
Abstract
Aims:
Morphine signaling via the μ-opioid receptor (MOR) is coupled to redox-dependent zinc release from endogenous stores. Thus, MOR activation stimulates the complex formed by RGSZ2 (a regulator of G protein signaling) and neural nitric oxide synthase (nNOS) to produce NO, and to recruit PKCγ and Raf-1 in a zinc-dependent manner. Accordingly, we investigated whether redox regulation of zinc metabolism was unique to the MOR, or if it is a signaling mechanism shared by G-protein coupled receptors (GPCRs).
Antioxid. Redox Signal.
17, 1163—1177.
Introduction
In neural tissue, nitric oxide synthase (nNOS) is primarily responsible for the production of nitric oxide (NO), which regulates a broad range of physiological functions. NO exerts most of action by reacting with cysteine Zn/S sites in zinc finger proteins, promoting the release of zinc and the S-nitrosylation of these thiol groups (32, 49). The nitrosylation of cysteines is readily reversible, typically via the S-nitrosoglutathione reductase or thioredoxin systems (31, 49).
Innovation
In the adult nervous system, the production of NO and the eventual release of zinc ions is mainly mediated by nNOS activity, under the control of the glutamate NMDAR. Here, we describe a new feature shared by several GPCRs, whereby receptor activation is coupled to NO production and RGSZ2-mediated release of zinc ions. This GPCR-mediated process converts NO signals into zinc signals, altering the function of Redox sensor proteins such as PKCγ and Raf-1. These findings may have important implications for understanding and treating neurodegenerative diseases and mood disorders, in which GPCRs and zinc metabolism are implicated.
While nNOS activation is typically linked to the NMDA glutamate receptor, the G protein-coupled μ-opioid receptor (MOR) also stimulates NO production by regulating nNOS (47). The MOR is associated with nNOS through a series of regulatory proteins and, for example, the RGSZ2 protein binds the PDZ binding motifs located in the N-terminal region of nNOS, thereby inhibiting NO production (14). RGSZ2 binding to the histidine triad nucleotide-binding protein 1 (HINT1) connects it to the MOR C-terminus (1, 18, 43). Indeed, the MOR agonist, morphine, activates GαGTP subunits, some of which bind to the RGSZ2-nNOS complex and provoke transient production of NO (43, 47). One of the main physiological consequences of MOR-mediated NO production is the release of zinc ions from endogenous stores. Subsequently, these ions can bind to cysteine-rich regions in PKCγ and Raf-1, promoting their simultaneous recruitment to the HINT1 protein at the MOR (45, 46). The role of zinc signals as intracellular messengers is dependent on their temporal and spatial compartmentalization. Notably, rather than protecting SH groups from oxidation, the strong binding of zinc to sulfur in cysteine thiol groups increases the vulnerability of thiolates to