
Abstract
Introduction
De-regulation in the homeostasis of iron and other metals and alterations in the expression of the proteins involved in iron metabolism are often observed in malignancies that may lead to a redox imbalance (294). There is also evidence from epidemiological, clinical, and experimental studies that suggests a link between iron and the development of neoplasia (297, 298). Owing to the complexity and heterogeneity of cancer, a clear-cut relationship between iron and carcinogenesis has yet to be established, and this is discussed as a part of this article. Additionally, it is clear that iron and other biological metals are important factors in cancer growth and progression that can be exploited by the development of anti-cancer drugs such as chelators (143). In this review, the mechanisms behind the action of these agents will be analyzed in detail along with an assessment of the investigations examining their preclinical and clinical efficacy.
The Perfect Balance: Normal Iron Metabolism and Regulation
Intestinal iron absorption and systemic trafficking
Iron is consumed via the diet in two main forms: nonheme/inorganic iron (e.g., from various legumes) and heme iron (e.g., from meat, fish, and poultry) (201). Intestinal iron absorption of both dietary iron forms occurs predominantly within the duodenum (Fig. 1) (201). Nonetheless, the pathways of inorganic and heme iron uptake from the intestinal lumen across the apical membrane of enterocytes are quite distinct (201). Inorganic iron is absorbed at the apical membrane of enterocytes via the divalent metal transporter 1 (DMT1) (88, 89, 110). Interestingly, DMT1 most efficiently transports metals in their divalent state (88, 89, 110), and, hence, inorganic iron(III) should first be reduced by an unidentified brush border ferric reductase (194). One such contender, duodenal cytochrome b (Dcytb; Fig. 1), was shown to possess ferric reductase activity, to be iron regulated and highly expressed at the duodenal brush border (194). However, a Dcytb-knockout in mice failed to reduce body iron stores, even in conditions of iron deficiency, illustrating that other compensatory mechanisms exist which are involved in the intestinal absorption of inorganic iron (111).

The uptake of heme in the intestine occurs via a different mechanism from that of inorganic iron (249, 268, 316). A potential heme transporter, heme carrier protein 1 (HCP1), has been identified (268). However, HCP1 also acts as a folate transporter (231). Therefore, both folate and heme may compete for absorption in the small intestine (164), or potentially other heme transporters could also exist. In fact, the exact role of HCP1 in heme absorption remains unclear and considering the importance of iron as a crucial nutrient, it is likely that there are several mechanisms involved in heme uptake in the gut (164). Once within enterocytes, heme is broken down into biliverdin and iron(II) by heme oxygenase-1 (HO-1; Fig. 1) (232).
Iron(II) is released from enterocytes into the circulation via the basolateral iron exporter, ferroportin 1 (Fpn1; Fig. 1) (1, 74, 195). Given that “free” iron can be deleterious, once within the circulation, it becomes bound to the serum protein, transferrin (Tf) (258), which specifically binds two iron(III) atoms (5). Copper-containing enzymes, including the transmembrane-bound protein, hephaestin, and the plasma protein, ceruloplasmin, are believed to oxidize iron(II) that is exported through the plasma membrane via Fpn1 to iron(III) for Tf binding (Fig. 1) (218, 307).
Iron(III) bound to Tf is delivered to tissues by the transferrin receptor 1 (TfR1) through receptor-mediated endocytosis via clathrin-coated pits (Fig. 2) (5). Once the endosome is acidified, the iron is released from Tf (5, 68) and exported out of the endosome into the cytosol via DMT1 (88, 89). Since Tf binds iron(III), whereas DMT1 transports iron(II), the iron should be reduced before endosomal export, probably by the ferrireductase six-membrane epithelial antigen of the prostate 3 (STEAP3) (213). After releasing iron, the Tf-TfR1 complex is recycled to the cell surface by exocytosis (Fig. 2), releasing apo-Tf back into the circulation (5, 68). Studies have also shown the existence of other iron uptake processes, including (i) a second transferrin receptor (TfR2) (150); (ii) a mechanism mediated by surface pinocytosis (242, 300); and (iii) other Tf-independent iron uptake mechanisms (176).
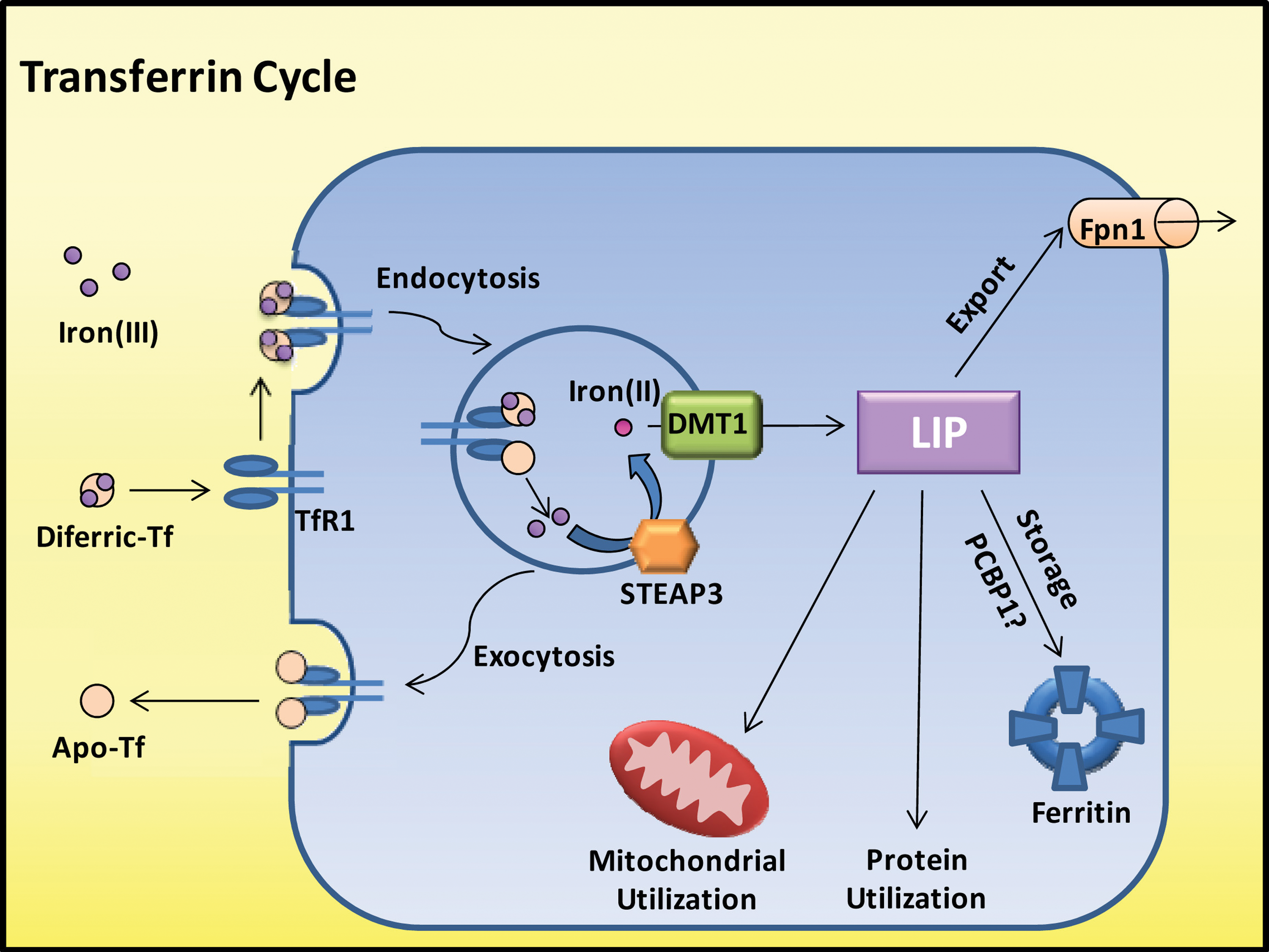
Once released from the endosome, iron is believed to enter the labile iron pool (LIP; Fig. 2) (107, 134) [for review see (160)]. Iron is proposed to exist in the LIP complexed with low-molecular-weight ligands, such as citrate, ATP, other inorganic irons, proteins, and sugars (142). This compartment remains poorly characterized (160), and investigators have questioned its existence (160, 308). It has also been suggested that iron is transferred directly from protein to protein or through organelle interactions (245, 269, 271). For instance, Sheftel et al. have proposed that iron is delivered from Tf-loaded endosomes to the mitochondria through direct interactions (269). However, the precise molecular mechanisms involved remain unclear. Recently, it has been suggested that the human poly (rC)-binding protein 1 and 2 (PCBP1 and 2) acts as a chaperone which facilitates the delivery of iron to ferritin for storage and also to hypoxia-inducible factor-1α prolyl hydroxylases (206, 271). Hence, PCBP1 and 2 could represent the first proteins identified to act as chaperones for intracellular iron transport.
Once iron is present within the cell, it can be (i) distributed to cytosolic ferritin for storage; (ii) incorporated into the active sites of proteins, such as ribonucleotide reductase (RR); (iii) distributed to the mitochondria for heme synthesis, iron-sulfur cluster generation, or storage in mitochondrial ferritin; or, alternatively, (iv) iron can be exported out of the cytoplasm via Fpn1 [for review see (127)].
Iron storage: ferritin
It is well established that cytosolic ferritin permits the storage of iron in a safe inert form, protecting cells from iron-induced oxidative damage and allowing for iron availability in times of need [for reviews see (175, 286)]. Cytosolic ferritin consists of 24 subunits including a mixture of two ferritin chain types, a heavy (H) chain (21,000 kDa), and a light (L) chain (19,000 kDa), which are present in different ratios depending on the tissue type (175, 286). The H-chain possesses ferroxidase activity that converts iron(II) to iron(III) in order to store iron in a safe inert form as ferroxyhydrite (175, 286). Each ferritin molecule can store up to 4500 iron atoms within its inner cavity (175, 286).
Apart from cytosolic ferritin, mitochondrial ferritin was discovered in 2001 as being specifically located within the mitochondrion and encoded by an intronless gene on chromosome 5q23.1 (171). Unlike its cytosolic counterpart, mitochondrial ferritin is localized to specific tissues, and its distribution does not directly correlate with mitochondrial content or iron levels (171). It has been suggested that mitochondrial ferritin may be involved in regulating heme synthesis within the mitochondrion of erythroid cells, storing excess mitochondrial iron when heme synthesis is impaired and making iron available again when heme synthesis resumes (171). Consequently, mitochondrial ferritin expression may be regulated by mitochondrial iron load (43, 171).
Iron homeostasis
The human body has no direct capacity to excrete iron and has adapted several mechanisms that maintain an appropriate iron balance (69). Iron homeostasis is highly regulated through transcriptional and post-transcriptional mechanisms (69). Post-transcriptional control of iron levels is mediated by the iron-regulatory proteins (IRPs) [for reviews, see (37, 252)]. IRPs bind to iron-regulatory elements that are found in the mRNA of molecules involved in iron trafficking (e.g., ferritin, Fpn1, TfR1, DMT1 etc.), thereby controlling their translation and regulating iron levels (37, 252). More recently, the liver-secreted iron-regulatory hormone, hepcidin, was discovered (221). Hepcidin regulates iron transport into the plasma by binding to the plasma membrane transporter, Fpn1, triggering its internalization and its degradation, resulting in the inhibition of iron release from iron-exporting cells, including enterocytes, macrophages, and hepatocytes, subsequently decreasing serum iron levels (207).
Deregulated Iron Balance in Cancer
It is clear that under normal conditions, iron levels are tightly controlled through various sophisticated pathways that maintain an ideal balance. Over the years, research has established that cancer cells do not completely conform to normal cellular pathways (116). For example, some of the hallmarks of cancer include self-sufficiency in growth signals, insensitivity to inhibitory signals, evasion of apoptosis, and unlimited proliferation (116). It is, therefore, no surprise that iron homeostasis is altered in malignant cells (294). At present, a complete understanding of how cellular homeostasis of iron and other metal ions is deranged in cancer cells is yet to be deciphered. However, it is clear from studies using a variety of chelators that neoplastic cells are differentially sensitive to iron depletion relative to their normal counterparts, demonstrating their altered metal metabolism (162, 158, 313, 331). Further investigations are ongoing to elucidate how metal trafficking and homeostasis are altered in neoplasia. Examples of perturbations of metal metabolism in malignancies are discussed in the next sections titled: “Perturbed iron homeostasis in cancer cells” and “Other biologically relevant metals in neoplasia.”
Depletion of iron in cells typically causes a G1/S arrest or apoptosis, demonstrating that this metal is required for cell-cycle progression and growth (165, 185). Iron is required for the activity of RR, which is responsible for the rate-limiting step of DNA synthesis that generates deoxyribonucleotides, the precursors to DNA, making DNA synthesis dependent on iron (155). Additionally, iron has been shown to regulate the expression of various proteins and modulate the signal transduction pathways involved in cellular proliferation and metastasis, including: cyclins, cyclin-dependent kinases (cdks), p53, p21, N-myc downstream regulated gene 1 (NDRG1), and so on [see review (328)]. In some cell types, iron depletion results in the up-regulation of the mRNA of the cdk inhibitor, p21CIP1/WAF1 , while, it conversely down-regulates its protein expression, which can lead to apoptosis (328). Furthermore, iron depletion has been shown to down-regulate cyclin D1, cyclin D2, and cyclin D3 (98). These studies illustrate that the expression of critical effectors of the cell cycle such as cyclins and cdks are affected by intracellular iron levels and, thus, iron levels can modulate cell-cycle progression (328).
Additionally, although iron is essential for many cellular processes, it can also participate in reactions that produce harmful free radicals. Iron has been implicated in the pathogenesis of several human diseases, including cardiovascular disease (63, 287), diabetes mellitus (82, 167), infections (141), neuro-degenerative disorders (34, 168, 219), and cancer (297). It is widely known that free radicals can react with DNA, resulting in mutations, inactivating tumor suppressor genes, and activating oncogenes (303). This can adversely affect the cell cycle and promote the development of transformed cells into overt malignancy (220). Therefore, iron has also been associated with cancer progression in both epidemiological and animal studies (297, 298).
