
Abstract
Introduction
Mixtures of superoxide and hydrogen peroxide are generated by the auto-oxidation of flavoenzymes, particularly the respiratory dehydrogenases (47). Most, if not all, organisms that use molecular oxygen as a terminal electron acceptor express enzymes that are responsible for scavenging these potentially damaging oxidants, notably superoxide dismutase, and the catalases and peroxidases that detoxify H2O2. Superoxide is also synthesized by the NADPH oxidase of phagocytic cells of the innate immune system, and the antioxidant defenses of some bacterial pathogens are therefore virulence determinants (30). Phagocytes also synthesize nitric oxide (NO), by the oxidation of the guanidino nitrogen of arginine catalyzed by NO synthase. Superoxide and NO react rapidly together to form the potently anti-bacterial peroxynitrite, which spontaneously converts to peroxynitrous acid, nitrogen dioxide, and hydroxyl radicals (Fig. 1). Recent evidence suggests that bacterial pathogens express enzymes capable of detoxifying peroxynitrite (67, 107). NO itself can also be detoxified by oxidation to nitrite, or reduction to N2O or ammonia. Some of the enzymes that scavenge reactive nitrogen species (notably the flavohemoglobin that oxidizes NO to nitrate) are also virulence determinants (8).
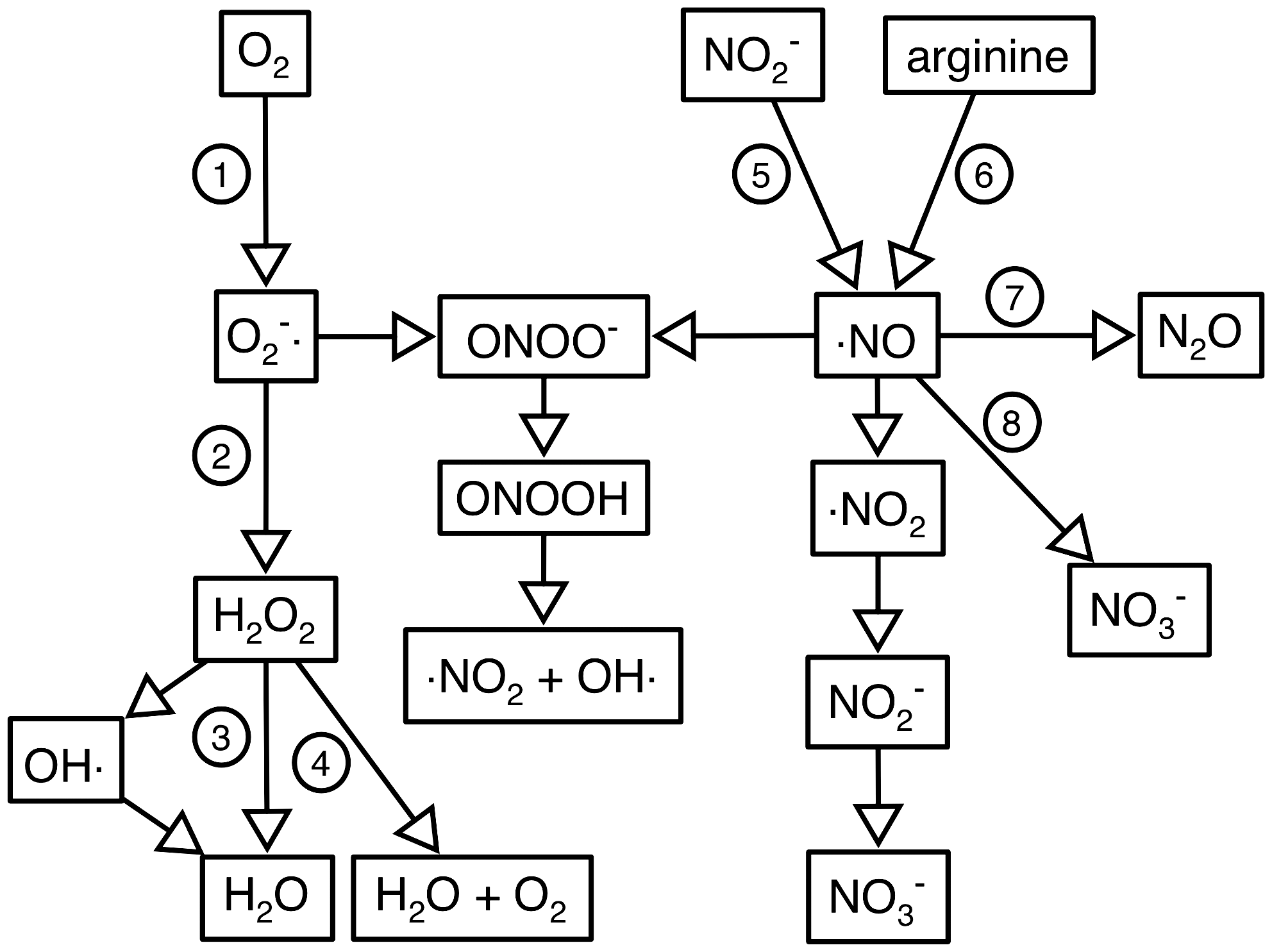
Superoxide and NO and the ROS and RNS derived from them have a wide variety of cellular targets, including metal centers in proteins, protein thiols and tyrosines, lipids, and DNA (30, 47). Expression of the genes encoding the enzymes that defend against ROS and RNS is typically substrate-inducible, transcription being regulated by activators or repressors that are sensitive to ROS and RNS. The same regulatory proteins may also coordinate other aspects of stress responses, such as the repair of damaged macromolecules, and the induction of genes encoding oxidant-resistant isoenzymes. This article will focus on the structures and functions of members of one subset of ROS and RNS responsive transcriptional regulators, those that contain a non-heme iron center. Specifically, these are the NO sensor NorR, the peroxide sensor PerR, and regulators of iron homeostasis (Fur, DtxR, and IdeR) in which the iron-sensing site might also be sensitive to ROS and RNS.
The Ferric Uptake Regulator
Function of Fur
The ferric uptake regulator (Fur) is a transcriptional regulator with a major role in iron homeostasis (34). Under iron-rich conditions, Fe2+ ions bind to Fur and stimulate its DNA binding activity. The Fe2+-containing form of Fur (Fur-Fe) acts either as a repressor or as an activator. On the one hand, Fur-Fe represses gene transcription by binding to sites close to the −35 and −10 promoter elements to block the access of RNA polymerase. On the other hand, Fur-Fe activates transcription by binding to several extended sites between 60 and 240 bp upstream of the transcription start site (14). Activation occurs by two different mechanisms: stimulation of transcription by recruitment of RNA polymerase (14, 25) or reversion of repression mediated by the histone-like nucleoid-associated protein, H-NS (71).
