
Abstract
The complexity of human DNA has been affected by aerobic metabolism, including endurance exercise and oxygen toxicity. Aerobic endurance exercise could play an important role in the evolution of Homo sapiens, and oxygen was not important just for survival, but it was crucial to redox-mediated adaptation. The metabolic challenge during physical exercise results in an elevated generation of reactive oxygen species (ROS) that are important modulators of muscle contraction, antioxidant protection, and oxidative damage repair, which at moderate levels generate physiological responses. Several factors of mitochondrial biogenesis, such as peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α), mitogen-activated protein kinase, and SIRT1, are modulated by exercise-associated changes in the redox milieu. PGC-1α activation could result in decreased oxidative challenge, either by upregulation of antioxidant enzymes and/or by an increased number of mitochondria that allows lower levels of respiratory activity for the same degree of ATP generation. Endogenous thiol antioxidants glutathione and thioredoxin are modulated with high oxygen consumption and ROS generation during physical exercise, controlling cellular function through redox-sensitive signaling and protein–protein interactions. Endurance exercise-related angiogenesis, up to a significant degree, is regulated by ROS-mediated activation of hypoxia-inducible factor 1α. Moreover, the exercise-associated ROS production could be important to DNA methylation and post-translation modifications of histone residues, which create heritable adaptive conditions based on epigenetic features of chromosomes. Accumulating data indicate that exercise with moderate intensity has systemic and complex health-promoting effects, which undoubtedly involve regulation of redox homeostasis and signaling. Antioxid. Redox Signal. 18, 1208–1246.
I. Introduction
It has been proposed that endurance running played a significant role in the evolution of Homo sapiens (46, 320), which allowed hominem, possibly from the time of Homo erectus (341), to hunt and obtain sufficient amounts of meat for physical development. Based on this hypothesis, early human hunters with extreme endurance capacity were undoubtedly highly successful (320). It can be suggested that on the basis of Darwin's fitness theory, greater capacity at endurance running improved the survival capacity of specific early humans. It appears that endurance capacity or at least one of the most significant markers of aerobic endurance, maximal oxygen uptake (VO2max), even in the XXI century, is important for survival.
There is a great deal of evidence that indicates that higher VO2max is associated with a decreased risk in the incidence of a number of lifestyle-related diseases, including breast, colon, and prostate cancer, cardiovascular diseases, type II diabetes, and Alzheimer's disease (35, 121, 174, 247, 312). In accordance with this, an extremely low level of oxygen uptake could have serious outcomes such as increased incidences of diseases that lead to early death (399). One could suggest that the level of endurance capacity plays an important role in survival (Fig. 1). This phenomenon nicely demonstrates that our dependence on oxygen and oxygen uptake capacity could have started with the evolution of the aerobic organism, through the development of H. sapiens. The question whether an endurance running-dependent hunting lifestyle played a role in the development of the antioxidant system in humans cannot be answered with full certainty. The fact that the specific activity of superoxide dismutase in different organs is higher in humans than in shorter-lived animal species suggests that man has better protection against reactive oxygen species (ROS) (401) than other mammals. Moreover, it is clear that the rate of ROS production is also attenuated in man compared to other species (217). Interestingly, enough regular physical exercise increases the level of antioxidant defense and decreases the rate of ROS production, which seems to happen as the result of the evolution of H. sapiens.
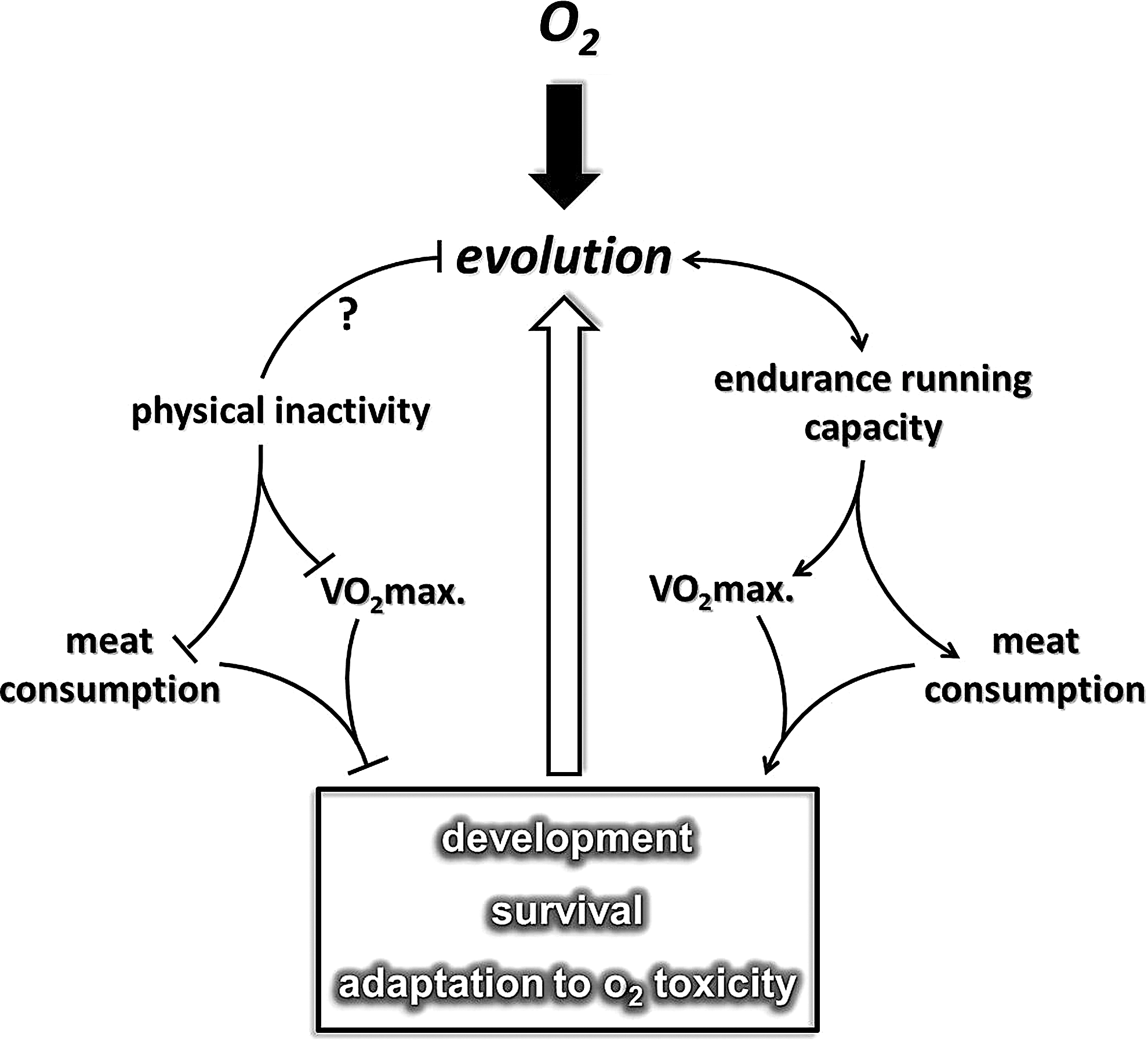
It has been suggested that the stone-age human spent about 4000 Kcal on physical activity daily, compared to the 400 Kcal utilized by the human male in the present century (40). The effects of physical exercise, or its converse, physical inactivity, are complex and systemic, and strongly affect redox homeostasis and the resistance to oxidative stress (309) (Fig. 2). Indeed, the increase in oxygen flux with exercise could be as high as 100-fold higher than resting values in the contracting skeletal muscle. This oxygen flux naturally results in an increased generation of ROS (77, 309, 356). In turn, this increased ROS production could result in a well-orchestrated adaptive response, which leads to the stimulation of preventive measures against oxidative stress-related diseases (158, 296). Data from human studies indicate that although endurance exercise showed elevated oxidative power of mitochondria, the efficiency of energy transfer (P/O ratio) did not change significantly (402). Moreover, endurance training, which increased the VO2max of subjects by 21% and the lactate threshold by 53%, did not significantly increase the sensitivity of mitochondrial oxidative function to ROS in skinned fibers (424).

Probably, as a result of evolution, humans have evolved to a state where exercise-induced ROS can stimulate elevation of the level of glucose transport to meet the need for increased metabolism, as well as other metabolic adaptations to the high demands of exhaustive exercise (346). Acute bouts of exercise, or exhaustive exercise, can elevate the oxidative damage to lipids, proteins, and DNA and disrupt ROS-modulated signaling, which together could jeopardize cell survival. Therefore, both acute and regular exercise can influence homeostasis, and then adaptation, which involves the enhanced oxygen uptake-related benefits, the risk, and consequences of increased oxygen uptake-dependent modulation of ROS production, and redox homeostasis. The present review aims to focus on the complex effects of physical exercise on oxygen utilization, metabolism, signaling, and redox-dependent adaptations, especially in the skeletal muscle. We will discuss the sources of ROS production, the adaptive response of antioxidant and oxidative damage-repairing systems. Moreover, we will evaluate the hormetic role of oxidative damage, which in a moderate degree could be important to membrane remodeling, protein turnover, specific gene expression, or chromatin opening. Sirtuins are redox-dependent metabolic sensors, and we will highlight the role of these histone deacetylases in exercise-associated adaptation. As well, we will touch upon the enigmatic role of nitric oxide (NO) in repair of skeletal muscle damage and how it could be involved in exercise-induced adaptation. Moreover, it will be demonstrated that ROS are important modulators of satellite cell proliferation in the skeletal muscle, and play a role in exercise-induced neurogenesis as well. Finally, the effects of exercise on hypoxia-inducible factors will be discussed, followed by the role of redox signaling in exercise-related epigenetical modifications.
II. General Adaptations to Exercise
Adaptation is dependent on the modulation of homeostasis. It is clear that homeostasis is a dynamic system with biological and functional/actual endpoints. Biological endpoints are signified by the point at which the system collapses. Exceeding these points is fatal. For example, a 43°C degree fever results in denaturation of proteins and eventually death.
Functional/actual endpoints mean the limit of individual tolerance, which is naturally below the biological endpoint and is a dynamic, variable value. The distance between optimal zone and biological endpoints represents the zone that can be targeted to induce adaptations to extend functional/actual endpoints. In the case of a high degree of adaptation, the distance between the biological endpoints and the functional endpoints can be narrowed. In other words, the distance between optimal zone and functional/actual endpoints can be increased (Fig. 3). It can be assumed that a larger range between optimal zone and functional/actual endpoints represents greater adaptive capability and better tolerance against stressors.
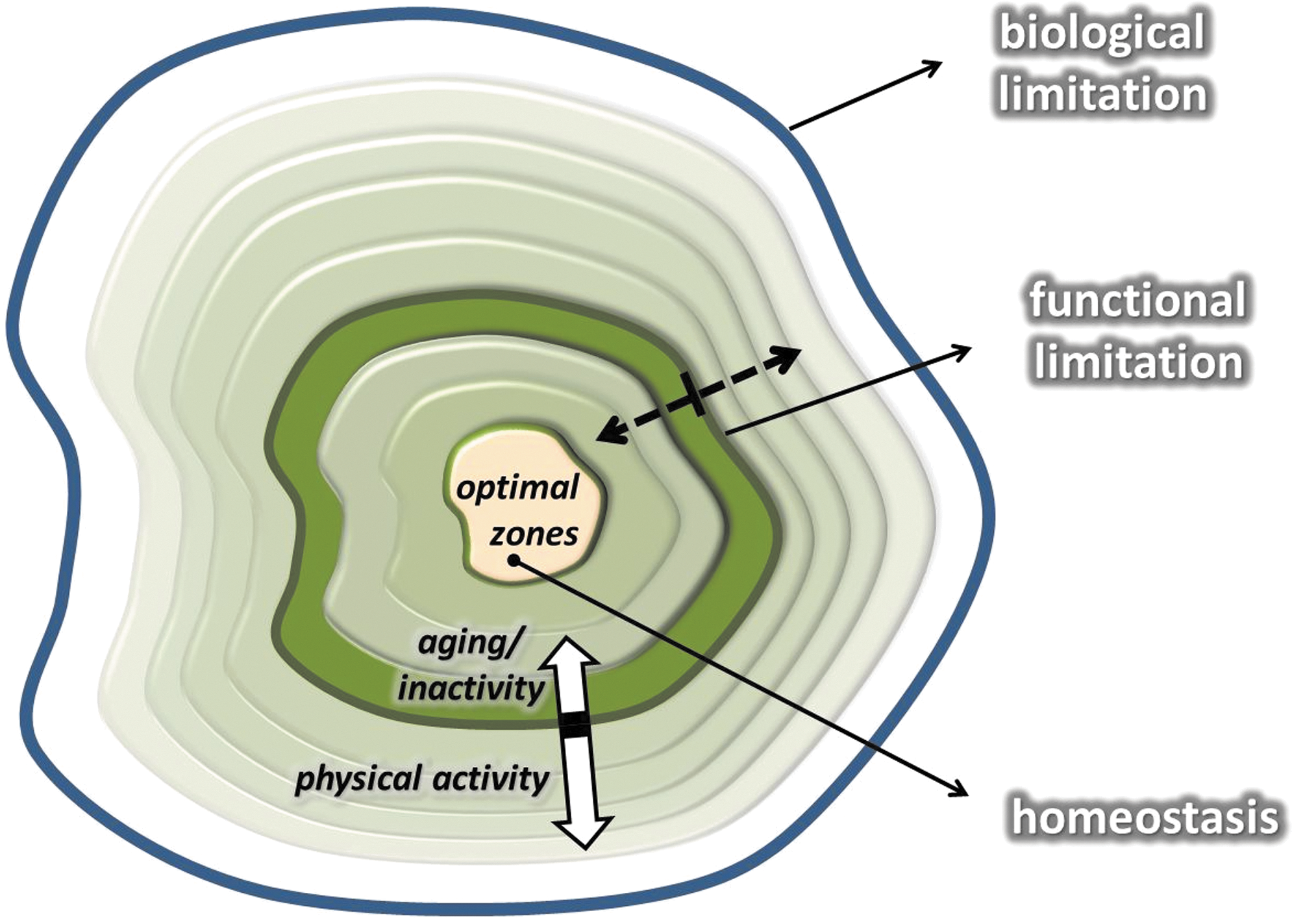
Two examples will suffice to exemplify this point. The resting heart rate of untrained individuals is around 70 beats/min, while the maximal heart rate is ∼220 minus the age of the individual. Therefore, the adaptive range is 130 beats for a 20-year old and just 50 beats for a centenarian. Well-trained endurance runners, on the other hand, have a significant decrease in resting heart rate, which could be as low as 35 beats per minute. If an individual has the same maximal heart rate as the 20-year old, trained, and untrained, the adaptive range increases from 130 to 165 with the extension of the functional endpoints. Another example is lactic acid tolerance. Both trained and untrained individuals have a resting blood lactic acid level around 1.5 mM/L. If they run until exhaustion on a treadmill, untrained individuals stop when their lactic acid levels reach ∼6–10 mM/L, while elite athletes can still continue until their lactic acid levels are over 20 mM/L.
A similar phenomenon is found for ROS handling. It is known that a single bout of exhaustive exercise results in elevated levels of lipid peroxidation, carbonylation of amino acid residues, and 8-Oxo-7,8 dihydroguanine (8-oxoG) in DNA (318). On the other hand, when a single bout of exhaustive exercise is given to well-trained subjects, the body responds without a large elevation in oxidative damage (318). In addition, regular exercise training-associated adaptation is a precondition against treatment with H2O2, which causes a significant degree of damage for untrained subjects (317). Moreover, when heart attacks or strokes are simulated in untrained and trained animals, the infarct size is significantly smaller in the trained groups (39, 81), showing that regular exercise acts as a preconditioning tool (317) by enhancing the adaptive zone, by narrowing the theoretical distance between functional and biological endpoints. A great deal of evidence exists that suggests that regular exercise-induced adaptations to ROS handling, through redox signaling, including antioxidant and oxidative damage repair systems, significantly contribute to the health-promoting effects of regular exercise. However, this phenomenon can also be interpreted in another way, namely that physical inactivity severely decreases the adaptive capacity, including redox processes, and this increases the incidence of a wide range of diseases, which generally leads to elevated mortality rate and decreases in mean lifespan. This phenomenon could be identified as being in opposition to aerobic endurance-dependent evolution.
