
Abstract
Introduction
Oxidative stress can be defined as an imbalance between the production of reactive species (RS, including both reactive oxygen species [ROS] and reactive nitrogen species [RNS]) and the ability to detoxify them and their intermediates/byproducts. Oxidative damage to lipids, proteins, and DNA has been detected in autopsies from individuals with PD (5, 6, 51). Interestingly, a decrease in the levels of the low-molecular-weight thiol antioxidant glutathione (GSH) is one of the earliest biochemical alterations detected in association with PD (84, 139). Since then, a number of research studies have addressed the role of GSH homeostasis in PD progression. GSH, a major antioxidant defense and the most abundant nonprotein thiol in mammalian cells, contains a free thiol group that mediates its redox functions (59, 60). Thiols are organosulfur compounds that contain a carbon-bonded sulfhydryl group. As part of the functional group of the amino acid cysteine, protein cysteines (protein thiols) are now thought to mediate redox signaling processes in response to oxidative stress. This has significantly changed researchers' interest from characterizing oxidative stress or damage to identifying specific redox signaling events involved in human disease progression. Cysteine residues serve numerous functions. Catalytic redox-active cysteines in the active sites of thiol oxidoreductases are involved in their catalytic activity and participate in oxidation, reduction, and isomerization of disulfide bonds, as well as in reactions that modulate their redox states. Regulatory cysteines present in proteins, such as transcription factors and phosphatases, alter protein activity by changing their redox state, without being catalytic themselves, and can be regulated by reversible intramolecular and intermolecular oxidative modifications. Structural cysteines participate in protein structure and folding via the formation of stable intramolecular and intermolecular disulfide bonds. Metal-coordinating cysteines coordinate metal ions and occur in the form of CxxC motifs. Catalytic non-redox cysteines participate in catalysis without alteration in their redox state and are present in various proteases. And finally, cysteines can act as sites of post-translational modifications, which are utilized for targeting proteins to membranes and as sites of post-translational modifications that influence protein activity, localization, and/or protein–protein interactions (57). An increasingly complex scenario of distinct thiol-dependent redox signaling pathways and enzymatic processes has emerged since GSH was found to participate in cellular redox homeostasis. In PD, a variety of thiol-based redox signaling events have been recently described as important triggers/regulators of dopaminergic neurodegeneration, and in this review, we aim to highlight the most important discoveries in this area.
PD Versus Experimental PD
A fraction of PD occurrence is related to mutations in genes such as those encoding α-synuclein (SNCA), DJ-1 (PARK7), PTEN-induced putative kinase 1 (PINK1), leucine-rich repeat kinase 2 (LRRK2), and Parkin (PARK2). Over 90% of PD occurs most commonly in a sporadic (idiopathic) form without a clearly defined genetic basis, and only a vaguely delineated pathogenesis likely linked to environmental causes. The major risk factor identified for PD is aging as its prevalence and incidence increase exponentially from ages 65 to 90. Thus, it is thought that PD arises from the convergence of genetic susceptibility, environmental exposures, and aging. However, the etiology of PD has yet to be clearly established. Cellular and animal disease models based on both genetic- and toxin-induced neurodegeneration have been used to understand PD pathogenesis. However, as reviewed by Horowitz and Greenamyre and Vance et al. (77, 177), not all experimental models recapitulate dopaminergic cell death, Lewy body formation, and/or symptoms observed in PD. Mitochondrial/environmental toxins, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or its active metabolite 1-methyl-4-phenylpyridine (MPP+), and pesticides rotenone and paraquat induce dopaminergic cell death in vitro and in vivo (24). The support for the use of MPTP as an experimental PD model originates in 1982 from the discovery of a group of young drug addicts, which developed a clinical picture almost indistinguishable from PD after self-administration of a synthetic heroin analog accidentally contaminated with MPTP. Epidemiological evidence shows an increase in the risk of developing PD upon exposure to environmental toxicants, such as the pesticides rotenone and paraquat (167). Several other toxicants, such as metals, maneb, organochlorine/organophosphorus pesticides, polychlorinated biphenyls, as well as early life inflammatory processes and occupational manganese exposure, have been implied to increase PD risk (62). Genetically engineered PD animal models have been developed for the overexpression of mutant autosomal-dominant genes (SNCA and LRRK2) and knockout/knockdown of autosomal-recessive genes (PARK2, PARK7, and PINK1). However, only marginal or null dopaminergic cell death has been observed in genetic-based animal models (144, 170), which suggests that genetic risk factors are not sufficient and that environmental exposures may also be required for nigral dopamine neuronal degeneration (gene–environment interactions). For example, high occupational pesticide exposure to paraquat and maneb in individuals carrying dopamine transporter (DAT) genetic variants increases the risk of developing PD (93, 146). New models studying gene–environment interactions have emerged since then. As reviewed by Gao and Hong (62), neurotoxins, such as paraquat and MPTP, as well as lipopolysaccharide (LPS)-induced inflammation exacerbate α-synuclein pathology. It is important to state that although many of the features of PD are observed in the current existing models of PD, none of them recapitulate the full spectrum of the disease. However, the use of both genetic- and toxin-based experimental PD models has helped us to identify important mechanisms regulating dopaminergic cell death and survival, and should keep helping us understand PD.
Oxidative Stress in Dopaminergic Cell Death and PD
Oxidative stress in PD has been primarily linked to mitochondrial dysfunction, as decreased activity of the mitochondrial electron transport chain (ETC) was evidenced in the substantia nigra of patients with PD (75, 157) (Fig. 1). Accordingly, toxins, such as MPTP, rotenone, and paraquat, act primarily by inhibition of complex I, a major component of the ETC, leading to an increased generation of RS (20, 159). Interestingly, recent reports suggest that complex I inhibition might not be the only mechanism involved in dopaminergic neuron death induced by rotenone, MPP+, or paraquat (34, 129, 132, 143). Additional sources for RS generation have been described in the mitochondria besides complex I (2) and differential contribution of mitochondrial respiratory chain complexes seen to mediate RS production in response to paraquat (25, 53).
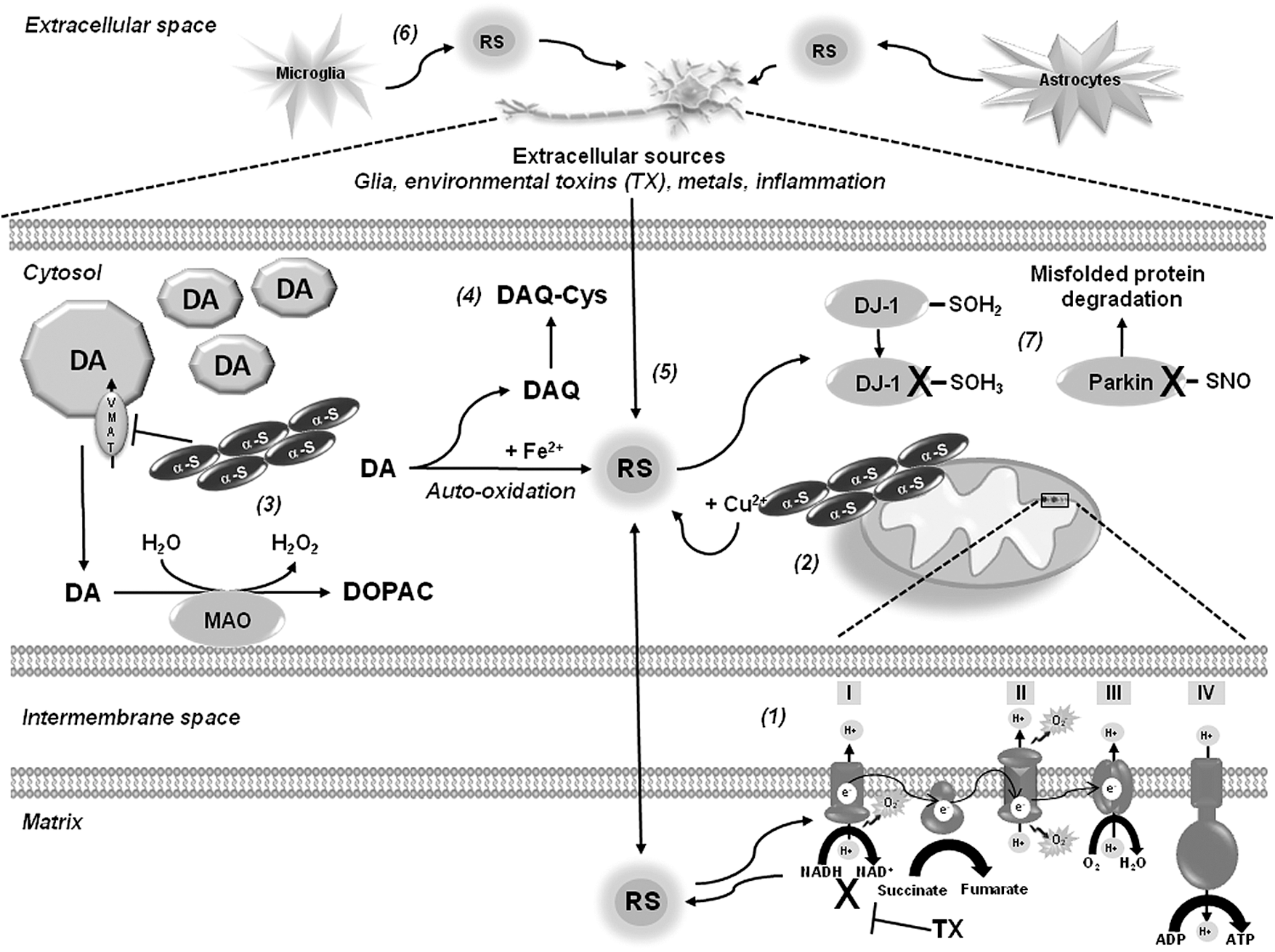
Accumulation of α-synuclein has been suggested to produce mitochondrial oxidative damage (49), while α-synuclein-copper (Cu2+) complexes have been shown to induce RS accumulation and dopamine oxidation (180). Metal-induced RS generation also contributes to oxidative damage in PD. Increased iron (Fe) deposition and increased free Fe levels were found in the substantia nigra of PD brains (162). Oxidative stress in PD is also associated with the pro-oxidant properties of dopamine (131). Mutant α-synuclein downregulates the vesicular dopamine transporter (VMAT2) augmenting cytosolic dopamine (111), which is either metabolized by monoamine oxidase (MAO) to generate hydrogen peroxide (H2O2), or auto-oxidized in the presence of Fe generating superoxide anion (•O2 −), H2O2, and dopamine-quinone species (DAQ) (1). Increased Fe concentrations have been found in the SNpc of PD brains, which might promote dopamine oxidation and generation of RS (194). Further, the neurotoxins rotenone, MPTP, and paraquat increase intracellular dopamine oxidation (89, 110, 112). Plasma membrane nicotinamide adenine dinucleotide phosphate (NADPH) oxidases have also been proposed as sources of intracellular (dopaminergic cells) and extracellular (microglial cells) ROS generation upon MPP+, rotenone, and paraquat treatment (43, 63, 187, 188).
