the active loop of IKK (52). Another interpretation of the observed lags could be membrane permeability or diffusion limitations: of note, the acting concentrations of TP are in the low nanomolar range and at least, one order of magnitude lower than those for andrographolide. The intracellular concentration of the fusion Neh2-luc protein synthesized (recalculated from luciferase activity as 9–10 nM fusion protein formed per hour for 30,000 cell/well density and 233-μ3 single-cell volume) is comparable with the concentration of TP added. However, the amount of the cell volume is four orders of magnitude less than the incubation medium volume, and thus there is a large excess of alkylating agent. If alkylation is irreversible, the amount of alkylating agent, and not its concentration, is in play, and the observed lag-periods must be ascribed to the mechanistic complexity. Despite the mechanistic details of triterpenoid action on the Keap1-Nrf2-Cul3 complex still await their elucidation, the compounds of this class (bardoloxone) are already in clinical trials (40). Since the alkylating potency of the TP with varied substitutions at the C17 carboxygroup remained unchanged, and the introduced substitutions improved BBB permeability, and new TP are expected to be far more advantageous over TP-224 and result in higher activation of the Nrf2 pathway (Figs. 1 and 3).
We previously reported activation of Nrf2-mediated gene transcription and associated neuroprotective effects of CDDO methyl amide (TP-224) in blocking MPTP-induced PD (48). However, when compared to TP-224, administration of either TP-319 or TP-500 at similar doses produced significantly higher levels of Nrf2 and its downstream ARE target hemeoxygensase 1 in N27 rat dopaminergic cells. These TP when compared to other prototypical Nrf2 activators such as TBHQ and sulforaphane were far more potent in dissociating the Nrf2/Keap1 complex (Fig. 1) or activate ARE-mediated gene transcription both in vitro (Supplementary Fig. S3) and in vivo (49). Here we found that nanomolar concentrations of both TP-319 and TP-500 in human neuroblastoma SHSY5Y cells and N27 rat dopaminergic cells, activated the Nrf2/ARE pathway, by inducing expression levels of Nrf2 and several downstream antioxidative and cytoprotective genes, including those involved in the glutathione biosynthetic machinery, to subsequently increase GSH levels. Importantly, decrements in the levels of GSH in nigral dopaminergic neurons in incidental Lewy body disease (also known as presymptomatic PD) may play an important role in the development of PD (7). Dose-dependent upregulation of GSH levels at low nanomolar concentrations by TP-319 and TP-500 in dopaminergic cells is consistent with their strong antioxidant actions. Unlike TP-224, both TP-319 and TP-500 were exceptionally potent activators of the Nrf2 pathway, leading to nuclear Nrf2 translocation as early as 1 h. Surprisingly, we found that both TP-319 and TP-500 resulted in Nrf2 migration that was slightly higher than its actual molecular weight, and that was abundant in the nuclear fractions (Fig. 2A) in comparison to the cytosol, or nuclear fractions treated with TP-224 (48). We reasoned this might be a phosphomodification of Nrf2 unique to TP-319 and TP-500 treatment, since it is postulated that phosphorylation of Nrf2 by upstream kinases, especially at serine 40, shuttles cytosolic Nrf2 to the nucleus to activate the Nrf2/ARE pathway (13, 28). Accordingly, we probed our cytosolic and nuclear fractions after triterpenoid treatment with an antiphospho Nrf2 (ser40) antibody. However, we failed to observe increased phosphorylated Nrf2 (ser40) after TP-319 and TP-500 treatment (data not shown). This may also be due to phosphorylation at alternate amino acid residues or other forms of post-translational modifications in Nrf2, yet to be identified. Significantly, the differences in Nrf2 migration patterns due to treatment with different TP were only observed in the liver and N27 rat dopaminergic cells, but not in striatum (Figs. 2C and Fig 5A, C), the reasons of which remain elusive. Despite these differences among various TP, our in vitro findings suggest that both TP-319 and TP-500 are equally superior activators of the Nrf2 pathway in comparison to TP-224 and other classical Nrf2 activators like TBHQ and sulforaphane.