III. Exercise, ROS, Antioxidant, and Housekeeping Systems
A. ROS and antioxidants
A great deal of epidemiological data has shown that exercise decreases the incidence of oxidative stress-associated diseases (174, 405). This beneficial effect is due to the fact that exercise-induced ROS production is necessary for oxidative stress-related adaptations. There are a number of ROS generating systems that increase blood flow to skeletal muscle during physical exercise, and more or less maintain blood flow to the brain, and significantly decrease oxygen supply to the liver and kidney (309). The mitochondrial electron transport chain is one main ROS generators found in skeletal muscle (295). As a result of exhaustive exercise, ROS production of complex I and III, with pyruvate/malate and succinate substrates, was increased by 187% and 138%, respectively, compared to the nonexercising group (342). Complex III has been suggested to yield superoxide on the cytoplasmic site of the mitochondrial membrane (47), which could be important for redox signaling. Moreover, mitochondria isolated from skeletal muscle after contraction showed significantly increased levels of H2O2 generation compared to noncontracting values (415). Sahlin et al. have shown that complex I is the major ROS generator in skeletal muscle of ultraendurance runners, as assessed by Amplex red reagents (343). However, it must be noted that the earlier estimation of about 1%–5% of the oxygen that enters mitochondria can be released as ROS (44) could be an overestimation, and the real value could be more than an order of magnitude lower (373). Upon this and related observations, it was suggested that mitochondria are not the main source of ROS production during exercise (298). On the other hand, a recent report suggests that mitochondria might be important sources of ROS, at complex I and III through peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) (19). In this particular study, a positive association was found between ROS production and mitochondrial respiration (19). The effects of the anabolic steroid, stanozolol, were investigated on mitochondrial ROS production, and it was found that complex I- and complex II-linked substrate generated H2O2 after exercise sessions, which was significantly attenuated by stanozolol (342). Therefore, despite the fact that the resting efflux of ROS is a magnitude lower than was suggested, results from the measurements of ROS/H2O2 efflux indicate that mitochondria are generating enhanced levels of ROS during intensive exercise. On the other hand, the release of ROS from each mitochondrial complex could differ significantly.
For example, at complex I, the iron–sulfur clusters, flavoprotein, and oxidoreductase, and at complex III, Q10 semiquinones are suggested to be the main sites of ROS generation (244, 373). Recently, a novel method has been developed, which allows detection of superoxide production in the mitochondrial matrix by the stochastic quantal events of superoxide (mitochondrial superoxide flashes [mSOFs]) (429). It has been shown that during muscle contraction, the activity of mSOF increases in mitochondria and is dependent on the activity of the electron transport chain and, furthermore, has a biphasic nature, showing an increase in brief and a decrease in longer tetanic contractions (429). This finding certainly does not rule out extracellular sources of ROS during contraction, which have been suggested as being important (304, 325, 448). Indeed, 5-lipoxygenase, cyclooxygenase, sarcolemmal NADPH oxidase, and xanthine oxidase (XO) have been implicated in superoxide generation in skeletal muscle (272, 295). NADPH oxidase is one of the major ROS generators during exercise (26, 297), since in the presence of ADP and Fe3+, NADPH oxidase catalyzes electron transfer from NADPH to molecular oxygen to form superoxide (21). XO is especially involved in ROS production during anaerobic exercise, and a linear relationship has been reported between XO and lactic acid levels (304). One of the reasons for this is the fact that XO activity is strongly dependent on the availability of substrate (437). During intense exercise due to the high rate of ATP degradation, AMP generation hypoxanthine and xanthine are formed and serve as substrates for XO, which uses molecular oxygen to generate ROS. Interestingly, we could detect increased XO activity in the liver 1 day after exhaustive acute exercise (305), and the protective effects of administered SOD derivatives showed that endothelium-associated XO is one source of ROS generation during intense exercise (304). Recently, myostatin has emerged as a potential ROS-inducing factor, especially during sarcopenia (372). It has been demonstrated that ablation of the myostatin gene resulted in attenuated loss of muscle mass with aging. Moreover, it turns out that myostation can induce ROS production through tumor necrosis factor-α (TNF-α) and NADPH oxidase (372). The role of exercise on myostation-mediated redox signaling is still unclear, and research is warranted on this topic. Activation of ryanodine receptor 1 (RyR1) in the sarcoplasmic reticulum of skeletal muscle is necessary for Ca2+ release and consequent generation of cross-bridge-related force production. With the aging of skeletal muscle, continuous Ca2+ leak were observed in RyR1 channels, which was associated with decreased tension, exercise capacity, and increased ROS production. Normalization of RyR1 function by pharmacological intervention that stabilized binding of calstabin1 to RyR1 significantly reduced Ca2+ leakage and increased endurance capacity (14).
It has been shown that a single bout of exercise and regular exercise modulate the ROS production in neutrophils differently; namely acute exercise caused apoptosis and reduction of mitochondrial membrane potential, while regular exercise increased the resistance against oxidative challenge (385). Muscle contraction generates heat, which has been shown to enhance ROS production (447). Needless to say, ROS production is an essential physiological process for muscle contraction, and it is estimated that the level of H2O2 can be increased by 100 nM during contractions (155). Indeed, it has been shown that low levels of exogenous H2O2 treatment increase, and the addition of catalase decreases, force production of the diaphragm (324). H2O2 was shown to modulate muscle contraction via Ca2+ channels (15). Administration of N-acetylcysteine (NAC) is often used to appraise the effects of ROS in muscle fatigue (70, 74, 229, 326), and it has been shown to decrease the rate of fatigue at 80% of VO2max, but not at higher intensities (74). NAC has been suggested to act through improved K+ regulation (229). Infusion of NAC appears to interact with some exercise-induced signaling pathways, as shown by attenuation of the exercise-induced activation of JNK. However, phosphorylation of p38 mitogen-activated protein kinase (MAPK), ERK1/2, or p65 subunit of nuclear factor-kappaB (NF-κB) was not affected (283).
Accumulating evidence suggests that ROS are generated during exercise and modulate signaling pathways and the level of muscle contraction, indicating that ROS content and the muscle function–dose relationship can be described by a hormesis curve (310). Low levels of ROS stimulate force production, while high levels do attenuate it.
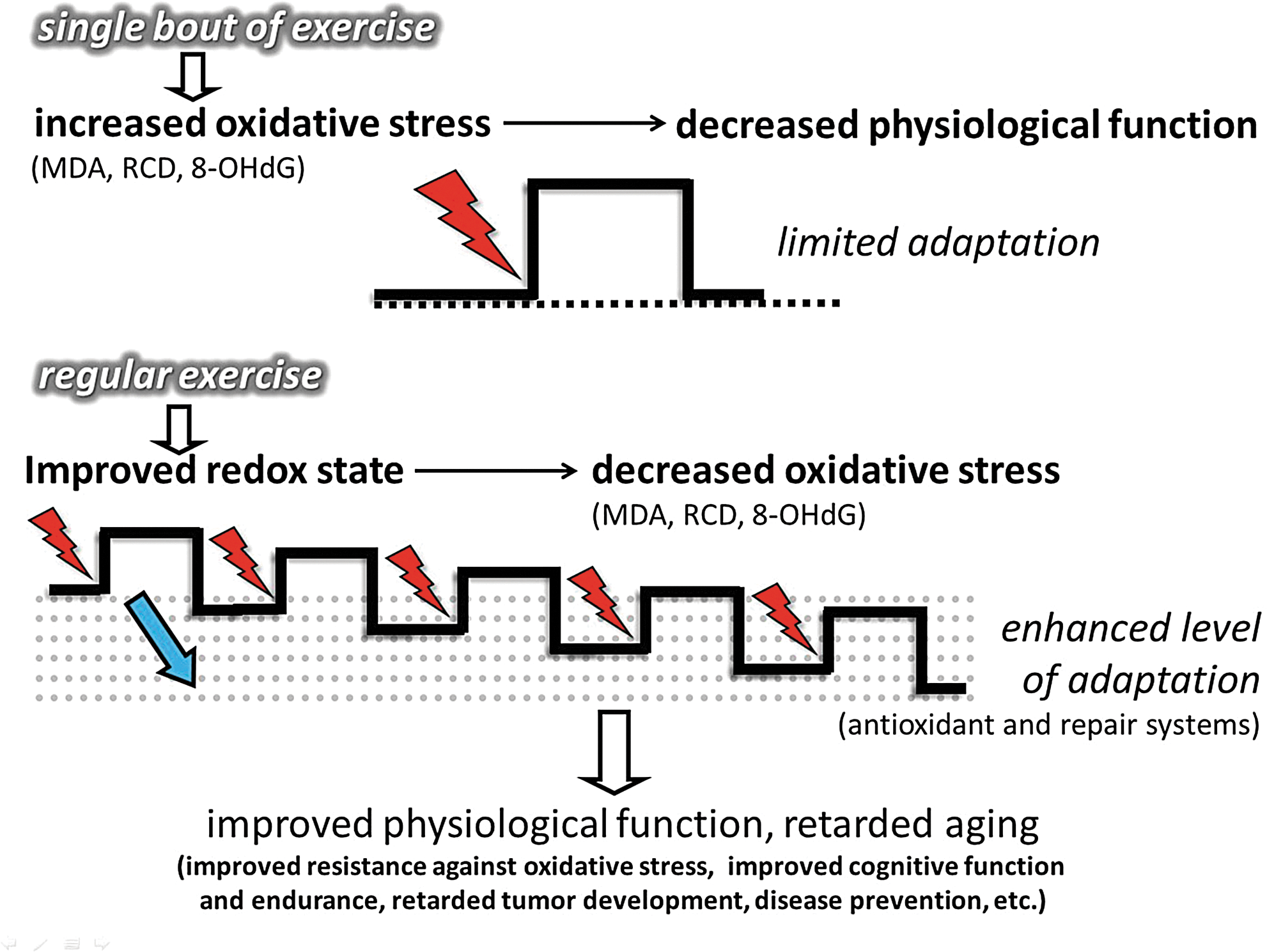
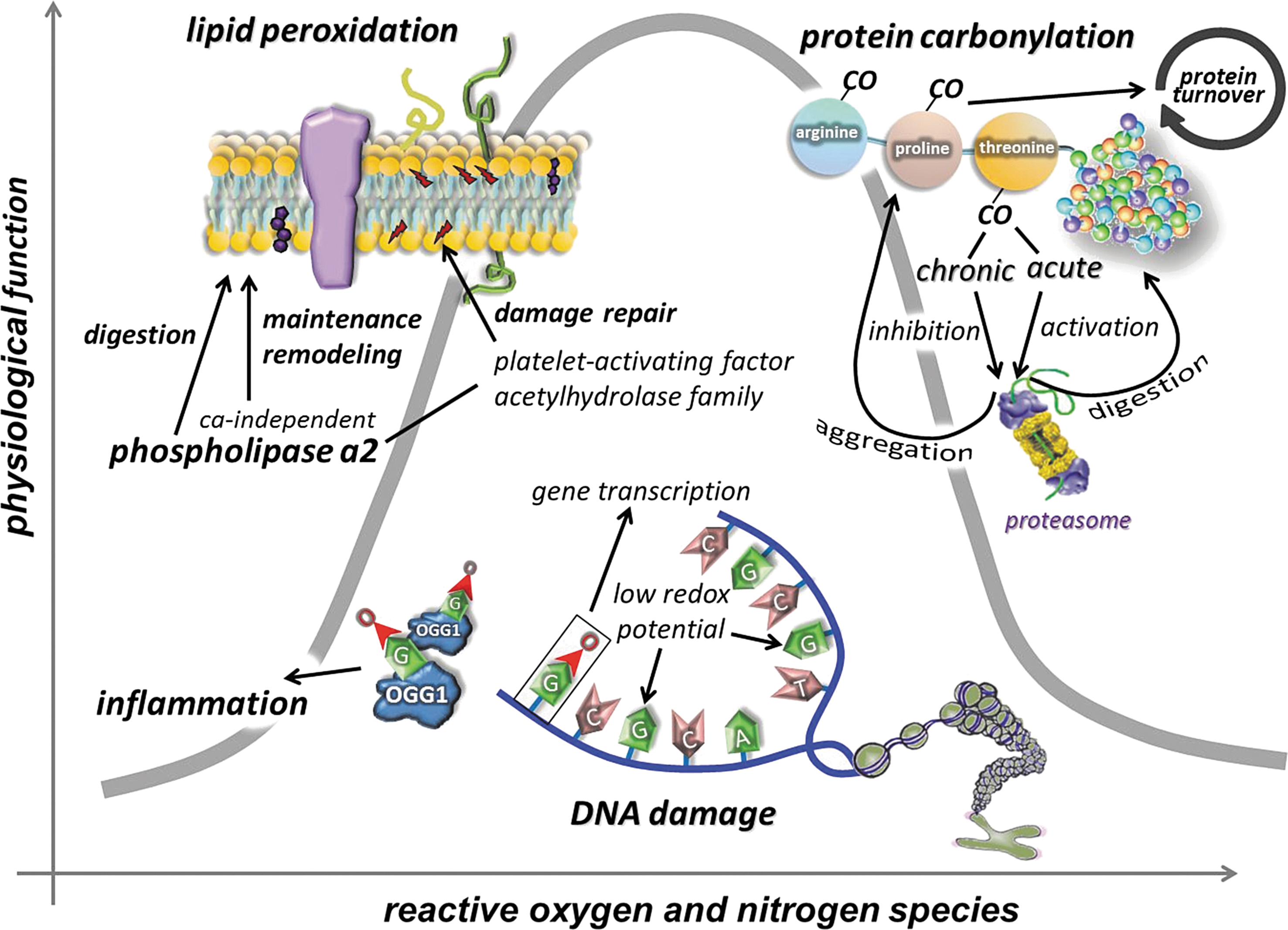
To curb the toxic and deleterious effects of ROS, cells are well equipped with enzymatic and nonenzymatic antioxidant systems. The exercise-induced ROS generation results in increased activity of enzymatic antioxidants, which then results in increased resistance to oxidative challenges, including a wide variety of oxidative stress-related diseases. One of the key issues of this adaptive response is the feature of intermittent exercise. Exercise-induced ROS production, in the recovery period, is mostly overcompensated by the upregulated antioxidant system (111, 112, 160). Chronic exposure to high levels of ROS can exhaust the nonenzymatic antioxidant system and lead to impaired cellular function, macromolecule damage, apoptosis, and necrosis. The intermittent nature of exercise gives a preconditioning role to exercise. After a bout of exercise that gives a metabolic and oxidative challenge, followed by a rest period, the system is allowed to adapt to the challenges caused by this stressor (318). Indeed, during resting periods, the body does not simply store more glycogen in the skeletal muscle, which would allow better performance, but also upregulates antioxidant and oxidative damage repair systems (Fig. 4) (111, 163). Subjects involved in regular exercise, due to an adaptive response, demonstrate higher levels of mitochondrial content, and produce lower levels of ROS at the given intensity than those who are untrained. Therefore, excess physical exercise is detrimental to poorly trained individuals, but progressive training allows the cells to more easily detoxify a larger amount of ROS, partly by the induced activity of the antioxidant systems. Moreover, it has to be mentioned that severe and extreme exercise regimens or intensities may pose serious threat of oxidative damage, even in healthy subjects, and those who are poorly trained and/or not well nourished are in higher risk to suffer oxidative stress.
For example, runners who often suffer from anemia, which could be associated with increased free-iron concentrations in the blood stream, are subjected to enhanced levels of oxidative damage. Moreover, exercise at high altitude also increases the risk of the accumulation of oxidative damage markers (83).
Chronic elevation of ROS is dangerous, as it could easily exhaust the system, while bouts of exposure, like preconditioning ischemia/reperfusion repeats, significantly increase the resistance to oxidative stress (128). Both acute and regular exercise could induce the activity of antioxidant enzymes, including manganese superoxide dismutase (Mn-SOD), copper–zinc superoxide dismutase (Cu,Zn-SOD), extracellular superoxide dismutase (EC-SOD), catalase, and glutathione peroxidase (GPX) (112, 138, 160, 269). The mechanism behind the induction of the antioxidant system complex most probably involves epigenetics, since it was shown that Cu,Zn-SOD knockdown results in decreased methylation of DNA (33). Therefore, it cannot be ruled out that exercise results in changes in DNA methylation and/or histone acetylation, which leads to enhanced activation of antioxidant enzyme genes. The exercise-induced ROS generation is naturally a powerful stimulus to activate the expression of antioxidant enzymes. Mn-SOD could be readily induced by ROS-mediated activation of TNF-α (428) or by NF-kB (440) among others.