Fenton chemistry
Iron is a transition metal that is able to readily alternate between two redox states, namely, iron(II) and iron(III). Hence, iron has the ability to donate or accept electrons from various molecules. This redox activity provides iron with the ability to catalyze numerous essential reactions, such as those involving catalase and cytochromes (296). However, this redox activity can also produce deleterious effects through the production of free radicals via the Fenton reaction (81). Several cellular reducing agents (e.g., ascorbate and glutathione) reduce iron(III) to produce iron(II) via Equation 1 (296). The oxidation of iron(II) is a one-electron process and, thus, produces superoxide,
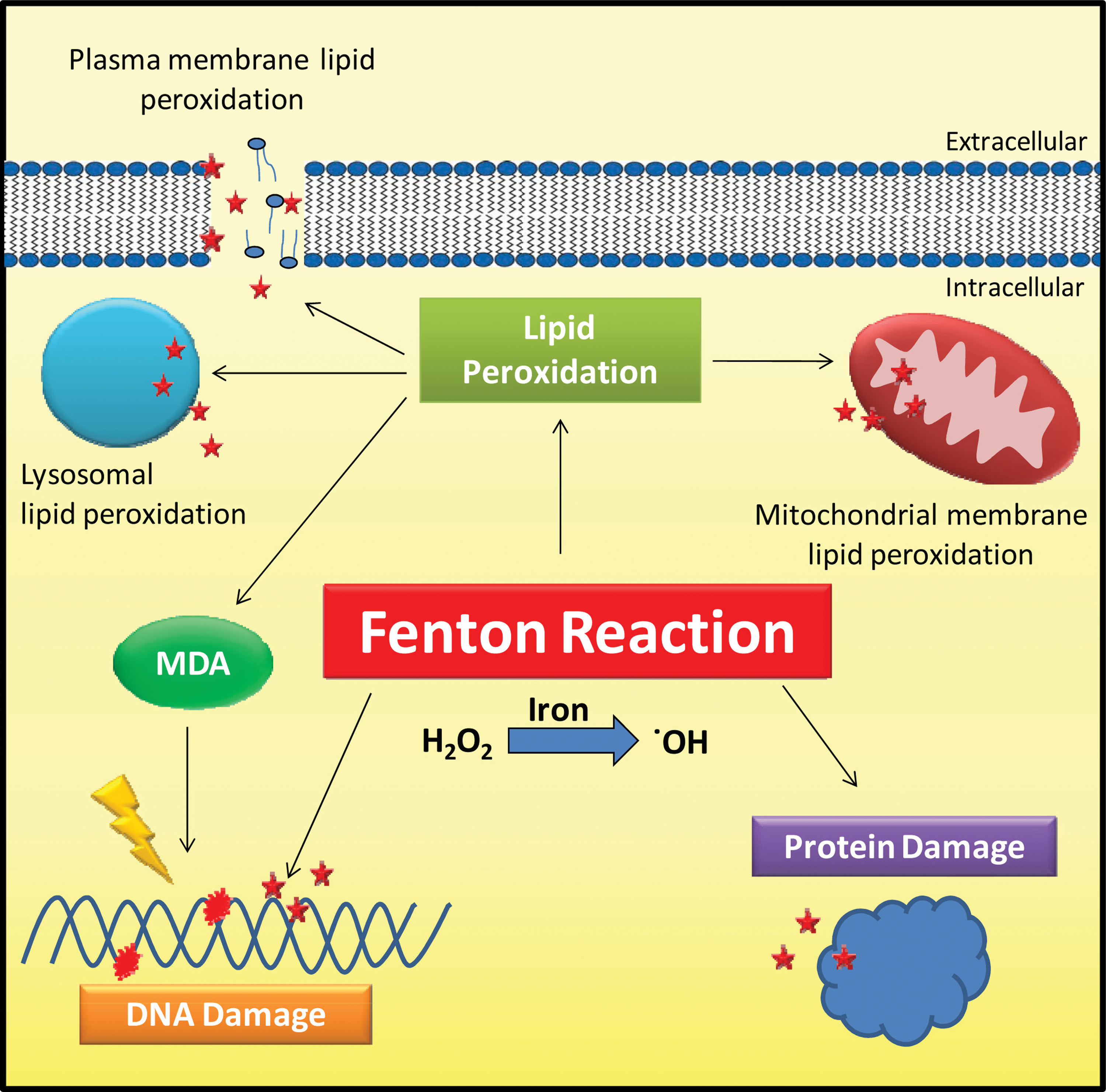
As a consequence of the iron-induced free radicals generated through these reactions, the human body possesses mechanisms that store iron in a safe form, via ferritin, and transport iron in a nontoxic bound form to the protein, Tf (244). The mammalian organism also possesses a number of oxidant defense mechanisms against oxidative stress, including superoxide dismutase and catalase (105). Despite these protective mechanisms, under certain metabolic circumstances and in the presence of “free” metal ions, iron-catalyzed free radicals participate in unwanted reactions that induce damage to nucleic acids, lipids, and proteins (Fig. 3). For instance, iron-induced free radicals can attack DNA, producing base or sugar modifications as well as strand breaks and cross links (177 –179, 196). Additionally, hydroxyl radicals can cause lipid peroxidation, resulting in the damage of the plasma, mitochondrial and lysosomal membranes, which may instigate cell death (113, 139, 281, 303). Lipid peroxidation also results in the formation of end products, some of which are mutagenic, such as malondialdehyde, which reacts with DNA bases and forms multiple adducts that are thought to be carcinogenic (191). Lastly, iron-catalyzed free radicals are also believed to damage and modify proteins through the oxidation of amino acids, resulting in inactive or less active proteins, which are required for normal cell functioning (102, 281, 285, 303). Therefore, collectively, these iron-mediated oxidative reactions damage normal cellular, tissue, and organ functioning, as evident in diseases where iron accumulation occurs, for instance, Friedreich's ataxia and β-thalassemia [for reviews, see (4, 261, 315)].
The relationship between iron and cancer development
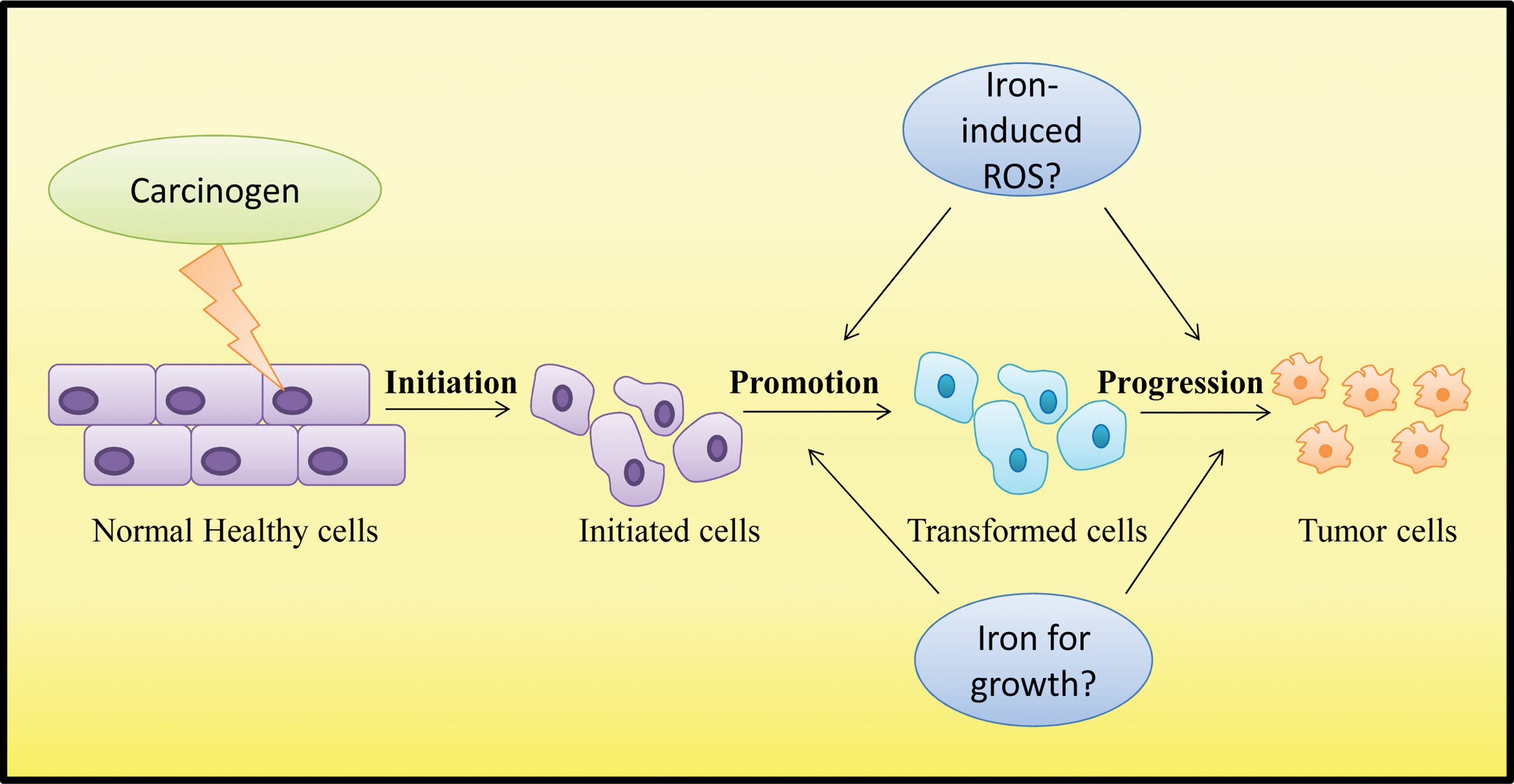
Iron has been implicated in the development of cancer (55, 296 –298). Although it is clear that iron can cause harmful effects through the Fenton reaction and that ROS are linked with cancer (303), it has been difficult to establish a cause-and-effect relationship. This is probably related to the fact that carcinogenesis is a multi-step process that involves various causative factors, including diet, lifestyle, and genetics, making it difficult to draw clear conclusions. Additionally, it is likely that iron is not a carcinogen by itself, but is instead a co-factor that promotes the progression of malignancies (Fig. 4).
A number of studies have demonstrated a link between iron and Kaposi's sarcoma (275), lung cancer (51, 250), colorectal cancer (153), and hepatocellular carcinoma (90, 126). For instance, investigations examining body iron stores have indicated that high body iron stores, as judged as levels exceeding a Tf saturation of 60%, have an elevated risk of developing cancer, particularly colorectal cancer (153). Moreover, blood donors have a significantly lower risk of developing nonhematological malignancies, such as liver, pancreas, lung, and stomach cancers, than control subjects (197). This may either be related to a decrease in iron stores in donors caused by repeated phlebotomy or may be linked to several other factors, including the concept that blood donors may have a healthier lifestyle (197). A high consumption of red meat and processed meats, which possess a high iron content, has been shown to be associated with an increased risk of developing colorectal cancer (209). Nonetheless, it is unknown whether this link is correlated to the intake of excess iron and the production of free radicals that damage the colonic mucosa or whether it is influenced by confounding factors such as the fat content in red meat or other dietary factors (209).
Interestingly, individuals with hemochromatosis, an iron overload disorder, are at a high risk of developing hepatocellular carcinoma and have a tendency to develop other extra-hepatic malignancies, including esophageal cancer and melanoma (90, 126). Studies performed by Kato et al. have demonstrated that treatment of hepatitis C patients with phlebotomy, performed until they reached mild-iron deficiency, combined with a low-iron diet, lowered their risk of developing hepatocellular carcinoma (148, 149). Hence, iron may be involved in the progression of cancer, potentially through the induction of ROS that predispose cells to cancer concomitantly with other carcinogens; or iron may simply stimulate tumor cell growth. Nevertheless, the studies just described are confounded by lifestyle, dietary, and other factors that cannot be controlled and, therefore, it is very difficult to establish a clear association.
Perturbed iron homeostasis in cancer cells
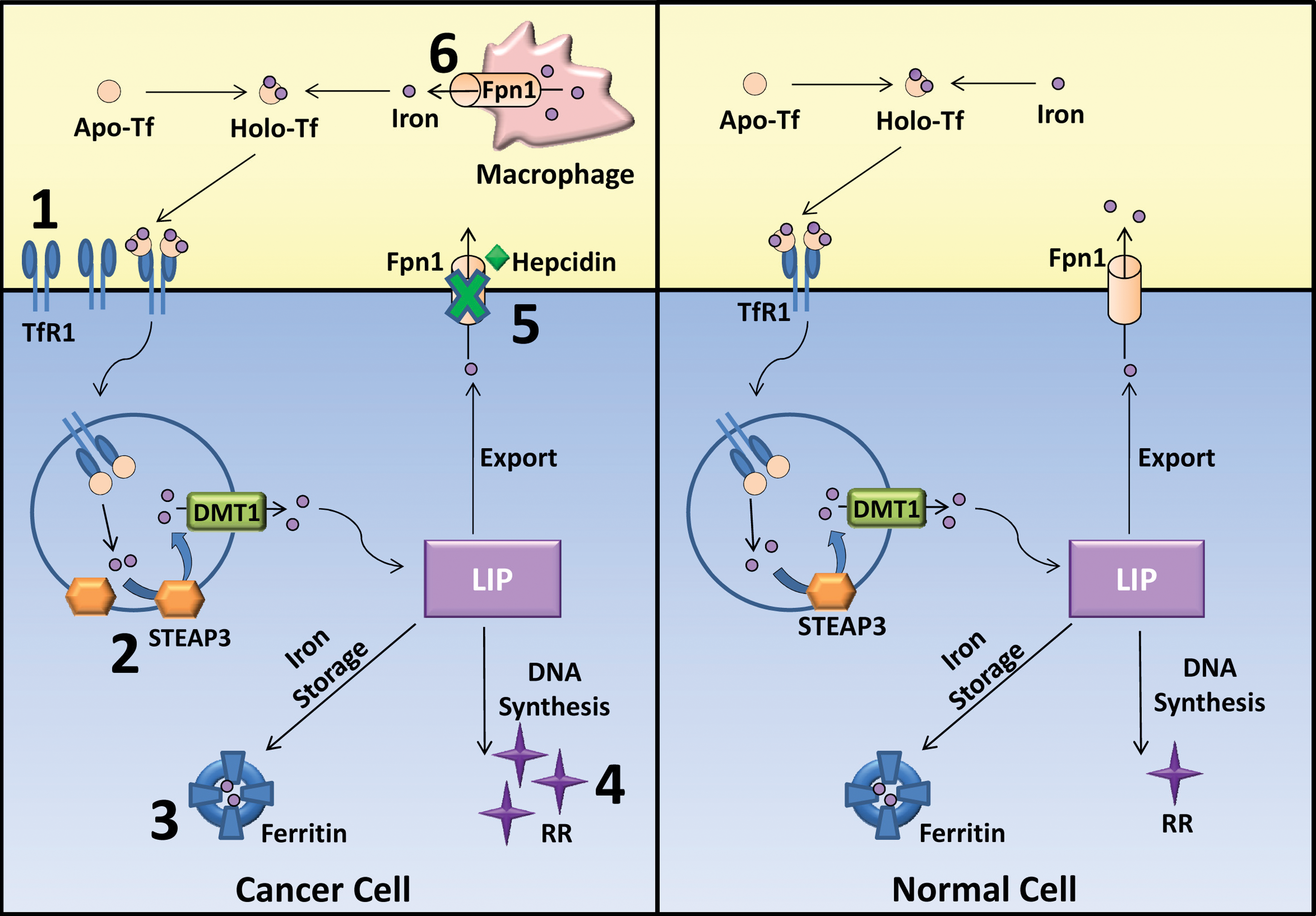
Several studies have demonstrated that there is a de-regulation of iron homeostasis in malignancies owing to the re-programming of gene expression (Fig. 5) (294). In fact, the cancer cell phenotype may provide a survival advantage either by stimulating proliferation or through the occurrence of genetic mutations via the production of cancer-promoting ROS (294). Nevertheless, cancer cells possess an extensive heterogeneity in genetic and epigenetic defects. Thus, not all cancer cells conform and possess an altered iron state. Additionally, it is difficult to attribute the exact mechanism that is responsible for the modulation of each iron-regulated protein in neoplastic cells. It is likely that there are several different molecular mechanisms which induce alterations of iron-regulated proteins depending on the neoplastic cells. For instance, it has been shown that c-myc, an aberrantly activated oncogene in certain cancers, decreases the expression of ferritin (308) and increases the expression of TfR1 (212). Examples are given next of the circumstances in which cancer cells possess altered iron trafficking and homeostasis through changes in the expression of iron-related proteins, which may generally confer an increased iron acquisition that could enhance tumor cell proliferation (294).