In vivo pharmacokinetic studies have shown that the addition of functional groups (ethylamide to TP-319 and trifluoroethylamide to TP-500) enhances brain penetration when compared to the methylamide TP-224, with TP-500 showing the highest penetration in the brain and the longest tissue half-life (Fig. 3) (27, 49). However, despite increased brain availability and the longer half-life for TP-500 compared to TP-319, the ability to induce the Nrf2 signaling in the nigrostriatal pathway or in the cortex of the mice was comparable for both compounds at the doses tested (Figs. 4 and 5). The reasons for these differences are unclear. Using two well-established neurotoxic paradigms of MPTP-induced PD, involving an acute 7-day and a subacute 21-day regimen of MPTP, we showed that oral administration of TP-319 and TP-500 dose-dependently attenuated MPTP-induced loss of striatal DA and its metabolites and the loss of TH-immunopositive neurons in the substantia nigra. At doses as low as 0.5 μmol, both TP-319 and TP-500 showed comparable, but significantly higher level of potency than TP-224 in ameliorating MPTP-neurotoxicity. Despite all the three TP showing similar potency in dissociating the Nrf2/Keap1 complex in our Neh2-luc assay, the BBB permeability for TP-319 and TP-500 far exceeded that of TP-224, and therefore were more potent in inducing neuroprotective effects against MPTP neurotoxicity.
MPTP administration leads to marked increases in oxidative damage, activation of glia, and levels of proinflammatory cytokines, resulting in the initiation and/or perpetuation of nigrostriatal dopaminergic neurodegeneration. Postmortem human PD brains accumulate elevated levels of oxidative stress markers such as oxidized proteins, lipids, and nucleic acids (24), and abnormally activated glia that secrete toxic cytokines, chemokines, and complement proteins (2). Both TP-319 and TP-500 significantly blocked accumulation of the oxidative stress marker 3-nitrotyrosine, numbers of reactive microglia, in the substantia nigra, and increases in proinflammatory genes, suggesting that activation of the Nrf2 pathway by these TP is beneficial in blocking oxidative stress and neuroinflammation associated with MPTP neurotoxicity. In fact, multiple studies have convincingly demonstrated that CDDO and its synthetic analogs (methylamide, eythylamide, trifluoroethylamide, and methyl ester) block markers of oxidative stress and neuroinflammation in several models of neurodegenerative diseases (36, 41, 48).
The most striking and compelling evidence for activation of the Nrf2 pathway by TP-319 and TP-500 and its direct association in ameliorating MPTP neurotoxicity came from our studies using WT and Nrf2 KO mice. Both TP-319 and TP-500 significantly attenuated MPTP neurotoxicity in the WT mice, but not in the Nrf2 KO mice. These effects directly correlated with the ability of these TP to induce selective activation of Nrf2/ARE genes in the WT, but not in the Nrf2 KO mice. Among the prominent Nrf2-regulated genes induced by TP-319 and TP-500 in the nigrostriatal pathway were NQO1and HO-1 and genes involved in glutathione-dependent functions. In the context of PD, NQO1 induction may be important to nigrostriatal dopaminergic neurons due its relevance in the reduction of oxidized catechol rings and ameliorating the detrimental effects of DA quinones. NQO1 is also involved in detoxification of protein-bound quinones and maintains alpha-tocopherol and coenzyme Q10 in their reduced antioxidant states (44). Induction of HO-1 may provide resistance to chronic oxidative stress in dopaminergic neurons induced by the accumulation of iron due to its involvement in the metabolism of the pro-oxidant heme to the antioxidant pigment biliverdin (11). Genes involved in modulating GSH-dependent functions like glutamyl cysteine ligases and glutathione reductase are critical for dopaminergic neurons to detoxify xenobiotics by synthesis and conjugation of GSH to a range of electrophilic substrates. Importantly, maintenance of glutathione levels is also crucial for nigrostriatal dopaminergic neurons, since conditional knockdown of the catalytic subunit of glutamyl cysteine ligase in nigral dopaminergic neurons leads to a decline in GSH levels, and the progressive loss of midbrain DA neurons in mice (5). Our studies conclusively demonstrate the remarkable ability of TP-319 and TP-500 to modulate crucial phase-2 antioxidant and cytoprotective genes that may be relevant for the survival of dopaminergic neurons in PD.
