
Abstract
Introduction
BAT is able to utilize glucose or fatty acids as fuel to produce heat in response to environmental stimuli such as cold temperature or diet (21, 38). Increased sympathetic nerve input to BAT, stemming from various upstream pathways in brain regions, including forebrain, hypothalamus, and brainstem (3, 28), also results in increased BAT proliferation and activity, largely due to secretion of the catecholamine neurotransmitter norepinephrine, which binds to adrenergic receptors on brown adipocytes. Adrenergic activation results in increased thermogenesis, as well as increased mitochondrial β-oxidation (37). To maintain fuel supply for these high-energetic demands, BAT is able to increase fatty acid uptake and as is now appreciated, BAT can engulf whole triglyceride particles via the fatty acid translocase CD36 (FAT/CD36) (1).
Innovation
The data described herein represent novel findings regarding the role of fatty acid uptake and catabolism in the regulation of mitochondrial activity of brown adipocytes. Specifically, we have shown that bone morphogenetic protein 7 (BMP7) is able to increase mitochondrial activity by increasing CPT1- and CD36-mediated fatty acid uptake and catabolism. These new findings offer the potential for BMP7-mediated energy expenditure as a means to combat obesity and related metabolic disorders, by increasing fatty acid utilization by brown adipose tissue.
Fatty acids are capable of activating UCP1 and also can become available for mitochondrial β-oxidation via a two-step process: (i) transporting fatty acids into the cell for storage, and then (ii) transport into the mitochondria for utilization. Once fatty acids enter the cell, they can be activated in the cytosol by chemical coupling to carnitine, and then can move across the mitochondrial membranes via the carnitine shuttle. Carnitine palmitoyltransferase 1 (CPT1), located on the outer mitochondrial membrane, is the first and rate-limiting step of this shuttle, followed by transport by CPT2 across the inner membrane (see model in Fig. 8) (42). Fatty acids are then oxidized via β-oxidation in the mitochondrial matrix, providing acetyl-CoA for the Krebs/TCA cycle, which then shuttles electrons through the electron transport chain (ETC), thereby producing ATP by oxidative phosphorylation. Mitochondrial activity through these pathways may be altered in brown adipocytes in response to energy demands of the cell.
Differentiation and activity of BAT are tightly controlled by a number of neuroendocrine and growth factors, such as norepinephrine, insulin, and thyroid hormone, as well as developmental regulators such as the bone morphogenetic proteins (BMPs). BMPs belong to the transforming growth factor-β superfamily, which regulates many aspects of organogenesis and patterning during embryonic development (7, 27). Only recently have the BMPs been implicated in the regulation of metabolism (15, 44, 49, 50, 56, 57, 61). Our laboratory has previously demonstrated that BMP7 is able to induce brown adipogenesis (52) and is able to drive primary adipose progenitors to differentiate to the brown fat lineage (43). BMP7 can rescue the differentiation defect of brown preadipocytes with impaired insulin signaling, suggesting that there is a crosstalk between these two signaling systems in the regulation of brown fat (62). Additionally, mice treated with BMP7 via adenoviral gene transfer display increased energy expenditure (50, 52). In this study, we demonstrate that BMP7 is able to increase mitochondrial activity in mature brown adipocytes, by enhancing cellular fatty acid uptake and catabolism. This increased mitochondrial activity is dependent upon the fatty acid transporters CPT1 and CD36. Collectively, these findings provide exciting evidence that the activity of BAT may be increased by facilitating fatty acid transport into the mitochondria, and BMP7 may be utilized to increase the activity of pre-existing depots of BAT, thereby providing a novel avenue to combat obesity.
Results
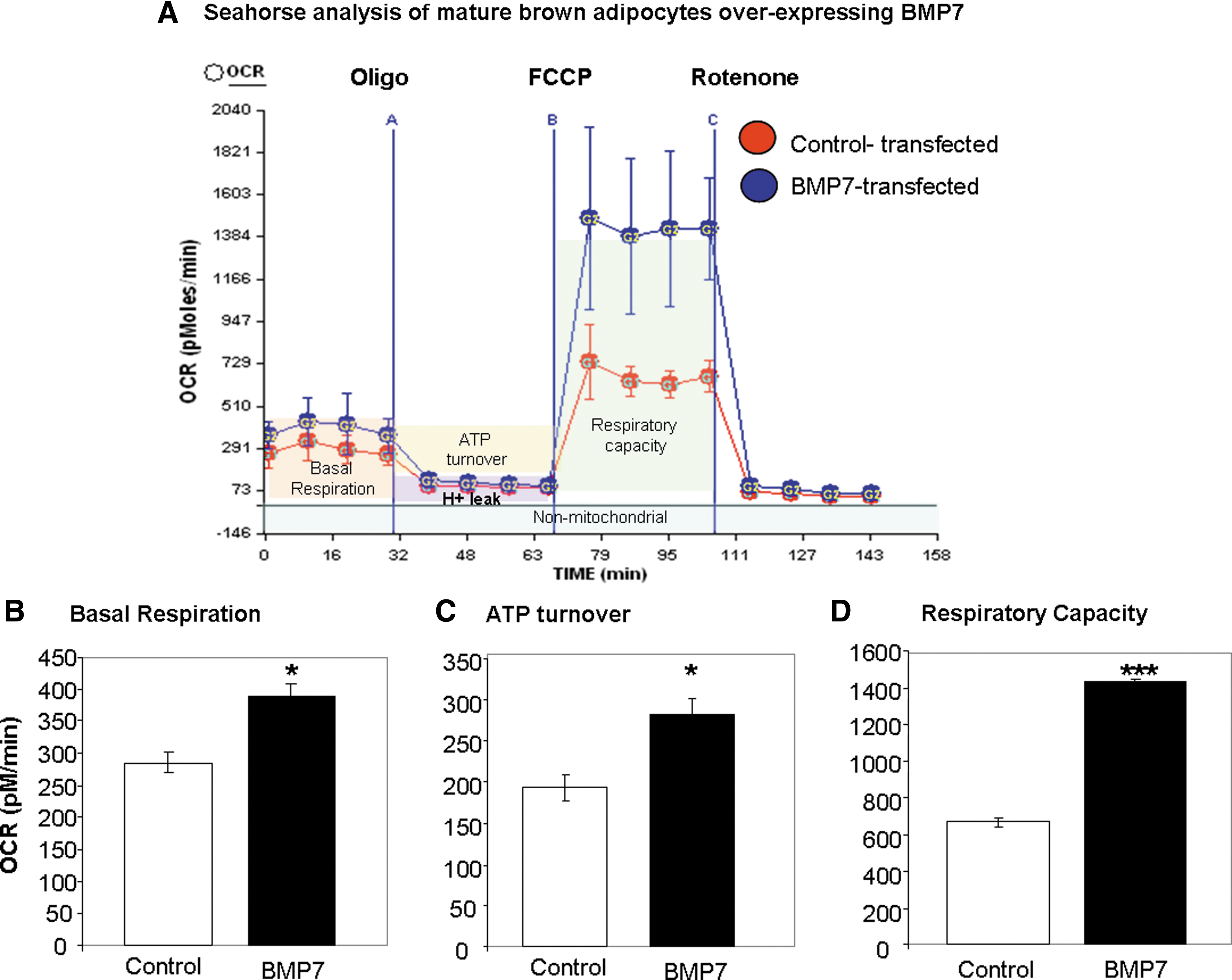
BMP7-transfected cells display increased basal respiration, ATP turnover, and respiratory capacity
We have previously demonstrated that BMP7 is able to induce differentiation of committed brown preadipocytes, even in the absence of normally required adipogenic induction cocktail (52). This process is accompanied by increased mitochondrial mass and activity. To determine if the enhanced mitochondrial capacity is a direct effect of BMP7 or secondary to differentiation, we first generated preadipocytes with stable overexpression of BMP7 or control vector, followed by a normal brown adipocyte differentiation procedure (62). Once the cells reached maturity and were multilocular, UCP1-positive brown adipocytes, the cells were analyzed for mitochondrial activity (respirometry) in a mitochondrial bioanalyzer, which is capable of measuring the oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR), representing the cell's oxidative phosphorylation and extracellular pH, respectively. By utilizing well-characterized mitochondrial toxins, one is able to generate a so-called bioenergetic profile. First, basal respiration is measured, followed by exposure to oligomycin, an inhibitor of ATP synthase, which allows measurement of ATP turnover. Then, the uncoupler carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone [FCCP] is added to measure the respiratory capacity, followed by the Complex 1 inhibitor rotenone, which prevents electron transfer activity and leaves only nonmitochondrial activity to be measured. Interestingly, the bioenergetic profile of BMP7-transfected mature brown adipocytes revealed significant increases in overall mitochondrial activity (Fig. 1A), including increases in basal respiration (Fig. 1B), ATP turnover (Fig. 1C), and respiratory capacity (Fig. 1D) compared to control cells.
