
Abstract
Many cellular functions involve cysteine chemistry via thiol–disulfide exchange pathways. The nucleophilic cysteines of the enzymes involved are activated as thiolate. A thiolate is much more reactive than a neutral thiol. Therefore, determining and understanding the pK as of functional cysteines are important aspects of biochemistry and molecular biology with direct implications for redox signaling. Here, we describe the experimental and theoretical methods to determine cysteine pK a values, and we examine the factors that control these pK as. Drawing largely on experience gained with the thioredoxin superfamily, we examine the roles of solvation, charge–charge, helix macrodipole, and hydrogen bonding interactions as pK a-modulating factors. The contributions of these factors in influencing cysteine pK as and the associated chemistry, including the relevance for the reaction kinetics and thermodynamics, are discussed. This analysis highlights the critical role of direct hydrogen bonding to the cysteine sulfur as a key factor modulating the equilibrium between thiol S–H and thiolate S−. This role is easily understood intuitively and provides a framework for biochemical functional insights. Antioxid. Redox Signal. 18, 94–127.
I. Introduction

The intrinsic pK a value for the free cysteine thiol–thiolate equilibrium in an aqueous solution is close to 8.6 (26, 39, 82, 89, 169). In folded proteins, this pK a can be shifted by the influence of the three-dimensional protein structure (13). Polar groups (charged or neutral) in the vicinity of a cysteine, and/or a different solvation environment compared to an aqueous solution, can influence the pK a of a cysteine thiol. Under physiological conditions (pH ∼7), a thiol with a pK a value below 7 will exist mostly as a more reactive thiolate, critical for catalysis (27). Note that physiological conditions are not always at pH ∼7, since different cellular organelles have different pH values. Lowered pK a values of catalytic cysteines influence the reaction kinetics and thermodynamics, and strongly influence the catalytic efficiency of an enzyme during thiol–disulfide exchange reactions. The effect of pK a lowering on reaction rate enhancement will in general be most significant when the pK a values are close to the solution pH (78). Perturbed pK a values also influence protein stability (189).
With accurate cysteine thiol pK a values, one gains insights into catalytic mechanisms and into the factors influencing the pK a values. It has long been known that the pK a values of catalytic cysteines in thiol–disulfide oxidoreductases of the thioredoxin (Trx) superfamily can adopt a wide range of values in proteins with a similar structural fold (19). Therefore, this superfamily of enzymes provides a paradigm to study the factors influencing and modulating the pK a of thiol groups (35, 43, 45, 49, 56, 61, 77, 117, 129, 132, 142, 179). A thorough compilation of experimentally measured pK a values for these enzymes (and related model systems) should be a helpful resource (Table 1).
Experimentally measured pK a values reported in the literature; these values are listed here with the same precision as in the original publication.
For clarity, only references to experimental (noncomputational) work are cited here; see main text for references to theoretical/computational work.
DsbA, disulfide-binding protein A; Grx, glutaredoxin; NA, not applicable; ND, not determined; NMR, nuclear magnetic resonance; PDB, Protein Data Bank; PDI, protein disulfide isomerase; Trx, thioredoxin.
II. pK a Determination Methods
The pK
a of a cysteine thiol group can be obtained from the equilibrium constant K
a for the deprotonation reaction:
with
in which [H+], [CysS−], and [CysSH] are the equilibrium concentrations given in mol/l.
The central equation for this equilibrium is the Henderson–Hasselbalch equation (Eq. 3), which is a convenient rearrangement of the Ka
equilibrium equation:
In this section, we briefly describe the experimental and calculation methods to obtain pK a values. We focus on the current widely used techniques to obtain thiol pK a values in proteins.
A. Experimental approaches
Traditional methods used to determine pK a values of cysteines in proteins are based on UV absorption spectroscopy, rate constant determination, microcalorimetry, and in some cases, nuclear magnetic resonance (NMR) spectroscopy. Raman spectroscopy (101), quantitative mass spectrometry (122), and potentiometric titrations (169) have also been used.
A well-established spectroscopic method is based on the greater absorption of ultraviolet light by the thiolate versus the thiol at 240 nm (A
red) (Fig. 2). The difference in the ultraviolet extinction coefficient of the thiol and the thiolate anion is about 4000 M
−1 cm−1 at 240 nm (14, 131), which has been used to determine the pK
a values to the thiols of disulfide-binding protein A (DsbA) (121) and in several wild-type and active-site mutants of Trx (40, 142, 166) and related proteins (Table 1). The protein in a buffer solution with high pH is titrated with HCl, and A
red is followed at decreasing pH during the titration. However, several corrections on A
red are needed. For correction of the varying protein concentration due to the increased volume during to addition of HCl, the absorption at 240 nm needs to be corrected for absorptions at 280 nm, the wavelength at which one can spectroscopically determine the protein concentration (A240red/A280red
). Mainly, the side chains of Phe, Tyr, and Trp will also absorb at 240 nm, and their absorbance might change due to changes in the environment during the titration. This source of variability on (A240red/A280red
), which is thiolate/thiol independent, can be corrected by measuring the absorption of the same protein in which the thiol group is absent (A240oxid/A280oxid
). Therefore, the same protein in which the cysteines are alkylated with iodoacetamide (IAM) can be used, which will not contribute to the thiolate absorbance at 240 nm. Alternatively, an engineered variant in which the cysteine is mutated to a serine can be used. In case of a protein with a −C-X-X-C- active-site motif, the oxidized form with a disulfide between the two cysteines can be used. To make each experimentally obtained absorption ratio (A240red/A280red
) pH independent, one normalizes by dividing the (A240red/A280red
) value of the thiolate form by the absorption (A240oxid/A280oxid
), where no thiol or thiolate is present. The (A240red/A280red
)/(A240oxid/A280oxid
) ratio is determined at varying pH by titration with HCl and plotted against pH (Fig. 2). The pK
a is obtained from a nonlinear least-square fitting with the rearranged Henderson–Hasselbalch equation:

in which A exp is (A240red/A280red )/(A240oxid/A280oxid ) for the experimentally determined value; ASH is the A 240/A 280 value for the protonated thiol form; and As- is the A 240/A 280 for the deprotonated thiolate form.
This rearranged equation has been deduced from the relation between the fraction A
exp and the absorption of the protonated and deprotonated form of the cysteine (Fig. 2), according to the following expression:
in which R is a fraction of the total absorption difference between totally deprotonated and protonated cysteine. By using equation (3), this fraction R can be written as:
which then finally results in the rearranged Henderson–Hasselbalch equation:
In some cases, the protein structural environment around the cysteine or its local dynamics is affected by the pH, which in turn influences the cysteine absorbance. In these situations, it is difficult to evaluate the transition between protonated and deprotonated cysteine at 240 nm.
Alternatively, thiol pK as in proteins and peptides can be determined based on the rates of alkylation as a function of pH (17, 83, 93, 106, 108), with IAM, with fluorescent IAM derivatives, with 5,5′-dithiobis(2-nitrobenzoate) (DTNB), with 2,2′-dipyridyl disulfide (2DPS), or with monobromobimane. The alkylation rates are evaluated over a certain pH range by evaluating the residual enzyme activity as a function of time. The pH-dependent degree of alkylation can also be used when there is no enzymatic assay to monitor direct changes in the reaction rate, typically when the cysteine of interest is not in an enzyme active site. Then, the amount of alkylated cysteine may still be determined as a function of time, for example, by chromatographic retention times or fluorescence of the alkylating agent linked to the cysteine. Nowadays, new superior fluorescent probes are being developed, which allows highly specific thiol labeling at low pH (127).
At each pH, the pseudo-first-order rate constants k obs are determined from the slope of the plots of ln (E0/Et ) versus time, with E0 the initial enzyme activity (or nonalkylated amount of protein) and Et the enzyme activity (or amount of alkylated protein) at time t.
The second-order kinetic constant (k 2) is then calculated by dividing k obs by the concentration of alkylating agent at each pH, and then fitting to the following equations to determine the best-fit pK app.
For a one-pK
a model:
or for a two-pK
a model (if another protein titratable side chain influences the reactivity of the nucleophilic cysteine):
with k′ and k′′, the two different second-order constants for the thiolate form.
This method has been applied for Trx (82), thiol/disulfide oxidoreductases (117), glyceraldehyde 3-phosphate dehydrogenase (108), yeast glutaredoxins (Grxs) (36), and peroxiredoxin 6 from Arenicola marina (106). For Mt_AhpE, an alkyl hydroperoxide reductase of Mycobacterium tuberculosis, the pK a of oxidation and overoxidation (Fig. 1) of the peroxidatic thiol group were measured by determining the rate constants of peroxynitrite and H2O2-mediated oxidation and over oxidation using the difference in intrinsic fluorescence (70). For highly reactive cysteines, kinetic measurements need to be carried out in a stopped-flow apparatus. Overall, the chemical modification method has been very successful with enzymes of the Trx superfamily, since in these enzymes, the main cysteine of interest is catalytically active and solvent exposed. This contributes to explain the wealth of pK a data on the Trx superfamily.
This method of chemical modification is however not trivial and certainly not applicable to all proteins, as it assumes that the cysteine side chain of interest is accessible for alkylation. Further, the pK a value of the cysteine is rightly assumed to be the principal determinant of its alkylation rate. However, this is only the case in an idealized situation, because some cysteine side chains are partially or fully protected by the protein structure; their alkylation rates can also be affected by steric accessibility, conformational stability, and dynamics. Protected cysteines can only react with alkylating reagents when they are transiently exposed to bulk solvent by global or local unfolding events. Because protected cysteines primarily (or only) react in transient unfolded (open) states, they will exhibit seemingly unperturbed pK a values (∼8.6). However, the actual pK a value of the cysteine in its protected (folded) state could be very different.
Another method that should be applicable to low-solubility proteins is isothermal titration microcalorimetry (ITC) (165). ITC has been used to infer pK
a values of reactive residues of enzyme–substrate complexes by measuring the substrate-binding enthalpy ΔHbinding as a function of pH and buffer composition (162). Tajc and coworkers (165) used ITC to monitor directly the covalent reaction of IAM with a thiolate to produce a thioether, as a function of pH. The ITC instrument is set up in a single-injection mode, and sufficient IAM (up to 300 mM) is introduced to alkylate all the thiolate groups. The produced heat change upon injection
with
NMR spectroscopy can also be used to determine the pK
a of a cysteine (38, 44, 77, 89, 112, 129). This method is expensive, requires a sophisticated instrumentation, and is labor intensive (resonance assignment). The presence of more than one cysteine in a protein does not limit in principle the determination of their pK
as by NMR; ideally, the pK
a values of all cysteine residues may be determined in the same NMR experiment. For the cysteine thiol ionization measured by 1D 1H NMR spectroscopy, the cysteine Cα and Cβ protons can be used as probes for the ionization state of the cysteine thiol group (89). A problem that might occur is that the resonances of the Cys Cα proton might not be followed over the entire pH range because of signal overlap or low intensity of the signal. Also, one potential complicating factor is that pH-dependent changes in a resonance chemical shift may occur due to pH-dependent changes in other surrounding residues. Because the chemical shift is particularly sensitive to the local environment, disentangling the effects of ionization at the monitored residue versus other changes in proximal residues can be nontrivial. With more expensive 13C-labeled material, the chemical shifts of the 13Cβ resonance can also be followed as a function of the pH (38). The obtained chemical shifts can then be plotted against pH and fitted, again with the Henderson–Hasselbalch equation:
with δ, the chemical shift of a resonance as function of pH, and δs- and δSH, the chemical shifts at high and low extremes of the pH, respectively. The pK a values obtained by NMR seem to agree well with those obtained by other spectroscopic methods (38, 77, 89).