B. Thiols and redox signaling in exercise
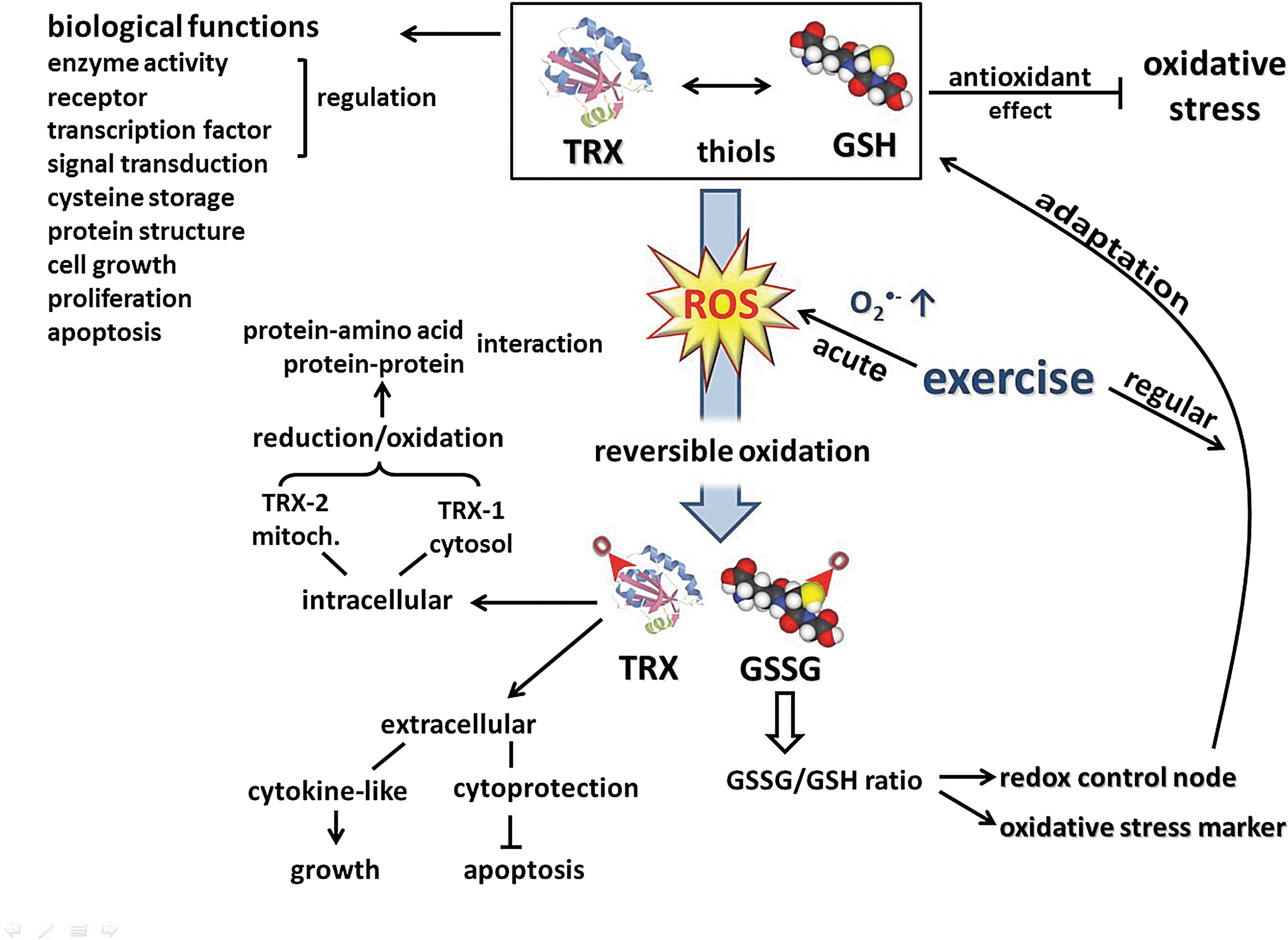
Current evidence indicates that in ROS-mediated signaling, redox-sensitive thiols play a critical role, coupling the intracellular changes in the redox state to regulation of cellular processes (34, 361). Thiols react faster than other amino acids with oxidizing species, and most of their oxidation products can be reversibly reduced. Therefore, reversible redox modification of active-site Cys residues provides a redox switch, an on-off mechanism for control of cellular signaling and function (167). On the other hand, like a rheostat, through allosteric regulation and by S-glutathione-derivatives, thiols can modulate protein function (177). Furthermore, cross-linking of proteins through disulfides yields mechanisms for protein aggregation and trafficking. The endogenous thiols, the glutathione (GSH) and thioredoxin (TRX) systems, play a central role in the antioxidant defenses that control cellular events and regulate protection against oxidative stress (361), a condition that alters normal redox control of cellular signaling, especially by disruption of thiol-redox circuits (166). In addition to their central role in supporting a large network of antioxidant defenses, GSH and TRX have various biological functions such as regulation of enzyme activity, receptors, transcription factors, and ultimately redox-sensitive signal transduction, short-term storage of cysteine, protein structure, cell growth, proliferation, and programmed cell death (358).
GSH (
Oxidation of thiols to disulfides has also been used as a sensitive marker of exercise-induced oxidative stress (203). Since 1985, a growing number of exercise studies have utilized GSSG levels and GSSG/GSH ratios as sensitive markers of oxidative stress, but the results are equivocal (109, 361). Major factors that may affect GSH oxidation response to acute exercise are the type and intensity of the exercise. For example, GSH oxidation is more prominent during exercise performed at higher intensities (362). Another factor may be the training status of the organism. In the trained individual, enhanced antioxidant defenses couple successfully with pro-oxidants to protect the organism against increased oxidative stress. As a result, GSH oxidation, which is a sensitive marker of oxidative stress, occurs less after exercise in trained individuals. Recently, we observed that in the regularly trained horse, with exercise or during recovery, there were no significant changes in the muscle GSSG- or the GSH-redox ratio, although postexercise free-radical amounts correlated positively with postexercise GSSG concentration (184). This can be explained by the well-known fact that regular exercise training of all types enhances GSH levels by inducing GSH synthesis and also by increasing GSH regeneration from GSSG (361).
Data on the role of endogenous GSH on exercise performance, especially endurance capacity, are limited. In one early study, endurance capacity for treadmill running in GSH-deficient rats was found to be reduced by half (360). Although there is a controversial report in the literature regarding the impact of GSH deficiency on prolonged swimming performance in mice (201), one may still postulate that endogenous GSH not only plays a critical role in circumventing exercise-induced oxidative stress but also determines exercise performance.
The TRX system, comprising NADPH, thioredoxin reductase (TrxR), and TRX itself, is critical for cellular redox balance and antioxidant function. TRX is a small multifunctional protein with a dithiol/disulfide active center and is the major cellular protein disulfide reductase. Located extracellularly, TRX shows a cytoprotective activity against oxidative stress-induced apoptosis. TRX has also a cytokine-like function acting as an autocrine growth factor (410). Intracellularly, TRX-1 in the cytosol and TRX-2 in the mitochondria are involved in the regulation of protein–protein or protein–nucleic acid interactions through the reduction/oxidation of protein cysteine residues. In response to a variety of cellular stresses, including oxidative stress, TRX translocates from the cytosol into the nucleus to regulate the expression of various genes. A growing number of transcription factors require TRX reduction for DNA binding (209). TRX and GSH are considered to be functionally and kinetically distinct systems. In both cells and plasma, GSH/GSSG redox is not equilibrated with other major thiol/disulfide couples (TRX and Cys/CySS), providing the basis to consider discrete redox circuits for signaling and control (167). Nevertheless, loss of cellular TRX-1 is known to result in elevated GSH levels (56). Furthermore, after an acute bout of exercise, during recovery, TRX-1 activity correlated negatively with the GSSG/TGSH ratio of oxidized GSH in skeletal muscle of horses (184). These results underline a cross-talk between two major thiol antioxidants in response to exercise-induced oxidative stress (Fig. 5).
In a setting of thiol antioxidant (alpha-lipoic acid [LA]) supplementation, we observed a negative correlation between the activity of TrxR at rest and postexercise levels of free radicals after LA supplementation (184). Acute exhaustive exercise induced TRX-1 mRNA levels (192), and 8 weeks of endurance training increased TRX-1 protein levels in the rat brain (193). In line with these results, a limited number of studies from other groups also provide evidence that TRX contributes to antioxidant defense against oxidative stress induced by acute exercise of variable intensity and duration. In mouse peripheral blood mononuclear cells, 30 min of swimming exercise induced TRX protein expression 12 and 24 h after exercise (381). Similarly, plasma TRX concentrations increased continuously during an ultramarathon race (228).
Thioredoxin-interacting protein (TXNip, also termed VDUP1 for Vitamin D-upregulated protein or TBP2 for thioredoxin-binding protein) has been identified as an endogenous inhibitor of TRX in a redox-dependent fashion (261). Redox-dependent interaction of TRX with TXNip only exists between the oxidized form of TXNip and the reduced form of TRX (277) and may modulate apoptosis signal kinase (ASK-1), redox factor 1, Forkhead box class O4 (FoxO4), and nod-like receptor proteins, which offers a novel mechanism of redox regulation (442). What is, to our knowledge, the only study in the literature to investigate the role of exercise on TXNip levels found that 8 weeks of exercise training did not influence TXNip levels in the rat brain despite an increase in TRX-1 protein. Interestingly, in the same set of experiments, streptozotocin-induced diabetes increased both TXNip levels and oxidative stress, measured as increased GSSG/TGSH ratio (193), suggesting a link between disturbed redox control and disease.
C. Oxidative damage and housekeeping
The term oxidative damage is often referred to as a condition where there is a large increase in the stable product of the interaction of ROS with lipids, proteins, and DNA. The deleterious effects of the oxidative damage on cell function are well known and have been reviewed elsewhere (6, 84, 215, 357, 400, 423). The extent of oxidative damage caused by an acute bout of physical exercise, even if it is exhausting, does not cause viability risking increase in oxidative damage, and it can be argued that this might be a necessary factor of adaptation. The fact that the most often measured macromolecule damage markers of oxidative stress, such as lipid peroxidation-derived aldehydes, protein oxidation byproducts, carbonyl groups, or 8-oxoG, are always detectable even with high antioxidant levels raises the possibility that these markers could play a significant physiological role, which, to date, has not been reported. Aging increases the level of oxidative damage, but the elevation could be dangerous after the last quarter of the life span, which can be attenuated by regular exercise (310). On the other hand, the extent of damage is easily measurable at a young age (Fig. 6).
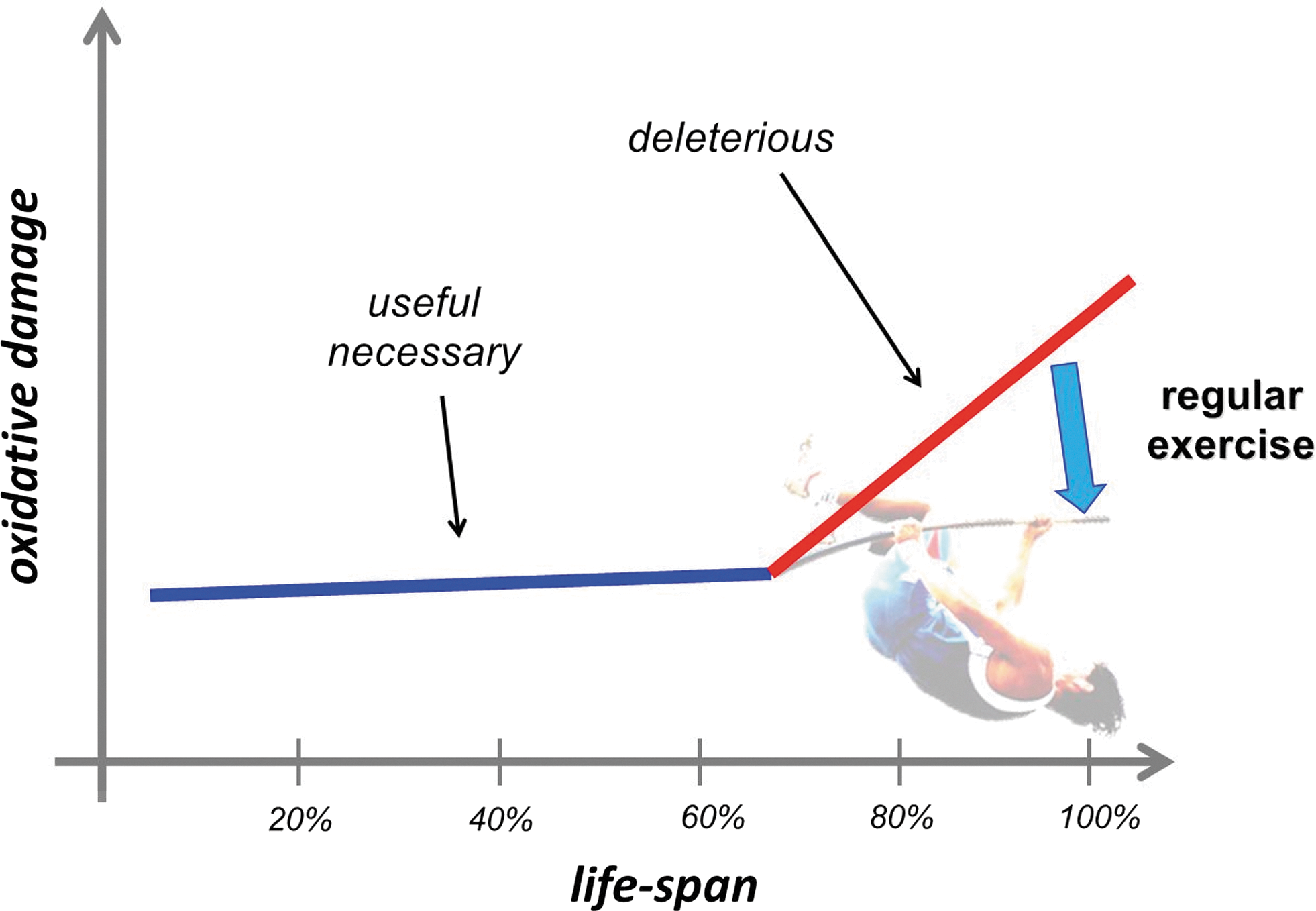
It is clear that ROS are crucial players in aging, although a number of other factors are also involved. In neurons, cardiomyocytes, skeletal myocytes, sensory cells, and other postmitotic cells, the role of ROS could be more pronounced in aging, due to the ROS-mediated damage and accumulation of damage and mutations that are believed to be important in loss of function in these cells. The substitution of aged molecules for mutated ones with loss-of-function molecules leads to a progressive deterioration of such tissues. The increase in oxidative damage in the last quarter of the lifespan is probably the combined effects of the increased level of ROS production (75, 155) and the failure of the oxidative damage repair systems (117, 189).
Lipid peroxidation appears to be an unavoidable process of aerobic life, and despite the well-developed antioxidant systems, certain products of lipid peroxidation are always detectable. Moreover, it has been shown that lipid peroxidation can be generated enzymatically by lipoxygenase. The physiological role of controlled lipid peroxidation is enigmatic, but it could be important in the physiological remodeling of cellular membranes (256). Moreover, targeted, controlled lipid peroxidation by lipoxygenase has been shown to be important for the degradation of mitochondrial membranes during the maturation of erythrocytes (322). Lipoxygenation or lipid peroxidation could modify whole membrane structures, the chemical composition, and the physicochemical properties of phospholipids, resulting in altered permeability, surface charge, and passive electric properties (191).
The phospholipase A2 (PLA2) enzyme family, which consists of more than 20 enzymes, hydrolyzes the sn-2 ester bond of glycerophospholipids and generates free fatty acids and lysophospholipids with physiological activity (349). Ca2+-independent phospholipase A2 (iPLA2-beta) appears to be one enzyme that could repair oxidized cardiolipin, a component of the mitochondrial membrane (185). However, most of the enzymes in the family of PLA2 are generators of oxidative stress and lipid peroxidation during prostaglandin synthesis from arachidonic acid, products of PLA2 reaction, rather than repair of lipid peroxidation (5). The levels of lipid peroxidation in various organs, including skeletal muscle and the brain (290, 384, 398), increase with aging. However, not all the lipid peroxidation products present to the same degree or at the same time. Lipid antioxidants, especially lipid-soluble vitamins and GSH, glutathione-S-transferases, and b-alanyl-
A large number of studies have demonstrated increased levels of lipid peroxidation products, including malondialdehyde and F2-isoprostane, after acute exercise bouts (159, 257, 318, 359). However, regular exercise training, in general, results in decreased levels of lipid peroxidation (159, 255, 259, 359). The regular exercise-associated attenuation of lipid peroxidation levels could be due to the increased capacity of the antioxidant systems, but the enhanced activity of lipid repair cannot be ruled out.