Tf receptor and RR expression
Numerous studies have shown that cancer cells, including colorectal cancer, breast cancer, bladder carcinoma, lymphoma, cervical cancer, and so on, express higher levels of TfR1 than do normal cells (33, 137, 180, 217, 265, 283, 310, 324). Additionally, a variety of studies have demonstrated that increased TfR1 expression is linked to tumor grade and stage, where higher expression is correlated with more advanced cancers (25, 115, 229, 265, 283). Moreover, studies have targeted the TfR1 for site-specific delivery of anti-cancer drugs due to its increased expression in malignant cells compared with normal tissue (230). To confirm that the elevated levels of TfR1 have an effect on cancer growth, it was demonstrated in vitro that tumor cell growth was inhibited on the administration of the anti-TfR1 monoclonal antibody 42/6, which sterically blocks Tf binding (301). Furthermore, it was demonstrated that the iron-dependent enzyme, RR, which catalyzes the production of deoxyribonucleotides, is up-regulated in cancer cells, facilitating the production of DNA precursors for DNA replication and tumor cell division (77, 290).
Ferroportin 1, hepcidin, and cancer
Hepcidin regulates the expression of Fpn1 by initiating its internalization and degradation and, thus, the expression of these proteins is inversely regulated (207). Recent studies have reported perturbed levels of Fpn1 and hepcidin in cancer cells and patients, resulting in iron accumulation (25, 124, 137, 146, 225). The expression of Fpn1 was shown to be decreased or absent at both the mRNA and protein levels in breast cancer cells and tumor samples when compared with normal breast epithelial cells, adjacent noncancerous tissue, or nonmalignant breast tissue from control individuals (137, 225). This reduction in Fpn1 occurred concomitantly with increased hepcidin expression in malignant cells relative to normal cells (225). Decreased Fpn1 was associated with a concurrent increase in LIP and a reduction in ferritin levels (225). Thus, decreased Fpn1 and increased hepcidin expression in malignant cells may cause increased cellular “free” iron, which subsequently becomes available for DNA synthesis and proliferation or DNA damage through ROS formation (225).
The levels of Fpn1 in other cancer types were also examined by Pinnix and colleagues using the Oncomine database [
In orthotopic breast cancer xenograft mouse models, mice with increased Fpn1 expression possessed reduced tumor growth and size, indicating that increased Fpn1 affects iron availability in cells and influences tumor growth (225). Interestingly, Fpn1 has been proposed to be a prognostic marker in breast cancer (225). Decreased Fpn1 expression in breast cancer patients was correlated with poor prognosis and metastasis, independent of other risk factors (225). Additionally, breast cancer patients with a combination of high Fpn1 and low hepcidin mRNA expression were shown to have a more favorable outcome with a 5 and 10 year distant metastasis-free survival rate of 95% and 91%, respectively (225). Thus, alterations in both Fpn1 and hepcidin expression may predict clinical outcomes and could potentially be a part of a novel oncogenic signaling mechanism.
Ferritin in neoplastic conditions
Ferritin expression has been shown to be up-regulated in tumor cells and patient tissue in a number of studies (25, 38, 124, 272, 291, 305), while others have shown that it is down-regulated (225, 272). High levels of ferritin mRNA in tissue from breast cancer patients were correlated with the presence of metastasis, lymph node involvement, and advanced clinical stage (321). There is also evidence that serum ferritin levels are elevated in patients with cancer (120, 152, 190, 224). Interestingly, the survival rate was lower in primary lung cancer patients with high levels of serum ferritin (200). Another study strengthened this link, demonstrating that advanced stage C colorectal cancer patients with either low or high levels of serum ferritin had a lower survival rate than those with normal levels (182). Additionally, elevated serum ferritin levels were shown to be a poor prognostic factor for neuroblastoma patients (117), and this was also associated with increased inflammation, iron deficiency and nonspecific tissue damage, which cancer patients often experience (120, 161). Hence, ferritin lacks the specificity that is necessary for a tumor marker. Contradictory and incomplete results complicate the relationship between ferritin and cancer and indicate that the relationship does not conform to one general pattern in all cases.
Ferritin expression was found to be decreased by c-myc, a proto-oncogene that is frequently aberrantly activated in malignancies, resulting in a shift from iron storage to “free” iron available for cell growth and DNA synthesis (317). Thus, it is likely that ferritin levels are partly controlled by the de-regulated molecular signals which feature during oncogenesis. Furthermore, ferritin was found to play a role in the promotion of angiogenesis (57). It was shown in vitro and in human prostate cancer xenografts that ferritin inhibits high-molecular-weight kininogen (HKa), an endogenous inhibitor of angiogenesis, and, consequently, impedes the anti-angiogenic activity of HKa, independently of the iron content of ferritin (57). Thus, alterations in ferritin levels may facilitate tumor progression.
STEAP and its relationship to tumorigenesis
Since it is increasingly evident that there is a link between iron and cancer, it is no surprise that the expression of the STEAP family is also altered in tumors. STEAP was shown to be over-expressed in all stages of prostate cancer, while exhibiting restricted expression in normal human tissues (95, 129, 320). STEAP was also reported to be expressed in a variety of other cancer types, such as colorectal (132), renal (10), lung (46), bladder cancer (10, 46), hormone-dependent murine mammary carcinoma (104), and in various human cancer cell lines, such as bladder, colon, ovarian, pancreatic, and Ewing sarcoma (129). STEAP3-expression was related to the increased intracellular iron storage of Raji Burkitt's lymphoma and HeLa cervical cancer cells compared with their control transfectants (132). Furthermore, STEAP3 over-expressing human Raji cells were shown to be resistant to iron deprivation-induced apoptosis (132). Additionally, STEAP3-over-expressing cells transplanted in mice were still able to grow even under hypo-ferric conditions compared with control mock transfectants, providing cancer cells with a survival advantage (132). Therefore, the discovery of STEAP3 is expected to enhance our understanding of the relationship between iron and cancer.
Tumor-associated macrophages
Cancers are composed of more than just neoplastic cells; they also consist of non-neoplastic stromal cells, including tumor-associated macrophages (TAMs) (282). TAMs have been shown to be involved in the promotion of tumor proliferation, angiogenesis, and metastasis (282). Macrophages play an essential role in iron homeostasis, recycling iron from the heme of hemoglobin (69). It is becoming evident that TAMs closely resemble the phenotype of alternatively activated (M2) macrophages (273). Studies have demonstrated that M2 macrophages have an altered iron-handling state, having beneficial implications for tumor cells (62, 238). In fact, M2 macrophages express lower levels of H chain ferritin as well as higher levels of TfR1, HO-1, Fpn1, and the macrophage scavenger receptor (CD163), which is responsible for hemoglobin uptake (62, 238, 259, 274). These macrophages were shown to possess enhanced iron uptake and release relative to control macrophages (62, 238). Furthermore, incubation of the human renal cell carcinoma cell line, RCC10, in M2 macrophage-conditioned media was shown to increase RCC10 proliferation, suggesting that M2 macrophages modulate cancer growth (238). Moreover, the media of M2-polarized macrophages from a patient with Fpn1 disease, characterized by a loss-of-function mutation in the Fpn1 gene, led to no significant growth changes in RCC10 cells (238). This may suggest that Fpn1 function, and, thus, iron efflux from macrophages was necessary for the promotion of tumor cell growth (238). Consequently, the distinct iron-handling phenotype of the M2 macrophages may allow them to supply iron to tumor cells, promoting tumor cell multiplication (238). A better understanding of TAMs and their interactions with cancer cells is required to design more efficacious cancer therapies based on iron metabolism.
Other biologically relevant metals in neoplasia
Similar to iron, other biological metals such as copper, zinc, and manganese are essential for metabolic processing (60, 292), and their levels are extensively regulated to prevent metal toxicity. For instance, copper can also cause oxidative damage through ROS formation via Fenton-like chemistry (292). “Free” copper is tightly controlled and is transported in the plasma bound to albumin and ceruloplasmin, while excess copper is stored by intracellular metallothioneins (292). As evident with iron, there is also support for alterations in the normal homeostasis of other biological metals in cancer (303, 326). Therefore, the link between copper, zinc, manganese and other metals in cancer is briefly reviewed next.
Copper
Copper is incorporated into proteins, such as cytochrome c oxidase and superoxide dismutase, which are involved in mitochondrial electron transport and anti-oxidant defense, respectively (292). Copper also appears to be a co-factor for angiogenesis, the process involved in the formation of new blood vessels (223) [see review (87)], and excess copper can produce ROS through a copper-catalyzed, Fenton-like reaction (140, 178). Similar to iron, copper is a redox-active metal that can contribute to lipid peroxidation as well as DNA damage (27).
Elevated serum and tissue copper levels have been discovered in several cancer types, including breast, colon, cervical, lung, ovarian, prostate, stomach cancer, and leukemia, in male and female patients, in different age groups, and in various geographical locations (112). Additionally, studies in breast cancer patients have also positively correlated high copper levels with advanced stages and progression of malignancy (163). Several copper chelators, which limit copper bioavailability and inhibit angiogenesis, have entered clinical trials, such as tetrathiomolybdate (TM) and penicillamine [for reviews, see (112, 284)]. In a phase I clinical trial in patients with metastatic solid tumors, TM treatment reduced copper levels with low toxicity and stabilized disease in five out of six patients (28). In phase II clinical trials in patients with advanced renal disease, TM was able to reduce copper levels and was well tolerated, although its anti-cancer activity was limited to the stabilization of disease for a median of 34.5 weeks (239).
Zinc
Zinc acts as a co-factor in enzymes, such as carbonic anhydrase, alcohol dehydrogenase, and carboxypeptidase, which are required for carbon dioxide regulation and the metabolism of alcohol and proteins, respectively (60). Zinc also serves as a structural ion of various transcription factors, such as the tumor suppressor p53 (60, 181). It is also well known that zinc is a metal ion that plays an important function in the active site of matrix metalloproteinases (MMPs), for example, MMP-2 and MMP-9, which play a crucial role in metastasis (103). Unlike iron and copper, zinc is not a redox-active metal, and, therefore, does not participate in Fenton-like reactions (60).
High concentrations of zinc are present within prostate epithelial cells (65, 91, 333). However, prostate cancer cells possess abnormally reduced zinc levels relative to nonmalignant prostate cells (84, 91, 114, 333). This may be a result of the epigenetic down-regulation of the human zinc uptake transporter (hZIP1) gene in primary prostate malignant cells (91). Under normal prostate zinc levels, zinc inhibits citrate oxidation, while in the zinc-deficient malignant state, citrate oxidation is increased and activates the Krebs cycle, resulting in a metabolic transformation that provides an increased energy source for cancer growth [see review (65)].
Zinc has also been shown to alter the activity of telomerase (208), an enzyme which is frequently activated in cancers that allows the unlimited proliferation of cancer cells (193). Zinc has been shown to enhance telomerase activity in renal cell carcinoma (NRC-12) and human prostatic cancer (DU145) cell lines, although the exact mechanisms of this activation remain unclear (208). Both an excess and a shortage of zinc results in p53 mis-folding, rendering the tumor suppressor dysfunctional, making p53 reliant on metal homeostasis (181). Furthermore, serum zinc levels were decreased in breast cancer patients, while iron and copper serum levels were significantly increased compared with control patients (163). These elements, specifically zinc, copper, selenium, and iron, were found to exist at higher levels within malignant tissue compared with adjacent normal tissue in cancer patients (163).
Manganese and other metals
Other trace elements, including manganese and cobalt, are essential for normal metabolic functioning (192, 303). In fact, one-third of all proteins require metals, whether it may be for structural or catalytic purposes (192). For instance, a well-known manganese(III)-containing enzyme, mitochondrial superoxide dismutase, catalyzes the dismutation of superoxide radicals into water and oxygen, making it an essential defense system against oxidative stress (192). Additionally, manganese is also found in several other metalloproteins that are necessary for normal reproduction, immune function, and energy metabolism (192). Furthermore, although cobalt is present in very small trace amounts, the element is a key constituent in vitamin B12 (251). In contrast to these vital metals, other heavy metals, such as arsenic, lead, and cadmium, are nonessential and are known to be carcinogenic (303).