In Escherichia coli, the targets for Fur regulation include genes and operons involved in iron acquisition, and these are repressed in conditions of iron sufficiency (4). Enteric bacteria express multiple iron storage proteins (ferritins and bacterioferritins) and these may be either upregulated or downregulated by iron sufficiency (71, 106). Fur is also a repressor of the gene encoding a small RNA, RyhB, which acts in trans to stimulate the degradation of target mRNAs (65, 66). Thus, under high iron conditions, Fur represses ryhB, and RyhB target mRNAs are stabilized. The targets for RyhB regulation include genes encoding nonessential iron-containing proteins, a mechanism that ensures that under iron-limiting conditions the available iron is directed towards iron proteins that are essential for growth (66). Fewer genes are directly activated by Fur-Fe, 10 such genes have been identified in Neisseria meningitidis (24) and 25 in Helicobacter pylori (29). In E. coli, Fur activates transcription of ftnA, encoding the major Fe storage protein ferritin, by silencing the repression mediated by H-NS. The use of two different modes of gene activation in E. coli by Fur-Fe (the RyhB route and H-NS anti-repression) has raised the question of the physiological role of each mechanism. An appealing model suggested by Nandal et al. (2010) is that the weak affinity of Fur-Fe for the extended sites in the ftnA promoter allows a very sensitive activation/repression of ftnA, and thus expression of ftnA will drop more rapidly with iron concentration than expression of the RyhB-dependent genes (71).
The apo-Fur protein also exhibits DNA binding activity and can both repress and activate gene expression. Examples of targets for apo-Fur include sodB and heat shock genes, but this mode of regulation appears to be restricted to H. pylori and N. meningitidis, and is not well understood (24, 29, 68).
Fur structure and mechanism
Mechanistically, Fur can simplistically be viewed as a regulator of transcription that uses iron as the co-factor. However, even the basic details of this model remain rather poorly understood (61). In addition to the expected one iron-binding site per ∼17 kDa monomer, there is a second binding site for zinc in Fur from E. coli, a site which has a structural role, since zinc is required to stabilize the dimeric form of the protein (3, 21, 77). The zinc in the structural site is coordinated by two cysteines (Figs. 2 and 3), and by nitrogen and oxygen ligands from amino acids that have not been identified (40, 49). Iron in the sensing site is coordinated by 2–3 histidine residues and 1–2 negatively charged residues, again the specific residues involved have not been identified (1). Amongst characterized Fur orthologs and homologs, some appear not to have the structural site, and some have an additional crystallographically defined low affinity site, which may not be occupied by a metal in vivo. As will become evident, the nomenclature used for the three sites is inconsistent and therefore rather confusing. Here, the three sites are referred to as the sensing, structural, and low affinity sites.

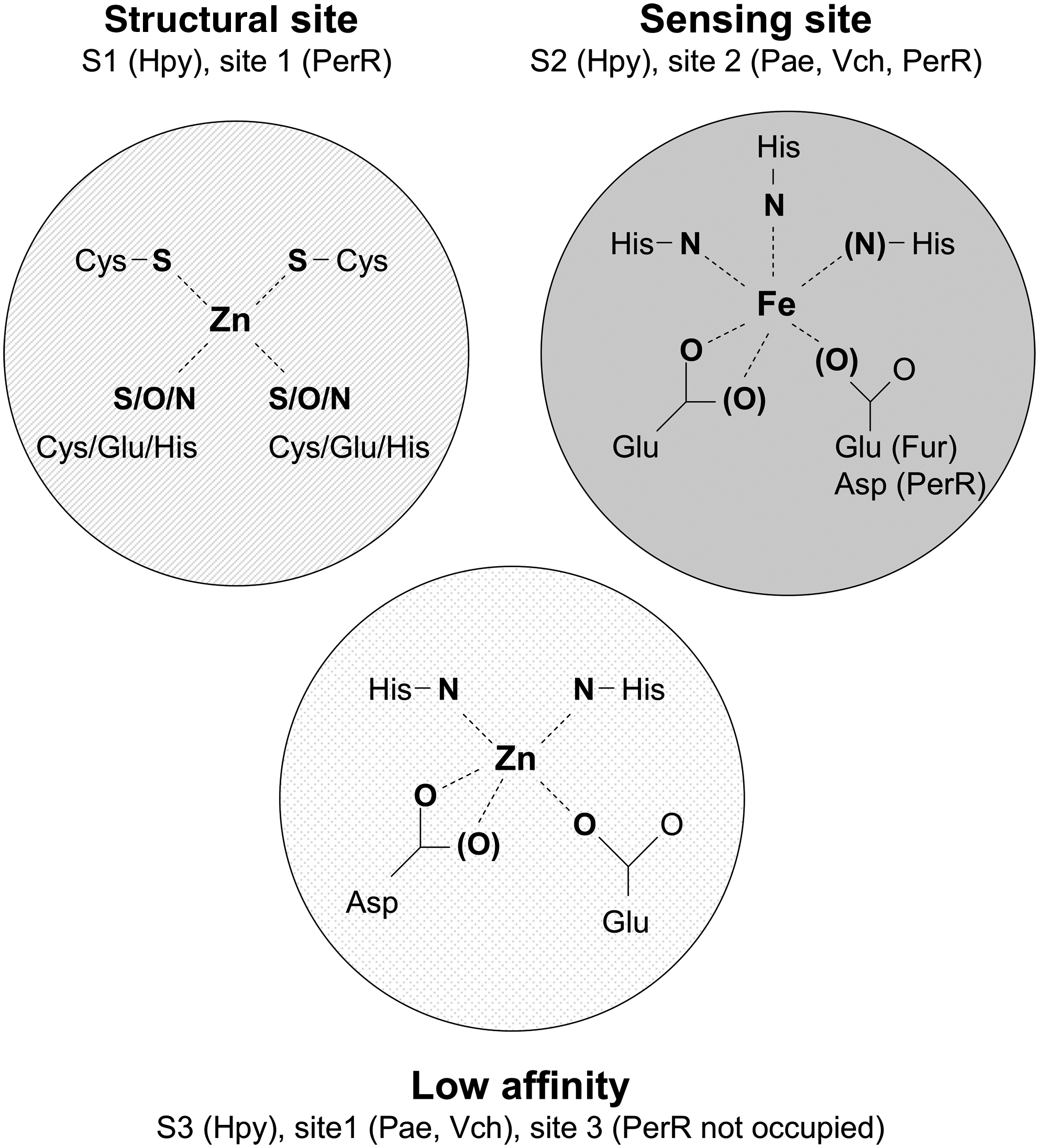
The first Fur structure to be solved was that of the Pseudomonas aeruginosa protein, which revealed two metal binding sites: Site 1 in the C-terminal dimerization domain, and Site 2 at the interface between the dimerization domain and the N-terminal DNA binding domain (78). On the basis of spectroscopic and metal exchange experiments, Site 1 was assigned as the regulatory iron-binding site, with Site 2 proposed to have a structural role (78). It was subsequently suggested that Site 1 in the P. aeruginosa protein is not physiologically relevant (i.e., is the low affinity site), and that Site 2 is the iron-sensing site (2, 61). There is biochemical evidence that the P. aeruginosa Fur does not contain a bound zinc (62), which is consistent with the absence of the structural site and with the suggestion that the low affinity site is not normally occupied. In the structure of the Vibrio cholerae Fur, Site 2 was also suggested to be the metal-sensing site, where iron (or zinc in the crystal structure) is coordinated by three histidine residues and a glutamate (93). In the structure of the Fur from H. pylori, all three sites are present and occupied by metal ions (26). In H. pylori Fur (Fig. 3), the three sites are designated S1 (structural), S2 (sensing), and S3 (low affinity). The low affinity site was shown to be dispensable for DNA binding (26). The zinc in S1 is coordinated by four cysteine residues, and is equivalent to the crystallographically-defined zinc site in PerR (50). This site is probably also similar to the structural site in the E. coli Fur, though the site in the E. coli Fur involves only two cysteine ligands (26, 40, 49). Overall, there is apparent diversity in the number and the structure (in terms of ligation) in Fur metal-binding sites. To summarize (Fig. 3): iron is sensed at a site in which it is coordinated by histidine and glutamate or aspartate residues (Site 2 in P. aeruginosa). It seems likely that this site is conserved in the E. coli protein (Fig. 2), although the iron ligands have not been identified conclusively. Zinc bound to the low affinity site is evident in the published structures of Fur proteins, the physiological role of this site is not clear, and it