All amino acid residues can be modified when attacked by ROS, generating oxidation of amino acid side chains. The level of oxidative damage to proteins is a magnitude higher than that observed in lipids and DNA (319). Oxidized proteins readily generate cross-linkages and aggregate in cells. The accumulation of this oxidative junk can jeopardize the cellular function and fate. However, oxidative damage to proteins renders them susceptible to degradation by increasing the hydrophobicity of the surface of proteins (376), which could serve as a tag for the proteasome system, and lead to degradation (375). This process does not consume energy and is done without ubiquitination (366). It has been shown that proteolytic susceptibility of proteins in relation to oxidative modifications shows a biphasic response, while at moderate oxidant concentrations, proteolytic susceptibility increases, and further oxidation leads to a decline in the proteolytic susceptibility, sometimes even below the basal degradation. Such behavior seems to be a common feature of all globular, soluble proteins with defined secondary and tertiary structures, independent of the origin of the proteins (49, 118, 170, 411).
The feature that carbonylated proteins are targeted for removal by proteasomes suggests that carbonylation may act as a signal ensuring that damaged proteins enter the degradation pathway rather than the chaperone/repair pathway, since carbonylation is an irreversible/unrepairable modification (264). Increased levels of protein carbonyls are linked to a variety of age-associated diseases and often correlate with the progress of disorders and diseases (375). Carbonyl derivatives are formed by a direct metal-catalyzed oxidative attack on the amino acid side chains of arginine, lysine, proline, and threonine. Moreover, carbonyl derivatives on cysteine, histidine, and lysine can be also generated by secondary reactions with reactive carbonyl compounds on sugars, lipids, and advanced glycation/lipoxidation end products (375). Methionine and cysteine residues are even more prone to oxidation than others due to the sulfur content of these residues. The oxidative modifications of these two residues are reversible, indicating a redox-dependent signaling of methionine and cysteine residues. Cysteine residues are converted to disulfide by oxidation and reduced back to cysteine under mild conditions such as in the presence of reducing agents such as NADH without enzyme reaction. Methionine residues are transformed to methionine sulfoxide, which in turn can be repaired by methionine sulfoxidereductases, which catalyze the reduction of methionine sulfoxide back to methionine residues (181). The role of methionine reductase in exercise studies remains to be evaluated. On the other hand, carbonylation, which is an irreversible modification, is very well studied in exercise studies. An acute bout of intensive exercise increases the rate and accumulation of carbonyl derivatives (36, 258, 316), and regular exercise either maintains or decreases carbonyl levels (45, 68, 114, 318). Increased activity of the proteasome system is important for protein remodeling and, of course, to prevent the accumulation of damaged proteins that could impair cell function. The effects of exercise on the proteasome system and carbonyl accumulation could be dependent on whether the exercise is acute or chronic and also the type of exercise (resistance or endurance) (230, 246, 293, 369). Mitochondria, the main site of ROS production, need a very efficient system to prevent the accumulation of oxidized proteins. The main quality control of mitochondrial proteins is mediated by Lon protease and HSP78 (42, 43, 253, 337, 411). Indeed, it has been shown that Lon expression can be readily induced by a moderate level of oxidative stress, which prevents the accumulation of oxidized proteins and protects cell viability (254). Ischemia, for instance, significantly induced the expression of Lon protease, indicating that endoplasmic reticulum-related stress could have an impact on mitochondrial biogenesis and the degradation of mitochondrial proteins (146). Moreover, it has been shown that silencing of Lon protease resulted in hepatic insulin resistance, and the restoration of Lon activity normalized insulin handling in the liver (199). Thus, Lon can be regarded as a stress protein (253).
Lon levels decline with age, which could result in the accumulation of oxidized, or dysfunctional proteins in the mitochondria (42, 43, 196), and perhaps in the loss of mitochondrial function. We have recently observed that regular exercise can prevent the age-related decline in Lon protease (187), and therefore rejuvenate proper quality control of proteins in older cells (411). We have also shown that exercise training induced HSP78 levels in both young and old animals (187), which indicates that regular exercise has beneficial effects on mitochondrial protein quality control.
8-oxoG is the most often measured form of oxidative damage to DNA and has been reported to increase with aging, ischemia/reperfusion, radiation, and a wide range of pathological disorders (37, 248, 260, 378). Human 8-oxoguanine-DNA glycosylase (OGG1) plays a major role in the base excision repair pathway by removing 8-oxoguanine base lesions generated by ROS. OGG1 activities are primarily regulated by p300/CBP-mediated acetylation, predominantly on Lys338/Lys341 (32, 148, 388). An increased deacetylase activity of sirtuins may lead to decreases in acetylation levels of proteins, which, in turn, would result in a decline in enzymatic activity of OGG1. We have recently shown that aging increased 8-oxoG content in the hippocampus, with increased levels of OGG1. However, the acetylation of OGG1 was very low in the aged animals (189). Physical activity, on the other hand, increases the acetylation of OGG1 and decreases the age-associated accumulation of OGG1 in human skeletal muscle (319). Indeed, we and others have shown that acute and regular exercise increases the activity of OGG1 (249, 303, 306, 313, 314, 348). Therefore, the upregulated DNA repair could be an important tool by which regular exercise maintains DNA purity and could curb the incidence of certain cancers (333).
It is highly possible that evolution of the biological system has selected guanine as the potential target of ROS. As a result of low redox potential, attractiveness to ROS is apparent and makes it the most oxidizable nucleic acid base, and this finding suggests that the generation of 8-oxoG could have a significant physiological role (307, 377), as other markers of oxidative damage, such as malondialdyde or protein carbonyls 8-oxoG, are always detectable in cells. An interesting observation made on OGG1 knockout animals is that lipopolysaccharide-induced inflammation was more tolerated than in the wild types (221). The lack of OGG1, hence, resulted in increased levels of 8-oxoG, and reduced removal of this damaged base, which actually provided better protection against inflammation. Thus, the inverse correlation between the activity of OGG1 in this inflammation model suggests that certain amounts of 8-oxoG might be more tolerable to the cells than the excised form that generates inflammation (221). It was recently suggested that oxidation of guanine could be important in the transformation of heterochromatin to euchromatin (307). In addition, it has been demonstrated that exposure of cells to estrogen increases 8-oxoG levels in DNA and recruits OGG1 and topoisomerase IIβ to estrogen-responsive DNA elements in the promoter region of 17β-estradiol (E2)-responsive genes (282). Therefore, 8-oxoG, besides its damaging affect on transversion base pairs, could be important for the generation of signals that are necessary for specific gene transcription. Indeed, it has been shown that the 8-oxoG-OGG1 complex can interact with Ras proteins and work as a guanine nuclear exchange factor (38). In addition, it could be very important to redox signaling that the 8-oxoG-OGG1 complex-mediated Ras activation results in phosphorylation of the mitogen-activated kinases MEK1,2/ERK1,2 and increasing downstream gene expression. This novel information on the interaction of free 8-oxoG, but not other oxidized bases such as 8-oxodG, FapyG, or 8-oxoA, with OGG1 leads to redox signaling and could partly explain why OGG1 knockout mice are more resistant to inflammation than wild types (221).
Data from exercise studies suggest that a moderate level of oxidative stress, which could slightly increase the levels of certain oxidative damage markers, might not actually jeopardize cell function, but rather could be necessary to initiate an adaptive response (Fig. 7). Physical exercise is a natural stimulator of ROS production and a mild oxidative stressor, as demonstrated by a moderate level of oxidative damage, which can lead to an enhanced physiological function.
IV. ROS, Metabolism, and Exercise
A. Sirtuins as redox-sensitive energy sensors of exercise
Nicotinamide adenine dinucleotide (NAD), with the ability to carry two electron equivalents, serves as an energy source, and at the same time, the cytoplasmic NAD/NADH ratio is a sensitive marker of the cellular redox state (235, 432, 441). NAD-dependent lysine deacetylases, sirtuins, control various cellular processes, including chromatin structure by the deacetylation of lysine residues of histones (271, 396), apoptosis and cell survival, by the interaction with p53 and FOXO transcription factors (233, 354, 444), mitochondrial biogenesis via deacetylation of PGC1a (252), lipid metabolism by the interaction with SREBP-1c among other proteins (291, 352, 390), insulin homeostasis (98, 205), DNA repair (171, 224, 231), inflammation by modulating the activity of NF-κB (344, 439), and oxygen sensing by deacetylating hypoxia-inducible factor-1a (HIF-1α) and HIF2α (66, 82, 210) among other mechanisms. Sirtuin-mediated deacetylation of these transcription factors would readily effect the expression of pro- and antioxidant genes, such as PUMA, NOXA, PIG3, GADD45, Nrf2, and Mn-SOD, and stress-activated protein kinases etc., which can readily regulate redox signaling. Besides the interaction between SIRT1 and poly(ADP-ribose) polymerase (PARP) to control metabolism (22), the latter is implicated in apoptosis (88), DNA repair (134), ischemia/reperfusion (403), diabetes (387), and inflammation (386).
Sirtuins (silent information regulator 2 [Sir2] proteins) belong to an ancient family of evolutionary-conserved NAD+-dependent enzymes with deacetylase and/or mono-ADP-ribosyltransferase activity. Seven Sir2 homologs, sirtuins (SIRT) 1 to 7, have been identified in mammals. The crystal structure of human SIRT1, which is a homolog of yeast Sir2, revealed a large groove that was intersected by a pocket lined with hydrophobic residues conserved with class-specific protein-binding sites of each Sir2 class (92). Enzymes from the sirtuin family are ubiquitously distributed. However, their level/activity has organ specificity. SIRT 3–5 are predominantly localized in the mitochondria.
The NAD/NADH ratio is changing readily during muscle contraction, due to the reduction of NAD to NADH that is catalyzed by glyceraldehyde 3-phosphate dehydrogenase and the conversion of NADH to NAD by the glycerol phosphate shuttle and lactate dehydrogenase (LDH). LDH facilitates the pyruvate uptake of mitochondria (52) that further influences the NAD/NADH ratio, and one can expect decreased SIRT1 activity right after intensive exercise bouts, due to the increased lactate/pyruvate ratio, which, in turn, decreases the NAD/NADH ratio. However, exercise bouts increase the activity of SIRT1 (188, 308, 383), indicating that within a certain range, SIRT1 activity is independent from the NAD/NADH ratio (9). On the other hand, significant oxidative stress could down-regulate SIRT1 (7).
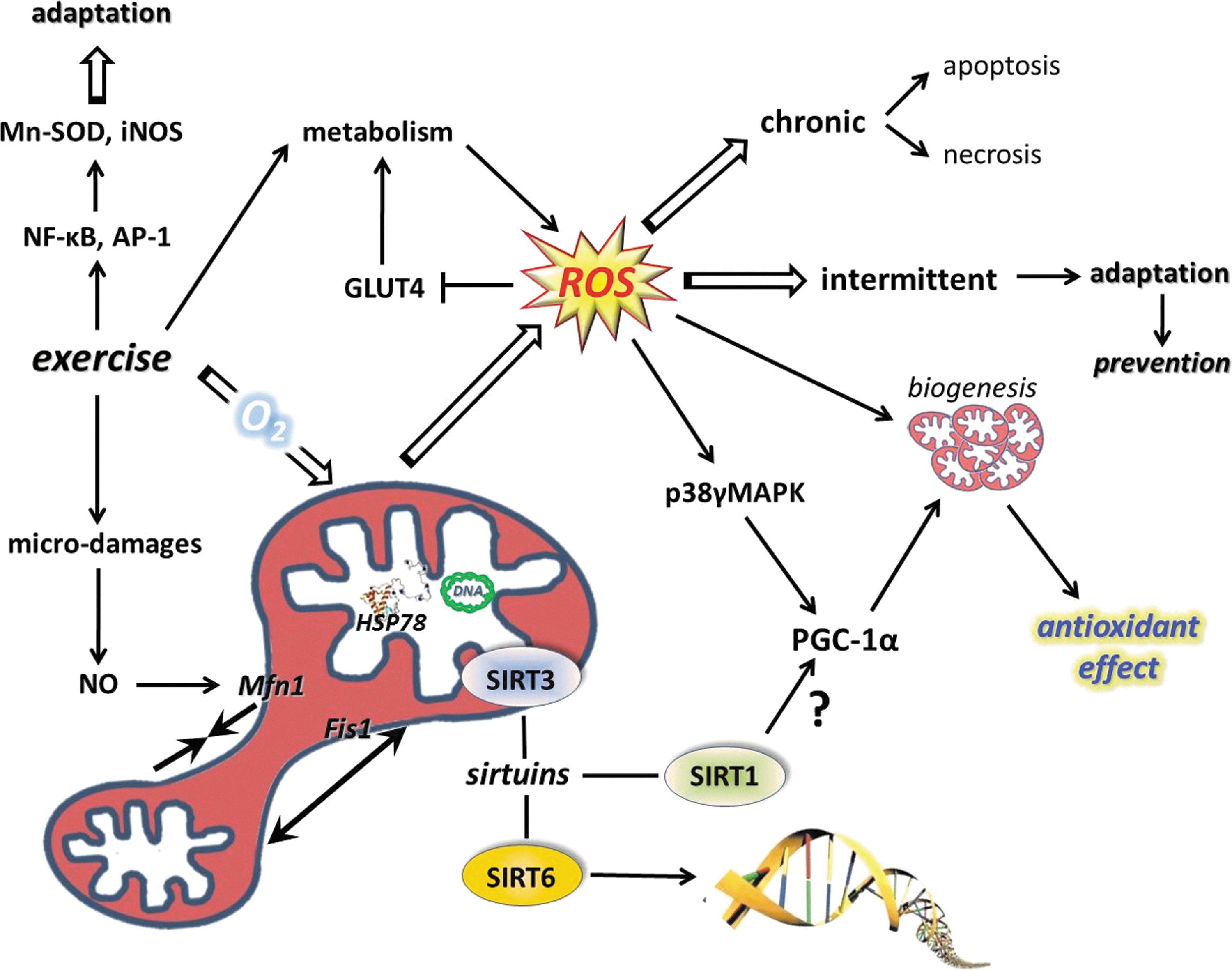
Sirtuins, especially SIRT1, are implicated in the aging process of different organisms (119, 391), and it has been shown that changes in SIRT1 activity by agents, such as caloric restriction (CR) or resveratrol or SRT501, increase lifespan via a wide range of processes, including suppressed apoptosis, and inflammation, or enhanced DNA repair (2, 292). Although, the beneficial effects of sirtuin activators on mammals are still unclear, some data indicate that sirtuins do play both protective and proaging roles (206). Accumulating evidence suggests that exercise alters the activity, content, and expression of different sirtuins (Fig. 8). Indeed, there are reports that have demonstrated an exercise-associated oxidative challenge that results in increased expression, content, and activity of SIRT1 in humans (73, 225, 308). It seems that besides AMPK, SIRT1 is a sensitive metabolic sensor in skeletal muscle (96), and the fact that histone lysine residues are one of the targets of SIRT1 implies that the redox-sensitive energy sensor SIRT1 links energy metabolism to transcriptional regulation. In addition to this, SIRT1 and p300/CBP have been shown to control myogenic transcription factor, which is associated with dynamic changes to the NAD/NADH ratio (99), and points out the role of redox signaling via SIRT1 in the development of skeletal muscle. SIRT1 is downstream in ROS signaling, but can be importantly upstream in regulating cellular levels such as activation of FOXO3 (164), muscle ring-finger protein 1 (MuRF1) (8), and protein kinase B (Akt) (382). Therefore, these NAD-dependent metabolic sensors are regulating redox signaling in a wide array (222).

Aging decreases the activity of SIRT1, which results in increased levels of lysine acetylation in the skeletal muscle and brain of rats (188, 189, 227). The effects of acetylation/deacetylation for the function of proteins have been extensively investigated in a number of laboratories. However, the mechanism is still not well understood. It has been shown that acetylation of lysine residues could affect ubiquitination of the same residue, and therefore could impact the stability of proteins. Mechanistically, larger degrees of acetylation, as we have observed with aging, could slow down ubiquitin-mediated degradation resulting in enhanced half-life and then larger accumulation of carbonylation, which could lead to an impaired physiological function. This hypothesis seems to be very attractive and concurs with some well-observed phenomena, such as age-associated increases in the half-life of proteins (116), decreased rates of protein degradation with aging (115), and impaired function as a result of aging. Exercise, depending on the intensity and duration, on the other hand, increases the activity of SIRT1, decreases the level of lysine acetylation, increases the turnover rate of proteins, decreases the accumulation of carbonyl groups, and improves cellular function (Fig. 9). It is important to note that the effects of a single bout of exercise and/or regular exercise could be completely opposite to oxidative stress, ubiquitination, and acetylation. The findings from a recent report indicate that aging does not significantly alter the expression or protein content of MuRF1, while patients with chronic heart failure showed increased expression and content of MuRF1, which was downregulated by 4wk of exercise training (107). Although this study did not examine the levels of sirtuins, it is clear that exercise training on healthy subjects and on patients with diseases would have different effects on the rate of ubiquitination and degradation. The stabilization of HIF-1α, in cardiac muscle during diseases, in the short term could be beneficial, but detrimental in the long term (144). Acetylation on protein stability varies significantly, and HIF-1α, for example, is ubiquitinated at normoxic conditions and degraded by proteasome (200), and acetylation of HIF-1α improves the interaction between ubiquitin ligase, and hence prepare protein for degradation. On the other hand, during hypoxia, SIRT-dependent deacetylation of HIF1α stabilizes the protein (200, 210).The fact that caloric restriction and physical exercise increase the activity of SIRT1 and attenuate the age-associated increase in lysine acetylation is intriguing, and needs further investigation. It is clear that the effects of lysine acetylation/deacetylation, in relation to SIRT1 and protein stability, result in a number of interesting findings.