The development of cancer has also been associated with aberrations in several metals. Studies have demonstrated that malignancies possess various changes in the levels and ratios of metals, including manganese, chromium, scandium, copper, iron, and zinc (100, 101, 267, 332). For instance, high levels of iron, nickel, chromium, zinc, cadmium, mercury, and lead were evident in breast cancer tissue compared with healthy controls (131). Moreover, a study demonstrated a reduction in the levels of manganese, nickel, zinc, titanium, selenium, and chromium in the serum of breast cancer patients compared with healthy controls, while in contrast, an elevation in iron and copper was evident (255). Consequently, an interrelationship between metal levels is likely to exist in cancer, particularly as certain metal pathways overlap and metals are able to “hijack” particular specialized transport pathways (292). For example, the duodenal enterocyte transporter, DMT1, will not only transport dietary iron, but may also transport manganese, copper, and certain nonessential metals, such as cadmium and palladium, which have no specific biological function (12, 192). Furthermore, metals, such as manganese(III) and aluminium(III), can form a complex with Tf, resulting in the cellular uptake of the metal (192). Therefore, it is no surprise that there is a crossover between metal metabolism in cancer. For example, copper and cobalt may replace zinc in the zinc-finger DNA-binding domain of the estrogen receptor, resulting in a malfunctioning protein and a signal transduction that may be carcinogenic (228). These metals can enter cells and compete for binding sites in a process termed molecular mimicry (12, 192). Thus, alterations in the levels of metals can result in changes in metal-dependent enzymatic activity, producing direct or indirect effects on tumor growth. Therefore, metal homeostasis within the mammalian organism is essential and is likely to be disrupted in cancer. For further information on metal homeostasis, the reader is referred to a recent review (192).
Chelators As Metal Disrupting Chemotherapeutic Agents
The de-regulation of iron and metal homeostasis in cancer cells has led to the discovery that compounds which can bind metal ions, namely chelators (Table 1), have anti-cancer activity (35, 36, 143, 220). Some of these ligands have gone through extensive structure-activity relationship studies in an effort to generate chelators with marked anti-tumor efficacy and low toxicity (16, 48, 64, 184, 247, 295, 313, 331). Several mechanisms are believed to be responsible for their cancer activity, including metal depletion or modulation, inhibition of RR, redox activity, apoptosis, and cell-cycle modulation. Next, an overview is presented on the evolution of iron chelators from desferrioxamine (DFO), one of the first iron chelators that demonstrates the inhibition of cancer cell growth, to the more potent thiosemicarbazone ligands.
Depending on the coordination of iron by deferiprone.
DFO, desferrioxamine; PIH, pyridoxal isonicotinoyl hydrazone; RR, ribonucleotide reductase; NF-κB, nuclear factor-κB; mTOR, mammalian target of rapamycin; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related protein; Dp44mT, di-2-pyridylketone 4,4,-di-methyl-3-thiosemicarbazone; NDRG1, N-myc downstream regulated gene 1; DpC, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone hydrochloride; Bp44mT, 2-benzoylpyridine 4,4-dimethyl-3-thiosemicarbazone.
Desferrioxamine
DFO (Desferal®; Fig. 6A) was the first chelator that was introduced for the widespread clinical treatment of iron-overload disorders, such as β-thalassemia (45). It is a naturally occurring chelator produced by the bacterium, Streptomyces pilosus (45). Although the ligand can bind to various metals, it has a particularly high affinity for ferric iron (45). This hexadentate chelator binds iron in a 1:1 ratio at its oxygen atoms, forming a stable complex (45), preventing the direct access of hydrogen peroxide (H2O2) or oxygen to the iron center (143). Therefore, this complex prevents iron from participating in the Fenton reaction, alleviating symptoms of oxidative damage evident in iron-overload diseases (143).
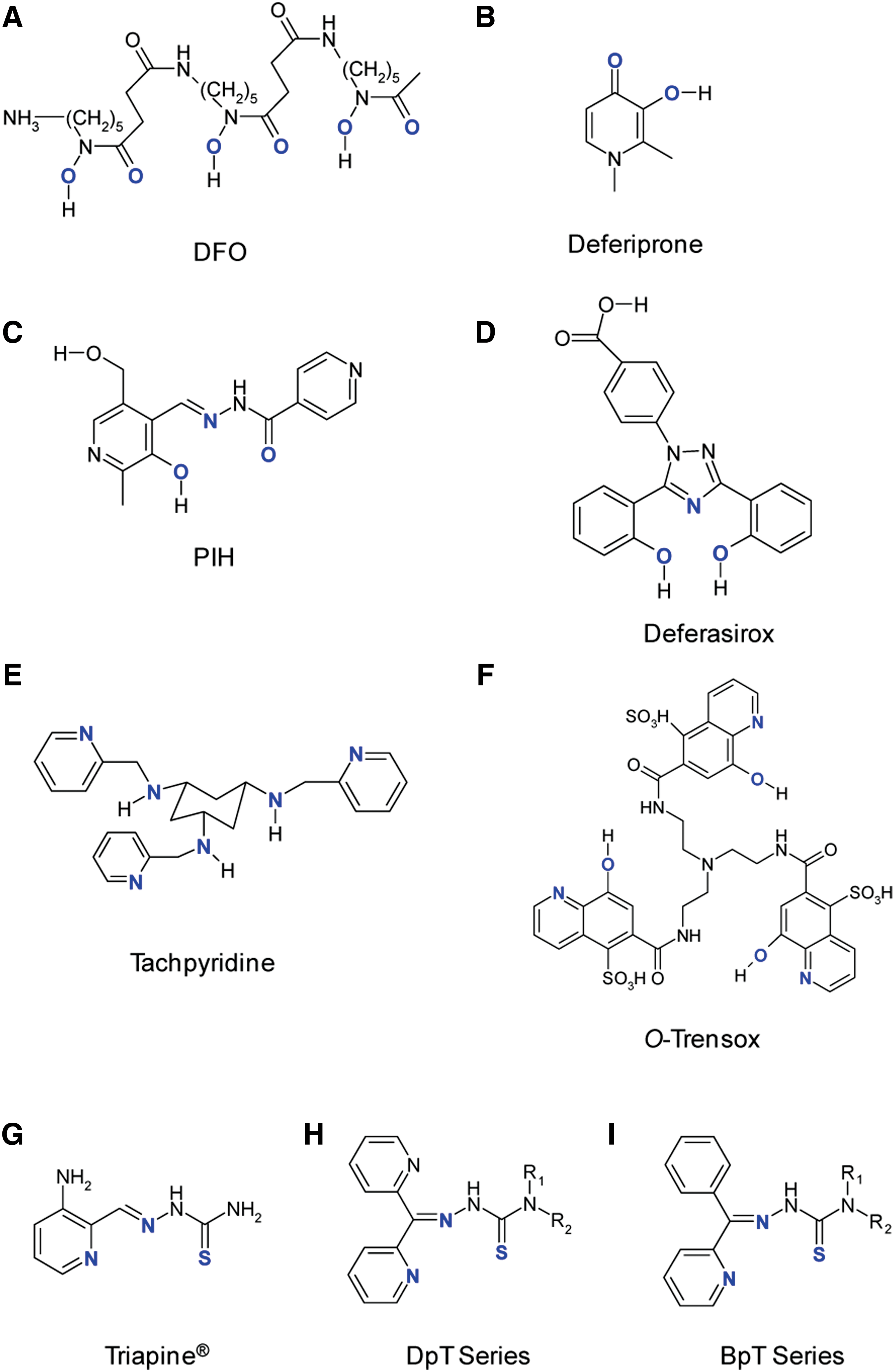
In addition to its clinical application in iron-overload disease, numerous studies have shown that DFO possesses anti-cancer activity in vitro, in animal models and in clinical trials. DFO has been shown to possess anti-proliferative activity in various cancer cell lines, including neuroblastoma, cervical carcinoma, ovarian, and human myeloid leukemia (17, 23, 26, 277). Additionally, the anti-proliferative activity of DFO was shown to be completely or, at least, partially reversible on co-treatment with iron salts or iron-saturated DFO, indicating that cellular metal binding is an important component responsible for its anti-tumor activity (17, 166). Furthermore, DFO caused an increase in TfR1 expression and a decrease in ferritin levels, demonstrating the presence of the well-known homeostatic regulatory mechanisms that attempt to maintain iron homeostasis (32). It has also been demonstrated that DFO causes a decrease in DNA synthesis, which was also reversed on the administration of iron salts (17, 277). DFO causes cell-cycle arrest at the G0-G1 phase (24, 32, 277), up-regulates p53 (172), down-regulates cyclin D1 (211), and induces apoptosis in ovarian cancer, Kaposi's sarcoma, cervical carcinoma cells (277), neuroblastoma, and leukemia cell lines (26, 80, 93, 278).
Studies examining DFO as a chemotherapeutic agent in animal models and in clinical trials showed promising anti-cancer effects (72, 73, 78, 118, 311). In a recent study conducted on 10 patients with chemo-resistant advanced hepatocellular carcinoma, DFO treatment produced a partial response in two of the patients and stabilized the disease in three patients (319). In one partially responsive patient, liver tumor and lung metastasis were no longer observed on CT scans 2 months after treatment, and the patient survived for a further 20.9 months (319). Conversely, DFO treatment was not beneficial to the remaining five patients, and their disease progressed (319).
Despite a number of positive results, several animal and human trials failed to produce a response to DFO treatment and have reported toxic side-effects (22, 151, 263). A number of limitations are thought to explain these inconsistent results. First, the highly hydrophilic nature of DFO results in poor membrane permeability and renders the drug orally inactive (39). Second, the short plasma half-life (t1/2=5–10 min) of DFO and its rapid metabolism leads to low efficacy (8, 215, 288). To overcome this latter problem, the administration of DFO requires inconvenient prolonged subcutaneous infusion (10–12 h/day, 5–6 times/week), but unfortunately, leads to poor patient compliance (45, 157). These disadvantages of the agent and the conflicting studies have encouraged the design of other, more potent chelators for clinical use.
Deferiprone
Deferiprone (l,2-dimethyl-3-hydroxypyrid-4-one, L1, CP20, Ferriprox®; Fig. 6B) is a synthesized ligand that was introduced as the first orally active iron chelator for the clinical treatment of iron-overload disease (15). Deferiprone chelates metal ions as a bidentate chelator, with a particularly high affinity for iron(III) (270). However, copper(II) and aluminium(III) ions can compete for the metal-binding site of the chelator (270). Deferiprone binds metals by two neighboring oxygen atoms, namely a carbonyl and hydroxyl group (15). Deferiprone is clinically available for the treatment of thalassemia major in Europe, Africa, South America, and Asia, but is not available in Canada and the United States due to conflicting scientific data and debatable side-effects (83). Deferiprone is an inexpensive alternative for patients who do not tolerate long subcutaneous infusions of DFO, and it demonstrates considerable activity (15, 156).
As a part of its ability to chelate iron, deferiprone has anti-proliferative and cytotoxicity activity in several human tumor cell lines, namely oral squamous cell carcinoma (HSC-2 and HSC-3) (322), Kaposi's sarcoma cells (278), neuroblastoma cell lines (CHP 100 and CHP 126) (24), HepG2 hepatocellular carcinoma (52), cervical carcinoma cells harboring human papilloma virus (HeLa and SiHa) (277), and promyelocytic leukemia (HL-60) cells (322). There is also evidence that deferiprone has a higher cytotoxicity in cancer cells compared with normal non-neoplastic cells such as human gingival fibroblasts, pulp cells, and periodontal ligament fibroblasts (322). Additionally, it appears that not all cancer cell types are sensitive to hydroxypyridone chelators. For instance, the human submandibular gland carcinoma is apparently resistant to the anti-proliferative effects of deferiprone, as well as other hydroxyketone chelators (322). Deferiprone was shown to induce apoptosis as assessed via flow cytometry, the terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay, and morphologically in Kaposi's sarcoma cells (278), HeLa and SiHa cells (277), caused DNA fragmentation in HSC-2 and HL-60 cells (322). This effect occurred more rapidly after deferiprone treatment than DFO, probably due to the greater lipophilicity and membrane permeability of the former agent (278). Additionally, it was demonstrated that deferiprone causes the activation of caspase-3, -8, and -9 in HSC-2 and HL-60 cells (322). It was also observed that similar to DFO, deferiprone causes cell-cycle arrest in human cancer cells (24, 52, 277), increasing the proportion of cells in the G0-G1 phase and decreasing the cells in the S-G2-M phase in neuroblastoma cells (24). Furthermore, deferiprone was observed to inhibit thymidine incorporation, and, therefore, inhibit DNA synthesis (52, 277). The addition of exogenous iron inhibited deferiprone-induced apoptosis (277, 278), cytotoxic activity (24, 322), cell-cycle arrest (24, 277), DNA fragmentation, and caspase activation (322).
Despite evidence of in vitro anti-cancer activity and iron-depletion, neuroblastoma (IMR-32 and JBN-1) xenografts in nude mice receiving deferiprone treatment possessed similar tumor sizes and growth relative to vehicle-treated mice (263). Furthermore, treatment with deferiprone did not inhibit the growth of cervical carcinoma xenografts in nude mice (276). Along with these disappointing in vivo results, several side-effects have been observed in patients receiving deferiprone treatment, including agranulocytosis, neutropenia, arthralgia, and gastrointestinal effects (44, 58) [see review (156)]. It was also reported that long-term deferiprone treatment in thalassemia patients aggravates liver fibrosis (216). However, the study reported limitations to their findings and indicated that patients within this group were older and more likely to experience fibrotic progression (216). Additionally, the possible inclusion of patients infected with hepatitis C within the study could have markedly affected the conclusion (216). Other long-term clinical trials did not observe the progression of liver fibrosis in deferiprone-treated patients (189, 312). Additionally, it has been suggested that deferiprone produces mutagenic responses in mouse lymphoma L5178Y cells, potentially favoring the formation of aggressive clones (314).
Several factors may contribute to the toxicity of deferiprone. For instance, high doses of deferiprone are necessary to compensate for the considerable fraction of deferiprone that undergoes rapid phase II metabolism through glucuronidation (280). Additionally, ROS generation may be involved in deferiprone toxicity. The major species formed at a pH of 7 at micromolar concentrations of deferiprone is the 3:1 deferiprone-iron complex that completely occupies the coordination sites of iron, preventing ROS formation (205). However, in very dilute solutions, this complex dissociates to form the 1:1 and 2:1 chelator-iron complexes, resulting in the incomplete coordination of iron that allows for ROS generation (66, 71, 205). These investigations on the redox activity of deferiprone have been extended to human studies. Indeed, patients with β–thalassemia/hemoglobin E treated with deferiprone demonstrate electron paramagnetic resonance (EPR) signals of the ascorbyl radical and DMPO-carbon-centered adduct in their sera, suggesting the incomplete coordination of iron by deferiprone in vivo, resulting in ROS generation (138). In contrast, other studies showed that the 2:1 deferiprone:iron(III) complex was more effective at scavenging ROS than the uncomplexed chelator, as it was effective at preventing hypoxia-reoxygenation injury (204). These variable results demonstrate that deferiprone acts as a pro-oxidant or protective anti-oxidant, depending on the ligand-to-iron ratio and the proportion of the species present.