may be not occupied by metal in vivo. The equivalent site in PerR (Site 3) is not occupied by metal ions in the crystal structure, and residues in this site in PerR site may instead have roles in modulating the metal selectivity of Site 2, via a network of hydrogen bonds (63). Specifically, Site 3 residues are located at the dimer interface, and are close to some of the residues in the sensing site (Site 2). Thus, it was suggested that Site 3 residues participate in a hydrogen-bonding network that modulates the metal binding affinity and specificity of Site 2 (63). Finally, the Fur proteins from E. coli and H. pylori (and PerR) have a structural zinc site, in which the metal is coordinated by four cysteines, or (in the E. coli protein) by two cysteines and two N/O ligands that serve to stabilize the dimeric structure.
Fur and oxidative stress
Iron metabolism and oxidative stress responses are inextricably linked (4, 17, 31, 47). Hydrogen peroxide and ferrous iron participate in the Fenton reaction, which generates hydroxyl radicals, the hydroxide anion, and ferric iron. Ferric iron can be reduced by cellular reductants, providing a mechanism for sustained Fenton chemistry (47). The hydroxyl radical reacts with many targets, including DNA, the latter being ultimately responsible for cell death. Since both superoxide and H2O2 can react with iron-containing proteins (47), elevated free iron is one consequence of oxidative stress (54), thus causing a feedback loop which exacerbates the production of hydroxyl radicals by Fenton chemistry. The expression of the fur gene is elevated during oxidative stress (114), and Fur regulation presumably helps to mitigate the elevated iron levels that occur during oxidative stress. For example, positive regulation by Fur-Fe of genes encoding iron storage ferritin-like proteins would lead to increased sequestration of iron within these proteins during oxidative stress (71, 106). In cells that are chronically stressed by an inability to remove H2O2, free iron levels are elevated, and are elevated still further in a strain in which fur expression is not induced by H2O2 (105). Since Fur is a highly abundant protein (∼5000 molecules/cell [114]), it may also function to directly bind excess iron during oxidative stress, which causes a ∼2-fold increase in Fur abundance (31, 114). Mutants lacking Fur have elevated free iron levels and are more sensitive to oxidative stress (99). Thus, Fur has a crucial role to play in coordinating the cell's response to oxidative stress. However, this does not necessarily imply that Fur is itself a target for oxygen radicals, as will be discussed below.
Is Fur a sensor of ROS?
Direct evidence for the sensitivity of E. coli Fur to H2O2 came from biochemical experiments. When purified in the absence of a reducing agent (DTT) and in the presence of EDTA, a proportion of Fur exists as a monomeric species containing two disulfide bonds (21). One of those disulfides involves Cys-92 and Cys-95, which are ligands in the structural zinc site (Figs. 2 and 3). Treatment with DTT and zinc caused a rapid monomer to dimer transition, and dimeric Fur bound to DNA with a ∼5 fold higher affinity than the monomeric form (21). Dimerization requires zinc ions, so the zinc site is required for stabilization of the active dimeric form of Fur (77). Treatment of dimeric Fur with diamide (a thiol oxidant) or H2O2 caused a dimer to monomer transition (with, presumably, a reduced DNA binding activity) although the rate of this reaction was slow, with a second-order rate constant k=0.04 M−1 s−1 for the reaction with 2 mM H2O2 (21). In contrast, the oxidation rates of the dedicated peroxide sensors OxyR and PerR are both approximately 105 M−1 s−1 (58, 60). Thus, there is some evidence for oxidant sensitivity of Fur in vitro, though the physiological significance of these observations is not clear. Persuasive evidence for the sensitivity of Fur to oxidative stress in vivo came from experiments with the Hpx- strain of E. coli that lacks peroxidase and catalase activities and so cannot consume H2O2. This strain accumulates approximately 1 μM H2O2 from endogenous sources (76). Fur-repressed genes are constitutively expressed in the Hpx- strain when cultured in defined medium, which is best explained by H2O2-mediated inactivation of Fur (105). However, derepression of the Fur regulon was abolished when the cells were supplemented with iron or grown in iron-rich medium (105). Results obtained in defined medium were best explained by the oxidation of “free” Fe2+ to Fe3+, thus leading to accumulation of apo-Fur. By contrast, in rich medium, the oxidation of iron is counterbalanced by iron uptake and the Fur regulon is fully repressed. Similar conclusions were reached with transcriptomic analyses of cells grown in iron-rich medium. In one of these experiment, Fur-regulated genes did not show a significant response to treatment with 1 mM H2O2, even in an oxyR mutant, which is less able to remove H2O2 than its wild-type parent (115). In cells treated with paraquat (used as a source of superoxide), the RyhB-regulated sodB gene was downregulated, which is consistent with inactivation of Fur (80). However, there were not widespread effects on other Fur-regulated genes in this experiment. Overall, these results indicate that Fur is not directly inactivated by ROS in vivo in iron replete conditions.