Since proteins tend to have a solubility minimum at their isoelectric point (pI), this might undermine the measurement of the pK a during the pH titration. In addition, proteins may unfold when a titration changes a protonation state important for the stability of the folded protein. When proteins start to aggregate, unfold, or precipitate, the experimental measurements become difficult and unreliable. These limitations and other considerations have emphasized the growing interest in the development of computational methods to calculate the pK as.
B. Computational methods
In proteins, there are many protonation sites, and in many cases, even several cysteine residues, complicating accurate experimental pK a measurements and interpretation. Therefore, there is an increasing interest in supplementing, guiding, and interpreting experimental approaches with computational methods for pK a rationalization/prediction (2, 98). Calculations can also help assign accurately measured pK as to particular residues.
The basis for pK a calculation techniques that can be used on large systems like proteins relies on estimates of energy terms, which influence the relative populations of the protonated and deprotonated states of the titratable groups. For the relevant chemical functionalities, those terms typically include the desolvation energy, the background interaction energy, and the site–site interaction energies. The desolvation energy is the energy change when transferring the titratable group from an aqueous solvent into the protein environment. The background interaction energy is the interaction energy between the titratable group and the protein, when all other titratable groups are considered as neutral entities. The main contribution to this term comes from hydrogen-bonding interactions between the titratable group and neutral polar groups. The site–site interaction energy describes the electrostatic interactions between pairs of charged titratable groups. In principle, one has to consider the interactions among all the charged groups simultaneously, a challenge when the titrations of several groups are coupled. These energy contributions can be addressed in various theoretical frameworks (2, 98), usually categorized as classical electrostatics with continuum solvation models, physics-based fully microscopic models, or more empirical approaches. All computational methods require a good-quality structural model of the protein, which determines for a large part the outcome of the calculations.
The Poisson-Boltzmann (PB) approach (4, 13, 29, 31, 35, 45, 68, 126, 161, 179, 188) is maybe the most representative and best-developed approach to address the pK a shifts in proteins in the framework of classical electrostatics (68, 154). The calculation of pK a shifts in proteins in the PB framework has been described in detail (13, 42). In summary, the pK a in a protein environment is calculated as a shift relative to the reference intrinsic pK a of the same residue free in aqueous solution. The reference pK a value is known from measurements on model systems, typically peptides in solution (bottom of Table 1). For the cysteine thiol, a reference unperturbed pK a of 8.3–8.6 is commonly accepted, with small variations in the corresponding measurements (17, 26, 39, 82, 89, 169). The range of cysteine pK as measured in peptides is much narrower than in folded proteins (Table 1). Yet, some pK a differences are observed in peptides, although the reasons for such spread are obscure, since there are no structures for these flexible peptides. One cannot say for sure if the differences between pK a values of cysteines in peptides can be rationalized by hydrogen-bonding differences. The differences in measured pK as also probably reflect different experimental protocols (169). Reference pK a values obtained recently with pentapeptides with neutral (blocked) termini (169) are of special interest, since they minimize the influence of secondary structure and hydrogen bonding, which can occur in larger peptides, and suggest a reference pK a value of 8.6 for cysteines. Note that the effect of the bulk aqueous solvent on the pK a is encapsulated in the reference pK a. Estimating the influence of the protein relative to such known reference pK a is the approach currently adopted with all methods.
The thiol of cysteines is treated like any other titratable group. Therefore, the pK a shift of a particular thiol is calculated by estimating the electrostatic interactions between the cysteine thiolate (and thiol) and the rest of the protein while taking into account the desolvation energies. In practical applications, neglecting electrostatic interactions between charged titratable groups which are far apart tends to be a valid working hypothesis (53). The desolvation corresponding to the transfer of a charge (e.g., a thiolate) from bulk water into the protein interior is calculated by representing the bulk solvent as a continuous high-dielectric-constant medium and the protein interior as a region of lower dielectric constant (13, 31, 55, 68). Such desolvation is largely electrostatic in nature. It destabilizes a charge, contributing to pK a shifts, for instance, by destabilizing the charged form of a titratable group. Thus, one expects reactive cysteines to be mostly solvent accessible because of steric requirements allowing encounters with the substrate, but also because proximity to the water contributes to stabilize their thiolate. On the other hand, cysteine residues are very vulnerable to oxidation, which might explain the limited occurrence of nonactive cysteine residues on protein surfaces (109).
Representing the protein interior by a single dielectric constant can only be an approximate, and sometimes crude, treatment. For extended discussions on this subject, see the following references (4, 31, 45, 55, 154, 158, 159). With a system comprised of different dielectric regions (solvent and solute interior), the straightforward use of Coulomb's law in its well-known, simplest form is not applicable, and trying to treat the system in this framework leads to a mathematical description too complex to solve. Therefore, one uses the PB equation instead to obtain the electrostatic properties (54, 154), usually in its linearized form (68). The physical parameters that need to be supplied by the user include the dielectric constant of the protein interior and of the aqueous solvent, the temperature, partial charges, and radii for the protein atoms, and the ionic strength. The atomic radii are critical, since they underpin the boundary between the dielectric regions. The ionic strength represents the screening of electrostatic interactions by counter ions, but the calculated pK a values tend not to be highly sensitive to this parameter. The general experience is that the calculated pK a values tend to be much more sensitive to the value of the protein dielectric constant. Calculations with the PB method are more time consuming than empirical methods (e.g., PROPKA); however, they are quite tractable on commodity computers; a PB pK a calculation on a medium-size protein takes less than a minute.
The best practices regarding PB-based pK a calculations are still an evolving field (2). To gain productive insights from such calculations, one requires a good grasp of the underlying method and approximations. Accordingly, no standard generic safe protocol can be recommended. Yet, the physical principles (desolvation, Coulombic interactions, and hydrogen bonds) underlying the PB calculations are intuitively easy to understand, and the PB framework should make the notion of pK a calculations accessible to the nonexpert, at least in terms of general ideas and as a basis for discussions. Of several software packages available, none has emerged as systematically more accurate or popular (2). Nevertheless, the software to perform PB pK a calculations is freely downloadable; see Lee and Crippen (98) and Fitch and Garcia-Moreno (42).
As far as we know, no PB-based study has focused on the calculations of cysteine pK as across proteins systematically. Instead, computational analyses of cysteine pK as were reported in case studies of individual enzymes. The PB methodology was used to calculate the cysteine pK a values of wild-type and mutant human Trx and Escherichia coli DsbA (49, 119). For the nucleophilic cysteine of wild-type Trx and DsbA, pK a values of, respectively, 7.1 and 2.6 were calculated, in good agreement with the experimental values of 6.9 and 3.4. Also for the -X1X2- mutants in the -Cys1-X1-X2-Cys2- active-site motif of Trx and DsbA, the calculated pK a values were close to their experimental counterpart (119). Only a slightly better agreement with the experimental pK a values was obtained after conformational relaxation of the flexible ionizable groups. For E. coli Grxs 1 and 3, as well as pig Grx (and its mutants), the PB methodology calculated the relevant cysteine pK a values in reasonable agreement with experiment (43, 44). In particular, the PB calculations account for the large downshift of the pK a of Cys1 in the -Cys1-Pro-Tyr/Phe-Cys2- motif of Grxs (pK a≤5) and correctly assign a significantly higher pK a to Cys2. Crucially, these studies provided a detailed theoretical analysis of the factors that lower the pK a of the catalytic cysteine. This analysis was strengthened by the formulation of true predictions that were subsequently tested, and experimentally supported (45). Small structural differences in the active site of enzymes of the Trx superfamily can largely rationalize the pK a variations for the catalytic cysteine across this superfamily (Fig. 3). In addition, these local interactions were found to be sensitive to the protein dynamics. This was already apparent in a PB-based study of the pK a of Cys32 in an NMR structure of E. coli Trx (35).
Importantly, the output of PB pK a calculations provides a decomposition of the energetic components contributing to a pK a shift, which allows for interpretation of the calculations. Such decomposition has provided much of the increasing evidence that the pK as of many titratable groups are primarily influenced by short-range polar interactions such as hydrogen bonds (6, 44, 45, 49, 103, 134, 141, 143). It strongly supports the view that the pK as are in general very sensitive to the details of the protein structure and dynamics (35, 43 –45, 134). The influence of local structural differences on the pK a calculations was also apparent when comparing pK as calculated with both X-ray and NMR structures of the same proteins (5, 86). Incidentally, there was no clear systematic improvement between calculated and measured pK as depending on the technique (X-ray or NMR) used to build the structural model. In addition, the raw X-ray or NMR coordinates may have to be refined before pK a calculations, to orient protein side-chain amide and imidazole groups, as well as conserved water molecules, according to the most likely hydrogen-bonding network (123, 126). Thus, a careful preparation of the protein structure is critical for pK a calculations, regardless of the pK a calculation method.
Preparing a relevant system for pK a investigations might be even more sophisticated, if the pK a depends on the binding between two or more molecular species. Thus, one may have to consider how the calculated pK a might be influenced by the binding of cofactors (ions and organic ligands) or by the formation of a protein–protein complex. Since low cysteine pK as are frequently involved in enzymes and their reactions with substrates, one can imagine situations where building a relevant enzyme–substrate complex may be a prerequisite to pertinent pK a calculations. Some of these aspects have been illustrated in a study of a covalent complex between Trx and arsenate reductase (ArsC) (141). Another interesting system to study a cysteine pK a influenced by complexation is DsbD. In the isolated C-terminal domain (gamma-domain), the pK a of the reactive Cys461 is unusually high, with a pK a of 9.3–10.5 (112, 161). Yet, there is indirect evidence that the pK a of Cys461 is lowered upon complexation with the substrate N-terminal domain of DsbD (113). A model in which substrate binding enhances the reactivity of the active-site cysteines has also been proposed for ResA (28). It has been proposed that changes of the relevant pK as during complexation allow controlled activation of reactive cysteines upon binding of cognate substrates, to restrict the reactivity toward those substrates (28, 113). Macromolecular crowding in the cell may also lead to nonspecific, but significant, interactions, which might alter protein structures, and therefore the pK a of reactive amino acids. However, these nonspecific effects are expected to be even more challenging to characterize than those arising in the formation of specific complexes.
Empirical pK
a calculation methods use rules derived from experimental observations to predict pK
as and have the advantage to be very fast (seconds per protein conformation). They also lend themselves to updates of the underlying model and associated terms, driven to maximize pragmatically the agreement between experiment and calculations without having to abide by a constraining or interpretable theoretical framework. An overview of the available empirical pK
a prediction methods is given in reference (98). A currently popular empirical method is PROPKA (12, 30, 88, 103, 111, 130, 133, 134, 149, 160), which is available (
The ΔrMc term includes desolvation effects, hydrogen bonding, and charge–charge interactions via empirical relations. The first version, PROPKA1, was developed using a test set of 314 experimental pK as, which contained only 12 cysteine pK as. For these cysteine residues, the root mean square deviation between experimental and calculated pK a values was 1.39. For oxidized (bonded) cysteine thiol groups, no pK a calculation is performed (flagged by returning a value of 99.99 instead of an estimated pK a value), with the exception of proteins from the Trx superfamily. Even when the cysteines of the conserved -Cys1-X1-X2-Cys2- motifs are in the oxidized form (i.e., when a disulfide bond is formed between Cys1 and Cys2), the pK a values of the reduced cysteines are evaluated.