oxidation (33). This results in redox-active zinc/cysteine coordination environments that respond to mild physiological oxidation by releasing zinc ions, such as that produced by NO. By contrast, zinc-free cysteines remain unaffected and ready to capture free zinc ions (20). When coupled with the reversibility of zinc binding upon reduction of the oxidized sulfur, this property makes cysteine-coordinated zinc fingers efficient redox sensors (33). Binding of HINT1 and RGSZ2 to cytosolic regions of GPCRs is not restricted to the MOR; we have also detected this interaction in the cannabinoid CB1 receptor, as well as to a lesser extent in the δ-opioid receptor (DOR) (10). As activation of the PLC/PKC and ERK1/2 pathways is shared by many GPCRs, we investigated whether regulation of nNOS and Redox release of endogenous zinc is a general feature of GPCR signaling in nervous tissue. Given the exquisite compartmentalization of nNOS regulation to the MOR's C terminus, we explored the possibility that the cysteine-rich domain (CRD) of the RGSZ2 N terminus could act as source of endogenous zinc ions released by GPCR-activated nNOS.
Results
Activation of different GPCRs promotes the release of endogenous zinc ions via a mechanism involving nNOS/NO
We sought to investigate whether the redox-induced mobilization of endogenous zinc was a signaling property shared by a range of neural GPCRs. First, we analyzed the capacity to mobilize zinc of agonists that act on GPCRs known to contain HINT1 at their corresponding C termini, such as MOR, CB1R, and DOR (10). As reported previously (45), incubation of mouse brain slices with morphine increased Newport Green fluorescence (a measure of the zinc ions that are being released from endogenous stores), a MOR-mediated effect that was attenuated by the opioid antagonist naloxone. Likewise, the zinc-mobilizing effect of deltorphin II, a DOR agonist, was attenuated by its selective antagonist, naltrindole and similarly, the effect of the CB1R agonist, WIN 55-212-2, diminished following LY320135 administration, a specific antagonist of this receptor (Fig. 1). Taken together, these findings demonstrate that metabotropic GPCRs for endogenous opioid peptides (MOR and DOR) and for the arachidonic acid derivative anandamide (CB1R) are all capable of provoking the release of endogenous zinc ions.
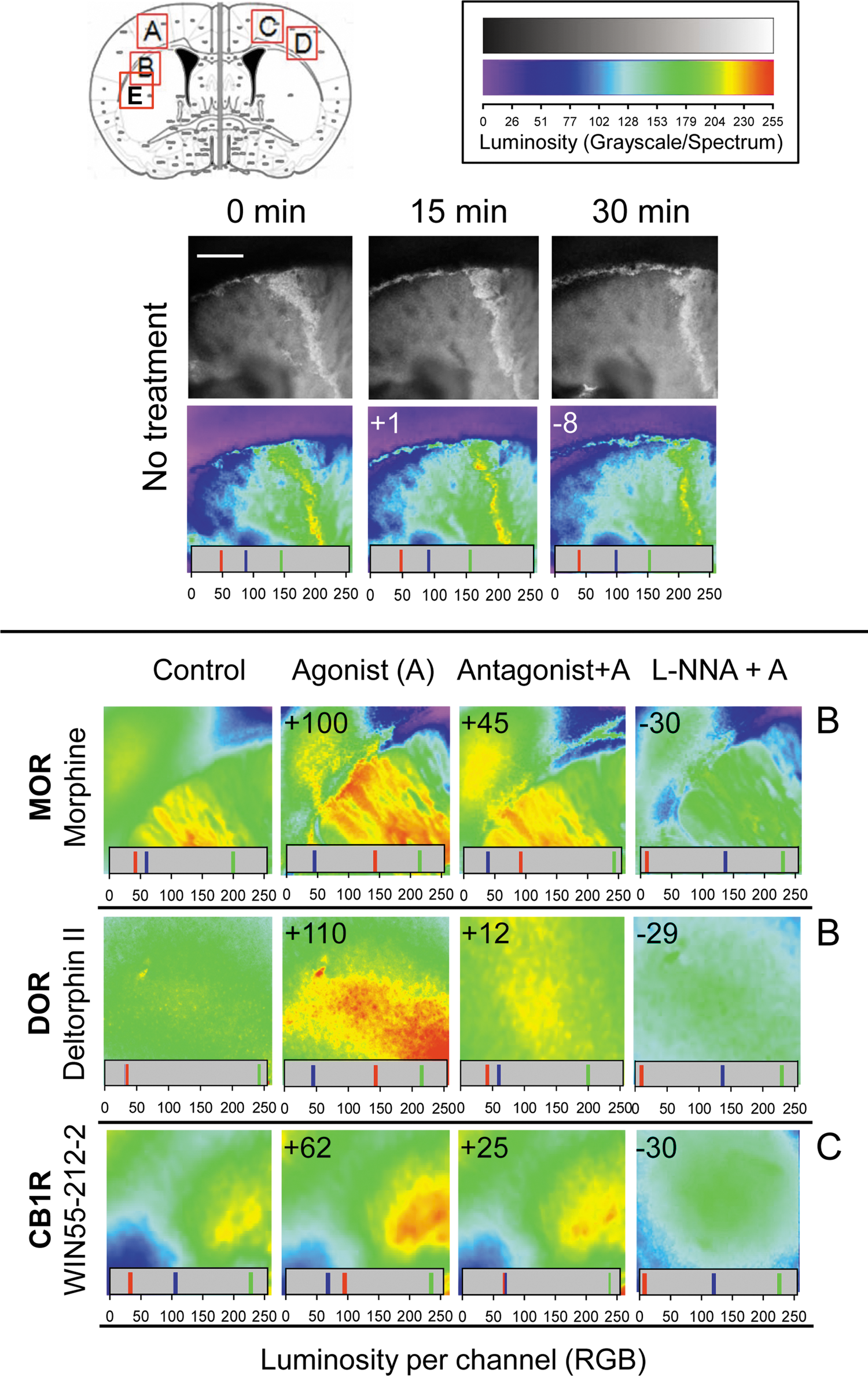
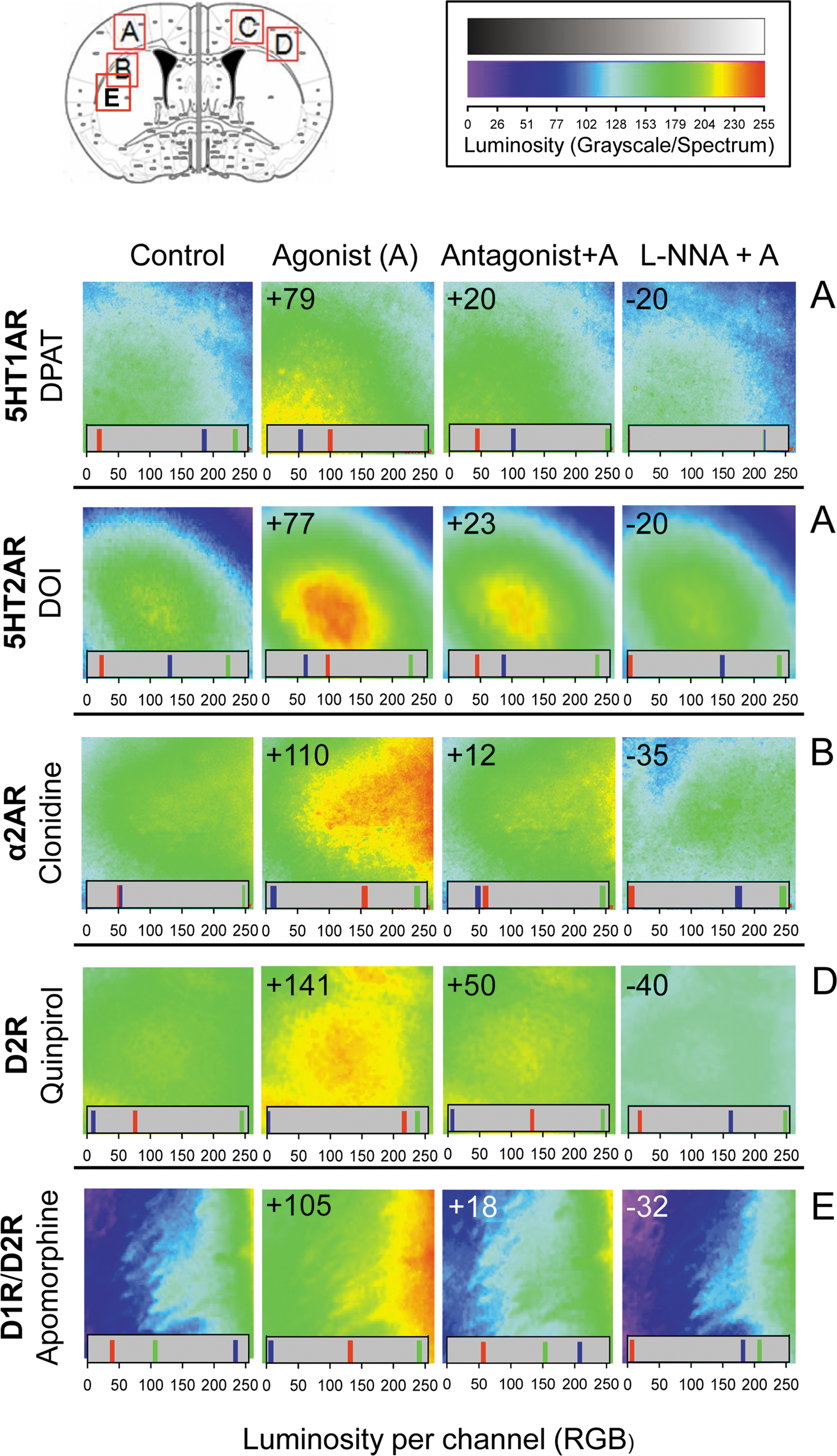
We found that several neural receptors for biogenic amines (5HT1A and 5HT2A serotonin receptors, the α2A noradrenegic receptor, and D1 and D2 dopamine receptors) mediated the mobilization of zinc ions in response to agonist stimulation (Fig. 2). Dopamine receptor-mediated zinc mobilization was promoted by quinpirole, a specific D2 agonist, and by apomorphine, which acts on both D1R and D2R types. These effects were attenuated by GPCR antagonism, and they were also prevented by NOS/NO inhibition.