GSH in Dopaminergic Cell Death and PD
As mentioned previously, a decrease in the GSH levels is one of the earliest biochemical alterations detected in association with PD as demonstrated by the observation that GSH loss occurs in incidental Lewy body disease, considered an asymptomatic precursor to PD (84, 139, 163). GSH itself regulates dopaminergic cell death through a wide variety of homeostatic processes. Because of space restrictions we will only highlight some major findings regarding the role of GSH in dopaminergic cell death and PD. However, recent reviews have covered this topic extensively (115, 196).
In experimental PD models, GSH depletion is linked to its oxidation in association with oxidative stress (52). Interestingly, GSH depletion induced by MPTP toxicity has also been associated with the impairment of neuronal cysteine uptake via the excitatory amino acid carrier-1 (EAAC1) (10) (Fig. 2). Intracellular GSH has a short half-life; thus, cells require the constant renewal of the GSH pool. Cysteine availability is rate limiting for the synthesis of GSH. However, because cysteine is unstable, the main extracellular source of cysteine is the dipeptide cystine (two conjugated cysteines via auto-oxidation). Blood plasma contains relatively high concentrations of cystine (100–200 μM), but only 10–20 μM of cysteine. Cystine uptake by a Na+-independent heteroexchange mechanism denominated that xc − operates as a 1:1 exchanger with intracellular glutamate, where cystine is transported inward in exchange for glutamate. By providing cystine to cells, system xc − has been shown to be an important regulator of intracellular GSH levels and antioxidant capacity of the brain (7). Interestingly, xc − deletion does not decrease GSH content in the striatum, nor it increases oxidative stress in striatum or substantia nigra. In contrast, it results in increased resistance to 6-hydroxydopamine (6-OHDA)-induced neurodegeneration, an effect connected to a decrease in the extracellular glutamate levels (116).
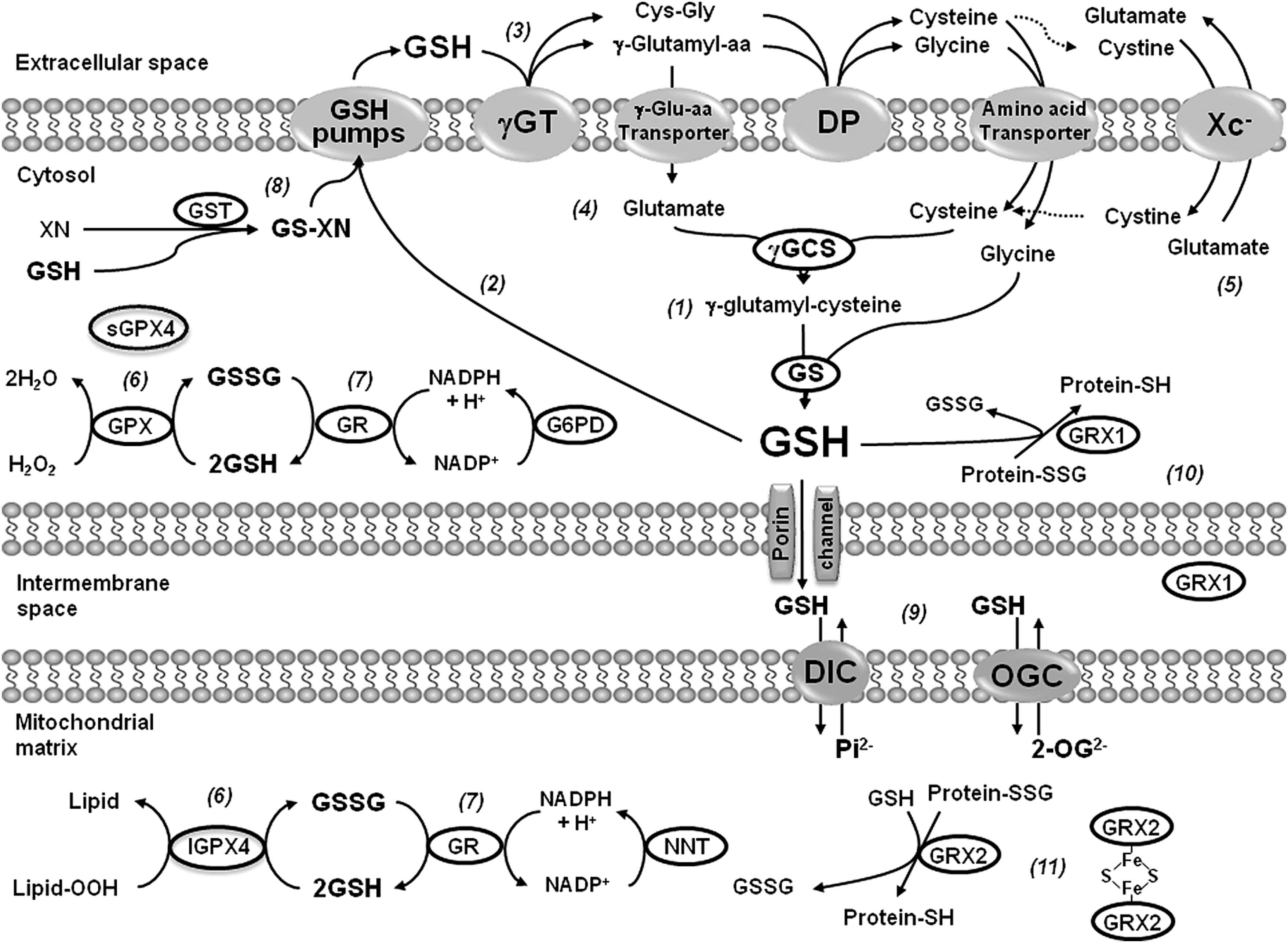
GSH depletion inhibits mitochondrial complex I activity in vitro and in vivo (28, 47, 86), and this has been associated with cysteine post-translational modifications (29, 47). Glutathione reductase (GR), which reduces glutathione disulfide (GSSG) back to GSH, requires reduced NADPH as the electron donor reductant, and glucose-6-phosphate dehydrogenase (G6PD) is indispensable for the regeneration of NADPH from NADP+. Transgenic mice overexpressing G6PD in dopaminergic cells are resistant to MPTP-induced toxicity (117). GSH-based antioxidant systems have also been reported to exert a protective effect against dopaminergic cell death, as mice deficient in glutathione peroxidase 1 (GPX1) exhibit an increased sensitivity to MPTP toxicity (98), while its overexpression protects against MPP+- and 6-OHDA-induced toxicity in vitro and in vivo (17, 88, 145, 169). GSH homeostasis is also modulated by PD-related genes. The protective effects of DJ-1 against oxidative stress have been associated with the upregulation of intracellular GSH content (109, 198). A recent randomized clinical trial failed to demonstrate a protective effect of intravenous GSH administration in PD patients (74). However, it is not clear how well GSH crosses the blood–brain barrier (BBB). Studies have demonstrated that GSH is transported across the BBB in the rat and guinea pig, while human cerebrovascular endothelial cells exhibit GSH uptake and efflux (90, 91, 201). In contrast, oral administration of N-acetyl-
GSH is compartmentalized within the mitochondria, endoplasmic reticulum (ER), and nuclei, and it is known that distinct GSH pools, particularly that of mitochondria and ER, are key regulators of cell death pathways. However, to date, there is little evidence regarding the distinct role of cytosolic versus mitochondrial GSH in dopaminergic cell death in spite of the clear importance of mitochondria in PD.