In summary, we have demonstrated that orally active, small-molecule synthetic TP such as TP-319 and TP-500 are extremely potent and direct activators of the Nrf2 pathway with robust neuroprotective properties in a toxin-induced preclinical model of PD. These results provide a strong rationale for further preclinical evaluation of such synthetic TP in more chronic models of PD harboring familial gene mutations. Collectively, these studies are expected to provide insights into how synthetic TP can be used most effectively for future clinical trials for the prevention and treatment of human PD.
Materials and Methods
Animals
Mice were housed and treated in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Institutional Animal Care and Use Committee of the Weill Medical College of Cornell University, New York. Mice were maintained in a pathogen-free facility and exposed to a 12-h light/dark cycle with food and water provided ad libitum. C57Bl6 mice were procured from Jackson laboratories, whereas Nrf2 KO mice used in the present study were on a pure C57Bl6 background (14). Age-matched WT and Nrf2 KO male mice used in the present study were obtained by crossing Nrf2 heterozygous mice.
N27 rat dopaminergic and human neuroblastoma SHSY5Y cell culture
Rat N27 cells were maintained in a medium containing RPMI 1640 (Invitrogen) with glutamine supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2. Cells were cultured up to 80% confluence in 12-well plates and treated with the respective doses of TP-319 and TP-500, indicated in the figure legends for real-time PCR, immunoblotting, and measurement of cellular GSH and GSSG levels. For nuclear translocation of Nrf2 from the cytosol, human neuroblastoma SHSY5Y cells were grown in the DMEM supplemented with high glucose and glutamine (Invitrogen) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were plated in 100-mm dishes, and when they were ∼70% confluent, were treated with 100 nM of TP-319 or TP-500 for 0, 1, or 2 h. The NE-PER Nuclear and Cytosolic extraction kit (Thermo Scientific) was used to fractionate nuclear and cytosolic fractions according to the manufacturer's instruction.
Neh2-luc reporter assay
TP (30 nM–1 μM) were tested in 96-well format white, flat-bottom plates in comparison with canonical Nrf2 activators TBHQ and sulforaphane (1–10 μM). All stock solutions and their serial dilutions were prepared in DMSO. SHSY5Y cells expressing the Neh2-luc reporter (HIF1 ODD-luc reporter was used as a control line) were plated at the density of 30,000 cell per well using a WellMate multichannel dispenser from Matrix (Thermo Fisher Scientific) and grown overnight on DMEM/F12+GlutaMAX (100 μl per well). Then, the compounds were added to each well, and the plates were incubated for a fixed time interval; the medium was removed; cells were lyzed; and luciferase activity was measured on a luminometer Lmax11384 (Molecular Devices) with BrightGlo™ reagent (Promega). The reporter activation was normalized to the background luminescence. Kinetics of reporter activation was measured by adding varied fixed concentrations of a studied compound at different time points followed by simultaneous cell lysis and activity measurement in the whole 96-well plate; this assay format minimizes experimental error originating from the well-known instability of the luciferase reagent. All experiments were performed in triplicate or quadruplicate.
Computer modeling
A 3D model of Keap1 was constructed using the crystal structure of FadR (PDB entry: 1e2x) as the template structure [as described in (47)]. Sequence alignment showed 14.6% and 36.3% of sequence identity and similarity, respectively. MODELER protocol (ver. 9v4) and Discovery Studio Software were used to generate 75 structural models of Keap1 IVR. The model with lowest objective function was selected for further study. Docking was performed by Discovery Studio Software using the LigandFit protocol with default parameters and CHARMm ligand minimization option. The ligand was treated as fully flexible, and the protein residues were kept restraint. Default scoring functions were enabled, and all poses were evaluated manually, and the best docking model was selected, respectively.