Several deferiprone analogs and pro-drugs have been produced in an effort to improve the problematic properties encountered with deferiprone (19, 59, 109, 123, 318). Certain derivatives have also been assessed for their anti-cancer activity (18, 96, 109). For instance, bicyclic deferiprone analogs were shown to possess anti-leukemic activity against BCR-ABL-expressing human leukemia cells (109). Additionally, a polyamine conjugate of deferiprone was shown to be 230 times more effective than deferiprone at inhibiting the growth of L1210 murine leukemia cells (18). It remains to be seen whether these deferiprone derivatives with an improved anti-proliferative efficacy will be useful for cancer therapy.
Pyridoxal isonicotinoyl hydrazone
The synthetic aroylhydrazone, pyridoxal isonicotinoyl hydrazone (PIH; Fig. 6C), was initially synthesized by Sah in 1954, and was stated to demonstrate anti-cancer activity against leukemia and mammary carcinoma in mice, although the results were not directly shown (253). PIH is a tridentate ligand synthesized via the Schiff base condensation of pyridoxal and isonicotinic acid hydrazide (226). In fact, PIH binds iron by the imine nitrogen, phenolic oxygen, and carbonyl oxygen donor atoms in a 2:1 ligand:iron ratio (29, 227). Similar to DFO, PIH has a high affinity for ferric iron, while a low affinity for ferrous iron (306). The binding of other metals by PIH has also been examined, with potentiometric titrations with magnesium(II), zinc(II), and calcium(II) demonstrating a markedly lower affinity than iron(III) (243).
Numerous in vitro investigations have examined PIH as an alternative chelator to DFO for the clinical treatment of iron-overload disease. PIH was shown to be far more effective than DFO at preventing cellular iron uptake from Tf and at increasing cellular iron release from prelabeled reticulocytes, fibroblasts, Chang cells, hepatocytes, and human neuroblastoma cells (11, 226, 235, 247). The ability of PIH to mobilize intracellular iron and decrease cellular iron uptake from Tf demonstrated the potential of PIH to act as an effective iron-chelating agent. The high iron-mobilizing activity of PIH was believed to be attributed to the lipophilic nature of the ligand, and, thus, its high permeability across the cell membrane in comparison to DFO, which is relatively hydrophilic (76, 247). These findings demonstrating the high membrane permeability of PIH were also explained by its pKa values, which demonstrated that a significant proportion of ligand (∼80%) is present as a neutral species at pH 7.4 (248). Additionally, the pKa values reveal that the ligand has a neutral charge predominately (∼100%) at pH 6 (248). Given that the small intestine has a pH range of 5–7, maximal absorption of the neutral ligand would occur through the intestine, allowing PIH to be orally administered (29, 248). These results indicated that PIH could be an orally effective candidate for replacing DFO, particularly with regard to the problematic subcutaneous administration route of DFO.
Interestingly, although PIH was shown to be more efficient at mobilizing iron, PIH exhibited lower anti-proliferative activity than DFO in the SK-N-MC neuroepithelioma cell line (PIH: IC50=75 μM; DFO: IC50=22 μM) (247). Consequently, these results demonstrated that the anti-proliferative activity of chelators does not depend solely on the ligand's ability to directly interact with iron. Hence, the low efficacy of PIH as an anti-cancer drug suggests that it had greater promise for the treatment of iron-overload disease than as a cytotoxic drug. This was further emphasized by the fact that PIH possessed anti-oxidant activity, a critical feature which is essential to alleviate the symptoms of iron overload (31, 143). In addition, PIH has also been shown to protect DNA against in vitro oxidative stress (121). This was considered to occur, because PIH binds iron, thereby decreasing the levels of “free” iron available to catalyze the Fenton reaction that mediates hydroxyl radical formation (121).
In vivo studies with PIH were also performed to ascertain whether effective iron mobilization could be observed in animal models. Oral and intraperitoneal treatment with PIH increased biliary, urinary, and fecal excretion of iron from rats (56, 125, 227). This occurred within 15 min of its intraperitoneal administration, and repeated treatment with PIH also resulted in a decrease of liver and kidney iron (56). Interestingly, PIH did not increase the excretion of copper or zinc despite forming in vitro complexes with these metals (56, 243). Furthermore, orally administered PIH induced iron excretion to an extent that was comparable to the intraperitoneal treatment of DFO (122). Consequently, the oral availability of PIH offers a more convenient route of administration than the painful subcutaneous method used for DFO. Additionally, no adverse effects were observed with PIH treatment during toxicity studies in rats and guinea pigs, which involved the assessment of blood counts, renal function, serum proteins, and liver enzymes (9, 30, 122).
Animal studies with PIH showed encouraging results, and, consequently, phase I clinical trials of PIH were performed. This chelator caused iron excretion with no evidence of toxicity, including no ophthalmologic abnormalities (30). Nevertheless, despite its success in terms of safety, PIH was not able to cause sufficient iron excretion in thalassemic patients, although it was suggested that the dosing regimens used were low and required optimization (30).
O-Trensox
O-Trensox (tris-N-(2-aminoethyl-[8-hydroxyquinoline-5-sulphonato-7-carboxamido])amine; Fig. 6F) is a hexadentate ligand that binds iron in both the ferric and ferrous oxidation states in a 1:1 iron:chelator ratio (264). The ligand binds metals with three “hard” phenolate oxygen donors and three “soft” pyridine nitrogen donor atoms (Fig. 7A). O-Trensox has a much greater affinity for iron(III) than any other biological metals (20). The chelator complexes most efficiently with metals in the following order: iron(III) >> copper(II)>zinc(II)>aluminium(III)>iron(II)>calcium(II) (20).
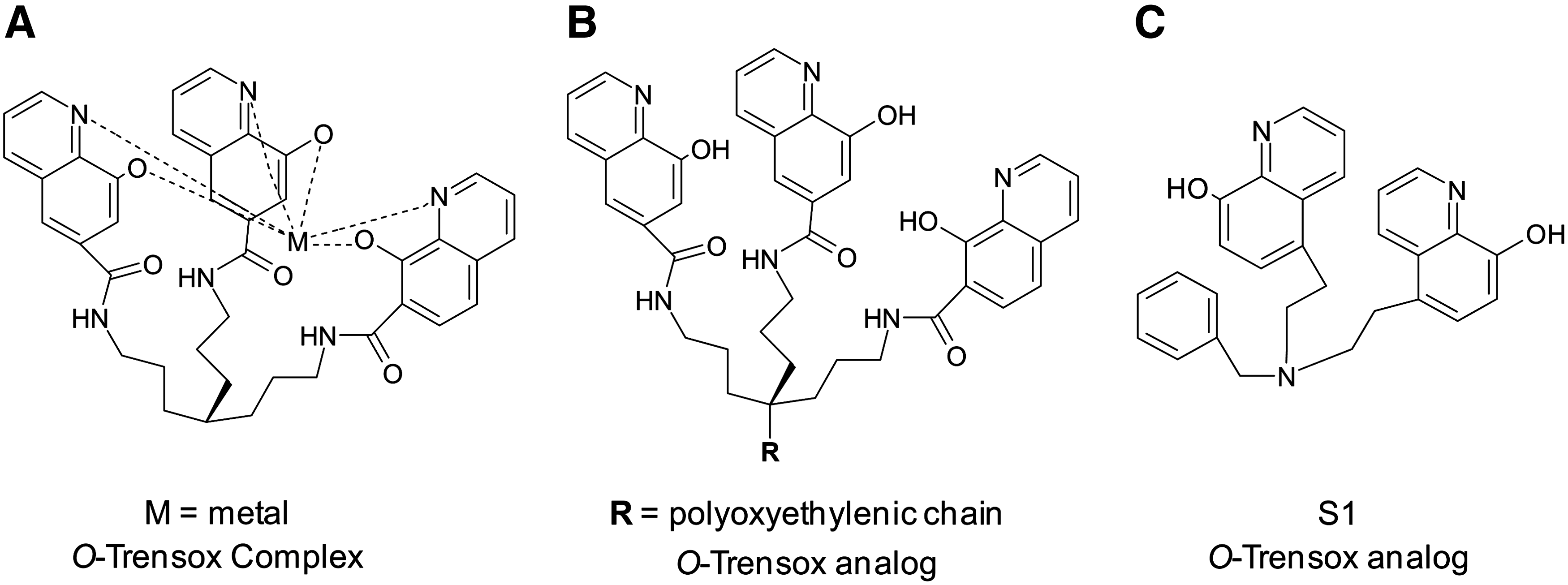
It has been shown that DFO is more effective than O-Trensox at mobilizing iron from ferritin in a cell-free system and from hepatocytes (233). On the contrary, O-Trensox was moderately more effective than DFO at decreasing hepatic iron levels in vivo in iron-overloaded rats (233). O-Trensox was able to protect hepatocytes from the toxic effects of iron, including decreasing lipid radical adduct generation (233). Additionally, O-Trensox reduced the levels of hydroxyl radicals in a cell-free system more efficiently than DFO, as monitored by EPR (233). However, the redox potential of the iron complex is 0.087 V, suggesting a tendency to be reduced in the range accessible to biological reductants (42). Nevertheless, the iron(III) complex of O-Trensox was not photo-reducible (42). While the positive controls, iron-EDTA and iron-citrate, induced marked radical damage to DNA in the presence of ascorbate, this was not observed with O-Trensox iron complexes (42). Thus, the O-Trensox complex, despite its redox potential, was protective against the toxic redox cycling of iron, preventing pro-oxidant-induced cell injury (42, 233).
The protective property of O-Trensox, along with its ability to mobilize hepatic iron, prompted research into its activity in liver cancer, a cancer type that is associated with iron overload (49, 133). O-Trensox inhibited cell proliferation and DNA synthesis in four cell culture models, as well as induced apoptosis through caspase activation, DNA degradation, and nuclear fragmentation in human hepatoblastoma HepG2 cells (97, 234). O-Trensox also increased the number of cells in the G1 and G2 phase and inhibited mitotic activity compared with untreated cells (234). These effects were reversible and could be inhibited on removal of the chelator, as well as with the addition of iron and zinc to the medium, suggesting that metal binding is significant to its cytotoxic activity, or, alternatively, the chelator complex is not membrane permeable (234).
Although O-Trensox was able to reduce DNA synthesis, induce apoptosis, and inhibit neoplastic growth to a greater extent than DFO (234), more recent chelators have since proved to be more effective agents (48). For example, desferasirox (see desferasirox section) has since been shown to possess superior anti-neoplastic activity relative to O-Trensox (48). Therefore, it is unlikely that O-Trensox will undergo further preclinical evaluation as an anti-tumor agent.
Appropriate chemical modifications have also been performed to O-Trensox in order to achieve a suitable hydrophilic-lipophilic balance, while maintaining affinity for iron(III) (130). A polyoxyethylenic chain of various lengths was inserted onto the scaffold of O-Trensox that connects the hydroxyquinolines (Fig. 7B) (130). However, the biological activity of these hydroxyquinoline analogs did not correlate with the hydrophilic-lipophilic balance (130). More recently, the analog, bis-N-(8-hydroxyquinoline-5-ylmethyl)benzylamine (S1; Fig. 7C) was synthesized with only two 8-hydroxyquinoline moieties, and its in vitro anti-neoplastic activity was assessed (170). S1 was shown to inhibit cellular proliferation and DNA synthesis in the presence of iron to a greater extent than O-Trensox in the human hepatoma HepaRG cell line (170). Consequently, S1 was deemed by the authors to be a superior anti-tumor ligand than its parent compound, O-Trensox (170). It remains to be seen whether these O-Trensox analogs will have in vivo anti-cancer activity and a role in future cancer chelation therapy.
Deferasirox
Deferasirox (Exjade®, Novartis; Fig. 6D) is a tridendate chelator that has a high affinity for ferric iron and chelates iron in a 2:1 ligand-to-iron ratio (173). Deferasirox received the U.S. Food and Drug Administration approval in 2005 for the treatment of iron overload caused by blood transfusions (173). Similar to Deferiprone, Deferasirox is orally active, has a half-life of 7- to 18 h, and is administered once daily (173). Consequently, deferasirox is becoming increasingly used for the clinical treatment of iron overload, as it provides an alternative to the conventional onerous and prolonged infusion of DFO (173).
Given that previous clinically employed iron chelators have been shown to possess anti-neoplastic activity, studies have also examined the efficacy of deferasirox in terms of its anti-tumor effects (48, 169, 214). Thus far, studies have demonstrated that deferasirox exerts growth inhibitory and apoptotic activity (48, 169, 198, 214). Deferasirox was shown to inhibit DNA replication and decrease cell viability in three leukemia cell lines, human HUH7 hepatoma cells, and rat FAO hepatoma cells (48, 169, 214). Deferasirox was also capable of inducing cell-cycle arrest in the G0-G1 phase, increasing the levels of fragmented DNA and caspase-3 activity, which is indicative of apoptotic cell death (48, 169, 214). Additionally, these apoptotic effects were not apparent after the iron saturation of the chelator, indicating that metal binding is essential for the induction of its anti-neoplastic effects (48, 169).
Furthermore, deferasirox caused a decrease in the levels of polyamines along with a concomitant reduction in mRNA levels of the enzymes responsible for polyamine biosynthesis, specifically ornithine decarboxylase and spermidine/spermine N-acetyltransferase (48, 169). There is also evidence that polyamines and their associated metabolic enzymes are required for proliferation to occur, and the inhibition of these proteins results in cell-cycle arrest and apoptosis (260, 262). Hence, deferasirox may exert its anti-cancer mechanisms through alterations in iron and polyamine metabolism (48, 169).
As evident with former chelators, a higher concentration of deferasirox was required to induce the same cytotoxic effects in normal primary cultured hepatocytes than in hepatoma cells, suggesting that deferasirox selectively targets cancer cells, resulting in less cytotoxicity to non-neoplastic cells (48). However, it is notable that based on the clinical profile of deferasirox and the side-effects of the drug (40, 41, 289), it is clear that deferasirox is not completely selective and is not exempt from nonspecific adverse drug interactions.