SIRT2 is mostly localized in neurons and nerves, and preferentially deacetylates tubulin and histone H4. Moreover, a recent finding suggests that SIRT2 is activating myelin formation in Schwann cells, thus controlling an essential polarity pathway during myelin assembly (25). The effects of exercise on SIRT2 are generally unknown.
Another isoform of the sirtuin family, SIRT6, is closely associated with chromatin and increases the resistance of DNA to oxidative DNA damage and facilitates base-excision repair (239). However, recent data suggest that SIRT6, besides its crucial role in chromatin structure, is a regulator of carbohydrate metabolism. SIRT6-deficient cells show nutrient-replete conditions and shift to lactic acid glycolysis, and transfer metabolism to the survival mode from normal aerobic conditions in the presence of oxygen (445). SIRT6 deficiency activates the gene of pyruvate dehydrogenase kinase and inhibits pyruvate dehydrogenase, resulting in impairment of pyruvate conversion to acetyl-CoA to fuel the TCA cycle (445). In accordance with this, we have observed decreased levels of SIRT6, after a single bout of exercise, in the skeletal muscle of young and old individuals (308). The expression of SIRT6 was not affected by aging in human skeletal muscle, but rat skeletal muscle increased the levels of SIRT6 protein, while regular exercise training attenuated this increase, suggesting that the age-associated changes in metabolism and/or in chromatin structure are normalized by an exercise-driven adaptation (188). SIRT6 appears to be differently regulated by exercise than SIRT1 (188), which could be due to the chromatin tightness, controlling the role of SIRT6. A single bout of exercise results in large metabolic and oxidative challenges to skeletal muscle, which, to regulate an adaptive response, must open chromatin to allow gene expression to cope with the exercise stressor (308). The downregulation of SIRT6 is in accordance with this hypothesis.
SIRT7 is the only mammalian sirtuin that is mainly localized in the nucleoli. When the nucleoli disintegrate, SIRT7 associates with condensed chromosomes (232). The molecular targets of SIRT7 have not been identified, but data obtained from SIRT7 knockout mice reveal that SIRT7 deficiency leads to progressive heart hypertrophy, accompanied by inflammation and decreased stress resistance (412). At least a part of this phenotype may be explained by the fact that SIRT7 deacetylates p53, leading to hyperacetylation of p53 in vivo along with various other changes in the signaling network of cardiomyocytes (412). The effect of exercise on SIRT7 remains to be explored.
Mitochondria, the powerhouse of ATP production, also host sirtuins. One is SIRT3, which could be present in the nuclei and mitochondria (351), and it has been suggested to control metabolism and oxidative stress of mitochondria (29). Overexpression of this enzyme positively regulates the oxidative capacity of mitochondria and attenuates the generation of ROS (190), indicating that SIRT3 plays a key function in redox regulation.
Moreover, it appears that the activation of Mn-SOD is dependent on SIRT3- (302) mediated deacetylation of 122 lysine residues (393). In addition, SIRT3 has a powerful deacetylase activity on histone H4 peptides and is a major regulator of mitochondrial deacetylation (216), affecting fatty acid oxidation (137) via the deacetylation of mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 (364). SIRT3 activates isocitrate dehydrogenase 2 and glutamate dehydrogenase by deacetylation, and these enzymes are involved in the production of mitochondrial NADPH. The elevated NADPH, in turn, is necessary for GSH reductase, which reduces GSSG to GSH, the cofactor necessary for mitochondrial GPX function (29).
We have found that aging increases SIRT3 levels in the rat hippocampus, and is attenuated by exercise training (188). On the other hand, exercise training and diet have been shown to increase the levels of SIRT3 (141, 274) in skeletal muscle and AMP-activated protein kinase (AMPK) as well as cAMP-response element-binding protein (CREB), indicating upstream modulators of SIRT3 (274). These data suggest that SIRT3 is a potent redox-dependent modulator of cellular metabolism and is readily modified by physical exercise.
SIRT4 is also localized in the mitochondria and has been implicated in the regulation of insulin secretion in beta-cells. Knockdown of SIRT4 increased fat oxidation in the liver and skeletal muscle (251), indicating that SIRT4, without significant deacetylase, but with high ADP-ribosyltransferase activity, also modulates cellular metabolism. The effects of exercise on SIRT4 are poorly investigated, but it appears that a single bout of exercise significantly decreases the mRNA expression of this enzymes in peripheral blood mononuclear cells (225).
SIRT5 is hosted by the mitochondria, and recent reports of this poorly studied sirtuin have revealed that SIRT5 is involved in the detoxification of ammonia by deacetylating carbamoyl phosphate synthetase 1, which is one of the key enzymes in the urea cycle, which catalyzes condensation of ammonia with bicarbonate to form carbamoyl phosphate (267). Ammonia is readily elevated during intensive exercise through deamination of AMP and branched-chain amino acids. Therefore, it is very likely that SIRT5 is modulated by exercise, and the adaptive response to regular exercise training involves SIRT5. Moreover, a recent report suggests that SIRT5, besides its deacetylase activity, also shows protein lysine desuccinylase demalonylase activity in the in vitro condition, implying that this post-translational modification might have a physiological role, controlled by redox signaling (86).
The available information strongly suggests that NAD-dependent sirtuins are very sensitive to metabolic challenges, and are modulated by redox signaling. Therefore, sirtuins are readily modified by physical exercise, and data suggest so far that sirtuins could play an important role in exercise-induced adaptations, including physical fitness, health promotion, and the attenuation of the progress of aging.
B. Redox signaling and biogenesis of mitochondria
It is clear that metabolic stress (in particular through AMPK/PGC-1α-dependent signaling processes) is one of the well-characterized ways to increase mitochondrial mass in skeletal muscle (123). However, it is also well characterized that increased metabolism is associated with changes in redox regulation (62). Therefore, oxidative stress has been suggested to be one of the causative signals of mitochondrial biogenesis (77), which is an important mechanism to reduce the exercise-induced leak of ROS from the mitochondrial network. For the same ATP production, more mitochondria can work at a lower respiratory capacity; hence, less ROS are produced, and therefore mitochondrial biogenesis can be regarded as part of the antioxidant system. Proteomic analyses yielded over 900 distinct mitochondrial proteins in human muscle and all, except the 13 encoded by mitochondrial DNA, are imprinted on nuclear DNA (273). In skeletal muscle fibers, the mass of mitochondria is dependent on the muscle subtype. The numbers and functions of mitochondria required to maintain fiber homeostasis are determined by regulatory circuits/programs that are under synchronized regulation between the mitochondrial and nuclear genes, to maintain function of respiratory electron transport complexes and proteins that are required for physiological functions of mitochondrial networks, including oxidative phosphorylation, ionic homeostasis, and mitochondrial uncoupling (thermogenesis).
In addition, skeletal muscle is equipped with two types of mitochondria: intermyofibrillar (IMF) and subsarcolemmal (SS). Although the location of mitochondria could be related to the level of ATP production, significant differences were not noted after oxidative stress or exercise training in the subpopulations (64, 65, 169). The findings of a recent study revealed that SIRT3 is present in IMF and SS, and chronic stimulation increased the content of IMF, SS, and SIRT3 in a parallel manner (122).
Mitochondrial gene expression is regulated by mitochondrial transcription factor A (TFAM), while the gene expression of nuclear DNA-encoded mitochondrial proteins is controlled by respiratory factors 1 and 2 (NRF1 and NRF2) (371). The coactivation of the transcription factors to promoter regions is carried out by PGC-1α, which is the master regulator of mitochondrial biogenesis (380). Overexpression of PGC-1α leads to a large increase in mitochondrial biogenesis, cytochrome oxidase II and cytochrome oxidase IV content, ATP synthase, myoglobin, and citrate synthase activity (212). Moreover, PGC-1α appears to have the capacity to increase mitochondrial electron transport activity while stimulating a broad range of anti-ROS mechanisms, including regulation of the expressions of Cu,Zn-SOD, GPX, catalase, uncoupling protein 2 (UCP2), and UCP3 (374). Indeed, knockout of PGC-1α leads to significant decreases in Cu,Zn-SOD, Mn-SOD, and GPX content, compared to those found in wild types of mice (202). Moreover, studies on PGC-1α knockout fibroblasts revealed significantly attenuated responses to H2O2 treatment by Mn-SOD, catalase, and GPX (374). Moreover, transgenic mice with overexpression of PGC-1α showed lower levels of loss in muscle mass as a result of aging, and the rate of oxidative damage was also attenuated, indicating that the exercise-associated upregulation of this transcription factor could cause similar health-promoting effects (431).
These observations confirm that PGC-1α actively is involved in antioxidant defense. Inflammation is associated with increased ROS production and oxidative damage, and the antioxidant role of PGC-1α is further supported by the anti-inflammatory effects of this coactivator. It has been shown that PGC-1α knockout mice, besides reduced endurance activity, exhibit increased levels of inflammation (129). HeLa cells were treated with ethidium bromide to deplete mitochondrial DNA, which resulted in increased oxidative stress and induction of TFAM and NRF-1, suggesting that ROS could be an important signal for mitochondrial biogenesis (236). Homocysteine-induced ROS production, which resulted in an increased expression of NRF-1 and TFAM, was attenuated by antioxidant treatments, again supporting the importance of ROS in mitochondrial biogenesis (281). Aging results in a chronic redox shift toward an oxidized milieu, which, in a significant way, can be attenuated by regular physical activity affecting a number of players in the mitochondrial biogenesis. Indeed, it seems that PGC-1α, TFAM, and NRF1 are all sensitive to redox balance and induced by ROS, leading to increased mitochondrial biogenesis and readily modified by exercise bouts (Fig. 10). Exercise resulted in increased ATP and ROS production by the mitochondria of skeletal muscle of subjects with normal glucose tolerance and impaired glucose tolerance by the induction of TFAM and NRF1 (106). Moreover, it was observed that aging and impaired glucose tolerance resulted in decreased ATP production and maintained levels of ROS (106).
Most of the protein components of the mitochondria are encoded by nuclear DNA, including RNAs, and are important to mitochondrial biogenesis. Recent data have revealed that mammalian polynucleotide phosphorylase (PNPase) is localized in the mitochondrial intermembrane space to regulate the translocation of RNA into mitochondria (425). Silencing the PNPase with RNA interference results in decreases in mitochondrial membrane potential, ATP synthesis, and increased production of lactic acid (63). PNPase-deficient cells accumulate 8-oxoG in cellular RNA, indicating that PNPase is involved in the antioxidant/damage repair process (436). Indeed, PNPase is bound in a higher degree to oxidized RNA (436). Overexpression of PNPase could also be harmful to cells, since it can cause increased production of ROS and inflammation (347). Thus, both up- and downregulation of PNPase could impair cell function (133). The available information on the effects of exercise on PNPase is very limited. Our unpublished observations suggest that aging decreases the mRNA levels of PNPase in human skeletal muscle, while exercise decreases it in young and increases the expression in old subjects (41). On the other hand, in skeletal muscle of rats, we have observed increased amounts of PNPase with aging, and this was attenuated with exercise training (187). These data suggest that the relationship between exercise and PNPase is very complex.
SIRT1 has been proposed to be one of the activators of PGC-1α during exercise-induced mitochondrial biogenesis, although a recent report showed no interaction between SIRT1 and PGC-1α (284). Moreover, besides acetylation, other post-translational modifications such as phosphorylation appear to represent an immediate mechanism to activate PGC-1α- and initiate PGC-1α-dependent gene expression (67). The results of a study with human subjects has revealed that NAC supplementation blocked exercise-associated activation of JNK, but the mRNA level of PGC-1α was not affected by the supplementation of this antioxidant (283). Moreover, it is not clear at the moment whether PGC-1α phosphorylation is significantly affected by a redox milieu.
GLUT4, the insulin-dependent carbohydrate transporter, can be modulated by ROS (136), which means a shift in the redox state toward an oxidative milieu suppresses carbohydrate metabolism and increases insulin resistance (48, 430). During exercise, the need for carbohydrate enhances carbohydrate uptake, but during rest, the upregulated antioxidant system provides excellent conditions for the insulin-dependent carbohydrate uptake (213) with increased concentrations of the GLUT4 transporter.
The regulatory role of ROS in mitochondrial biogenesis was suggested many years ago, and a significant body of reference material has accumulated supporting this idea. Moderate levels of ROS appear to stimulate mitochondrial biogenesis via PGC-1α, SIRT1, and TFAM, while the increased levels of mitochondria decrease ROS production, in addition to the antioxidant role of PGC-1α and PNPase.
V. Exercise-Induced Inflammation and ROS
Inflammation is a protective process that is necessary for healing, damage repair, and fighting foreign bodies. Chronic inflammation is generally associated with increased levels of ROS and proinflammatory cytokines. Skeletal muscle is a powerful generator of some myokines, especially interleukin-6 (IL-6). The observation that Vitamin C and E administration decreases the exercise-induced generation of IL-6 suggests that ROS are stimulators of IL-6 production in skeletal muscle (94). Recently, it has been suggested that IL-6 acts as an energy sensor, exhibiting local and endocrine metabolic effects (278, 279). IL-6 and other cytokines such as IL-8, cyclooxygenase2, and TNF-α are significantly activated after unaccustomed muscle contractions, which cause microdamage to sarcomeres (54, 208). The repair of the damage by satellite cells is crucial, as these cells divide and fuse to form myotubes. The differentiation of myotubes to myonuclei then can be inhibited by NF-κB. Indeed, NF-κB has been suggested to be a regulator of muscle differentiation, because the RelA/p65 heterodimer complex can interact with the transcriptional regulator of YinYan, leading to inhibition of myogenesis (426). In addition, TNF-α, which can activate NF-κB, has been shown to cause proliferation of myoblasts, but to inhibit differentiation (95). Indeed, TNF-plus-interferon-gamma-induced activation of NF-κB resulted in significant losses in MyoD mRNA levels in C2C12 myocytes and inhibited differentiation (126). On the other hand, Li and Schwartz (207) demonstrated on C2C12 myoblasts that TNF-α mediated signaling to NF-κB, and serum response factor is important in the early phase of differentiation, indicating that TNF-α- NF-κB signaling on myoblast differentiation and on satellite cell-mediated repair of sarcomeres is dose dependent.
It has been shown that repeated bouts of exercise cause activation of NF-κB and activator protein-1 (AP-1), which might be important for exercise-mediated adaptation (53). The redox-dependent activation of NF-κB and AP-1 has been extensively reviewed (78, 157, 204), and here we briefly focus on the exercise-related mechanism. A single bout of exercise has been shown to activate DNA binding of NF-κB and AP-1, which facilitates the mRNA expression of Mn-SOD (142, 161). Allopurinol-mediated inhibition of XO decreases NF-κB activation, which has been suggested to attenuate the adaptive response to exercise training in skeletal muscle (110). On the other hand, it appears that regular exercise attenuates the age-associated increase in NF-κB activity at least in the liver (311). The findings from human lymphocytes and skeletal muscle of patients with chronic heart failure support the exercise-related activation of NF-κB (3, 76, 420). It seems to be well accepted that activation of the NF-κB-signaling cascade is important to the gene expression Mn-SOD and inducible nitric oxide synthase (iNOS) (162). Table 1 shows the list of some of the key ROS-dependent transcription factors.
T, transcription factor; B, biological function; E, exercise response; ROS, reactive oxygen species; VO2max, maximal oxygen uptake; ↑, increase.
NO, mediated in redox signaling, is out of the scope of the present article, since it deserves an independent review, based on the importance of this signaling molecule. Because the NO-dependent damage repair of skeletal muscle is often overlooked, we will briefly summarize the current understanding of this phenomenon.