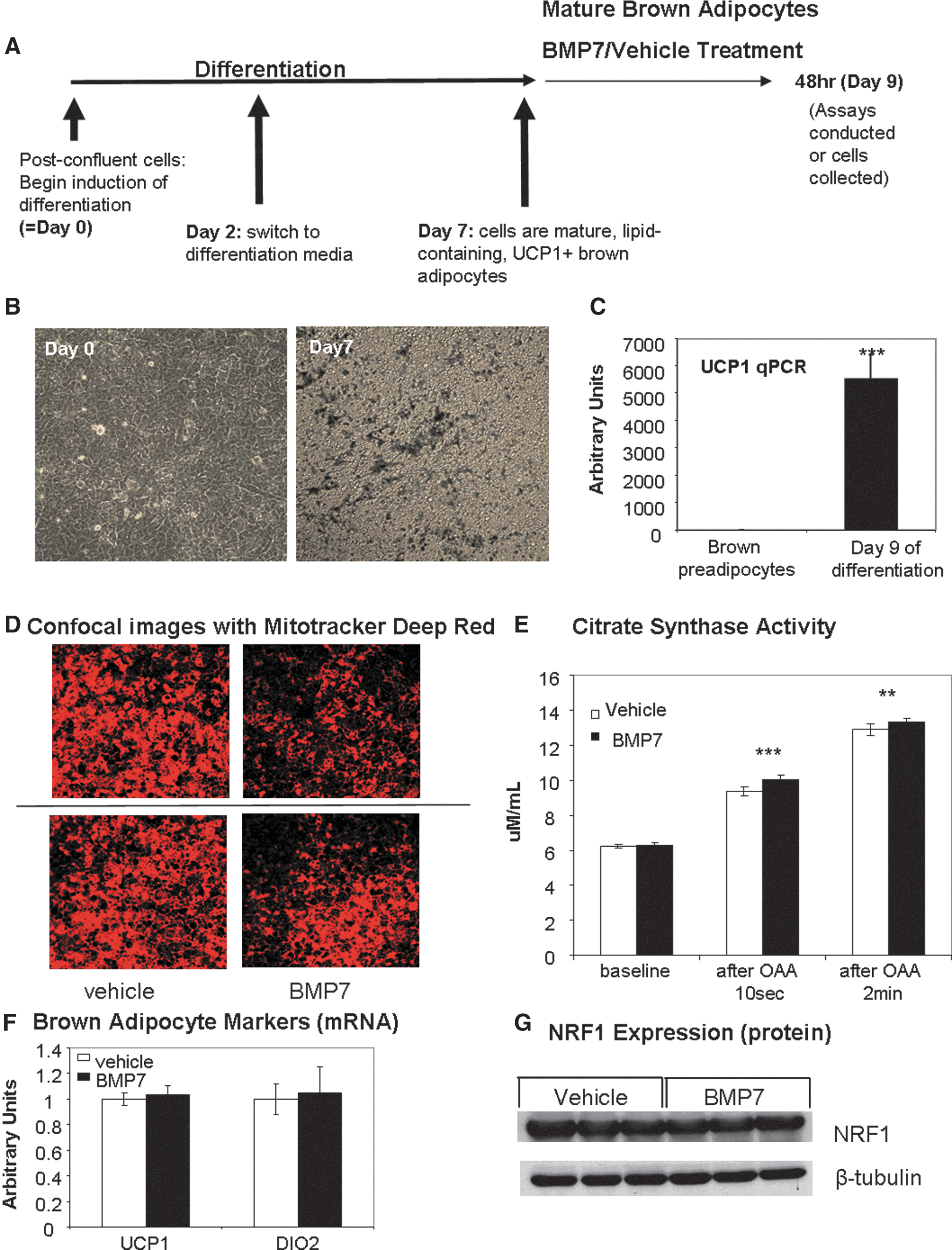
BMP7 increases mitochondrial activity in mature brown adipocytes without increasing the mitochondrial number
To continue to analyze BMP7's effects on mitochondrial activity in mature brown adipocytes, we utilized an immortalized murine brown preadipocyte cell line (19, 53), which was differentiated for 7 days, at which point the cells were mature, lipid laden, and UCP1 positive (Fig. 2A–C). We began BMP7 treatment at day 7 of differentiation, followed by analyses. We first measured the ability of BMP7 to affect uptake of Mitotracker Deep Red, a cell-permeable fluorescent mitochondrial dye that accumulates depending on membrane potential and is retained after fixation. We observed a consistent decrease in Mitotracker Deep Red accumulation in BMP7-treated cells after 48 h by either flow cytometric analysis (Supplementary Fig. S1A; Supplementary Data are available online at
To determine whether BMP7 treatment affected the cell number or the mitochondrial number in mature brown adipocytes, we first measured total DNA content in cells collected from culture plates after 48 h of BMP7 treatment and saw no differences, indicating that the cell number was the same after treatment (Supplementary Fig. S2A). Additionally, we measured the mtDNA copy number at 48 h and also found no significant difference (Supplementary Fig. S2B). As the mtDNA copy number does not directly reflect mitochondrial number because each mitochondrion can have various copies of mtDNA, we utilized electron microscopy to quantify the mitochondrial number and area. Neither measurement was affected by BMP7 treatment (Supplementary Fig. S2C, D).