Thiol-Based Oxidoreductases in Dopaminergic Cell Death and PD
Thioredoxins
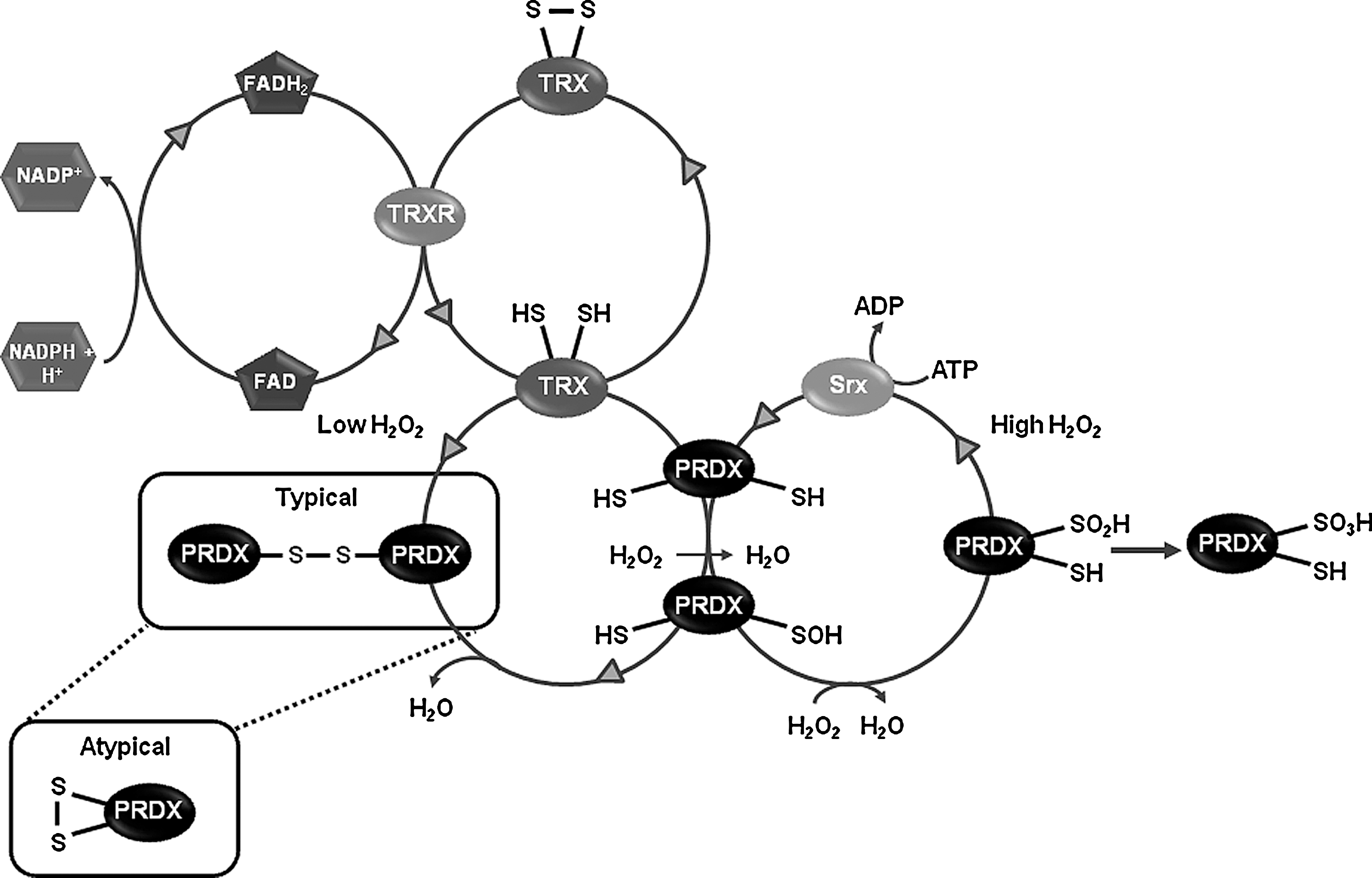
Thioredoxins (TRXs) are dithiol-redox proteins with a conserved Cys-Gly-Pro-Cys catalytic site that reduces or binds to proteins modifying their activity. The TRX redox system depends on thiol-disulfide exchange reactions at the active site. Thioredoxin reductase (TRXR), a homodimeric selenium containing flavoprotein, transfers reducing equivalents from NADPH to TRXs reducing them. While the TRX1/TRXR1 system is localized in the cytoplasm, the TRX2/TRXR2 couple is mitochondrial. In addition, TRXs provide reducing equivalents for the peroxide scavenging activity of peroxiredoxins (PRDXs) (Fig. 3). TRX1 has also been shown to reduce and activate a number of transcription factors. In the substantia nigra both TRX1 and TRX2 mRNA have been detected (107, 150). At the protein level, recent immunohistochemical analyses have demonstrated the strong expression of both TRX1 and TRX2 in the substantia nigra of mouse and rat brain, while weaker but consistent detection of TRXR1 and TRXR2 was also found (9, 66). Overexpression and administration of TRX1 protect against MPP+- and paraquat-induced toxicity in dopaminergic cells (12, 190). However, oxidation of TRX2 but not TRX1 induced by rotenone and MPP+, was reported in a different study. Contradictory results exist regarding the ability of paraquat to induce either TRX1 or TRX2 oxidation (141, 147). TRX1 also functions as a chaperone, and this seems to be independent of its redox activity. Expression of the Parkin-associated endothelin receptor-like receptor (Pael-R), a substrate of the E3 ubiquitin ligase Parkin, causes selective age-dependent degeneration of Drosophila dopaminergic neurons. Interestingly, overexpression of redox-inactive mutants of Drosophila TRX homologs, which remained active as chaperones, prevented neuronal loss by Pael-R expression (174).
Peroxiredoxins
PRDXs are ubiquitous thiol peroxidases that scavenge peroxides in cells (Fig. 3). Catalysis of H2O2 is initiated by the reaction of their active site cysteine residue with H2O2, and recent studies have demonstrated that the reaction rate for PRDXs with H2O2 is in many cases comparable to that of catalase (42, 71). PRDXs are broadly distributed and highly abundant proteins comprising up to 1% of soluble cellular protein. Mammals have six PRDXs, with PRDX1, 2, and 6 found in the cytoplasm, PRDX4 in the ER, PRDX3 in the mitochondria, and PRDX5 in various compartments within the cell, including peroxisomes and mitochondria. In the substantia nigra, dopaminergic neurons express PRDX2–5 at very low levels, being PRDX5 the isoform with the lowest expression. Interestingly, non-dopaminergic neurons of the substantia nigra show higher expression levels of PRDX3 and PRDX5, while PRDX1 and PRDX6 are only expressed in glial cells (9, 67). An increase in PRDX2 and a decrease in PRDX3 and PRDX6 levels are observed in postmortem PD brains (99). PRDX2 oxidation has also been reported in PD brains (55). Paraquat induces oxidation of both PRDX3 and PRDX1 (147). Overexpression of PRDX1, PRDX2, and PRDX4 protects against 6-OHDA-induced dopaminergic cell death (79, 104), whereas silencing mitochondrial PRDX3 and PRDX5 increases sensitivity to MPP+ (48). Interestingly, PRDX2 protective effects seem to be mediated by modulation of TRX1 oxidation and apoptosis signal-regulating kinase-1 (ASK-1) (Fig. 4). In addition, mutations in LRRK2 are associated with autosomal dominant PD and a recent report shows that in vitro and in postmortem human samples of PD patients, mutant LRKK2 increases the inhibition of PRDX3 by phosphorylation (8).