PROPKA has been updated twice. In PROPKA1, no pK a shifts due to ligands, ions, and structural water molecules were considered. These effects were incorporated in PROPKA2 (12). In PROPKA3 (130), residues are no longer classified as either buried or surface residues, but an interpolation between these two extremes is used. This results in a burial ratio by which Coulomb interactions are no longer strictly either turned off (surface residues) or turned on (buried residues). A linear interpolation between the two extremes is made via a position-dependent weight function that depends on the number of heavy atoms within a sphere of 15 Å around the charge center.
A benchmark study (110) of PROPKA on cysteine residues for several Trx and ArsC proteins revealed a fair correlation (R2=0.74 with an average deviation from experimental value of 0.88 pK a units) with experimental pK as (Table 2). Before pK a calculation, the raw X-ray structures were energy minimized with CHARMM. For comparison, Table 2 reports the PROPKA2 values for nonminimized structures, giving in some cases a notably degraded performance of PROPKA. This strengthens the notion that a careful preparation of the protein structure is critical for pK a calculations. The performance of PROPKA3 with cysteines appears to be less accurate than the performance of PROPKA2 (Table 2). The advantage of PROPKA is its balance between speed and performance, and especially its Web-based ease of use. Therefore, PROPKA is a valuable tool to give initial insights in the protonation state of a cysteine. PROPKA illustrates how the development and successive adjustments of empirical models rely on augmented training sets of experimental data. Indeed, the continued experimental determination of pK a values for cysteines is very valuable for an improved calibration of empirical pK a calculators.
See (110).
pK a value obtained from E. coli Trx2.
pK a value obtained from C15A/C10S/C82A Sa_ArsC.
pK a value obtained from oxidized C15A Sa_ArsC.
ArsC, arsenate reductase; NPA, natural population analysis.
The observation that pK as in proteins may be primarily influenced by short-range interactions (35, 43 –45, 49, 141) suggests that one may not need to include the full protein in the calculations. This has led to the development of a pK a calculation protocol in which calculations of site–site electrostatic interaction energies were omitted for pairs of titratable groups beyond a distance cut-off value (125). The immediate environment of the titratable group is treated in detail, while the rest of the protein is described less accurately, which lowers the calculation time significantly without apparently degrading the quality of the calculated pK a values. These findings suggest that protein active sites might be treated as independent units with respect to pK a calculations. It opens the possibility to apply computationally demanding quantum mechanical methods to the calculation of pK as in their protein microenvironment. A possibility is to use a model in which only the titratable group plus its directly interacting protein environment is represented. The rest of the protein is represented by a bulk dielectric constant or treated by molecular mechanics in a combined quantum mechanical (QM)–molecular mechanics approach. Alternatively, the bulk protein around the region of interest can be neglected altogether, without being included in the calculations; examples can be found in Li et al. (102), Zheng et al. (195), and Ullmann et al. (170). The combination of a model system for the protein site of interest and a surrounding dielectric constant was used by Roos et al. (141). The pK a values were predicted via the linear relationship between the natural population analysis (NPA) charge calculated quantum mechanically on the sulfur atom of the deprotonated thiolate, and compared with the experimental pK a values. The more negative the NPA charge on the sulfur atom, the higher the tendency to bind a proton and the more basic (higher pK a) the thiol is. For Trx and ArsC systems, this linear relation works very well (Fig. 4 and Table 2) (141).

C. Future perspective for pKa calculations applied to cysteines
The continued development of computational methods has produced powerful tools to calculate pK
a values for each titratable group in a protein. When used wisely, such tools help to formulate specific working hypotheses about the pK
a values of interest and the factors influencing these pK
as. It provides a basis for a productive interplay with experimental approaches, including mutagenesis. However, theoretical pK
a prediction methods still suffer from a number of limitations (2, 30, 98). Large errors can be encountered for residues with unusual pK
a values (2, 31, 98, 126, 157), which tend to be of particular interest, because they are often involved in catalytic activities. Ionizable side chains partially or fully sequestered from bulk solvent by burial in the protein core are particularly prone to marked discrepancies between calculated and measured pK
as. In a recent blind calculation of 77 pK
as with undisclosed experimental values, by 12 groups using a range of methods (
One avenue would be to develop more physically complete models with all atomic details at all stages of the calculations. For instance, one could aim to treat the aqueous solvent with discrete and dynamic water molecules rather than with a continuum dielectric. One can also envisage an increasing role for reliable quantum chemistry to quantify interactions. At the other extreme, one could may be improve empirical models by increasing the training sets on which they are based. There is also room to improve the current theoretical models, for instance, with respect to the protein structural relaxation and dynamics, or the underlying force fields. Polarizable force fields may improve the electrostatic energies when switching between neutral and charged state for titratable groups (23).
A common issue with pK a calculations is the assumption that a protein structure is rigid and identical to a representative crystal structure. For example, the same structure is frequently used to evaluate the energetics of both the charged and neutral form of a titratable group, although one would expect the microenvironment of this group to relax according to its charge state. This suggests that different conformers and tautomers should be included in the pK a calculations (51, 91, 192). Sampling of the hydrogen locations in response to protonation states already yields improvements (91). With the multiconfigurational continuum electrostatic method (MCCE) (51), the sampling was extended to the side-chain orientations. The MCCE approach gives pK a values in better agreement with experiment than the single-configurational continuum electrostatic method. Another option is to precede the pK a calculation with a substantially long molecular dynamics (MD) run (43 –45, 90, 161, 173). There is no recipe to determine the appropriate length of a simulation, but at least tens of nanoseconds (and ideally more) appear required, for example, to sample the conformations of long, flexible, charged side chains. In addition, MD simulations have proved of special interest to refine NMR structural models before the pK a calculations (43). Using the MD snapshots as input, the pK as can be calculated for regularly spaced snapshots, and then averaged (since the Boltzmann weighting of conformers is in principle implicitly contained in the simulated populations). This does yield not only the averaged calculated pK a values but also the instantaneous pK a fluctuations along the simulation. Examination of the instantaneous pK a values and their variation can help interpret the structural factors, which affect the calculated pK a, with full microscopic details. It may reveal that the averaged pK a (equivalent to its measured counterpart) is a composite of distinct pK a per structural microstates. Such microstates may be associated to different reactivities and may lead to mechanistic insights. When the MD runs are too short, it may produce a bias that can sometimes strongly influence the relative occurrence of the different populations and consequently the outcome of the pK a calculations (90), for instance, when the pK a of a semiburied residue depends on the motion of flexible charged side chains at the protein surface (43). Indeed, MD simulations do not always improve subsequent pK a calculations (125, 185). For instance, MD simulations are still typically performed while keeping the initially assigned protonation states constant, in which case those states might be overstabilized in the MD conformational ensemble, compared to alternative protonation states. This is being addressed with the development of MD simulations at constant pH (85). Alternatively, one can perform simulations at various predefined relevant protonation states (128).
Another way to improve pK
a calculations is the collection of additional reliable experimental reference data. That may be particularly the case for cysteine residues for which there are still only limited experimentally measured pK
as, and not enough structural information for redox active cysteines at the various stages of their redox cycles. Crystallizing the reduced form of some enzymes has proved particularly problematic (72), as the nucleophilic cysteines have the tendency to oxidize to sulfenic, sulfinic, and sulfonic acids (144). This heterogeneous population of partially oxidized proteins may hamper crystallization (10). Therefore, additional good-quality experimental data on thiol pK
as in proteins combined with deeper insights in the factors controlling pK
a values (e.g., regarding protein dynamics) should pave the way toward more accurate pK
a predictors. Meeting these challenges will require genuine collaborative efforts. In this light, the pK
a cooperative was founded to improve pK
a calculations with proteins (
III. Factors That Control the pK a Values of Cysteine Thiols in Proteins
The propensity of a chemical functionality to ionize depends on its environment, in particular on factors that affect the stability of the charged form of the functionality. Therefore, the pK a of a cysteine thiol can be strongly influenced by its microenvironment in a protein. Charge–charge interactions, hydrogen bonds, aqueous desolvation, and helix–dipole effects are generally invoked to rationalize perturbed pK as of cysteine residues (62, 124). However, it has become increasingly clear that residues in the immediate environment of a titratable group play a prominent role (45, 153). In this section, we discuss the limited role of charged side chains and long-range electrostatic interactions on the cysteine pK as in oxidoreductases and the direct link between thiol pK a and hydrogen bonds. These ideas were initially inspired by studies of the catalytic cysteine in enzymes of the Trx superfamily (45). However, since these notions emerged from analyses rooted in physical chemistry, one expects that they can be generalized to a good extent (103, 134), as confirmed by inspection of a variety of systems (section III.D).
A. Limited role of charged side chains and long-range electrostatics
It has long been known that many enzymes have a thiolate cysteine in their active site with a pK a clearly lower than the pK a of free cysteine in an aqueous solution (Table 1). For example, proteins of the Trx superfamily have a recurrent -Cys1-X1-X2-Cys2- active-site motif within a conserved structural framework, where the pK a of Cys1 is dramatically lowered. Considerable efforts have been dedicated to characterize enzymes of this superfamily, which includes DsbA, Grx, and Trx, and many variants (Table 1). In these enzymes, the pK a of Cys1 covers a broad range of values, from ∼3.5 in DsbA to ∼7.1 in Trx, to >8.0 in atypical Trxs such as ResA (35, 38, 43, 45, 46, 49, 56, 61, 77, 82, 117, 129, 142, 179). Mutagenesis studies have shown that the pK a of Cys1 is strongly influenced by the X1-X2 residues (56, 117).