As we have described, oxidative stress potentially exerts two opposing effects on Fur. On the one hand, oxidative stress may inactivate Fur, which would lead to upregulation of Fur-repressed genes. On the other hand, both superoxide and H2O2 elevate Fur expression (114), a response which causes downregulation of the Fur regulon, and may act to mitigate the elevated free iron levels caused by oxidative stress. The upregulation of fur by H2O2 depends on direct activation of the fur promoter by the thiol-based sensor OxyR (105, 114), and this cross-talk between the OxyR and Fur regulons presumably provides a means to coordinate the responses to oxidative stress-elevated free iron. Increased Fur abundance during oxidative stress is probably the dominant effect that acts specifically to counterbalance the damage done to the protein under these conditions (105). On the whole, these experiments argue that Fur is not, in wild-type strains, a sensitive and physiological sensor of superoxide and H2O2, but is still able to regulate iron metabolism under stress conditions. However, the possibility cannot be excluded that Fur is a sensor of ROS during severe oxidative stress, and that Fur inactivation under these conditions is adaptive, by causing upregulation of sodA, and increased uptake of Fe2+.
Fur and RNS
The metal centers of Fur are potential targets for RNS. Cysteine-liganded zinc sites are susceptible to RNS, most likely by S-nitrosation of protein thiols (92), and iron can be modified by nitrosylation. NO reacted with purified Fur-Fe to form a product with spectroscopic features indicative of an Fe-NO complex, and the NO-treated protein was inactive for DNA binding (22). Whole cell electron paramagnetic resonance (EPR) spectroscopy with a strain overexpressing Fur revealed that Fur could be nitrosylated in vivo to generate the same complex that was observed in vitro (22). NO-treated Fur was later shown to contain a mixture of dinitrosyl iron complexes (DNIC), a major (75 %) and a minor (25 %) species, with two different electronic configurations (20). The reaction of NO with Fur-Fe proceeds by reaction of Fe2+ with two molecules of NO to form a diamagnetic dinitrosyl-iron core with a {Fe(NO)2}8 electronic configuration that has been tentatively assigned to the minor species. A third NO molecule may act as a reductant to generate the characteristic paramagnetic DNIC species associated with a g=2.03 EPR signal and a {Fe(NO)2}9 electronic configuration, which is the major product of the reaction (20). The in vitro experiments indicate that Fur-Fe(NO)2 complexes are rather stable in solution, and that NO binding cannot be reversed by addition of excess of myoglobin as a competitor (22). However, inactivation of Fur in vivo is not irreversible and growth recovery after NO challenge is correlated with restoration of Fur activity (22). It has been shown that L-cysteine in combination with oxygen is able to remove dinitrosyl-iron complexes from proteins in vitro and in vivo, in the absence of new protein synthesis (88, 112). However, the significance of this repair process for Fur is not clear, given that the recovery of Fur activity was monitored in anaerobic cultures (22). Thus, the mechanism of repair of Fur-Fe(NO)2 remains an open question.
That the reaction of Fur with NO also happens in vivo was first suggested by the observation that a gene regulated by Fur was de-repressed by exposing cultures to NO (22). Subsequent transcriptomic experiments have not presented a consistent pattern (95). Several researchers have observed that the expression of members of the Fur regulon is perturbed by NO or sources of RNS (S-nitrosoglutathione or acidified nitrite) in a manner that is consistent with inactivation of Fur (53, 69, 70, 82, 90, 96). However, in other experiments, there was no effect of NO or RNS on members of the Fur regulon (33, 46). One possible explanation for these discrepancies is that iron availability in the different growth media used varied, such that, in some cases, cells were somewhat iron limited despite being cultured in iron-rich medium (33). Under iron-limited conditions, and in the absence of NO, the Fur regulon will be partially de-repressed, and the fold increase in gene expression caused by NO will be less than that seen when comparing a wild-type strain to a fur mutant. In contrast, under iron replete conditions, NO-inactivation of Fur might cause almost complete de-repression of Fur targets, resulting in an apparently larger effect of NO. On the other hand, high intracellular iron concentrations might help to rapidly reverse the inactivation of Fur by NO. As suggested earlier by the reversibility of Fur inactivation by NO, if Fe(NO)2 complexes could be extracted from Fur, leaving an apo protein, then apo-Fur could be reloaded with iron to regenerate Fur-Fe. Thus, the relationship between iron status and NO exposure is potentially complex, and the effect of NO on genes regulated by Fur might be highly dependent upon growth conditions.
Is the inactivation of Fur by NO a physiologically significant adaptive response? In B. subtilis, S. aureus, Burkholderia multivorans, H. pylori as well as in E. coli, a fur mutant is more sensitive to NO than the wild-type parent, indicating that constitutive expression of the Fur regulon exacerbates the bacteriostatic effect of NO (22, 69, 70, 84, 90, 113). These observations tend to argue against an adaptive role for Fur in the response to NO. This point can be illustrated by contrast to NsrR, an NO-sensitive repressor, removal of which causes an NO-resistant phenotype (94, 109). Some authors have proposed that the inactivation of Fur by NO is a deleterious effect of NO rather than an adaptive response (90). Other groups have postulated that the constitutive de-repression of Fur targets in a fur mutant does not mimic the benefits of temporary induction (70), perhaps because the fur mutant accumulates a high level of iron and then forms toxic Fe–NO complexes upon NO challenge (84). Furthermore, not all Fur-regulated genes are responsive to NO in E. coli. An example is fiu, which is not sensitive to NO despite being constitutively transcribed in a fur mutant (22). The differential effects of NO on different Fur-regulated promoters may be related to the strength of Fur repression at each promoter; NO may finely tune the DNA binding activity of Fur through dinitrosylation of the iron site. An alternative or additional possibility is that the genes that are regulated by apo-Fur (that are not sensitive to NO but differentially regulated in a fur mutant) may have a critical role to play in the response to NO.
Another way to think about the question of physiological relevance is to consider whether the Fur regulon includes genes whose products have a role to play in the response to NO. The answer to this question is, possibly, yes, since in E. coli, in addition to iron uptake and storage systems, Fur also regulates the sufABCDSE operon, the products of which are involved in the synthesis and repair of iron–sulfur clusters (73). Since some iron–sulfur clusters are sensitive targets for NO, it would make sense for cluster repair machinery and iron uptake systems to be upregulated by NO exposure. Furthermore, since [Fe–S] proteins are apparently the main source of dinitrosyl-iron complexes in NO stressed cells (56), the formation of Fur-bound dinitrosyl-iron complexes may mimic the reaction of NO with [Fe–S] proteins. It is tempting to suggest that Fur is not a monitor of NO but of NO-induced damage to [Fe–S] proteins. Having said that, very little is known about the repair of nitrosylated clusters, and the role that the suf gene products may play in that process. Further, the suf operon is regulated by the [Fe–S] protein IscR, which is also somewhat sensitive to NO (46, 82), so the contribution that Fur makes to the NO-induction of suf operon expression is not known.