Anderson (13) made an interesting observation on the role of NO on satellite cells in skeletal muscle. After mechanical damage of NOS-I knockout or NO inhibited mice, very different repair processes were noted, and NO ablation or inhibition significantly attenuated the repair. It was evident that NO facilitates the activation of satellite cells, which are located in the basal lamina of skeletal muscle and are necessary for repair (13). The beneficial function of NO in damage repair is restricted not only to satellite cell proliferation and differentiation but also to fusion. Moreover, NO and cGMP activate follistatin, which is the antagonist of myostatin (287). NO-driven follistatin activation could be an important anabolic signal that is necessary for remodeling and muscle hypertrophy as well. Interestingly, treadmill running-related overuse of tendon results in increased NO production, which has been suggested to play a role in the repair process (389). The induction of mechanical damage to gastrocnemius muscle resulted in increased NO formation and is believed to initiate the signaling process to damage repair (178).
The muscle contraction-associated increase in IL-6 levels is, at least in part, dependent on ROS concentration, and is a necessary signal for metabolic processes and inflammation. NF-κB and AP-1 are redox-sensitive transcription factors that control inflammation, which in a chronic phase is associated with increased ROS production, but also is involved in antioxidant defense by the regulation of Mn-SOD. Through the activation of satellite cell proliferation, NO is important in damage repair/remodeling in the skeletal muscle, which might be important in delayed muscle soreness (315).
In addition to the role of NO in the repair of skeletal muscle, it should be mentioned that a great deal of exercise-related adaptation is mediated by NO in the vascular system, especially in the endothelium. Due to shear stress, NO seems to be the mechanism responsible for the beneficial impact of exercise training on vascular function and provides a potent physiological stimulus to adaptation in endothelial function and vascular remodeling (183). Exercise stimuli, which result in activation of endothelial nitric oxide synthase (eNOS) and related agonists, such as bradykinin and acetylcholine, significantly increase blood flow causing vasodilation (404). The half-life of NO is relatively short in the circulation, around 0.1 s, (176) and it is rapidly oxidized to nitrate and nitrite. However, alternative pathways can also be present to generate bioactive NO from nitrate and nitrite (10) and are involved in a wide range of processes, such as cellular signaling, regulation of blood flow, cellular metabolism, and hypoxic response (218). Reaction of NO to superoxide generates the highly toxic peroxinitrite that could readily cause DNA damage and impair the activity of DNA repair enzymes, including OGG1 (156). Despite the exercise-induced increase in eNOS and neuronal nitric oxide synthase (nNOS) protein content in skeletal muscle (417), it appears that exercise does not significantly increase the formation of peroxinitrite, since moderate levels of exercise increase OGG1 activity (306).
VI. The Crucial Role of Redox Regulation on New Cell Formation in Skeletal Muscle and Brain
Skeletal muscle cells and neurons are nondividing cells, but it is clear that stem cells after differentiation can lead to new cell formation even in postmitotic tissues.
In skeletal muscle, resident adult myogenic precursor cells referred to as satellite cells are mitotically quiescent, but can be readily activated by a single bout of exercise (59) and a moderate dose of H2O2 supplementation (237). Satellite cells can be divided into low-proliferative and high-proliferative subpopulations (321). It has been shown that the proliferating capacity of these satellite cells is dependent on mitochondrial membrane potential, ATP balance, and ROS generation (336), and high-proliferating capacity favors an oxidized cellular milieu. On the other hand, when satellite cell cultures were challenged by relatively high H2O2 concentrations (1 mM), it turned out that the proliferation reduced significantly, while the viability of the cells did not change to any great extent (327). Aging seems to impair the proliferating capacity of satellite cells and is associated with reduced antioxidant capacity and increased Ca2+ levels (100). The repair of skeletal muscle after injury involves high-mobility group box 1, which is an intracellular protein that translocates to the nucleus to bind to DNA and activates gene expression and stem cells. It appears that a reduced cellular milieu may be important to maintain the bioactivity of high-mobility group box 1, since oxidation abolishes the muscle stem cell migration to the response of high-mobility group box 1 (419). The results of a recent study revealed that angiopoietin-2, which is a powerful factor of angiogenesis, is present in precursor cells of skeletal muscle, and is induced by H2O2 in primary cell culture (237). As described earlier, redox-sensitive TNF-α and NF-κB could impair differentiation of myoblasts to myotubes, and a recent study suggests that IL-1α, along with TNF-α, also has the capability to act upon transforming growth factor-β-activated kinase-1 (406). The signaling pathway of these cytokines is a distinct route that involves the p38α-mediated recruitment of polycomb-repressive complex 2 to the Pax7 promoter in satellite cells (242). The effects of cytokines released upon muscle damage are dependent of the extent of dose, and it is clear that at low concentrations, they stimulate repair, while in large doses, the adverse effects are more pronounced (243).
These data are important to understand why and how precursor cells in the skeletal muscle can be involved in redox-associated angiogenesis, which is important to exercise-induced adaptation and/or damage repair. The available information on satellite cells suggest that redox homeostasis is an important regulatory factor of proliferation; although the results are not unequivocal, it can be suggested that a fine tuning of satellite cells by ROS, depending on the dose, can cause activation or inhibition.
Besides satellite cells, neuronal precursor cells are also sensitive to redox milieu. It is well established that neuronal precursor cells in the dentate gyrus are able to proliferate throughout life and differentiate, and their progenity can lead to neurogenesis (89). Observations suggest that progenitor cells can readily respond to changes in energy homeostasis. Therefore, ischemia/reperfusion, aging, and metabolic pathology or even physical exercise can change the rate of neurogenesis. Indeed, precursor cells exhibit high mitotic and multipotentiality potential, and ROS are an important signals that control their ability to divide and differentiate (211). One of the reasons for this is that precursor cells are very sensitive to oxygen levels that are suggested to be around 2% in the brain (368). Lowering the level of oxygen concentration by transient middle cerebral artery occlusion on the rat brain leads to increases in neurogenesis (17). It has been shown that neuronal precursor cells exhibit about four-times higher ROS levels than other cell types, and the concentration of ROS, which is dependent on the density of precursor cells, is associated with the rate of proliferation (211). The fine redox tuning, therefore, is a necessary modulator of the proliferation of neuronal progenitor cells, and the bell-shaped dose–response represents the relation of ROS and neurogenesis (Fig. 11) (262).
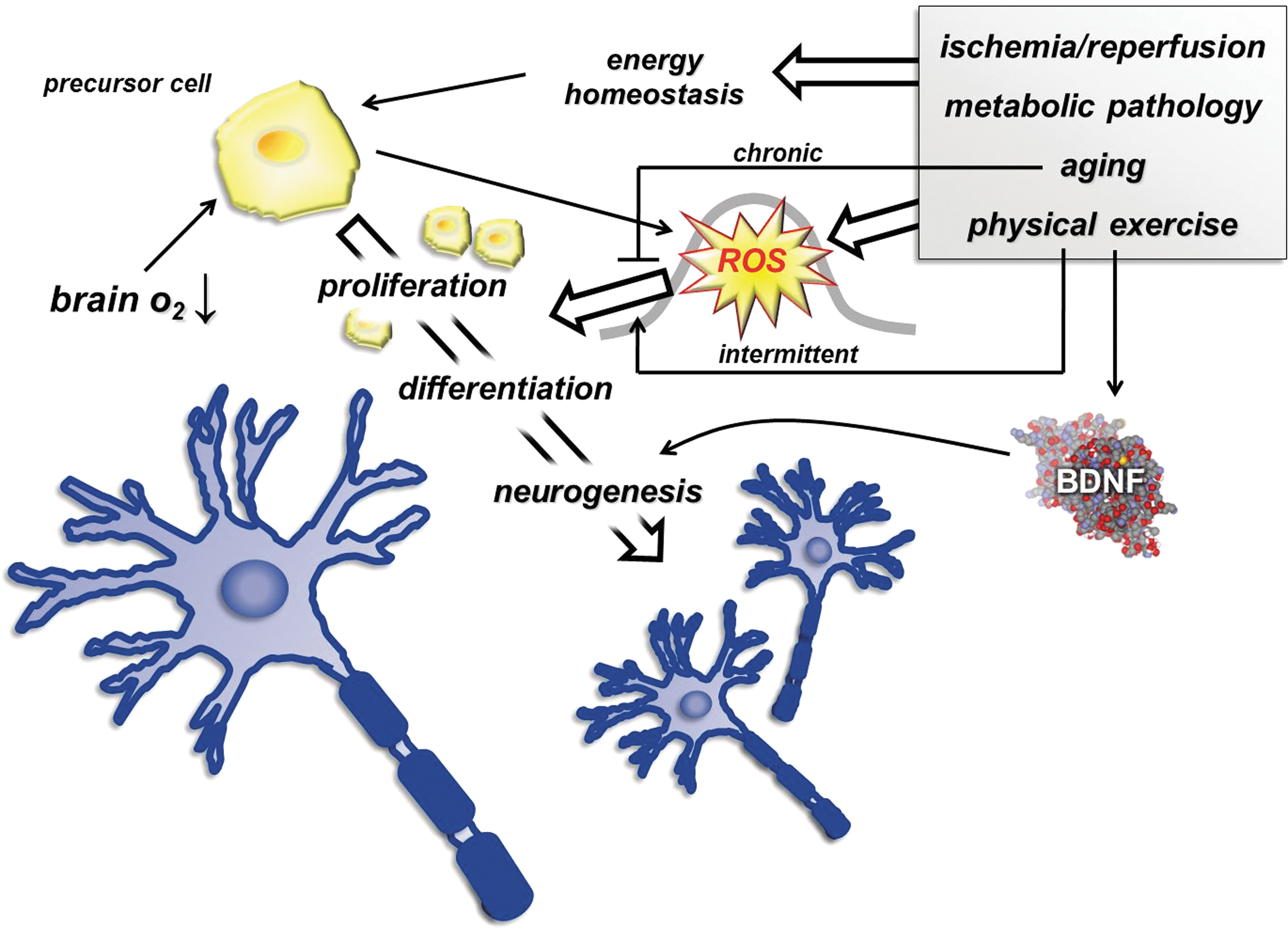
The original article that reported that exercise results in neurogenesis has been confirmed by a number of articles that have demonstrated that the newly formed neurons are functional (413, 414). A direct relationship between ROS and exercise-induced neurogenesis has not been reported, but in our recent study, we have observed an age-related decline in neurogenesis, which was associated with increased levels of 8-oxoG (189). The age-related attenuation of neurogenesis has been suggested to be due to blood-borne factors (421), and hence the age-dependent chronic oxidative stress has not been ruled out as a negative regulator. The question whether the exercise-induced neurogenesis is ROS dependent needs to be addressed. Brain-derived neurotrophic factor (BDNF) has been implicated in neurogenesis (27) and shown to be sensitive to redox state (31, 367, 427). Exercise is a known inducer of both, BDNF and neurogenesis. Hence, it cannot be completely ruled out that the exercise-related change in the redox milieu is one of the reasons that exercise stimulates BDNF levels. BDNF expression was elevated after focal ischemia in the cerebral cortex (147), and it has been shown that oxidative damage interplays with BDNF (435). Indeed, oxidative damage has been implicated in brain function.
VII. Exercise, ROS and Oxygen Sensing, HIF, and Vascular Endothelial Growth Factor
Intracellular redox balance, which includes oxidizing and reducing powers, is essential for normal, physiological function. ROS could be produced by both an oxidative and a reduced milieu (83) and physical exercise. However, an abnormal elevation of reducing power could occur with the interruption of electron flow at the electron transport chain, which easily can lead to deleterious effects on cells (151). Indeed, it appears that hypoxia can lead to reductive stress, which also results in increased ROS production by the mitochondrial electron transport system (238). It is believed that ROS are generated at complex I and complex III of the electron transport chain. During hypoxia, less O2 is available to be reduced to H2O at cytochrome oxidase, thus causing accumulation of reducing equivalents within the mitochondrial respiratory sequence. This accumulation is known as reductive stress, and this reaction leads to ROS formation by the auto-oxidation of one or more mitochondrial complexes, such as the ubiquinone–ubiquinol redox couple. Changes in cellular redox state activate one of the most important sensors of oxygen, namely HIF-1α.
A. Hypoxia in skeletal muscle
A gradient of oxygen partial pressure (pO2) exists within the body, due to ∼160 mmHg in inspired gas, ∼115 mmHg in arterial blood, and ∼38 mmHg in capillary blood (328). Within tissues, the pO2 gradient depends on the distance of cells from the closest O2-supplying blood vessels and the metabolic activity and consequent O2 consumption of the resident cells (395). For example, pO2 in intramyocellular cells falls to as low as 3.1 mmHg under normoxia and 2.1 mmHg under hypoxia, respectively (328). Mitochondria house the final biochemical steps in the production of reducing equivalents that react at the terminal oxidases of the respiratory chain, with molecular oxygen provided by the respiratory system from the environment. In mitochondria, O2 finally disappears, and oxygen partial pressure goes to zero (145). Mitochondria thus are an effective oxygen sink, and this allows organisms to use all of the available oxygen partial pressure of the actual environment to drive the respiratory cascade from the lungs through circulation to the mitochondria (394).
The transcription factor, HIF-1α, acts as a master regulator for the expression of genes involved in the hypoxic response of most mammalian cells (355, 365). Namely, HIF-1α is hydroxylated and degraded in normoxia, but is stable in hypoxia and translocates into the nucleus to form an active complex with HIF-1β, which is constitutively expressed (154, 355). Nevertheless, it is clearly expressed in various tissues such as the brain, kidney, liver, heart, and skeletal muscle in mice kept in normoxic conditions (21% O2), whereas it is undetectable in the lungs (379). Further, when mice were kept in extreme hypoxia (6% O2), the expression levels of HIF-1α in the brain, kidney, liver, heart, and skeletal muscle were significantly increased, while expression in the lungs was not affected (379, 443). These findings indicate that there is a clear pO2 gradient between the lungs and other tissues, and that with the exception of relatively well-oxygenated lungs, low pO2 and HIF-1α play physiologically essential roles in homeostasis of various tissues. The proposed explanation might be rather simplistic as factors as oxygen uptake and supply and extraction by individual organs could play an important role in determining the HIF response. It cannot be ruled out that pO2-dependent HIF response after induction is often bell-shaped, and results from both HIF mRNA expression induction and HIF protein stabilization in the absence of oxygen.
Mounier et al. (240) have shown, in untrained human skeletal muscle under normoxic conditions, a higher HIF-1α protein expression in predominantly oxidative muscles than in predominantly glycolytic muscles. The HIF-1α mRNA expression pattern however was not in agreement with the HIF-1α protein level. Interestingly, none of the HIF-1α target genes, such as the most studied angiogenic factor involved in muscle angiogenesis and vascular endothelial growth factor (VEGF), exhibited a muscle fiber-specific related mRNA expression at rest in normoxia. However, soleus presented a significantly higher VEGF protein content than vastus lateralis and muscles of triceps. Taken together, such findings indicate that there are muscle-specific differences in HIF-1α and VEGF expression within human skeletal muscle at rest in normoxic conditions.
Thus, the following section focuses on the significance of various conditions, such as hypoxia, oxidative stress, and physical exercise, in the HIF-1α- and VEGF-signaling pathways, with emphasis on skeletal muscle.
B. HIF-1α-independent regulation of VEGF and angiogenesis
Peroxisome proliferator-activated receptor (PPAR)-γ coactivator 1α (PGC-1α) regulates mitochondrial biogenesis in multiple cell types through coactivation of key transcription factors, including NRF-1 and NRF-2, estrogen-related receptor-α (ERRα), Gabpa/b, PPARs, and the thyroid hormone receptor (16, 265). PGC-1α is a mediator of signaling in response to deprivation of nutrients and oxygen, and it strongly regulates VEGF and other angiogenic factors to elicit neovascularization in vivo. The regulation of VEGF in response to hypoxia is thought to be mediated primarily through the well-known HIF factors (355). Surprisingly, the novel PGC-1α/ERR-α pathway (16) is apparently independent of the HIF pathway. PGC-1α−/− mice are viable, suggesting that PGC-1α is not essential in embryonic vascularization (16). Angiogenesis in the adult occurs in both physiological and pathological contexts (57). The robust induction of vascularization by PGC-1α and its critical function in the response to limb ischemia strongly implicate PGC-1α in the angiogenic response to ischemia, providing protection against further ischemic insults (16).
On the other hand, PGC-1α is coupled to HIF signaling through the regulation of intracellular oxygen availability, allowing cells and tissues to match increased oxygen demand after mitochondrial biogenesis with increased oxygen supply, because of intracellular hypoxia (265). PGC-1α-dependent induction of HIF target genes under physiological oxygen concentrations is not through transcriptional coactivation of HIF or upregulation of HIF-1α mRNA, but through HIF-1α protein stabilization.