To determine if BMP7-treated cells have increased activity of the Krebs cycle, which would provide substrate to the ETC, we measured citrate synthase activity. Citrate synthase is the first step of the Krebs/TCA cycle, catalyzing the condensation of acetyl-CoA (produced by β-oxidation and glycolysis) and oxaloacetic acid (OAA) to form citric acid. For this assay, OAA is added to initiate the reaction, and citrate synthase activity was measured 10 s and 2 min after OAA addition. BMP7-treated brown adipocytes displayed a small, but significant, increase in citrate synthase activity at both time points (Fig. 2E), indicating that BMP7 allows more substrate to be metabolized before the ETC. This is similar to findings in vivo that demonstrate that cold exposure increases citrate synthase activity of brown fat (26).
Because increased mitochondrial activity in brown adipocytes also could be due to increased expression of functional brown adipocyte markers, we measured expression of classic BAT markers UCP1, deiodinase 2 (DIO2) (Fig. 2F), and CIDEA (Supplementary Fig. S1B), as well as cytochrome C (Supplementary Fig. S1B) and the protein level of mitochondrial transcription factor nuclear respiratory factor 1 (NRF1; Fig. 2G). There were no differences with BMP7 treatment. Thus, BMP7 treatment to mature adipocytes is not further differentiating the cells, nor does it appear to dedifferentiate the cells. Together, these data indicate that the cells treated with BMP7 have increased mitochondrial activity and possibly also increased uncoupling, but do not display differences in markers of mitochondrial mass or brown adipocyte genes. Without an increase in UCP1 gene expression, increased uncoupling may be due to increased activation of UCP1 by fatty acids.
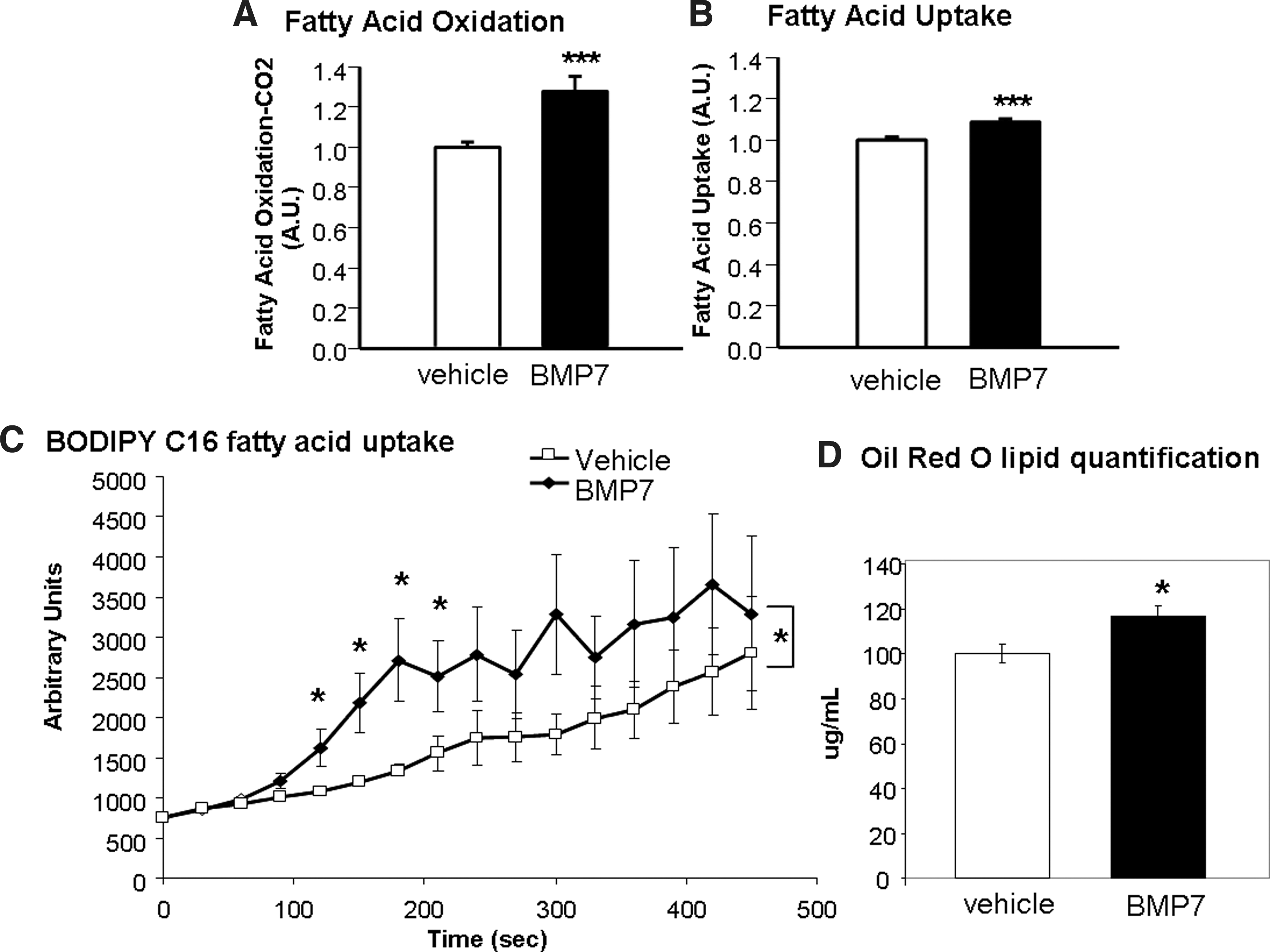
BMP7-treated brown adipocytes display increased fatty acid uptake and oxidation
Since brown adipocytes rely heavily on fatty acids as fuel, we measured whether BMP7-treated cells had increased fatty acid uptake and oxidation, which may account for the increased citrate synthase activity observed in Figure 2E. Indeed, BMP7-treated cells displayed a significant increase in fatty acid oxidation, as measured by conversion of 14C-labeled palmitic acid into CO2 (Fig. 3A), as well as increased fatty acid uptake as measured by both 14C-labeled palmitic acid uptake (Fig. 3B) and uptake of a fluorescent BODIPY-labeled C16 long-chain fatty acid (Fig. 3C). In accordance with this, BMP7-treated cells also exhibited increased Oil Red O staining for lipid content, which was quantified after propranolol extraction of the dye (Fig. 3D). Together, these data indicate that while BMP7-treated brown adipocytes do not appear to increase mitochondrial mass or expression of classic brown adipocyte genes, each mitochondrion appears to have higher metabolic activities as evidenced by increased fatty acid uptake, oxidation, and citrate synthase activity.
Basal OCR is increased in response to fatty acids, but not pyruvate, in BMP7-treated cells
Measurement of mitochondrial activity in the respirometer allows us to analyze many different aspects of mitochondrial activity, by manipulating the components of the serum-free assay medium (i.e., altering fuels such as carbohydrate and lipid) as well as injecting various compounds that alter mitochondrial activities. With 10 mM glucose in the assay medium, BMP7-treated cells consumed similar levels of oxygen as the vehicle-treated cells, and the OCR was not changed even when pyruvate was added (pyruvate delivered through Port A in Fig. 4A). However, after the addition of the uncoupler FCCP, BMP7-treated cells exhibit an increase in respiratory capacity (Fig. 4A, quantified in Fig. 4B). Under these conditions, there were no differences in the ECAR between vehicle- and BMP7-treated cells (Fig. 4C).