In some Neisseria species, Fur-Fe is an activator of the genes encoding nitrite reductase (aniA or pan1) and NO reductase (norB) and the coordinated regulation of those genes with NsrR may help counteract NO poisoning, which is consistent with an adaptive role for the inhibition of Fur-Fe DNA binding activity by NO (25, 74).
DtxR and IdeR
DtxR (diphtheria toxin repressor) and IdeR (iron-dependent regulator) are related iron-dependent repressors from Corynebacterium and Mycobacterium spp., respectively. These proteins regulate iron acquisition and metabolism and so are functionally equivalent to Fur, although not similar in sequence. DtxR and IdeR contain two metal-binding sites per monomer, it is thought that occupation of one site stabilizes the structure of the monomer, while the second site is the iron sensing site that controls the DNA binding activity (19). A Mycobacterium smegmatis ideR mutant is sensitive to oxidative stress (28), which reflects the close relationship between iron homeostasis and oxidative stress responses and does not necessarily implicate IdeR as a target for ROS. There is a report that the Corynebacterium diphtheriae DtxR protein can be inactivated by NO both in vivo and in vitro (64), although the data supporting these observations have not been published. Data from transcriptomics experiments suggest that the M. tuberculosis IdeR is sensitive to both H2O2 and NO (108). One caveat is that the concentrations of H2O2 and NO used in these experiments were rather high, although the up-regulated genes overlap with those up-regulated during infection of activated macrophages, arguing that the responses are physiologically significant (108).
The Peroxide Regulon Repressor, PerR
PerR is a major regulator of the peroxide inducible peroxide stress response. PerR is a metal-dependent transcriptional regulator, a member of the Fur family (Fig. 2), which was originally described as a Mn2+- and Fe2+-dependent repressor of the peroxide stimulon in B. subtilis (11, 43). The B. subtilis (Bsu) PerR acts as a negative regulator, such that its target genes are de-repressed in response to H2O2 challenge. PerR regulated genes include katA, encoding a heme-catalase; ahpCF, encoding an alkyl hydroperoxidase and its reductase partner; mrgA, encoding a Dps-like protein (DNA protection during starvation protein) that is able to sequester metal ions and protect DNA from metal catalyzed oxidation (MCO) reactions; hemAXCDBL, an operon encoding proteins involved in heme biosynthesis, and zosA, encoding a Zn2+ uptake system (10, 16, 27, 36). B. subtilis perR mutants have a severe growth defect. PerR is a repressor of fur, and the growth defect can be explained by elevated expression of fur leading to iron starvation, which is exacerbated by heme sequestration by the constitutively expressed heme catalase KatA (32). The mechanism of H2O2 sensing by Bsu PerR has been elucidated, and two crystal structures of Bsu PerR have been reported recently, as will be discussed below.
Metal-dependent activation of PerR
Bsu PerR contains a Zn2+ ion that has a role in the stabilization of the dimeric structure (100), as is also the case for the E. coli Fur (21, 77). As a member of the Fur family, PerR displays metal-dependent DNA binding activity. The regulatory metal binds to a second site in PerR, the regulatory site (Fig. 3). The crystal structure of PerR with Mn2+ in the regulatory site and Zn2+ in the structural site (denoted PerR-Zn-Mn) revealed that Mn2+ is coordinated by five protein ligands (His-37, Asp-85, His-91, His-93, and Asp-104) that are conserved among PerR as well as Fur proteins (26, 50). The binding of Mn2+ in the regulatory site induces a conformational change from a “flat” to a “caliper-like” closed conformation; and converts PerR into a DNA binding protein allowing transcriptional repression in vivo (50, 100).
H2O2 sensing mechanism of PerR
The Zn2+ ion of PerR is coordinated by four cysteine residues that can be oxidized by H2O2. However, the rate of this reaction is slow, and it may not contribute to the in vivo sensitivity of PerR to ROS (59). Rather, the physiological sensing mechanism depends upon the MCO of histidine residues in the Fe2+-occupied regulatory site (60). The proposed mechanism relies on reduction of H2O2 by Fe2+, and formation of a hydroxyl radical (HO•) that reacts rapidly with surrounding amino acids. The reaction of H2O2 with PerR-Zn-Fe results in the formation of 2-oxo-histidines by selective oxygen incorporation into His-37 and/or His-91, which coordinate the Fe2+ (60). Although His-37 is more sensitive to oxidation, the MCO reaction at either or both sites triggers a rearrangement toward the flat conformation, leading to dissociation of the PerR DNA complex, and de-repression of PerR targets (101).
The biochemical studies of the PerR sensing mechanism led to the striking conclusion that the true physiological signal for PerR is not H2O2 per se but HO•. Given the extremely short half-life of HO• (42), it seems unlikely that the ambient concentration of HO• can be sensed by any protein sensor. Thus, bacteria have evolved a mechanism that combines both Fe2+ and H2O2 sensing and restricts the Fenton reaction to a protein microenvironment. The local generation of HO• in the PerR regulatory site has not been directly demonstrated, and other mechanisms are still possible, such as the formation of an iron-oxo intermediate. Nevertheless, PerR appears to act as a sensor of “potential” HO• formation. But is it true that the PerR regulon is only upregulated under intracellular conditions that favor the Fenton reaction? The PerR regulon is also upregulated under iron starvation conditions, presumably by accumulation of PerR-Zn-apo (7, 35, 43). Nevertheless, the highest level of induction of PerR regulated genes such as katA is reached in the presence of H2O2 and high concentrations of Fe2+ (43), supporting the notion that PerR is adapted to sensing H2O2 in conditions that would likely favor the generation of HO•. Moreover, the resistance of PerR to oxidation when Mn2+ is in the regulatory site instead of Fe2+ indicates a mechanistic specificity for Fe2+ and further supports the role of PerR as a sensor of HO• (43, 60).
There is no known enzymatic activity capable of repairing 2-oxo-histidine. PerR is therefore considered to be a ‘sacrificial’ regulator that is irreversibly oxidized (60). This implies that upregulation of the PerR regulon will persist as long as no new PerR synthesis occurs.