The RGSZ2 protein is a redox sensor that releases zinc ions in response to NO
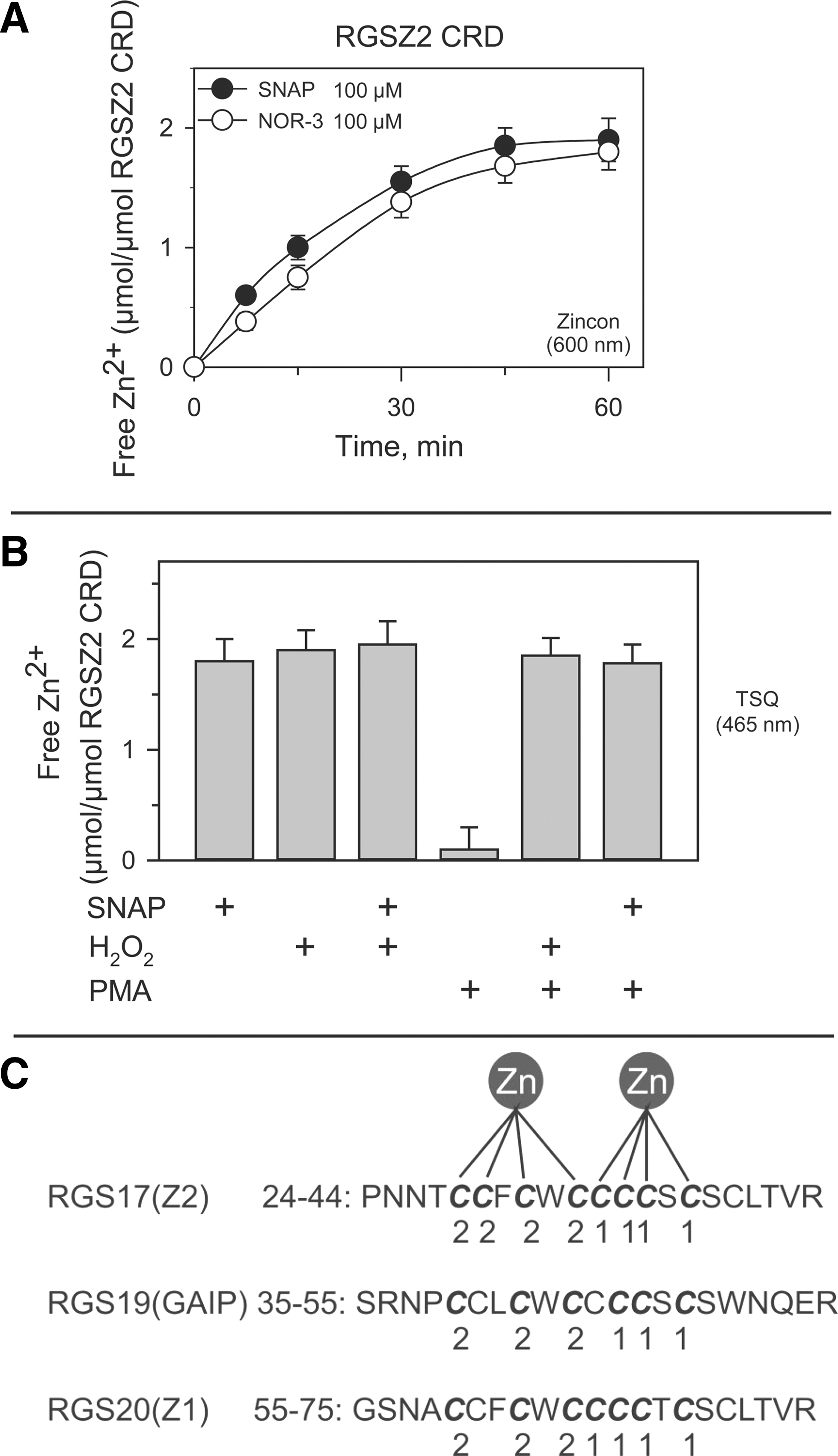
The RGS17 protein, also known as RGSZ2, belongs to the Rz subfamily of RGS proteins (21, 30). Immediately upstream of the RGS domain, RGSZ2 contains a series of PDZ binding motifs that bind to the PDZ domain in nNOS (residues 61–64 MESI, 75–78 ADEV, and 76–79 DEVL). In the MOR, the RGSZ2–nNOS interaction serves to regulate NO production and the release of zinc from intracellular stores in response to morphine binding (14). Importantly for NO signaling, the amino terminus of RGSZ2 features a cysteine-rich domain (CRD: 28 to 40) that contains 9 cysteine residues out of a total of 13.
To determine whether RGSZ2 CRD could bind zinc ions and release them in response to NO donors, the N terminal region on RGSZ2 (1–80) containing the CRD was exposed to the NO generators SNAP and NOR-3 (100 μM), which released about two molar equivalents of zinc (Fig. 3A). A similar level of zinc release was observed in response to oxidation induced by 2.5 mM hydrogen peroxide, a concentration that induces maximal release of zinc ions from PKC zinc fingers (24, 50), or by a combination of SNAP and H2O2. Phorbol-12-myristate-13-acetate (PMA) produced no detectable release of zinc ions from the RGSZ2 CRD (Fig. 3B). Computational analysis revealed a linear arrangement of double zinc motifs in the RGSZ2 zinc finger [metal binding sites:
Impairment of RGSZ2 protein expression markedly attenuates GPCR-induced zinc release from endogenous stores
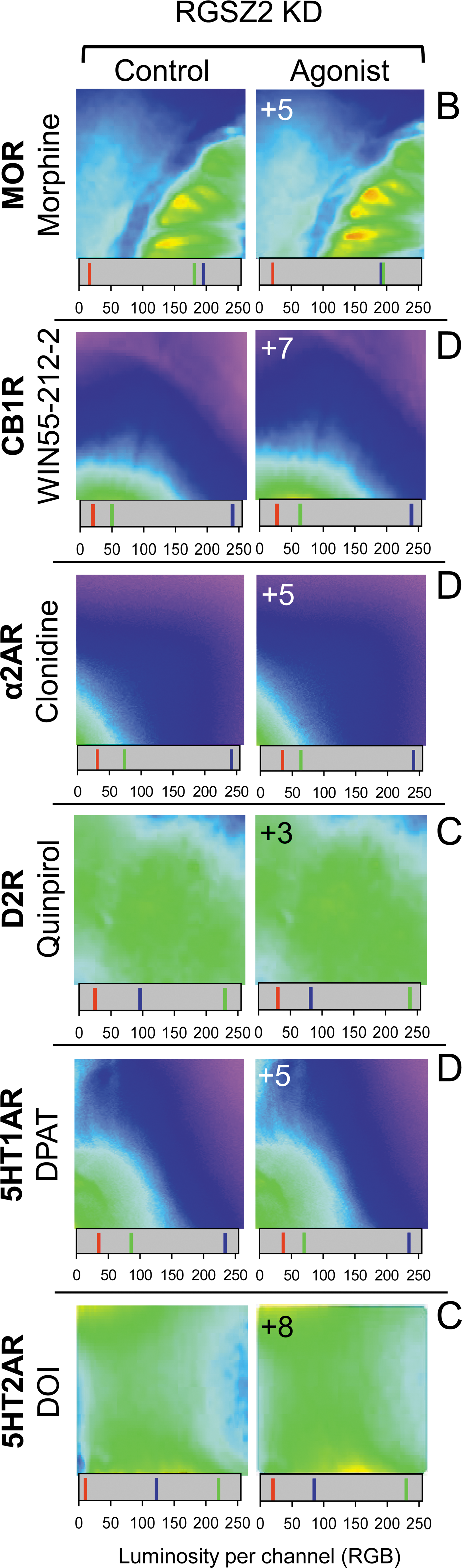
The RGSZ2 protein appears to play an essential role in cellular homeostasis and, indeed, targeted deletion of its gene is lethal. Thus, the relevance of this protein was determined by reducing its expression in adult mice (11, 14). In brain slices from mice with impaired RGSZ2 expression, GPCR agonists induced the release of nondetectable to moderate levels of endogenous zinc (Fig. 4).
GPCRs associate with RGSZ2 and HINT1 proteins in brain synaptosomes
The association of the RGSZ2/nNOS signaling complex with the HINT1 protein at the MOR C terminus appears necessary for morphine to regulate the production of NO and the subsequent release of zinc from endogenous zinc finger proteins (1, 14, 46). Therefore, we explored whether the GPCRs of interest were also associated with these proteins in brain synaptosomes. Antibodies recognizing external epitopes on the GPCRs were used to immunoprecipitate the corresponding receptor (Fig. 5A), thereby avoiding the blockage of their cytosolic sequences that may occur due to interaction with third partner proteins, such as G proteins, RGS proteins, or others. The ex vivo assays revealed that all GPCRs studied co-precipitated RGSZ2, nNOS, and HINT1 proteins (Fig. 5B). This finding indicates that, in addition to regulating G proteins, GPCRs may also promote the activation of nNOS/NO pathways.