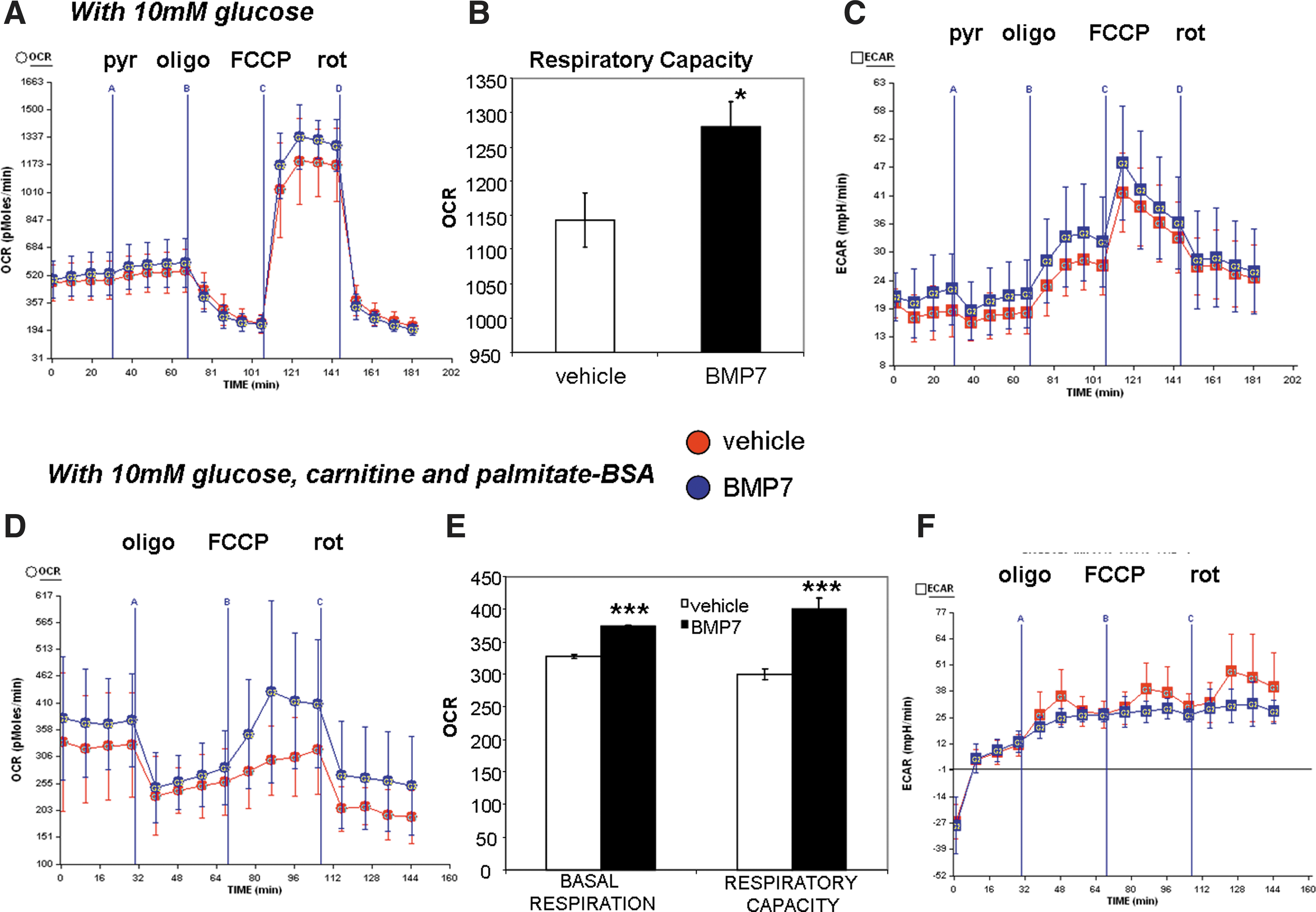
By contrast, when the cells are given 10 mM glucose plus carnitine and the long-chain fatty acid palmitate (conjugated to bovine serum albumin [BSA]), BMP7-treated cells display an increase in basal oxygen consumption as well as respiratory capacity (Fig. 4D, quantified in Fig. 4E). Similarly, these conditions also did not alter the ECAR between groups. Together, these data indicate that BMP7 treatment renders brown adipocytes more responsive to fatty acids versus pyruvate or glucose alone as a fuel source. Indeed, while BMP7-treatment increased the respiratory capacity regardless of substrate in the assay medium, only fatty acids were able to increase basal respiration in BMP7-treated cells.
In addition, BMP7 treatment increased oxidative phosphorylation (as measured by OCR), but not pyruvate utilization (as measured by ECAR), which was unchanged in both conditions. In separate experiments, we found that BMP7 treatment led to a small increase in insulin-stimulated glucose oxidation, but had no effects on lipogenesis (Supplementary Fig. S3A, B). Therefore, BMP7 treatment likely increased fatty acid uptake, but not lipogenesis, to provide fuel for oxidative phosphorylation, and while BMP7-treatment does not increase OCR in the presence of glucose as fuel, the BMP7-treated cells are still able to oxidize glucose more than vehicle-treated cells.
Increased fatty acid uptake and catabolism after BMP7 treatment are due to the fatty acid transporters CPT1 and CD36
If BMP7-treated brown adipocytes increase their mitochondrial activity via increased availability of fatty acids in the mitochondria as a fuel source, then BMP7 must trigger changes that allow increased fatty acid uptake and utilization. CPT1, the mitochondrial fatty acid transporter, displayed a trend to be increased by BMP7 treatment at the protein level at 48 h (Fig. 5A, quantified in Fig. 5B). Interestingly, we also observed a significant decrease in levels of phospho-ACC2, which acts to inhibit CPT1, both at the protein level at 6 h (Fig. 5E) and ACC2 mRNA at 24 h (Supplementary Fig. S4A). However, ACC1, which is inhibited by adenosine monophosphate activated-kinase (AMPK) and is involved in fatty acid synthesis, was not changed at either the mRNA (Supplementary Fig. S4B) or the protein levels (phosphorylated total ACC in Supplementary Fig. S5A).