Protein Cysteine Oxidation in Dopaminergic Cell Death and PD
To understand the exact regulatory role of oxidative stress in the signal transduction events leading to dopaminergic cell death, we must determine the substrate specificity and biomolecular alterations caused by RS. Recent efforts have been undertaken to understand the molecular mechanisms by which oxidative stress regulates dopaminergic cell death associated with PD. Protein, lipid, and nucleic acid oxidation has been demonstrated to have an active role in dopaminergic cell death, but the exact mechanisms remain unclear (5, 6, 11, 46, 50). Specific amino acids, such as cysteine, methionine, tryptophan, and tyrosine residues, are prone to oxidative modification. By regulating protein structure and activity, oxidative post-translational modifications regulate a variety of physiological processes. An increase in protein oxidation has been reported in brains from PD patients as evidenced by the accumulation of oxidative and nitrative protein modifications (5, 46). An important cellular target or sensor of RS is the thiol group of the amino acid cysteine. Redox-sensitive cysteines undergo reversible and irreversible thiol modifications in response to ROS or RNS, thereby modulating protein function, activity, or localization. Oxidation and reduction of cellular thiols are thought to be one of the major mechanisms by which oxidative stress is integrated into cellular signal transduction pathways. Almost all physiological oxidants react with thiols (140, 186). Distinct RS, such as •O2 − and peroxides (H2O2, and peroxynitrite [ONOO−]), mediate one- and two-electron oxidation of protein cysteines, respectively, leading to the formation of reactive intermediates, including protein sulfenic acids (PSOH) and protein thiyl radicals (PS•) (Fig. 5). Once formed, a PSOH can lead to the formation of additional oxidative modifications that act as signaling events regulating protein function. The reaction of PSOH with either a neighboring cysteine or GSH will generate a disulfide bond (PSSP) or a glutathionylated residue (PSSG). Both disulfide products can be reduced back to the thiol by the action of the TRX/TRXR and the glutaredoxin (GRX)/GSH systems, respectively. Additionally, PSOH can undergo further reaction with H2O2 to irreversibly generate protein sulfinic (PSO2H) and sulfonic (PSO3H) acids. To prevent overoxidation of critical cysteine residues, PSOH may be converted to disulfides or PSSG (138, 140, 186). A variety of oxidative cysteine modifications have been reported in PD and experimental PD models (Table 1).
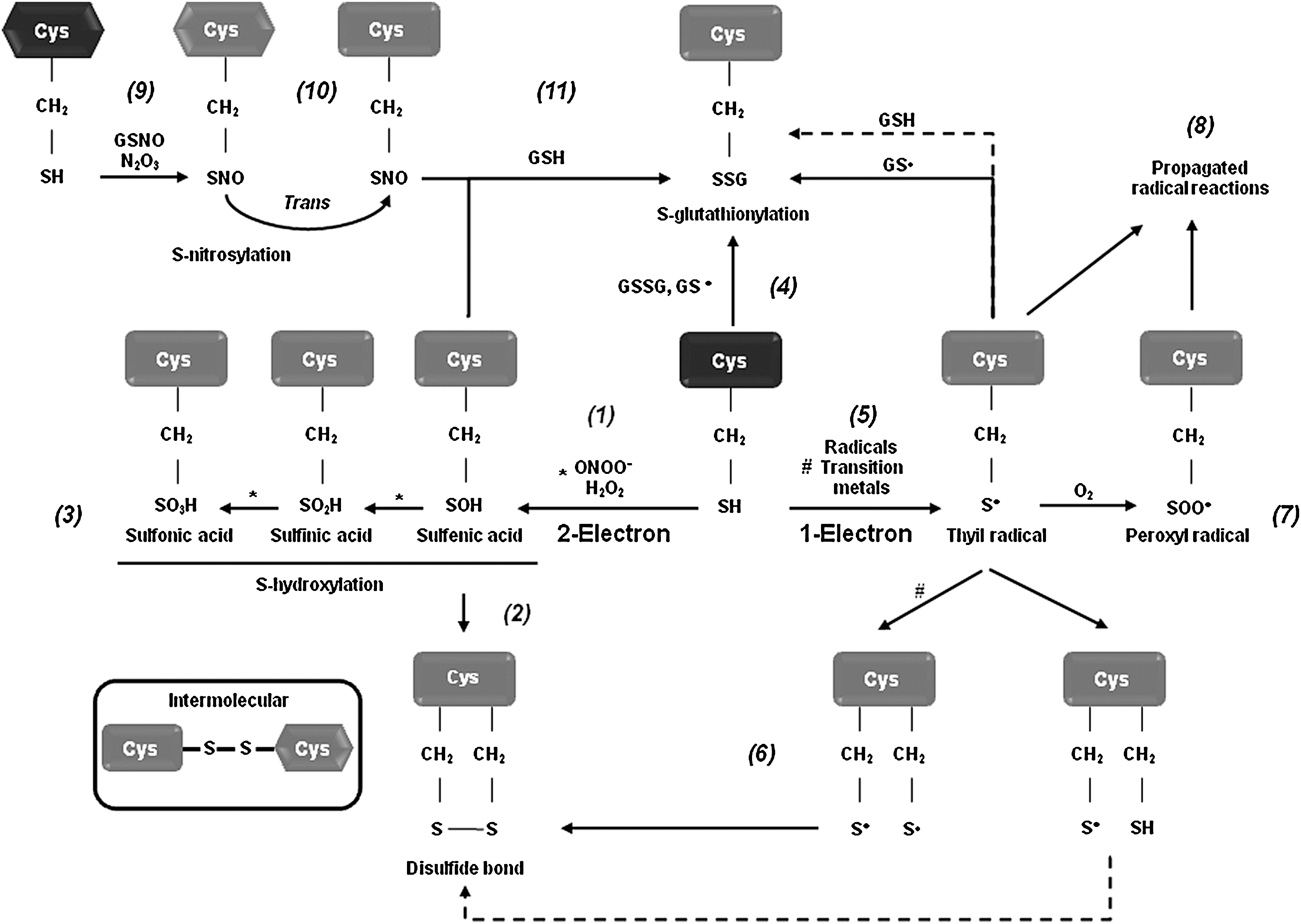
DLBD, diffuse Lewy body disease; MPP+, 1-methyl-4-phenylpyridine; 6-OHDA, 6-hydroxydopamine; PD, Parkinson's disease.
Recent studies have demonstrated the importance of protein cysteine oxidation in the regulation of dopaminergic neurodegeneration and mitochondrial activity in PD (Table 1). DJ-1 mutations are associated with early onset PD. DJ-1 protects against oxidative stress and dopaminergic cell death induced by mitochondrial/environmental toxins. PSO2H formation at Cys106 in DJ-1 has been detected upon treatment with MPP+ and paraquat, as well as in erythrocytes of PD patients (4, 23, 156), and this oxidation has been proposed to regulate ROS scavenging properties and chaperone function of DJ-1 (185, 200) (Fig. 1). Cysteine oxidation to PSO2H/PSO3H inhibits the E3 ligase activity of the autosomal recessive PD gene Parkin, which regulates ubiquitination-mediated degradation of misfolded proteins (120). Cysteine sulfonation of the deubiquitinating enzyme ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1), whose mutation is linked to early onset PD, was also found in PD brains (Table 1) (32).
DAQ byproducts from dopamine oxidation have been reported to bind to nucleophilic sulfhydryl groups on protein cysteines (73, 131) (Fig. 1). DAQ binding to protein thiols is thought to inactivate proteins, and thus replenishment of intracellular GSH pools prevents dopamine-induced toxicity by binding to DAQ (72). Inactivation of the Na+-K+-ATPase has been observed in the presence of dopamine (96). A recent proteomic analysis demonstrates the susceptibility of a variety of proteins to be covalently modified by dopamine, including the PD-associated proteins UCH-L1 and DJ-1 (176), but the exact amino acid residues involved were not identified.