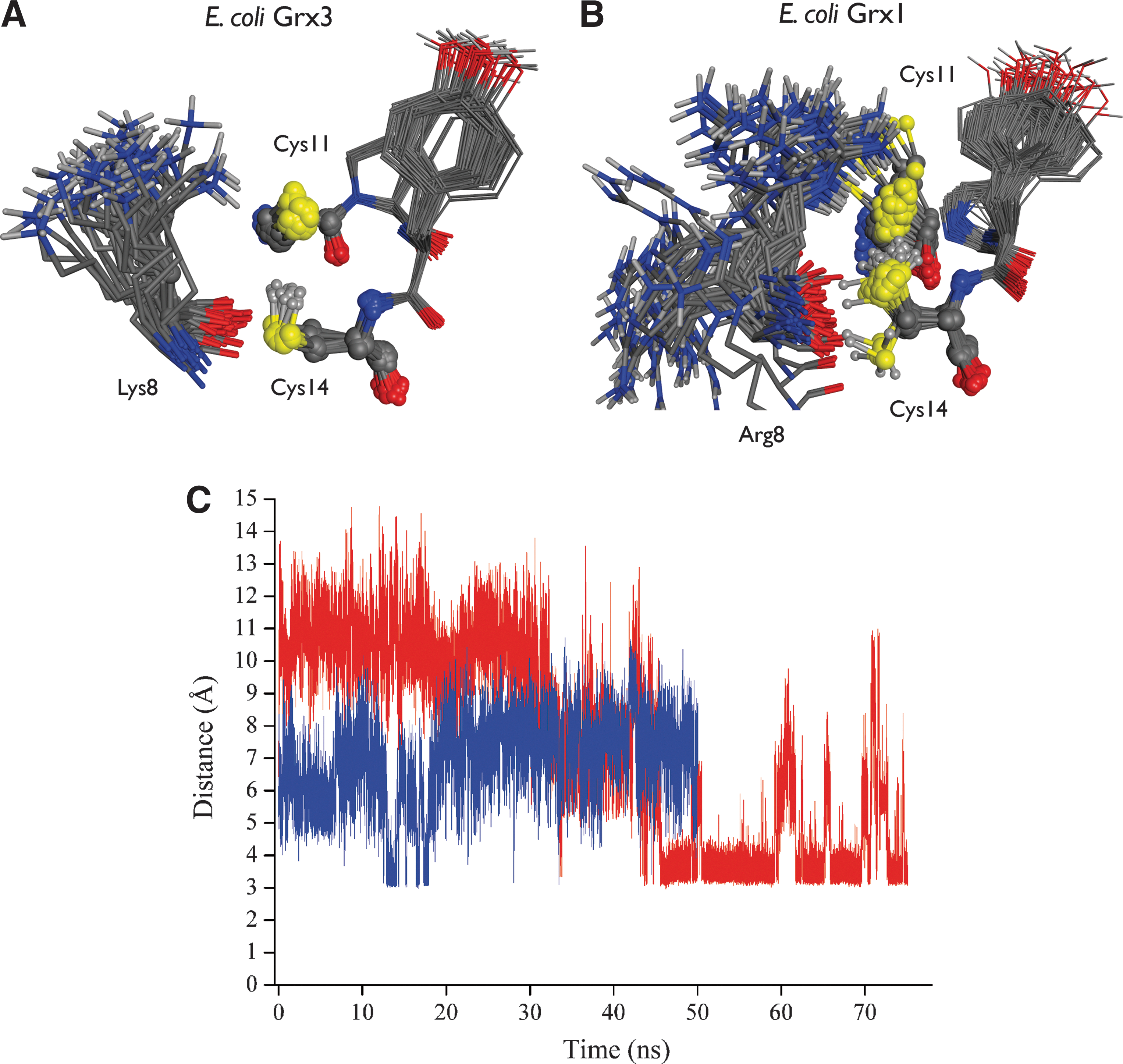
The discovery of the very low pK as of Cys1 initially fuelled the speculation that this pK a would probably be stabilized by positively charged side chains. This hypothesis has now been tested with a number of systems for which it has been mostly dispelled. However, it formed the basis for much mutagenesis work, for example, with pig Grx (190), E. coli Grx3 (45, 129), E. coli DsbA (56, 66, 117), and human Grx1 (75). Early work suggested that Arg26 in pig Grx may be the key for a direct stabilization of the thiolate of Cys22, but structural work has since shown that formation of a salt bridge between these two side chains is sterically precluded (44). In reduced E. coli Grx3, His15 was mutated to Val (129), assuming that His15 would be positively charged and thereby stabilizing the thiolate of Cys11. Yet, His15 was subsequently shown to be neutral at physiological pH and cannot stabilize the thiolate via long-range electrostatics (45). The other candidate basic side chain for stabilization of the thiolate in E. coli Grx3 is Lys8. In MD simulations of E. coli Grx3 (43, 45), the side chain of Lys8 was found to be very flexible with its amino group spending most of the time solvated by water, away from the thiolate of Cys11 (Fig. 5). In this situation, water screens the electrostatic interactions between the thiolate and the lysine side chain, and the long-range Coulomb interaction between these groups was found to be negligible for the thiolate stabilization (43, 45). The amino group of Lys8 comes occasionally into contact with the thiolate in the computer simulations, reflected in a transient further drop in the calculated pK a of Cys11 of E. coli Grx3. However, the formation of this salt bridge was short-lived and predicted to contribute little to the overall stabilization of the thiolate (45), as subsequently confirmed experimentally (45). Similar arguments probably also explain why Lys19 has only a marginal influence on the pK a of Cys22 in human Grx1 (75).
The extensive mutagenesis work with E. coli DsbA also supports the notion of a limited influence of the surrounding charged side chains on the pK a of its catalytic Cys30. There are many mutants of residues X1 and X2 in the -Cys30-X1-X2-Cys33- active-site motif of DsbA (56, 66, 117), for which the pK a of Cys30 has been measured (Table 1). In the majority of the DsbA mutants, the pK a of Cys30 remains below 5.0 (Table 1 and (56, 117)), suggesting that the side chains of residues X1 and X2 are not the main factors decreasing the pK a of Cys30. Indeed, including a basic arginine in mutant -C30-T31-R32-C33- gave a pK a of 4.76 for Cys30 (56), higher than the pK a of ∼3.5 for the wild-type sequence -C30-P31-H32-C33-. This supports the view that positioning basic side chains in the vicinity of a cysteine is not sufficient to lower its pK a. It is consistent with the observation that the pK a of Cys30 is virtually insensitive to the E37Q and E38Q mutations in the surrounding of the DsbA active site (66). In fact, elimination of all charged residues in the neighborhood of the active site of DsbA had a negligible impact on the pK a of its nucleophilic cysteine (74). Instead, in DsbA as well as in other members of the Trx superfamily, there is evidence that interactions between the thiolate and the backbone of residues X1 and X2 are critical for modulation of the thiolate pK a (45).
In general, computational and experimental results indicate that the surrounding charged side chains do not primarily control the thiolate pK a in the Trx superfamily. An interesting possible exception, however, has been uncovered with Arg8 in reduced E. coli Grx1 (43). A comparative study of homologous E. coli Grx1 and Grx3 by MD and pK a calculations convincingly suggested that Arg8 forms a salt bridge (including a hydrogen bond) with Cys11 relatively frequently in Grx1 (43), in contrast to Lys8 and Cys11 in Grx3 (Fig. 5). This could be explained in terms of subtle differences between the conformational dynamics of Arg8 versus Lys8. Therefore, it was proposed that Arg8 contributes to lower the pK a of Cys11 in Grx1 (43), but not Lys8 in Grx3. These computational insights have recently received indirect independent experimental support (155). Thus, peripheral side chains can sometimes influence the stability of the catalytic thiolate, and such mechanism may play a role in other systems (83, 96, 100). The extracytoplasmic atypical Trx ResA provides another example, for which it has been reported that Glu80, located close to the active site, but not forming hydrogen bonds with the active thiolates, plays a role in controlling the acid–base properties of both active-site cysteines (100). Another interesting case is that of protein disulfide isomerase (PDI), where the pK a values of cysteine residues play a crucial role during oxidative protein folding (83). In PDI, Arg120 was reported to lower the pK a of the C-terminal cysteine from 9.2 to 8.6 (96). A movement of Arg120 in the active site was reported, but not described in details (83), so it is unclear if its effect on the C-terminal cysteine is mediated by a direct hydrogen bond or longer-range interactions. Overall, however, the role of peripheral side chains for thiolate stabilization in redox enzymes of the Trx superfamily appears limited.
If the charged side chains form hydrogen bonds with the thiolate, it stabilizes the thiolate. Apart from Grx1, this was also demonstrated in human peroxiredoxin, where a conserved arginine residue (Arg127) is hydrogen bonded to the redox-active cysteine Cys51 and strongly diminishes the proton affinity of the thiolate form of Cys51 (16). The proton affinity is related to the Cys51 pK a. Another conserved arginine molecule (Arg150), which is also a part of the active site with Cys51, but is not hydrogen bonded to Cys51, has a much smaller influence on the Cys51 proton affinity. So, hydrogen bond contacts seem important to mediate the influence of charged side chains on cysteine pK as.
Considering the enzymatic functional requirements, it is maybe not surprising that nature has not selected flexible charged side chains as the main mechanism for thiolate stabilization in the Trx superfamily and its solvent-exposed active site. First, peripheral side chains have to be long (Arg and Lys), and therefore flexible, to reach to the thiolate. Thus, a stable ionic contact with the thiolate would incur an entropic cost. Second, a side chain approaching the thiolate on its solvent-exposed side may occlude the active site and sterically prevent the approach between the thiolate and its substrate. Third, when charges are exposed to a high-dielectric medium such as water, the electrostatic interaction between charges is screened and strongly diminished. Therefore, water molecules in a solvent-exposed active site could easily disrupt the salt bridge. Fourth, the stabilization of the thiolate by charged side chains would expose the enzymatic activity to the influence of ionic strength. In contrast, this ionic strength effect is minor when the thiolate is stabilized by hydrogen bonds with neutral groups, for example, with backbone amides (44, 116). In addition, controlling a cysteine pK a by local hydrogen bonds means that the peripheral ionized side chains can evolve independently of the maintenance of this pK a (44). That leaves the peripheral side chains free to evolve under different selection pressures, guided may be instead, by substrate recognition. Also, hydrogen bonds allow a much more precise molecular control of the enzyme chemistry, for instance, by discriminating between the active-site N-terminal and C-terminal cysteines in the -C-X-X-C- active-site motif of enzymes (Fig. 3). Indeed, it is difficult to imagine how long-range electrostatic interactions with flexible peripheral charged side chains would discriminate between the active-site N-terminal and C-terminal cysteines, since these two cysteines are very close in space. Instead, very directional localized hydrogen bonds can dramatically decrease the pK a of the N-terminal cysteine without affecting the neighboring C-terminal cysteine, as discussed in the next section.
B. The strong influence of direct hydrogen bonds on the pKa of cysteines
Before discussing how hydrogen bonds perturb the pK a of cysteine side chains, it is helpful to review the information pertaining to hydrogen bonds involving sulfur. Hydrogen bonds are energetically favorable interactions formed between a donor group (D−H) and an acceptor atom (A). D is an electronegative atom that polarizes the Dδ− −Hδ+ bond, resulting in a partial positive charge on the hydrogen atom. The hydrogen positive charge interacts with the (partial) negative charge of the acceptor atom, resulting in a favorable electrostatic interaction. Thus, hydrogen bonds are largely electrostatic interactions in nature, resulting in the well-known pattern: Dδ− −Hδ+–A(δ)−.
Since sulfur is less electronegative than oxygen or nitrogen, the role of sulfur in hydrogen bonding has been a matter of debate. Compared to oxygen, sulfur has a lower electronegativity and a larger radius, which reduce the ability of sulfur to participate in hydrogen-bonding interactions, as a donor or as an acceptor. However, there is now evidence that the thiol group can act as a moderately strong hydrogen bond donor or acceptor, and the thiolate is a hydrogen bond acceptor (1, 37, 39, 45, 57, 84, 129, 137, 183, 196). Hydrogen bonds with sulfur are longer than those with nitrogen or oxygen because of the size of the sulfur atom and its more diffuse electron cloud (57, 196). A statistical analysis on more than 500 high-resolution protein crystal structures indicated a 5:1 donor:acceptor ratio for sulfur in protein thiol groups (196), suggesting that sulfur in (neutral) thiols has a greater propensity to donate a hydrogen bond than to accept one. Yet, some QM calculations have suggested that sulfur may, at least sometimes, be almost as strong a hydrogen-bond acceptor as oxygen (137, 182). In the reduced -Cys1-X1X2-Cys2- motif of the Trx superfamily, the cysteine thiols act as hydrogen bond donors and acceptors (39, 45, 129).