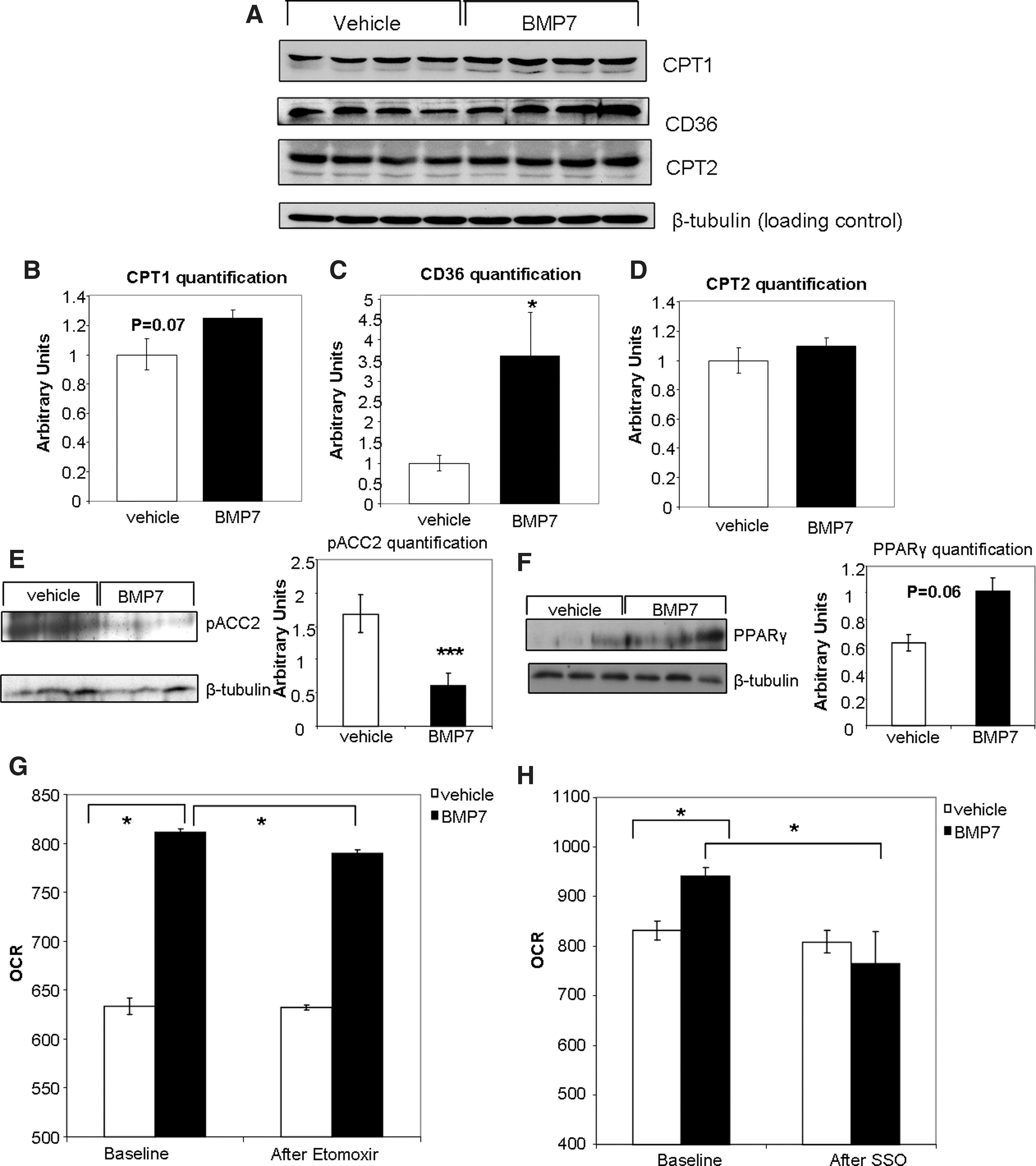
Additionally, we observed a significant increase in protein levels of CD36, which transports fatty acids into the cell (Fig. 5A, quantified in Fig. 5C). Interestingly, levels of peroxisome proliferator-activated receptor gamma (PPARγ), a transcription factor that regulates CD36 gene expression (1, 2), were significantly increased at the protein level at 6 h (Fig. 5F), and also displayed a consistent increasing trend at 2, 4, and 24 h at the mRNA level (Supplementary Fig. S4C). Together, our findings indicate that in mature brown adipocytes, BMP7 can increase fatty acid transport into the cell possibly via increased PPARγ and CD36, and into the mitochondria possibly through decreased ACC2 and increased CPT1.
To determine if CPT1 or CD36 is required for the observed increase in mitochondrial activity, we utilized specific inhibitors of these transporters coupled with respirometry measurements. When measuring mitochondrial activity in the presence of fatty acid as fuel, but in the absence of any inhibitors, BMP7 treatment resulted in increased OCR versus vehicle-treated cells, as seen previously (Fig. 5G, H). After addition of the CPT1 inhibitor etomoxir (Etom), there was a small reduction of OCR in BMP7-treated cells (Fig. 5G). Similarly, the increase in OCR by BMP7 treatment was abolished after exposure to the CD36 inhibitor sulfosuccinimidyl oleate (SSO; Fig. 5H). Therefore, inhibition of fatty acid transport into the cell (CD36, inhibited by SSO) or fatty acid transport into the mitochondria (CPT1, inhibited by etomoxir) impairs oxidative phosphorylation in the BMP7-treated cells more so than vehicle-treated cells, which may still be utilizing mainly glucose for respiration.
BMP7 utilizes the p38-ATF2 and SMAD1/5/8 pathways to increase mitochondrial activity
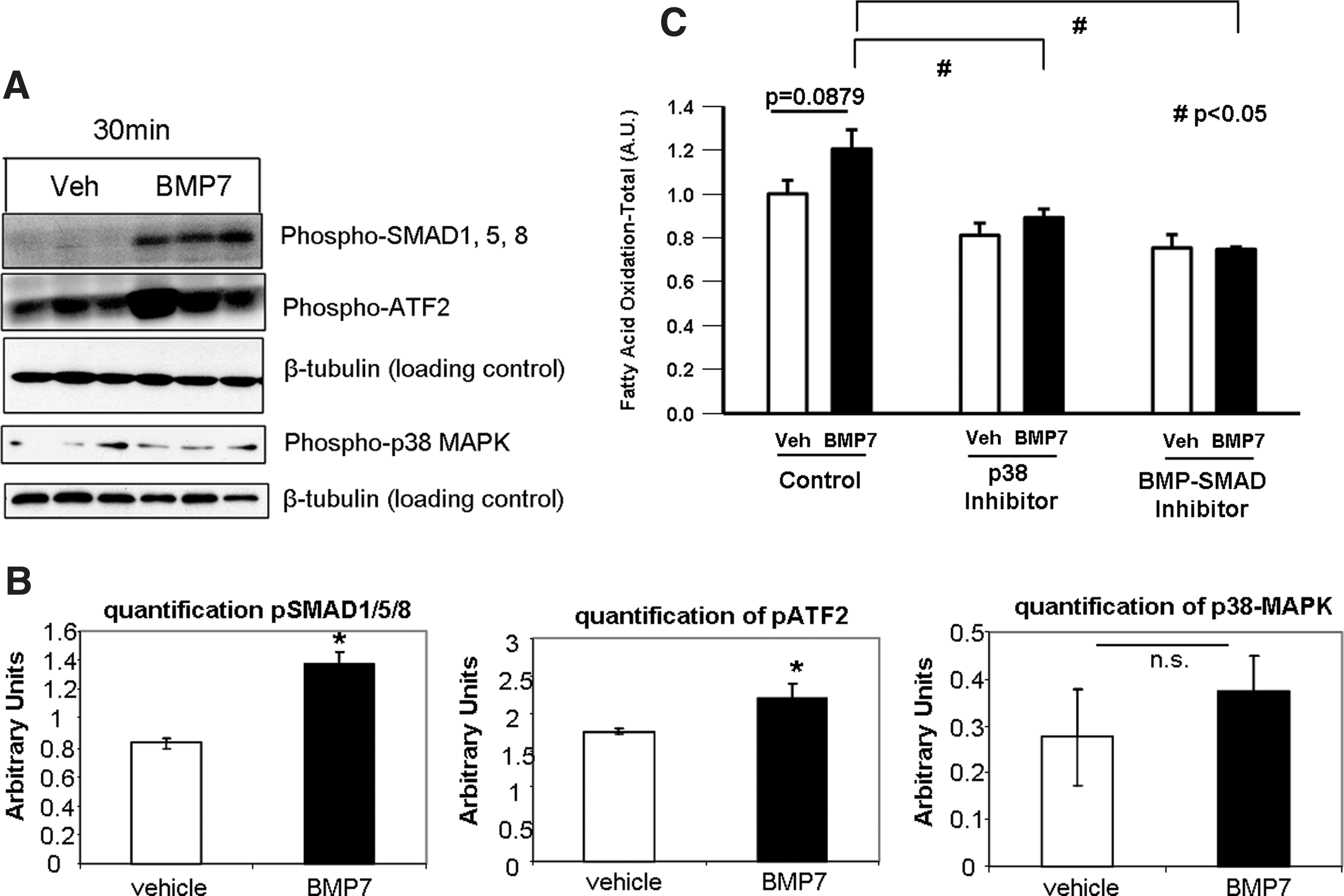
To determine which signaling pathways mediate the effects of BMP7 on fatty acid uptake and mitochondrial activity, we measured activation of signaling pathways known to be activated by BMPs as well as pathways known to be involved in fatty acid metabolism. BMPs signal through a canonical SMAD1/5/8 pathway and also commonly signal through the p38 mitogen-activated protein kinase (p38-MAPK)-ATF2 pathway. Mature brown adipocytes treated with BMP7 for 30 min displayed a robust increase in phosphorylation of SMAD1/5/8, as well as an increase in phospho-ATF2 (Fig. 6A). Despite potential crosstalk between the insulin