Protein glutathionylation and GRXs in PD
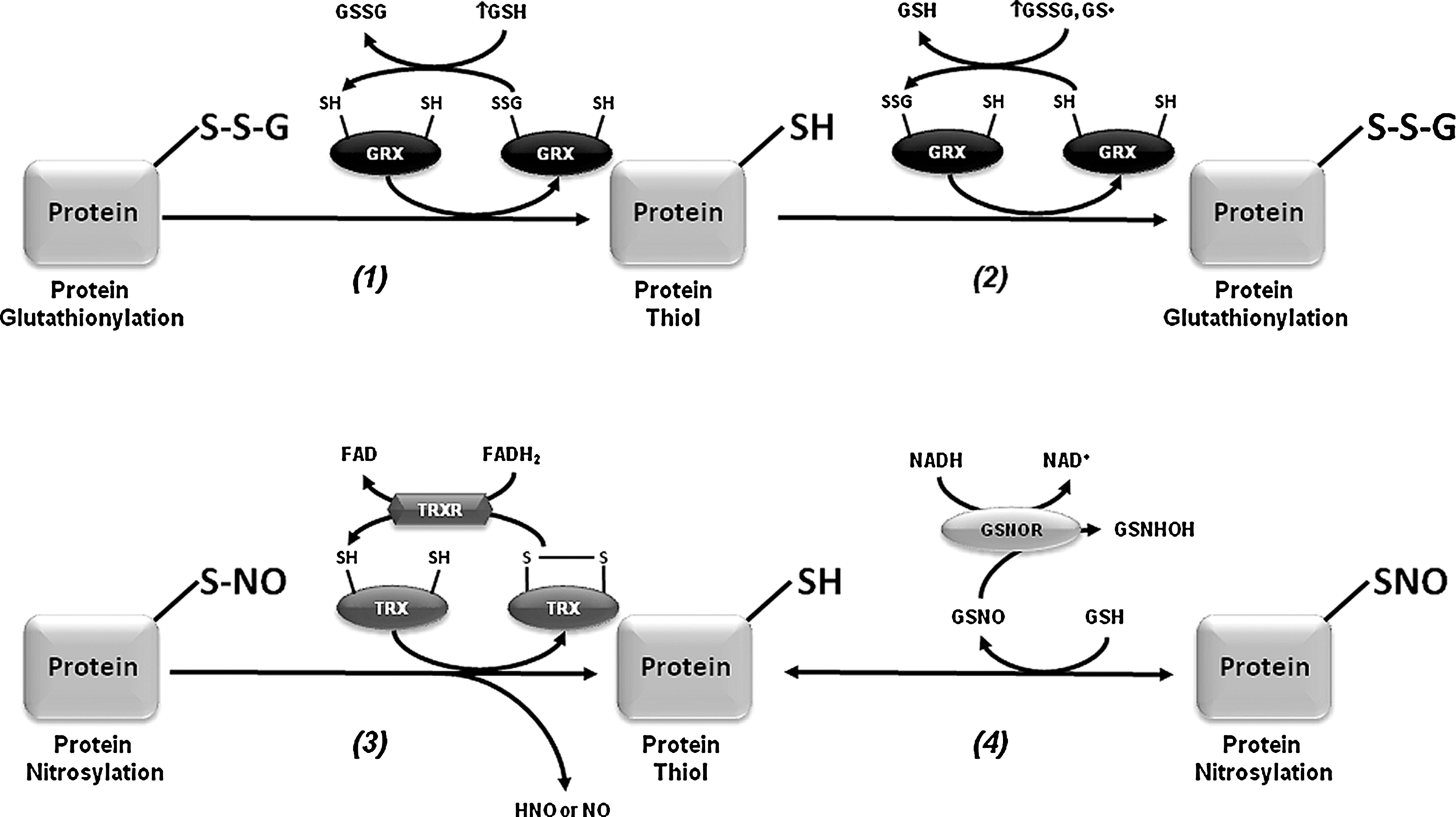
Protein glutathionylation (PSSG) is defined as the reversible formation of mixed disulfides between GSH and protein thiols involved in the redox regulation of protein function. PSSG has been demonstrated to occur by mechanisms other than the reaction of GSH with PSOH (122) (Fig. 5). For example, PSSG formation might occur by thiol exchange reactions between protein cysteines and GSSG, which requires an unusually high redox potential only reported for a limited number of protein targets (122). Nitrosylation of protein cysteines and GSH generates protein nitrosylation (PSNO) and nitrosoglutathione (GSNO), and biochemical studies demonstrate the potential of GSNO to promote PSSG (122). Previous studies have demonstrated the occurrence of PSSG/deglutathionylation in dopaminergic cells and experimental PD models. However, very few protein targets susceptible to PSSG have been identified in dopaminergic cells, which are restricted to the mitochondrial compartment. Mitochondria PSSG has been reported in response to oxidative stress in dopaminergic cells (130). In contrast, a recent report demonstrates that mitochondrial NADP(+)-dependent isocitrate dehydrogenase (IDPm), which supplies NADPH for GSH production, is inactivated by glutathionylation in response to oxidative stress. Interestingly, mice treated with MPTP exhibit an increased level of IDPm-SSG (Table 1) (97). On the other hand, deglutathionylation of mitochondrial proteins and impairment of complex I activity have also been associated with cell death in dopaminergic neurons (130).
PSSG linkages are removed by changes in the intracellular GSH/GSSG balance and/or the activity of GRXs (Fig. 6). GRXs are oxidoreductases that utilize the reducing power of GSH to catalyze protein deglutathionylation. However, GRXs can induce both PSSG and deglutathionylation depending on the GSH/GSSG ratio and the presence of thiyl radicals (GS•) (15, 122). GRX1 is expressed at lower levels in the substantia nigra and striatum compared with hippocampus and cerebral cortex (9, 13). GRX1 is localized primarily in the cytosol although recent reports also demonstrate its presence in the intermembrane space (134). GRX1 exerts a protective effect against dopamine and MPTP toxicity in neuronal cells (44, 45, 95). Interestingly, incidence of PD is lower in women than in men, and it has been demonstrated that GRX1 is highly expressed in female compared with male mouse midbrain and striatum regions, which correlates with a decreased sensitivity to MPTP toxicity (94). Downregulation of GRX1 induces the loss of complex I activity, mitochondrial membrane potential, and DJ-1, as well as translocation of the death-domain-associated protein (DAXX), which is associated with dopaminergic cell death (95, 153, 154). The dopamine precursor levodopa (
Mitochondrial GRX2 is primarily localized in the matrix and is abundantly expressed in dopaminergic cells of the substantia nigra (9). GRX2 contains a Fe-S cluster that bridges two GRX2 monomers and serves as a redox sensor for its activation during conditions of oxidative stress (Fig. 2). Downregulation of GRX2 disrupts Fe-S center biogenesis (decreased Fe incorporation into mitochondrial proteins) and complex I activity in dopaminergic cells (92, 102), while its overexpression protects against MPTP-induced toxicity (92).
PSNO in dopaminergic cell death and PD
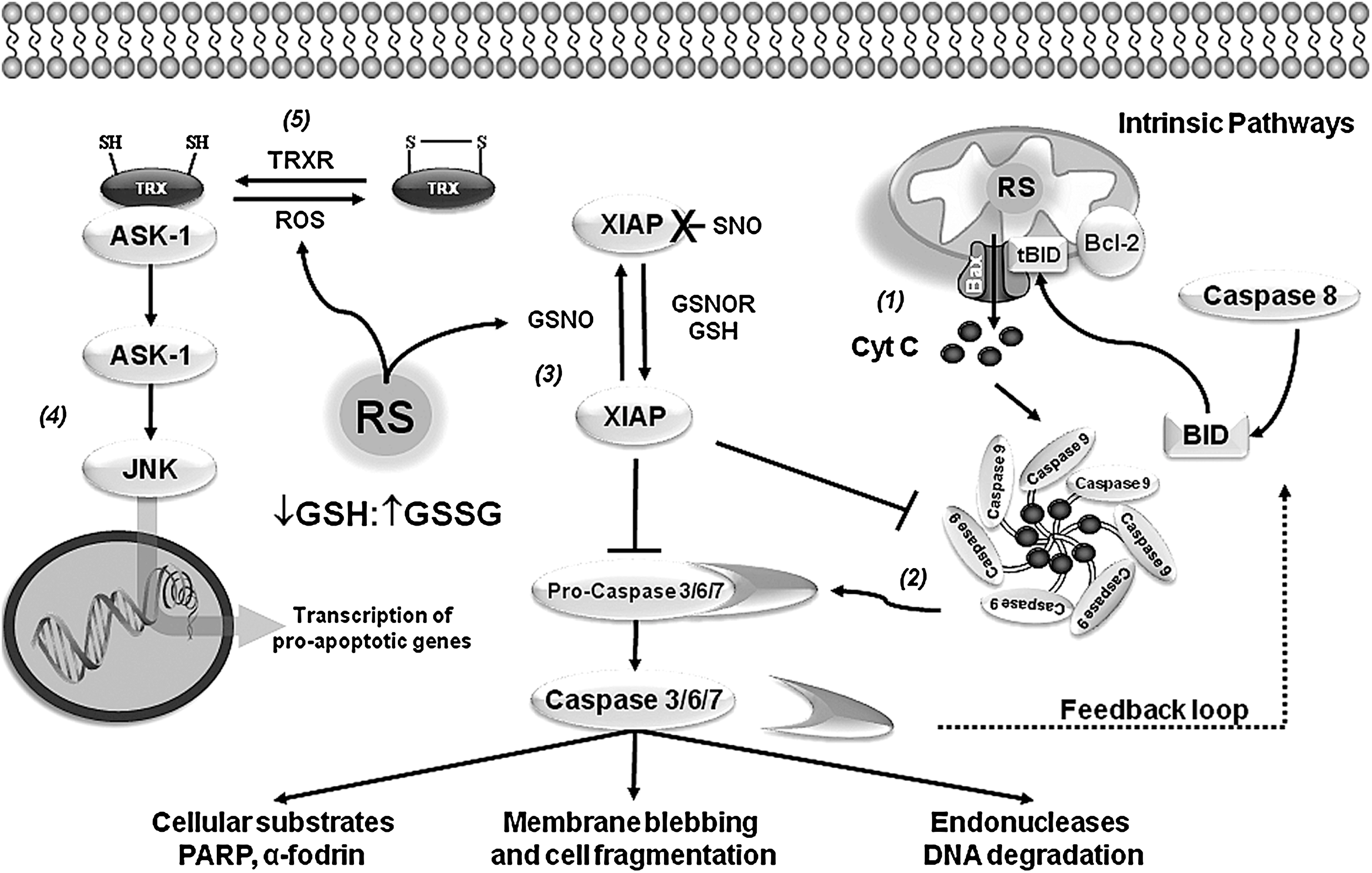
PSNO (also named cysteine nitrosation) refers to the reversible covalent adduction of a nitroso (NO) group to a protein cysteine resulting in the formation of PSNO. PSNO formation is regulated by nitric oxide synthase (NOS) expression and activity, and by endogenous NO-mediated nitrosylating agents, such as dinitrogen trioxide (N2O3), or by transition-metal-catalyzed addition of NO. Transfer of NO groups between PSNO and GSNO (transnitrosation) has been reported as one of the major mechanisms mediating PSNO. GSNO is formed during the oxygen-dependent oxidation of nitric oxide (•NO) in the presence of GSH, and as a minor byproduct from the oxidation of GSH by ONOO− (58) (Figs. 5 and 6). Increased PSNO levels of distinct substrates have been reported in PD and, in most cases, this modification is linked to the impairment of enzymatic function. Parkin–SNO has been shown in PD brains and in mice treated with MPTP, which inhibits its ubiquitin E3 ligase activity (35, 192) (Fig. 1 and Table 1). Similarly, protein disulfide isomerase (PDI), an ER enzyme that catalyses thiol-disulfide exchange reactions facilitating disulfide bond formation and protein folding, is PSNO in PD brains, which also inhibits its enzymatic activity leading to the increased accumulation of misfolded proteins and concomitant activation of the unfolded protein response (173). In addition, increased PSNO of PRDX2 and X-linked inhibitor of apoptosis protein (XIAP) is also found in PD brains (55, 172). PRDX2-SNO is associated with loss of its peroxide scavenging activity (55), while XIAP-SNO impairs its ability to inhibit caspase 3 (172). Interestingly, XIAP-SNO formation was shown to involve the transfer of the NO group from the executioner caspase 3 to XIAP (transnitrosation) (Fig. 4 and Table 1) (129). Progressive dopaminergic degeneration by LPS-induced neuroinflammation was recently shown to be preceded by PSNO of mitochondrial complex I, which was prevented by NOS inhibitors (31). Protein denitrosylation can be regulated by TRXs and also through the metabolism of GSNO via GSH-dependent formaldehyde dehydrogenase or class III alcohol dehydrogenase, also known as GSNO reductase (GSNOR) (Fig. 6) (16). However, neither a role for TRXs nor for GSNOR has been reported in PSNO formation in association with PD.