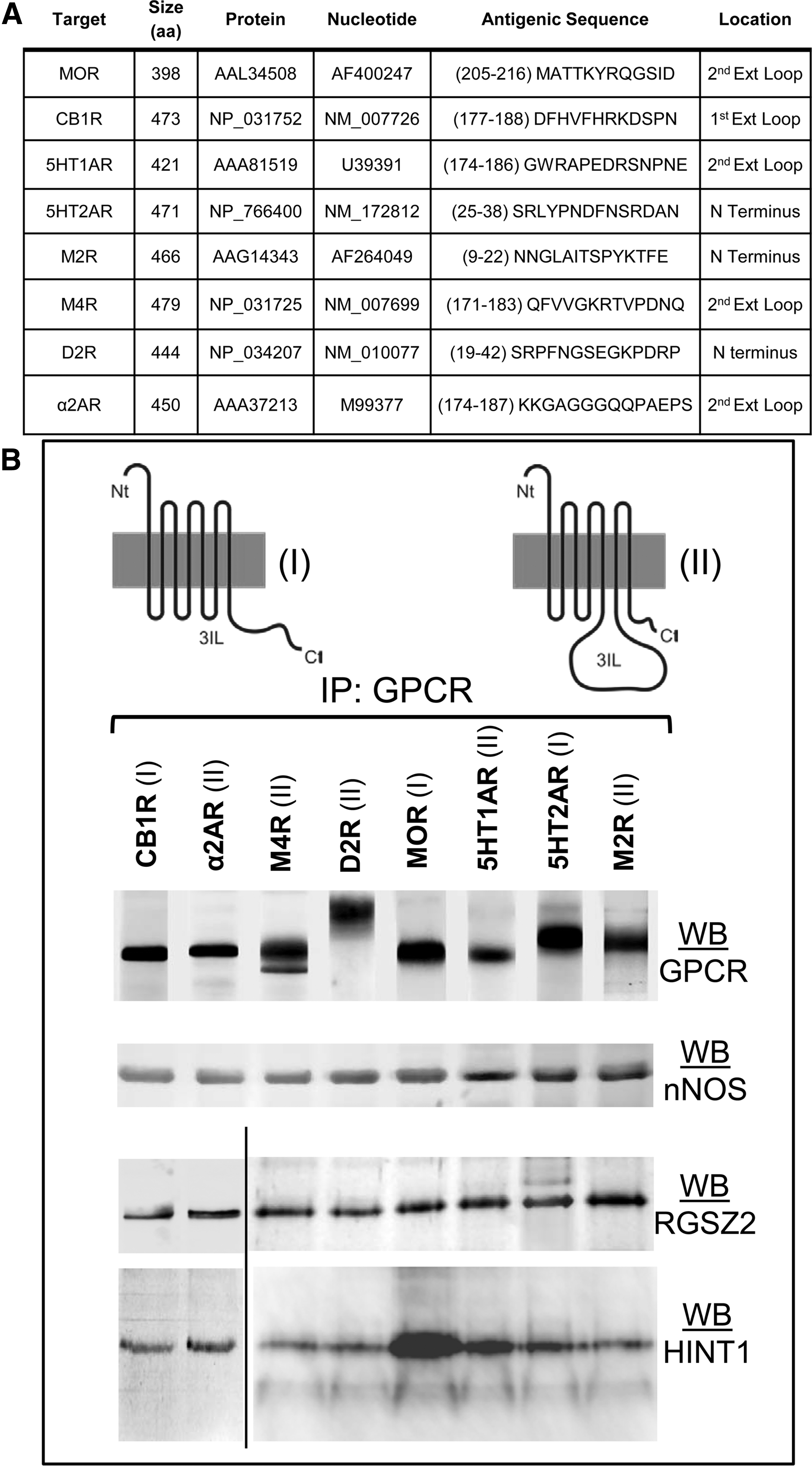
RGSZ2 physically interacts with the cytosolic C terminus or the third internal loop of GPCRs
GPCRs exhibit notable differences in the length of the C terminus and third internal loop, regions that are essential for their interaction with signaling proteins and the regulation of their activities. When we studied the characteristics of these sequences for the GPCRs of interest (Fig. 5B), a short C terminal sequence was usually balanced by a long third internal loop and vice versa. Therefore, apart from the regulation of G proteins depending on the class of GPCR, cytosolic interactions probably occur via either the C terminus or the third internal loop.
The MOR C terminus requires the HINT1 protein to recruit RGSZ2 (43), although RGSZ2 can also interact directly with other GPCRs. The formation of RGSZ2–GPCR complexes in vivo was supported by the results the bimolecular fluorescence complementation (BiFC) analysis (Fig. 6). Chinese hamster ovary (CHO) cells were transfected with a mix of RGSZ2 coupled to VC155 (1:1) and the GPCR of interest coupled to the C terminus of VN173. The physical association of RGSZ2 with the GPCR allows the VC155 and VN173 fragments to form a stable fluorescent complex, which could be detected in several cells.
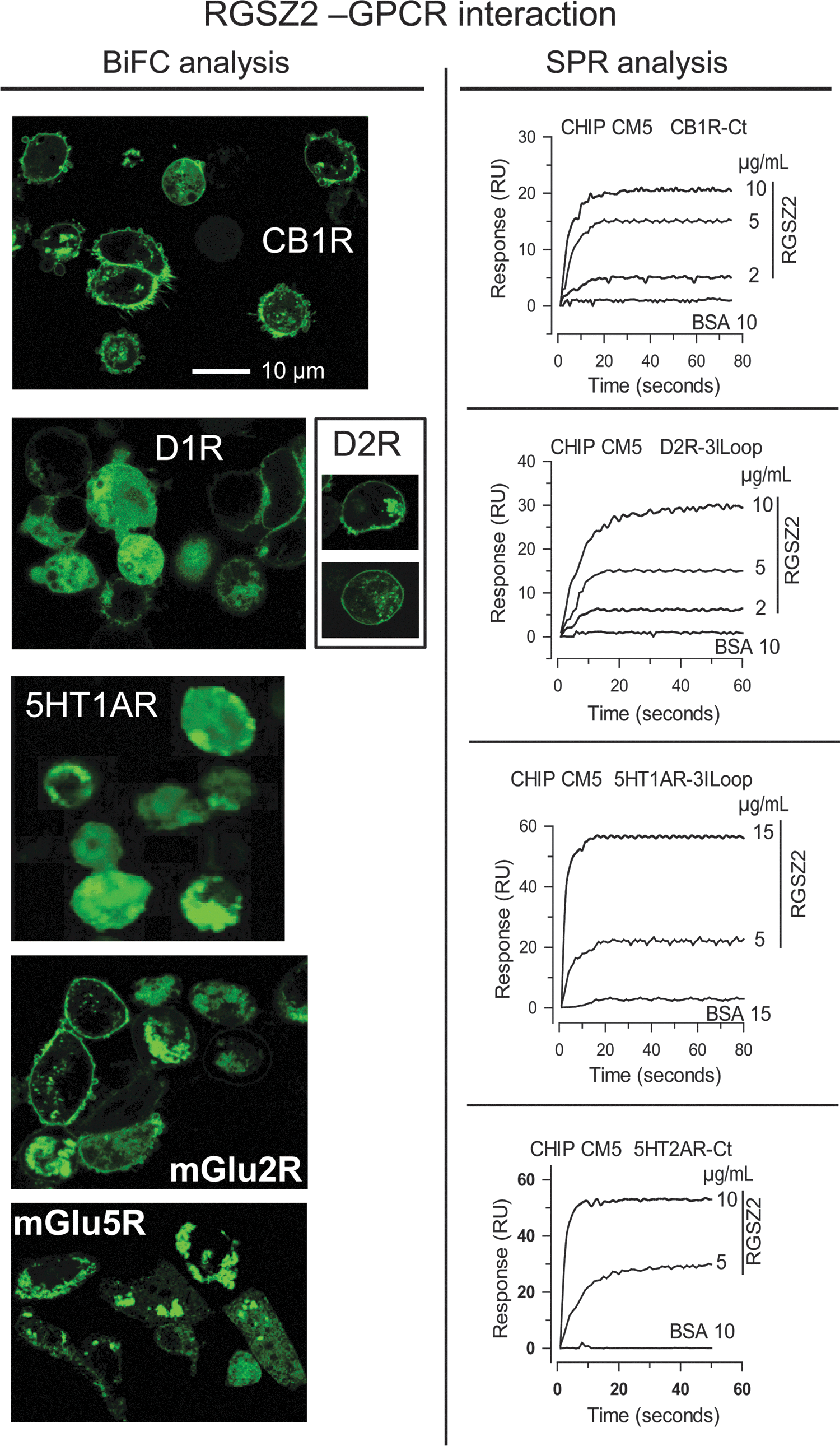
The participation of amino acid sequences of the C terminal or the third internal loop in the GPCR–RGSZ2 interaction was determined by surface plasmon resonance analysis. The C terminal sequences of CB1 and 5HT2A, and the third internal loop sequences of α2A, M2, M4, D2, and 5HT1A, were covalently coupled to a CM5 sensor chip. Real-time interactions were studied by passing a solution containing the RGSZ2 protein over the sensor surface, and RGSZ2 was seen to interact directly with the C terminus or third cytosolic loop of these receptors. These findings demonstrate that RGSZ2 can be recruited to the GPCR environment either directly or through its binding to the HINT1 protein.