and BMP pathways (62), BMP7 did not activate Akt phosphorylation (Supplementary Fig. S5A). Additionally, although the AMPK pathway is known to regulate fatty acid metabolism, BMP7 did not activate this pathway, either at the protein levels of phospho-AMPK (Supplementary Fig. S5A) or AMPK subunit activity (Supplementary Fig. S5B). We also did not observe any effects on protein expression of total phospho-ACC (Supplementary Fig. S5A). Therefore, we focused on SMAD and p38-MAPK-ATF2 as candidate pathways regulating the observed effects on mitochondrial activity. When measuring the ability of BMP7 to induce fatty acid uptake, we found that both a BMP-SMAD inhibitor and a p38-MAPK inhibitor were able to blunt BMP7-induced fatty acid oxidation (Fig. 6B), further supporting a role for these signaling pathways in the observed phenotype after BMP7 treatment.
In vivo delivery of BMP7 increases energy expenditure, body temperature, and BAT protein expression of CD36
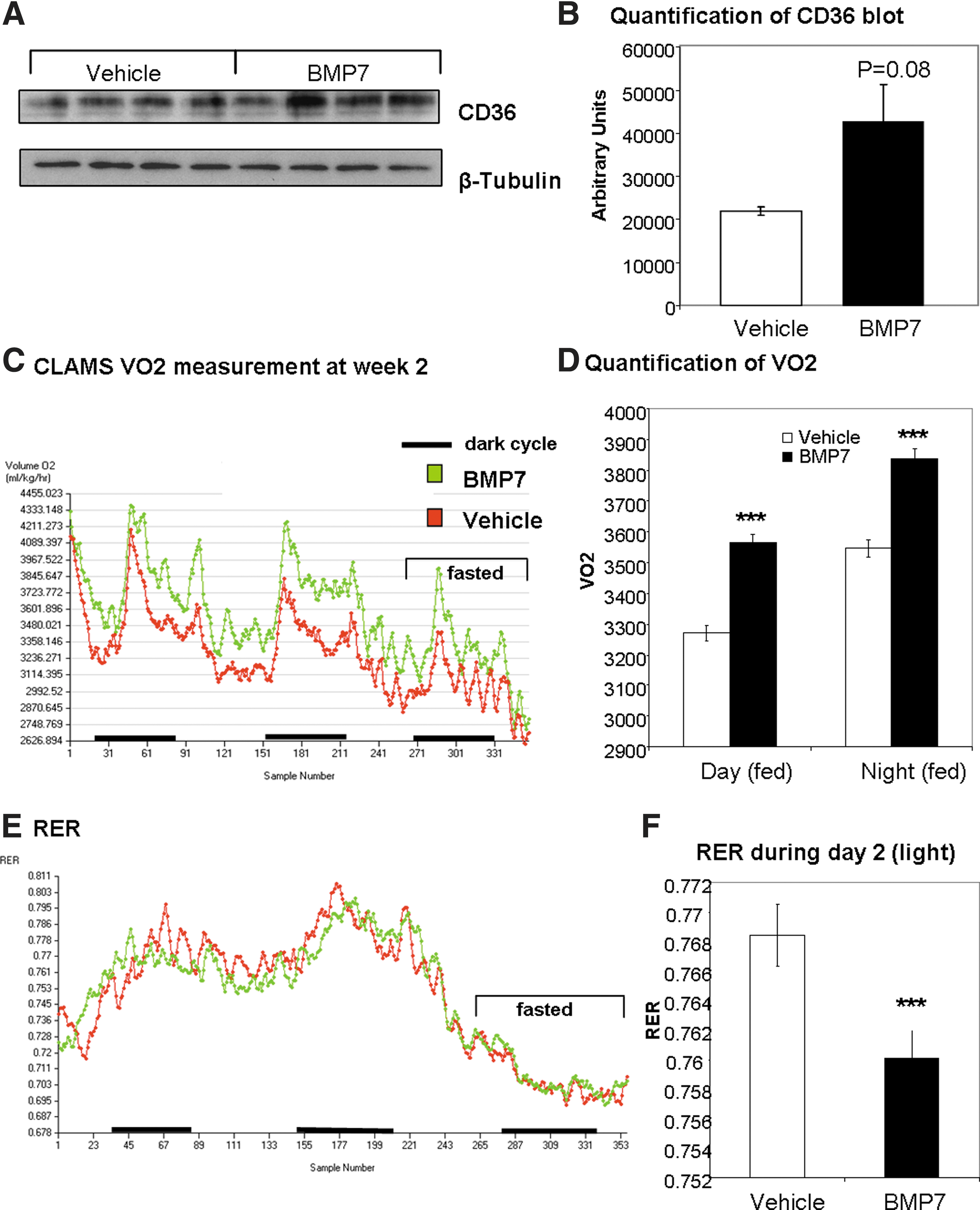
Next, we sought to determine if a similar mechanism to increase fatty acid uptake and oxidation was employed by BMP7 in vivo. We have previously shown that systemic delivery of BMP7 to chow-fed mice increases energy expenditure and body temperature (52), and that systemic delivery of BMP7 to high-fat-fed mice results in increased energy expenditure, decreased food intake, and reversal of diet-induced obesity (50). To determine if BMP7 is affecting CD36 in BAT in vivo, which may account for the observed increase in energy expenditure, we delivered rhBMP7 via a subcutaneous osmotic pump for 4 weeks. While it is possible that systemic BMP7 may be affecting the central or sympathetic nervous system activity to increase BAT function, or may be inducing brown adipogenesis, we specifically assessed the ability of BMP7 to affect BAT expression of the fatty acid transporter CD36. Indeed, similar to our in vitro observations, we observed an induction of CD36 protein expression in BMP7-treated mice (Fig. 7A; quantified in 7B). During this time, mice displayed increased energy expenditure as measured by oxygen consumption (Fig. 7C, D) and a decreased respiratory exchange ratio (RER; Fig. 7E; quantified in 7F), indicating greater metabolic activity and more utilization of fatty acids as fuel, respectively. Future studies will determine whether CD36 in mouse BAT is actively transporting more fatty acids into the cells for fuel, in response to BMP7 administration.