RNA isolation and real-time RT-PCR
Total RNA from N27 cells, mouse liver, striata, ventral midbrain, and cortex was isolated according to the manufacturer's protocol using TRIzol reagent (Invitrogen). About 2 μg of total RNA was reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (Invitrogen). cDNA was diluted, and 100 ng was used to amplify in an ABI prism 7900 HT Real-time PCR system (Applied Biosystems) for various genes using primers (Table 2) and Fast SYBR® Green Master Mix (Invitrogen). Cycling parameters were 95°C for 10 s, followed by 60°C for 1 min. Relative expression was calculated using the ΔΔCt method (25). Values are expressed as a fold of control reaction and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression.
Cd68, cluster of differentiation 68; Gapdh, glyceraldehyde-3-phosphate dehydrogenase; Gclc, glutathione cysteine ligase regulatory subunit; Gclm, glutathione cysteine ligase modulatory subunit; Gsr, glutathione reductase; Ho-1, hemeoxygenase 1; Ccl2, (Mcp1) monocyte chemotactic protein-1; Nqo1, NADPH quinone oxidoreductase 1; Nrf2, nuclear factor E2-related factor 2; Tnf, tumor necrosis factor.
Western blot analysis
Protein extracts were prepared from N27 rat dopaminergic cells, human neuroblastoma SHSY5Y cells, mouse ventral midbrain, striata, and liver tissues by homogenization in a TNE buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 1× Complete protease inhibitor cocktail [Roche], 1× phosphatase inhibitor cocktail I and II [Sigma]). Protein concentration was determined by BCA method (Pierce Biotech), and 25 μg of protein was resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose, and probed using appropriate primary antibodies (anti-Nrf2, [1:2000 Epitomics]; anti-Nrf2, [1:2000, C-20; Santa Cruz Biotechnology, Inc.]; anti-NQO1 [1:1000; Abcam], anti-glutathione reductase [1:2000; Abcam], anti- GCLC [1:10,000], anti-GCLM [1:10,000], anti-hemeoxygenase-1 [1:2500; Epitomics], anti-aldolase [1:1000; Abcam], anti-PARP1 [1:1000; Santa Cruz Biotechnology, Inc.], and anti-GAPDH [1:5000; Fitzgerald]). Densitometric analyses were performed using Quantity One Software (Bio-Rad).
MPTP and triterpenoid administration in mice for neuroprotective studies
Acute and subacute MPTP intoxication paradigms were used to test the neuroprotective effects of TP using male C57Bl6 and/or Nrf2 KO and their age-matched WT littermates.
Acute MPTP
In this protocol, 10-week-old C57Bl6 mice (n=8 per group) were divided into four different groups consisting of (i) a control group treated with saline alone; (ii) a group treated with MPTP alone; (iii) a group treated with either TP-319 or TP-500 alone; and (iv) groups treated with TP-319 or TP-500 in combination with MPTP. TP-319 and TP-500 were dissolved in 1:4 ethanol:neobee oil (a derivative of coconut oil). MPTP 10 mg/kg free base was administered intraperitoneally three times a day every 2 h or at 15 mg/kg free base administered intraperitoneally four times a day every 2 h. Animals with TP-319 or TP-500 in combination with MPTP were administered TP by oral gavage with 4, 2, 1, and 0.5 μmol of the respective drugs in 100-μl volume once a day for 4 days before MPTP and once a day for 3 days after MPTP. On the day of MPTP, no drugs were administered to mice. Animals belonging to MPTP and control groups received the vehicle (ethanol:neobee oil, 1:4), whereas the drug-alone group received respective doses of triterpenoid drugs at the same frequency. All animals were sacrificed on the 7th day after MPTP. For neuroprotective studies in WT and Nrf2 KO mice, 10-week-old male Nrf2 KO and age-matched WT littermates were treated with 2 μmol of TP-319 or TP-500 at a similar frequency and volume described above. MPTP 10 mg/kg free base was administered four times a day every 2 h. All animals were sacrificed on the 7th day after MPTP.