Structural determinants of the H2O2 sensing mechanism of PerR
An important question is: what makes PerR much more sensitive than Fur to H2O2? The crystal structure of PerR with Mn2+ in the regulatory site (PerR-Zn-Mn) revealed that Mn2+ is five-coordinated in a distorted square pyramidal environment, leaving an open position presumably to allow H2O2 binding (50). An X-ray absorption spectroscopic study of PerR with Fe2+ in the regulatory site (PerR-Zn-Fe) indicates a similar open shell pattern for coordination of the Fe2+ ion (50). Interestingly, the coordinating residues in PerR are conserved in Fur proteins, and a similar open shell pattern has been described for the E. coli Fur-Fe by X-ray absorption spectroscopy (26, 51), and for the H. pylori (chain B), V. cholerae, and P. aeruginosa Fur proteins crystallized with a Zn2+ ion in the regulatory site (26, 70, 83). Thus, the regulatory sites in PerR and Fur proteins both display an open shell that cannot explain the differential sensitivity of PerR to H2O2. The difference between PerR and Fur is probably linked to more subtle differences that have not yet been identified.
Is the essential role of PerR in virulence linked to H2O2 sensing?
PerR has been identified as a critical regulator for virulence in a number of pathogens, including Streptococcus pyogenes, S. aureus, N. gonorrhoeae, Campylobacter jejuni, and Listeria monocytogenes (9, 41, 55, 75, 81, 83, 86, 87, 89, 110, 111). Surprisingly, although perR mutants are highly resistant to H2O2 in culture (due to constitutive expression of the PerR regulon), the virulence of perR mutants in animal infection models is fully compromised. At first glance, these results suggest that H2O2 resistance is not a critical factor contributing to virulence, which is probably a false assumption as virulence is generally impaired in strains lacking H2O2 resistance enzymes such as KatA, AhpCF, or MrgA (9, 72, 75). More interestingly, these results suggest that the physiological role of PerR is more complex than previously thought. Transcriptomic analyses in S. pyogenes (41) and C. jejuni (75) have partially unraveled the complexity of the PerR regulatory network in pathogens. Both studies identified genes apparently regulated by PerR that are not directly involved in H2O2 resistance, including genes involved in sugar utilization, motility, fatty acid biosynthesis, and DNA repair. The transcriptomics data also implicate PerR as both a negative and positive regulator, and identify some genes that are regulated by PerR but that are not sensitive to H2O2 (41, 75). Thus, the virulence defect of PerR mutants may not be directly related to H2O2 metabolism. Further, the perR null mutant may not be a good model for studying the role of PerR in response to H2O2 during infection. In B. subtilis and L. monocytogenes, it has been shown that the perR mutant accumulates second site mutations in an as yet unidentified locus (15, 87). These variants do not display the same level of resistance to H2O2 as an otherwise isogenic perR null mutant. Therefore, the behavior of the perR mutant in an infection model may reflect both the absence of PerR and the phenotype(s) associated with second site mutations. In conclusion, PerR is critical for the ability of some pathogens to mount an H2O2-inducible defense response, but other aspects of the function of PerR probably also contribute to its important role in virulence.
PerR and NO
Two different studies have reported the de-repression of the PerR regulon by NO in B. subtilis (44, 69), potentially by nitrosylation of the Fe2+ site of PerR as was described for Fur (22), or perhaps indirectly by a nitro-oxidative process leading to NO-dependent H2O2 production. However, the physiological advantage of NO-dependent upregulation of the PerR regulon is not clear, since enzymes involved in H2O2 resistance are not necessarily important for NO defense. One possible underlying reason is that NO potentially induces iron starvation-like conditions that inactivate PerR, thus activation of the PerR regulon may be seen as a consequence of iron restriction. Furthermore, the NO induction of the PerR regulon appears to be species specific, since it was not observed in S. aureus (44, 90). Nitrosative stress evidently causes iron starvation in S. aureus (90), therefore it is not clear why PerR is resistant to RNS in this organism.
The NO Reductase Regulator, NorR
Function of NorR
The denitrifying bacteria express a membrane-associated respiratory NO reductase, which reduces NO to N2O, with the electrons required for this reduction originating either from the quinone pool or from small soluble periplasmic electron carriers. In Ralstonia eutropha, the respiratory NO reductase is a single subunit quinol-dependent enzyme (18) encoded by a gene designated norB, which is co-transcribed with norA. The function of the product of the norA gene (variously designated ytfE, scdA, and dnrN in other organisms) is not well understood, but it has been implicated in the repair of damaged iron–sulfur clusters and/or in somehow buffering the cytoplasmic concentration of NO (52, 98). The R. eutropha norAB genes are upregulated by exposure of cultures to a source of NO, this induction requires a transcriptional activator NorR, which is encoded by the norR gene that is adjacent to and divergently transcribed from norAB (79). The R. eutropha NorR protein interacts with three cis-acting sites in the norR-norAB intergenic region (12).
In E. coli and Salmonella enterica serovar typhimurium, one of the enzymes responsible for detoxifying NO is the flavorubredoxin (FlRd or NorV), a member of the flavo-diiron family of proteins. FlRd is encoded by the norV gene, which is co-transcribed with the norW gene that encodes an NADH-dependent FlRd reductase. The complex thus couples NADH oxidation to the reduction of NO to N2O (38, 39). The norVW genes are induced by exposure to a source of NO or other RNS, induction requiring the product of the norR gene, which is adjacent to and divergently transcribed from norVW (37, 45, 70). NorR-mediated activation of norVW in response to NO requires three NorR binding sites in the norR-norVW intergenic region; occupation of these three sites also causes negative autoregulation of norR expression (45, 102, 103).
Genome context analysis and the distribution of NorR binding sites suggest that there are other targets for NorR regulation in different organisms (91). Predicted NorR targets include genes encoding the flavohemoglobin, Hmp (which oxidizes NO to nitrate), and the hybrid cluster protein, Hcp and its reductase Hcr (which have poorly understood roles in the response to NO). It has been confirmed that the NorR (FhpR) of P. aeruginosa activates the flavohemoglobin gene (hmp or fhp) in response to NO (5). Some Vibrio species are predicted to encode two NorR proteins; genome context information suggests that one activates the hcp-hcr genes, and the other the norVW genes (91). This regulatory pattern has not been confirmed experimentally.
The roles of NorR and its targets in the virulence of bacterial pathogens have not been extensively tested. It has been reported that the flavorubredoxin of Salmonella enterica serovar typhimurium is not a virulence determinant in a mouse model (8), though the lack of a phenotype may be due, in part, to the redundancy of NO detoxification mechanisms. NorR is known or predicted to regulate other genes in some organisms, for example hmp (5, 91), and it will be interesting to test the roles of these genes in virulence, for example in P. aeruginosa.