Other candidates are mentioned for the HIF-1α-independent regulation of VEGF and angiogenesis. Overexpression of suppressor of cytokine signaling 3 (SOCS3), a well-established negative regulator of signal transducer and activator of transcription 3 (STAT3), a mediator of cytokine signaling, which is a crucial factor for myogenesis, enhances the mRNA expression of downstream targets of STAT3, c-FOS, and VEGF, despite inhibition of STAT3 phosphorylation, and is correlated with enhanced mRNA expression of genes associated with muscle maturation and hypertrophy (55). The mechanism by which SOCS3 may mediate these responses however is unknown and will require further investigation. The decline in the regenerative capacity of skeletal muscle with age may be linked to inefficient STAT3/SOCS3 signaling. Actually, a single bout of maximal leg-extension exercise (resistance exercise) markedly increased the STAT3 signaling, including the SOCS3 gene in human skeletal muscle, accompanied by significant increases in levels of mRNAs for downstream genes of STAT3, c-FOS, JUNB, c-MYC, and VEGF, but probably not via HIF signaling (407). In addition, the levels of HIF-1 mRNA subunits did not change in response to short-term one-legged exercise training, suggesting no change in HIF-1 mRNA transcript levels in the regulation of training-induced VEGF expression (124).
Increased production of ROS, such as H2O2, also induces the expression of VEGF-A, the prototype VEGF ligand, by an HIF-independent and Sp1-dependent mechanism, in which ligation of VEGF-A to VEGF receptor 2 (VEGFR2) results in signal transduction leading to tissue vascularization. Such ligation generates H2O2 via an NADPH oxidase-dependent mechanism (1). That the function of one mitogen, VEGF, largely depends on signaling driven by another mitogen, H2O2, generates a novel paradigm with major therapeutic implications for a variety of angiogenesis-related disorders (338). Furthermore, oxygen–glucose deprivation generates ROS in cerebral endothelial cells, and in turn induces VEGF signaling via its receptor, Flk-1 (VEGFR2), and activates extracellular signal-regulated kinase (ERK) 1/2, thereby suggesting that VEGF acts via ERK 1/2 through oxidative-stress-dependent mechanisms, showing that ERK 1/2 can be considered a molecular target for stroke therapy (250). Interestingly, high glucose blunts VEGF response to hypoxia in immortalized rat proximal tubular cells, this effect being mediated by the oxidative stress-regulated HIF–hypoxia-responsible element pathway (173).
C. p38γ MAPK/PGC-1α signaling regulation by physical exercise and its significance for mitochondrial biogenesis and angiogenesis in skeletal muscle
p38γ MAPK, but not p38α MAPK or p38β, is required for PGC-1α upregulation in mitochondrial biogenesis and angiogenesis in response to voluntary wheel running and nerve stimulation in mice. None of the p38 MAPK isoforms was required for endurance exercise-induced IIb-to-IIa fiber-type transformation in these mice (213). Meanwhile, calcineurin (CnA) nuclear factor of activated T-cell (NFAT) regulatory axis controls fiber-type transformation in endurance exercise-induced skeletal muscle adaptation (288). Collectively, endurance exercise on one hand induces activation of the Ca2+-dependent CnA-NFAT pathway in control of fiber-type transformation, and on the other hand activates p38γ MAPK, which promotes PGC-1α activity and expression in control of mitochondrial biogenesis and angiogenesis (213, 288).
An acute bout of sprinting exercise that increased ROS production, mainly through a nonmitochondrial enzyme, XO, stimulated PGC-1α expression and other key proteins in the mitochondrial biogenic pathway, such as NRF-1 and TFAM. This nuclear-initiated signaling event was accompanied by activation of p38 MAPK and CREB phosphorylation. Inhibition of XO by allopurinol severely attenuated exercise activation of the PGC-1α signaling pathway, thus providing strong evidence that mitochondrial biogenesis in skeletal muscle is controlled, at least in part, by a redox-sensitive mechanism (143, 172). Moreover, stretch-stimulated glucose uptake in skeletal muscle is mediated by an ROS- and p38 MAPK-dependent mechanism that appears to be AMPKα2- and phosphatidylinositol 3-kinase (PI3-K)-independent (60). This novel signaling mechanism is distinct from canonical contraction- and insulin-stimulated signaling that increases glucose uptake, probably providing an alternative pathway for the development of novel therapeutic drugs to overcome insulin resistance. MAPK activation is also involved in the contraction-induced secretion of VEGF protein from skeletal muscle, which is partially mediated via adenosine acting on A2B adenosine receptors (140).
D. Two-faced ROS in HIF activation
ROS are widely believed to cause or aggravate several human pathologies such as neurodegenerative diseases, cancer, stroke, and many ailments. Antioxidants are assumed to counteract the harmful effects of ROS and therefore prevent or treat oxidative stress-related diseases (93). On the other hand, there is growing evidence that ROS have deleterious or beneficial effects; this dual nature of ROS means that ROS act as intracellular signaling molecules as defense mechanisms against microorganisms (108). For instance, the mechanisms by which ROS and cytosolic H2O2 levels are involved in stabilization/activation and eventual degradation/silencing of HIF-1α and other redox-sensitive transcription factors, such as MAPK and PI-3K/Akt, are discussed (179).
Mitochondria and mitochondrion-derived ROS were required for the stabilization of HIF-1α by MAPK and by exposure of hepatoma cells to 6 h of hypoxia (1.5% O2) (61). This hypoxic stabilization of HIF-1α in hepatoma cells was blocked by the overexpression of catalase, which catalyzes dismutation of H2O2 into H2O and O2, and is an inhibitor of the mitochondrion super anion channel. Also, HIF-1α and VEGF overexpression in pulmonary artery smooth muscle was induced by CoCl2, which mimics the hypoxic response, via ROS (28). Such overexpression was inhibited by Vitamin C and by overexpression of GPX and catalase. In contrast, the addition of H2O2 in the absence of a hypoxic stimulus causes stabilization of HIF-1α (61). Anoxia, on the other hand, may stabilize HIF-1α essentially through depletion of the cosubstrate dioxygen of prolyl-hydroxylases, indicating that increases in cytosolic H2O2 in consequence of hypoxia-driven ROS production may cause HIF-1α accumulation in hypoxia through reduction of its hydroxylation (145, 12). Actually, during the acute increase phase of HIF-1α (between 0 and 2 h), 8-hydroxy-2′-deoxyguanosine (8-OHdG) was positively correlated with HIF-1α during sustained hypoxia in humans (285). Moreover, ROS-activated signaling events involving MAPK and PI-3K/Art have been implicated in the modulation of the transcriptional activity of HIF-1α (234). Further, angiotensin II in vivo and in vitro upregulates renal VEGF expression by a mechanism that involves HIF-1 activation and oxidative stress (345).
Thus, it now seems possible that ROS have important roles in the regulation of cell signaling, although these substances are potentially cell damaging. For example, there appears to be a potential feedback loop in which PGC-1α regulates antioxidant mechanisms via regulation of mitochondrial respiration and ROS. In turn, ROS might regulate antioxidant defense genes through activation of redox-sensitive transcription factors, supporting the view that ROS are important signaling molecules in adaptations to exercise (350) (Fig. 12). While increasing evidence suggests that mitochondria play a less-important role in oxidant production in contracting skeletal muscles than was previously predicted, and that the primary site of contraction-induced ROS production in muscle fibers remains unclear (294, 297), PGC-1α is transiently induced by acute exercise (20, 143). This induction has been suggested to occur in response to altered energy demands and because of PGC-1α interacting with the metabolic enzyme AMPK (130). PGC-1α has also been proposed to be a key mediator of long-term adaptations to exercise (130). Indeed, basal levels of PGC-1α gene expression increased approximately twofold after a period of endurance training in humans (339).

Intriguingly, and in conflict with the free radical theory of aging (131), longevity and health-promoting effects of caloric restriction and specifically reduced glucose metabolism may be due to increased formation of ROS within the mitochondria. This causes an adaptive response that culminates in subsequently increased stress resistance, assumed to ultimately cause a long-term reduction of oxidative stress, which is an adaptive response more specifically named mitochondrial hormesis or mitohormesis (330). Molecular mediators of endogenous ROS defense (Cu,Zn-SOD, Mn-SOD, and GPX) are also induced by exercise, and this effect is blocked by antioxidant supplementation (331). Consistent with the concept of mitohormesis, exercise-induced oxidative stress ameliorates insulin resistance and causes an adaptive response promoting endogenous antioxidant defense capacity. As expectedly or unexpectedly, supplementation with antioxidants may preclude these health-promoting effects of exercise in humans. Taken together, physical exercise induces numerous molecular regulators of insulin sensitivity and antioxidant defense, most of which are almost completely inhibited by antioxidant pretreatment in healthy young men (329 –331).
E. Effect of exercise on HIF and VEGF signaling
The study on acute exercise by Ameln et al. (11) was the first to show that several components of the HIF-1 pathway, involving VEGF and erythropoietin, are activated in response to acute changes in oxygen demand in healthy human skeletal muscle, due to a concurrent decrease in von Hippel-Lindau tumor suppressor protein (VHL) levels. This finding suggests that oxygen-sensitive pathways could be relevant for adaptations to physical activity by increasing capillary growth in human skeletal muscle, and supports the general importance of the HIF pathway (11). VEGF mRNA is further increased when blood flow to the exercising leg is restricted.
Several studies have revealed increased levels of the HIF-1 pathway in human skeletal muscle after acute exercise (125, 220). The positive response of mRNAs for HIF-1α and HIF-2α with acute exercise however is blunted with training (220). Moreover, a positive correlation was found between exercise-induced changes in VEGF mRNA on one hand, and those in HIF-1α mRNA, HIF-1β mRNA, and lactate on the other, while no significant differences between the restricted and nonrestricted groups were observed (125), unlike the results found in the study of Ameln et al. (11). Gustafsson et al. (125) noted that blood flow restriction, made possible by application of external pressure 50 mmHg higher than atmospheric pressure, caused about a 20% decrease in blood flow at same work load, creating a condition that allowed the study of the interaction between HIF and VEGF during exercise. This lack of a further increase in VEGF mRNA when flow restriction was added may be explained by the smaller further reduction in oxygen saturation and oxygen tension compared with the rest-to-exercise transition, even if this difference was large enough to induce a greater lactate concentration increase in the restricted condition. Another explanation could be that even if there was no significant VEGF mRNA difference before exercise between the nonrestricted and restricted conditions, a strong influence of the pre-exercise value of VEGF mRNA on the exercise-induced increase makes a difference between the two conditions more difficult to detect.
Exercise induces several pathways that are known to be important for regulating angiogenesis. VEGF is critical for basal and activity-induced regulation of skeletal muscle capillarization, whereas the importance of other growth factor pathways remains to be elucidated. Although several signaling pathways have been identified (104), significant work remains before the intracellular signaling pathways and transcription factors involved in basal and exercise-induced angiogenesis are fully understood.
For example, in addition to AMPK, exercise activates several signaling pathways that may be the links between metabolism and capillarization, including calcium (Ca2+)-regulated pathways (104). Large, but transient, increases in intracellular Ca2+ occur during exercise as a result of neural activation and muscle cell depolarization. Ca2+ is intimately involved in actin–myosin cross-bridge formation and is responsible in part for exercise-induced improvements in insulin sensitivity that occur, in part, through increases in glucose transporter 4. Increasing intracellular Ca2+ leads to the activation of several downstream signaling proteins, including calmodulin-dependent kinase and CnA, possibly resulting in increased expression of VEGF mRNA in primary human myotubes in vitro. This suggests that Ca2+ may be involved in muscle VEGF regulation (104), although much more work needs to be done before these studies are finalized. In addition, PPAR-β, which is also known as PPAR-δ and is involved in muscle development and metabolism, increases VEGF and promotes muscle angiogenesis through a CnA-dependent pathway (103).
Moreover, NO is involved in muscle VEGF regulation (104, 438). nNOS-mediated NO regulates, in part, muscle mitochondrial respiration, whereas endothelial NO is involved in blood flow regulation. Disrupting normal NO production (via L-NAME) eliminates the increase in collateral blood flow induced by training, but does not disturb the increase in muscle capillarity within the active muscle. Similarly, inhibiting VEGFR kinase activity eliminates the increase in collateral-dependent blood flow, and lessens, but does not eliminate, angiogenesis within the calf muscle, illustrating distinctions between the processes influencing angiogenesis and arteriogenesis (Fig. 13) (438).
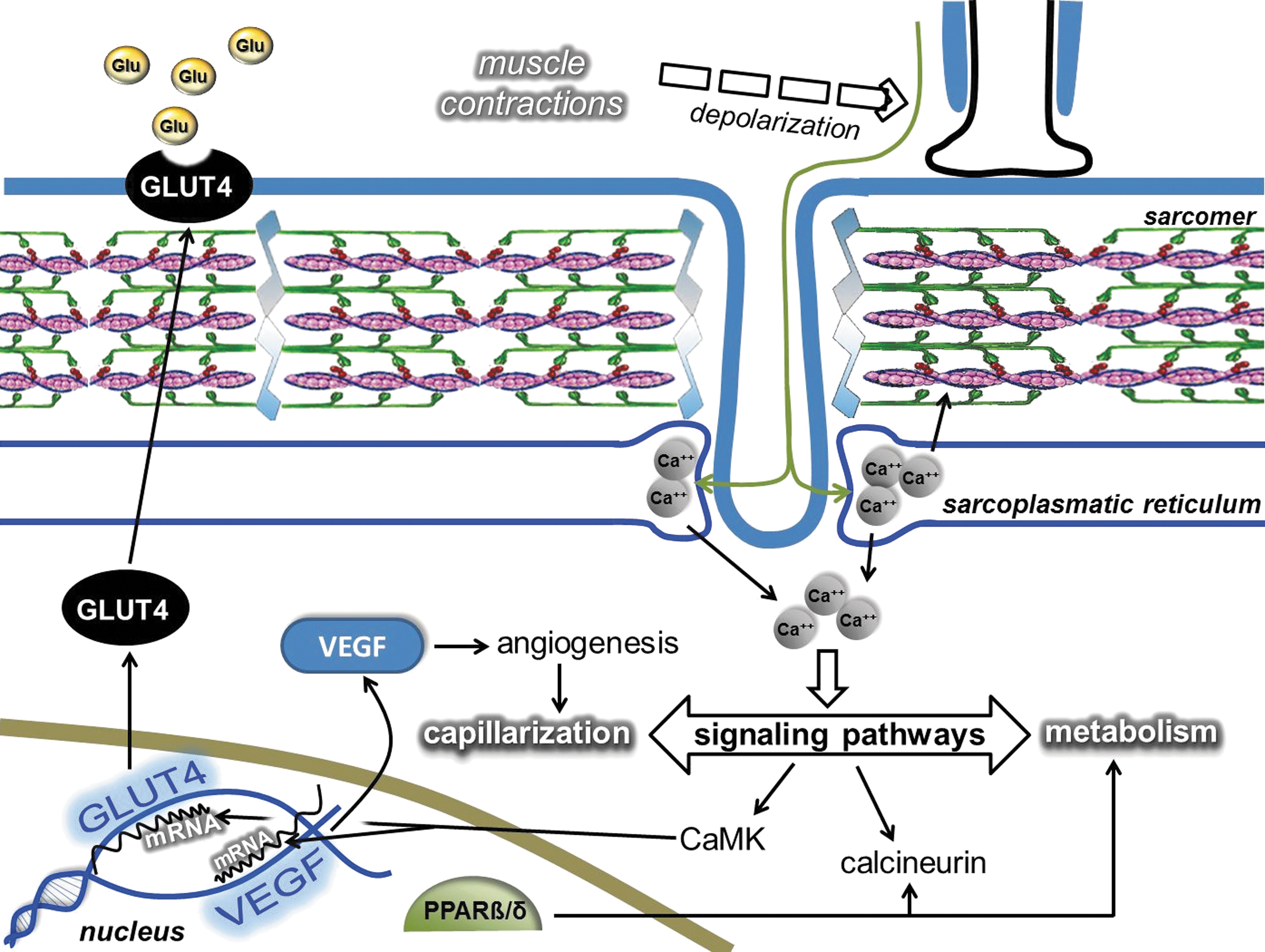
In general, the effects of endurance training on the activity of the HIF pathway in human skeletal muscle under hypoxic conditions appear to be definitely higher than those under normoxic conditions (219, 422, 446), although there are several exceptions (97, 241), indicating that combining hypoxia with exercise training appears to improve some aspects of muscle O2 transport and/or metabolism (Fig. 14) (219). Although there were no significant changes in the levels of mRNAs for HIF-1α or VEGF in human skeletal muscle after endurance training under normoxic conditions, the changes in HIF-1α mRNA expression correlated well with those in VEGF mRNA expression (270), suggesting that HIF-1α influences the training-induced VEGF gene expression, or alternatively that HIF-1α and VEGF expression is coregulated at the transcriptional level in human skeletal muscle. Taken together, it is envisioned that cumulative effects of transient changes in transcription during recovery from successive bouts of exercise may represent the underlying kinetic basis for the cellular adaptations associated with endurance training. Furthermore, no plasma VEGF contents in overweight men aged 50–60 years were affected by long-term endurance exercise under normoxic conditions (51). Likewise, low- or high-intensity-resistance exercise did not significantly alter serum VEGF content in humans (334).