Discussion
Obesity is currently a global pandemic. Genetic and environmental factors combined with a modern lifestyle lead to an imbalance of the energy equation, resulting in an excess of calories being stored in adipose tissue. Eventually, this increase in lipid storage leads to dysfunction of adipose tissue, which is accompanied by local inflammation and systemic insulin resistance, and leads to several comorbidities of obesity such as type 2 diabetes and cardiovascular disease. Therefore, current biomedical research is saddled with the goal of preventing and reversing these detrimental effects of caloric excess. In opposition to WAT, which stores this excess energy in lipid droplets, BAT is tasked with increasing energy expenditure in the form of mitochondrial activation and thermogenesis. If properly activated, BAT is believed to be able to assist with excess lipid in states of obesity, by utilizing the fuel for its own energy expenditure.
The increased metabolic activity of BAT requires a plentiful supply of substrate, and as a result, BAT is a site of rapid fatty acid uptake and turnover. For example, in response to stimulation from cold exposure, BAT is able to increase lipogenesis, cellular fatty acid uptake, and lipid oxidation (6, 13). Gene expression studies have also indicated that cold exposure increases both fatty acid oxidation and fatty acid synthesis in brown adipocytes (60). In fact, the high rate of lipid clearance by BAT is even able to reverse the hyperlipidemia caused by apolipoprotein-AV deficiency (1). The process of thermogenesis relies heavily on stored lipid as a substrate (31). Some of the lipid is broken down to release free fatty acids for activation of UCP1 (4), while others are combusted in the mitochondria, where UCP1 uncouples oxidative phosphorylation from ATP production, releasing heat.
The ability to increase the metabolic activity of BAT is an enticing potential therapeutic option for combating metabolic disorders such as obesity. Our laboratory has previously described BMP7 as a growth factor able to induce BAT development and brown adipogenesis, and to increase energy expenditure and reduce appetite and body weight (43, 50, 52). We now demonstrate that BMP7 is able to increase metabolic activity of brown adipocytes, independent of increasing cell or mitochondrial number, or of altering differentiation state. This occurs, at least in part, by increasing fatty acid transport into the cell and into the mitochondria, by the fatty acid transporters CD36 and CPT1, respectively. The resulting increase in availability of fatty acids is accompanied by increased fatty acid oxidation, increased citrate synthase activity, and increased uncoupling in brown fat cells treated with BMP7.
Cells overexpressing BMP7 throughout differentiation display increased basal respiration, ATP turnover, and respiratory capacity, after differentiation when the cells have reached maturity. This indicates that BMP7 has specific effects on mitochondrial activity, independent of the differentiation state. BMP7 treatment of mature brown adipocytes results in increased oxygen consumption (a surrogate for oxidative phosphorylation) and respiratory capacity, which is further increased when the cells are exposed to glucose, carnitine, and palmitate, but not to glucose and pyruvate together. Interestingly, brown fat has the ability to actively combust glucose in the form of pyruvate, only when UCP1 is activated by free fatty acids (31, 45). Thus, our data suggest that BMP7 is able to increase fatty acid uptake and utilization in brown adipocytes by enhancing fatty acid transport mechanisms and activating UCP1.
One of the earliest changes we observe in BMP7-treated brown adipocytes is a slight increase of CPT1 expression at 4–6 h post-treatment. CPT1 is the rate-limiting step of the carnitine shuttle in the mitochondrial membrane, and triggers fatty acid transport into the mitochondrial matrix through the outer mitochondrial membrane (see diagram of shuttle in Fig. 8). Phosphorylated ACC2 is able to convert acetyl-CoA to malonyl-CoA, which inhibits CPT1 activity. Therefore, the decrease in ACC2 observed at the gene and protein levels presumably activates the already increased levels of CPT1. This increase in CPT1 is likely the initial event leading to increased fatty acid uptake in the cells, as observed later by multiple fatty acid uptake assays. Interestingly, increased CPT1 expression or activity is associated with a phenotype whereby WAT undergoes browning, or development of a BAT-like phenotype (48), indicating that this rate-limiting step in fatty acid transport into the mitochondria is an important factor for the phenotype of BAT. Indeed, in various tissues examined in Sprague-Dawley rats, BAT has the highest CPT1 activity and palmitate oxidation rate (11). Retinoic acid delivery also triggers WAT remodeling, and is accompanied by changes in both muscle- and liver-type CPT1 in adipose tissue (24). Furthermore, decreased CPT1 activity is observed in BAT mitochondria of streptozotocin-diabetic mice (14), and carnitine is necessary to maintain the phenotype and function of BAT (33). In this context, CPT1 appears to be an important and necessary component of brown adipocytes, including the brown fat cells appearing in white fat depots.

Recently, FAT/CD36 has been implicated in whole-particle triglyceride uptake by BAT (1). CD36 mainly transports long-chain fatty acids across membranes of various cell types, including adipocytes (22). Interestingly, we observed an increase in CD36 protein in brown fat cells at 48 h after BMP7 treatment, a time when there is increased fatty acid uptake and oxidation. Since this increase is not observed at earlier time points, like CPT1 is, we believe that the increase in CD36 expression and transport is triggered at later time points in response to the increased demand for substrate after increased CPT1 transport has already provided additional fuel for active β-oxidation in the mitochondria. Additionally, BMP-signaling pathways have been shown to induce expression and activities of PPARγ (34, 47), which is known to increase transcription of CD36 (1, 2). Indeed, we saw a consistent trend for increased PPARγ gene expression at several early time points of BMP7 treatment, and increased PPARγ protein at 6 h, suggesting that BMP7 may modulate CD36 expression via a PPARγ-dependent pathway.
CD36 has already been implicated in increased mitochondrial activity. For example, CD36 is the dominant fatty acid transporter in WAT (9), and CD36 expression is increased in the interscapular WAT located above BAT of cold-exposed mice (54). Without CD36, mice cannot fight the cold and turn on thermogenesis (1). CD36 variants have also been associated with changes in the body–mass index in humans (20). In addition, CD36 may also mediate adipocyte–macrophage crosstalk and may regulate inflammation and atherosclerosis (17, 58), and therefore could be involved in the pathogenesis of the metabolic syndrome (18). Interestingly, CD36 has been observed to be localized to the outer mitochondrial membrane, in addition to the cell surface, and may interact with CPT1 (41, 46), but it is currently unclear if this occurs in other cell types or in brown adipocytes specifically. Nevertheless, the fact that pharmacological inhibition of CPT1 or CD36 in brown adipocytes abolished the BMP7-induced mitochondrial activity indicates that these molecules play an essential role in the regulation of mitochondrial metabolism in energy-dissipating brown adipocytes.