Zinc supports the translocation of signaling proteins to the GPCR environment in vitro and ex vivo
The interaction through which the RGSZ2 protein binds to the HINT1 protein was not affected by adding zinc to the medium. However, as purified recombinant zinc-binding proteins contain zinc, the RGSZ2-HINT1 interaction was also evaluated after experimental removal of this metal ion, which did not affect this association (Fig. 7A). Similarly, the binding observed between recombinant HINT1 and PKCγ was not affected by exogenously added zinc, although HINT1-PKCγ binding was clearly impaired after removing protein-bound zinc. In these circumstances, the addition of free zinc ions to the incubation media promoted a stronger interaction than that observed prior to zinc removal (Fig. 7A).
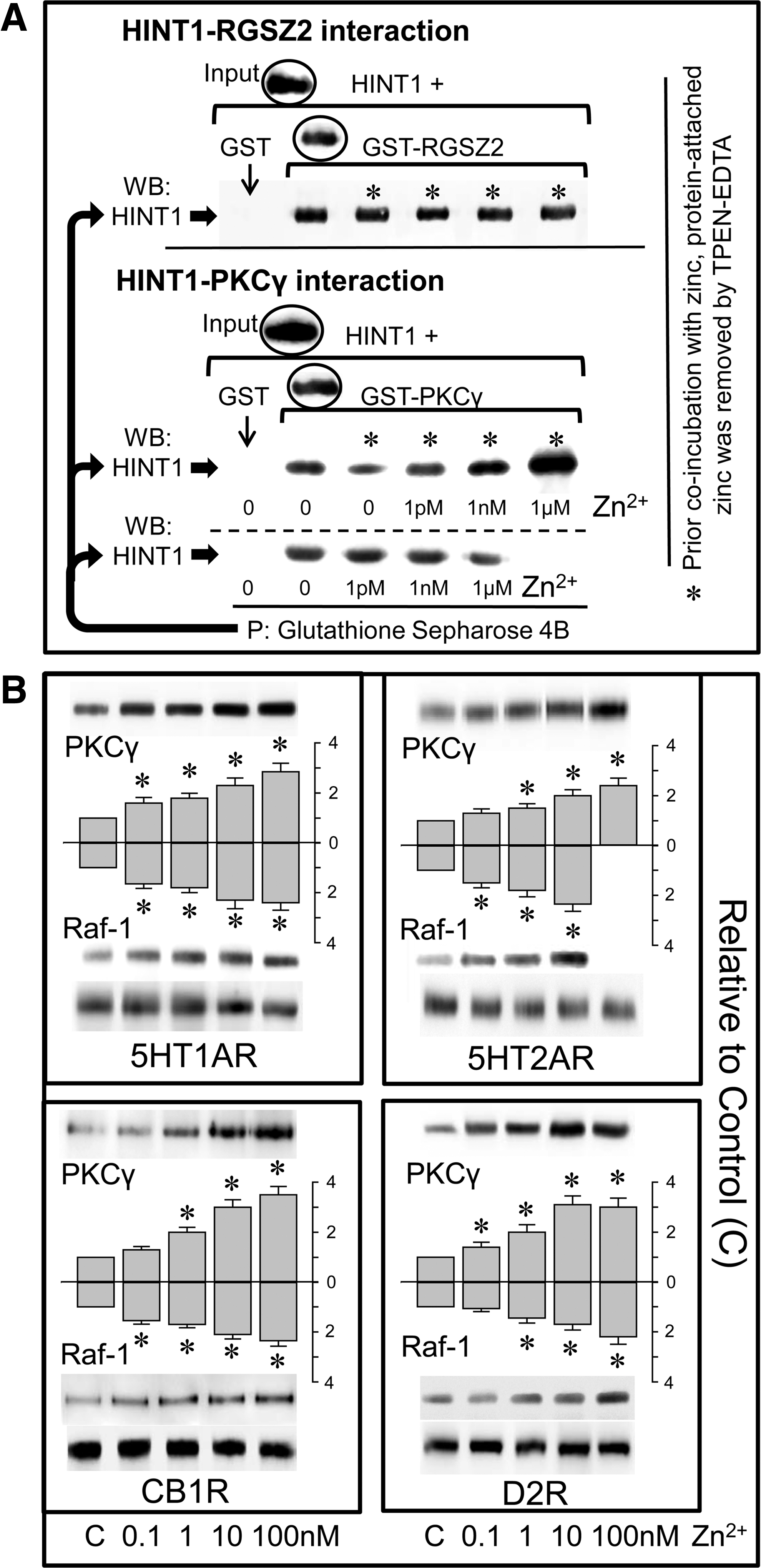
I
Besides the PKCγ CRD, that of Raf-1 also exhibits a zinc-dependent association with the MOR (45). Using membranes derived from mouse brain tissue, we determined the capacity of added zinc ions to promote the association of these signaling proteins with various GPCRs. The membranes were exposed for 4 h to increasing concentrations of zinc at 4°C before the GPCR was immunoprecipitated, and in the presence of low nM zinc, 5HT1A, 5HT2A, CB1, and D2 associated with Raf-1 and PKCγ (Fig. 7B). Moreover, the NO donor SNAP enhanced the association of GPCRs with PKCγ in these membrane preparations, although this interaction was attenuated in the presence of the heavy-metal chelator, TPEN (Fig. 8A). Phorbol esters and calcium are cofactors that contribute to the activation of the conventional PKC isoforms. The binding of DAG or phorbol esters (PMA) to the regulatory domain of PKC causes the release of some of the associated zinc ions (23, 24, 50), as well as promoting the release of PKC from the GPCR. In our membrane assays, we also observed that PMA diminished the association of PKCγ with all the GPCRs evaluated (Fig. 8B). In vitro, PMA disrupted the association between recombinant HINT1 and PKCγ proteins mediated by zinc, and promoted the activation of the PKCγ released (Fig. 8C).