The perception of hydrogen bonds by sulfur is probably dominated by crystallographic observations of distances and angles between sulfur atoms and potential donor or acceptor groups (1, 37, 57, 84, 196). A crystallography-based view of the geometric parameters of various types of hydrogen bonds involving sulfur in proteins was recently presented (196). To consider hydrogen bonds that stabilize thiolate anions, we concentrate on the geometric parameters of sulfur as a hydrogen bond acceptor. In protein crystal structures of NH–S systems, the distance between the donor nitrogen and the acceptor sulfur ranged from 3.25 to 3.55 Å, with deviations from linearity of the NH–S angle up to 25° (1, 37). An analysis of 151 high-resolution protein X-ray structures identified a mean distance of 3.54 Å and a mean angle of 138° for hydrogen bonds with cysteine sulfur atoms as hydrogen bond acceptors (196). Table 3 summarizes the mean distances and angles for cysteine sulfur accepting hydrogen bonds from different conventional donor types (196). In principle, one should also consider nonconventional, weaker, C−H hydrogen bonds to the sulfur. Indeed, there is now ample evidence that the C−H bond can be polarized enough to form interactions with a marked hydrogen bond character (33, 59, 167, 174). In proteins, examples of C−H hydrogen bond donors include the backbone Cα−H group, and aromatic C−H vectors provided by aromatic side chains (18, 71, 168, 181). Although the C−H groups are weaker hydrogen bond donors than conventional donors, they could still contribute to tune the pK a of some thiols. One such example has been proposed with pig Grx (Fig. 6), for which a detailed analysis (44) suggested that an aromatic C−H from a phenylalanine contributes to stabilize the thiolate. However, favorable interactions between sulfur acceptor and nonconventional C−H hydrogen bond donors have barely been explored. Thus, our analysis concentrates on conventional hydrogen bonds to the sulfur.

Geometric mean with standard deviation in brackets.
d (S–H): distance between the acceptor S and donor H atom. d (S–X): distance between the acceptor S and the donor X atom. θ (S–H–X): angle between sulfur, hydrogen, and donor.
Part of the data used in this table are reproduced from reference (196).
For redox cysteines, the geometric hydrogen-bonding parameters are likely to depend whether the accepting sulfur is neutral or in thiolate form. A thiolate acceptor may allow a broader range of angles for interaction with a hydrogen bond donor. In addition, one has to consider the situation where sulfur receives a hydrogen bond from another thiol, which lengthens the hydrogen bond distance. Thus, pragmatically, the following geometric criteria are frequently suitable to capture hydrogen bonds between sulfur and a donor atom: S–D distance <4 Å and S–H−D angle >90° (141, 196). In the following paragraphs, we discuss some examples that demonstrate the influence of hydrogen bond interactions on thiol pK a in redox enzymes of the Trx superfamily.
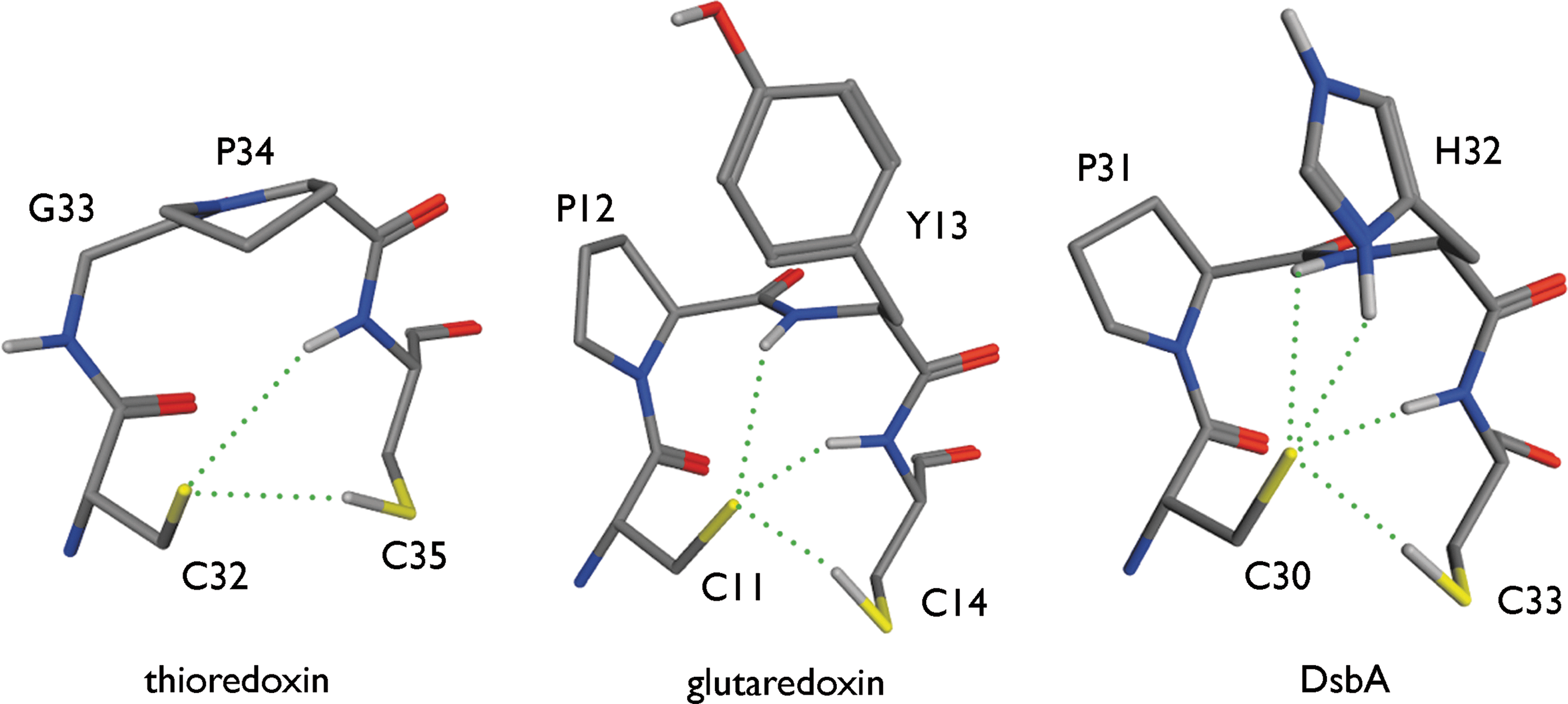
For the conserved -Cys1-X1-X2-Cys2- motif of proteins in the Trx superfamily, observations link the pK a of Cys1 to the number of hydrogen bonds received by the acceptor sulfur of Cys1. Structural analysis revealed that in reduced Trx, two hydrogen bonds are formed with Cys1; three in reduced Grx; and four in reduced DsbA (45) (Fig. 5). This is consistent with the pK a of 3.5 for Cys1 in DsbA, of 4.0 to 5.0 in Grx, and ranging from 6.3 to 7.1 in Trx (38, 46, 77, 82). Note the importance of the hydrogen bonds with the backbone N–H groups, which may also rationalize the largest pK a variations across the DsbA X1-X2 mutants. The predominant role of direct hydrogen bonding in stabilizing the thiolate in the Trx superfamily is consistent with the lack of effect of increased ionic strength (0.05–2 M) on the pK a of Cys22 of human Grx1 (44, 116). Using PB-based calculations, the pK a of Cys1 in Grx was estimated with the hydrogen bonds to the thiolate formed or not, depending on the conformation adopted by the side chain of Cys1 (45). Simply changing the rotamer of Cys1 can switch the hydrogen bonds to the sulfur on or off, which showed that disrupting the hydrogen bonds clearly increases the calculated thiol pK a. In general, the more hydrogen bonds to the sulfur that were present, the lower the pK a of the thiol in PB calculations (35, 43, 45). The influence of hydrogen bonds on the pK a of Cys1 is also seen in calculations with PROPKA1 (103). PROPKA1 calculates a Cys1 pK a of 3.4, 4.4, and 5.5 in, respectively, DsbA (Protein Data Bank [PDB] entry 1DSB), human protein disulfide isomerase (hDPI, PDB entry 1MEK), and E. coli Grx 3 (PDB entry 3GRX). PROPKA1 attributes these low pK as to hydrogen bond interactions between the thiolate form of Cys1 and surrounding residues (103).
There is also evidence that controlling the thiol pK as with hydrogen bonds plays a role during thiol–disulfide exchange reaction mechanisms, as suggested for Trx and Staphylococcus aureus pI258 arsenate reductase (Sa_ArsC) (Fig. 7). Sa_ArsC is one of the endogenous substrates of the powerful reductase Trx (8, 21, 115). Trx reduces oxidized ArsC via the nucleophilic attack of Cys29 of Trx (Cys1 in Cys1-X1-X2-Cys2) on the ArsC disulfide (Cys82–Cys89), leading to the release of Cys82 and formation of the Trx-ArsC Cys29–Cys89 mixed disulfide (115). In a subsequent step, this mixed disulfide needs to be reduced to release reduced ArsC (141). In this process, Cys32 of Trx (Cys2 in Cys1-X1-X2-Cys2) attacks Cys29 of Trx in the mixed disulfide, and Trx becomes oxidized (Cys29–Cys32 disulfide formed). Thus, the dissociation of the Trx-ArsC mixed disulfide proceeds via the nucleophilic attack of Cys32 of Trx on Cys29 of the Cys29Trx–Cys89ArsC disulfide. In isolated, reduced Trx, Cys32 has a high pK a (pK a>9), and is present in its thiol form. In the mixed disulfide, the resolving cysteine Cys32 needs to be activated to its nucleophilic thiolate form. A detailed study of the Trx-ArsC mixed disulfide complex (141) suggested that two hydrogen bonds between the Cys32 sulfur and backbone amides of Cys29 and Trp28 of Trx stabilize the thiolate form of Cys32. In the presence of these hydrogen bonds, the pK a of Cys32 drops to ∼7.7 (141), which activates Cys32 for its nucleophilic attack on Cys29. Formation of these hydrogen bonds to Cys32 was uncovered by MD simulations after localized conformational rearrangements around Cys32 (141). This illustrates how hydrogen bonds control the reactivity of a thiol via small, but precise, structural rearrangements.

The above examples, and others (see section III.D), support the notion that hydrogen bonds may be the primary factors modulating thiol pK as. It follows that a first indication of a decreased thiol pK a could be gained from structural information, simply by counting the hydrogen bonds to sulfur atoms in the structural model. Since the influence of hydrogen bond interactions is at short range, they are very sensitive to the details of the protein structure and dynamics (43 –45). Consequently, the details of the structural model used for visual interpretation and pK a calculations are important, and apparently, minor structural changes may strongly affect the calculated pK a values of interest (3, 35, 43 –45, 192). Hence, a degree of manipulation of the raw X-ray coordinates may be required to orientate protein side-chain amide and imidazole groups, as well as conserved water molecules, according to the most likely hydrogen-bonding network (123, 126). Also, NMR structures present special challenges, since their details can be prone to uncertainties, which affect the outcome of pK a calculations (35, 43, 134).
Apart from hydrogen bonds, another putative influence on the pK a needs further consideration. In the Trx superfamily active sites, Cys1 is located at the N-terminus of an α-helix. Helices have long been perceived to decrease the pK a of residues at their N-terminus. For example, a pK a decrease of the N-terminal cysteine of, respectively, 1.8 and 2.0 units was measured in rhodanese (151) and human Trx (46). The measured pK a of a N-terminal aspartate in a helical dodecapeptide was suppressed by 0.6 units (81). Earlier quantum chemical studies on papain have shown that a helix near the active site facilitates the proton transfer from the N-terminal cysteine to the histidine residue of the catalytic dyad (171). The origin of this effect is explored in the following section.