It is apparent that the anti-cancer activity of chelators does not occur solely through their ability to cause iron mobilization, but is also reliant on their complex effects on multiple intracellular targets that are critical for cellular proliferation. Deferasirox has been shown to up-regulate or down-regulate various genes associated with apoptosis and cell-cycle regulation in K562 human leukemia cells (214). It is of interest that deferasirox increased the expression of regulated in development and DNA damage response 1 (REDD1) at the mRNA and protein levels, resulting in a concurrent down-regulation of the mammalian target of rapamycin (mTOR) in leukemia cell lines (214). Notably, the mTOR pathway is known to be abnormally activated in cancers, resulting in the de-regulation of cell growth and proliferation, and mTOR inhibitors are considered to be therapeutically beneficial (47, 75, 79, 128). Thus, inhibiting the mTOR pathway by using deferasirox may, in part, lead to the inhibition of cancer progression.
Other studies have shown that this agent can also influence nuclear factor-κB (NF-κB) activity (198). NF-κB activation occurs when bound inhibitory subunits, known as inhibitors of κB (IκBs), are degraded [see review (99)]. The NF-κB complex subsequently translocates from the cytoplasm to the nucleus, where it regulates the transcription of genes that are involved in tumor promotion, angiogenesis, and metastasis (99). Deferasirox was shown to inhibit NF-κB activity through the sequestration of the complex within the cytoplasm (inactive state) in two leukemia cell lines, K562 and HL60 (198). Furthermore, the addition of the iron complex, ferric hydroxyquinoline, to K562 cells did not affect deferasirox-induced NF-κB inhibition, demonstrating that the effect of deferasirox on NF-κB activity occurs through mechanisms which are independent of iron binding (198). Moreover, another study demonstrated that tribbles homolog 3 (TRIB3), the negative regulator of NF-κB, was up-regulated in K562 leukemia cells after the incubation with desfersirox (214). Taken together, these results suggest that deferasirox affects the NF-κB pathway which is frequently aberrantly active in many cancers (99). Thus, NF-κB inhibition by deferasirox could be considered a valid therapeutic approach.
Considering that many chemotherapeutics are used in combination, studies have shown that the preincubation of K562 cells with deferasirox and subsequent incubation with the chemotherapeutic agent, etoposide, lead to an increase in the number of apoptotic cells in comparison to both drugs alone (198). These findings suggest that deferasirox enhances the activity of chemotherapeutics and has a potential beneficial application in combinational cancer therapy.
Currently, the anti-cancer activity of deferasirox has only been examined in a small number of in vivo studies. In a leukemia mouse model, deferasirox was shown to suppress tumor growth and induce apoptosis without causing any adverse effects (214). More recently, deferasirox was shown to induce complete remission in a patient with chemotherapy-resistant acute monocytic leukemia (94). This latter investigation was the first which demonstrated that the compound may possess anti-cancer activity in humans. Consequently, the full potential for deferasirox as an anti-tumor agent remains important to be further validated.
Tachpyridine
Tachpyridine (N,N',N''-tris(2-pyridylmethyl)cis,cis-1,3,5-triaminocyclohexane; Fig. 6E) is a hexadentate chelator with six nitrogen donor atoms that has undergone preclinical evaluation as an anti-cancer agent (295). Tachpyridine has been shown to bind iron(II), zinc(II), copper(II), calcium(II), magnesium(II), and manganese(II) (295). However, in aqueous solutions, magnesium, manganese, and calcium complexes are labile, whereas iron, zinc, and copper bind more strongly (295). In the presence of oxygen, once tachpyridine binds iron(II) in a 1:1 ratio, the ligand undergoes iron-mediated oxidative dehydrogenation, resulting in an inseparable mixture of mono- and diimino iron(II) complexes (Fig. 8A) (222, 334). Tachpyridine can be further completely oxidized by H2O2 into tris(imino) iron(II) complexes (222). Unfortunately, at present, very little is known about the anti-tumor activity of tachpyridine in vivo and, in particular, in hypoxic conditions of malignant tumors. Under these latter conditions, it can be speculated that the oxidative dehydrogenation reactions (222, 334) may not be as rapid. However, further studies under a hypoxic environment assessing the form of tachpyridine present in tumors after the administration of the ligand to cancer xenograft-bearing mice would be of interest.
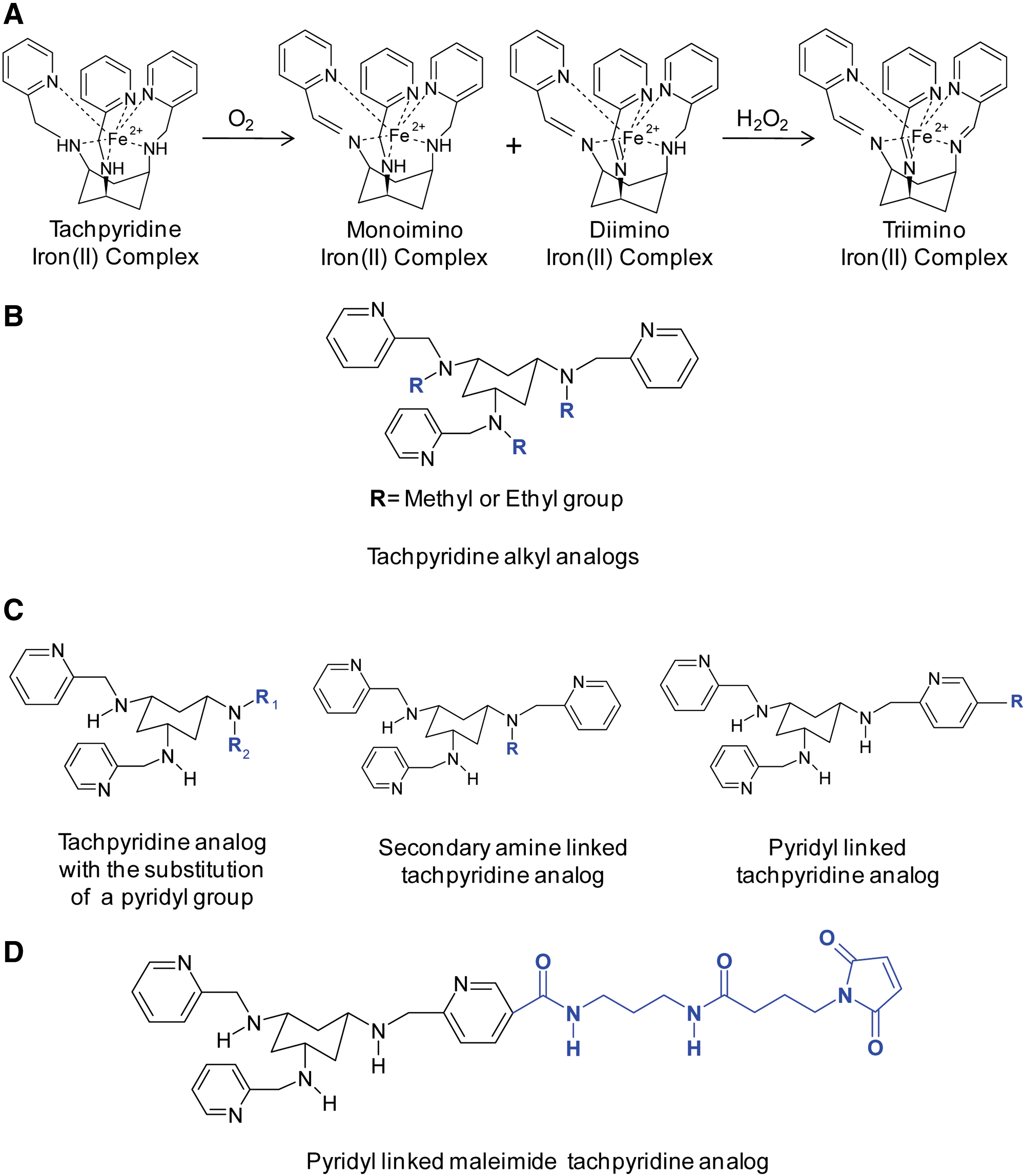
Tachpyridine can also bind iron(III) and reduce it to yield iron(II), as well as remove iron(III) from an iron(III) (ATP)3 complex, producing the same two iron(II) imino complexes (222). Additionally, the tachpyridine-induced reduction of bound iron(III) to iron(II) facilitates the production of ROS through the Fenton reaction (254). Therefore, unlike other chelators that provide protection against metal-mediated oxidative damage, the tachpyridine-iron complex is redox active, a consequence of its “soft” donor nitrogen atoms, rendering the complex pro-oxidative (254).
Tachpyridine exerts apoptotic activity, as assessed by cellular morphology, nuclear segmentation, and chromatin condensation and has greater anti-proliferative activity against several human cancer cell lines (IC50: 4.6–6 μM) relative to the modest activity of DFO (IC50: 70 μM) (2, 295, 323). Furthermore, a sevenfold higher concentration of tachpyridine was necessary to produce a similar toxic effect in normal MRC-5 fibroblasts, demonstrating that slow proliferating cells are less sensitive to the drug (295). The anti-cancer activity of the tachpyridine complexes was also examined (295). In these latter studies, the zinc, copper, and iron complexes were not cytotoxic, while the calcium, magnesium, and manganese complexes maintained the same cytotoxicity as the ligand alone (295). These results were consistent with the various complexes of calcium, magnesium, and manganese dissociating in aqueous solution (295, 334). In contrast, zinc, copper, and iron remain bound to tachpyridine and are present at detectable levels on incubation of the ligand alone in vitro (295, 334). These results also suggest that tachpyridine principally targets iron, copper, and zinc, and this binding is necessary for cancer cell cytotoxicity (295). Supporting these findings, it should be noted that the preincubation of cells with either zinc or iron inhibited tachpyridine-induced cytotoxicity (334). Additionally, an alkylated derivative of tachpyridine (Fig. 8B) with hindered metal binding did not possess cytotoxic activity, suggesting that intracellular metal binding was crucial for the anti-proliferative efficacy of this agent (295). Moreover, tachpyridine inhibits ferritin synthesis in bladder cancer cells, similar to DFO, without a generalized inhibition of protein synthesis, confirming its interaction with cellular iron (295).
Studies aimed at understanding the mechanisms underlying the anti-cancer activity of tachpyridine revealed similarities and differences between various structurally unrelated chelators. Unlike the majority of iron chelators that cause G1/S phase arrest (32, 277), tachpyridine triggered cell-cycle arrest at the G2 phase, whereas N-methyl tachpyridine, a derivative with hindered metal binding, did not mediate cell-cycle arrest (302). This demonstrated that metal binding was mandatory for tachpyridine-induced G2 phase arrest (302). Further studies suggested that the mechanism responsible for this arrest was through the activation of ataxia telangiectasia mutated/ataxia telangiectasia and Rad3-related protein (ATM/ATR), which subsequently phosphorylates p53 and the cell-cycle checkpoint kinases (CHK), CHK1 and CHK2 (302). However, tachpyridine was equally effective at inhibiting growth, causing G2-cell cycle arrest and inducing apoptosis in cancer cells with null, mutant, and wild-type p53 (2, 302). In MCF-7 breast cancer cells, tachpyridine caused an increase in p53 protein, although it did not lead to a concomitant increase in p21 expression (2). Considering that p21 is a downstream target of p53, it can be suggested that the augmented increase in p53 may not be transcriptionally competent (2). Of direct relevance, similar observations have been found with other iron chelators (172). Collectively, these studies indicate that tachpyridine triggers cell death through a p53-independent apoptotic mechanism (2). Importantly, these studies indicate the potential clinical usefulness of tachpyridine, particularly considering that most tumors have p53 mutations which render certain common chemotherapeutic agents ineffective (2).
Additionally, since tachpyridine induced apoptosis in various cell lines (2), its influence on the well-known apoptotic pathways were studied. Tachpyridine was found to induce apoptosis in HeLa cells through the activation of caspases-3, -8, and -9 with the instigation of the mitochondrial caspase pathway being more vital for cell death than the Fas-associated protein with the death domain and the death receptor signaling pathway (108). Notably, preincubation with zinc or copper or N-alkylated tachpyridine was unable to induce cell death and activate caspases, confirming that apoptosis induction by tachpyridine was dependent on metal binding (108, 334). This chelator was also able to increase the sensitivity of HTC 116 human colon carcinoma cells to ionizing radiation in a manner independent of cellular p53 status (302). In contrast, tachpyridine did not augment the sensitivity of noncancer cells (302).
Analogs of tachpyridine have also been synthesized in order to produce superior cytotoxic agents (53, 54). Several tachpyridine derivatives have incorporated a linker to allow the conjugation of a monoclonal antibody to the chelator, in order to enhance the selective targeting of cancer cells (53, 54). This linker was designed to incorporate a maleimide group that can undergo Michael addition with a thiol group and allow the conjugation of thiolated monoclonal antibodies (53, 54). In an effort to create this linker analog, a pyridyl group of tachpyridine was substituted with an alternative side chain, for example, a propyl group or hydrogen (Fig. 8C) (53, 54). This resulted in a reduction in cytotoxicity as compared with tachpyridine, demonstrating that the pyridyl ring was necessary for the anti-cancer activity of the tachpyridine analogs (53, 54, 323). Furthermore, the replacement of the secondary amine for an alternative side chain similarly reduced cytotoxicity relative to tachpyridine (54). Thus, both the pyridyl group and the secondary amine that directly participate in metal binding are vital for the maintenance of the cytotoxic activity of these analogs (54, 240). However, the addition of a side chain onto the pyridyl ring had little effect on the cytotoxic activity as compared with tachpyridine (54). Among these analogs, the tert-butoxycarbonyl-protected derivative possessed the greatest cytotoxic activity, and a maleimide linker was created to allow for the conjugation of antibodies without compromising cytotoxicity (Fig. 8D) (54). An important aspect, which has not been reported thus far, is the validation of the therapeutics potential of tachpyridine or its analogs as anti-cancer drugs using tumor animal models.
Thiosemicarbazones
In an effort to develop increasingly active anti-cancer agents, the thiosemicarbazone class of iron chelators was synthesized (7, 143, 202, 256). These chelators have a high affinity for a variety of transition metals, such as iron(II), copper(II), cobalt(II), gallium(III), zinc(II), and manganese(II) (7, 143, 202, 256). Similar to the aroylhydrazones, thiosemicarbazones bind metal ions in a tridentate manner. Although there is an abundance of literature available on a variety of thiosemicarbazones, this section will only focus on the investigation of relevant α-pyridyl thiosemicarbazones as anti-neoplastic agents. These tridentate thiosemicarbazones bind metals through a sulfur and two nitrogen donor ligating atoms and can form an octahedral iron complex in a 2:1 manner (327).