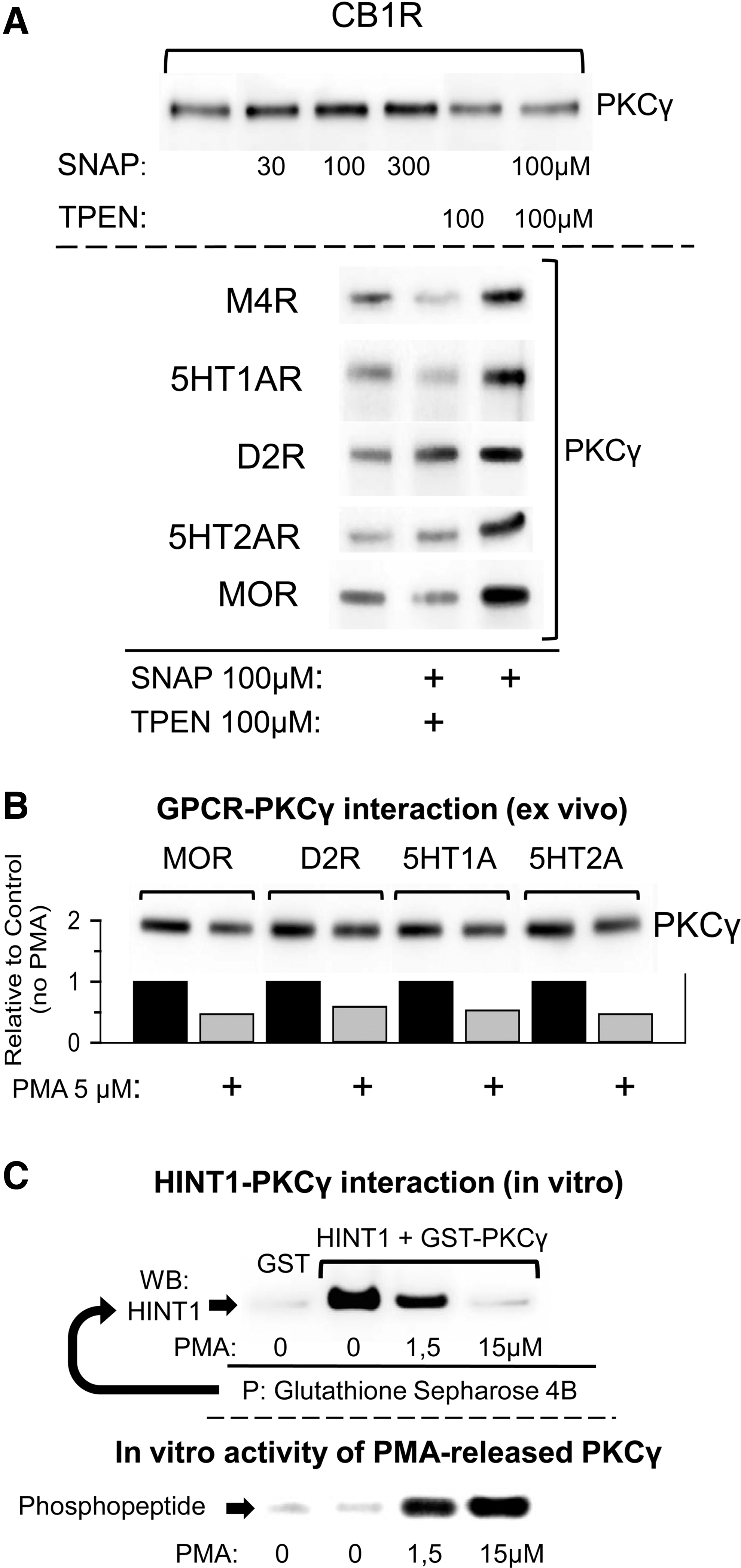
Discussion
In the present study, the regulation of NO production and the subsequent release of zinc ions from intracellular stores by neuronal GPCRs have been described. In the light of this data, the RGSZ2 protein emerges as a potential redox transducer that can be regulated by a variety of GPCRs. Zinc ions released from the RGSZ2 zinc finger domain by the action of NO can facilitate the coupling of redox sensor proteins such as PKCγ or Raf-1 to the C terminal or third internal loop of these metabotropic receptors. Thus, redox signaling represents a process used commonly by the most abundant subfamily of cell surface receptors, mobilizing zinc ions from intracellular stores to successfully integrate their signals into adaptive neuronal processes.
The GPCRs analyzed all associate with the RGSZ2 zinc finger protein and with the HINT1 protein, which recruits a series of regulatory proteins to the GPCR's environment in a NO and zinc-dependent manner, including PKCγ and Raf-1. The RGSZ2 and HINT1 proteins are widely distributed throughout the CNS (11, 28, 30), and they co-localize with GPCRs in the cell membrane where they contribute to the propagation of extracellular signals (15). The GPCR-mediated activation of PKCγ and ERK1/2 kinases is part of the negative regulation of certain GPCRs, such as the MOR and CB1R (9, 13). Thus, NO signaling participates in GPCR desensitization, although it is also required for the GRK- and β-arrestin-dependent internalization of these receptors. The agonist-induced production of NO separates β-arrestin from NOS binding and it targets the released β-arrestin to clathrin, initiating internalization of the GRK-phosphorylated GPCR (41). These are just a few examples of the relevance of redox processes and zinc mobilization in GPCR-mediated neuronal signaling.
The link between the glutamate NMDAR, NO production, and nitrosative signaling has been well documented. Thus, in postsynaptic neuron nNOS binds to PDZ binding motifs in the PSD-95 scaffold protein via its N terminal PDZ domain. NMDAR activation then leads to calcium permeation and the formation of intracellular calcium–calmodulin complexes that activate nNOS to produce NO, as well as producing other effects. NMDAR signaling is propagated by NO-mediated S-nitrosylation of the cysteine residues of many proteins, such as N-ethylmaleimide sensitive factor and stargazing, which directly regulate the function of the AMPAR, and that of the NMDAR indirectly (49). The mechanisms underlying GPCR-induced NO/zinc signaling have principally been analyzed for the MOR, identifying the fundamental role of HINT1. The HINT1 protein is a member of the HIT family of proteins that display conserved sequence identity in a broad range of organisms including mycoplasma, plants, and humans. HINT1 exists as a homodimer that specifically binds zinc ions without affecting its tertiary structure (25, 43), suggesting that zinc participates in HINT1's interaction with third party proteins. Interestingly, HINT1 was initially named as a PKC inhibitory (PKCi) protein, as it binds to the regulatory domain of conventional PKC isoforms and stabilizes this kinase in its inactive form (25, 35). Like the PSD-95, the RGSZ2 protein contains PDZ binding motifs that recruit nNOS, and these RGSZ2–nNOS complexes couple to the cytosolic C terminus of the MOR via the HINT1 protein (1, 14, 18, 43). The interaction of HINT1 with RGSZ2 does not require zinc ions, although binding of PKCγ to the HINT1 protein is clearly dependent on this metal ion. The binding of zinc ions to the PKCγ C1 regulatory domain promotes its folding into the inactive conformation of the kinase (50) that can interact with the HINT1 protein. Accordingly, zinc-depleted PKCγ displays poor affinity for the HINT1 protein, while this interaction was strongly enhanced in the presence of zinc ions. This observation indicates that zinc also participates in this intermolecular interaction probably trough is simultaneous binding to HINT1 histidines and PKCγ zinc finger cysteines.