Glutathione-S-Transferases in PD
Although polymorphisms in glutathione-S-transferases (GSTs) have been associated with PD (119, 184), the exact role that these enzymes play in dopaminergic degeneration is far from understood. Loss-of-function mutations of GST-S1 enhance dopaminergic degeneration induced by Parkin mutants in Drosophila, while overexpression of GST-S1 prevents neuronal cell loss (183). Interestingly, inhibition or downregulation of GST-pi, which is found highly expressed in PD brains compared with controls (160), increases sensitivity to MPTP and dopamine toxicity (81, 165). However, there is no clear evidence of whether the protective mechanism of GST-pi against dopaminergic cell death is associated with its catalytic function in GSH conjugation to electrophilic substrates (Fig. 2), its ability to directly regulate signal transduction pathways through protein–protein interactions, or its recently proposed role in protein glutathionylation, reviewed in this Forum and in the article by Tew et al. (168).
Antioxidant Gene Transcription
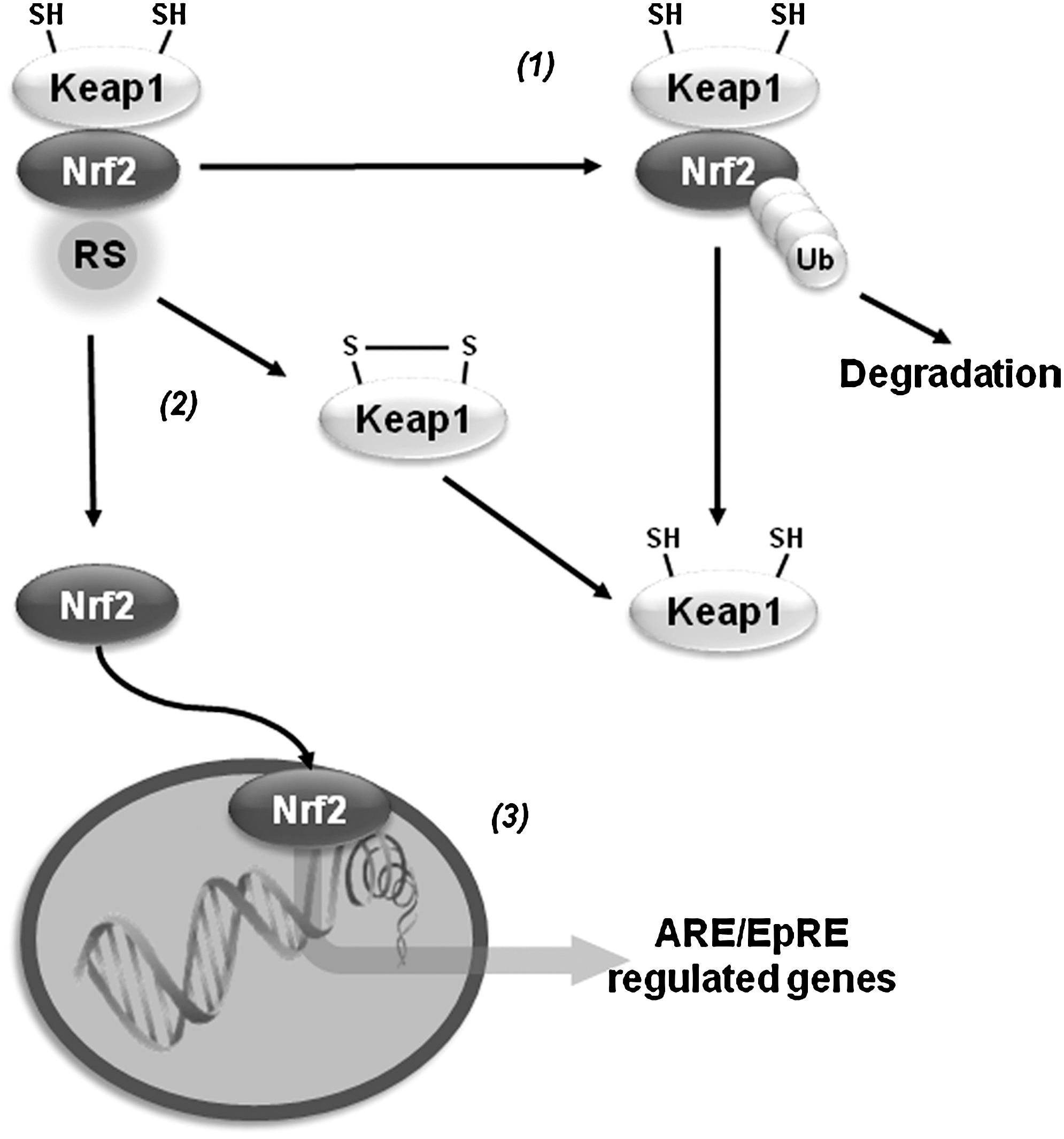
Oxidative stress activates the transcription of a variety of antioxidant genes through cis-acting sequences known as antioxidant response elements (AREs) or electrophile-responsive elements (EpREs). Members of the Cap-N-Collar family of transcription factors, including the nuclear factor (erythroid-derived 2)-like 1 (Nrf1) and 2 (Nrf2), have been identified to bind ARE. Nrf2 regulates the expression of proteins that directly or indirectly exert antioxidant functions and that also enhance GSH synthesis and regeneration. Under resting conditions, Nrf2 is kept in a cytosolic complex with the kelch-like ECH-associated protein 1 (Keap1, also called inhibitor of Nrf2 or INrf2), where it is subjected to proteasomal degradation. Oxidants or electrophiles induce a conformational change in Keap1 that allows the translocation of Nrf2 to the nucleus for transduction. Keap1 contains zinc (Zn)-coordinated Cys273 and Cys288 that are sensitive to alkylation. Initial thiylation of cysteines is thought to be followed by thiol-disulfide exchange to form internal disulfides within Keap1. However, recent evidence suggests that distinct activators act on different sets of cysteines in Keap1, which translates into specific biological effects (cysteine code). Transcription of γ-glutamylcysteine synthetase (γ-GCS), G6PD, GPXs, PRDXs, TRX/TRXR, sulfiredoxin (SRX), and GSTs by Nrf2 synergizes to improve the capacity of the GSH/thiol-based antioxidant systems described previously to counteract oxidative damage (21). Similar to Nrf2, Nrf1 has also been shown to regulate the expression of γ-GCS (18). A protective Nrf2 haplotype associated with increased transcriptional activity was linked to decreased risk of PD (179). Initial reports indicated that DJ-1 affects the transcriptional function and stability of Nrf2 by preventing association of Nrf2 with Keap1 (40). However, a separate study was not able to confirm these results (61). Overexpression of Nrf2 protects against α-synuclein toxicity in Drosophila (14). Paraquat induces a decrease in the expression of Nrf2, which is paralleled by a decrease in γ-GCS and intracellular GSH levels (191), while 6-OHDA and maneb induce Nrf2 activation (113, 147). Nrf2 activators protect against MPTP and 6-OHDA (83, 164), while Nrf2 knockout mice show exacerbated neurodegeneration and inflammation induced by MPTP (22, 27, 149) (Fig. 7).