C. Reinterpretation of the helical effect on the pKas of cysteines
The decreased pK as of residues at the N-terminus of helices have been attributed for a long time to a helix–macrodipole effect, which would originate from the vector sum of the microdipole moments of the individual peptide units, and would be oriented along the helix-axis (67, 135, 150, 156, 176, 180). However, recent PB calculations on several model helices stressed that the helix dipole depends on the geometry and on the solvent exposure of the helix termini (152). Therefore, the helix macrodipole is not a simple vector sum of the individual dipoles. In vacuum, the helix dipole increases with the helical length, but in transmembrane helices in which both helical termini are solvent exposed, the helical dipole was reported to decrease with the helical length (152). For solvent-exposed helices, the effective dipole is strongly dependent on the orientation of the helix relative to the aqueous medium (152). When aqueous solvent is present, the helix macrodipole is counteracted by the solvent reaction field, which drastically reduces the long-range effects of the helix macrodipole. So, in many situations, there is effectively no helix macrodipole at work. Thus, although the older helix macrodipole hypothesis has been widely influential (19), there is strong evidence that the origin of the so-called helical effect can be explained and reinterpreted without needing to invoke a helical macrodipole. These new insights came from several studies, both computational and experimental (6, 45, 49, 66, 89, 103, 143, 147).
Early new insights into the helical effect on pK as came from computational studies on papain, for which more than half of the helical effect was attributed to hydrogen bonds with the backbone rather than to the macrodipole (147). This emphasized localized short-range interactions in addition to long-range electrostatic interactions. With sulfate-binding protein, Åqvist et al. (6) calculated the electrostatic contribution to the free energy of interaction between the helix and the substrate SO4 2− bound at its N-terminus, and concluded that the charge-stabilizing effect of the α-helix can be best explained by short-range interactions with individual peptide bond N–H dipoles at the N-terminus of the helix. They found that the first two helical turns account for 95% of the overall helical effect. The minimal influence of the helix macrodipole was supported by investigating the effect of introducing a positive charge by mutation at the helical C-terminus. Such charge would have neutralized the dipole charge, but its introduction had a negligible effect according to the calculations (6).
It has also been possible to address the helical effect experimentally, although it is more difficult to control the presence and geometry of a helix experimentally. DsbA offered an opportunity to study experimentally the effect of the helix dipole on the pK a of its catalytic Cys30 by manipulating a kink in the relevant helix (66). Mutants designed to alter the helical kink were expected to affect the overall helix dipole; however, only a minor effect on the pK a of Cys30 at the N-terminus of this helix was observed.
Kortemme and Creighton explored experimentally and systematically the nature of the helical effect (89), by monitoring the pK a of a cysteine at the N- or C-terminus of model α-helical peptides. They observed that a thiol pK a at the N-terminus of a peptide with high helical content was decreased by up to 1.6 pK a units (pK a values from 7.20 to 7.63) relative to a normal thiol pK a measured in an unfolded peptide. The interpretation was that a combination of electrostatic charge–helix dipole and hydrogen bonding interactions contribute to the pK a-lowering effect (89). Yet, several observations were consistent with the particular importance of hydrogen bonds and local effects. Thus, variation of the (neutral) amino acids at the peptide N-terminus had an impact on the thiol pK a at this N-terminus, pointing to the importance of local conformational effects and geometries, compatible with the pK a being decreased by hydrogen bonds. The same study varied the ionic strength to distinguish between stabilization of the thiolate by hydrogen bonds or by a more-diffuse electrostatic interaction between the thiolate and a helix macrodipole. Hydrogen-bonding interactions should be much less sensitive to screening by salt than charge–helix dipole interactions. Indeed, the thiol pK a was only weakly affected by changes in ionic strength, suggesting a dominant role for hydrogen bonding. Further evidence for a very weak interaction between the thiolate charge and a helix macrodipole came from pK as measured for thiols at the C-terminus of peptides with high helical content (89). Such pK a increased by only 0.2 pK a units relative to a normal cysteine pK a. The asymmetry of the magnitude of the pK a shifts observed at the N- versus C-terminus is difficult to reconcile with a strong overall helix macrodipole. However, this asymmetry can be explained in terms of local hydrogen bond interactions, since the helical N-terminus, but not the C-terminus, presents amide N–H donor groups for interactions with a negative charge.
Recent work showed that the main influence on the pK a of Cys1 of the -Cys1-X1-X2-Cys2- motif located at the N-terminus of an α-helix in proteins of the Trx superfamily is from hydrogen bond interactions. In the PB study of E. coli Grx3 (45), the effect of the entire helix on Cys1 (Cys11) was investigated by turning off incrementally the charges on the peptide bond dipoles one after another, starting with the amide groups closest to the cysteine of interest (Fig. 8). This revealed that the first two helical amide groups decreased the Cys11 pK a by ∼1.5 units each; the third and fourth amide groups decreased the pK a by ∼0.5 and ∼0.2 units, respectively; with the sixth amide group, a decrease of only 0.09 units was found. So, the pK a value of Cys1 in the -Cys1-X1-X2-Cys2- motif could be predicted in satisfactory agreement with an experiment without invoking the helix–macrodipole effect (45). This result was confirmed during the development of PROPKA (103), when it was found that pK a values can be reproduced based on the numbers of hydrogen bonds formed with the cysteine sulfur without intervention of a helix macrodipole (there is no such parameter in the PROPKA model).

Further evidence came from a computational quantum mechanical study on the effect of the helix length on the pK a of a cysteine thiol located at the N-terminus of model helices (143). Both 310- and α-helices of increasing lengths were tested, made of (neutral) alanine residues (143). The pK as were calculated with the NPA method (see section II.B). An initial decrease of the cysteine pK a with the first helical turns was found, but this effect weakened quickly after a few added residues. This pK a decrease was accompanied by the increase of the backbone amide–SγCys hydrogen bond strength. The first Ala residue decreases the cysteine pK a by 1.5 or 0.6 units in an α- or 310-helix, respectively. For the second residue, an extra decrease of, respectively, 0.2 or 0.1 pK a units was found in α- or 310-helices. The third residue was responsible for an extra decrease of 0.1 units (Table 4). As such, the pK a decrease diminished for every additional helical residue. Thus, once the hydrogen bonds to the sulfur are formed (typically associated with the first helical residues i.e., cysteine plus one alanine), the next residues strengthen these hydrogen bonds, but this has only a secondary effect on pK a perturbation. These QM calculations (143) pointed out that the lowering of the pK a of the N-terminal cysteine of an α- or 310-helix is largely due to the hydrogen bonds formed between the cysteine sulfur and the helical N-terminus.
Helix–macrodipole, NPA charge and pK a of N-terminal SγCys obtained in aqueous solution in 310-helices and in α-helices.
Table adapted from reference (143).
Overall, the above-mentioned studies consistently show that direct hydrogen bonds between the cysteine sulfur and the helical N-terminus are essentially sufficient to account for the thiol pK a downshifts. To explain such pK a shifts, the notion of helix macrodipole is not required. This further supports the idea developed in the previous section that hydrogen bonds to the cysteine sulfur are the main factors influencing the pK a values of the corresponding thiol groups.
D. How general are the mechanisms modulating the pKa of cysteines?
Apart from their function in the redox biochemistry, cysteines play important roles in the catalytic processes of a wide variety of enzymes (80, 109, 110) like proteases, transferases, kinases, phosphatases, and isomerases. Cysteine residues coordinate metallic redox centers as in iron–sulfur clusters. Coordination of metals by cysteines can also play structural roles, such as zinc coordination in Zn-finger domains (94). In addition, the structural organization and oxidative folding of proteins rely on disulfide bonds. One can surmise that the structural and physical principles modulating the cysteine thiol pK a in redox enzymes, as discussed in the previous sections, are likely to be largely transferable to cysteines in other protein families.
For example, the reactive cysteine (Cys106) in human DJ-1 [a protein linked to Parkinson's disease and member of the class I glutamine amidotransferase-like superfamily (107)] has a decreased pK a of 5.4. A marked contribution to this pK a shift has been attributed to a hydrogen bond interaction with a conserved protonated glutamic acid Glu18, accounting for a pK a decrease of 1.0 unit (184). On the other hand, the sulfate anion at 5.9 Å of the reactive cysteine only increases the thiol pK a by 0.4 units (184). This further illustrates the limited role of long-range electrostatics and identifies hydrogen bonding as a key factor determining the pK a of Cys106. Another example of hydrogen bonds determining a cysteine pK a is found in human muscle creatine kinase. In this enzyme, the pK a of cysteine 282 was measured to be 5.8 (177). QM calculations pointed out that the main determinants of this low pK a are the hydrogen bonds between Cys282 and the −OH group of a serine side chain and a backbone amide (120, 177). Each hydrogen bond lowers the pK a by, respectively, 0.8 and 1.5 units (120).
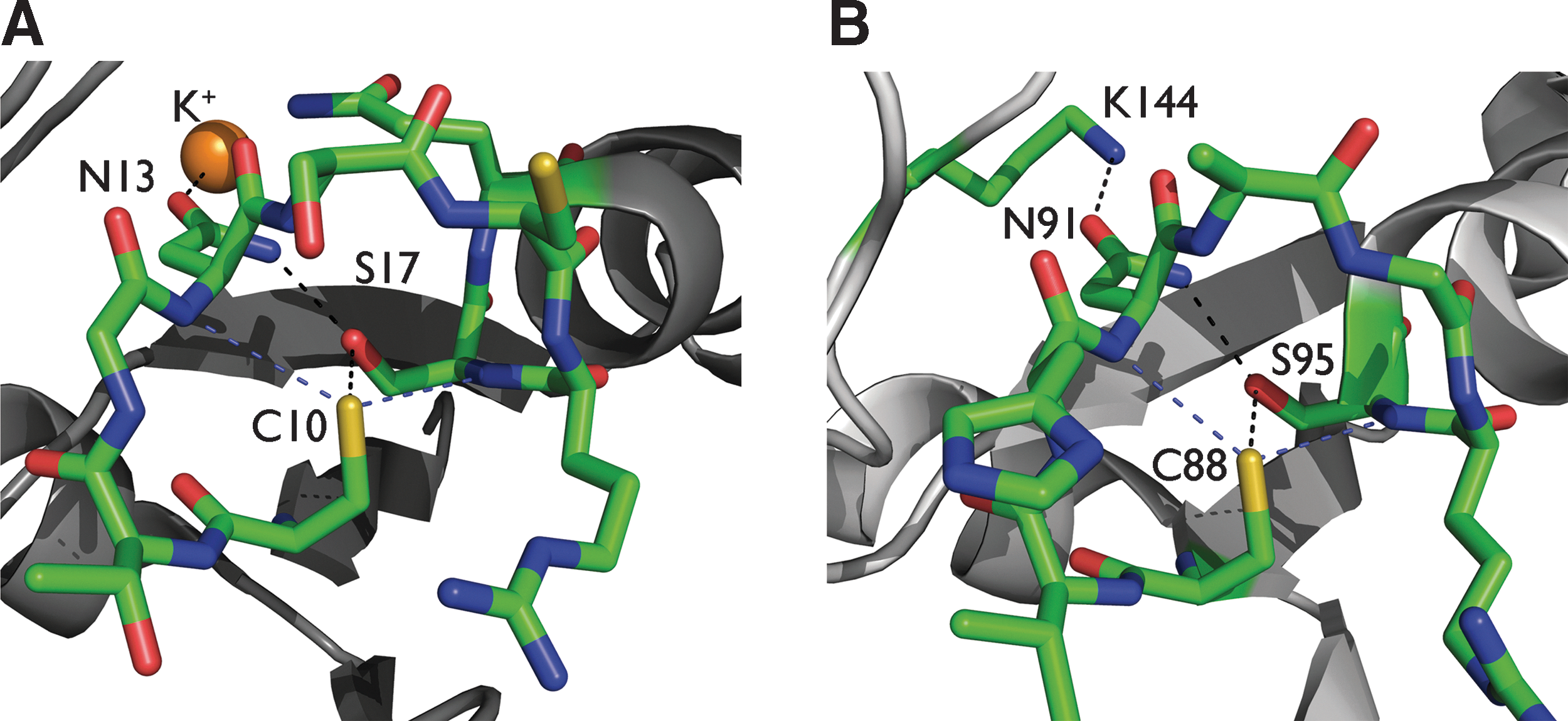
In human α-antitrypsin, the pK a of the active Cys323 was measured to be 6.9 (58). QM studies indicated that a hydrogen bond with the amide group of a neighboring Leu residue decreases the pK a of Cys323 by 1 (120). In ArsCs with a low-molecular-weight tyrosine phosphatase fold from S. aureus (145) and Corynebacterium glutamicum (175), a hydrogen bond network decreases the pK a of the nucleophilic cysteine (Fig. 9). Another example is found in Tn501 mercuric ion reductase, the key enzyme involved in reducing Hg2+ to Hg0 in bacteria (11). Its N-terminal heavy-metal-associated domain contains two cysteines, Cys11 and Cys14, with decreased pK as of 7.7 and 7.2, respectively (97). The authors invoke the position of both cysteines at the N-terminus of an α-helix as main factor lowering the pK as. This helical effect is not further characterized, that is, the authors do not give evidence for a helical dipole effect nor mention hydrogen bonds. Based on section III.C, we propose that the so-called helical effect may be attributed to hydrogen bond interactions between Cys11 or Cys14 and N-terminal helical residues. This is supported by an examination of the structure with the PDB code 2KT2 (97). Based on this NMR structure, the following hydrogen bonds are found between the thiol side chain of Cys14 and backbone amides: Cys14Sγ–NHTyr10: 3.4 Å and Cys14Sγ–NHCys11: 3.7 Å, while no hydrogen bonds formed with Cys11 could be observed. This is consistent with the authors' statement that the lower pK a found for Cys14 compared to Cys11 may be attributed to the more precise positioning of Cys14 in the first turn of the helix compared to Cys11 located in the more flexible loop preceding the helix (97). Further, the −OH group of Tyr62 accounts for an extra pK a decrease of 0.8–1 pK a units for both cysteines, while the electrostatic interaction with the positively charged His8 side chain influences the pK a of both cysteines by only 0.2 to 0.4 pK a units (97). Again, it suggests that the decreased pK as of Cys11 and Cys14 can be attributed to hydrogen bonding rather than to electrostatic interactions between charged residues.