Thus, a series of concurrent processes position the inactive PKCγ in the MOR environment where its activation can be tightly regulated to prevent the unwanted spreading of its effects. In this domain, MOR-activated PLCβ provides DAG to activate the HINT1-bound PKCγ. By interacting with the histidines of the PKC regulatory domain, this DAG lipid signal releases part of the zinc content and unfolds the kinase into its active conformation (23, 24, 50). Moreover, by acting on HINT1 histidines, DAG would also provoke the release of the zinc ions required to support PKCγ-HINT1 inhibitory association. This action weakens the intermolecular bridging and disorders the surface of PKCγ involved in its binding to HINT1, favoring the release of the active kinase. Similarly, MOR-released zinc ions also promote the binding of inactive Raf-1 to the HINT1 protein, facilitating PKCγ-mediated activation of the Raf-1-ERK1/2 pathway (45). PKC can also be activated by reactive oxygen species (ROS); however, ROS do not enhance the activation promoted by cofactors such as DAG and calcium but rather, they fix the kinase in a persistent, autonomously active form (19, 22). Significantly, NADPH oxidase/ROS production and the sustained activation of PKCγ triggered by opioids are essential to develop and maintain NMDAR-mediated tolerance to the analgesic effects of morphine (8, 44). Thus, the action of PKCγ on NADPH oxidase subunits promotes the production of ROS (3), which remove the DAG-resistant zinc from the regulatory domain of PKCγ and fix the kinase in its active form (22).
The concept of a biological redox switch, or more specifically, redox zinc switch, involves motifs in peptides or proteins that utilize the redox chemistry of cysteine thiol groups to bind and release zinc ions (32, 39). Controlling the availability of zinc and the cysteine ligand-based redox reactions is essential to modulate certain transient heterologous protein-protein interactions (quinary structure) (32). Lipid second messengers such as DAG, as well as redox action and zinc ions, dynamically regulate PKC activity by switching the kinase between its unfolded active and folded inactive state, the latter binding more readily to the HINT1 protein at the GPCRs. Both activating agents, lipid second messengers and ROS, release zinc ions from PKC (22, 50) and within a given kinase activation cycle, zinc must remain in close proximity to facilitate the recovery of the kinase for the next cycle (23). These requirements, coupled with the compartmentalization of GPCR redox signaling, suggest that the NO-sensitive source of zinc ions should be incorporated into this regulatory machinery. As RGSZ2 exerts an inhibitory influence on NO production, which is lifted upon MOR activation (14, 47), our results indicate that the regulation of the zinc finger of RGSZ2 by mild oxidation of nNOS/NO could be coupled to PKC, providing the kinase with the zinc ions necessary for it to reach its inactive state and bind to the HINT1 protein in the surroundings of the GPCR (Fig. 9). The release of zinc ions appears not to compromise other functions of RGSZ2 and thus, the RGSZ2 redox zinc switch would appear to act as a redox transducer (32) that releases zinc (“zinc signal”), which can in turn modulate the activity of PKCγ and Raf-1, among other targets. By contrast, PKC behaves as a redox sensor, whereby the ROS-induced release of zinc ions alters its activity (kinase activation).

The excess generation of NO and NO-derived reactive nitrogen species leads to nitrosative stress and altered cell metal ion metabolism, both of which have been implicated in the pathophysiology of neural disorders (17, 37). Typically, NMDAR overactivation at least partially contributes to neurological diseases such as Alzheimer's and Parkinson's diseases, multiple sclerosis, and amyotrophic lateral sclerosis (27), as well as mood disorders such as schizophrenia and depression (29, 36). Nonetheless, the link between GPCR signaling, NO production, and zinc metabolism raises the possible role of metabotropic receptors in certain neural disorders. The GPCR-associated and nNOS-coupled RGSZ2 protein is implicated in human cognitive ability, while the genome wide association database links this gene to Alzheimer's disease, narcolepsy, and panic disorder (https://gwas.lifesciencedb.jp/cgi-bin/gwasdb/gwas_gene.cgi?name=RGS17 accessed on Jul 2, 2012). Moreover, where the RGSZ2 gene resides, locus 6q25, is one of the most relevant loci for schizophrenia-susceptibility on this chromosome (26). Impaired RGSZ2 expression also induces overstimulation of the nNOS/NO pathway, as well as rapid and durable loss of GPCR function (14). The HINT1 protein has also been implicated in certain mental disorders, including schizophrenia and bipolar disorder (2). A disruption in zinc homeostasis is increasingly associated with the pathophysiology of affective disorders and interestingly, depression has been correlated with serum hypozincemia. Indeed, zinc exhibits antidepressant-like activity in preclinical animal models. Moreover, the efficacy of antidepressants that block serotonin reuptake and normalize serum zinc levels suggests that this effect is mediated by GPCRs (5, 38). Dysfunction of aminergic receptors, such as 5HT1A and 5HT2A, is typically implicated in depression. Thus, impaired coupling of these serotonin receptors to redox signaling (e.g., nNOS regulation and NO-dependent release of zinc ions) may diminish their signaling capacity. In such cases, the strength of the zinc signals produced by these GPCRs can be improved by increasing agonist input, as occurs when serotonin reuptake is inhibited by antidepressants. Given the implication of zinc homeostasis in brain disorders, several therapies based on metal ion chelation, redistribution and supplementation are currently in development (16).
In summary, we found that a range of neuronal GPCRs that respond to endogenous ligands of diverse chemical nature all physically couple with the RGSZ2-nNOS signaling complex. Activation of these receptors increases NO production and releases zinc ions from redox zinc switches, such as the RGSZ2 protein. These findings demonstrate that GPCR signaling depends on redox processes and zinc mobilization for successful integration into neuronal metabolism.
Materials and Methods
Zinc microfluorescence imaging in CD1 mouse striatal slices
To image intracellular Zn2+, brain slices (200 μm thick) were preloaded for 1 h with 50 μM of cell-permeable Newport Green DCF diacetate (N7991, Invitrogen), 0.1% pluronic acid (P3000MP, Invitrogen), and 0.5% dimethyl sulfoxide, as described previously (45). Saline, agonists, and antagonists were added to the wells, and the contribution of NO to zinc signals was determined using the NOS inhibitor NG-nitro-L-arginine (L-NNA). Images were obtained by confocal microscopy on a Leica DMIII 6000 CS confocal fluorescence microscope equipped with a TCS SP5 scanning laser (excitation, 488 nm; emission, 498–520 nm) and using a 10×0.4 HC PL APO objective. See also Supplementary Materials and Methods (Supplementary data are available online at
In vitro determination of zinc release from rRGSZ2
Free zinc ions were detected as described previously (45). The N-terminal (Nt) region of RGSZ2 was obtained as a GST fusion protein and this RGSZ2 Nt protein was incubated for 30 min at RT with 100 μM ZnCl2. Free zinc was removed by extensive washing and centrifugation in an Amicon Ultra-4 centrifugal filter device (UCF8 01024), followed by buffer exchange in PD-10 gel chromatography columns (GE Healthcare 17-0851-91). GST-RGSZ2 Nt was concentrated (20 μM) and then exposed at the intervals indicated to 100 μM of the NO donors SNAP and NOR-3, or to 2.5 mM hydrogen peroxide (all in HEPES buffer; 25 mM, pH 7.8). Complexing was initiated by adding the zinc chelator Zincon (40 μM; Sigma #96440) and the optical density was read at 600 nm. Another set of assays was performed by adding TSQ to the protein samples at a final concentration of 10 μM. The RGSZ2 Nt-containing samples were excited at 334 nm and fluorescence emission was recorded at 465 nm in a spectrofluorimeter. A standard zinc solution (Sigma 39059) was used to calibrate the assay systems.