Metallothioneins in PD
Alterations in metal homeostasis are associated with PD. More specifically, Zn and Fe are found increased, while Cu levels are decreased in the substantia nigra of PD brains. In the cerebrospinal fluid and blood of PD patients, an increased level of Cu has been reported (19, 78, 135). Interestingly, higher intake of Zn reduces the risk of PD (123). In contrast, occupational exposure to Cu increases the risk to develop PD (68). Metallothioneins (MTs) are a family of small cysteine-rich proteins with free radical scavenging and heavy metal binding properties (Fig. 8). GSH acts as a reducing agent for free-radical-induced MT oxidation (171). GSH alone stabilizes Zn binding to MTs and the presence of GSSG (or any other oxidizing agent) results in a release of Zn that is synergistically increased by GSH (87). There are four mammalian MT isoforms, MT-I to MT-IV. MT-I (encoded by seven genes) appears to be a superior scavenger for free radicals compared with MT-II (100). Within the brain, the most abundant isoforms are MT-I/II produced primarily by astrocytes (36). Brain MTs are single polypeptides of 61–68 amino acids, including 20 highly conserved cysteines and no disulfide bonds. MTs usually bind 7 divalent metal ions (Zn2+) and up to 12 monovalent Cu+ ions, partitioned into 12 metal-thiolate clusters located in separate protein domains (182). In PD brains and in vivo PD models, expression levels of MTs are reduced (126, 148). In addition, increased MT-I/II expression in reactive astrocytes was found in PD brains (121). MT gene expression is regulated by AREs. Dopamine exposure increases MT-I/II expression, which is associated to Nrf2 binding to ARE of MT genes (125). In addition, MT-I and -II have been reported to protect against DAQ-induced toxicity (124). MT-I/II transgenic mice are resistant to MPTP-induced toxicity, which is linked to increased levels of complex I activity and coenzyme Q10 (158), while their knockout increases susceptibility to MPTP (54). MT-III is highly expressed in the normal human brain, where it is present in neurons and astrocytes as well as the extracellular space. 6-OHDA downregulates MT-III mRNA levels in the basal ganglia (126), and those of MT-I in the striatum (161). Cu2+ binds to α-synuclein and accelerates its aggregation (103, 142, 175). MT-III prevents cell death induced by 6-OHDA (80) and hydroxyl radical (•OH) generation from α-synuclein-Cu2+ complexes through removal of Cu2+ (69, 118). The ability of MTs to protect against oxidized dopamine products has been reported to involve direct scavenging by the formation of covalent adducts (64).
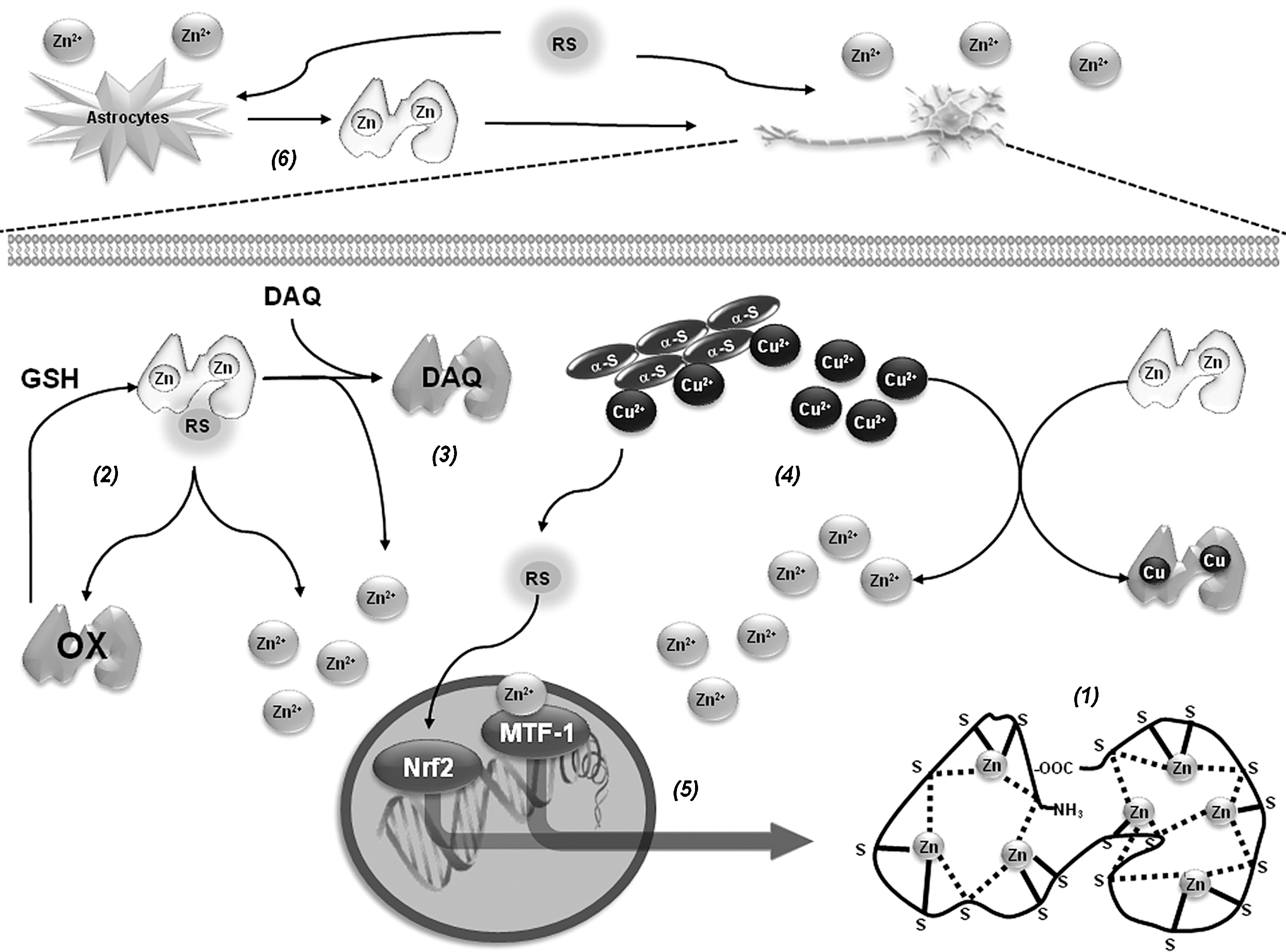
MT-I and -II are highly inducible in response to metals, oxidative agents, inflammation, and stress. Metal-induced synthesis of MTs is mediated through cis-acting DNA sequences known as metal-responsive elements (MREs), which are present in the promoter region (Fig. 8). The metal-responsive transcription factor-1 (MTF-1), also termed MRE-binding transcription factor-1 or metal regulatory transcription factor-1, is a pluripotent transcriptional regulator involved in cellular adaptation to heavy metal exposure and oxidative stress, and is the main inducer of MT expression (70). Double-mutant flies for Parkin and MTF-1 genes are not viable, while overexpression of MTF-1 of Drosophila MT dramatically augments life span and protects mitochondrial structure (155). People who abused methamphetamine or other amphetamine-like stimulants have been found to be more likely to develop PD. Methamphetamine induces dopamine release and oxidative damage in dopaminergic cells. Overexpression of MT-II protects against methamphetamine-induced cell death (3). (+/-)3,4-Methylenedioxymethamphetamine (MDMA or “ecstasy”) also increases MT-I and MT-II mRNA levels and double knockout mice were more vulnerable to MDMA toxicity (189).