The originally proposed thiosemicarbazone molecular target: RR
Traditionally, the anti-proliferative activity of thiosemicarbazones was attributed to the inhibition of RR (3, 256). However, the exact mechanism responsible for the inhibition of enzyme activity remains unclear. Initial investigations proposed that thiosemicarbazones inhibited RR through the binding of iron at the active site in the R2 subunit of the enzyme, preventing enzyme activity (203, 257). Conversely, additional reports have suggested that the inhibition of RR occurs due to the depletion of iron pools required to supply iron for enzyme activity (61, 106). Interestingly, it has been demonstrated that the thiosemicarbazone-metal complexes are more potent inhibitors of RR than the ligand alone, suggesting that alternative mechanisms other than iron-depletion are responsible (266). Considering this, it has been suggested that redox-active metal complexes of thiosemicarbazones induce ROS that subsequently inhibit RR activity (266). Furthermore, recent studies have proposed that in addition to the mechanisms just mentioned, the inhibition of RR may occur indirectly as a result of changes in thiol-related, anti-oxidant systems induced by ROS generated by thiosemicarbazone-iron and copper complexes (329).
Early in vivo studies and clinical trials with thiosemicarbazones
It has been shown that isoquinoline-1-carboxaldehyde thiosemicarbazone (IQ-1; Fig. 9A) is a potent inhibitor of RR (203, 256). IQ-1 demonstrates anti-neoplastic activity against lymphosarcoma, leukemia, Lewis lung carcinoma, adenocarcinoma, and Ehrlich ascites carcinoma in animal models (21, 67, 92). However, IQ-1 was insoluble in aqueous media, making drug administration to patients difficult and thereby discouraging further investigation (70).

The first of the pyridyl thiosemicarbazone compounds that reached clinical trials was 5-hydroxy-2-formylpyridine thiosemicarbazone (5-HP; Fig. 9B) (70). In contrast to IQ-1, 5-HP was prepared as a salt, which is soluble in aqueous media, facilitating its administration (70). 5-HP demonstrated anti-tumor activity in a variety of solid tumors in dogs and mice and was able to prevent the incorporation of 3H-thymidine into DNA in human leukemic and sarcoma 180 ascites cells, demonstrating its ability to inhibit DNA synthesis (3, 21, 67, 70). In spite of the positive preclinical activity of 5-HP, disappointing results were obtained in clinical trials due to the rapid metabolism of 5-HP in patients attributable to drug glucuronidation and urinary excretion (70). While some transient anti-leukemic activity was evident in three of the patients in this latter study, treatment with 5-HP did not result in remission or effective anti-neoplastic activity (70). Additionally, gastrointestinal toxicity, myelosupression, and hemolysis were present in certain patients treated with 5-HP, limiting its therapeutic value (70). Owing to the shortcomings of 5-HP, new thiosemicarbazones were synthesized, as discussed next.
Triapine.®
Triapine (Vion Pharmaceuticals; Fig. 6G) is a more potent RR and cancer cell growth inhibitor than the anti-neoplatic drug, hydroxyurea (HU) (266). Triapine increases IRP-RNA-binding activity, demonstrating that the chelator is effective at causing the chelation of intracellular iron pools (85, 143). The oxidative activity of Triapine has also been documented through studies demonstrating (i) the increased oxidation of ascorbate in the presence of iron(III); (ii) the increased hydroxylation of benzoate in the presence of H2O2 and iron(II); (iii) the marked plasmid DNA degradation in the presence of iron(II); and (iv) the ability of Triapine to deplete reduced glutathione in cells (50). These results led to the conclusion that Triapine not only chelates iron, but also generates cytotoxic ROS (50). These findings were further substantiated by a subsequent investigation by Shao et al. (266), who demonstrated that the iron(II)-Triapine complex reduces O2 to produce ROS.
Triapine was found to inhibit the growth of both HU-sensitive and -resistant cancer cell lines, including L1210 leukemia and KB nasopharyngeal carcinoma cells (64, 85, 86). Furthermore, Triapine had pronounced anti-cancer effects without marked toxicity in L1210 leukemia, M109 lung carcinoma, and A2780 ovarian carcinoma xenografts in mice (85). Triapine was also shown to traverse the blood-brain barrier in a mouse model of brain-inoculated L1210 leukemia cells and killed more than 90% of leukemia cells, making the drug a potential candidate for the treatment of brain tumors (85). Of interest, Triapine-associated inhibition of DNA synthesis was reversed quicker in normal cells of the intestinal mucosa and bone marrow in comparison to cancer cells (85). This discovery indicates that Triapine exhibits an important clinical chemotherapeutic property whereby preferential recovery of normal tissue is possible during the treatment-free periods of chemo-regimens (85). Finch et al. (85) also demonstrated that Triapine acted in synergy with other chemotherapeutic agents, such as etoposide, cisplatin, and doxorubicin (DOX), to cause tumor regression in mouse tumor models.
Following the success of Triapine in cellular and animal models, Triapine began evaluation in phase I clinical trials. A phase I trial of 21 patients with advanced or metastatic cancer treated intravenously with Triapine demonstrated modest clinical activity in patients with advanced or metastatic cancer (309). In these studies, patients showed a decrease in serum tumor markers or a prolonged stabilization of disease (309). Additionally, Triapine doses of 120 mg/m2/d were well tolerated in most patients, whereas doses of 160 mg/m2/d resulted in dose-limiting toxicities, such as neutropenia, hyper-bilirubinemia, nausea, or vomiting (309). Additional phase I studies examined the administration of Triapine in combination with gemcitabine, a clinical chemotherapeutic that acts as an inhibitor of DNA synthesis, topoisomerase I, and RR (325). The combination therapy demonstrated anti-tumor activity in patients with a range of metastatic or advanced cancer types (325). In fact, 3 out of 22 patients showed an objective partial response, while 1 patient had evidence of tumor regression (325). In refractory acute leukemia and aggressive myeloproliferative disorders, the administration of Triapine followed by treatment with the nucleoside analog, fludarabine, resulted in a complete response rate of 17%, suggesting that Triapine may be an effective drug for hematological malignancies (147).
Further investigations with Triapine progressed to phase II clinical trials after signs of promising clinical activity in phase I studies. A phase II clinical trial of Triapine in patients with metastatic renal cell carcinoma illustrated some of the deleterious side-effects of the drug when administered at 96 mg/m2 during a 2 h infusion repeated daily for a 4 day period (154). This treatment schedule was repeated fortnightly. Some adverse effects evident in patients included fatigue, nausea, neutropenia, methemoglobinemia, hypoxia, and hypotension (154). Consequently, only approximately half of the patients received at least 90% of the planned Triapine dose (154). Due to the limited efficacy of treatment, the study was terminated before completion (154). More recent studies also mirrored these findings. Several trials were terminated as patients failed to respond to the Triapine regimen (186, 188, 299). In addition, these studies demonstrated that Triapine could cause methemoglobinemia, making the drug toxic, problematic, and impractical (186, 188, 299).
The di-2-pyridylketone thiosemicarbazone class of chelators
More recently, an even more effective series of tridentate chelators, namely the di-2-pyridylketone thiosemicarbazone (DpT) series (Fig. 10), were synthesized (331). These ligands were initially designed as hybrids of the di-pyridyl ketone isonicotinoyl hydrazone (16) and 2-hydroxy-1-napthylaldehyde thiosemicarbazone series (184). In an initial in vitro study, di-2-pyridylketone 4,4,-di-methyl-3-thiosemicarbazone (Dp44mT), di-2-pyridylketone 4-ethyl-3-thiosemicarbazone (Dp4eT), di-2-pyridylketone 4-allyl-3-thiosemicarbazone (Dp4aT), and di-2-pyridylketone 4-phenyl-3-thiosemicarbazone (Dp4pT) all showed very high anti-proliferative activity in SK-N-MC neuroepithelioma cells, MCF-7 breast cancer cells, and SK-Mel-28 melanoma cells compared with DFO and Triapine (331). Dp44mT was deemed the most potent of these DpT analogs with an IC50 of 0.03 μM in SK-N-MC cells (331). Thus, studies progressed to assess the future prospects of Dp44mT as a potential anti-cancer agent. It was demonstrated that Dp44mT had potent anti-proliferative activity in vitro in more than 28 cell lines (210, 236, 293, 313, 331). Furthermore, Dp44mT was shown to be more effective than the clinically used cytotoxic agent DOX (average IC50: 0.62 μM) in 26 of the 28 cell lines and was markedly more effective than Triapine (average IC50: 1.41 μM) across all cell lines (313). With the emergence of multi-drug resistant cancer cells being a significant clinical problem, it is important to note that Dp44mT was equally or more effective in suppressing the proliferation of etoposide-resistant breast cancer cells (MCF-7/VP) and vinblastine-resistant epidermoid carcinoma (KB-V1) when compared with their wild-type chemo-sensitive cells (313). In addition, Dp44mT was able to inhibit the growth of the PC-3 prostate cancer cell line, as well as sensitize the cells to a concurrent course of radiotherapy under normoxic and hypoxic conditions (293). Consequently, Dp44mT may be a valuable agent for patients who develop drug- and radiotherapy-resistant cancers.
Importantly, a greater concentration of Dp44mT was required to achieve growth inhibition in noncancerous normal cells, for instance, in human MRC-5 fibroblasts (331), mammary epithelial cells (236), peripheral blood mononuclear cells (210), and prostate epithelial cells (293), demonstrating that the chelator potentially possesses a substantial therapeutic index which is crucial for clinical applications. Therefore, investigations proceeded to explore whether the administration of Dp44mT could selectively inhibit the growth of tumors in animal models. The first in vivo study revealed that Dp44mT could induce apoptosis and inhibit the growth of mouse M109 lung carcinoma xenografts to 47% of the vehicle-treated control after only 5 days of treatment (0.4 mg/kg) in mice (331). This anti-tumor activity was not accompanied by any marked weight loss or changes in leukocyte cell count compared with control mice (331). However, a slight decrease in hemoglobin, hematocrit, erythrocyte, and platelet count were observed at the maximum tolerated dose (MTD) of 0.4 mg/kg, but not at lower doses (331). Yuan et al. also demonstrated that Triapine showed substantial anti-tumor activity when used at its MTD (6 mg/kg), which was a dose 15 times greater than that of Dp44mT (0.4 mg/kg) (331). However, greater toxicity was evident in Triapine-treated mice than in those treated with Dp44mT, including a substantial decrease in animal weight, hemoglobin, hematocrit, erythrocyte, and leukocyte cell counts (331).
In complementary studies by Whitnall et al., Dp44mT markedly inhibited the growth of human lung carcinoma, neuroepithelioma, and melanoma tumor xenografts in nude mice (313). For instance, after 7 weeks of treatment with Dp44mT at a dose of 0.4 mg/kg per day via an intravenous administration, the SK-Mel-28 melanoma xenografts were 92% smaller than the control tumors treated with vehicle (313). A 30-fold higher dose of Triapine (12 mg/kg per day) was required to induce similar anti-tumor activity in the tumor xenografts tested (313). In contrast to Triapine, Dp44mT at a dose of 0.4 mg/kg per day produced less alterations in biological and hematological indices, as well as no reduction in animal weight in comparison to the control group (313). Interestingly, Dp44mT induced cardiac fibrosis in a dose-dependent manner, which was evident at a high nonoptimal dose of 0.75 mg/kg per day (313). Therefore, Dp44mT possessed a dose-limiting toxicity that was similar to the clinically used anthracycline, DOX, which is thought to induce cardiotoxicity through the generation of ROS (279). Paradoxically, Dp44mT has been tested as a cardio-protective drug that protects against the cardiac toxicity of DOX (119, 237). Both studies demonstrated that Dp44mT was ineffective at attenuating DOX-induced cardiotoxicity (119, 237). Recently and importantly, Dp44mT was shown to reduce the incidence and growth of metastatic breast cancer cells in a metastasis mouse model without evidence of toxicity (174).
Several studies have highlighted the potential cytotoxic mechanisms of Dp44mT (Fig. 11). To demonstrate that it was the chelator's ability to bind metals that caused its anti-cancer activity, the compound, di-2-pyridylketone 2-methyl-3-thiosemicarbazone (Dp2mT), was synthesized (331). Significantly, Dp2mT is unable to bind metals as a result of the presence of a methyl group at the 2-position that prevents electron delocalization which is vital for metal ion binding (331). This chelator demonstrated no anti-proliferative activity (IC50>25 μM), which indicated that metal chelation was important for the anti-neoplastic activity of these ligands (331). Indeed, while Dp44mT acted as an effective chelator that inhibits iron uptake from Tf and increases cellular iron efflux from prelabeled cells, Dp2mT was no more effective than control medium alone (331). Considering this latter result, it was of interest that Dp44mT did not cause iron depletion in the tumor xenografts of mice despite inhibiting tumor growth (313). This indicated that the mechanism of anti-tumor activity of Dp44mT in vitro in cell cultures was different from that found in vivo.

A significant finding in terms of its mechanism of action was that the Dp44mT-iron complex was redox active not only within the “test tube” but also in cells (246, 331). This was confirmed in later studies where the cytotoxic activity of Dp44mT was shown to be dependent on the generation of cytotoxic ROS (183, 293). For example, the incubation of cells with the anti-oxidant, GSH, or its precursor, N-acetylcysteine, protected PC3 prostate cancer cells, SK-N-MC neuroepithelioma, and DMS-53 lung carcinoma cells against Dp44mT-mediated toxicity (183, 293, 329). Conversely, cellular GSH depletion using buthionine sulfoximine enhanced the cytotoxicity of Dp44mT, illustrating the protective role of this anti-oxidant against redox stress generated by the Dp44mT-iron complex (183, 293).
Interestingly, both the iron and copper complexes of Dp44mT have been shown to be redox active and can induce hydroxyl radical formation that is involved, at least in part, in the cytotoxic activity of the chelator (135, 136, 183, 246, 293, 331). Notably, the copper complexes of Dp44mT inhibited the growth of cancer cells to a greater extent than either the ligand alone or the iron complex (136, 183). Therefore, copper binding and the redox activity of the copper-Dp44mT complex may be a key component of the anti-cancer efficacy of Dp44mT. Interestingly, recent investigations have shown that due to its lysosomotropic properties, Dp44mT accumulates within lysosomes, forming a redox-active complex with copper that induces oxidative stress, resulting in lysosomal membrane permeabilization (183). This consequently leads to the release of cathepsins from the lysosomes into the cytosol, resulting in the activation of the mitochondrial apoptotic machinery (183).