Structure and mechanism of NorR
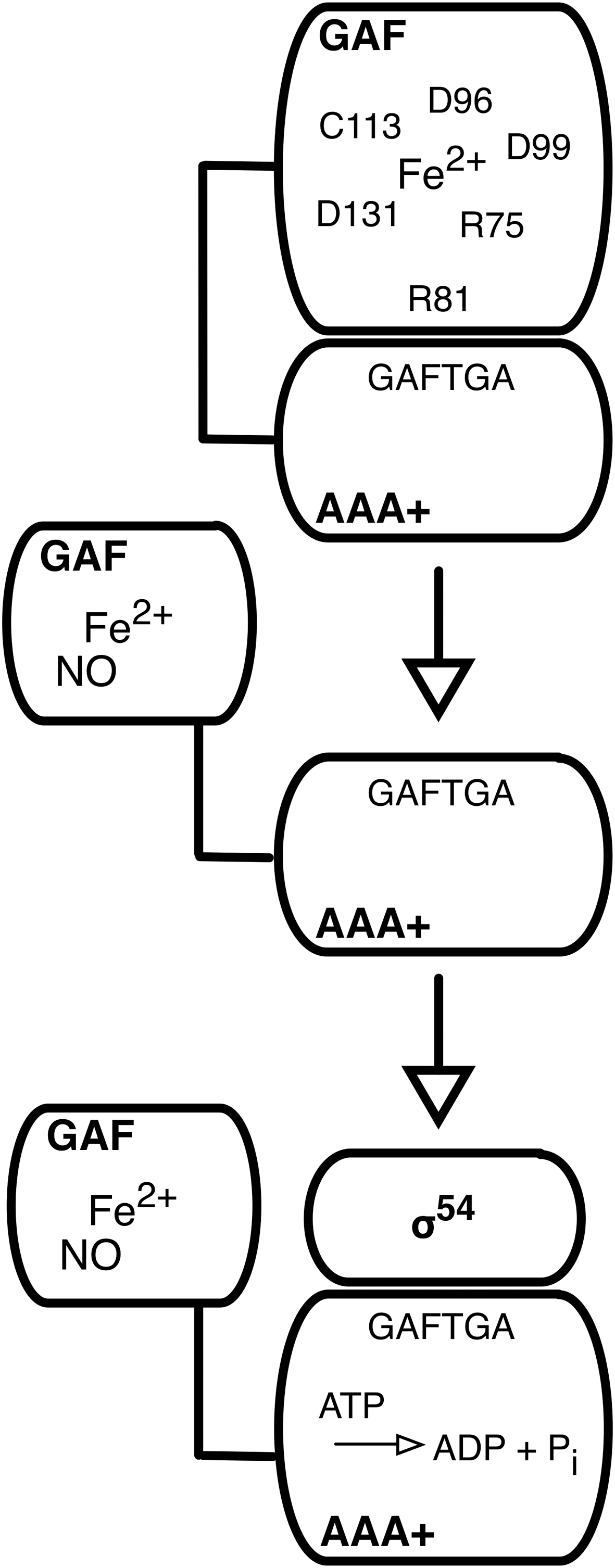
NorR is a member of the bacterial enhancer binding protein (EBP) family; EBPs are required for the initiation of transcription catalyzed by RNA polymerase containing the alternative sigma factor, σ54 (85). NorR has a three-domain structure that is typical for the EBPs, with a C-terminal DNA binding domain, a central domain from the AAA+ family (ATPases associated with diverse cellular activities) that has ATPase activity and interacts with σ54, and an N-terminal regulatory domain. In the EBPs, the N-terminal domain controls the activity of the AAA+ domain, either positively or negatively, in response to environmental signals. In the ‘on’ state, the AAA+ domain couples ATP hydrolysis to the remodeling of the σ54-RNA polymerase, which then undergoes the isomerization reaction from closed to open complex (85). The NorR N-terminal domain is from the GAF (found in cGMP phosphodiesterase, adenylyl cyclase, and FhlA) family (6), and acts negatively, since removal of the GAF domain generates a NorR derivative that is persistently active in the absence of NO (21, 37, 79). EPR spectroscopic analyses of cells expressing NorR and exposed to NO revealed spectra suggestive of the formation of a mono-nitrosyl species, the product of the reaction of one molecule of NO with Fe2+. Purified NorR (or its GAF domain alone) could be reconstituted with one equivalent of Fe2+, and when treated with NO, similar EPR signals were observed. Full-length NorR reconstituted with Fe2+ and treated with NO could interact productively with σ54-RNA polymerase and stimulate the isomerization reaction. The binding of NO to NorR-Fe is reversible with a dissociation constant estimated to be around 50 nM (23). These observations led to a model (Fig. 4), in which the GAF domain of NorR coordinates a mononuclear non-heme ferrous iron. In the ‘off’ state, the GAF domain acts as an intramolecular inhibitor of the activity of the AAA+ domain. Reaction with NO generates a mono-nitrosyl complex at the Fe2+ center, with an accompanying (though as yet uncharacterized) conformational change that relieves inhibition (23). As mentioned above, NorR is an NO sensor with a sensitivity in the low nM range, which may correspond to a threshold for NO toxicity. The reversible binding of NO further suggests that induction of NorR target genes ceases rapidly as NO is removed by the enzyme encoded by those genes.
NorR is relatively resistant to oxygen in vitro (the iron does not readily oxidize), and NorR targets can be induced by NO sources in aerobic cultures (37, 45, 70). The isolated GAF domain is more prone to oxidation than full-length NorR (104), perhaps suggesting that the Fe2+ center is solvent exposed in the isolated domain, and protected by the interaction with the AAA+ domain in the full-length protein. Some amino acid substitutions in the GAF domain increase the sensitivity of NorR to oxygen, and the wild-type protein can be oxidized by H2O2 (104). The physiological significance, if any, of the latter observation is not known. There is a report that the fhp gene of P. aeruginosa can be induced by H2O2 and other ROS in a FhpR (NorR)-dependent fashion (57). However, the effect of ROS seems to be rather small, and potentially complicated by the contribution of other regulators of the fhp promoter (57). Another study found no effect of H2O2 on the abundance of the norV mRNA in E. coli (70). The balance of evidence points to NorR being a specific and sensitive sensor of NO, and, possibly, related RNS.
Subsequent publications have confirmed and extended the details of the NorR mechanism. A combination of site-directed mutagenesis, in vivo activity assays, EPR and magnetic circular dichroism (MCD) spectroscopies, iron binding assays, and structural modeling implicated five residues (three aspartates, an arginine, and a cysteine) as potential ligands to the iron in the GAF domain (104). Substitutions of these residues produced diverse outcomes in terms of NorR activity, including inactive and constitutively active (in the absence of NO) variants. Indeed, for one residue (Cys-113), both phenotypes were observed, since glycine and alanine substitutions led to constitutive activity, while a serine substitution inactivated NorR (104). Based on site-directed mutagenesis and iron content assays, the equivalent five residues of the R. eutropha NorR were also proposed to function as iron ligands (56). However, in this case all substitutions inactivated the protein. In fact, where the amino acid substitutions are the same, there is no disagreement between the two studies. And in both cases, substitutions of the putative cysteine ligand had the most severe effect on the ability of NorR to bind iron (56, 104). In both studies, a number of other conserved residues in the GAF domain were substituted with a range of outcomes (inactive, constitutively active, no effect). Some of these substitutions may identify residues with roles in other steps of the NorR mechanism, for example the interaction of the GAF domain with the AAA+ domain (see below).