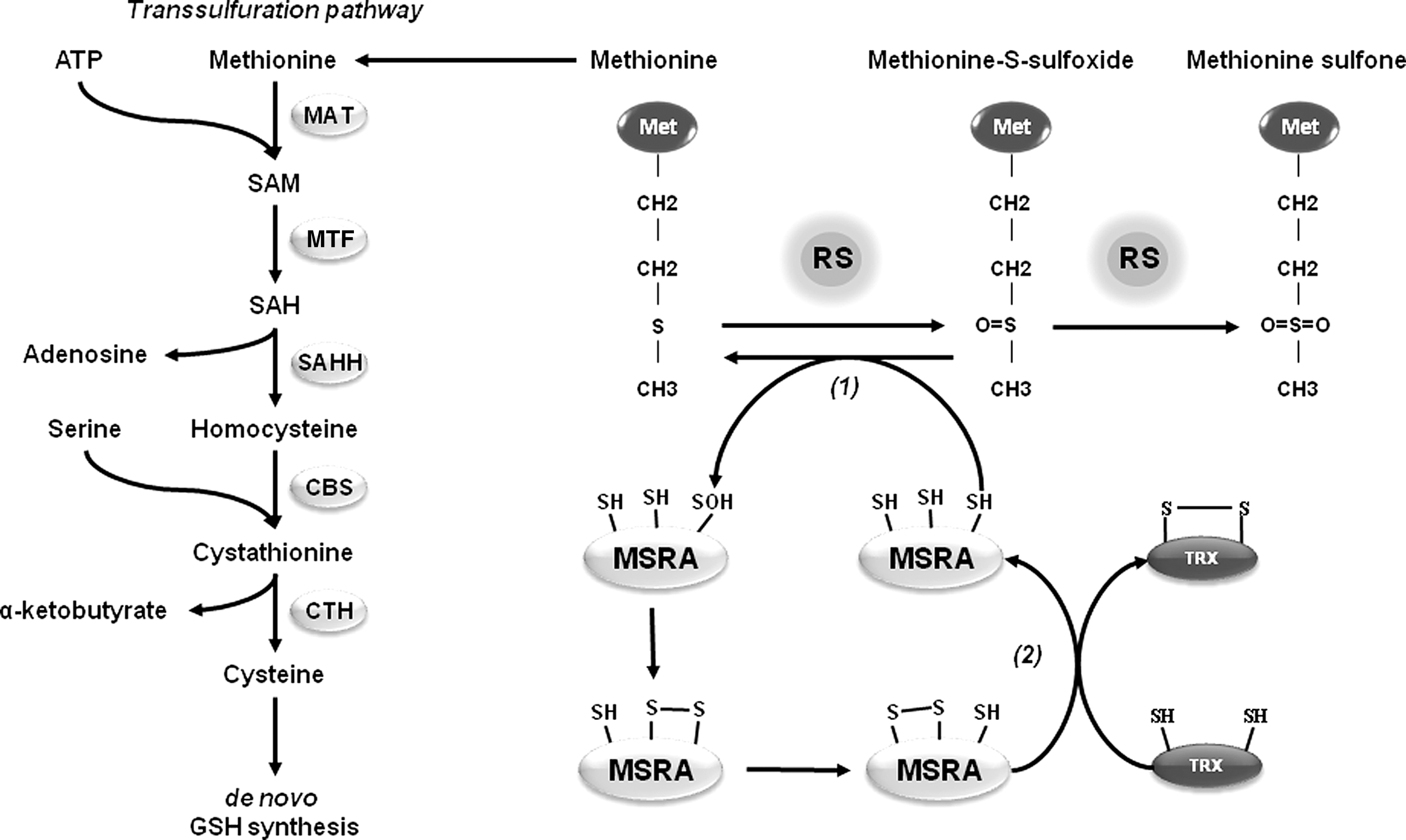
Methionine Sulfoxide Reductases in PD
Methionine is one of the most vulnerable amino acid residues to oxidation by RS. Addition of an extra oxygen atom oxidizes methionine to methionine sulfoxide (MetSO), and a strong oxidant might further oxidize MetSO to methionine sulfone (MetSO2) (Fig. 9). Two different stereoisomers of methionine sulfoxide,
Potential Therapeutic Approaches
As described above, distinct thiol-based redox systems have been reported to protect against experimental PD. Thus, it could be envisioned that GSH replenishment or gene transfer of genes involved in de novo GSH synthesis and GSH-dependent antioxidant systems, such as GPXs, could represent a potential therapeutic approach against oxidative stress associated with PD. Accordingly, cell-permeable GSH analogs or NAC (30, 38, 128, 136, 137, 169), and overexpression of G6PD and GPX1 protect dopaminergic cells from experimental PD in vivo (17, 117, 145). The main goals of gene therapy in PD are to restore the ability of the brain to produce dopamine and to stop progressive neurodegeneration. Gene delivery using adenoviral vectors has been the most commonly utilized approach for gene therapy and a number of phase I and II clinical trials using viral vectors as means to transport enzymes to the striatum of PD patients are currently being tested. Unfortunately, no available treatment has yet proven to have a definitive neuroprotective effect for patients with PD (76). The knowledge generated in the last years supports a protective role of thiol-redox systems against PD. Recently, lentiviral delivery of PRDX2 has been shown to protect against 6-OHDA-induced dopaminergic cell death in vivo (79). Thiol-based antioxidants function as integrated coupled systems, where the activity of enzymes, such as GPX, GRX, and PRDX, depends on the reducing capacity of their reducing counterparts GSH/GR and TRX/TRXR. Thus, it is important to consider that simultaneous gene transfer of thiol-redox systems might have an increased protective effect compared with the overexpression of single enzymes.
Oxidative stress in PD is associated with mitochondrial dysfunction. Thus, mitochondrial-targeted antioxidants also represent a preemptive approach to counteract oxidative damage. Overexpression of mitochondrial manganese superoxide dismutase (MnSOD or SOD2) has been shown to protect against MPTP-induced toxicity (106). Mitoquinone or mitochondria-targeted ubiquinone (MitoQ)—which comprises the lipophilic triphenylphosphonium cation covalently linked to ubiquinone, the active antioxidant moiety of CoQ10, enabling MitoQ to cross membranes and to accumulate several-hundred fold within mitochondria—has been recently described as a potent mitochondrial antioxidant. However, in a double-blind placebo-controlled study, MitoQ did not slow PD progression (166). Other sources of RS formation that have been suggested to participate in PD are metal complexes and dopamine metabolism. In this regard, MT-III prevents ROS formation induced by α-synuclein-Cu2+ complexes in vitro, but this needs to be translated into experimental PD models in vivo (118). The monoamine oxidase inhibitors (MAO-Is) are used clinically as PD therapeutic agents (85). By inhibition of dopamine metabolism, MAO-Is decrease the formation of H2O2 (41, 114). Furthermore, MAO-Is also protect against α-synuclein toxicity (26).
Transcriptional regulation of antioxidant genes by Nrf2 might also represent an approach to prevent dopaminergic cell death as demonstrated by its protective effect against α-synuclein toxicity in vivo (14). However, mitochondrial dysfunction in PD is not only expected to generate oxidative stress but also energy deficiency. The proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) is a transcription co-activator for nuclear receptors that coordinates a number of gene expression programs to promote mitochondrial biogenesis and oxidative phosphorylation, as well as antioxidant gene expression (39). Overexpression of PGC-1α protects against neuronal cell death induced by MPTP, rotenone, and α-synuclein (127, 197). However, viral-mediated overexpression in vivo of PGC-1α induces dopaminergic cell loss, suggesting that PGC-1α levels cannot be increased for therapeutic purposes above physiological levels (37).
Conclusions and Perspectives
Thiol-redox signaling has emerged as a central mechanism by which the generation of RS and oxidative stress are integrated into the activation of distinct signaling cascades regulating diverse homeostatic processes and pathological outcomes. Dopaminergic cell death associated with PD is largely linked to oxidative stress, and recent studies demonstrate the importance of thiol-redox signaling regulating neuronal loss. GSH depletion is one of the earliest biochemical alterations described during the progression of PD, and GSH-based antioxidants, such as GPX1, have been demonstrated to exert protective effects against dopaminergic cell death in experimental PD. Thiol-based oxidoreductases, such as PRDXs and TRXs, have also been demonstrated to mediate protective effects by scavenging peroxides and regulating the activation of pro-apoptotic signaling cascades, respectively. On the other hand, oxidative post-translational protein modifications in cysteines, including cysteine hydroxylation, glutathionylation, and nitrosylation, have been shown to regulate the function of PD genes (DJ-1 and Parkin), mitochondrial activity (complex I), and anti-apoptotic proteins (XIAP). The results summarized here demonstrate the relevance of thiol-redox signaling cascades in PD and guarantee further interest into elucidating novel therapeutic approaches against PD by regulation of thiol-redox homeostasis. In this regard, potential therapeutic approaches against oxidative stress and redox imbalance in PD include: (i) replenishment of the intracellular GSH pool and/or gene transfer of thiol-based antioxidants systems, such as GPX(GRX)/GR/NADPH and PRDX/TRX/TRXR; (ii) the use of mitochondrial-targeted antioxidants, MTs and/or MAO-Is to counteract oxidative stress in PD, which is mainly associated with impairment of mitochondrial function, RS formation by metal complexes, and dopamine oxidation; and (iii) transcriptional regulation of both antioxidant and gene expression programs to promote mitochondrial biogenesis and oxidative phosphorylation by Nrf2 and PGC-1α, in order to target both oxidative stress and energy failure due to mitochondrial dysfunction in PD.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health Grant P20RR17675 Centers of Biomedical Research Excellence (COBRE), and Interdisciplinary Grant from the Research Council and the Life Sciences Grant Program of the University of Nebraska-Lincoln. LZF received a Post-doctoral Fellowship from the National Council of Science and Technology (CONACYT 211456) of Mexico.