Several studies have demonstrated the ability of Dp44mT to induce tumor cell apoptosis (210, 331). For instance, Dp44mT was demonstrated to cause apoptosis in NB4 leukemia cells to a greater extent than DFO (210). Additional studies have been performed that examine the pathways involved in the induction of apoptosis by Dp44mT. In fact, the incubation of Dp44mT with neoplastic cells caused the efflux of holo-cytochrome c (331), an increase in the expression of caspase-3 (210, 331), caspase-8, and −9 protein (331) and a decrease in mitochondrial membrane integrity (210, 331). Furthermore, Dp44mT caused a decrease in the expression of the anti-apoptotic protein Bcl-2, while increasing Bax expression (pro-apoptotic) (331) and inducing G1 arrest in leukemic and breast cancer cells (210, 236). Dp44mT was also shown to inhibit neoplastic cell clonogenic growth, induce DNA double-strand breaks, activate DNA damage and cell-cycle checkpoints, and inhibit DNA topoisomerase IIα in p53 mutant MDA-MB-231 breast cancer cells (236).
More recently, in an attempt to improve its therapeutic window, novel analogs of Dp44mT were synthesized to generate the novel second-generation DpT chelator, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC; Fig. 10B) (158). In three out of four of the pancreatic cell lines examined, Dp44mT and DpC were more effective at inhibiting tumor cell growth in vitro than the current chemotherapy drugs, gemcitabine and 5-fluorouracil (158). It was also shown that Dp44mT and DpC alter the expression of a range of different molecular targets and can up-regulate the expression of NDRG1 and the cdk inhibitor p21CIP1/WAF1, while reducing cyclin D1 levels (158). A particularly notable result was that the chelators induced the phosphorylation of NDRG1 at Ser-330 and Thr-346, which is important for the activity of this metastasis and growth suppressor against the tumor (NDRG1 is discussed in greater detail in the next section titled “Challenges and Opportunities”). In vivo studies also demonstrated that DpC inhibited the growth of pancreatic cancer xenografts of nude mice to a greater extent than Dp44mT or the “gold standard” chemotherapeutic for pancreatic cancer treatment, gemcitabine (158). Importantly, DpC was also shown to be less toxic than Dp44mT, with no evidence of marked alterations in normal tissue histology (158). Importantly, DpC did not induce cardiac fibrosis, making it the most selective and effective chelator of the series (158). Considering the very poor prognosis of pancreatic cancer patients and the marked activity of this agent in vivo, DpC awaits further preclinical evaluation and may prove to be a potent and selective anti-cancer agent.
The 2-benzoylpyridine thiosemicarbazones class of chelators
As a consequence of the cardiac fibrosis induced by Dp44mT treatment in nude mice, the 2-benzoylpyridine thiosemicarbazone (BpT; Fig. 12) series of ligands was synthesized in order to minimize adverse side-effects (145). The DpT analogs were modified to generate the BpT series through the addition of an aromatic phenyl ring in the place of the 2-pyridyl group (145). This alteration was performed to increase chelator lipophilicity and reduce the electron-withdrawing effects of the pyridyl nitrogen. Theoretically, these alterations could have marked effects on not only ligand permeability, but also the affinity of metal binding, and, thus, may influence chelator efficacy.

In vitro cellular proliferation studies using SK-N-MC neuroepithelioma cells demonstrated that the BpT analogs (IC50: 0.002–0.005 μM) had greater activity than DFO (IC50: 4.51 μM), Triapine (IC50: 0.26 μM), and most of the DpT series (IC50: 0.002–0.027 μM) with the exception of Dp44mT (IC50: 0.001 μM) (145). The treatment of SK-N-MC cells with the BpT series identified 2-benzoylpyridine 4-ethyl-3-thiosemicarbazone (Bp4eT) (IC50: 0.002 μM) as having the highest anti-proliferative efficacy within this series (145). These ligands were also shown to exert greater anti-proliferative activity in cancer cells relative to normal cells (145). In fact, the IC50 values in normal MRC-5 fibroblasts were ∼1500-times higher than in SK-N-MC neuroepithelioma cells (145). Similar to Dp44mT, the chelators, 2-benzoylpyridine 4,4-dimethyl-3-thiosemicarbazone (Bp44mT), Bp4eT, and 2-benzoylpyridine 4-allyl-3-thiosemicarbazone (Bp4aT), possessed higher anti-proliferative activity in vinblastine-resistant KB-V1 epidermoid cells compared with nonresistant KB3-1 cells, suggesting that these ligands may be useful in overcoming drug resistance (330).
Furthermore, it was shown that BpT analogs were able to increase iron efflux from prelabeled cells and decrease iron uptake from Tf by SK-N-MC cells, although in the majority of cases, these series were less effective than their DpT counterparts (145). Additionally, the redox activity of the BpT series was examined using the ascorbate oxidation and benzoate hydroxylation assays (145). These studies demonstrated that the iron complexes of the BpT analogs, excluding 2-benzoylpyridine 4-phenyl-3-thiosemicarbazone (Bp4pT), had greater redox activity in comparison to those of the DpT series (145). It was suggested that this may explain, in part, the potent anti-proliferative activity evident in the BpT series in vitro in contrast to the DpT chelators (145). In terms of the mechanism of action, Bp4eT, similar to Dp44mT, inhibited RR activity, and it was suggested that this could occur by iron deprivation and via modulation of thiol-related anti-oxidant systems (329).
In vivo investigations have also been performed on chelators that were deemed the most effective of the BpT series (330). Due to poor solubility, Bp4eT and Bp4aT could not be intravenously administered at sufficient doses to induce anti-tumor activity in DMS-53 lung carcinoma xenografts in nude mice (330). However, Bp44mT inhibited DMS-53 tumor growth via both the oral and intravenous route (330). A toxicological examination of the Bp44mT-treated mice demonstrated that the chelator caused a slight reduction in red blood cell count, a small decrease in hemoglobin, mild anemia, a decrease in hepatic and splenic iron levels, a decrease in albumin levels, and an increase in serum liver enzymes (330). In spite of this, Bp44mT-administration in mice neither caused significant weight loss nor induced cardiac fibrosis, in marked contrast to Dp44mT (330). Additionally, several of the toxicological changes were reversed on the removal of Bp44mT-treatment, and, hence, the agent was well tolerated (330). Interestingly, iron levels were not significantly decreased in tumor xenografts, confirming previous findings documented with Dp44mT (313, 330). On assessment of the tumor xenografts, Bp44mT was shown to result in a significant decrease in TfR1 expression and an increase in ferritin H chain expression, demonstrating that tumor iron pools had been augmented after treatment with the ligand (330). Conversely, total copper and zinc levels in organs and tumor xenografts remained unaltered (330). Additionally, Bp44mT-treatment induced a decrease in cyclin D1 and p21CIP1/WAF1 expression in tumor xenografts relative to control tumors, demonstrating that Bp44mT perturbed the cell cycle (330). This latter investigation was the first study which showed that Bp44mT can be given orally with potent anti-tumour efficacy. Clearly, the oral administration of a novel and effective chemotherapeutic agent provides the benefits of convenience for chronic dosing regimens and is an important property for this series of compounds.
Anti- Versus Pro-Oxidative Actions of Chelates and/or Their Respective Metal Complexes Versus Iron Deprivation
Understanding the mechanism of the action of chelators in cells and their cytotoxic activity is challenging. In some cases, once complexed, the iron center may work as an anti-oxidant (O-Trensox) (42, 233), while in other cases, the metal complex acts as a pro-oxidant, for example, for Triapine or Dp44mT (246, 266, 331). For some ligands, for example, DFO, the metal is bound and causes iron deprivation which is cytotoxic, while the iron complex itself is not toxic (143).
The differences between the production of ROS by the metal complex and the production of hydroxyl radicals via Fenton chemistry catalyzed by “free” iron(II) are critical to address. It is important to understand both the biology and chemistry involved. Under physiological conditions, almost all iron within the organism is bound and sequestered rather than being “free” and unbound, so as to protect against oxidative stress (244). For example, this occurs when iron is bound to Tf or ferritin. Under these circumstances, the iron atom is within a coordination environment in a protein that prevents the generation of ROS. In contrast, the binding of iron or copper by synthetic thiosemicarbazone chelators from cellular molecules leads to a coordination sphere where the ability of the metal to undergo redox reactions to generate ROS is facilitated. This difference is crucial to recognize, as it is dictated by the chemistry of the ligating atoms and can be synthetically manipulated by the medicinal chemist to generate chelators that form iron complexes which are either redox active (e.g., Triapine or Dp44mT) or redox inactive (e.g., DFO). This aspect of ligand design has been previously reviewed in detail [see (144)].
Challenges and Opportunities
Challenges faced in the development of chelators, as well as many other drugs, are off-target side-effects and pharmacokinetic problems. For instance, some detrimental adverse effects that are already evident in the clinical trials of 5-HP include myelosuppression, hemolysis, and gastrointestinal toxicity (70). Additionally, the side-effects of Triapine administration include fatigue, nausea, neutropenia, hypoxia, and methemoglobinemia (154, 309). These are particularly undesirable for cancer patients who are already in a compromised condition. Nonetheless, Triapine is still undergoing a phase II clinical trial (ClinicalTrials.gov Identifier: NCT00941070) despite the dose-limiting toxicities and the lack of anti-tumor activity evident in certain studies, which has prevented this agent from entering phase III trials.
Interestingly, a phase I trial that assessed Triapine and cisplatin in combination with pelvic radiation therapy for the treatment of 10 cervical cancer patients demonstrated encouraging results with all of them attaining complete clinical response and no disease progression at an average follow-up period of 18 months (162). However, another patient from the same investigation who suffered recurrent uterine sarcoma was not successfully treated with Triapine (162). Notably, in the same study, in the patients administered intravenous Triapine (25 mg/m2 thrice weekly), no significant symptomatic methemoglobinemia was reported, with the agent being well tolerated (162). Based on the success of this latter work, a follow-up phase II clinical trial (ClinicalTrials.gov Identifier: NCT00941070) is currently underway for patients with cervical and vaginal cancer. It is anticipated that this study will be completed shortly and will determine whether Triapine will play a specific role in cervical or vaginal cancer.
In general, chelators have also been shown to possess their fair share of pharmacological problems. For example, DFO and 5-HP are rapidly metabolized and possess a short plasma half-life, making it crucial to administer DFO and 5-HP via an inconvenient long subcutaneous infusion and at higher doses, respectively (39, 70). As a result of these issues, new more potent chelators are being synthesized in an attempt to reduce further nonspecific side-effects and pharmacological problems.
Another challenge is to understand more comprehensively the difference in iron metabolism between normal and neoplastic cells. In the last 20 years, there has been an “explosion” of information regarding the identification of new molecules involved in the metabolism and trafficking of iron within cells. However, little progress has been made while assessing differences in their molecular expression profiles between normal and neoplastic cells. Intriguingly, it has been recently suggested that alterations in the regulation of iron metabolism characterize the malignant state, with an iron regulator gene signature predicting outcome in breast cancer (199). These studies demonstrated that high levels of TFRC, encoding TfR1, and low levels of HFE (hemochromatosis gene), a negative regulator of TfR1 and hepcidin, producing a “high intracellular iron” phenotype, were associated with a poorer prognosis in breast cancer (199). These results are in agreement with the authors' previous findings, which examined an interplay between low levels of Fpn1 and high levels of hepcidin, providing a “high intracellular iron” phenotype, which conferred a poorer prognosis in breast cancer (225) (see section titled Ferroportin 1, hepcidin and cancer).
A more detailed understanding of the differences in the metabolism of iron between the normal and neoplastic state could lead to more specific iron-targeting strategies. Certain potent chelators, in particular those of the thiosemicarbazone class, have been shown to increase the expression of the iron-regulated metastasis suppressor, NDRG1 (158, 313). This is a critical finding in terms of understanding the anti-tumor activity of iron chelators. NDRG1 has been found to be down-regulated in various cancer types, including prostate, pancreatic, breast, and colon cancer (6, 13, 14, 304). Its down-regulation correlates to poorly differentiated advanced cancers [for review, see (159)]. Additionally, NDRG1 up-regulation has been shown to inhibit the growth and metastasis of colon, prostate, breast, and pancreatic cancer (159). Recently, it was demonstrated that NDRG1 interacts with the Wnt receptor, low-density lipoprotein receptor-related protein 6 (LRP6), and, subsequently, inhibits the Wnt signaling pathway, increasing the degradation of β-catenin, thereby suppressing metastasis (174).
Considering that metastasis is a critical problem in cancer treatment that leads to poor patient prognosis and death, up-regulating NDRG1 expression through chelation therapy may provide a novel avenue of treatment. One such chelator, Dp44mT, was able to up-regulate NDRG1 and significantly suppress metastasis in vitro and in a human bone metastatic breast cancer mouse model without apparent toxicity (174). In addition to Dp44mT, the chelator DpC alters NDRG1 expression and is of particular interest due to its potent anti-cancer activity and minimal adverse effects (158). DpC is able to up-regulate the expression of NDRG1 and also its phosphorylation state that is crucial for anti-metastatic activity (158). Preclinical in vitro and in vivo investigations have demonstrated that DpC has potent in vitro and in vivo anti-cancer activity against pancreatic cancer (158). Therefore, the up-regulation of NDRG1 using DpC may be a novel mechanism of targeting a highly lethal and belligerent disease such as pancreatic cancer. This presents a novel opportunity to increase the positive outcomes from chemotherapy and drive a major change in cancer outcomes by targeting a novel molecule.
Conclusions
Several studies have convincingly shown the de-regulation of the expression of iron-related proteins in malignancies. Furthermore, it is clear that iron and other biological metals are important factors in cancer growth and progression that can be exploited in the development of novel anti-cancer drugs, such as chelators. Indeed, recent studies have demonstrated that a number of ligands, in particular those of the thiosemicarbazone class, show marked anti-tumor activity. The ability of some of these agents to be given by the oral route provides a convenient route for drug dosing, demonstrating the potential of chelators to be implemented in the clinics.
Footnotes
Acknowledgments
A.M.M. is grateful to the Sydney Medical School of The University of Sydney for a Bob and Nancy Edwards Postgraduate Ph.D. Scholarship. D.R.R. was supported by a NHMRC Senior Principal Research Fellowship and Project grants and an ARC Discovery grant. D.S.K. was supported by a Cancer Institute New South Wales Early Career Development Fellowship and Innovation Grant.