Consistent with the in vitro findings, we observed that systemic administration of BMP7 increased protein expression of CD36 in BAT, which was accompanied by an increase in energy expenditure and greater fatty acid utilization as indicated by RER. However, it is important to recognize that systemic BMP7 may have myriad effects, including (i) direct activation of fatty acid uptake, mitochondrial activity, and thermogenesis in BAT; (ii) increased brown adipogenesis; (iii) effects on the central nervous system to increase sympathetic innervation of BAT (43, 52). By using a brown adipocyte cell line, we can directly measure changes in mitochondrial function that are due to cell intrinsic properties of the brown adipocyte itself, without confounding variables due to other cell types found in brown fat in vivo. Therefore, these in vitro findings offer great promise for a better understanding of in vivo brown fat function. Taken together, BMP7 or other treatments that can exploit the utilization of excess lipid for fuel in mitochondrial processes are appealing candidates for treatment of lipid-overload situations, such as obesity.
Materials and Methods
Cell culture
An immortalized, murine brown preadipocyte cell line created by our laboratory was differentiated to mature brown adipocytes following the schema in Figure 2, with or without pretransfection with BMP7 or the control vector. Briefly, the cells were grown to postconfluence, followed by 2-day induction with a medium containing insulin, T3, dexamethasone, IBMX, and indomethacin, as described previously (52). Then, this medium was removed, and cells were given a growth medium with insulin and T3 for an additional 5 days. At this point (day 7), cells appear as mature, lipid-laden, and UCP1-positive brown adipocytes, and were then treated with BMP7 (R&D Systems; 3.3 nM) or vehicle for 48 h or other time periods. In some experiments, cells were treated with inhibitors of p38 signaling (Sigma SB202190) or BMP-SMAD signaling (LDN-193189; a generous gift of Dr. Paul Yu at the Brigham and Women's Hospital) (59).
Seahorse bioanalyzer
The Seahorse XF24 (Seahorse Bioscience,
Mitotracker deep red: flow cytometry and confocal microscopy
Cells were washed, incubated with Mitotracker Deep Red (Invitrogen) for 20 min, and washed again, followed by fixation. The vehicle- and BMP7-treated cells were compared either by flow cytometric analysis or confocal microscopy.
Citrate synthase assay
Citrate Synthase was measured from whole-cell extracts utilizing a commercial kit (Sigma; Cat #CS0720), followed by quantification of fluorescence in a plate reader before and after addition of the substrate oxaloacetate (OAA). Calculations were based on the manufacturer's instructions.
Fatty acid uptake and fatty acid oxidation assays
Fatty acid uptake and oxidation were determined by measuring both 14C-labeled palmitic acid uptake and conversion of 14C-labeled palmitic acid into CO2. Briefly, the culture medium was removed, and cells were incubated with DMEM containing 4% albumin, 0.5 mM palmitic acid, and 0.2 μCi/ml [1-14C]-palmitic acid for 1 h. The DMEM was transferred to a vial containing acetic acid (1 M), capped quickly, and allowed to sit 1 h for CO2 gas to be released. 14CO2 released was absorbed by hyamine hydroxide, and activity was counted. Fatty acid oxidation was calculated from CO2 generated. To measure fatty acid uptake, cells were rinsed twice with PBS and lysed after incubation with [1-14C]-palmitic acid. Lipids were extracted using a chloroform–methanol mixture (2:1), and 14C counts were determined in the organic phase. Fatty acid uptake was calculated as the total of 14C lipids in the cells and 14CO2 generated.
BODIPY assay
Cells were treated with BMP7 or vehicle after which fatty acid uptake was measured using the fluorescent-labeled long-chain fatty acid, 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-hexadecanoic acid (BODIPY FL C16; Invitrogen, hereafter referred to as BODIPY), which was conjugated to BSA. Assays were conducted in the presence of the quenching agent trypan blue, as described elsewhere (40). After excitation at 490 nm, fluorescence at 520 nm was determined using a fluorescent plate reader. Because trypan blue cannot enter live cells, the only fluorescence detected was intracellular.
Quantitative polymerase chain reaction and mtDNA copy number
Total RNA and total DNA were extracted using the All Prep Kit (Qiagen) or the gDNA kit (Zymo). Quantitative polymerase chain reaction (qPCR) was conducted with SyBr green as described previously (52). The mtDNA copy number was measured by comparing COXII (mtDNA) to β-tubulin (nuclear DNA). All primer sequences can be found in (16).
Oil Red O staining
Cells were stained in six-well dishes with Oil Red O (Sigma) for 20 min, followed by washing, photographic imaging, and drying of the plates. Oil Red stain was then extracted from each well with propranolol, and absorption was read in a plate reader at 510 nm for quantification of the lipid content.
Western blot
Cells were lysed in a RIPA buffer and homogenized in a Bullet Blender (except for the p-p38 blot, where cells were collected in the Sorenson buffer and disrupted by sonication). Equal protein concentrations (determined by Bradford Assay) were loaded on a 10% Tris–HCL gel for PAGE. Proteins were then transferred to a PVDF membrane and probed with antibodies for CPT1-L (Adi), CPT2 (Abcam/MitoSciences), CD36 (Santa Cruz), ACC2 (Epitomics), PPARγ (Abcam), or signaling components such as SMAD1/5/8, p38, or AMPK (all from Cell Signaling), followed by incubation with appropriate secondary antibodies and visualization with enhanced chemiluminescence substrate. Band densities were quantified using ImageJ software.
Mouse model
Four weeks after consuming a diet with 60% kcal from fat (Research Diets), C57BL/6 mice (Taconic) were implanted with subcutaneous osmotic minipumps (Model 2006; Alzet) containing either recombinant human BMP7 (gift from Stryker) or vehicle. Mice received 0.72 μg of BMP7 per day at a constant flow rate for 4 weeks.
Statistical analysis
Comparisons were made by Student's t-test or analysis of variance with Fisher post hoc assessment using the StatView program. In all experiments, each group had n=3–12 replicates. *p<0.05; **p<0.01; ***p<0.001
Footnotes
Acknowledgments
The authors wish to thank the following people for technical advice: Cecile Vernochet, Andre Kleinridders, Chris Cahill, Kristina Kriauciunas, Carly Cederquist, Stephane Gesta and C. Ronald Kahn (Joslin Diabetes Center), and Zhiyong Cheng (Children's Hospital). We thank Lindsay McDougall (Joslin Diabetes Center) for technical assistance, Eric Widmaier (Boston University) and Paul Yu (Brighm and Women's Hospital) for reagents, and Tim Schulz (Joslin Diabetes Center) for critical reading of the manuscript. We acknowledge Stryker Regenerative Medicine (Hopkinton, MA) for the generous gift of recombinant BMP7 for the in vivo experiments. This work was supported in part by the NIH grants R01 DK077097 (Y.-H.T.), Joslin Diabetes Center's Diabetes Research Center (P30 DK036836 from the NIDDK), and a research grant from the Eli Lilly Research Foundation and by funding from Harvard Stem Cell Institute (to Y.-H.T.). KLT was supported by NIH F32 DK091996.
Author Contributions
K.L.T. wrote the manuscript and designed, conducted, and analyzed the experiments. D.A. and L.G. conducted the fatty acid uptake and oxidation, lipogenesis, and glucose oxidation experiments. M.D.L. ran the BODIPY experiments, some of the western blots, and some qPCR. T.L.H. ran the some of the qPCR and western blots. H.Z. conducted the bioinformatics analyses. Y.H.T. wrote the manuscript and designed experiments.
Author Disclosure Statement
The authors do not have anything to disclose.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.