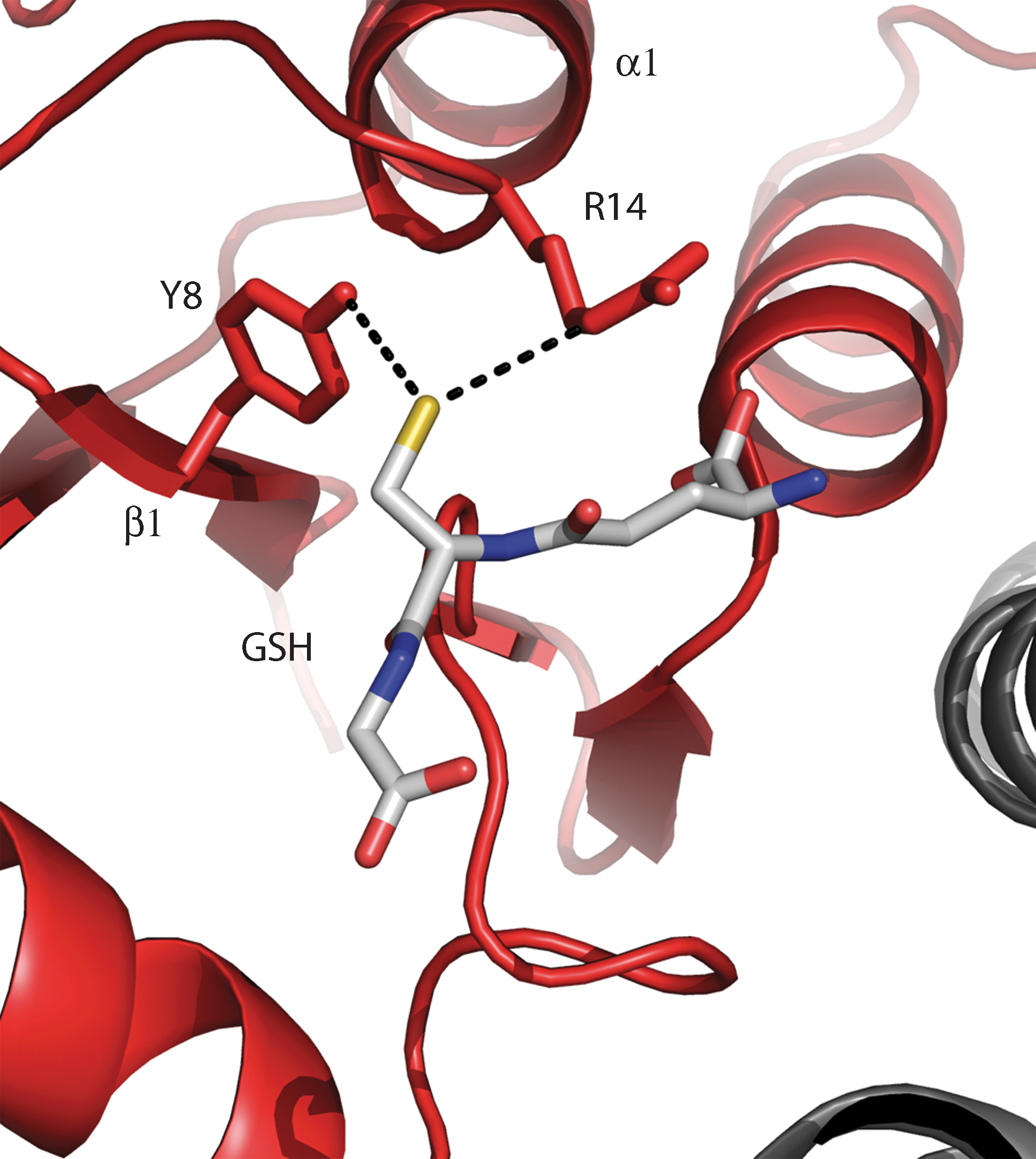
The lowering of the pK a of the cysteine in glutathione (GSH) is seen in GSH S-transferases. GSH S-transferases are involved in cellular detoxification in a wide variety of organisms and catalyze the conjugation of GSH to electrophilic substrates by lowering the pK a of the cysteine of GSH. In relation to this mechanism, the alpha, mu, pI, sigma, and theta GSH S-transferase classes are the best documented (7). GSH S-transferases are homodimers with a hydrophilic subunit interface and each polypeptide chain consists of two domains, an N-terminal domain with a Trx-fold and a C-terminal α-helical bundle. The N-terminal domain contains the active-site functional group, the hydroxyl group of a tyrosine or serine residue (79, 87, 105, 178), believed to activate the cysteine of GSH. The reactive species of GSH in the binary complexes is most probably the thiolate anion, which accepts a hydrogen bond from the seryl or tyrosyl hydroxyl group (E-OH–SG) and gathers additional stabilization from a positive charge of an arginine in the class α enzymes (Fig. 10). In some mu-class GSH S-transferases extra stabilization of the GSH thiol comes from a second sphere of electrostatic effects in which the π-electron cloud of the tyrosine is involved (PDB code 6GST) (187). Hydrogen bonding and other electrostatic effects lower the pK a of the GSH thiol from ∼9 to ∼6 in the enzyme–GSH complex (20, 105), so that it is predominantly present as thiolate at physiological pH, and more nucleophilic.
It is not surprising that the factors modulating cysteine pK a values also modulate pK a values of other titratable groups in proteins (103). Local interactions were also proposed to be the main pK a determinants for aspartate and glutamate residues in turkey ovomucoid third domain (OMTKY3) (102), since calculated and experimental pK a values were in agreement when considering only interactions in the immediate vicinity (4–5 Å) of Asp or Glu. The developers of PROPKA generalized these conclusions regarding the pK as of Asp and Glu residues by identifying hydrogen bond interactions as the main source for their pK a perturbations (103). It is known that Asp and Glu residues at the N-terminal of an α-helix usually have lower pK a values (133). As with cysteines at helical N-termini, this effect has long been attributed to the helical macrodipole (67, 176). However, recent analyses concluded that hydrogen bonds between helical backbone amides and Asp and Glu residues are the main contributors to their decreased pK a values (133), instead of a helical macrodipole. These conclusions echo those obtained for cysteines (45, 143).
IV. Functional Properties Influenced by the Cysteine pK as
The previous sections summarized techniques for determining the pK as of cysteines and gaining insight into the factors that modulate those pK as. This was largely illustrated based on the active-site cysteines of the redox proteins of the Trx superfamily. During a catalytic cycle, those cysteines undergo oxidation and reduction. The enzymatic activity of these proteins is determined by their reaction kinetics and redox potentials. In this section, we discuss how these properties are influenced by the relevant cysteine pK as. This underlines the functional relevance of the pK a of cysteines involved in enzymatic catalysis.
Lowered pK a values of catalytic cysteines influence the reaction kinetics and thermodynamics, and strongly influence the catalytic efficiency of an enzyme. This is especially true for thiol–disulfide exchange reactions, which are characterized by a Brøndsted coefficient 0<βnuc<1 for the nucleophilic cysteine (17, 78). The β-coefficients (βnuc , βlg , and βc) determine the slope of the plot of the logarithm of the second-order rate constant versus the pK a: [log(k s−)=βnuc×pKa(nuc)+βlg×pKa(lg)+βc×pKa(c)+C, with C a constant applicable for a specific thiol–disulfide exchange reaction, and βlg and βc, the Brøndsted coefficients for respectively the leaving group thiol and the central thiol when a thiolate attacks an unsymmetrical disulfide] (Fig. 11). Brøndsted coefficients characterize the sensitivity of the reaction to the pK a. The coefficient establishes the change in atomic charge as the reaction proceeds from the ground state to the transition state (Fig. 11). Complete proton transfer to the nucleophile gives a value for βnuc of 1, and no transfer a value of 0. At those extreme values, changing the pK a has no influence on the reaction rate. For nucleophilic thiols with a pK a below the solution pH, an increased concentration of the thiolate is less significant than the decreased nucleophilicity resulting from electron withdrawal. The most significant effect in thiol–disulfide exchange reactions comes from the decrease of the pK a of the leaving group. As the pK a of the leaving thiolate is decreased by electron-withdrawing substituents or electrostatic effect, the rate constants for the reaction increases with a factor of 3.2 to 5 for each unit decrease in the pK a of the leaving thiolate (βlg=−0.5 to −0.7) (164). Decreasing the pK a of the leaving thiolate from 8.5 to 4.5 should increase the rate for thiol–disulfide exchange by ∼100-fold to 630-fold (52). Also, the electron withdrawal on the central thiol (βc) will accelerate the thiol–disulfide exchange with a factor of ∼2 for each unit decrease in the pK a (βc=−0.3) (164). All together, the decrease of the thiolate pK a in proteins should be more important in the function of the thiolate as a leaving group than in the function of the thiolate as a nucleophile.