Subacute MPTP
In this protocol, 12-week-old C57Bl6 mice (n=8 per group) were divided into the four different groups described above. MPTP 30 mg/kg free base was administered intraperitoneally once daily for 5 days. Animals with TP-319 or TP-500 in combination with MPTP were gavaged with 2 μmol of the respective triterpenoid in 100-μl volume once a day for 4 days before MPTP, again daily once 2 h before MPTP, and once a day for 3 days after MPTP. All animals were sacrificed 21 days after the last dose of MPTP.
Triterpenoid administration in mice for assessment of Nrf2-dependent genes
To test activation of the Nrf2/ARE pathway, in vivo male mice (C57Bl6 or Nrf2 KO and their age-matched WT littermates) were administered 4 μmol of TP (TP-319 or TP-500) in 100 μl of vehicle (ethanol:neobee oil, 1:4) by oral gavage twice 12 h apart. Control groups of mice received vehicle (ethanol: neobee oil, 1:4) at the same volume and frequency as TP. Mice were sacrificed, and the whole brain, striata, ventral midbrain, cortex, and liver were collected at 6, 9, 24, and 48 h after the last dose of each triterpenoid.
Measurement of tissue levels of TP by HPLC–mass spectrometry
Brain and liver (about 200–400 mg) tissue were homogenized on ice in 0.5 ml acetonitrile using Tissue Miser (Fisher Scientific). The homogenates were vortexed and then centrifuged at 21,000 g, at 4°C, for 10 min, and the supernatant was transferred and diluted 1:1 with 20 mM ammonium acetate pH 7.4. Diluted buffered acetonitrile extracts were centrifuged at 20,817 g, 4°C, for 5 min. Supernatants were loaded on a Waters XTerra MS C18 5-μm column, with initial column conditions 46% acetonitrile, 10 mM ammonium acetate, pH 7.4. The column was subjected to an 8-min gradient from 46% to 94% acetonitrile and then washed with 94% acetonitrile for 6 min. Compounds were detected using a Waters ZQ mass spectrometer with an electrospray ionization probe, positive ionization mode. Analysis was performed using the Waters MassLynx software package. Quantitation was against standard curves generated by adding dilutions of compounds to the acetonitrile extract from control solid tissues and expressed as amount per wet weight of tissue (36).
Measurement of striatal levels of catecholamines and MPP+ by HPLC
Striatal levels of DA and its metabolites DOPAC and HVA were measured after sonication and centrifugation in chilled 0.1 M perchloric acid (PCA, 100 μl/mg tissue) as previously described (48). Briefly, 15 μl supernatant was isocratically eluted through an 80-×4.6-mm C18 column (ESA, Inc.) with a mobile phase containing 0.1 M LiH2PO4, 0.85 mM 1-octanesulfonic acid, and 10% (v/v) methanol and detected by a 2-channel Coulochem II electrochemical detector (ESA, Inc.). Concentrations of DA, DOPAC, and HVA are expressed as ng per mg protein. The protein concentrations of tissue homogenates were measured according to the BCA assay (Pierce Biotech). For MPP+ measurement, TP-319 or TP-500 was administered at a dose of 4 μmol once daily for 4 days followed by a dose on the 5th day, 30 min before MPTP. Striatal tissues were sonicated and centrifuged in 0.1 M PCA, and an aliquot of supernatant was injected onto a Brownlee aquapore×03-224 cation exchange column (Rainin). Samples were eluted isocratically with 20 mM boric acid–sodium borate buffer, pH 7.75, containing 3 mM tetrabutylammonium hydrogen sulfate, 0.25 mM 1-heptanesulfonic acid, and 10% isopropanol. MPP+ levels were detected with a fluorescence detector set by excitation at 295 nm and emission at 375 nm (48).