A subsequent study examined the mechanism by which the GAF domain of NorR inhibits the activity of the AAA+ domain in the absence of NO (13). Constitutively active NorR variants were isolated with substitutions in the AAA+ domain. Three of these substitutions were within a highly conserved GAFTGA motif, which is within a predicted loop structure that in other EBPs makes contact with σ54 and drives the formation of open complex by RNA polymerase (85). A substitution of the second glycine of the GAFTGA motif (G266D) generated a protein that was fully active in the absence of NO; this substitution had small or no effects on the DNA binding, oligomerization, and ATPase activities of NorR, and on its ability to drive open complex formation. The authors concluded that the GAF domain targets the σ54 interaction surface of the AAA+ domain to inhibit NorR activity (13). The earlier study (104) had implicated Arg-81 in the GAF domain of NorR as potentially having a role in the repressive interaction with the AAA+ domain. R81L was shown to be a suppressor of the G266D phenotype, that is GAF-mediated repression was restored in the doubly substituted protein, which had a low activity in the absence of NO and a wild type response to NO (13). Thus, these residues may be located on the interacting surfaces of the GAF and AAA+ domains (Fig. 4).
Clearly, much remains to be learned about the mechanism of NorR, in terms of both the NO sensing mechanism, and how the reaction with NO is coupled to transcription activation. Structural information for NorR in the ‘on’ and ‘off’ states would, of course, be enormously helpful. Progress with a structural understanding of the EBPs has until recently been rather slow, but the number of solved structures is now growing (85), so obtaining structural information for NorR is a realistic goal.
Conclusions
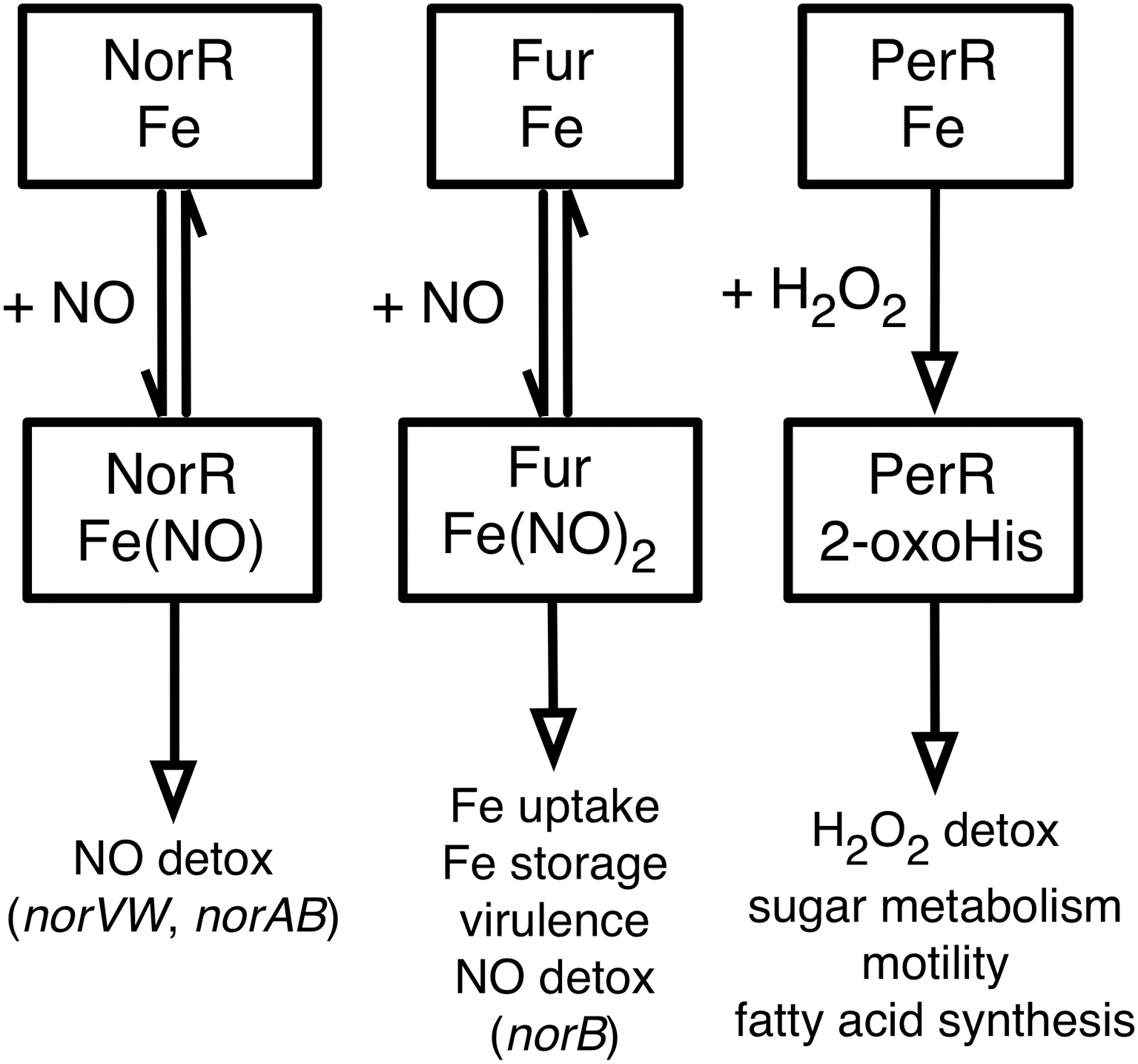
The reactions of Fur and NorR with NO, and of PerR with H2O2 and the regulatory roles of the modified proteins that have been discussed in this review are summarized in Figure 5. The reactions of Fur with H2O2 and of PerR with NO are of less certain physiological significance and so they are omitted from the figure, as are the DtxR and IdeR proteins. While a great deal has been learned about the role of non-heme iron proteins in coordinating responses to ROS and RNS, there is clearly much that remains to be understood. For example, one important question that cuts across all of these regulatory systems (and others not described herein) is that of specificity—what structural and chemical features of a particular protein determine its specificity for a particular ligand or reactant? The Fur and PerR proteins illustrate this problem especially well, since they are related in sequence and have similar metal centers, but differ dramatically in their sensitivities to H2O2 and NO. In many cases (such as NorR) the conformational changes that are driven by interaction with a specific ligand are also very poorly understood. We anticipate with excitement the light will doubtless be shed on these and other questions in the coming years.
Footnotes
Author Disclosure Statement
The authors disclose no conflicts of interest.