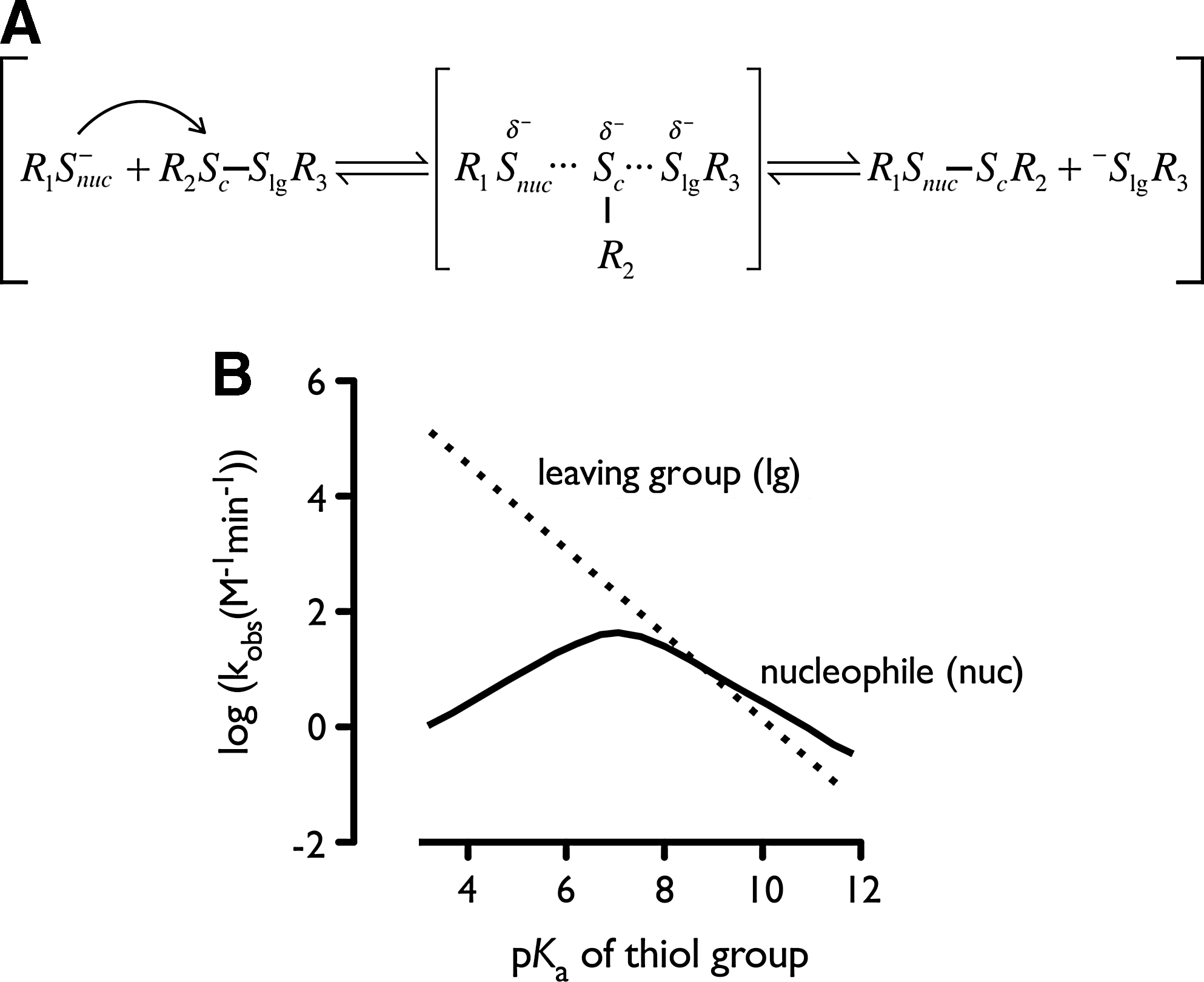
Another example on how the pK a determines reaction rates is the correlation between a cysteine pK a and the rate constant of the H2O2-induced cysteine oxidation to sulfenic acid. Thiolates react more rapidly with H2O2 than thiols (183); here again, a low pK a means a high reaction rate. Ferrer-Sueta et al. refined this overall view (41). Features that act to decrease the pK a may also decrease the nucleophilic character of the thiol, and hence make it less reactive. The effect of lowering pK a on rate enhancement will in general be most significant when pK a values are close to solution pH. A small increase in the concentration of the thiolate resulting from a further reduction of pK a will be undermined by the corresponding decreased nucleophilicity resulting from electron withdrawal (Fig. 11).
In the Trx superfamily, the pK a of the nucleophilic cysteine is related not only to reaction rates but also to the disulfide reduction potential. When this pK a decreases, the associated disulfide/thiol reduction potential increases, that is, the enzyme becomes a stronger oxidant. This was shown for Trx and DsbA active-site mutants for which a linear correlation between pK a and reduction potential was found (69, 142). Empirical relations between the cysteine pK a and the disulfide/thiol reduction potential have been proposed for Trx-type oxidoreductases (118). Here, we show a correlation for Trx-fold enzymes of E. coli (Fig. 12). The redox potential increases with ∼54 mV for each decreasing pK a unit. The only exception is seen for the gamma-domain of DsbD, which has an unusual high pK a of more than 9 [9.3 (161) and 10.5 (112)] and a redox potential of −241 mV (25). This makes the nucleophilic cysteine (Cys461) poorly reactive toward the disulfide of DsbDα and prevents its nonspecific oxidation (32). However, formation of the complex between DsbDγ (C-terminal domain) and DsbDα (N-terminal domain) seems to lower the pK a of Cys461, allowing the transfer of electrons between these two DsbD domains (112, 113).
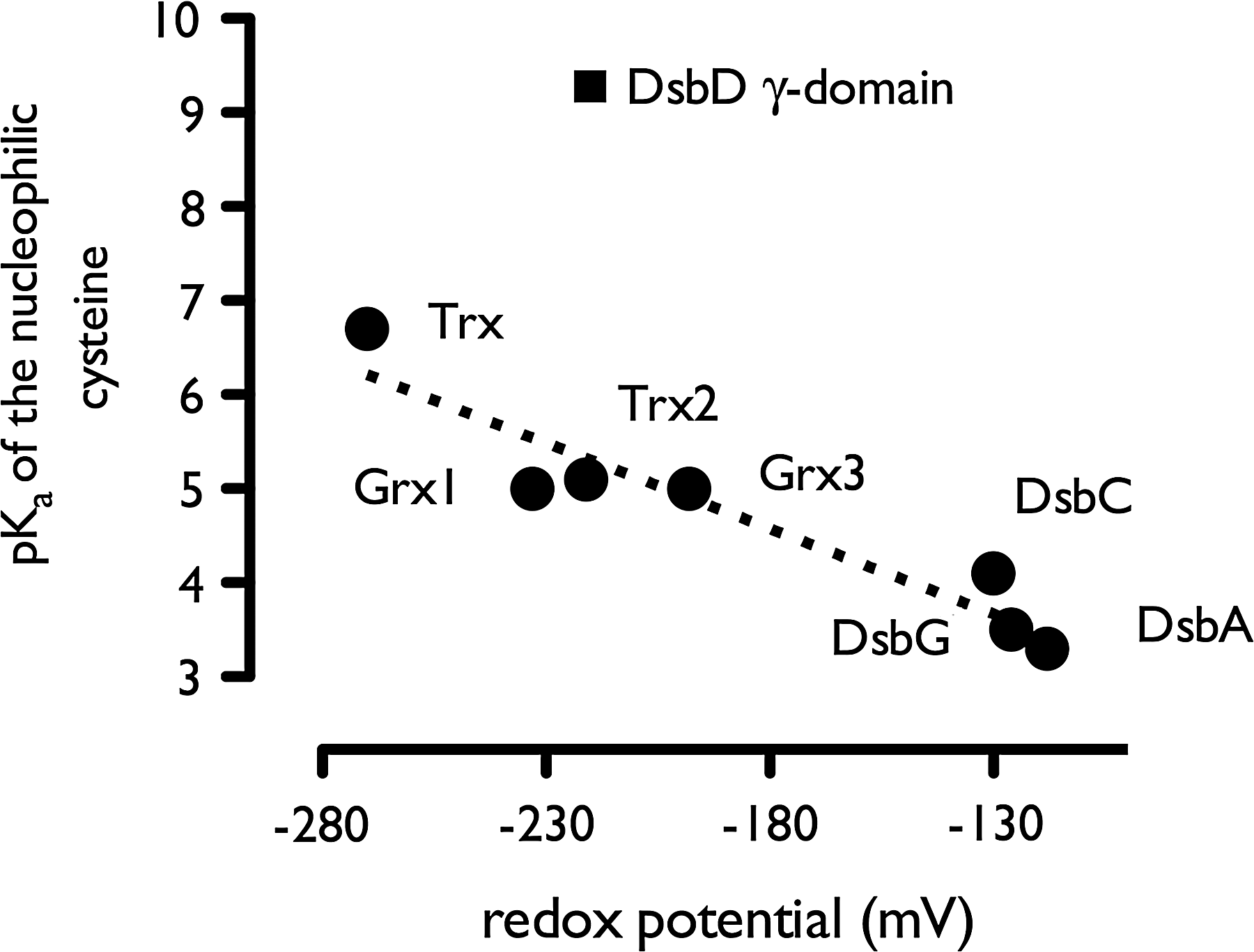
The importance of pK as with respect to disease and health can be gathered from many examples. For example, Trx proteins, the function of which is largely depended on the pK a of the cysteines in the -Cys1-X-X-Cys2- motif, are involved in the regulation of diverse proteins like peroxiredoxin, transcription factors, and ribonucleotide reductase. As such, Trxs are involved in maintaining the redox homeostasis of the cell (22, 24, 63, 78), antioxidant defense, apoptosis and DNA synthesis, and repair. All these factors are involved in many diseases like diabetes, cardiovascular diseases, cancer, Alzheimer's, and Parkinson's disease. For example, proteins involved in protein folding, like PDI, have a direct impact on heart and kidney failure (34). In addition, GSH S-transferases, which catalyze the conjugation of GSH to a broad range of xenobiotic substrates by lowering the pK a of GSH, are well known to influence the metabolism of a number of drugs (65, 148). Thus, there is ample evidence that cysteine pK as are critical to many biomolecular mechanisms essential for cellular functions, and are also involved in pharmacological processes. Therefore, understanding the molecular factors that control these pK as will help future research to understand the molecular basis of some disease conditions.
V. Conclusions
Empirical and case-by-case determination of the pK a values of catalytic cysteines remains very valuable, since it provides insights into the chemistry of individual enzymatic reactions. Also, determining additional reliable experimental pK a data is very important to strengthen the foundations for the development of theoretical predictive methods. However, there are limits to what one can expect from time-consuming experimental measurements. It is probably unrealistic to hope to be able to measure all interesting pK a values. Indeed, many relevant pK a values will adjust transiently during molecular encounters and complex reaction mechanisms (141). In these situations, it is already clear that computational approaches will become the methods of choice. However, for those theoretical methods to be used routinely and confidently, their performances need to be further tested and improved. The results of accurate calculations will provide not only the pK a values themselves but also precise insights into the factors modulating the pK as and the associated functional properties. Such improvements will require method developments, which would immensely benefit from close collaborations between computational and experimental scientists.
Footnotes
Acknowledgments
JM is group leader Redox Biology at the Vlaams Instituut voor Biotecnologie (VIB), and thanks NF and GR for this fruitful collaboration. GR thanks the Fund for Scientific Research Flanders (FWO) for a postdoctoral fellowship. We would like to thank all the 10 reviewers for their valuable comments, which have enormously improved this review.