Measurement of cellular GSH and GSSG levels by HPLC
N27 rat dopaminergic cells were sonicated in chilled 0.1 M PCA and centrifuged. Briefly, 15 μl supernatant was isocratically eluted through a 4.6-×150-mm C18 column (ESA, Inc.) with a mobile phase containing 50 mM LiH2PO4, 1.0 mM 1-octanesulfonic acid, and 1.5% (v/v) methanol and detected by a 2-channel Coulochem III electrochemical detector (ESA, Inc.), set with a guard cell potential 950 mV, Channel 1 potential for GSH detection and Channel 2 potential 880 mV for GSSG detection (48). Concentrations of GSH and GSSG are expressed as nmol per mg protein.
Immunohistochemistry and morphometric analysis
Mice were anesthetized with sodium pentobarbital, transcardially perfused with 0.9% saline, followed by 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), pH 7.4. Brains were dissected out, postfixed in 4% paraformaldehyde for 24 h, and cryopreserved in 30% sucrose/PBS for 48 h. Snap-frozen brains were coronally sectioned at 40-μm thickness, encompassing the substantia nigra using a cryostat. Briefly, sections were rinsed in PBS and incubated in 3% hydrogen peroxide/10% methanol solution for 10 min to quench endogenous peroxidase activity. Sections were permeabilized/blocked in 10% normal goat serum (NGS)/0.1% Triton X-100/PBS for 1 h at room temperature. Sections were incubated overnight at 4°C with the following primary antibodies in PBS containing 2% NGS/0.01% Triton X-100: rabbit polyclonal anti-TH (1:1000; Novus Biologicals), rat monoclonal anti-CD11b (1:500; Serotec), and rabbit polyclonal anti-3-nitrotyrosine (1:100; Calbiochem). Biotinylated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) were used appropriately after incubation with streptavidin ABC solution (Vector Laboratories). Immunostaining was visualized by diaminobenzidine (Sigma) chromogen. Sections were mounted on glass, dehydrated, and cover-slipped with cytoseal (Thermo Scientific). Digital images were captured with a Coolpix 5000 Nikon Camera. TH-immunostained sections were counterstained with thionin before dehydration and cover-slipped with cytoseal. Nissl (thionin)-stained and TH-positive neuronal counts were estimated within the substantia nigra by Stereoinvestigator software (Microbrightfield) as previously described (39). CD11b-positive microglia were quantified on images captured from three coronal sections per mouse encompassing the substantia nigra. Quantitative analysis (cell number per square millimeter) was determined using NIH ImageJ software (50). SNpc 3-nitrotyrosine immunostaining was quantified by expressing the area occupied by the precipitate of 3-nitrotyrosine staining within the defined fields from three coronal sections per mouse encompassing the substantia nigra (45).
Statistical analysis
Results were expressed as means±standard error of the mean. Significance was determined by one-way or two-way analysis of variance followed by the Student–Newman–Keuls test or a two-tailed unpaired Student t-test. Significance was set at p≤0.05. All statistical analyses were performed using Prism software (GraphPad).
Footnotes
Acknowledgments
This work was supported by grants from the NIH R21 NS062165 (BT), R01 NS060885 (BT), R01 ES017295 (MFB), R01 CA78814 (MBS), and Michael J. Fox Foundation for PD grant (BT and MFB), Department of Defense grant (MFB), Thomas Hartman Foundation for PD (IGG), Winifred Masterson Burke Relief Foundation (IGG), and Reata Pharmaceuticals Irving Texas. NAK is a PD Foundation postdoctoral fellow. We thank Dr. Terrance Kavanagh (University of Washington) for GCLC and GCLM antibodies and Dr. Curt Freed (University of Colorado) for rat dopaminergic 1RB3AN27 (N27) cells.
Author Disclosure Statement
All authors declare no conflicts of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.