Preparation and solubilization of the synaptoneurosome-enriched fraction: GPCR immunoprecipitation and co-precipitation of associated proteins
All procedures involving mice were performed in strict accordance with the guidelines of the European Community for the Care and Use of Laboratory Animals (Council Directive 86/609/EEC), and Spanish Law regulating animal research (RD 1201/2005). The experimental protocols were reviewed and approved by the Committee for Animal Experimentation at the CSIC.
For the immunoprecipitation studies, the cerebral cortex from 8 CD1 mice (Charles River, Spain) were pooled, and the assays were repeated at least twice with samples obtained from different mice. The methods used to prepare the PAG synaptosomal fraction have been described previously (46). The affinity-purified IgGs against the extracellular domains of GPCRs were labeled with biotin (Pierce 21217 and 21339), and immunoprecipitated from solubilized membranes as described (12, 42). The immunoprecipitated proteins were resolved by SDS/polyacrylamide gel electrophoresis (PAGE) in 10 cm×10 cm×1.5 mm gel slabs (7%–14% total acrylamide concentration, 2.6% bisacrylamide cross-linker concentration), and the separated proteins were then transferred to 0.2 μm PVDF membranes and probed with the selected antibodies in DecaProbe chambers (PR 150, Hoefer-GE, Barcelona, Spain). The primary antibodies were detected using the corresponding secondary antibodies and visualized by ECL (RPN2132, Amersham Biosciences).
Bimolecular fluorescence complementation analysis
The pPD49.83 plasmid was used to generate two cloning vectors for BiFC analysis (44). The constructs containing the N-terminal fragment of Venus truncated at residue 173 (VN173) or the C-terminal fragment of Venus starting at residue 155 (VC155) were a gift from Dr Chang-Deng Hu at Purdue University (USA). Chinese hamster ovary (CHO) cells were transfected using Lipofectamine 2000 (Invitrogen) and incubated for 24 h prior to testing for transgenic expression. Samples were visualized on glass bottom plates (MatTek Co, MA) using a Leica DMIII 6000 CS confocal fluorescence microscope equipped with a TCS SP5 scanning laser.
Surface plasmon resonance analysis
Interactions were determined using a BIACORE X (GE) as described previously (43). The GPCRs (50 μg/mL) were coupled to a CM5 sensor chip (GE, BR-1000-14) by amine coupling, and sensorgrams were collected at 25°C at a flow rate of 5 μL/min after passing RGSZ2 (75 μL) over the sensor surface. Increasing analyte concentrations were studied and the results were plotted using the BIAevaluation software (v 4.1).
In vitro interaction between RGSZ2/PKCγ and HINT1 proteins: The effects of zinc ions and PMA
The interaction of recombinant HINT1 (200 nM) with either GST-RGSZ2 (100 nM) or GST-PKCγ (100 nM; Abnova #P4759) was studied. Zinc was first removed from the recombinant proteins by incubating them for 60 min at room temperature (RT) in a buffer containing 10 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM EDTA, 200 μM DTT, and 1 mM N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine (TPEN) (48). The samples were diluted in 10 mM HEPES (pH 7.5), 150 mM NaCl, and concentrated in centrifugal filter devices (10 kDa nominal molecular weight limit: Amicon Microcon YM-10 #42407, Millipore). After zinc removal with TPEN, the influence of added zinc on the association of the recombinant proteins was evaluated by incubating them either alone (negative control) or together with the GST tagged protein in 400 μL of a buffer containing 10 mM HEPES (pH 7.4), 150 mM NaCl, and 0.05% Tween20. Recombinant HINT1 was incubated for 30 min with rotation at RT with GST-RGSZ2 or GST-PKCγ in the presence of increasing amounts of zinc chloride: 1 pM, 1 nM, and 1 μM (Puratronic, Alfa Aesar 231-592-0). Subsequently, 40 μL glutathione Sepharose 4B (GE#17 0756 01) was added to the protein mixture, which was then recovered by centrifugation, washed three times, solubilized in 2X Laemmli buffer, and the presence of HINT1 was analyzed in Western blots. In another set of assays, endogenous zinc was first removed from the HINT1 and GST-PKCγ proteins, and they were then incubated in the presence of 1 μM zinc chloride for 30 min at RT before the free zinc was removed using 10 kDa nominal molecular weight limit centrifugal filter devices (Amicon Microcon YM-10 #42407, Millipore). Parallel samples were used to evaluate the disrupting effect of PMA (Calbiochem #524400) on the PKCγ/HINT1 association, as well as on the resulting enzymatic activity of this PKCγ. The latter was determined using the Pep Tag protein kinase C assay (Promega, Madison, WI). Samples containing the HINT1/PKCγ complexes that had been exposed to PMA were incubated for 30 min at 30°C with the specific substrate in the presence of an activating solution. The reaction was stopped at 95°C for 10 min and the PKCγ-induced phosphorylation of the peptide substrate was determined.
In vitro effect of zinc ions on the recruitment of signaling proteins to GPCRs: Influence of zinc chelation and NO generators
The effect of increasing concentrations of zinc on the association of PKCγ and Raf-1 with various GPCRs was studied in cortical synaptosomal membranes. Membranes were incubated with zinc chloride (Puratronic, Alfa Aesar 231-592-0) for 4 h at 4°C before the GPCRs were immunoprecipitated. Subsequently, free zinc ions were removed by centrifugation and extensive washing. The synaptosomal membranes were then solubilized in Nonidet P-40 buffer as described previously. The solubilized membranes were incubated overnight at 4°C with approximately 3 μg of a biotin (Pierce #21217 and 21339) conjugated primary antibody (affinity-purified IgGs) raised against extracellular sequences in 5HT1AR, 5HT2AR, CB1R, and D2R. The GPCR-associated proteins were then separated by SDS-PAGE and analyzed by Western blotting. The samples were incubated overnight at 4°C with the NO generator SNAP (Tocris Bioscience #0598) and the anti-GPCRs IgGs, and the procedure was continued as described above. The heavy metal ion chelator TPEN (Fluka WA16827) was added to the solubilized samples together with SNAP.
Statistical analyses
ANOVA, followed by the Student-Newman-Keuls test (SigmaStat, SPSS Science Software, Erkrath, Germany) was used for the statistical analyses. The significance was defined as p<0.05.
Footnotes
Acknowledgments
This research was supported by FIS PS09/00332 (JG), PI11-01704 (PSB). We would like to thank Beatriz Fraile and Gabriela de Alba for their excellent technical assistance.
Author Disclosure Statement
The authors declare that, excluding income received from our primary employer “Ministerio de Ciencia e Innovación, e Instituto de Salud Carlos III” no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional services, and that there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.