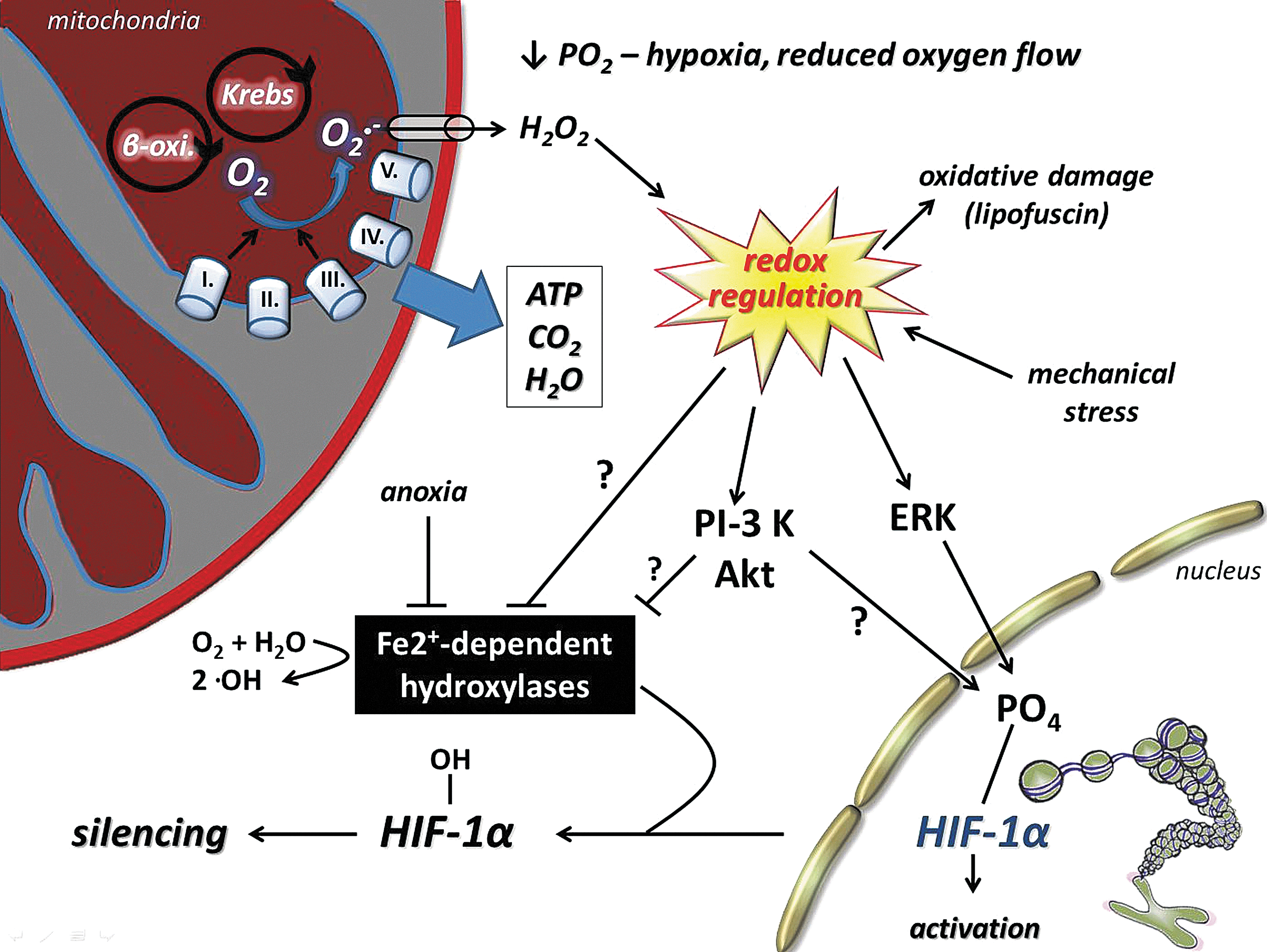
VIII. Exercise, ROS, and Epigenetics
Environmental and life-style-dependent factors can result in heritable changes in phenotypes through the methylation of DNA and reversible post-translational modification of amino acid residues of histone proteins without altering the basic structure of DNA. Histone modifications and DNA methylation predominantly occur at cytosine/guanine dinucleotide sites (132) by DNA methyltransferases (DNMT). Recently, it was reported that DNMT1, one of the key enzymes of DNA methylation, interacts with SIRT1, which causes deacetylation of DNMT1, which impairs cell cycling at G2/M transition (280). As we stated earlier, SIRT1 activity is redox dependent, and hence it is not surprising that the nature of DNA methylation and post-translational modification of histones are more dynamic than thought before (69).
It has been demonstrated that excessive levels of ROS could lead to genomic instability through an increase in DNA damage and chromosome degradation (24). ROS have been implicated in double-strand breaks that result in chromosome breaks (175). However, we do not intend to further explore the deleterious effects of ROS on chromosomes, rather we would emphasize the regulatory role of ROS in epigenetics.
Histone acetyl transferases play a key role in inserting acetyl groups on lysine residues of histones, and among them, p300/CBP is regulated by the p38/MAPK pathway and coactivates a number of transcription factors, including NF-κB and AP-1 (397). In addition, MAPK/ERK, IGF-1, and other growth factors, as well as cytokines, generate ROS for signaling and show a negative intraspecific relationship with longevity (335). The p38/MAPK-activated p300/CBP acetylates OGG1 (32, 388) and enhances the repair of this enzyme. According to these observations, a low dose of radiation has been shown to facilitate growth (153) via ROS generation, and it can cause DNA hypomethylation, which is related to DNA repair (289), since DNA repair processes incorporate cytosine, but not methylcytosine. This is how radiation-associated DNA repair can cause DNA demethylation and activation of silenced genes.
Interestingly, dietary restriction, which is generally associated with decreased accumulation of oxidative damage, including carbonyl groups, increases the accumulation of histone carbonylation in aged rats (363). Carbonylation of histone proteins increases the charge of basic amino acid residues, resulting in a more compact chromatin structure that would make transcription and repair of DNA less active. This is what happens in aging. Hence, it cannot be ruled out that controlled carbonylation of amino acid residues on histone proteins is involved in epigenic regulation.
Hypomethylation of DNA has been reported immediately after a single bout of exercise in the skeletal muscle samples of healthy subjects at the promoter regions of PGC-1α, pyruvate dehydrogenase kinase isoenzyme 4, and PPAR-δ (23).
It has been reported that exercise results in phosphoacetylation of histone H3 and induction of c-Fos gene in dentate gyrus (71). The excellent work from the laboratory of Gomez-Pinilla has shown that the well-known activating effect of exercise on BDNF includes epigenetic factors. Exercise influences DNA methylation at the BDNF gene promoter region modifying acetylation of H3 (113). It has also been shown that exercise during pregnancy results in increased BDNF expression, enhanced hippocampal cell survival, neurogenesis, and better memory in the offspring (180, 198). These studies did not evaluate the levels of ROS, but it is known that ROS can facilitate BDNF expression and neurogenesis (195, 367).
Regular exercise can increase mean life span, and it was suggested that nearly all models that lead to life extension show resistance against stressors, including ROS (226). One excellent example is from rats bred with intrinsic aerobic endurance running capacity. Low-running-capacity rats suffer from insulin resistance, cardio-vascular problems, and obesity when compared to high-running-capacity rats (HCR) (353, 433), and have a shorter maximal life span (186). Importantly, HCR generate higher levels of ROS, but because of the higher activity of antioxidant and DNA repair enzymes, HCR are more resistant to oxidative stress (409). In addition to this, we observed from the 22nd generation of these selectively bred rats that HCR have significantly higher concentrations of mitochondria and HSP78 content (Hart et al., unpublished observation). We have measured p300/CBP expression levels from the skeletal muscle of young, old, sedentary, and physically active individuals and found age-associated increases (308). These observations fit well with exercise-ROS-dependent changes in epigenetics (Fig. 15).
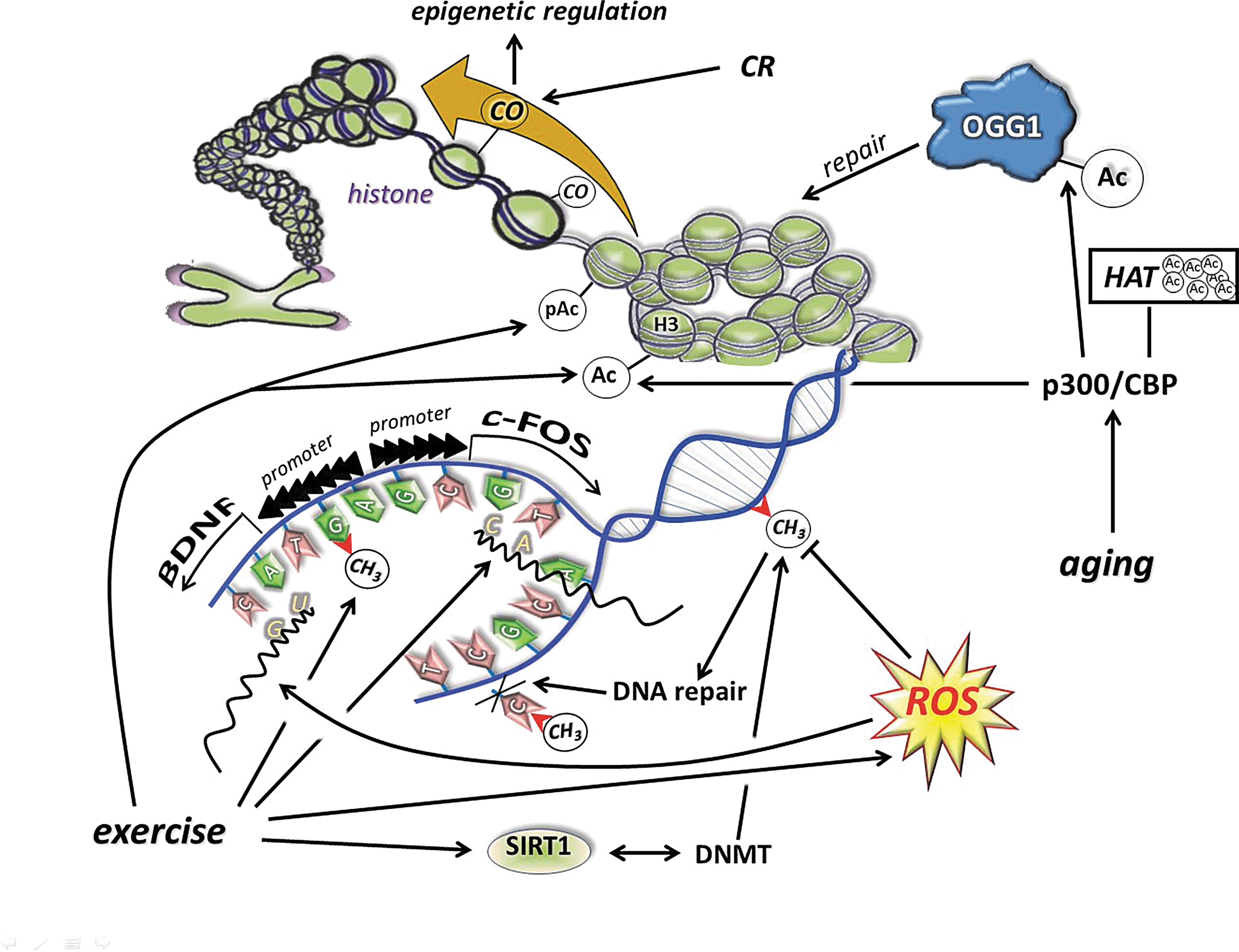
The interaction between genes and their surroundings to produce epigenetics could lead to changes in the phenotype. In the skeletal muscle, the differences in antioxidant and DNA repair capacity between slow- and fast-twitch fibers are strongly dependent on metabolism and ROS production capacity (152, 268, 313, 370). The exercise-associated ROS dependence of epigenetics is still not well studied, but the available limited data suggest that regular exercise could alter DNA methylation and the post-translational modification of histone residues by redox-mediated adaptation.
IX. Conclusion
High oxygen consumption and endurance running-associated evolution have developed a dynamic multifunctional redox homeostasis, with a strong relationship to metabolic function. The regular exercise-associated metabolic challenge is paired with oxidative challenges resulting in a wide range of adaptive responses. On the other hand, modern lifestyle increases in physical inactivity have suppressed the adaptive capacities, both in metabolic and redox homeostasis, leading to increased incidences of ROS-associated diseases and deaths. The increased ROS generation at moderate levels of exercise facilitates muscle contraction and then leads to muscle fatigue, which is an important protective mechanism of the body to avoid cellular damage. ROS are crucial for the activation of the key transcription factors, coactivators to induce adaptive responses to higher metabolic demand, including mitochondrial biogenesis, improved capillarization, enhanced antioxidant, and oxidative damage-repairing systems. We have pointed out in this review that moderate levels of oxidative damage could facilitate remodeling of cell membranes and proteins, while 8-oxoG could be necessary for specific gene activation and the opening of chromosomes. The role of PGC-1a in the antioxidant system is very specific, since moderate ROS could induce this coactivator, which enhances the expression of antioxidant enzymes and could induce the number of mitochondria, which at the same ATP demand would result in decreased levels of ROS. However, other players of the mitochondrial biogenesis fusion and fission team are sensitive to redox balance. Without question, redox homeostasis is crucial for the biogenesis of mitochondria. Sirtuins are NAD+-dependent histone deacetylases and sensitive sensors of metabolic and redox alterations. The target proteins of sirtuin are involved in metabolism, inflammation, DNA repair, apoptosis, and redox regulation. Furthermore, sirtuins are readily modified by exercise-induced changes in metabolism and redox signaling. It has been shown that redox-sensitive inflammatory transcription factors, such as NF-κB and AP-1, are members of the exercise-mediated signaling cascades, by which exercise modulates inflammation, aging, and apoptosis. We have evaluated the possibility that NO is an important mediator of the repair process, after unaccustomed exercise, by the activation of satellite cells in skeletal muscle. Interestingly, ROS could play a similar role in the brain, since neuronal precursor cells are very sensitive to oxygen/ROS availability. HIF is not just an important redox-sensitive oxygen sensor, but an important regulator of exercise-induced adaptation.
Moreover, regular exercise has an impact on the reversible structure of chromosomes, which can transfer adaptive changes to offspring, and redox signaling is involved in this process. Therefore, during compensated physical activity, the level of exercise-induced ROS production does not seriously jeopardize cellular or organ function, but brings about a wide range of health-promoting adaptations to the organism.
In summary, the present review intended to emphasize that (i) aerobic and redox signaling were important for evolution; (ii) regular exercise increases the efficiency of antioxidant and oxidative damage repair systems; (iii) moderate levels of oxidative damage, which can be caused by a single bout of exercise, can be crucial for membrane remodeling, protein turnover, and transcriptional regulation; (iv) sirtuins are NAD+-dependent modulators of a wide range of metabolic and redox processes, and are readily modified by both a single bout and regular exercise; (v) mitochondrial biogenesis is associated with redox signaling and is a key component of antioxidant defense; (vi) exercise-induced activation of stem and precursor cells in skeletal muscle and the brain can be redox dependent; (vii) HIF is an exercise-sensitive oxygen sensor and a redox regulator; and (viii) exercise-induced changes in epigenetics could be redox dependent.
Footnotes
Acknowledgments
The present work was supported by Hungarian grants from ETT 38388, TéT JAP13/02, OTKA (K75702), and TAMOP-4.2.2/B-10/1-2010-0013 awarded to Z. Radák. The authors acknowledge the assistance of Professor AW. Taylor in the preparation of this article.
Finally, we are grateful to the peer reviewers for their constructive comments, which helped us to improve this review.